

Abstract
Obesity, a major risk factor for the development of diabetes mellitus, cardiovascular diseases and certain types of cancer, arises from a chronic positive energy balance that is often due to unlimited access to food and an increasingly sedentary lifestyle on the background of a genetic and epigenetic vulnerability. Our understanding of the humoral and neuronal systems that mediate the control of energy homeostasis has improved dramatically in the past few decades. However, our ability to develop effective strategies to slow the current epidemic of obesity has been hampered, largely owing to the limited knowledge of the mechanisms underlying resistance to the action of metabolic hormones such as leptin and ghrelin. The development of resistance to leptin and ghrelin, hormones that are crucial for the neuroendocrine control of energy homeostasis, is a hallmark of obesity. Intensive research over the past several years has yielded tremendous progress in our understanding of the cellular pathways that disrupt the action of leptin and ghrelin. In this Review, we discuss the molecular mechanisms underpinning resistance to leptin and ghrelin and how they can be exploited as targets for pharmacological management of obesity.
Obesity is associated with increased morbidity and mortality owing to the many medical conditions that are caused by excess fat accumulation1. Many factors, such as genetic predisposition, ready availability of calorie-dense food and a sedentary lifestyle, have contributed to the rise in obesity prevalence. The regulatory systems that control body weight homeostasis also often promote positive energy balance, which might also contribute to weight gain and fat accumulation in individuals with obesity. Indeed, whereas a robust biological response is triggered to restore homeo stasis when body fat stores are endangered (such as in times of starvation), excess of adiposity is associated with absence of significant response. The regulatory pathways controlling energy balance become dysfunctional and are unable to defend the body against excess energy stores in obesity.
A prototypic defect that occurs in obesity is the development of resistance to the action of key metabolic hormones such as leptin and ghrelin. Leptin and ghrelin have emerged as important players in the neuroendocrine control of energy homeostasis (FIG. 1). These hormones communicate information to the central nervous system (CNS) about the current levels of energy reserves and nutritional status. In this Review, we discuss the molecular signalling pathways and the dietary and nutritional interventions that interfere with the actions of leptin and ghrelin. We also discuss the cross-talk between the ‘classic’ leptin-resistance phenomenon and the novel concept of ghrelin resistance.
Figure 1 |. Physiological functions of leptin and ghrelin.
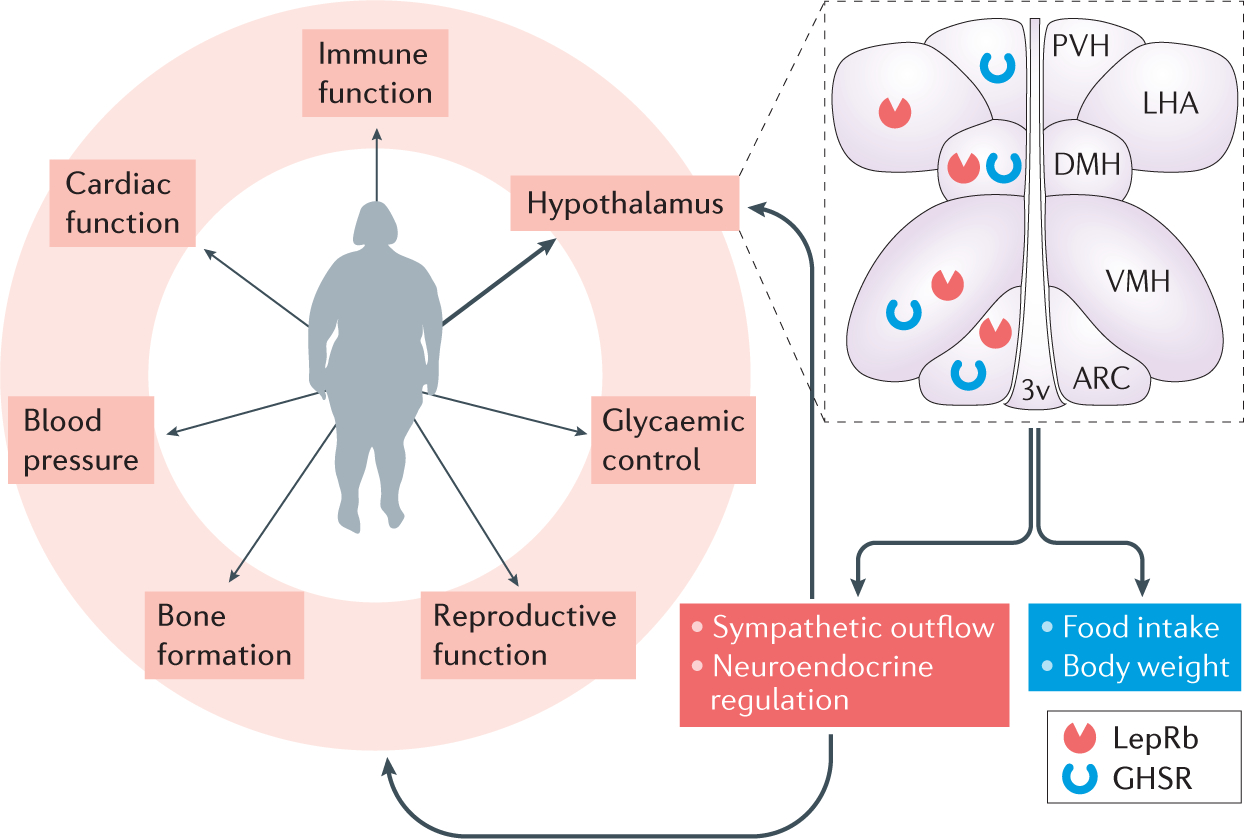
Leptin and ghrelin are secreted from adipose tissue and from the stomach, respectively, enter the circulation and affect a wide range of physiological processes. In addition to direct peripheral targets, these hormones exert their actions in different regions of the brain, including several nuclei of the hypothalamus that are important for energy homeostasis, where both the ‘long’ form of the leptin receptor (LepRb) and the ghrelin receptor growth hormone secretagogue receptor (GHSR) are broadly expressed. The actions of the two hormones on their receptors regulate food intake and body weight, sympathetic nervous system tone and neuroendocrine responses, which in turn regulate physiological function of peripheral organs to coordinate homeostasis. 3v, third ventricle; ARC, arcuate nucleus of the hypothalamus; DMH, dorsomedial nucleus of the hypothalamus; LHA, lateral hypothalamic area; PVH, paraventricular nucleus of the hypothalamus; VMH, ventromedial nucleus of the hypothalamus.
Leptin
Leptin, discovered in 1994 by positional cloning2, is a 16 kDa cytokine that is produced predominantly by adipose tissue: it is released into the bloodstream and circulates in proportion to body fat mass3. Consequently, levels of leptin convey information about the energy reserves of the body to the centres that regulate energy homeostasis4–6. Increased adipose depots that are associated with positive energy balance increase leptin production and its circulating levels, which typically trigger a response to reduce feeding and promote energy expenditure7–14. Conversely, a fall in circulating levels of leptin, associated with negative energy balance in the body (such as during calorie restriction and/or excessive exercise) triggers a strong motivation to eat and conserve energy15,16.
Leptin has also been implicated in the regulation of the immune system, autonomic and cardiovascular regulation, reproductive function and bone formation (FIG. 1). For example, in humans and rodent models, leptin deficiency reduces the immune system response, decreases sympathetic nervous system activity and blood pressure, delays puberty and can lead to infertility, in addition to reducing bone density17–22. Importantly, the role of leptin in these processes is largely independent of its role in the regulation of body weight, which implies a broader role of this hormone beyond energy homeostasis.
Leptin receptor signalling
The leptin receptor (LepR) was cloned from the mouse choroid plexus by affinity-based screening assay and was found to be a single transmembrane spanning receptor of the class I cytokine receptor family23. In mice, six LepR isoforms have been identified and have been classified as secreted (LepRe), short (LepRa, LepRc, LepRd and LepRf) and long (LepRb) forms24,25. These isoforms arise from the single Lepr gene by alternative mRNA splicing or post-translational cleavage24. LepRb is primarily responsible for leptin signalling and seems to mediate most physiological actions of leptin. For example, the phenotypes of mice that lack the LepRb isoform, such as db/db mice, are very similar to the phenotypes of mice lacking leptin, such as the ob/ob mice or those lacking all isoforms of LepR, such as the db3J/db3J mice26.
LepRb is expressed at high levels in areas of the brain that are involved in the regulation of feeding and energy expenditure27. In the hypothalamus, LepRb is expressed in a subset of neurons in nuclei that are important for metabolic regulation, such as the arcuate nucleus (ARC), ventromedial nucleus of the hypothalamus (VMH) and dorsomedial nucleus of the hypothalamus, and in the lateral hypothalamic area27 (FIG. 1). Similar to other cytokine receptors, LepRb signals via a non covalently associated kinase, Janus kinase 2 (JAK2)28–30. Upon binding of leptin to its extracellular domain, homodimerized LepRb undergoes a conformational change that enables the activation of JAK2, which then phosphorylates other tyrosine residues within the LepRb–JAK2 complex to activate downstream signalling cascades31,32 (FIG. 2). Three tyrosine phosphorylation sites, Tyr985, Tyr1077 and Tyr1138, have been identified within the carboxy-terminal tail of LepRb33. Signal transducer and activator of transcription 3 (STAT3) is recruited to Tyr1138 and phosphorylated by JAK2; this enables STAT3 translocation to the nucleus and transcriptional initiation of target genes, including suppressor of cytokine signalling 3 (SOCS3), which is an inhibitor of LepRb–JAK2 signalling. Likewise, STAT5 is recruited to Tyr1077 and is also phosphorylated and activated by JAK2 (REFS 34,35). Finally, protein tyrosine phosphatase non-receptor type 11 (PTPN11; also known as SHP2) is recruited to Tyr985 and activates extracellular signal-regulated kinase (ERK), which mediates some of the downstream effects of LepRb 36. Notably, SOCS3 is also recruited to Tyr985 and provides feedback inhibition to suppress LepRb signalling36 (FIG. 2). Stimulation of LepRb also activates the insulin receptor substrate (IRS)– phosphoinositide 3-kinase (PI3K) pathway; however, the exact mechanism involved is unclear18,23. Mechanistic target of rapamycin complex 1 (mTORC1) and its downstream effectors, such as ribosomal S6 kinase (S6K) and 40S ribosomal protein S6, are important targets of the LepRb–PI3K axis and mediate the physiological actions of leptin37,38.
Figure 2 |. LepRb signalling and the molecular mechanisms contributing to leptin resistance in obesity.
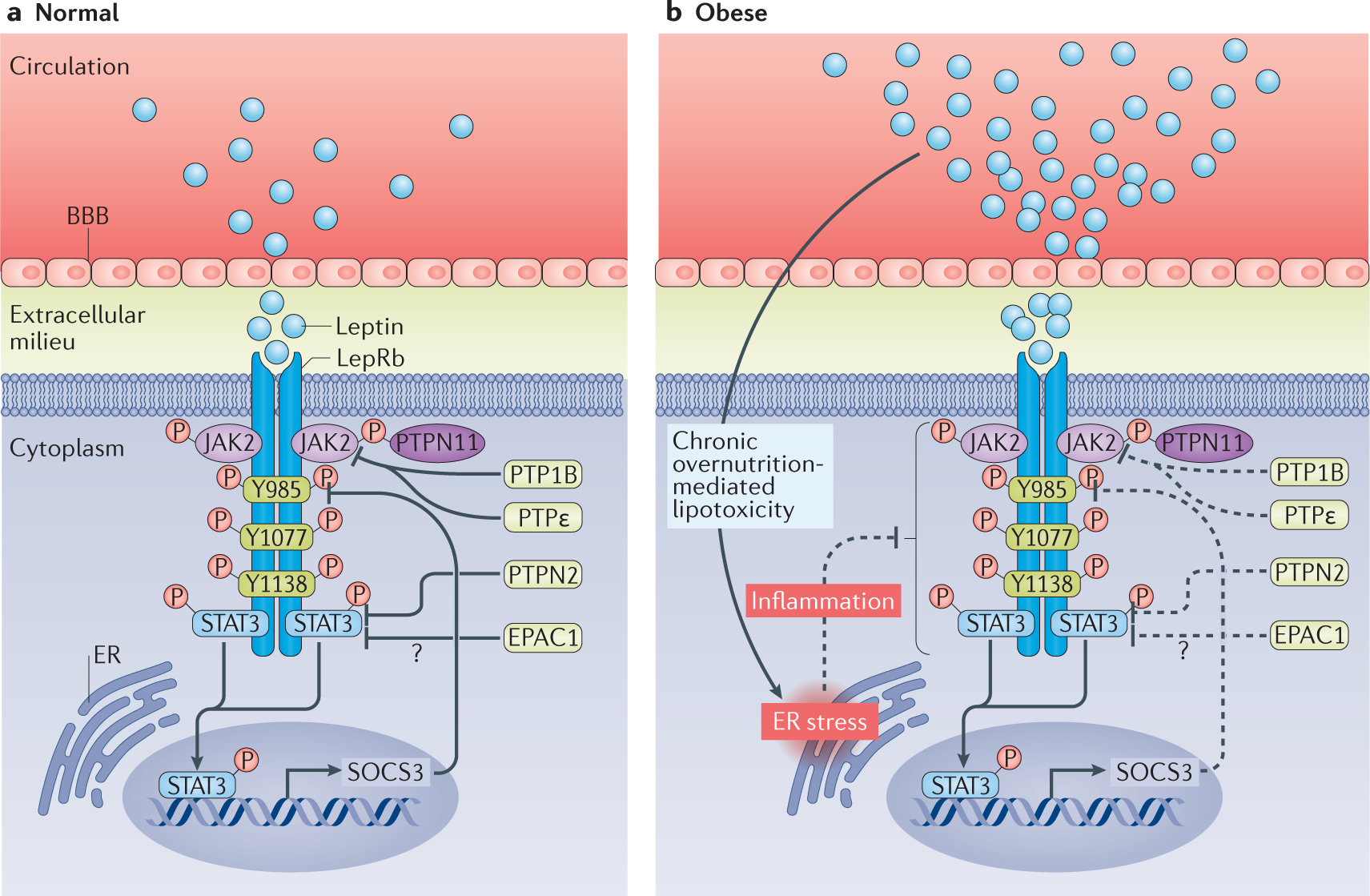
a | In individuals with normal body weight, circulating leptin crosses the blood–brain barrier (BBB) and binds to the ‘long’ form of the leptin receptor (LepRb), which induces phosphorylation of Janus kinase 2 (JAK2) and of multiple tyrosine residues in the LepRb intracellular domain. LepRb also receives inhibitory signals from multiple negative feedback loops (such as suppressor of cytokine signalling 3 (SOCS3), protein tyrosine phosphatase 1B (PTP1B), PTP non-receptor type 2 (PTPN2), PTPe and exchange protein directly activated by cyclic AMP 1 (EPAC1)), ensuring that activation of LepRb does not go beyond a physiologically necessary point. b | In obesity, circulating levels of leptin increase, which is associated with diminished leptin transport across the BBB and activation of the inhibitory negative feedback systems that eventually lead to diminished LepRb signalling. Increased free fatty acids and chronic overnutrition cause lipotoxicity and endoplasmic reticulum (ER) stress, and trigger inflammatory responses that might contribute to a blunted physiological response to leptin in obesity. STAT3, signal transducer and activator of transcription 3.
