

Abstract
Platelet metabolism is linked to platelet hyper- and hypoactivity in numerous human diseases. Developing a detailed understanding of the link between metabolic shifts and platelet activation state is integral to improving human health. Here, we show the first application of isotopically nonstationary 13C metabolic flux analysis to quantitatively measure carbon fluxes in both resting and thrombin activated platelets. Metabolic flux analysis results show that resting platelets primarily metabolize glucose to lactate via glycolysis, while acetate is oxidized to fuel the tricarboxylic acid cycle. Upon activation with thrombin, a potent platelet agonist, platelets increase their uptake of glucose 3-fold. This results in an absolute increase in flux throughout central metabolism, but when compared to resting platelets they redistribute carbon dramatically. Activated platelets decrease relative flux to the oxidative pentose phosphate pathway and TCA cycle from glucose and increase relative flux to lactate. These results provide the first report of reaction-level carbon fluxes in platelets and allow us to distinguish metabolic fluxes with much higher resolution than previous studies.
Keywords: blood platelets, metabolic flux analysis, metabolomics, thrombin
INTRODUCTION
Platelets are anucleate cells that play a vital role in hemostasis, the physiological process of blood clot formation in response to vascular injury that occurs over the timescale of minutes. As such, much of what is known about platelet metabolism is derived from studies focused on signaling in response to acute exposure to agonists (Lepropre et al., 2018) and platelet interactions with other blood and vascular cells (Jaffe, 1987; Semple and Freedman, 2010). There have also been significant efforts in identifying the metabolic cause and effect of platelet storage lesion in transfusion medicine (Murphy, 1995; Murphy et al., 1992). Beyond hemostasis, platelets are increasingly recognized for their various roles in innate immunity, diabetes, hyperglycemia, atherosclerosis, kidney disease, and coronary artery disease (Burkhart et al., 2014; Kramer, 2014; van der Meijden and Heemskerk, 2019). Recent studies in these chronic disorders suggest that changes in platelet metabolism play a role in their pathophysiology and can potentially be used as a biomarker (Davizon-Castillo et al., 2019; McDowell et al., 2020).
Previous studies on platelet metabolism have deduced metabolic pathway trends as a result of storage (Paglia et al., 2014), old age (Davizon-Castillo et al., 2019), activation (Ravi et al., 2015), and in response to specific reaction or pathway inhibitors (Ju et al., 2012). Primarily, these studies have monitored metabolic markers that indicate changes in whole metabolic pathways. Such techniques include radiometric labels (Cohen and Wittels, 1970), respirometry (Sowton et al., 2018), and extracellular flux analysis (Fidler et al., 2017). For example, Seahorse extracellular flux (XF) analyzer measures oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), which are interpreted as indirect measurements of mitochondrial oxidative phosphorylation and glycolysis, respectively (Nayak et al., 2018). While informative, these measurements can be challenging to interpret, in part due to the existence of multiple acidification mechanisms and the technique’s reliance on sequentially administered inhibitors to probe the cellular utilization of glucose (Kholmukhamedov and Jobe, 2019). Metabolic pathways are complex, and attenuation of an individual reaction within a pathway does not necessarily result in attenuation of the whole pathway, further complicating the pursuit of interpreting extracellular flux measurements.
Metabolomics has been performed extensively on platelets during storage (Jóhannsson et al., 2018; Paglia et al., 2015; Paglia et al., 2014) and after chemical treatment (Abonnenc et al., 2016; Ju et al., 2012) to identify changes in metabolite pool sizes. While this technique can provide a global view of changes in metabolism, how metabolite pool sizes change remains unknown. Quantitative measurement of reaction fluxes is needed to identify how metabolism was rewired to lead to the changes in metabolite concentration. Isotope assisted metabolic flux analysis (13C-MFA) is the current gold standard to quantitatively measure carbon fluxes in a cell by tracking a stable isotopic tracer, in this case 13C, through metabolism (Crown and Antoniewicz, 2013b; Wiechert, 2001). Experimental data is combined with computational metabolic models to deconvolute the data and determine intracellular fluxes. Enzymes rearrange the atoms in metabolites, and 13C-MFA uses isotope labels to measure the rearrangements of carbon atoms; these labeling patterns, or isotopomers, can then be mathematically fit with established stoichiometric metabolic models and known atom transitions to obtain intracellular fluxes (Cheah and Young, 2018). This technique provides experimentally derived fluxes, as well as the ability to validate metabolic network models that include compartmentalization, parallel pathways, and cyclic reaction sets (Antoniewicz, 2015) and has been successfully implemented to measure fluxes in a variety of organisms including bacteria and yeast (Schwechheimer et al., 2018), plants (Dersch et al., 2016) and mammalian cells (Mueller and Heinzle, 2013).
