

Abstract
The role of the gut microbiome in the development of renal stone diseases has not been well characterized. This study focused on the taxonomic and functional profiles of gut microbiomes according to the prevalence and incidence of nephrolithiasis. Stool samples from 915 Korean adults were collected at baseline. Participants were followed for a median of 4.0 years. We evaluated the biodiversity of the gut microbiota and taxonomic profiles associated with nephrolithiasis status, using 16S rRNA gene sequencing. Nephrolithiasis status was categorized into three groups: control (no-stone at both baseline and follow-up visits), incidental nephrolithiasis, and prevalent nephrolithiasis. Compared to the control and incidental nephrolithiasis, the prevalent nephrolithiasis showed a reduced evenness in alpha diversity. Nephrolithiasis was associated with a reduced abundance of some key taxa involved in short-chain fatty acid production. Moreover, the abundance of Bifidobacterium, which possess oxalate-degrading ability, was higher in the control. Conversely, there was no significant difference in the bacterial composition between the incidental and prevalent nephrolithiasis. In our study with repeated nephrolithiasis measurements, prevalent renal stones were associated with an altered gut microbiota composition compared to the control. Besides the known oxalate degradation pathway, other functional pathways inferred in this study require further investigation.
Subject terms: Renal calculi, Microbiology
Introduction
Nephrolithiasis, also known as kidney stone disease, is a common disease worldwide, with a prevalence of 5–10%. With a continuous increase in its prevalence and incidence, nephrolithiasis poses a considerable global health and economic burden1–3. Supersaturation of urine by constituents such as calcium, oxalate, or uric acid is the most widely accepted mechanism of nephrolithiasis4,5. However, its pathogenesis and risk factors are not yet fully understood.
Recently, contribution of the gut bacteria to the pathophysiological gastrointestinal-renal axis has received increasing attention. Gastrointestinal microbiota has been suggested to play a role in oxalate metabolism, one of major proportion of nephrolithiasis, possibly by affecting the intestinal absorption of oxalate and renal stone formation6–8. However, research on this topic has mainly focused on Oxalobacter formigenes that utilizes oxalate as a principal energy substrate9. Its existence in feces has been correlated with a diminished renal excretion of oxalate and a decreased risk of stone recurrence7. Multiple previous studies have failed to identify a significant role of O. formigenes in nephrolithiasis, because isolation of O. formigenes from feces is not feasible9. However, recent analytical methods, such as 16S ribosomal RNA gene sequencing and shotgun metagenomics, have facilitated delineation of the gastrointestinal microbiome. At present, only five human studies, which have shown inconsistent findings, have evaluated the association between nephrolithiasis and the gastrointestinal microbiome10–14. All five of these studies included patients with symptomatic renal stone disease and were limited by small sample sizes and cross-sectional associations with single-time measurements.
The present study aimed to examine the association between the prevalence and incidence of renal stones and the gut microbiota in a relatively large-scale study of 915 participants.
Methods
Study participantss
This prospective cohort study enrolled participants who underwent comprehensive annual or biennial examinations at the Kangbuk Samsung Hospital Healthcare Screening Center in South Korea15 between June 2014 and September 2014. A total of 1463 Korean adults aged 23 to 78 years who agreed to participate in this study and provided stool samples at Kangbuk Samsung Hospital Healthcare Screening Center16 were included. Fecal samples were obtained only at baseline. Abdominal ultrasonography is a routine part of a health checkup program.
Participants who met any of the exclusion criteria were not included in the analysis (Fig. 1). We excluded 548 participants based on the following criteria: missing data (n = 7); use of antibiotics within 6 weeks prior to enrollment or probiotics within 4 weeks prior to enrollment (n = 72); history of cancer (n = 52); current use of antacid medication (n = 32); use of diabetes medication (n = 53); history of liver cirrhosis or identification of liver cirrhosis on an ultrasonography (n = 3); gout (n = 24); chronic kidney disease (n = 9); and samples with less than 5000 sequences (see below for further details) (n = 90). As the changes in the renal stone status over time were the focus of this study, participants with no follow-up visits by December 31, 2017, were also excluded (n = 302). None of the patients had a history of stomach, colon, kidney, or bladder surgery. Some individuals met more than one of the exclusion criteria. In the final analysis, a total of 915 participants were included. The characteristics of the study participants and the selection process are provided in detail in the Supplementary Methods. Dietary consumption was assessed using a 103-item self-administered food frequency questionnaire (FFQ) developed for use in Korea17. This validated FFQ was designed to measure usual food consumption during the previous year. Data were collected during the health checkup.
Figure 1.
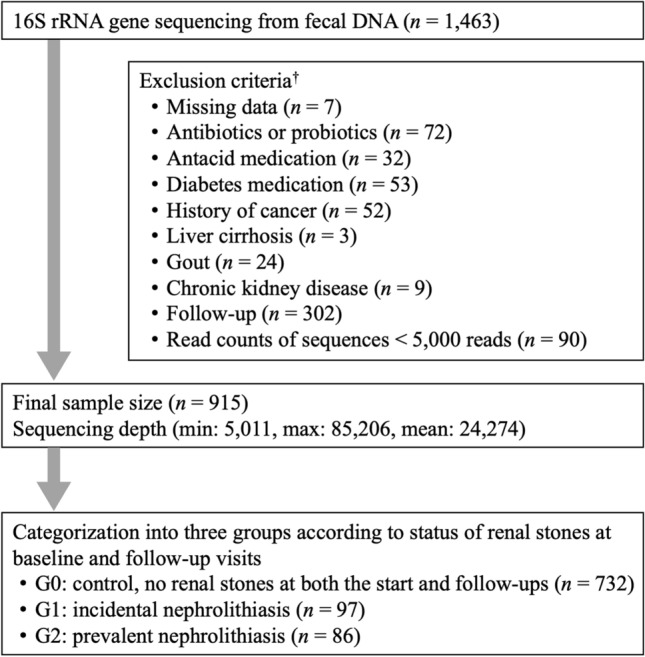
Inclusion and exclusion criteria for study participants. †Some individuals met several exclusion criteria.