Leptin resistance in obesity
Differential resistance to endogenous versus exoge- nous leptin.
Obesity is associated with leptin resistance, a phenomenon that is similar to insulin resistance in patients with type 2 diabetes mellitus4. Leptin resistance is associated with elevated circulating levels of leptin, as well as with the inability of exogenous leptin to decrease food intake and body weight. However, some data using LepR antagonists suggest that resistance to endogenous leptin does not contribute to high-fat diet (HFD)-induced obesity. In these data, intraperitoneal or intracerebroventricular administration of a LepR antagonist has the same effect on food intake and body weight gain in hyperleptinaemic mice with diet-induced obesity (DIO) and in lean mouse controls39. These findings indicate that the action of endogenous leptin is intact in mice with DIO despite hyperleptinaemia and resistance to exogenous leptin, thus challenging the view that leptin no longer suppresses food intake and reduces body weight in obesity. Interestingly, in clinical studies, even individuals with obesity (average BMI: 33 kg/m2) feel strong hunger in response to moderate (10%) weight loss, and administration of leptin (0.08–0.14 mg per kg of fat mass; which restored plasma concentrations of leptin to those before weight loss) can mitigate this hunger40. Although these observations suggest that LepR signalling might be a therapeutic target for treating obesity, how energy imbalance and obesity develop when endogenous leptin is functional is still puzzling.
Selective leptin resistance.
In obesity, leptin resistance seems to be selective, in a manner similar to selective insulin resistance41. This concept was derived from observations in mouse models that exogenous leptin could cause renal sympathetic nerve activation and increase blood pressure but was unable to exert an anorectic or weight-reducing action42,43. These findings also highlight that the cardiovascular sympathetic effects of leptin can be dissociated from its effects on energy homeostasis. Similar to ob/ob mice, patients with obesity who lack leptin have low-to-normal sympathetic nerve activity and blood pressure, which highlights the importance of leptin in driving sympathetic overactivity and hypertension in the context of obesity44. Currently, the proposed mechanisms of selective leptin resistance include specific brain region and/or cell type actions, as well as differentially regulated intracellular signalling cascades downstream of LepR, or indeed any combination of these factors45, but these mechanisms need to be further investigated.
Potential mechanisms of leptin resistance
Leptin and blood-brain barrier.
Leptin circulates as either a free protein or in an inactive form that is bound to the circulating form of the receptor (LepRe)46. Interestingly, in contrast to lean individuals in whom up to 65% of circulating leptin is bound to LepRe, individuals with obesity predominantly have the active free form of circulating leptin (~85% of total leptin)47,48 Consequently, individuals with obesity might have chronically elevated levels of active free leptin in the brain, which desensitize LepRb and increase leptin resistance. Indeed, levels of leptin in the cerebrospinal fluid (CSF) strongly correlate with plasma levels of leptin and increased BMI49. Whereas absolute amount of leptin in the CSF of individuals with obesity might be higher than in the CSF of lean individuals, the efficiency of movement of leptin from plasma to the CSF (measured as the CSF:plasma leptin ratio) diminished by as much as 80% in individuals with obesity49. This finding is further supported by the fact that intravenous administration of leptin caused a dose-dependent increase in CSF levels of leptin in lean mice, but not in mice with DIO50. Assessment of leptin transport rate across the blood– brain barrier (BBB) using multiple-time regression analysis and in situ brain perfusion showed that obese mice have reduced leptin passage across the BBB51,52. These findings might explain why systemic administration of exogenous leptin to suppress food intake and reduce body weight is blunted in individuals with obesity53,54.
Astrocytes have emerged as potential mediators of leptin resistance in obesity. Astrocytes, which express different isoforms of LepR, participate in the transport of leptin across the BBB52. A HFD causes rapid activation of astrocytes, thus inducing inflammation, and hyperleptinaemia induced by a long-term HFD further activates astrocytes and inflammation, reducing delivery of leptin to the brain55,56. Moreover, binding of leptin to its receptors in astrocytes interferes with the action of leptin in neurons, and astrocyte-specific deletion of Lepr ameliorates leptin resistance in mice with DIO57. Tanycytes have also been involved in obesity-associated leptin resistance and, in the median eminence, form a diet-responsive neurogenic niche for newborn neurons that express LepRb and can integrate the functional circuit later in their life58. Tanycytes in the hypothalamus seem to be a conduit for leptin into the brain59. Blood-borne leptin is first taken up by tanycytes in the median eminence and then released into the mediobasal hypothalamus, a process that is disrupted in mice with DIO59. The passage of leptin through tanycytes is colchicine sensitive, indicating the involvement of intracellular vesicular trafficking and suggesting that the release of leptin by tanycytes requires LepRb–ERK signalling59. Importantly, treatment with epidermal growth factor, which activates ERK signalling, can rescue impaired tanycytes-mediated transport of leptin in mice with DIO and increase energy expenditure and locomotor activity, facilitating weight loss59. These findings indicate that leptin released from tanycytes has functional consequences on energy balance. Surprisingly, ablation of tanycytes-derived newborn neurons by CT-guided irradiation protects mice from weight gain during HFD feeding60. With our current limited knowledge, targeting median eminence tanycytes to treat obesity might not be a realistic therapeutic approach, but developing a long-lasting leptin analogue that can cross the BBB might help to circumvent obesity-associated reduction in leptin transport in the brain61.
Defective LepRb trafficking.
Impaired trafficking of LepRb to the membrane in neuronal subpopulations of the hypothalamic nuclei that control energy homeostasis has emerged as a novel mechanism of leptin resistance. Bardet–Biedl syndrome (BBS) is a highly pleiotropic autosomal recessive human disorder in which obesity is a common manifestation and that is associated with leptin resistance and energy imbalance62. Mice with global loss or mutation of Bbs genes, which are used as a model of BBS, are obese and have leptin resistance62. In addition, targeted deletion of the Bbs1 gene in the nervous system leads to obesity in mice57. Interestingly, this phenotype can be reproduced by selective deletion of Bbs1 in the mediobasal hypothalamus or in LepRb-expressing cells in the brain, but not in adipocytes, thus excluding a contribution of adipocyte Bbs genes to obesity63. Mice that are deficient in BBS proteins show impaired trafficking of LepRb to the plasma membrane, which leads to leptin resistance, but this resistance is independent from obesity63,64. BBS1 directly interacts with LepRb, which might explain how BBS proteins mediate the trafficking of LepRb63,64. Moreover, a 36–41% reduction in the number of leptin binding sites, as determined by autoradiography, occurs in the hypothalamic nuclei of obesity-prone rats65, which suggests that defects in LepRb trafficking are also present in common forms of obesity.
Suppression of LepRb signalling in obesity
Many molecules and signalling pathways contribute to obesity-associated leptin resistance. Our enhanced understanding of LepRb signalling has highlighted certain molecules as candidates that might restore sensitivity to leptin in obesity.
SOCS3.
SOCS3, a member of a large family of cytokine-inducible inhibitors of signalling, was discovered by an expression cloning approach in mouse monocytic leukaemic M1 cells that respond to cytokines such as IL-6 (REF. 66). SOCS3 can be rapidly induced by IL-6 and in turn suppresses cytokine signal transduction in a negative feedback loop66,67. Expression of Socs3, but not of Socs1 or Socs2, is rapidly induced in the hypothalamus of ob/ob mice in response to peripheral administration of leptin, and overexpression of Socs3 blocks leptin-induced signal transduction in cultured mammalian cell lines68. These observations suggest that SOCS3 mediates cellular leptin resistance in obesity68. Indeed, mice heterozygous for Socs3 (REF. 69) or lacking SOCS3 specifically in the brain70 have increased sensitivity to exogenous leptin-mediated suppression of food intake and reduction of body weight, and are resistant to DIO. These findings implicate SOCS3 as a negative regulator of LepR signalling in vivo. However, two other studies in mice have shown that deletion or overexpression of Socs3 in LepRb-expressing cells does not affect HFD-induced weight gain, which questions the causative role of SOCS3 in the development of cellular leptin resistance71,72.
PTPs.
LepRb signalling transduction relies on initial activation of JAK2 and subsequent phosphorylation of tyrosine residues within LepRb and STAT3 (FIG. 2). The overall protein phosphorylation in a cell depends on the dynamic balance of phosphorylation by kinases and dephosphorylation by PTPs. Consequently, any phosphatase that targets JAK2 and STAT3 could suppress LepRb signalling. Several PTPs have been implicated in LepRb signalling, including PTPN11, which binds to Tyr985 to activate the RAS–mitogen-activated protein kinase (MAPK) pathway; PTP1B (also known as PTPN1), which dephosphorylates JAK2; PTPN2 (also known as TCPTP) and PTPε, which dephosphorylate STAT3; and phosphatase and tensin homologue (PTEN), which dephosphorylates phosphatidylinositol-3,4,5-trisphosphate (PIP3) to regulate PI3K signalling pathway.
When PTP1B was first described, it was found to negatively regulate insulin signalling by dephosphorylating IRS73–75 but was also found to suppress LepRb signalling in vivo by directly dephosphorylating JAK2 at Tyr1007 and Tyr1008 (REF. 76). Ptp1b-knockout mice, which are lean and hypersensitive to leptin, display increased energy expenditure and resistance to DIO77–80 These effects can be recapitulated by ablation of PTP1B in neurons81, presumably those expressing LepRb82,83 but not in muscle or liver81. Targeting PTP1B activity is a potential therapeutic intervention in obesity84. For example, a small molecule inhibitor of PTP1B, trodusquemine, acts in the CNS to promote weight loss in both mice with DIO and genetic mouse models of obesity85–88. Ptp1b-directed antisense oligonucleotides yielded promising results in a phase II clinical trial89.
Levels of PTPN2 are increased by as much as twofold in the hypothalamus of mice with DIO, and mice that lack neuronal PTPN2 have increased sensitivity to leptin (indicated by the exaggerated leptin-induced decrease in food intake and body weight and by activation of hypothalamic STAT3) and, on a HFD, remain leptin sensitive and gain less weight than control mice90. The mechanisms of action of PTPN2 seem to implicate a direct dephosphorylation of nuclear STAT3 that causes its export from the nucleus90. Importantly, intracerebroventricular administration of a specific PTPN2 inhibitor in wild-type mice increased leptin-induced STAT3 phosphorylation, resulting in decreased body weight and increased energy expenditure, which suggests that targeting this molecule for pharmacological treatment of obesity might be feasible90. However, PTPN2 seems to be redundant in LepRb signalling, as Ptpn2 deletion in a subset of leptin-sensitive neurons expressing pro-opiomelanocortin (POMC) did not promote leptin-induced STAT3 phosphorylation or suppression of food intake and body weight loss91. However, expression of PTP1B and PTPN2 in a subset of POMC neurons in the ARC (with some POMC neurons co-expressing both PTP1B and PTPN2) might have differential but synergistic effects on systemic energy metabolism91. Single deletion of Ptp1b or Ptpn2 from POMC neurons revealed that PTP1B, but not PTPN2, mediates leptin sensitivity91, whereas PTPN2, but not PTP1B, seems to be involved in insulin signalling. Notably, ablation of both PTP1B and PTPN2 in POMC neurons leads to a synergistic enhancement of the actions of leptin and insulin that promotes energy dissipation through browning of white adipose tissue91.