Platelets present unique challenges for developing metabolic models and obtaining experimental 13C-MFA measurements. First, unlike other cells that 13C-MFA has been applied to, platelets are anuclear. This means they do not replicate and are unable to respond to environmental cues by altering mRNA abundance. Moreover, their lifetime is limited to 7–10 days both inside and outside of the body (Quach et al., 2018). Additionally, platelets are known to display unique phenotypes across storage lengths (Paglia et al., 2014), which potentially limit the timescale for experimental measurements, especially during steady state labeling experiments, which often require incubation with isotope tracers for several days (Dai and Locasale, 2016).
In this work, we describe the first application of isotopically nonstationary metabolic flux analysis (INST-MFA) to measure carbon fluxes in resting and activated platelets. To our knowledge, this is the first report using 13C-MFA to measure metabolic flux in an anuclear cell. We establish metabolic steady state in platelets, define a metabolic model to track atom transitions throughout platelet central metabolism, and quantitatively measure metabolic fluxes of resting and thrombin activated platelets.
RESULTS
Metabolic modeling suggests glucose and acetate tracers
The workflow for implementing INST-MFA in platelets requires experimental, analytical, and computational approaches (Figure 1). It is common to start on the computational model, which can aid in experimental design. The metabolic network of platelet central metabolism used is shown in Figure 2 and was constructed in the isotopomer network compartmental analysis (INCA) software, a MATLAB-based software package for INST-MFA (Young, 2014). The experimental tracers used for 13C-MFA determines the