The present prospective cohort study was conducted according to a protocol approved by the Institutional Review Board of Kangbuk Samsung Hospital (KBSMB 2013-01-245-12 and 2020-09-003). Written informed consent was obtained from all participants included in this study. Demographic, laboratory, and other clinical data, along with stool samples, were collected according to the ethical guidelines compliant with the Declaration of Helsinki.
Renal stone and group definition
Abdominal ultrasonography was performed during the first and follow-up visits by experienced radiologists who were blinded to the study aim using a Logic Q700 MR with a 3.5 MHz transducer (Logic Q700 MR; GE, Milwaukee, WI, USA; LOGIQ E9, GE, Madison, WI, USA). All participants underwent abdominal ultrasonography in the supine or right-lateral position for both kidneys. Nephrolithiasis was diagnosed when hyperechoic structures causing acoustic shadowing were observed in the collecting system on ultrasonography18. Based on the renal stone status at the initial and subsequent visits, patients were categorized into the following groups: G0, no renal stones (control), those without renal stones at the initial and subsequent visits; G1, incidental renal stones, those without renal stones initially but with renal stones at the follow-up visits; and G2, prevalent renal stones, those with renal stones at the beginning of the experiment. The median follow-up period was 4.0 years (interquartile range, 2.0–5.0 years, maximum 5.5 years); each participant was followed from baseline until the development of renal stones or the last follow-up examination, whichever came first.
Sample collection, DNA extraction, and 16S rRNA gene sequencing
Fresh fecal samples were collected from each volunteer, in a sterile container, immediately frozen at –20 °C, and stored at –70 °C within 24 h until further manipulation. Total DNA was extracted from the stool samples within 1 month of storage using the MOBio PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA) according to the manufacturer’s instructions.
To amplify and sequence the V3–V4 hypervariable region of the 16S rRNA gene, specific fusion primers were used (Illumina, San Diego, CA, USA). Libraries were pooled for sequencing, using the full complement of Nextera XT indices, and sequenced on the Illumina MiSeq platform (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions19.
Microbial profiling and statistical analysis
Demultiplexed sequences were processed using DADA2, which is a plugin of the QIIME2 package (version 2017.11, https://qiime2.org )20 and the low-quality regions of the sequences and chimeras were removed. After denoising with DADA2, amplicon sequence variants (ASVs) were generated and regarded as 100% operational taxonomic units. Then, we constructed a feature table containing the counts of each unique sequence in each sample. We filtered out features present in only one sample, based on the suspicion that they did not represent actual biological diversity, and were attributed to polymerase chain reaction or sequencing errors.
The biodiversity of the samples (⍺-diversity) was calculated using the number of ASVs observed in each sample, the Shannon index, which accounts for both evenness and richness, Faith’s phylogenetic diversity21, and Pielou’s evenness. As some of the variables were not normally distributed, the Kruskal–Wallis test was used to compare the differences among groups. The dissimilarity between samples (β-diversity) was calculated using the UniFrac distance22 to estimate dissimilarities among group members by incorporating the phylogenetic distances between ASVs. Unweighted and weighted UniFrac distances were calculated to determine the presence or absence and abundance of ASVs, respectively. Non-phylogenetic β-diversity indices, such as the Bray–Curtis dissimilarity index23 were also used for the abundance data. We used pairwise permutational multivariate analysis of variance with 999 random permutations to test the significance of the differences among groups. The microbial community composition was depicted using a principal coordinate analysis plot. All tests were corrected for multiple tests. Basic statistical analyses were performed using R Studio (version 1.3.1073; R Foundation for Statistical Computing, Vienna, Austria), and microbial diversity plots were plotted using the “ggplot2” R package (version 3.3.2).
Taxonomy was assigned to ASVs using a pre-trained naïve Bayes classifier and the q2-feature-classifier plugin against the Silva database (version r138.1) of the 16S rRNA sequence database in the QIIME2 package (version 2020.8, https://qiime2.org). To investigate any significant differences in the relative taxa abundances from the phylum to genus levels among groups, we used two statistical tools from R (version 4.0.2): analysis of composition of microbiomes (ANCOM, v2.1)24 and generalized linear models implemented in multivariate association with linear models (MaAsLin2). After adjusting for age, sex and BMI, we compared the abundance of taxa among G0, G1, and G2 in a pairwise manner. ANCOM compares the relative abundance of taxa among groups by the log-ratio of the abundance of each taxon to that of the remaining taxa, one at a time. The final significance was expressed in the empirical distribution of W at each taxonomic level. We used the taxa-wise false discovery rate (FDR) option with the significance level set to FDR < 0.05 to generate W statistics and a threshold of 0.6 for declaring a significant association. For the MaAsLin2 models, G0 was set as the reference group and compared with G1 and G2. The coefficient values and p-values that passed the significance threshold (Benjamini–Hochberg FDR p-value < 0.05) are represented in the MaAsLin2 analyses. Linear discriminant analysis effect size (LEfSe) was used to detect potential nephrolithiasis-specific bacterial markers25. LEfSE utilizes a two-step statistical analysis including the nonparametric Kruskal–Wallis sum-rank test to identify bacterial taxa, which differ significantly in abundance between groups. This is followed by linear discriminant analysis to estimate the effect size of each differentially abundant feature. Only taxa with a linear discriminant analysis score (log10) greater than three (p < 0.05) were considered significantly enriched.