PTPε, which negatively regulates LepRb signalling, exists in two forms: a soluble form and a transmembrane receptor-type form (RPTPε)92. PTPε has been implicated in ERK signalling by inhibiting ERK1 and ERK2 kinase activity and by interfering with their downstream events93. PTPε also suppresses JAK2–STAT3 signalling through its association with and dephosphorylation of JAK2 (REF. 92). PTPε and RPTPε are co-expressed with LepRb in hypothalamic nuclei such as the ARC, and treatment with leptin induces phosphorylation of RPTPε in the hypothalamus, an effect that is pronounced in mice with DIO93. Moreover, female, but not male, mice that lack both RPTPε and PTPε have increased sensitivity to leptin and are protected from fat accumulation induced by HFD, ageing and ovariectomy93. However, as male mice are more susceptible to DIO than females, the observation of a female-specific effect of PTPε on leptin sensitivity and DIO has diminished the enthusiasm for its development for therapeutic intervention. Nonetheless, these findings might enhance our understanding of sexual dimorphism of body energy homeostasis.
Exchange proteins directly activated by cAMP pathway.
cAMP mediates the actions of hormones by activating protein kinase A (PKA) and exchange proteins directly activated by cAMP (EPACs)94. The role of PKA in regulating energy balance has been well documented95, but only recently has EPAC, which acts as a cAMP-regulated guanine nucleotide exchange factor for the small G protein RAP1, been found to function as a negative regulator of LepRb signalling96,97. In ex vivo hypothalamic organotypic cultures, elevated levels of cAMP were found to impair the signalling cascades that are activated by leptin, including STAT3 and S6K pathways, independently of the activation of PKA97. By contrast, activation of EPACs is sufficient to impair LepRb signalling with concomitant induction of SOCS3 and also blunts leptin-induced depolarization of hypothalamic POMC neurons. Moreover, intracerebroventricular administration of an EPAC activator blunted the anorexigenic effect of leptin. Taken together, these findings suggest an important role of EPAC in the negative regulation of LepRb signalling97.
This conclusion is further supported by the observation that mice lacking Epac1, which encodes one of the two isoforms of EPAC in mammals, are more resistant to DIO and have improved glucose tolerance and heightened central leptin sensitivity, as measured through STAT3 activation, compared with control mice98. Furthermore, inhibition of EPAC1 using a selective chemical inhibitor (ESI-09) can reduce plasma levels of leptin in vivo and resulted in increased leptin sensitivity in organotypic hypothalamic slices98.
Inflammation and endoplasmic reticulum stress.
Endoplasmic reticulum stress has emerged as a key factor in obesity-associated inflammation and leptin resistance. HFD consumption increases the expression of pro-inflammatory cytokines in the hypothalamus, such as IL-1, IL-6 and tumour necrosis factor (TNF), which activate inflammatory pathways99. Surprisingly, only 1 day of HFD feeding, or a single central administration of fatty acid, is sufficient to induce hypothalamic inflammation in rodents100–102. A HFD activates nuclear factor-κB (NF-κB) and its upstream regulator inhibitor of NF-κB kinase-β (IKKβ) by increasing endoplasmic reticulum stress in the hypothalamus, which might cause leptin resistance102 (FIG. 2). A lack of TNF receptor or brain infusion of a TNF antibody improves leptin resistance in HFD-fed mice103,104. Furthermore, amelioration of hypothalamic endoplasmic reticulum stress using chemical chaperones such as 4-phenylbutyric acid (4-PBA; which enhances protein folding) or genetic approaches such as expression of a constitutively active IKKβ improves leptin resistance and protects mice from DIO102,105–108. POMC neurons seem to be crucial for this effect, as targeted overexpression of XBP1, a transcription factor that modulates endoplasmic reticulum stress response, in this neuronal population is sufficient to protect animals from DIO108. 4-PBA has been used in the clinic for the treatment of urea cycle disorders in children, sickle cell disease, thalassaemia and cystic fibrosis109, and is clinically safe with few adverse effects110. 4-PBA might therefore be a suitable candidate drug for treating the leptin resistance that is induced by endoplasmic reticulum stress.
Celastrol, a small molecule found in the roots of the Tripterygium wilfordii plant, has been identified as a candidate to improve sensitivity to leptin in obesity by ameliorating endoplasmic reticulum stress107. Systemic, intraperitoneal (100 μg/kg) or oral (10 mg/kg) delivery of celastrol can dramatically decrease body weight (up to 45%) in mice with DIO but not in leptin-deficient ob/ob mice, db/db mice or lean control mice107. A subsequent independent study has shown that celastrol-induced weight loss and energy expenditure involve heat shock factor protein 1 (HSF1) and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α)-mediated stimulation of mitochondrial and thermogenic gene programmes, leading to the activation of brown and beige adipose tissues and increasing muscle endurance111. However, we need a deeper understanding of the molecular mechanisms underlying the effects of celastrol on leptin resistance, and further studies are needed to clarify whether celastrol can be used in humans to combat obesity.
Ceramide-induced lipotoxicity might also cause hypothalamic endoplasmic reticulum stress and leptin resistance in obesity112. Reduction of leptin resistance, leading to weight loss, increases brown adipose tissue thermogenesis and improves metabolic condition, and glucose homeostasis can be achieved by amelioration of endoplasmic reticulum stress by overexpressing 78 kDa glucose-regulated protein (GRP78)112, an endoplasmic reticulum chaperone that facilitates proper protein folding. This mechanism is also pertinent to LepR-deficient Zucker rats that are characterized by elevated hypothalamic levels of ceramide112. Manipulation of GRP78 function in the VMH can induce weight loss in obese Zucker rats112, demonstrating the potent effects of targeting ceramide-induced lipotoxicity and endoplasmic reticulum stress even in the absence of LepRb signalling. However, this finding raises the possibility that the beneficial effects of endoplasmic reticulum stress inhibition might be independent of leptin signalling.
Neonatal programming.
Excess nutrition and growth during prenatal and/or postnatal life might contribute to the aetiology of obesity and related diseases in later life113,114. A classic model of neonatal nutritional programming is the manipulation of rat litter size in the first days of life115–117. Rats raised in small litters gain more weight than those raised in litters of a normal size. In addition, rats from small litters have hyperphagia, increased adiposity, hyperleptinaemia, hyperinsulinaemia, impaired glucose tolerance, elevated triglycerides and increased systolic blood pressure115–118. This effect can be explained by the increased milk intake of individual rats in small litters. Importantly, overfed rats in this model maintain the obese phenotype and the associated metabolic disturbances throughout their lifespan115–118.
Leptin resistance in overfed neonatal rats results in impaired sensitivity to leptin of hypothalamic neurons, and the responsiveness to leptin in VMH and ARC neurons is different in rats raised in small litters. Whereas leptin increases the firing rate of VMH neurons of rats raised in normal-sized litters, it reduces the discharge rate of VMH neurons of rats raised in small litters119. Conversely, leptin inhibits neurons in the ARC of rats raised in normal-sized litters but has no effect on ARC neurons of small-litter rats120. This programmed leptin resistance of overfed neonatal rats is age dependent: in juvenile (24 days old) rats raised in small litters, expression of LepRb is decreased by ~45% in the hypothalamus115. These data are also consistent with the increase in expression of agouti-related protein (AgRP) and neuropeptide Y (NPY) in the ARC of rats raised in small litters, despite their marked hyperleptinaemia115,116. However, overfed adult (60 days old) rats do not show any change in the hypothalamic expression of LepRb or of any other LepR isoform116. Interestingly, adult rats raised in small litters have abnormally low levels of leptin in the CSF compared with controls, despite increased plasma levels of leptin, suggesting that the rate of leptin influx into the CNS is compromised in these animals116.
Ghrelin
Ghrelin was identified in 1999 as the endogenous ligand of the growth hormone secretagogue receptor (GHSR) from extracts of rat stomach121–124. Although first described as a growth hormone (GH) secretagogue121,125–127, the association of ghrelin with food intake, adiposity and metabolism regulation rapidly became the main focus of ghrelin function-related research122,123,128–130. However, the pleiotropic actions of ghrelin have implicated this hormone in different physiological processes (FIG. 1). Ghrelin has also been considered as a target to treat obesity131,132 and the metabolic disorders that are associated with cachexia133, sarcopenia134 and myopenia135.
GHRL encodes a precursor peptide that is 117 amino acids in length: preproghrelin, which is post-translationally processed into at least five products. Among them, acyl-ghrelin and desacyl-ghrelin have emerged as the most biologically relevant121,124. Acyl-ghrelin accounts for ~10% of the total amount of ghrelin and is acylated at Ser3 in the cytoplasm before secretion121. This process is catalysed by ghrelin O-acyltransferase (GOAT) and occurs in the endoplasmic retictulum136–138. GOAT expression and activity are modulated by nutrient availability, particularly by the availability of medium-chain fatty acids, which are used as acylation substrates and promote acyl-ghrelin production and secretion139. Acylation of ghrelin is required for its binding to GHSR and for its endocrine, metabolic and orexigenic actions121,124,140. The role of desacyl-ghrelin is still controversial, which is quite remarkable, given that it accounts for 90% of the circulating levels of ghrelin124,141, with new actions ascribed to this molecule that could be mediated by GHSR or an unidentified mechanism124,140,142,143. Obestatin is also translated from the GHRL transcript and was first described as a hormone with anorexigenic actions on appetite and body weight144; however, follow-up studies have so far failed to confirm the proposed anorexigenic actions of this molecule145–148.
Ghrelin synthesis and secretion
When nutrient availability is low, levels of ghrelin increase, and, after consumption of a meal, ghrelin levels are decreased, as measured by gene expression, hormone secretion from the stomach and circulating levels in both rodents and humans149–152. These findings are consistent with the idea that ghrelin is metabolically more active during negative energy balance than during positive energy balance153,154. Ghrelin has an inverse relationship with BMI: it is upregulated in under-nourished states, such as anorexia nervosa, and is downregulated in states of positive energy balance, such as obesity155–157. One exception is Prader–Willi syndrome, which is characterized by obesity, severe hyperphagia, GH deficiency and hypogonadism142 Patients with this syndrome have high circulating levels of ghrelin that could explain the hyperphagia and the consequent obesity158,159. However, it should be noted that individuals with Prader–Willi syndrome display hyperghrelinaemia in infancy, before the manifestation of hyperphagia160,161, and do not lose weight or appetite when the levels of ghrelin are decreased through treatment with octreotide, a somatostatin analogue162.
Strikingly, the actual physiological role of ghrelin is still unclear. For example, pharmacological doses (up to 5 pmol/kg/min; intravenously) of ghrelin increase food intake in humans and rodents109,163–166, but whether the orexigenic effect of ghrelin and its chronic obesogenic effects are coupled is unclear. Although ghrelin might provide an acute hunger signal in the preprandial period, little evidence exists to support a role for high levels of ghrelin in inducing feeding. Whereas in humans and rodents plasma levels of desacyl-ghrelin are increased upon long-term starvation, there are conflicting data about the changes in acyl-ghrelin139,167. Furthermore, neither mice deficient in ghrelin, GHSR or GOAT nor transgenic mice overexpressing ghrelin and/or GOAT have impaired feeding compared with wild-type animals139,168,169. Consistent with this finding, selective ablation of ghrelin-producing cells in adult mice using diphtheria toxin does not promote changes in feeding, body weight or sensitivity to a HFD, despite the substantial reduction (80–95%) in plasma levels of ghrelin. However, this ablation of ghrelin-secreting cells was associated with severe hypoglycaemia, consistent with a role for the hormone in the control of blood glucose during caloric deficit170. Ghrelin-induced stimulation of food intake requires a 10-fold to 100-fold increase in its circulating levels compared with its physiological levels171. However, anti-ghrelin vaccines, which were originally developed as antiobesity drugs, did not have chronic anorexigenic properties132. The preprandial surge of ghrelin might prepare the organism for incoming food to metabolize and store energy efficiently, and indeed ghrelin activation is highly influenced by dietary lipids124,139,171. For example, ghrelin might signal to the brain that abundant calories are available. This notion is consistent with the fact that ghrelin also stimulates expression of genes that are involved in lipogenesis129,172,173, and ghrelin signalling in the hypothalamus is totally dependent on neuronal fatty acid metabolism130,174,175. However, new assays show that total levels of ghrelin increase during fasting, specifically the levels of desacyl-ghrelin, whereas the levels of acyl-ghrelin remain unchanged139, which indicates that, although extensively studied, the mechanism of ghrelin secretion is still not well understood124,176.