confidence of calculated flux results (Metallo et al., 2009), therefore care was taken during this step to select a mixture that produced unique transient labeling behavior and several isotopomers per metabolite that remained enriched over the course of the experiment. Furthermore, parallel labeling experiments with complementary labeled carbon substrates can improve flux resolution (Crown and Antoniewicz, 2013a). Constraints derived from the iAT-PLT-636 model (Thomas et al., 2015) were used with the INCA tracer simulation function to identify glucose and acetate as viable carbon sources for probing the glycolysis/pentose phosphate pathway and the tricarboxylic acid (TCA) cycle, respectively. Acetate is an oxidative fuel that enters the TCA cycle via acetyl-CoA (Gulliksson, 1993; Guppy et al., 1990). Simulations with a variety of readily available isotopic tracers revealed that mixtures of [1,2-13C2]glucose, [U-13C6]glucose, [1-13C]acetate, [2-13C]acetate, and their unlabeled counterparts provided favorable steady state labeling distributions (Supplemental Fig. 1).
Figure 1. Summary of the workflow for performing 13C-MFA in platelets.
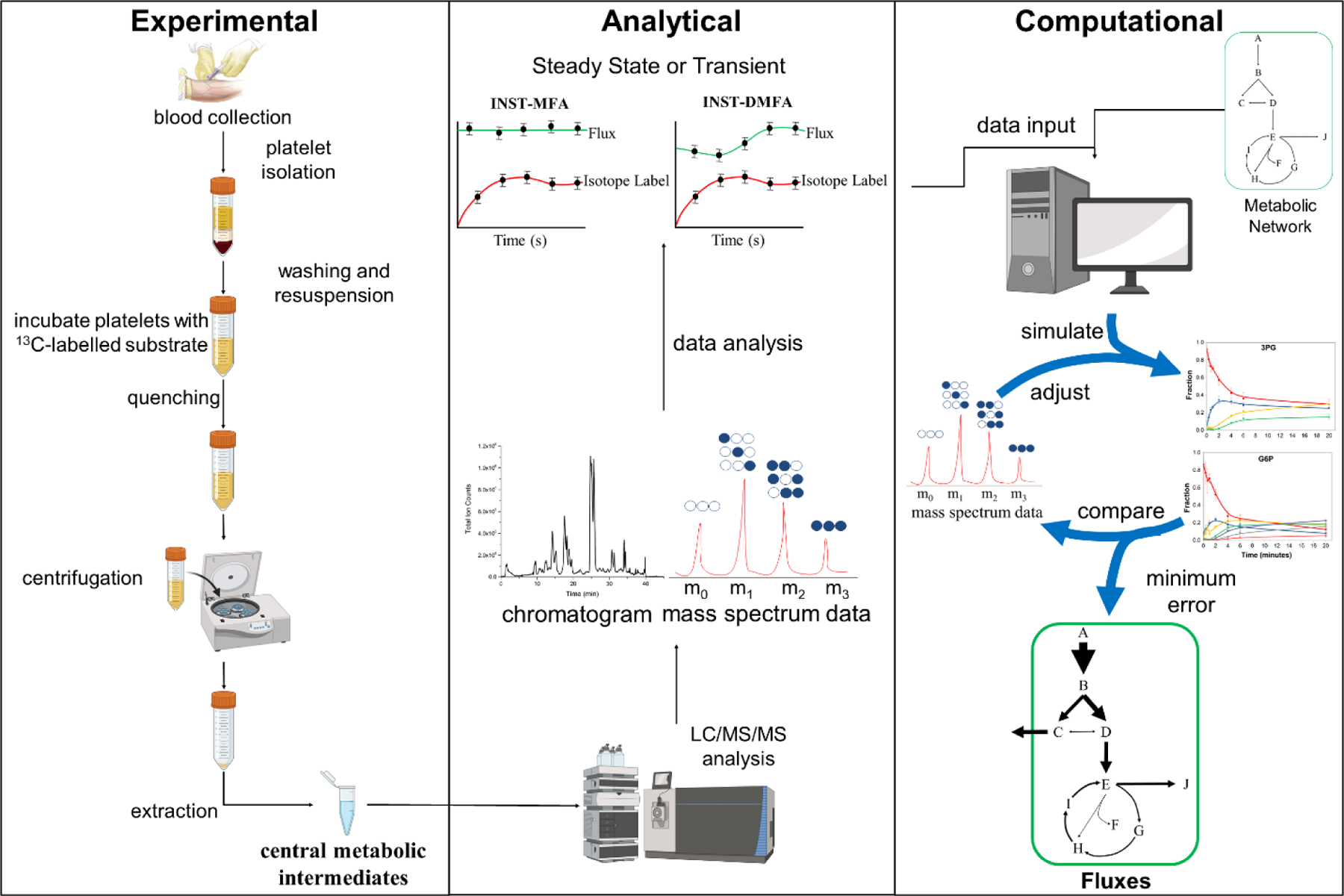
Washed platelets isolated from whole blood are incubated with 13C-glucose or 13C-acetate and then rapidly sampled, quenched, and intracellular metabolites extracted. The metabolite profile is analyzed using LC-MS/MS to obtain mass isotopomer distributions (MIDs). The analytical data is then paired with the modeled reaction network and respective atom transitions; fluxes are determined through iterative parameter adjustment to minimize the error between the simulated and measured isotopomer profiles. Figure adapted from (Sake et al., 2019). [please use color for figure in print]
Figure 2. Platelet central metabolic network.