Prediction of metagenome functions
For functional inferences of the microbial community, we conducted a Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) (v2.2.0-b)26 analysis of the ASVs according to the instructions published at https://github.com/picrust/picrust2/wiki. PICRUSt2 predictions were based on the gene family enzyme classification (EC) numbers (as of January 21, 2016). We generated a metabolic pathway database (MetaCyc) using pathway abundance predictions from the EC-based gene family predictions27. We also inferred Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways levels by using the last publicly available mapping of KEGG orthologs to pathways (which is from around 2011)28,29. The predicted functional pathways were compared among the groups using statistical analysis of taxonomic and functional profiles (STAMP) version 2.1.330. Statistical differences in the pathways were tested using Welch’s t-test with a Benjamini–Hochberg FDR correction (q < 0.05) to adjust for multiple tests.
Results
Participants demographics
Baseline characteristics of the enrolled participants according to renal stone status are shown in Table 1. Of the 915 participants (mean age: 40.21 years, men 64.70%), 732 were in no renal stone (G0) group, 97 in incidental renal stone (G1) group, and 86 in the prevalent renal stone (G2) group. The G2 group was the oldest on an average and had the highest prevalence of hypertension among the three groups. There were no significant differences in morbidities, including diabetes, obesity, metabolic syndrome, and nutritional intake (Supplementary Table S1) among the three groups.
Table 1.
Demographic and clinical characteristics of the study participants according to renal stone status.
| Characteristics | Overall | Stone category | P | ||
|---|---|---|---|---|---|
| G0 (No-stone) | G1 (Incidental stones) | G2 (Prevalent stones) | |||
| Number (male %)§ | 915 (64.7%) | 732 (64.34%) | 97 (67.01%) | 86 (65.12%) | 0.872 |
| Age at baseline (year)† | 40.21 (± 7.89) | 39.77 (± 7.79) | 40.89 (± 6.87) | 43.19 (± 9.16) | < 0.001 |
| BMI (kg/m2)† | 23.57 (± 3.08) | 23.56 (± 3.15) | 23.69 (± 3.14) | 23.50 (± 2.44) | 0.907 |
| Waist circumference (cm)† | 82.24 (± 8.80) | 82.17 (± 9.02) | 82.42 (± 8.52) | 82.58 (± 7.08) | 0.900 |
| Systolic BP (mmHg)† | 109.41 (± 13.45) | 109.26 (± 13.53) | 109.68 (± 14.41) | 110.38 (± 11.63) | 0.747 |
| Diastolic BP (mmHg)† | 70.97 (± 10.16) | 70.76 (± 10.14) | 71.92 (± 11.35) | 71.70 (± 8.89) | 0.453 |
| Glucose (mg/dL)† | 94.17 (± 11.94) | 93.91 (± 10.84) | 94.23 (± 16.38) | 96.33 (± 14.67) | 0.208 |
| Total cholesterol (mg/dL)† | 198.11 (± 32.97) | 197.95 (± 32.96) | 196.23 (± 32.55) | 201.56 (± 33.74) | 0.529 |
| LDL-C (mg/dL)† | 120.35 (± 29.71) | 120.21 (± 29.58) | 119.16 (± 31.43) | 122.85 (± 29.03) | 0.681 |
| HDL-C (mg/dL)† | 56.92 (± 14.10) | 56.78 (± 14.05) | 56.73 (± 14.77) | 58.35 (± 13.84) | 0.614 |
| Triglycerides (mg/dL)‡ | 100.00 (± 72.00) | 102.50 (± 73.00) | 95.00 (± 61.00) | 96.50 (± 64.00) | 0.912 |
| hsCRP (mg/dL)† | 0.91 (± 1.89) | 0.92 (± 1.79) | 0.82 (± 1.55) | 1.01 (± 2.90) | 0.807 |
| Current smoker (N, %)§ | 157 (17.16%) | 115 (15.71%) | 24 (24.74%) | 18 (20.93%) | 0.024 |
| Alcohol intake (> 20 mg; N, %)§ | 210 (22.95%) | 174 (22.75%) | 22 (22.68%) | 22 (25.58%) | 0.801 |
| Hypertension (N, %)§ | 107 (11.69%) | 77 (10.52%) | 12 (12.37%) | 18 (20.93%) | 0.017 |
| Diabetes (N, %)§ | 22 (2.64%) | 16 (2.19%) | 2 (2.06%) | 4 (4.65%) | 0.359 |
| Obesity (N, %)§ | 242 (26.45%) | 199 (27.19%) | 23 (23.71%) | 20 (23.26%) | 0.598 |
| Metabolic syndrome (N, %)§ | 115 (12.57%) | 92 (12.57%) | 12 (12.37%) | 11 (12.79%) | 0.996 |
BMI, body mass index; BP, blood pressure; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; and hsCRP, high-sensitivity C-reactive protein.
Participants were compared according to renal stone status, using an analysis of variance (ANOVA) for continuous variables or a χ2 test for categorical variables.
† Mean (SD).
‡ Median (interquartile range).
§ ≥ 20 g of ethanol per day.