Ghrelin receptor signalling
The effect of ghrelin on feeding is mediated by GHSR, as the orexigenic effect of this hormone is blunted in Ghsr-knockout mice177. GHSR is highly expressed in the hypothalamic cell populations that regulate feeding and body weight, such as ARC AgRP-expressing and NPY-expressing neurons and VMH neurons expressing AMP-activated protein kinase (AMPK) and fatty acid synthase178–181 (FIG. 3).
Figure 3 |. Hypothalamic ghrelin signalling.
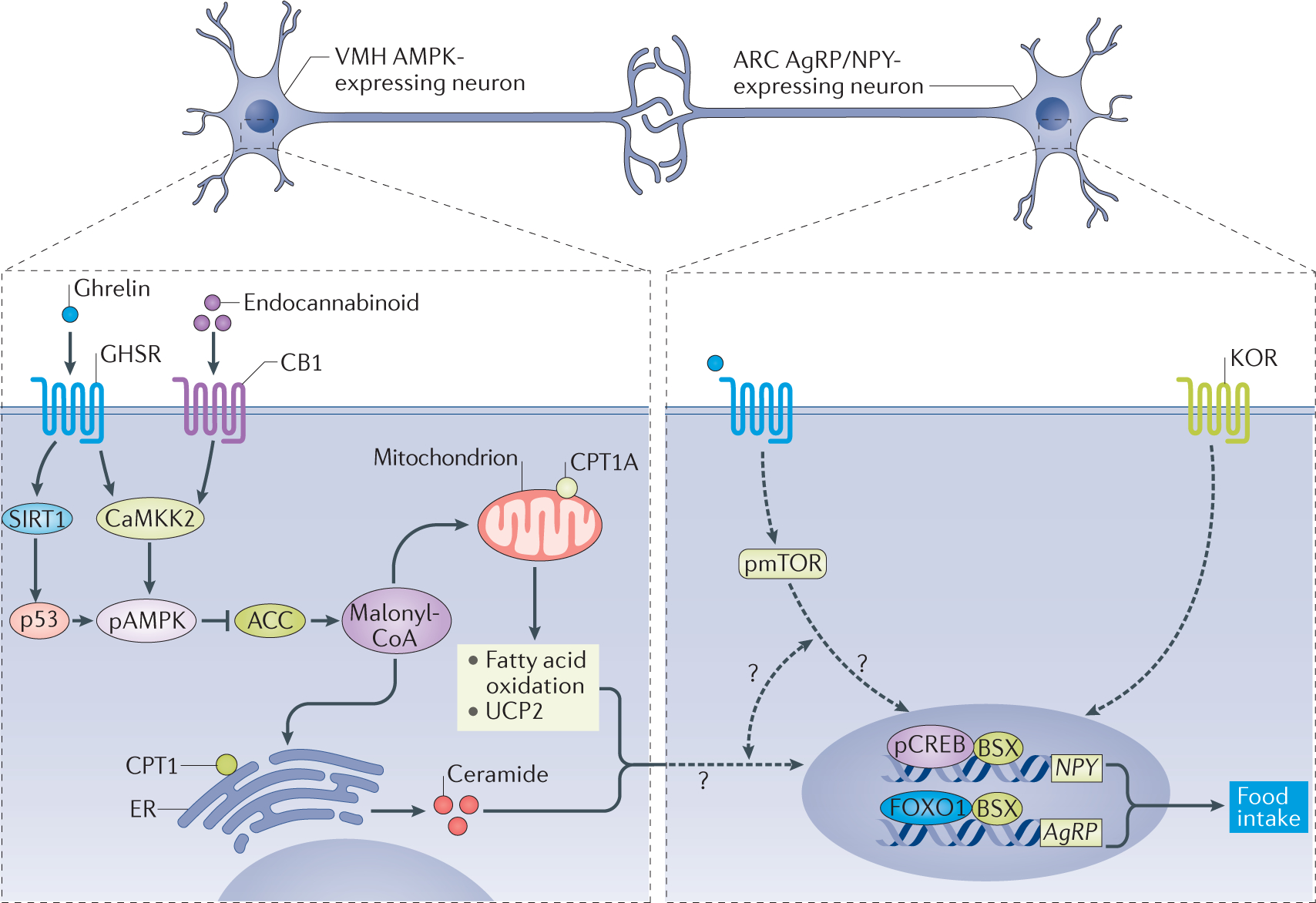
Ghrelin binds to growth hormone secretagogue receptor (GHSR) and stimulates hypothalamic sirtuin 1 (SIRT1)–p53 and calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2)–AMP-activated protein kinase (AMPK) axes in the ventromedial nucleus of the hypothalamus (VMH). This action requires cannabinoid receptor 1 (CB1). Consequently, hypothalamic levels of malonyl-CoA, the physiological inhibitor of the enzyme carnitine palmitoyltransferase 1 (CPT1) isoforms A and C, are elevated. This effect promotes disinhibition of CPT1A, increases fatty acid oxidation, alters the levels of reactive oxygen species, increases expression of UCP2 and promotes the CPT1C-mediated increase in levels of ceramide. These metabolic changes activate the nuclear transcription machinery by increasing expression and/or activity of key transcription factors, such as cyclic AMP-responsive element-binding protein (CREB) and its phosphorylated isoform, pCREB, FOXO1, and brain-specific homeobox protein homologue (BSX) in the arcuate nucleus (ARC), increasing mRNA expression of AgRP and NPY, which induces feeding. Mechanistic target of rapamycin (mTOR) and k-opioid receptor (KOR) also mediate the effects of ghrelin in the ARC; however, the association between these two events is unclear. ACC, acetyl-CoA carboxylase; AgRP, agouti-related protein; ER, endoplasmic reticulum; NPY, neuropeptide Y; pAMPK, phosphorylated AMPK; pmTOR, phosphorylated mTOR.
Ghrelin and hypothalamic fatty acid metabolism.
The binding of ghrelin to GHSR increases intracellular levels of PIP3 and Ca2+ via induction of phospholipase C and PKC182,183. The rise in intracellular Ca2+ activates hypothalamic calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2), which activates the cellular energy sensor AMPK130,174,184–187. Ghrelin also specifically activates a hypothalamic sirtuin 1–p53 pathway that promotes AMPK phosphorylation188,189. Activation of AMPK in the hypothalamus modulates fatty acid metabolism. When activated by ghrelin, phosphorylated AMPK inactivates acetyl-CoA carboxylase, leading to a decrease in the levels of malonyl-CoA, which disinhibits carnitine palmitoyl transferase 1A (CPT1A)130,174,175,185–187,190–192. In addition to the ghrelin-induced phosphorylation of AMPK catalytic subunit-α (AMPKα), AMPKγ2 is involved in the orexigenic action of ghrelin193. Chronic activation of AMPK, due to a mutation in the gene encoding AMPKγ2, promotes ghrelin-dependent hyperphagia and obesity in mice and humans193. The overall outcome of such effect is increased fatty acid oxidation and accumulation of reactive oxygen species, which are buffered by UCP2 (REF. 174). In addition, hypothalamic CPT1C, a brain-specific isoform that is located in the endoplasmic reticulum194, was implicated in the orexigenic action of ghrelin, as indicated by the lack of effect of ghrelin on food intake in CPT1C-null mice195. This pathway involves an increase in hypothalamic ceramide synthesis195. Genetic or pharmacological inhibition of CaMKK2, AMPK, CPT1A, CPT1C or UCP2, as well as increased concentrations of malonyl-CoA or decreased levels of ceramide in the hypothalamus, prevents ghrelin-induced feeding130,174,184,186,187,195.
Furthermore, ghrelin caused a ~2-fold upregulation in mTORC1 signalling in the ARC, and pharmacological or genetic central inhibition of the mTORC1 pathway decreases the orexigenic action of ghrelin and ghrelin-mediated induction of AgRP and NPY expression196,197. This evidence indicates that ghrelin induces feeding by acting on AMPK and mTORC1 in two different hypothalamic nuclei, the VMH and the ARC, respectively130,175,187,196,197. However, whether these two networks act in concert or independently is currently unclear196,198.
The effects of ghrelin on hypothalamic neuropeptides.
Ghrelin ultimately increases the potential firing of AgRP-expressing and NPY-expressing neurons174, and activates transcriptional events in the ARC by increasing expression and/or activity of key transcription factors, such as cAMP-responsive element-binding protein (CREB) and its phosphorylated isoform, pCREB; FOXO1 and its phosphorylated isoform, pFOXO1; and brain-specific homeobox protein homologue (BSX)185,199. These changes underlie, at least in part, the rise in orexigenic AgRP and NPY neuropeptides in the ARC123,130,185,200,201. Interestingly, AMPK and its direct downstream targets, as well as the orexigenic neuropeptides, are located in different neuronal populations: whereas AgRP-expressing and NPY-expressing neurons are located in the ARC, AMPK-expressing neurons, which send synaptic projections to ARC neurons202, are located in the VMH130,175. The binding of ghrelin to GHSR increases intracellular levels of Ca2+ through the mobilization of Ca2+ from intracellular stores and through the opening of Ca2+ channels203. Ghrelin signalling increases presynaptic AMPK activity by promoting intracellular Ca2+ release and the subsequent activation of CaMKK2184. The action stimulates the firing rate of AgRP-expressing and NPY-expressing neurons and inhibits the firing activity of POMC neurons by increasing inhibitory GABAergic neurotransmission through postsynaptic inputs204. Consequently, the anorectic effect of α-melanocyte-stimulating hormone (αMSH; the cleaved product of POMC) is inhibited, which further enhances orexigenic drive. However, if feeding continues, leptin then stimulates POMC neurons, which exert an anorectic action, and promotes the release of opioids to inhibit AMPK in presynaptic neurons through k-opioid receptors202,205 (FIG. 3).
Food intake is induced through activation of cannabinoid receptor 1 (CB1) and stimulation of AMPK by both exogenous and endogenous cannabinoids206 (FIG. 3). A functional CB1 is needed for ghrelin to affect AMPK activity and food intake; these findings are based on studies using CB1-null animals and CB1 antagonists, which do not respond to or block, respectively, ghrelin orexigenic actions or ghrelin-induced AMPK activation191,207,208 Conversely, the effect of ghrelin on GH release does not require CB1 signalling207,208. The increased intracellular levels of Ca2+ that are induced by binding of ghrelin to GHSR activate diacylglycerol lipase-α, the enzyme that synthesizes 2-arachidonoylglycerol (an endogenous cannabinoid), directly or through activation of PKC, resulting in increased 2-arachidonoylglycerol synthesis and release in the hypothalamus191. This mechanism was implicated in ghrelin-mediated inhibition of glutamate excitatory inputs, originating from ARC neurons, onto parvocellular neurons of the paraventricular nucleus of the hypothalamus that promote feeding204,209,210.
In addition to the hypothalamus, ghrelin exerts its orexigenic effect in other regions of the brain, such as the ventral tegmental area (VTA), in which the motivational dopaminergic system is located211,212. A negative energy balance potentiates the orexigenic effect of ghrelin; for example, the induction of AgRP and NPY expression in response to ghrelin is increased in fasted rats102. Furthermore, refeeding, as well as hormonal signals of positive energy balance (insulin and leptin), suppresses ghrelin responsiveness, potential firing rates and Agrp and Npy gene expression in the ARC181,213–215. Therefore the fact that the effects of ghrelin are increased by fasting is probably due to the reduced levels of leptin and insulin that characterize such a state.
Ghrelin resistance in obesity
The role and relevance of ghrelin resistance in obesity are unclear. Ghrelin resistance has been proposed as a mechanism to protect against the higher body-weight set point that is established during times of food availability and to maximize energy reserves during a time of food scarcity154. In such cases, inhibition of ghrelin sensitivity might prevent the rebound in body weight that follows dietary restriction-induced weight loss in patients with obesity216. Supporting this hypothesis is the observation that ghrelin-null mice with DIO have reduced body-weight regain after calorie-restricted weight loss216.
Effects of dietary obesity.
Unexpectedly for an orexigenic hormone, a positive energy balance induces ghrelin resistance (FIG. 4) in humans and rodent models, and obesity is associated with reduced secretion and plasma levels of ghrelin155–157,217. Moreover, circulating levels of ghrelin do not fall in response to a meal in humans with obesity218. These changes in ghrelin release might result from ghrelin-secreting cells in the stomach that do not respond to noradrenaline-mediated stimulatory actions of the sympathoadrenal system on β1-adrenergic receptors in obesity219. Furthermore, mice lacking β1-adrenergic receptors in ghrelin-expressing cells have diminished ghrelin release upon severe caloric deficit220. Ghrelin-producing cells from mice with DIO also have a blunted response to the inhibitory actions of glucose on ghrelin release219. Furthermore, ghrelin transport through the BBB and the expression of Ghsr are reduced in obese mice221,222, leading to reduced sensitivity to ghrelin217,223, which might promote hypothalamic ghrelin resistance. Consistently, central and peripheral injections of ghrelin in obese mice fail to induce AgRP and NPY expression and secretion221,224, raising the possibil that impaired response of AgRP-expressing and NPY-expressing neurons contributes to ghrelin resistance in the hypothalamus.
Figure 4 |. Hypothalamic ghrelin resistance.