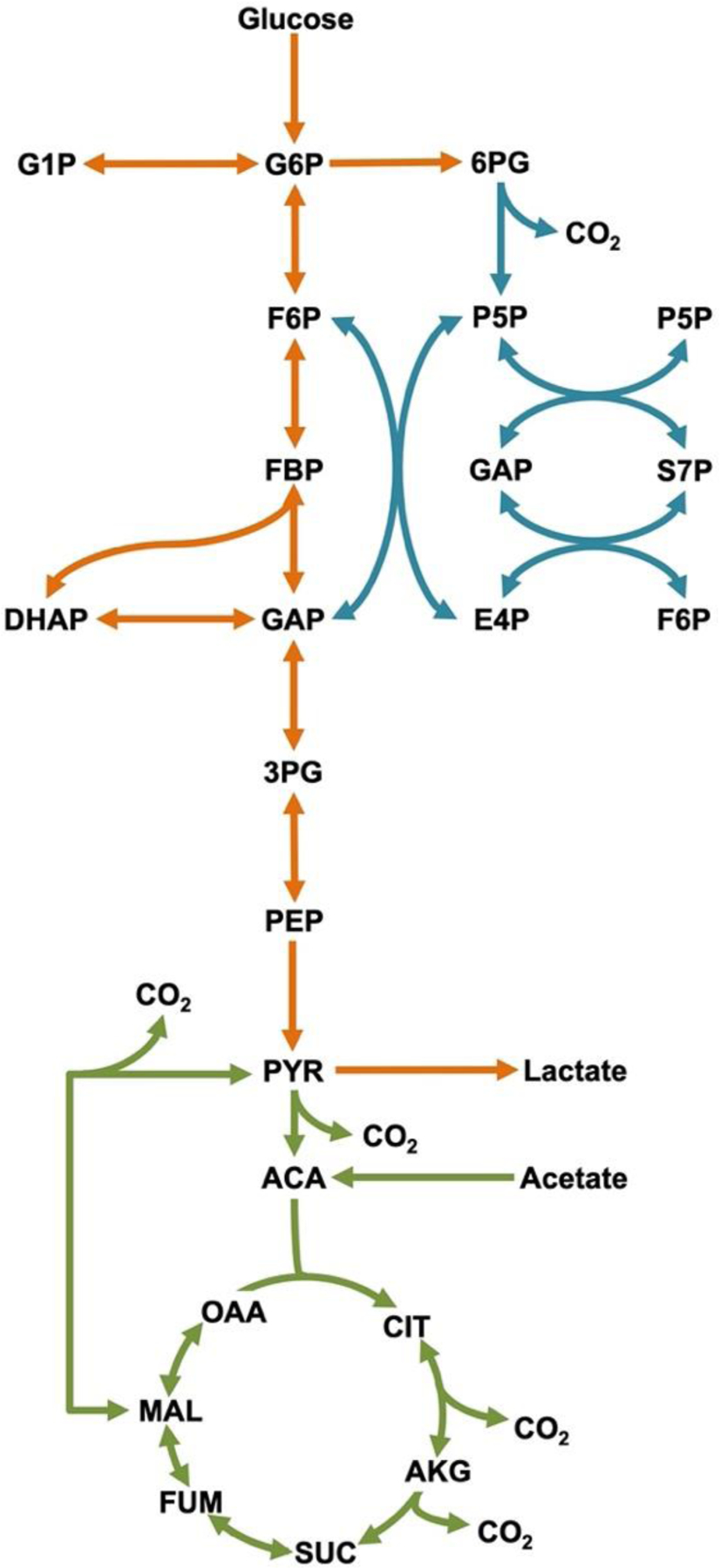
Reactions that are reversible are shown with two-headed arrows. Metabolic pathways are color coded to represent glycolysis (orange), pentose phosphate pathway (blue), and the tricarboxylic acid (TCA) cycle (green). [please use color for figure in print]
Washed platelets maintain short-term metabolic steady state
A key assumption of 13C-MFA is that cellular metabolism remains approximately at steady state throughout the course of the labeling experiment (Noh et al., 2006), which occurs when the pool size of metabolites remains unchanging with time. For conventional metabolomic studies in bacteria, metabolic steady state is assumed while the culture is in the exponential phase of growth (Noh et al., 2006). Similarly for mammalian cell studies, steady state can typically be assumed for continuous cultures (Wiechert and Noh, 2005). To confirm metabolic steady state is a reasonable assumption over the timescale of our experiments, we tracked the total pool size of each targeted metabolite for one hour following washing and a 90-minute resting period. Representative metabolites from resting platelets with a vehicle control and platelets activated with 1 U/mL thrombin are shown in Figure 3. Separate studies of metabolite pool size in washed platelets across multiple donors showed similar results (Supplemental Fig. 2).
Figure 3. Metabolite pool size of representative metabolites over 60 minutes.
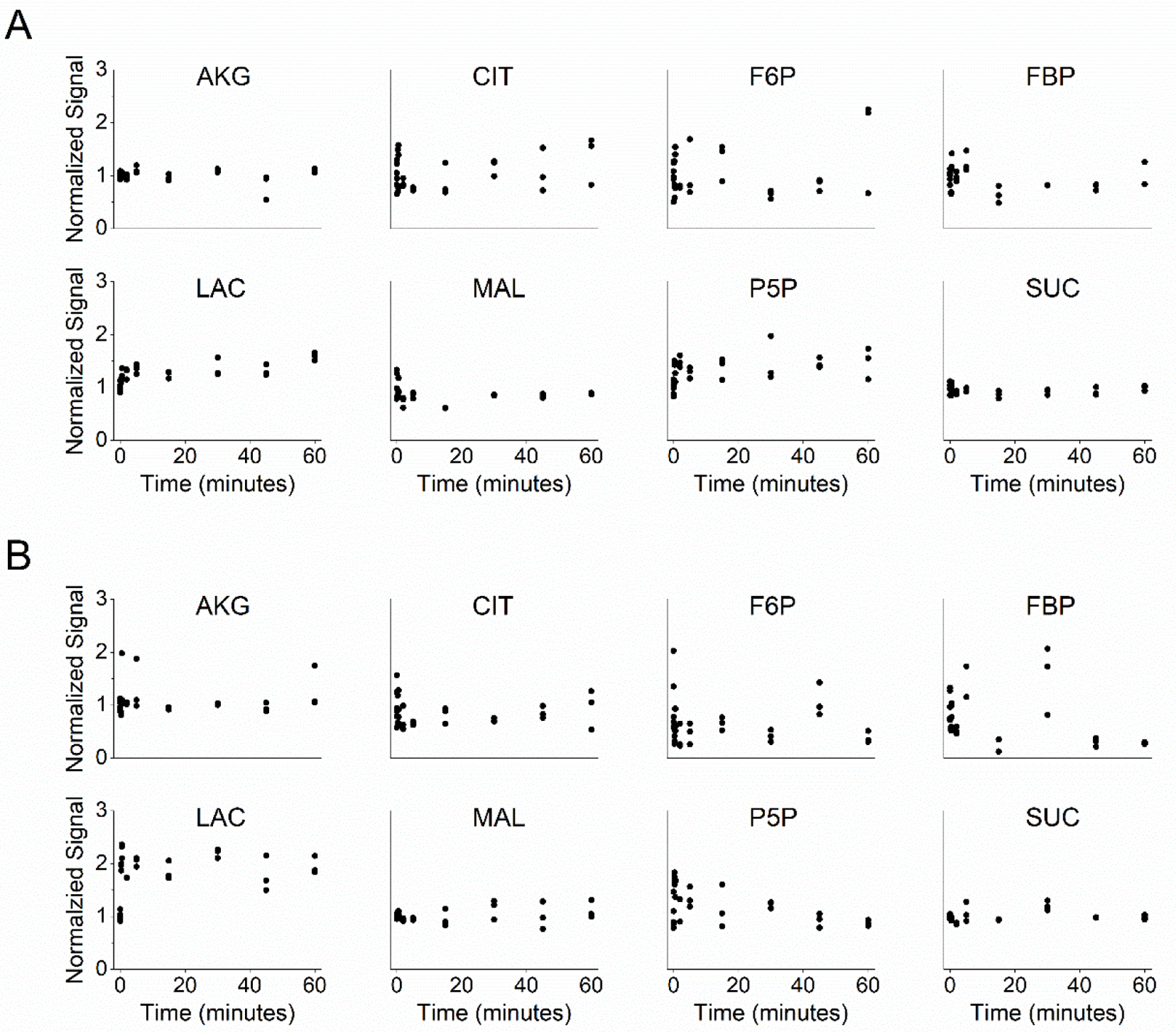
Pool size for (A) resting (vehicle = ethanol) and (B) thrombin activated (1 U/mL) platelets is represented by the sum of the MS signal for each metabolite’s isotopomer, displayed normalized to the mean at time = 0 min. Repeats at each time represent replicates from a single donor. See also Supplemental Fig. 2.