Comparison of microbial diversity among the renal stone groups
After rarefying the feature tables to 5011 reads per sample, the rarefaction curves showed that all groups tended to plateau, indicating that the biodiversity was adequately covered by the applied sequencing depth (Supplementary Fig. S1). We found a significant decrease in the microbial evenness in G2 compared with that in G0 (pairwise Kruskal–Wallis, q = 0.041) and G1 (q = 0.041) (Kruskal–Wallis in all three groups, p = 0.040, Fig. 2D). However, considering the richness, the number of observed features (p = 0.498, Fig. 2A), phylogenetic diversity (p = 0.479, Fig. 2B), and Shannon’s index (p = 0.103, Fig. 2C) were not significantly different among the three groups, although all indices showed the lowest median in G2. For the β-diversity analysis, there were no significant differences in the fecal bacterial communities among the three groups (Supplementary Fig. S2).
Figure 2.

Alpha diversity of the microbiota among the renal stone groups. Boxplots representing alpha diversity include the observed features (A), Faith's phylogenetic diversity (B), Shannon's index (C), and Pielou's evenness (D) of the three groups. Statistics were calculated using a pairwise Kruskal–Wallis test among the three groups. The p-values were corrected using the Benjamini–Hochberg false discovery rate (FDR) (*p < 0.05). Boxes indicate the interquartile range (IQR) of the 25th to 75th percentiles. The median value is shown as a line within the box, and the whiskers extend to the most extreme value within 1.5 × IQR. Possible outliers are shown as dots. G0: no renal stones (control); G1: incidental renal stones; and G2: prevalent renal stones.
Abundance of fecal bacterial communities according to the renal stone groups
To better understand how the microbial community composition fluctuated with renal stone incidence, we examined which organisms were present at different taxonomic levels and their relative abundances.
We first used ANCOM, which has a low false-positive rate and allows covariate adjustment24,31, and then used MaAsLin to ensure reproducibility of the results (Table 2). After adjusting some of the covariates, including sex, age, and BMI, we found that G1 had a significantly reduced abundance of the phylum Actinobacteria [ANCOM, W = 14; MaAsLin, exponentiated (exp) coefficient = 0.74; p = 0.01], the classes Actinobacteria [W = 15; exp(coefficient) = 0.80; p = 0.03] and Coriobacteriia [W = 14; exp(coefficient) = 0.81; p = 0.01], the order Bifidobacteriales [W = 32; exp(coefficient) = 0.80; p = 0.03], and the family Bifidobacteriaceae [W = 54; exp(coefficient) = 0.80; p = 0.03] compared to G0. According to the ANCOM results (W = 14), the class Fusobacteriia was more abundant in G2 than in G0. In addition, the MaAsLin results showed that Fusobacteriia was more abundant in G1 [exp(coefficient) = 1.16; p = 0.04] and G2 [exp(coefficient) = 1.25; p = 4.6 × 10–3] than in G0. At the genus level, the relative abundances of Dorea, Incertae sedis, Bifidobacterium, uncultured genus belonging to Lachnospiraceae, and Faecalibacterium were reduced by 10% (coefficient = –0.11) to 22% (coefficient = –0.25) in G1 compared to those in G0. We also found that the abundances of the genera Eubacterium eligens group, Dialister, Phascolarctobacterium, and Erysipelatoclostridium differed significantly between G0 and G2 (W > 170). The Eubacterium eligens group and Dialister were less abundant, while Phascolarctobacterium was more abundant in G2 than in G0. We observed that there were no significant differences in the composition of gut microbiota between G1 and G2. Notably, compared to G0, there were no shared significant taxa between G1 and G2, except for the class Fusobacteriia, in the MaAslin results. Figure 3 shows the relative abundance of the taxa represented in Table 2.
Table 2.
Comparison of the microbial relative abundances between pairwise groups according to renal stone status at the phylum, class, order, family, and genus levels.
| Taxonomic assignment | Wb (coefficientsc) | ||
|---|---|---|---|
| G0† vs. G1 | G0† vs. G2 | G1† vs. G2 | |
| p__Actinobacteriota | 14 (–0.30*) | 1 (–0.22) | 0 |
| p__Actinobacteriota; c__Actinobacteria | 15 (–0.22*) | 0 (–0.16) | 0 |
| p__Actinobacteriota; c__Coriobacteriia | 14 (–0.21*) | 0 (–0.15) | 0 |
| p__Fusobacteriota; c__Fusobacteria | 7 (0.15*) | 14 (0.22**) | 0 |
| p__Actinobacteriota; c__Actinobacteria; o__Bifidobacteriales | 32 (–0.23*) | 0 (–0.17) | 0 |
| p__Actinobacteriota; c__Actinobacteria; o__Bifidobacteriales; f__Bifidobacteriaceae | 54 (–0.23*) | 0 (–0.17) | 0 |
| p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae; g__[Eubacterium]_eligens_group | 0 (–0.05) | 230 (–0.33**) | 0 |
| p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae; g__Dorea | 210 (–0.25**) | 0 (–0.14) | 0 |
| p__Firmicutes; c__Clostridia; o__Oscillospirales; f__Ruminococcaceae; g__Incertae_Sedis | 206 (–0.25**) | 0 (0.03) | 0 |
| p__Firmicutes; c__Negativicutes; o__Veillonellales-Selenomonadales; f__Veillonellaceae; g__Dialister | 0 (0.05) | 201 (–0.28*) | 0 |
| p__Actinobacteriota; c__Actinobacteria; o__Bifidobacteriales; f__Bifidobacteriaceae; g__Bifidobacterium | 190 (–0.23*) | 0 (–0.17) | 0 |
| p__Firmicutes; c__Negativicutes; o__Acidaminococcales; f__Acidaminococcaceae; g__Phascolarctobacterium | 0 (0.13) | 176 (0.31*) | 0 |
| p__Firmicutes; c__Bacilli; o__Erysipelotrichales; f__Erysipelatoclostridiaceae; g__Erysipelatoclostridium | 0 (0.02) | 171 (0.20*) | 0 |
| p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae; g__uncultured | 170 (–0.22**) | 0 (0.00) | 0 |
| p__Firmicutes; c__Clostridia; o__Oscillospirales; f__Ruminococcaceae; g__Faecalibacterium | 159 (–0.11*) | 0 (–0.07) | 0 |
p__: phylum, c__: class, o__: order, f__: family, g__: genus, s__: species, G0: no renal stone group, G1: incidental renal stone group, G2: prevalent renal stone group.