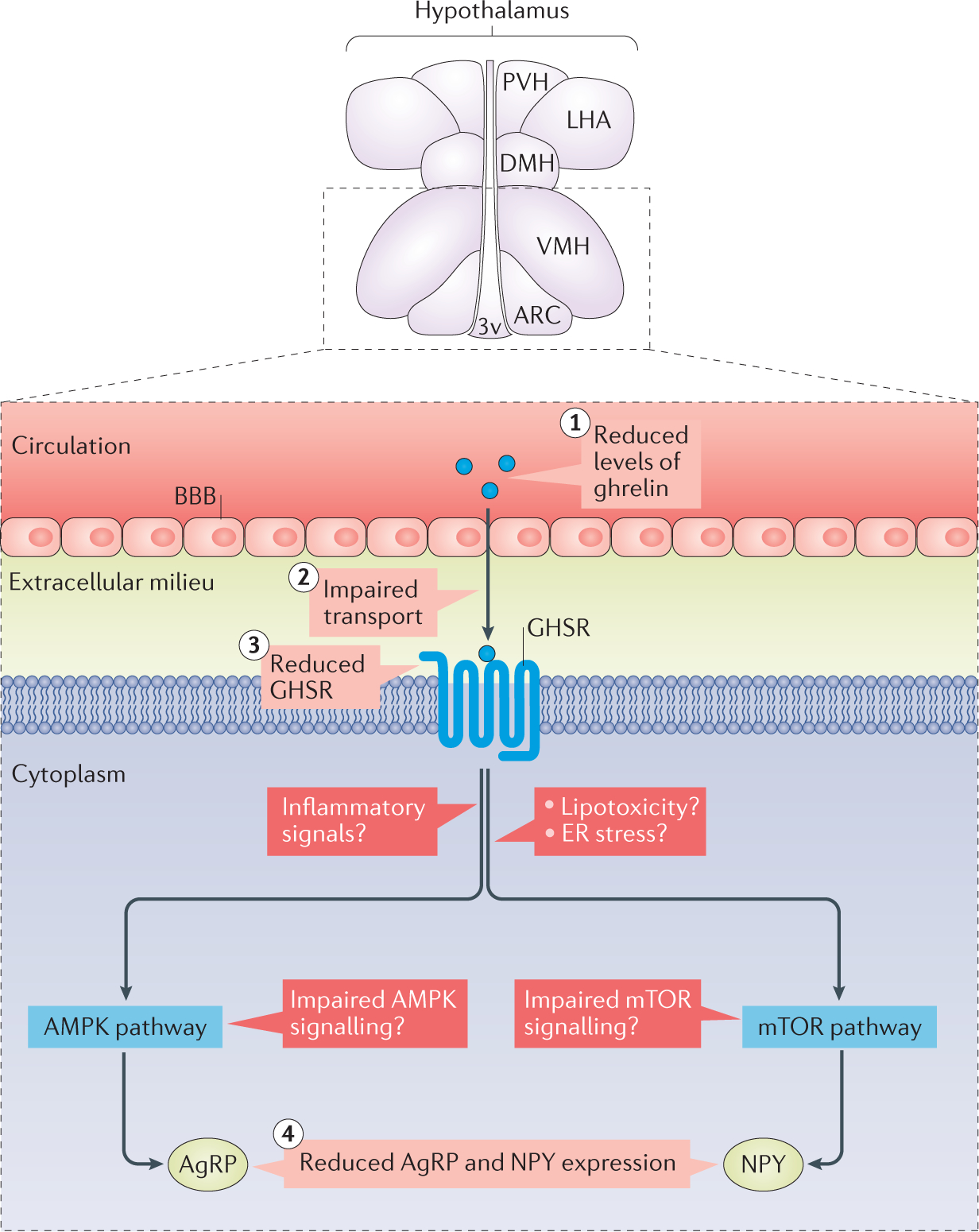
Obesity-associated ghrelin resistance might develop via different mechanisms, such as decreased circulating levels of ghrelin (1); impaired transport of ghrelin through the blood–brain barrier (BBB) (2); reduced expression of growth hormone secretagogue receptor (GHSR) (3); and reduced expression of agouti-related protein (AgRP) and neuropeptide Y (NPY) (4), which reduces the orexigenic action of ghrelin. The molecular mechanisms leading to the reduction of neuropeptide expression are unclear, but possible candidates include hypothalamic inflammation, lipotoxicity, endoplasmic reticulum (ER) stress and impaired AMP-activated protein kinase (AMPK) or mechanistic target of rapamycin (mTOR) pathways. 3v, third ventricle; ARC, arcuate nucleus of the hypothalamus; DMH, dorsomedial nucleus of the hypothalamus; LHA, lateral hypothalamic area; PVH, paraventricular nucleus of the hypothalamus; VMH, ventromedial nucleus of the hypothalamus.
Interestingly, ghrelin resistance in response to a HFD can occur rapidly and almost independently from the length of the nutritional intervention. Indeed, short-term (12 h) exposure to a HFD is enough to alter the orexigenic effects of ghrelin173. Notably, the capacity of ghrelin to modulate lipogenesis in white adipose tissue is unaffected by a HFD, indicating that different neuronal circuitries mediate ghrelin-specific regulation of food intake and lipid metabolism173.
Neonatal programming.
Blocking ghrelin in neonatal mice can increase αMSH and AgRP projections from the ARC to the paraventricular nucleus of the hypothalamus and can promote increases in body weight, visceral fat and blood levels of glucose and a decrease in sensitivity to leptin225. In addition, chronic administration of ghrelin postnatally (from day 4 to day 12) impairs the normal development of ARC projections, leading to metabolic dysfunction. Chronic exposure to ghrelin also led to attenuation of leptin-induced STAT3 activation in ARC neurons225. These results indicate that ghrelin has an inhibitory role in the development of hypothalamic neural circuits and that ghrelin action during neonatal life is required for optimal metabolic regulation.
Small-litter mice have lower levels of total ghrelin and acyl-ghrelin in the serum and decreased expression of Ghrl mRNA in the stomach226. Notably, normalization of the levels of ghrelin by chronic neonatal injection in small-litter mice does not improve the metabolic phenotype. This finding indicates that central ghrelin resistance in mice raised in small litters might arise from altered transport in the mediobasal hypothalamus226.
Leptin and ARC neuropeptides.
Leptin has been implicated in ghrelin resistance, as indicated by the antagonistic physiological roles of leptin and ghrelin systems227,228. Anatomical data show that GHSR and LepR are co-expressed in more than 90% of neurons in the ARC229 This notion is also exemplified by leptin-mediated inhibition of AgRP and NPY expression, an effect opposite to that of ghrelin230–232. Furthermore, ob/ob mice have normal responses to ghrelin, and these responses are suppressed after leptin administration224. Notably, ob/ob mice fed a HFD remain sensitive to ghrelin, further supporting the idea that hyperleptinaemia, instead of obesity or a HFD, causes ghrelin resistance224.
Inflammation and endoplasmic reticulum stress.
Hypothalamic inflammation has been suggested as a potential mechanism of ghrelin resistance100,233,234. Obese ob/ob mice and lean mouse controls fed a HFD have similar responses to ghrelin, despite the ob/ob mice having a higher level of gliosis, a marker of central inflammation, which indicates that hypothalamic gliosis is not a cause of ghrelin resistance224. However, ghrelin resistance was linked to local inflammation in nodose ganglia and to the subsequent dysregulation of vagus afferents234. This is based on the observation that upregulation of macrophage and/or microglia markers and of inflammatory cytokines in nodose ganglia of mice with DIO was associated with decreased expression and signalling of nodose ganglion GHSR, and a blunted ghrelin-induced decrease in vagal afferent nerve activity234. Another proposed mechanism of ghrelin resistance in obesity relates to endoplasmic reticulum stress102,105,234. In mice, treatment with ghrelin increases hypothalamic concentration of ceramide via modulation of CPT1C in the endoplasmic reticulum195, and, as described earlier in the text, ceramide synthesis is induced by obesity, promoting hypothalamic lipotoxicity and endoplasmic reticulum stress112. However, as the levels of ghrelin are decreased in obesity, this hormone is not likely to have a role in the production of ceramides in this context. In addition, ghrelin can reduce hypothalamic expression of endoplasmic reticulum stress markers such as CCAAT/enhancer-binding protein-ε (CEBPε) and phosphorylated eukaryotic translation initiation factor 2α (pEIF2α)185. Thus, the relationship between endoplasmic reticulum stress, ghrelin and ghrelin resistance in obesity remains unclear, warranting further research.
AMPK.
Despite the evidence suggesting that AgRP and NPY mediate the development of ghrelin resistance in obesity, the underlying molecular mechanism is unclear. AMPK in the hypothalamus, which does not respond to leptin in mice with DIO, might mediate this effect. For example, contrary to what happens in normal chow-fed mouse controls, in mice with DIO, leptin does not inhibit AMPK activity in the ARC, which might contribute to leptin resistance235,236. Whether the ghrelin-induced increase in hypothalamic AMPK is also blunted by a HFD is currently unclear, but is an interesting possibility.
Ghrelin resistance in extra-hypothalamic regions.
Ghrelin resistance is also found in areas of the brain other than the hypothalamus, such as the VTA, where ghrelin acts on dopaminergic neurons to modulate feeding reward responses211,212. Notably, in mice with DIO, although ghrelin retains its orexigenic properties when administered specifically in the VTA237, it fails to induce the same reward response when injected intraperitoneally238. These data indicate that obesity differentially affects the homeostatic (hypothalamic-based) and motivational (VTA-based) actions of ghrelin on food intake. However, these mechanisms in the extra-hypothalamic regions remain to be defined.
Mutations in GHSR.
Several mutations and singlenucleotide polymorphisms of GHSR have been identified in humans that contribute to obesity and short stature239 For example, the nonsense GHSR mutation Ala204Glu, which prevents the normal constitutively active GHSR from functioning, is associated with familial short stature syndrome, which can be partially reversed with GH treatment240,241. Functional analyses of mutated GHSR have shown multiple defects, including altered cell surface expression and changes in ligand binding and basal and stimulated signalling239–243. These defects might be considered as a state of ghrelin resistance, although currently the physiological relevance to obesity in humans is unclear.
Conclusions
Resistance to the metabolic actions of leptin and ghrelin is thought to contribute to the development and maintenance of obesity. However, recent evidence challenges the notion that DIO-related leptin resistance exists39. The reasons behind the differential resistance to exogenous versus endogenous leptin remain to be determined. Nonetheless, in our opinion, the definition of leptin resistance should be revisited so as to refer to the inability of exogenous leptin to affect food intake and body weight.
Unfortunately, the progress in deciphering the molecular processes underlying leptin and ghrelin resistance in obesity has not yet translated into novel and effective obesity treatments, which highlights the need for better understanding of the mechanisms of obesity-associated leptin and ghrelin resistance. The pleiotropic actions of ghrelin and leptin (FIG. 1) have led to an interest in understanding the consequences of resistance to these hormones beyond metabolic disorders. Indeed, several pathological conditions, including cardiovascular and reproductive dysfunction, might result from defects in the actions of these hormones and in their signalling pathways. For example, substantial evidence suggests that selective leptin resistance underlies obesity-related hypertension and sympathetic nervous system overactivity244. This notion is supported by the ability of leptin to increase blood pressure and cardiovascular sympathetic outflow in obesity in spite of the resistance to the anorexigenic effects of this hormone. Despite this finding, the molecular and cellular bases of selective leptin resistance remain elusive. Similarly, the current knowledge about ghrelin resistance is scarce. Several possibilities have been proposed, but more work is needed to fully understand the molecular basis and physiological significance of ghrelin resistance. Decoding the mechanisms underlying selective resistance to metabolic hormones will greatly enhance our understanding of the diseases that are commonly associated with obesity.
Key points.