Isotope labeling dynamics and flux measurements
Uptake and excretion rates are parameters that constrain 13C-MFA models. We measured the uptake of both exogenous carbon sources, glucose and acetate, as well as the production of lactate over the course of our experiments (Figure 4). When treated with 1 U/mL thrombin, the measured glucose consumption of washed platelets increased by 4.5-fold from 50±16 to 222±17 nmol/1010 platelets/min and lactate production by 3.7-fold from 141±17 to 553±26 nmol/1010 platelets/min, suggesting that the increase in glucose consumption fuels additional pathways outside glycolysis and lactate production. Acetate uptake remained stable with and without treatment with thrombin, and on a carbon atom basis, was approximately equal to the glucose uptake of resting platelets. These uptake and excretion fluxes were used to constrain the INST-MFA model alongside isotopic labeling measurements. Additionally, glycogen was quantified to estimate the contribution of this endogenous pool as a carbon source. Resting platelets were found to contain 0.122±0.048 μg glycogen/106 platelets. A 47% reduction in glycogen was measured from thrombin activated platelets, which were measured at 0.065±0.025 μg glycogen/106 platelets.
Figure 4. Measured uptake and excretion fluxes for resting (orange) and thrombin activated (green) platelets.
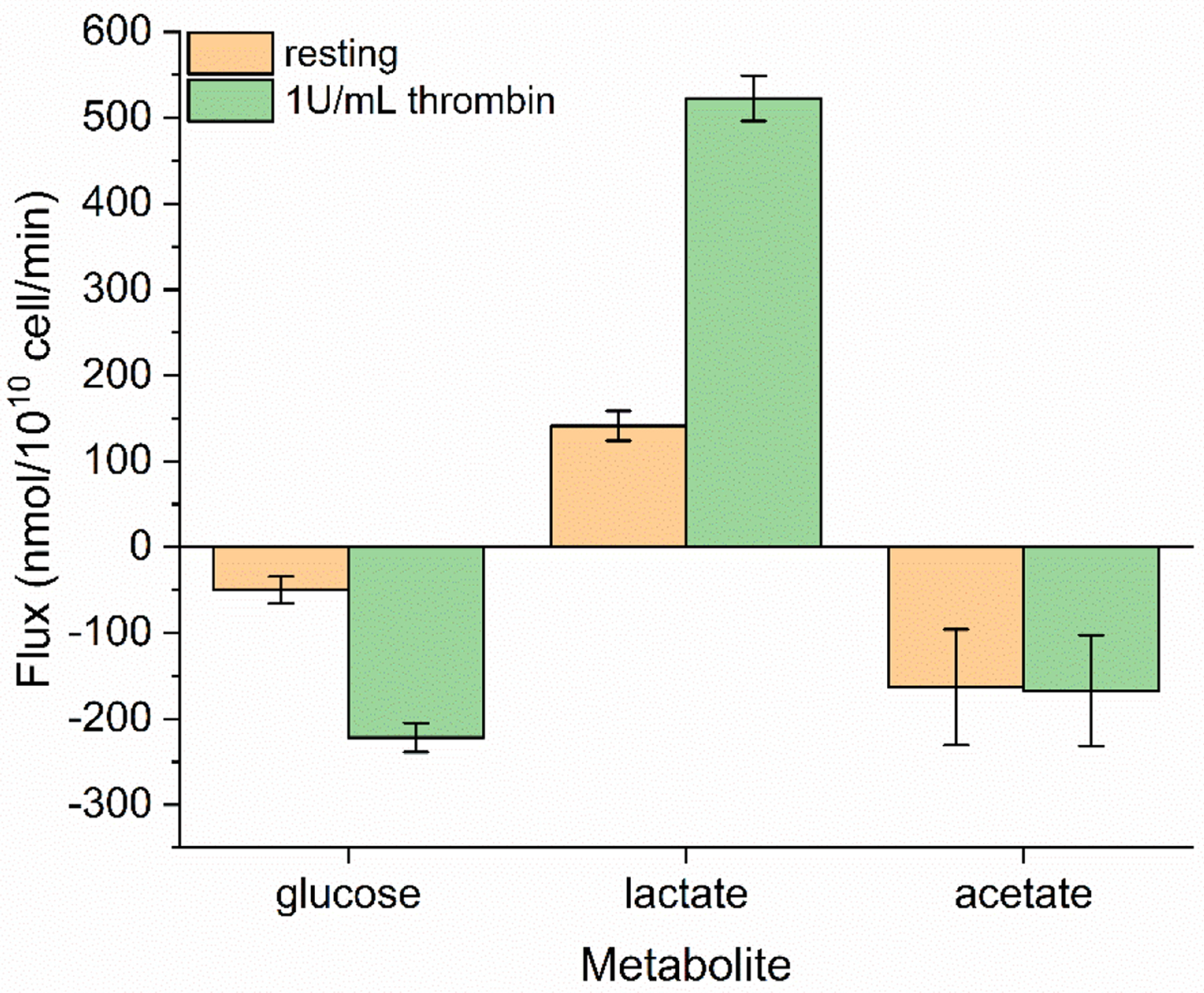
Glucose, lactate, and acetate fluxes are shown as negative for uptake/carbon source and positive for excretion/carbon sink. Extracellular metabolite concentration was measured from the reserved carbon labeling experiment supernatants. The reported flux represents the slope of a linear regression to the measured extracellular concentrations using a least squares method. Error bars represent standard error. [please use color for figure in print]