aNumber of phyla, 15; classes, 23; orders, 50; families, 89; genera, 260.
bW = X for taxon k, then H0k is rejected X times. The W statistics for the significantly different taxa relative to over 60% of the other taxa at each taxon level are represented in bold.
cCoefficients for the log-transformed relative abundance of each taxon in the linear model adjusted for age, sex, and BMI using MaAsLin2 on pairwise comparisons between groups. * p < 0.05, ** p < 0.01.
†Group used as the reference group in the regression model.
Figure 3.

Bar plot showing the relative abundance of the taxa that differed significantly among the renal stone groups at the (A) phylum, (B) class, (C) order, (D) family, and (E) genus levels. Upper error bars represent the standard deviation. G0: no renal stones (control); G1: incidental renal stones; and G2: prevalent renal stones.
Based on the results of diversity and taxonomic analysis, we performed LEfSe analysis to detect potential bacterial markers that most likely explain the differences among the renal stones groups by coupling standard tests for statistical significance with additional tests encoding biological consistency and effect relevance25. Comparisons of G0 vs. G1 and G0 vs. G2 identified 10 and 11 taxa with linear discriminant analysis scores of > 3, respectively (Fig. 4). The LEfSe results confirmed the significant enrichment of Actinobacteria, Bifidobacteriales, Dorea spp., and Bifidobacterium spp. in G0 compared to that in G1 (Fig. 4A). We also found that Fusobacteria and Catenisphaera spp. were more abundant in G1 than in G0. A significantly higher abundance of the genera Phascolarctobacterium, Fusobacterium, and Micrococcus was noted in G2 than in G0 (Fig. 4B). The significantly higher abundance of the genera Eubacterium eligens group and Dialister in G0 than in G2 was also confirmed in the LEfSe results.
Figure 4.

Linear discriminant analysis effect size (LEfSe) analysis identified the most differential microbiota between G0 and G1 (A), and G0 and G2 (B). The LDA score also indicates the effective size and ranking of each differentially abundant taxon (LDA score > 3.0; alpha value p < 0.05). Plots were depicted using LEfSe from the Galaxy platform of the Huttenhower lab. G0: no renal stones (control); G1: incidental renal stones; and G2: prevalent renal stones.
Predicted metabolic pathways
For a better understanding of the function of renal stone-associated bacteria, we inferred the predictive pathways using the MetaCyc and KEGG databases of PICRUSt2. Figure 5 shows the statistically significant (FDR q < 0.05) and suggestive (FDR q < 0.25) metabolic pathways and how they contributed differently to each of the three groups according to renal stone status. Among the pathways, a MetaCyc pathway related to allantoin degradation IV (anaerobic) and three KEGG pathways related to styrene degradation, cyanoamino acid metabolism, and proteasome exhibited significant differences between G0 and G1, with decreased pathways in the G1 compared to those in G0 (Fig. 5A, B). A KEGG pathway related to the metabolism of xenobiotics by cytochrome P450 was significantly less abundant in G2 than in G0 (Fig. 5C). There was no statistically significant difference in the pathways between the G1 and G2 groups.
Figure 5.

Abundances of the predicted functional pathways were different among the renal stone groups. Extended error bar plots show the significantly different MetaCyc (A) and KEGG (B) pathways between G0 and G1. Metabolism of xenobiotics by cytochrome P450 of the KEGG pathway was less abundant in G2 compared to that in G0 (C). Bar plots on the left display the mean proportion of each pathway. Dot plots on the right show differences in the mean proportions between the two indicated groups using the p-value. In STAMP, differences in the abundances between the two groups were compared using White's non-parametric t-test. The p-values corrected using the Benjamini–Hochberg FDR are shown on the right. G0: no renal stones (control); G1: incidental renal stones; and G2: prevalent renal stones.
Discussion
In this study of 915 participants with repeated ultrasonography examinations, the incidental and prevalent stone groups showed an altered gut microbiota composition and functionality, compared with no stone group at both the initial and follow-up visits. While most previous studies comparing groups with and without nephrolithiasis have predominantly focused on O. formigenes, our findings indicate that other gut microbiota might be involved in nephrolithiasis risk. Moreover, our study differentiated incident from prevalent stones, with relatively large sample sizes in each group, and explored the differences in the gut microbiome and metabolic pathways.
Gut microbiome alpha-diversity has been linked to human health, with lower levels of diversity associated with several chronic diseases32. A reduction in diversity is called microbial imbalance or dysbiosis. We found that the gut microbiome of the prevalent stone groups showed lower evenness than those of the other groups, reflecting changes in the abundance of some specific taxa.