The anorexigenic hormone leptin and the orexigenic hormone ghrelin are crucial for metabolic regulation and energy homeostasis
Obesity-associated resistance to leptin and ghrelin promotes adiposity and might contribute to the diseases that are associated with this condition beyond metabolic disorders
Resistance to leptin and ghrelin is a multifactorial process that involves changes at several levels: from disturbed hormonal production to altered receptor trafficking and signalling in the brain
Several molecules and signalling pathways associated with leptin and ghrelin receptors have been identified as potential targets to overcome resistance to these hormones, but none has reversed the energy imbalance in the long term
The identification of novel molecular targets and pathways that can be modulated to enhance sensitivity to leptin and ghrelin and restore energy homeostasis is necessary for the development of efficient pharmacological treatments for obesity
Acknowledgements
H.C. is funded by the US National Institutes of Health (HL127673 and MH109920). M.L.’s research is funded by the European Community’s Seventh Framework Programme (FP7/2007–2013) under grant agreement number 281854; the ObERStress project: Xunta de Galicia (2015-CP079); MINECO co-funded by the FEDER Program of EU (SAF2015–71026-R and BFU2015–70454-REDT/Adipoplast). Centro de Investigación Biomédica en Red (CIBER) de Fisiopatología de la Obesidad y Nutrición is an initiative of the Instituto de Salud Carlos III (ISCIII). K.R.’s research is supported by the US National Institutes of Health (HL084207); the American Heart Association (14EIA18860041); the University of Iowa Fraternal Order of Eagles Diabetes Research Center; and the University of Iowa Center for Hypertension Research.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Stewart ST, Cutler DM & Rosen AB Forecasting the effects of obesity and smoking on U.S. life expectancy. N. Engl. J. Med. 361, 2252–2260 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y et al. Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Friedman JM & Halaas JL Leptin and the regulation of body weight in mammals. Nature 395, 763–770 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Frederich RC et al. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat. Med. 1, 1311–1314 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Considine RV et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 334, 292–295 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Havel PJ et al. Relationship of plasma leptin to plasma insulin and adiposity in normal weight and overweight women: effects of dietary fat content and sustained weight loss. J. Clin. Endocrinol. Metab. 81, 4406–4413 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Vaisse C et al. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat. Genet. 14, 95–97 (1996). [DOI] [PubMed] [Google Scholar]
- 8.Schwartz MW, Seeley RJ, Campfield LA, Burn P & Baskin DG Identification of targets of leptin action in rat hypothalamus. J. Clin. Invest. 98, 1101–1106 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen P et al. Selective deletion of leptin receptor in neurons leads to obesity. J. Clin. Invest. 108, 1113–1121 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhillon H et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Balthasar N et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42, 983–991 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Leinninger GM et al. Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab. 14, 313–323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rezai-Zadeh K et al. Leptin receptor neurons in the dorsomedial hypothalamus are key regulators of energy expenditure and body weight, but not food intake. Mol. Metab 3, 681–693 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dodd GT et al. The thermogenic effect of leptin is dependent on a distinct population of prolactinreleasing peptide neurons in the dorsomedial hypothalamus. Cell Metab. 20, 639–649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leal-Cerro A et al. Serum leptin levels in male marathon athletes before and after the marathon run. J. Clin. Endocrinol. Metab. 83, 2376–2379 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Maffei M et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1, 1155–1161 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Lago R, Gomez R, Lago F, Gomez-Reino J & Gualillo O Leptin beyond body weight regulation — current concepts concerning its role in immune function and inflammation. Cell. Immunol. 252, 139–145 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Lam QL & Lu L Role of leptin in immunity. Cell. Mol. Immunol. 4, 1–13 (2007). [PubMed] [Google Scholar]
- 19.Haynes WG, Morgan DA, Walsh SA, Mark AL & Sivitz WI Receptor-mediated regional sympathetic nerve activation by leptin. J. Clin. Invest. 100, 270–278 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahmouni K, Haynes WG & Mark AL Cardiovascular and sympathetic effects of leptin. Curr. Hypertens. Rep. 4, 119–125 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Elias CF & Purohit D Leptin signaling and circuits in puberty and fertility. Cell. Mol. Life Sci. 70, 841–862 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen XX & Yang T Roles of leptin in bone metabolism and bone diseases. J. Bone Miner. Metab. 33, 474–485 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Tartaglia LA et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 83, 1263–1271 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Tartaglia LA The leptin receptor. J. Biol. Chem. 272, 6093–6096 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Chua SC Jr et al. Fine structure of the murine leptin receptor gene: splice site suppression is required to form two alternatively spliced transcripts. Genomics 45, 264–270 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Bates SH & Myers MG Jr. The role of leptin receptor signaling in feeding and neuroendocrine function. Trends Endocrinol. Metab. 14, 447–452 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Scott MM et al. Leptin targets in the mouse brain. J. Comp. Neurol. 514, 518–532 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ihle JN & Kerr IM Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 11, 69–74 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Taniguchi T Cytokine signaling through nonreceptor protein tyrosine kinases. Science 268, 251–255 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Kloek C et al. Regulation of Jak kinases by intracellular leptin receptor sequences. J. Biol. Chem. 277, 41547–41555 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Devos R et al. Ligand-independent dimerization of the extracellular domain of the leptin receptor and determination of the stoichiometry of leptin binding. J. Biol. Chem. 272, 18304–18310 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Couturier C & Jockers R Activation of the leptin receptor by a ligand-induced conformational change of constitutive receptor dimers. J. Biol. Chem. 278, 26604–26611 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Allison MB & Myers MG Jr. 20 years of leptin: connecting leptin signaling to biological function. J. Endocrinol. 223, T25–T35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hekerman P et al. Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS J. 272, 109–119 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Gong Y et al. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J. Biol. Chem. 282, 31019–31027 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Bjorbak C et al. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J. Biol. Chem. 275, 40649–40657 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Cota D et al. Hypothalamic mTOR signaling regulates food intake. Science 312, 927–930 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Harlan SM, Guo DF, Morgan DA, Fernandes-Santos C & Rahmouni K Hypothalamic mTORC1 signaling controls sympathetic nerve activity and arterial pressure and mediates leptin effects. Cell Metab. 17, 599–606 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ottaway N et al. Diet-induced obese mice retain endogenous leptin action. Cell Metab. 21, 877–882 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE & Leibel RL Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J. Clin. Endocrinol. Metab. 87, 2391–2394 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Mark AL, Correia ML, Rahmouni K & Haynes WG Selective leptin resistance: a new concept in leptin physiology with cardiovascular implications. J. Hypertens. 20, 1245–1250 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Correia ML et al. The concept of selective leptin resistance: evidence from agouti yellow obese mice. Diabetes 51, 439–442 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Rahmouni K, Morgan DA, Morgan GM, Mark AL & Haynes WG Role of selective leptin resistance in diet-induced obesity hypertension. Diabetes 54, 2012–2018 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Simonds SE et al. Leptin mediates the increase in blood pressure associated with obesity. Cell 159, 1404–1416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mark AL Selective leptin resistance revisited. Am. J. Physiol. Regul. Integr. Comp. Physiol 305, R566–R581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinha MK et al. Evidence of free and bound leptin in human circulation. Studies in lean and obese subjects and during short-term fasting. J. Clin. Invest. 98, 1277–1282 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Magni P et al. Free and bound plasma leptin in normal weight and obese men and women: relationship with body composition, resting energy expenditure, insulin-sensitivity, lipid profile and macronutrient preference. Clin. Endocrinol. 62, 189–196 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Houseknecht KL et al. Evidence for leptin binding to proteins in serum of rodents and humans: modulation with obesity. Diabetes 45, 1638–1643 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Schwartz MW, Peskind E, Raskind M, Boyko EJ & Porte D Jr. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat. Med. 2, 589–593 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Morgan DA, Thedens DR, Weiss R & Rahmouni K Mechanisms mediating renal sympathetic activation to leptin in obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol 295, R1730–R1736 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banks WA & Farrell CL Impaired transport of leptin across the blood–brain barrier in obesity is acquired and reversible. Am. J. Physiol. Endocrinol. Metab 285, E10–E15 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Pan W et al. Astrocyte leptin receptor (ObR) and leptin transport in adult-onset obese mice. Endocrinology 149, 2798–2806 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caro JF et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet 348, 159–161 (1996). [DOI] [PubMed] [Google Scholar]
- 54.Banks WA Leptin transport across the blood–brain barrier: implications for the cause and treatment of obesity. Curr. Pharm. Des. 7, 125–133 (2001). [DOI] [PubMed] [Google Scholar]
- 55.de Git KC & Adan RA Leptin resistance in diet-induced obesity: the role of hypothalamic inflammation. Obes. Rev. 16, 207–224 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Koga S et al. Effects of diet-induced obesity and voluntary exercise in a tauopathy mouse model: implications of persistent hyperleptinemia and enhanced astrocytic leptin receptor expression. Neurobiol. Dis. 71, 180–192 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Jayaram B et al. Astrocytic leptin-receptor knockout mice show partial rescue of leptin resistance in diet-induced obesity. J. Appl. Physiol. 114, 734–741 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pierce AA & Xu AW De novo neurogenesis in adult hypothalamus as a compensatory mechanism to regulate energy balance. J. Neurosci. 30, 723–730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balland E et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 19, 293–301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee DA et al. Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nat. Neurosci. 15, 700–702 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang C et al. Tat-modified leptin is more accessible to hypothalamus through brain–blood barrier with a significant inhibition of body-weight gain in high-fat-diet fed mice. Exp. Clin. Endocrinol. Diabetes 118, 31–37 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Guo DF & Rahmouni K Molecular basis of the obesity associated with Bardet–Biedl syndrome. Trends Endocrinol. Metab. 22, 286–293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo DF et al. The BBsome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. PLoS Genet. 12, e1005890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seo S et al. Requirement of Bardet–Biedl syndrome proteins for leptin receptor signaling. Hum. Mol. Genet. 18, 1323–1331 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Irani BG, Dunn-Meynell AA & Levin BE Altered hypothalamic leptin, insulin, and melanocortin binding associated with moderate-fat diet and predisposition to obesity. Endocrinology 148, 310–316 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Starr R et al. A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921 (1997). [DOI] [PubMed] [Google Scholar]
- 67.Minamoto S et al. Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) family: SSI-2 and SSI-3. Biochem. Biophys. Res. Commun. 237, 79–83 (1997). [DOI] [PubMed] [Google Scholar]
- 68.Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE & Flier JS Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1, 619–625 (1998). [DOI] [PubMed] [Google Scholar]
- 69.Howard JK et al. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat. Med. 10, 734–738 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Mori H et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 10, 739–743 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Pedroso JA et al. Inactivation of SOCS3 in leptin receptor-expressing cells protects mice from diet-induced insulin resistance but does not prevent obesity. Mol. Metab 3, 608–618 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reed AS et al. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 59, 894–906 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ahmad F, Li PM, Meyerovitch J & Goldstein BJ Osmotic loading of neutralizing antibodies demonstrates a role for protein-tyrosine phosphatase 1B in negative regulation of the insulin action pathway. J. Biol. Chem. 270, 20503–20508 (1995). [DOI] [PubMed] [Google Scholar]
- 74.Maegawa H et al. Thiazolidine derivatives ameliorate high glucose-induced insulin resistance via the normalization of protein-tyrosine phosphatase activities. J. Biol. Chem. 270, 7724–7730 (1995). [DOI] [PubMed] [Google Scholar]
- 75.Seely BL et al. Protein tyrosine phosphatase 1B interacts with the activated insulin receptor. Diabetes 45, 1379–1385 (1996). [DOI] [PubMed] [Google Scholar]
- 76.Myers MP et al. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 276, 47771–47774 (2001). [DOI] [PubMed] [Google Scholar]
- 77.Elchebly M et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 283, 1544–1548 (1999). [DOI] [PubMed] [Google Scholar]
- 78.Cook WS & Unger RH Protein tyrosine phosphatase 1B: a potential leptin resistance factor of obesity. Dev. Cell 2, 385–387 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Zabolotny JM et al. PTP1B regulates leptin signal transduction in vivo. Dev. Cell 2, 489–495 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Klaman LD et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 20, 5479–5489 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bence KK et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 12, 917–924 (2006). [DOI] [PubMed] [Google Scholar]
- 82.Tsou RC, Zimmer DJ, De Jonghe BC & Bence KK Deficiency of PTP1B in leptin receptor-expressing neurons leads to decreased body weight and adiposity in mice. Endocrinology 153, 4227–4237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsou RC, Rak KS, Zimmer DJ & Bence KK Improved metabolic phenotype of hypothalamic PTP1B-deficiency is dependent upon the leptin receptor. Mol. Metab. 3, 301–312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.He RJ, Yu ZH, Zhang RY & Zhang ZY Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 35, 1227–1246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lantz KA et al. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obes. (Silver Spring) 18, 1516–1523 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Ahima RS et al. Appetite suppression and weight reduction by a centrally active aminosterol. Diabetes 51, 2099–2104 (2002). [DOI] [PubMed] [Google Scholar]
- 87.Takahashi N, Qi Y, Patel HR & Ahima RS A novel aminosterol reverses diabetes and fatty liver disease in obese mice. J. Hepatol. 41, 391–398 (2004). [DOI] [PubMed] [Google Scholar]
- 88.Zasloff M et al. A spermine-coupled cholesterol metabolite from the shark with potent appetite suppressant and antidiabetic properties. Int. J. Obes. Relat. Metab. Disord. 25, 689–697 (2001). [DOI] [PubMed] [Google Scholar]
- 89.Isis Pharmaceuticals, Inc. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. PRNewswire www.prnewswire.com/newsreleases/isis-pharmaceuticals-reports-positivephase-2-data-for-isis-113715-in-patients-with-type-2-diabetes-65238987.html (2009).