To further probe platelet metabolism, labeling experiments were performed with glucose as the primary tracer for labeling glycolysis and the pentose phosphate pathway and in parallel, with acetate as the tracer for labeling TCA cycle intermediates. Generally, glutamine is suggested as an optimal tracer for probing the TCA cycle of mammalian cells (Metallo et al., 2009) and has been used successfully in parallel labeling experiments (Ahn and Antoniewicz, 2013); however there is uncertainty regarding whether glutamine metabolism in platelets is complete (Murphy, 1995; Murphy et al., 1992). We previously ruled out glutamine as a tracer after independent experimental trials suggested poor glutamine utilization by platelet cells. Labeling experiments with [U-13C5]glutamine showed poor enrichment and slow labeling dynamics in TCA metabolites (Supplemental Fig. G1). Analysis of the supernatants from these independent experiments showed no detectible uptake of glutamine (Supplemental Fig. G2). Together, these results indicate that platelets do not readily utilize glutamine as a significant carbon source under these conditions. Therefore, we removed glutamine from further experiments and instead opted for acetate as the secondary tracer, which has been shown to be a significant oxidative carbon source for platelets (Gulliksson, 1993; Jóhannsson et al., 2018). The dynamic glucose and acetate labeling measurements and INST-MFA model fits of select metabolites representing all three major central metabolism pathways are shown in Figure 5. Fits for remaining metabolites are shown in Supplemental Figs. 3–5. The data do not indicate significant differences in labeling speed during thrombin activation but do show a higher overall 13C-enrichment (total fraction of carbon enriched with 13C) for glycolysis metabolites.
Figure 5. Experimentally measured mass isotopomer abundances (symbols) and INST-MFA model fits (lines).
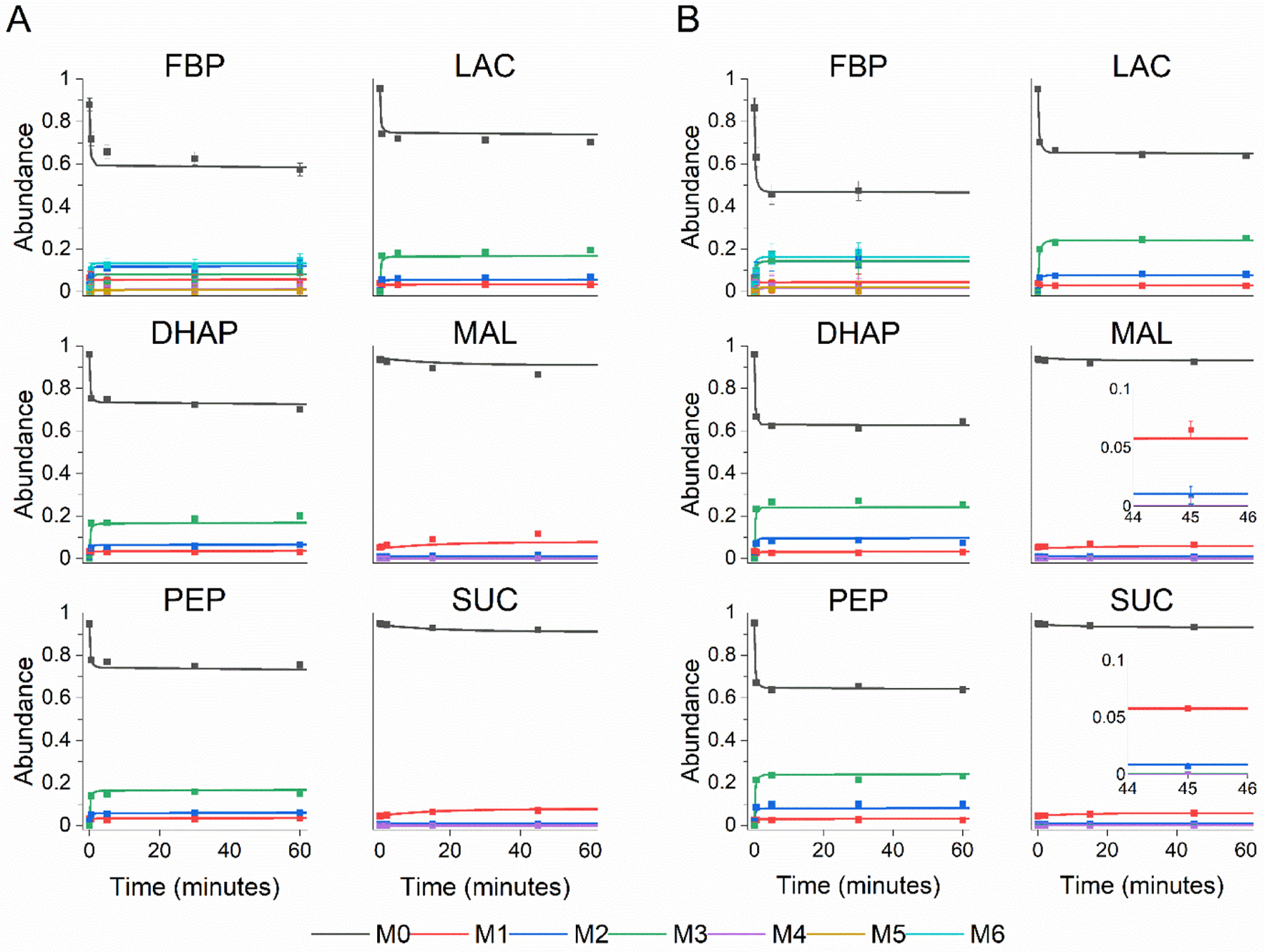
Each panel shows the 13C labeling trajectories as fractional abundance of fructose bisphosphate (FBP), dihydroxyacetone phosphate (DHAP), phosphoenolpyruvate (PEP), lactate (LAC), malate (MAL), and succinate (SUC) for (A) resting and (B) thrombin activated platelets. Labeling for FBP, DHAP, PEP, and LAC originate from the glucose tracer and labeling for MAL and SUC come from the acetate tracer. Raw mass isotopomer abundances are shown without correction for natural abundance. Error bars represent standard measurement error. [please use color for figure in print]