Despite the essential role of O. formigenes in oxalate formation, recent human studies using 16S or metagenomic techniques have reported no differences in the gut species between healthy control groups and kidney stone groups10–14. Our results were consistent with those of the previous reports that found significant differences in the abundances of other taxa as opposed to Oxalobacter between the control group and the renal stone group. We found that Bifidobacterium was more abundant in no stone group than in incidental stone group. Bifidobacterium is also known to effectively degrade oxalate, albeit somewhat less effectively (11–68%) than O. formigenes (98%)33. Tavasoli et al. suggested that although some Lactobacillus and Bifidobacterium strains have oxalate-degrading potential, they may not be among the major oxalate-degrading bacteria of the gut microbiome. Recently, Denburg et al. reported that oxalate-degrading bacterial taxa, including Eggerthella lenta and several Lactobacillus species, were decreased in kidney stone formers34.
We also found that the abundances of Dorea, Incertae sedis, and Faecalibacterium were lower in incidental stone group than in no stone group. Additionally, compared with no stone group, the incidental stone group showed high abundances of Fusobacteria, Phascolarctobacterium, and Erysipelatoclostridium, and lower abundances of Eubacterium eligens group and Dialister. The association between these taxa and kidney stones has been reported in previous studies10–12,14. These observations imply that the gastrointestinal-renal axis may not solely rely on a single player or a limited number of species11,14. Ticinesi et al.14 suggested that oxalate-degrading activity may be shared by various taxa, influencing each other in a complex metabolic network.
Interestingly, we observed a lower abundance of Faecalibacterium in incidental stone group than in no-stone group, while there was no difference in abundances between prevalent stone and no-stone groups. A notable reduction in Faecalibacterium prausnitzii abundance has been reported in kidney stone groups in previous studies11,14,35. This bacterium is a principal manufacturer of short-chain fatty acids (SCFAs) within the human intestinal microbiome11,14. Recently, Liu et al.36 reported on the relationship between the gut microbiota and SCFAs regarding nephrolithiasis. They found that the abundance of pathways involved in SCFA production was positively correlated with the level of bacteria in the gut microbiota in the no-stone control group, but not in groups with kidney stones, which indicated that the gut microbiota of no-stone controls may tend to produce more SCFAs than that of nephrolithiasis patients. Moreover, the proportion of renal crystals was reduced after the administration of acetate, propionate, or butyrate in a rat model, which indicated that SCFAs could effectively prevent the formation of renal calcium oxalate crystals36. On the other hand, Dialister, which is also an SCFA-producing bacterium, was only depleted in prevalent stone group. We observed that there were no bacteria that showed significant differences in both the incidental stone and prevalent stone groups, when compared to no-stone group, and were associated with either incidental stone or prevalent stone group. This may be due to differences in the lifestyles of the incidental stone and prevalent stone groups. It is possible that patients with prevalent renal stones can alter their lifestyle behaviors, which may affect the gut microbiota.
The potential gastrointestinal-renal axis in patients with nephrolithiasis was recently proposed by Ticinesi et al.37. These authors placed O. formigenes at the heart of a bacterial complex that was engaged in the breakdown of oxalate and avoidance of hyperoxaluria. In contrast to the more straightforward axial theory, which emphasizes the role of O. formigenes alone, these authors postulated the involvement of additional taxa that had oxalate-metabolizing properties, together with taxa that were triggered by the presence of oxalate and worked synergistically to break down salt, thus diminishing oxalate absorption.
Interestingly, the 16S data suggested that several metabolic pathways were related to renal stone formation. As allantoin is the final covert material created by uricase, allantoin degradation could be related to solubility. Therefore, allantoin degradation could increase uric acid contents38. Regarding styrene degradation based on microbiological activity, the citrate cycle can be affected39. Indeed, hypocitraturia or low urinary citrate excretion is a common feature in patients with nephrolithiasis. The pathway involved in the proteasome includes an interaction between calcium oxalate crystals and renal tubular epithelial cells40. Furthermore, the metabolism of xenobiotics by cytochrome P450 can be affected by the human gut microbiome, contributing to various diseases, including kidney stones41.
This study incorporated a large population and investigated the association between the gastrointestinal microbiome and nephrolithiasis. However, the relevance of these results to current or potential clinical practices is not immediately apparent. In view of the multifaceted nature of the gut microbiome, further research is necessary to demonstrate the effectiveness of interventions aimed at restricting the absorption of oxalate across the intestinal barrier. Our study did not incorporate 24 h urine analysis or stone chemical analysis. Information on stone size and specific stone type, determined by detailed image or composition analyses, was also unavailable. We used abdominal ultrasonography to diagnose nephrolithiasis, although unenhanced computed tomography (CT) is considered a more accurate reference method. A recent study comparing ultrasonography and CT reported a fair sensitivity of 70.0% and specificity of 94.4% for detecting nephrolithiasis42. Even though ultrasonography can be both appropriate and feasible in a large population study without radiation hazard, we cannot exclude misclassification of nephrolithiasis determined using ultrasonography. Furthermore, seasonal variations were not considered in this study. It is possible that participants in the incidental stone group had prior stone episodes but no stones on the baseline ultrasonography during the same season. Moreover, among the prevalent stone patients (n = 86), 76 (88.4%) had at least one follow-up visit, 50 (65.8%) were diagnosed with nephrolithiasis, and 26 (34.2%) had no stones detected at follow-up. Those with prevalent nephrolithiasis without stones at follow-up may represent a false positive result for nephrolithiasis or stone passage during follow-up. Unfortunately, detailed information on stone treatment or stone passage was not available, limiting our ability to differentiate between the two conditions. Additionally, subjects who had passed stones spontaneously and had no stones visible on ultrasound might have been misclassified as no stone group. Thus, no differentiation between symptomatic and asymptomatic stones and misclassification of stone formers might limit the implication of our findings. Another limitation of our study is that we only obtained the baseline fecal samples of participants without microbiome data at follow-up, which restricted our ability to examine the relationship between changes in the fecal microbiome and renal stone status over time. Further analysis including serial data of gut microbiota during follow-up periods is required to elucidate the longitudinal association between microbiome and renal stone changes over time. Although diet is a key factor in shaping the gut microbiota, the gut microbiome profile in individuals susceptible to renal calculi does not appear to be affected by diet in our study. Ticinesi et al.14 reported that dietary habits are apparently not involved in the nephrolithiasis-associated abnormalities of the gut microbiota composition, with the only possible exception being calcium intake. Moreover, additional elements may alter the constitution of the bacteria, which in turn may have a bearing on the pathophysiology of renal stone disease. Finally, our study was based on 16S rRNA gene sequencing, which provides limited information about bacterial genes and their functions. Whole metagenomic sequencing could expand our understanding of the strains associated with renal stones, their genes and functions, and metabolic pathways. Additionally, concordant analysis including the urinary and fecal microbiome could explain the relationship between the gut microbiome and nephrolithiasis more clearly43,44. Additional studies are required to discern the way in which the composition and purpose of the microbiome may impact therapy outcomes and influence renal stone disorders.