- 90.Loh K et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 14, 684–699 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dodd GT et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 160, 88–104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rousso-Noori L et al. Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner. Cell Metab. 13, 562–572 (2011). [DOI] [PubMed] [Google Scholar]
- 93.Toledano-Katchalski H et al. Protein tyrosine phosphatase ε inhibits signaling by mitogen-activated protein kinases. Mol. Cancer Res. 1, 541–550 (2003). [PubMed] [Google Scholar]
- 94.Schmidt M, Dekker FJ & Maarsingh H Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol. Rev. 65, 670–709 (2013). [DOI] [PubMed] [Google Scholar]
- 95.McKnight GS et al. Cyclic AMP, PKA, and the physiological regulation of adiposity. Recent Prog. Horm. Res. 53, 139–159 (1998). [PubMed] [Google Scholar]
- 96.Almahariq M, Mei FC & Cheng X Cyclic AMP sensor EPAC proteins and energy homeostasis. Trends Endocrinol. Metab. 25, 60–71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fukuda M, Williams KW, Gautron L & Elmquist JK Induction of leptin resistance by activation of cAMP–Epac signaling. Cell Metab. 13, 331–339 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yan J et al. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Mol. Cell. Biol. 33, 918–926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.De Souza CT et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 146, 4192–4199 (2005). [DOI] [PubMed] [Google Scholar]
- 100.Thaler JP et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 122, 153–162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Posey KA et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab 296, E1003–E1012 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang X et al. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Romanatto T et al. Deletion of tumor necrosis factor-α receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J. Biol. Chem. 284, 36213–36222 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 104.Milanski M et al. Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes 61, 1455–1462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ozcan L et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 9, 35–51 (2009). [DOI] [PubMed] [Google Scholar]
- 106.Hosoi T et al. Endoplasmic reticulum stress induces leptin resistance. Mol. Pharmacol. 74, 1610–1619 (2008). [DOI] [PubMed] [Google Scholar]
- 107.Liu J, Lee J, Salazar Hernandez MA, Mazitschek R & Ozcan U Treatment of obesity with celastrol. Cell 161, 999–1011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Williams KW et al. Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 20, 471–482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Perlmutter DH Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 52, 832–836 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Carducci MA et al. A Phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin. Cancer Res. 7, 3047–3055 (2001). [PubMed] [Google Scholar]
- 111.Ma X et al. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1–PGC1α transcriptional axis. Cell Metab. 22, 695–708 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Contreras C et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 9, 366–377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Contreras C et al. Effects of neonatal programming on hypothalamic mechanisms controlling energy balance. Horm. Metab. Res. 45, 935–944 (2013). [DOI] [PubMed] [Google Scholar]
- 114.Duque-Guimaraes DE & Ozanne SE Nutritional programming of insulin resistance: causes and consequences. Trends Endocrinol. Metab. 24, 525–535 (2013). [DOI] [PubMed] [Google Scholar]
- 115.Lopez M et al. A possible role of neuropeptide Y, agouti-related protein and leptin receptor isoforms in hypothalamic programming by perinatal feeding in the rat. Diabetologia 48, 140–148 (2005). [DOI] [PubMed] [Google Scholar]
- 116.Lopez M et al. Perinatal overfeeding in rats results in increased levels of plasma leptin but unchanged cerebrospinal leptin in adulthood. Int. J. Obes. 31, 371–377 (2007). [DOI] [PubMed] [Google Scholar]
- 117.Lucas A Programming by early nutrition: an experimental approach. J. Nutr. 128, 401S–406S (1998). [DOI] [PubMed] [Google Scholar]
- 118.Morris MJ, Velkoska E & Cole TJ Central and peripheral contributions to obesity-associated hypertension: impact of early overnourishment. Exp. Physiol. 90, 697–702 (2005). [DOI] [PubMed] [Google Scholar]
- 119.Davidowa H & Plagemann A Different responses of ventromedial hypothalamic neurons to leptin in normal and early postnatally overfed rats. Neurosci. Lett. 293, 21–24 (2000). [DOI] [PubMed] [Google Scholar]
- 120.Davidowa H & Plagemann A Decreased inhibition by leptin of hypothalamic arcuate neurons in neonatally overfed young rats. Neuroreport 11, 2795–2798 (2000). [DOI] [PubMed] [Google Scholar]
- 121.Kojima M et al. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402, 656–660 (1999). [DOI] [PubMed] [Google Scholar]
- 122.Tschop M, Smiley DL & Heiman ML Ghrelin induces adiposity in rodents. Nature 407, 908–913 (2000). [DOI] [PubMed] [Google Scholar]
- 123.Nakazato M et al. A role for ghrelin in the central regulation of feeding. Nature 409, 194–198 (2001). [DOI] [PubMed] [Google Scholar]
- 124.Muller TD et al. Ghrelin. Mol. Metab 4, 437–460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Seoane LM et al. Ghrelin elicits a marked stimulatory effect on GH secretion in freely-moving rats. Eur. J. Endocrinol. 143, R7–R9 (2000). [DOI] [PubMed] [Google Scholar]
- 126.Peino R et al. Ghrelin-induced growth hormone secretion in humans. Eur. J. Endocrinol. 143, R11–R14 (2000). [DOI] [PubMed] [Google Scholar]
- 127.Takaya K et al. Ghrelin strongly stimulates growth hormone release in humans. J. Clin. Endocrinol. Metab. 85, 4908–4911 (2000). [DOI] [PubMed] [Google Scholar]
- 128.Wren AM et al. Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86, 5992 (2001). [DOI] [PubMed] [Google Scholar]
- 129.Theander-Carrillo C et al. Ghrelin action in the brain controls adipocyte metabolism. J. Clin. Invest. 116, 1983–1993 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.López M et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. 7, 389–399 (2008). [DOI] [PubMed] [Google Scholar]
- 131.Foster-Schubert KE & Cummings DE Emerging therapeutic strategies for obesity. Endocr. Rev. 27, 779–793 (2006). [DOI] [PubMed] [Google Scholar]
- 132.Zorrilla EP et al. Vaccination against weight gain. Proc. Natl Acad. Sci. USA 103, 13226–13231 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Muller TD, Perez-Tilve D, Tong J, Pfluger PT & Tschop MH Ghrelin and its potential in the treatment of eating/wasting disorders and cachexia. J. Cachexia Sarcopenia Muscle 1, 159–167 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lainscak M, von Haehling S, Doehner W & Anker SD The obesity paradox in chronic disease: facts and numbers. J. Cachexia Sarcopenia Muscle 3, 1–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.von Haehling S, Morley JE & Anker SD From muscle wasting to sarcopenia and myopenia: update 2012. J. Cachexia Sarcopenia Muscle 3, 213–217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gutierrez JA et al. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc. Natl Acad. Sci. USA 105, 6320–6325 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yang J, Brown MS, Liang G, Grishin NV & Goldstein JL Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132, 387–396 (2008). [DOI] [PubMed] [Google Scholar]
- 138.Gonzalez CR, Vazquez MJ, López M & Dieguez C Influence of chronic undernutrition and leptin on GOAT mRNA levels in rat stomach mucosa. J. Mol. Endocrinol. 41, 415–421 (2008). [DOI] [PubMed] [Google Scholar]
- 139.Kirchner H et al. GOAT links dietary lipids with the endocrine control of energy balance. Nat. Med. 15, 741–745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Delhanty PJ, Neggers SJ & van der Lely AJ Mechanisms in endocrinology: ghrelin: the differences between acyl- and des-acyl ghrelin. Eur. J. Endocrinol. 167, 601–608 (2012). [DOI] [PubMed] [Google Scholar]
- 141.Delhanty PJ, Neggers SJ & van der Lely AJ Should we consider des-acyl ghrelin as a separate hormone and if so, what does it do? Front. Horm. Res. 42, 163–174 (2014). [DOI] [PubMed] [Google Scholar]
- 142.Toshinai K et al. Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology 147, 2306–2314 (2006). [DOI] [PubMed] [Google Scholar]
- 143.Heppner KM et al. Both acyl and des-acyl ghrelin regulate adiposity and glucose metabolism via central nervous system ghrelin receptors. Diabetes 63, 122–131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang JV et al. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science 310, 996–999 (2005). [DOI] [PubMed] [Google Scholar]
- 145.Seoane LM, Al Massadi O, Pazos Y, Pagotto U & Casanueva FF Central obestatin administration does not modify either spontaneous or ghrelin-induced food intake in rats. J. Endocrinol. Invest. 29, RC13–RC15 (2006). [DOI] [PubMed] [Google Scholar]
- 146.Nogueiras R et al. Effects of obestatin on energy balance and growth hormone secretion in rodents. Endocrinology 148, 21–26 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Gourcerol G, St Pierre DH & Tache Y Lack of obestatin effects on food intake: should obestatin be renamed ghrelin-associated peptide (GAP)? Regul. Pept. 141, 1–7 (2007). [DOI] [PubMed] [Google Scholar]
- 148.Kobelt P et al. Peripheral obestatin has no effect on feeding behavior and brain Fos expression in rodents. Peptides 29, 1018–1027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tschop M et al. Post-prandial decrease of circulating human ghrelin levels. J. Endocrinol. Invest. 24, RC19–RC21 (2001). [DOI] [PubMed] [Google Scholar]
- 150.Cummings DE et al. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50, 1714–1719 (2001). [DOI] [PubMed] [Google Scholar]
- 151.Drazen DL, Vahl TP, D’Alessio DA, Seeley RJ & Woods SC Effects of a fixed meal pattern on ghrelin secretion: evidence for a learned response independent of nutrient status. Endocrinology 147, 23–30 (2006). [DOI] [PubMed] [Google Scholar]
- 152.Seoane LM et al. Sensory stimuli directly acting at the central nervous system regulate gastric ghrelin secretion. an ex vivo organ culture study. Endocrinology 148, 3998–4006 (2007). [DOI] [PubMed] [Google Scholar]
- 153.Briggs DI & Andrews ZB Metabolic status regulates ghrelin function on energy homeostasis. Neuroendocrinology 93, 48–57 (2011). [DOI] [PubMed] [Google Scholar]
- 154.Zigman JM, Bouret SG & Andrews ZB Obesity impairs the action of the neuroendocrine ghrelin system. Trends Endocrinol. Metab. 27, 54–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tschop M et al. Circulating ghrelin levels are decreased in human obesity. Diabetes 50, 707–709 (2001). [DOI] [PubMed] [Google Scholar]
- 156.Otto B et al. Weight gain decreases elevated plasma ghrelin concentrations of patients with anorexia nervosa. Eur. J. Endocrinol. 145, 669–673 (2001). [PubMed] [Google Scholar]
- 157.Otto B et al. Postprandial ghrelin release in anorectic patients before and after weight gain. Psychoneuroendocrinology 30, 577–581 (2005). [DOI] [PubMed] [Google Scholar]
- 158.Cummings DE et al. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat. Med. 8, 643–644 (2002). [DOI] [PubMed] [Google Scholar]
- 159.DelParigi A et al. High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader–Willi syndrome. J. Clin. Endocrinol. Metab. 87, 5461–5464 (2002). [DOI] [PubMed] [Google Scholar]
- 160.Feigerlová E et al. Hyperghrelinemia precedes obesity in Prader–Willi syndrome. J. Clin. Endocrinol. Metab. 93, 2800–2805 (2008). [DOI] [PubMed] [Google Scholar]
- 161.Kweh FA et al. Hyperghrelinemia in Prader–Willi syndrome begins in early infancy long before the onset of hyperphagia. Am. J. Med. Genet. A 167A, 69–79 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.De Waele K et al. Long-acting octreotide treatment causes a sustained decrease in ghrelin concentrations but does not affect weight, behaviour and appetite in subjects with Prader–Willi syndrome. Eur. J. Endocrinol. 159, 381–388 (2008). [DOI] [PubMed] [Google Scholar]
- 163.Neary NM et al. Ghrelin increases energy intake in cancer patients with impaired appetite: acute, randomized, placebo-controlled trial. J. Clin. Endocrinol. Metab. 89, 2832–2836 (2004). [DOI] [PubMed] [Google Scholar]
- 164.Schmid DA et al. Ghrelin stimulates appetite, imagination of food, GH, ACTH, and cortisol, but does not affect leptin in normal controls. Neuropsychopharmacology 30, 1187–1192 (2005). [DOI] [PubMed] [Google Scholar]
- 165.Druce MR et al. Ghrelin increases food intake in obese as well as lean subjects. Int. J. Obes. Relat. Metab. Disord. 29, 1130–1136 (2005). [DOI] [PubMed] [Google Scholar]
- 166.Druce MR et al. Subcutaneous administration of ghrelin stimulates energy intake in healthy lean human volunteers. Int. J. Obes. 30, 293–296 (2005). [DOI] [PubMed] [Google Scholar]
- 167.Liu J et al. Novel ghrelin assays provide evidence for independent regulation of ghrelin acylation and secretion in healthy young men. J. Clin. Endocrinol. Metab. 93, 1980–1987 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sun Y, Ahmed S & Smith RG Deletion of ghrelin impairs neither growth nor appetite. Mol. Cell. Biol. 23, 7973–7981 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pfluger PT et al. Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am. J. Physiol. Gastrointest. Liver Physiol 294, G610–G618 (2008). [DOI] [PubMed] [Google Scholar]