Metabolic flux analysis does not indicate significant amino acid metabolism
In addition to central carbon metabolites, labeling profiles were measured for four amino acids: alanine, aspartate, glutamine, and glutamate (Supplemental Figs. 3–5). Alanine and glutamine showed enrichment close to zero in all experiments. The labeling profile of glutamate was similar to that of malate. Aspartate was enriched from the acetate tracer during the resting experiment but was enriched from both glucose and acetate during the thrombin experiment (Supplemental Fig. 5). Aspartate labeling proved particularly useful for verifying the fate of glucose, as 13C-enrichment in aspartate rose to 5% with added thrombin when glucose was the tracer (Supplemental Fig. 5).
Metabolic flux analysis reveals glycolytic rerouting upon thrombin stimulation
Mass isotopomer abundances for 19 metabolites and six extracellular flux measurements (three for each parallel labeling experiment) were used to fit the INST-MFA model for each condition. Both resting and thrombin conditions were fitted with the sum-of-squared residuals between computational and experimental measurements falling within the expected ranges for a 95% confidence interval; SSR = 610.7 with range [518.0 651.8] for vehicle control and SSR = 586.0 with range [521.8 656.0] for thrombin. Measured metabolic fluxes for resting and thrombin activated platelets are presented in Figure 6. Washed, resting platelets in the presence of glucose and acetate display balanced, yet fragmented metabolism between glycolysis and the TCA cycle. Glucose is stoichiometrically converted to lactate via glycolysis. A significant portion of glucose metabolism (37% of the flux through G6P) is directed through the oxidative pentose phosphate pathway. Our results show that acetate is the preferred substrate for oxidative metabolism in resting platelets, not glucose.
Figure 6. Flux maps of platelet metabolism.