Compared with healthy controls, the incidental and prevalent renal stone groups exhibited an altered gut microbiota composition that could contribute towards nephrolithiasis physiopathology. Besides the known oxalate degradation pathway, other functional pathways inferred from taxonomic profiles have been suggested, which require further investigation in the future.
Supplementary Information
Acknowledgements
We are thankful for the computing resources provided by the Global Science experimental Data hub Center (GSDC) Project and the Korea Research Environment Open NETwork (KREONET) of the Korea Institute of Science and Technology Information (KISTI). The funding sources had no role in the study design, data analysis, interpretation of the results, or writing of the manuscript.
Author contributions
Study concept and design: J.H.K., Y.C., S.R. Acquisition of data: H.L.K., Y.C., S.R. Analysis and interpretation of data: H.N.K., D.Y., J.H.K. Drafting of the manuscript: H.N.K., J.H.K. Critical revision of the manuscript for important intellectual content: K.J.J., Y.-S.C., H.J.P. Statistical analysis: H.N.K., D.Y. Obtaining funding: H.N.K., J.H.K. Administrative, technical or material support: H.L.K. Supervision: J.H.K. Other: None.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2020R1A2C1012931) and by the Medical Research Funds from Kangbuk Samsung Hospital. This study was also supported by Soonchunhyang University Research Fund.
Data availability
The datasets in the current study are available from the corresponding author upon reasonable request, and the raw 16S rRNA gene sequence data can be found in a public repository: the Clinical & Omics Data Archive (CODA) at the Korea National Institute of Health (accession number R000635; http://coda.nih.go.kr/coda/coda/search/omics/genome/selectSearchOmicsGenomePop/R000635.do).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Han-Na Kim and Jae Heon Kim.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-07796-y.
References
- 1.Abufaraj M, et al. Prevalence and trends in kidney stone among adults in the USA: analyses of national health and nutrition examination survey 2007–2018 data. Eur. Urol. Focus. 2020 doi: 10.1016/j.euf.2020.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, et al. Epidemiology of urolithiasis in Asia. Asian J. Urol. 2018;5:205–214. doi: 10.1016/j.ajur.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan SR, et al. Kidney stones. Nat. Rev. Dis. Primers. 2016;2:16008. doi: 10.1038/nrdp.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ratkalkar VN, Kleinman JG. Mechanisms of stone formation. Clin. Rev. Bone Miner. Metab. 2011;9:187–197. doi: 10.1007/s12018-011-9104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang D, et al. Urinary stone composition analysis and clinical characterization of 1520 patients in central China. Sci. Rep. 2021;11:6467. doi: 10.1038/s41598-021-85723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robijn S, Hoppe B, Vervaet BA, D'Haese PC, Verhulst A. Hyperoxaluria: a gut-kidney axis? Kidney Int. 2011;80:1146–1158. doi: 10.1038/ki.2011.287. [DOI] [PubMed] [Google Scholar]
- 7.Siener R, et al. The role of Oxalobacter formigenes colonization in calcium oxalate stone disease. Kidney Int. 2013;83:1144–1149. doi: 10.1038/ki.2013.104. [DOI] [PubMed] [Google Scholar]
- 8.Sadaf H, Raza SI, Hassan SW. Role of gut microbiota against calcium oxalate. Microb. Pathog. 2017;109:287–291. doi: 10.1016/j.micpath.2017.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Siva S, et al. A critical analysis of the role of gut Oxalobacter formigenes in oxalate stone disease. BJU Int. 2009;103:18–21. doi: 10.1111/j.1464-410X.2008.08122.x. [DOI] [PubMed] [Google Scholar]
- 10.Stern JM, et al. Evidence for a distinct gut microbiome in kidney stone formers compared to non-stone formers. Urolithiasis. 2016;44:399–407. doi: 10.1007/s00240-016-0882-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suryavanshi MV, et al. Hyperoxaluria leads to dysbiosis and drives selective enrichment of oxalate metabolizing bacterial species in recurrent kidney stone endures. Sci. Rep. 2016;6:34712. doi: 10.1038/srep34712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang R, et al. 16S rRNA gene sequencing reveals altered composition of gut microbiota in individuals with kidney stones. Urolithiasis. 2018;46:503–514. doi: 10.1007/s00240-018-1037-y. [DOI] [PubMed] [Google Scholar]
- 13.Tavasoli S, et al. Association of intestinal oxalate-degrading bacteria with recurrent calcium kidney stone formation and hyperoxaluria: a case-control study. BJU Int. 2020;125:133–143. doi: 10.1111/bju.14840. [DOI] [PubMed] [Google Scholar]
- 14.Ticinesi A, et al. Understanding the gut-kidney axis in nephrolithiasis: an analysis of the gut microbiota composition and functionality of stone formers. Gut. 2018;67:2097–2106. doi: 10.1136/gutjnl-2017-315734. [DOI] [PubMed] [Google Scholar]