- 170.McFarlane MR, Brown MS, Goldstein JL & Zhao TJ Induced ablation of ghrelin cells in adult mice does not decrease food intake, body weight, or response to high-fat diet. Cell Metab. 20, 54–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nishi Y et al. Ingested medium-chain fatty acids are directly utilized for the acyl-modification of ghrelin. Endocrinology 146, 2255–2264 (2005). [DOI] [PubMed] [Google Scholar]
- 172.Sangiao-Alvarellos S et al. Central ghrelin regulates peripheral lipid metabolism in a growth hormone-independent fashion. Endocrinology 150, 4562–4574 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Perez-Tilve D et al. Ghrelin-induced adiposity is independent of orexigenic effects. FASEB J. 25, 2814–2822 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Andrews ZB et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 454, 846–851 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gao S, Casals N, Keung W, Moran TH & Lopaschuk GD Differential effects of central ghrelin on fatty acid metabolism in hypothalamic ventral medial and arcuate nuclei. Physiol. Behav. 118, 165–170 (2013). [DOI] [PubMed] [Google Scholar]
- 176.Al MO et al. Review of novel aspects of the regulation of ghrelin secretion. Curr. Drug Metab. 15, 398–413 (2014). [DOI] [PubMed] [Google Scholar]
- 177.Sun Y, Wang P, Zheng H & Smith RG Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc. Natl Acad. Sci. USA 101, 4679–4684 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Guan XM et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res. Mol. Brain Res. 48, 23–29 (1997). [DOI] [PubMed] [Google Scholar]
- 179.Tannenbaum GS, Lapointe M, Beaudet A & Howard AD Expression of growth hormone secretagogue-receptors by growth hormone-releasing hormone neurons in the mediobasal hypothalamus. Endocrinology 139, 4420–4423 (1998). [DOI] [PubMed] [Google Scholar]
- 180.Willesen MG, Kristensen P & Romer J Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 70, 306–316 (1999). [DOI] [PubMed] [Google Scholar]
- 181.Nogueiras R et al. Regulation of growth hormone secretagogue receptor gene expression in the arcuate nuclei of the rat by leptin and ghrelin. Diabetes 53, 2552–2558 (2004). [DOI] [PubMed] [Google Scholar]
- 182.Garcia A, Alvarez CV, Smith RG & Dieguez C Regulation of pit-1 expression by ghrelin and ghrp-6 through the gh secretagogue receptor. Mol. Endocrinol. 15, 1484–1495 (2001). [DOI] [PubMed] [Google Scholar]
- 183.van der Lely AJ, Tschop M, Heiman ML & Ghigo E Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr. Rev. 25, 426–457 (2004). [DOI] [PubMed] [Google Scholar]
- 184.Anderson KA et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 7, 377–388 (2008). [DOI] [PubMed] [Google Scholar]
- 185.Lage R et al. Ghrelin effects on neuropeptides in the rat hypothalamus depend on fatty acid metabolism actions on BSX but not on gender. FASEB J. 24, 2670–2679 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Varela L et al. Ghrelin and lipid metabolism: key partners in energy balance. J. Mol. Endocrinol. 46, R43–R63 (2011). [DOI] [PubMed] [Google Scholar]
- 187.López M, Nogueiras R, Tena-Sempere M & Dieguez C Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat. Rev. Endocrinol. 12, 421–432 (2016). [DOI] [PubMed] [Google Scholar]
- 188.Dietrich MO et al. Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: roles for Sirt1 in neuronal firing and synaptic plasticity. J. Neurosci. 30, 11815–11825 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Velasquez DA et al. The central Sirtuin 1/p53 pathway is essential for the orexigenic action of ghrelin. Diabetes 60, 1177–1185 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Andersson U et al. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 279, 12005–12008 (2004). [DOI] [PubMed] [Google Scholar]
- 191.Kola B et al. The orexigenic effect of ghrelin is mediated through central activation of the endogenous cannabinoid system. PLoS ONE 3, e1797 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sangiao-Alvarellos S et al. Influence of ghrelin and GH deficiency on AMPK and hypothalamic lipid metabolism. J. Neuroendocrinol. 22, 543–556 (2010). [DOI] [PubMed] [Google Scholar]
- 193.Yavari A et al. Chronic activation of γ2 AMPK induces obesity and reduces β cell function. Cell Metab. 23, 821–836 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sierra AY et al. CPT1C is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 283, 6878–6885 (2008). [DOI] [PubMed] [Google Scholar]
- 195.Ramírez S et al. Hypothalamic ceramide levels regulated by CPT1C mediate the orexigenic effect of ghrelin. Diabetes 62, 2329–2337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Martins L et al. Hypothalamic mTOR signaling mediates the orexigenic action of ghrelin. PLoS ONE 7, e46923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Stevanovic D et al. Ghrelin-induced food intake and adiposity depend on central mTORC1/S6K1 signaling. Mol. Cell. Endocrinol. 381, 280–290 (2013). [DOI] [PubMed] [Google Scholar]
- 198.Martinez de Morentin PB et al. Hypothalamic mTOR: the rookie energy sensor. Curr. Mol. Med. 14, 3–21 (2014). [DOI] [PubMed] [Google Scholar]
- 199.Sakkou M et al. A role for brain-specific homeobox factor Bsx in the control of hyperphagia and locomotory behavior. Cell Metab. 5, 450–463 (2007). [DOI] [PubMed] [Google Scholar]
- 200.Seoane LM et al. Agouti-related peptide, neuropeptide Y, and somatostatin-producing neurons are targets for ghrelin actions in the rat hypothalamus. Endocrinology 144, 544–551 (2003). [DOI] [PubMed] [Google Scholar]
- 201.Nogueiras R et al. Bsx, a novel hypothalamic factor linking feeding with locomotor activity, is regulated by energy availability. Endocrinology 149, 3009–3015 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yang Y, Atasoy D, Su HH & Sternson SM Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 146, 992–1003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kohno D, Gao HZ, Muroya S, Kikuyama S & Yada T Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes 52, 948–956 (2003). [DOI] [PubMed] [Google Scholar]
- 204.Cowley MA et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649–661 (2003). [DOI] [PubMed] [Google Scholar]
- 205.Romero-Pico A et al. Hypothalamic κ-opioid receptor modulates the orexigenic effect of ghrelin. Neuropsychopharmacology 38, 1296–1307 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kola B et al. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J. Biol. Chem. 280, 25196–25201 (2005). [DOI] [PubMed] [Google Scholar]
- 207.Lim CT et al. Ghrelin and cannabinoids require the ghrelin receptor to affect cellular energy metabolism. Mol. Cell. Endocrinol. 365, 303–308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kola B et al. The CB1 receptor mediates the peripheral effects of ghrelin on AMPK activity but not on growth hormone release. FASEB J. 27, 5112–5121(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Olszewski PK, Grace MK, Billington CJ & Levine AS Hypothalamic paraventricular injections of ghrelin: effect on feeding and c-Fos immunoreactivity. Peptides 24, 919–923 (2003). [DOI] [PubMed] [Google Scholar]
- 210.Olszewski PK, Billington CJ, Grace MK & Levine AS α-Melanocyte stimulating hormone and ghrelin: central interaction in feeding control. Peptides 28, 2084–2089 (2007). [DOI] [PubMed] [Google Scholar]
- 211.Abizaid A et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Invest. 116, 3229–3239 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Romero-Pico A et al. Central manipulation of dopamine receptors attenuates the orexigenic action of ghrelin. Psychopharmacology 229, 275–283 (2013). [DOI] [PubMed] [Google Scholar]
- 213.Hewson AK, Tung LY, Connell DW, Tookman L & Dickson SL The rat arcuate nucleus integrates peripheral signals provided by leptin, insulin, and a ghrelin mimetic. Diabetes 51, 3412–3419 (2002). [DOI] [PubMed] [Google Scholar]
- 214.Kohno D et al. Leptin suppresses ghrelin-induced activation of neuropeptide Y neurons in the arcuate nucleus via phosphatidylinositol 3-kinase- and phosphodiesterase 3-mediated pathway. Endocrinology 148, 2251–2263 (2007). [DOI] [PubMed] [Google Scholar]
- 215.Scott V, McDade DM & Luckman SM Rapid changes in the sensitivity of arcuate nucleus neurons to central ghrelin in relation to feeding status. Physiol. Behav. 90, 180–185 (2007). [DOI] [PubMed] [Google Scholar]
- 216.Briggs DI et al. Calorie-restricted weight loss reverses high-fat diet-induced ghrelin resistance, which contributes to rebound weight gain in a ghrelin-dependent manner. Endocrinology 154, 709–717 (2013). [DOI] [PubMed] [Google Scholar]
- 217.Perreault M et al. Resistance to the orexigenic effect of ghrelin in dietary-induced obesity in mice: reversal upon weight loss. Int. J. Obes. Relat. Metab. Disord. 28, 879–885 (2004). [DOI] [PubMed] [Google Scholar]
- 218.English PJ, Ghatei MA, Malik IA, Bloom SR & Wilding JP Food fails to suppress ghrelin levels in obese humans. J. Clin. Endocrinol. Metab. 87, 2984 (2002). [DOI] [PubMed] [Google Scholar]
- 219.Uchida A et al. Altered ghrelin secretion in mice in response to diet-induced obesity and Roux-en-Y gastric bypass. Mol. Metab. 3, 717–730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Mani BK, Osborne-Lawrence S, Vijayaraghavan P, Hepler C & Zigman JM β1-adrenergic receptor deficiency in ghrelin-expressing cells causes hypoglycemia in susceptible individuals. J. Clin. Invest. 126, 3467–3478 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Banks WA, Burney BO & Robinson SM Effects of triglycerides, obesity, and starvation on ghrelin transport across the blood–brain barrier. Peptides 29, 2061–2065 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Briggs DI, Enriori PJ, Lemus MB, Cowley MA & Andrews ZB Diet-induced obesity causes ghrelin resistance in arcuate NPY/AgRP neurons. Endocrinology 151, 4745–4755 (2010). [DOI] [PubMed] [Google Scholar]
- 223.Gardiner JV et al. The hyperphagic effect of ghrelin is inhibited in mice by a diet high in fat. Gastroenterology 138, 2468–2476 (2010). [DOI] [PubMed] [Google Scholar]
- 224.Briggs DI et al. Evidence that diet-induced hyperleptinemia, but not hypothalamic gliosis, causes ghrelin resistance in NPY/AgRP neurons of male mice. Endocrinology 155, 2411–2422 (2014). [DOI] [PubMed] [Google Scholar]
- 225.Steculorum SM et al. Neonatal ghrelin programs development of hypothalamic feeding circuits. J. Clin. Invest. 125, 846–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Collden G et al. Neonatal overnutrition causes early alterations in the central response to peripheral ghrelin. Mol. Metab. 4, 15–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Coll AP, Farooqi IS & O’Rahilly S The hormonal control of food intake. Cell 129, 251–262 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Williams KW & Elmquist JK From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat. Neurosci. 15, 1350–1355 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Perello M et al. Functional implications of limited leptin receptor and ghrelin receptor coexpression in the brain. J. Comp. Neurol. 520, 281–294 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Stephens TW et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature 377, 530–532 (1995). [DOI] [PubMed] [Google Scholar]
- 231.Hahn TM, Breininger JF, Baskin DG & Schwartz MW Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1, 271–272 (1998). [DOI] [PubMed] [Google Scholar]
- 232.Elias CF et al. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron 23, 775–786 (1999). [DOI] [PubMed] [Google Scholar]
- 233.de Morentin PB & López M “Mens sana in corpore sano”: exercise and hypothalamic ER stress. PLoS Biol. 8, e1000464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Naznin F et al. Diet-induced obesity causes peripheral and central ghrelin resistance by promoting inflammation. J. Endocrinol. 226, 81–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Martin TL et al. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J. Biol. Chem. 281, 18933–18941 (2006). [DOI] [PubMed] [Google Scholar]
- 236.Dagon Y et al. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 16, 104–112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Lockie SH, Dinan T, Lawrence AJ, Spencer SJ & Andrews ZB Diet-induced obesity causes ghrelin resistance in reward processing tasks. Psychoneuroendocrinology 62, 114–120 (2015). [DOI] [PubMed] [Google Scholar]
- 238.Finger BC, Dinan TG & Cryan JF Diet-induced obesity blunts the behavioural effects of ghrelin: studies in a mouse-progressive ratio task. Psychopharmacology 220, 173–181 (2012). [DOI] [PubMed] [Google Scholar]
- 239.Wang W & Tao YX Ghrelin receptor mutations and human obesity. Prog. Mol. Biol. Transl Sci. 140, 131–150 (2016). [DOI] [PubMed] [Google Scholar]
- 240.Pantel J et al. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J. Clin. Invest. 116, 760–768 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Pantel J et al. Recessive isolated growth hormone deficiency and mutations in the ghrelin receptor. J. Clin. Endocrinol. Metab. 94, 4334–4341 (2009). [DOI] [PubMed] [Google Scholar]
- 242.Liu G, Fortin JP, Beinborn M & Kopin AS Four missense mutations in the ghrelin receptor result in distinct pharmacological abnormalities. J. Pharmacol. Exp. Ther. 322, 1036–1043 (2007). [DOI] [PubMed] [Google Scholar]
- 243.Inoue H et al. Identification and functional analysis of novel human growth hormone secretagogue receptor (GHSR) gene mutations in Japanese subjects with short stature. J. Clin. Endocrinol. Metab. 96, E373–E378 (2011). [DOI] [PubMed] [Google Scholar]
- 244.Rahmouni K Obesity-associated hypertension: recent progress in deciphering the pathogenesis. Hypertension 64, 215–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]