- 15.Chang Y, et al. Metabolically healthy obesity and development of chronic kidney disease: a cohort study. Ann. Intern. Med. 2016;164:305–312. doi: 10.7326/M15-1323. [DOI] [PubMed] [Google Scholar]
- 16.Kim HN, et al. Correlation between gut microbiota and personality in adults: a cross-sectional study. Brain Behav. Immun. 2018;69:374–385. doi: 10.1016/j.bbi.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Ahn Y, et al. Validation and reproducibility of food frequency questionnaire for Korean genome epidemiologic study. Eur. J. Clin. Nutr. 2007;61:1435–1441. doi: 10.1038/sj.ejcn.1602657. [DOI] [PubMed] [Google Scholar]
- 18.Schepens, D., Verswijvel, G., Kuypers, D. & Vanrenterghem, Y. Images in nephrology. Renal cortical nephrocalcinosis. Nephrol. Dialy. Transpl. Off. Publ. Eur. Dial. Transp. Assoc. Eur. Renal Assoc.15, 1080–1082, doi:10.1093/ndt/15.7.1080 (2000). [DOI] [PubMed]
- 19.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolyen E, et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018;2:e27295. doi: 10.7287/peerj.preprints.27295v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faith DP, Baker AM. Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol. Bioinform. Online. 2007;2:121–128. [PMC free article] [PubMed] [Google Scholar]
- 22.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bray JR, Curtis JT. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 1957;27:325–349. doi: 10.2307/1942268. [DOI] [Google Scholar]
- 24.Mandal S, et al. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015;26:27663. doi: 10.3402/mehd.v26.27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Douglas GM, et al. PICRUSt2: an improved and extensible approach for metagenome inference. BioRxiv. 2020 doi: 10.1101/672295. [DOI] [Google Scholar]
- 27.Caspi R, et al. The MetaCyc database of metabolic pathways and enzymes. Nucl. Acids Res. 2018;46:D633–D639. doi: 10.1093/nar/gkx935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucl. Acids Res. 2016;44:D457–462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiss S, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickard JM, Zeng MY, Caruso R, Nunez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017;279:70–89. doi: 10.1111/imr.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lieske JC. Probiotics for prevention of urinary stones. Ann. Transl. Med. 2017;5:29. doi: 10.21037/atm.2016.11.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denburg MR, et al. Perturbations of the gut microbiome and metabolome in children with calcium oxalate kidney stone disease. J. Am. Soc. Nephrol. 2020;31:1358–1369. doi: 10.1681/ASN.2019101131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stanford J, Charlton K, Stefoska-Needham A, Ibrahim R, Lambert K. The gut microbiota profile of adults with kidney disease and kidney stones: a systematic review of the literature. BMC Nephrol. 2020;21:215. doi: 10.1186/s12882-020-01805-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu, Y. et al. The relationship between gut microbiota and short chain fatty acids in the renal calcium oxalate stones disease. 34, 11200-11214 (2020). [DOI] [PubMed]
- 37.Ticinesi A, Nouvenne A, Meschi T. Gut microbiome and kidney stone disease: not just an Oxalobacter story. Kidney Int. 2019;96:25–27. doi: 10.1016/j.kint.2019.03.020. [DOI] [PubMed] [Google Scholar]
- 38.Wiederkehr MR, Moe OW. Uric acid nephrolithiasis: a systemic metabolic disorder. Clin. Rev. Bone Miner. Metab. 2011;9:207–217. doi: 10.1007/s12018-011-9106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Leary ND, O'Connor KE, Dobson AD. Biochemistry, genetics and physiology of microbial styrene degradation. FEMS Microbiol. Rev. 2002;26:403–417. doi: 10.1111/j.1574-6976.2002.tb00622.x. [DOI] [PubMed] [Google Scholar]
- 40.Chiangjong W, Sinchaikul S, Chen ST, Thongboonkerd V. Calcium oxalate dihydrate crystal induced changes in glycoproteome of distal renal tubular epithelial cells. Mol. Biosyst. 2011;7:1917–1925. doi: 10.1039/c1mb05052d. [DOI] [PubMed] [Google Scholar]
- 41.Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016;14:273–287. doi: 10.1038/nrmicro.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanno T, et al. The efficacy of ultrasonography for the detection of renal stone. Urology. 2014;84:285–288. doi: 10.1016/j.urology.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Dornbier RA, et al. The microbiome of calcium-based urinary stones. Urolithiasis. 2020;48:191–199. doi: 10.1007/s00240-019-01146-w. [DOI] [PubMed] [Google Scholar]
- 44.Zampini A, Nguyen AH, Rose E, Monga M, Miller AW. Defining dysbiosis in patients with urolithiasis. Sci. Rep. 2019;9:5425. doi: 10.1038/s41598-019-41977-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets in the current study are available from the corresponding author upon reasonable request, and the raw 16S rRNA gene sequence data can be found in a public repository: the Clinical & Omics Data Archive (CODA) at the Korea National Institute of Health (accession number R000635; http://coda.nih.go.kr/coda/coda/search/omics/genome/selectSearchOmicsGenomePop/R000635.do).