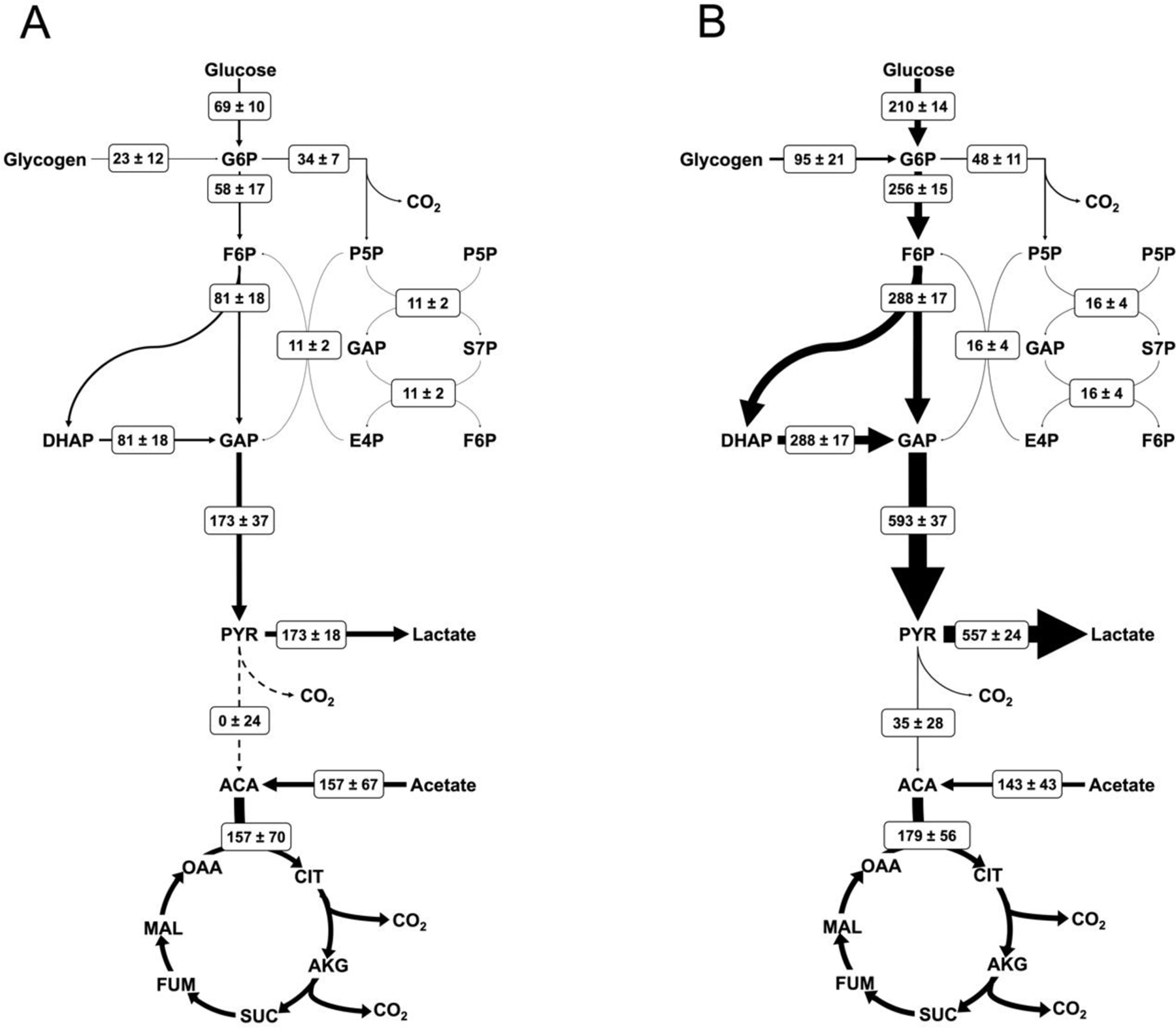
Net fluxes are shown for (A) resting and (B) thrombin activated conditions in the form M±SE where M is the calculated flux and SE is the standard error of the 95% confidence interval between upper and lower bounds with units of nmol/1010 platelets/min. Arrow thickness is linearly proportional to the net flux, with dashed lines representing zero net flux. All calculated net fluxes and their 95% confidence interval are listed in Supplemental Table 1.
Upon activation with thrombin, platelet metabolism undergoes global increases. Flux through the oxidative pentose phosphate pathway increases 45% and TCA cycle flux increases 14%. The greatest augmentation occurs through glycolysis, where the flux increases between 3.4- and 4.4-fold, resulting in significant increases in lactate production. This follows the trends observed with increased glucose uptake and lactate excretion in thrombin activated platelets (Figure 4). Importantly, thrombin activation causes an absolute increase in glucose oxidation via the TCA cycle (Figure 6). Interestingly, when fluxes are normalized to resting platelets by their carbon uptake through G6P, the relative flux through the TCA cycle decreases by approximately 67% (Figure 7). All CO2 producing pathways show an increase in flux with thrombin stimulation leading to an overall 26% increase in CO2 production from 349±190 to 441±72 nmol/1010 platelets/min.
Figure 7. Normalized fluxes of activated platelet metabolism.
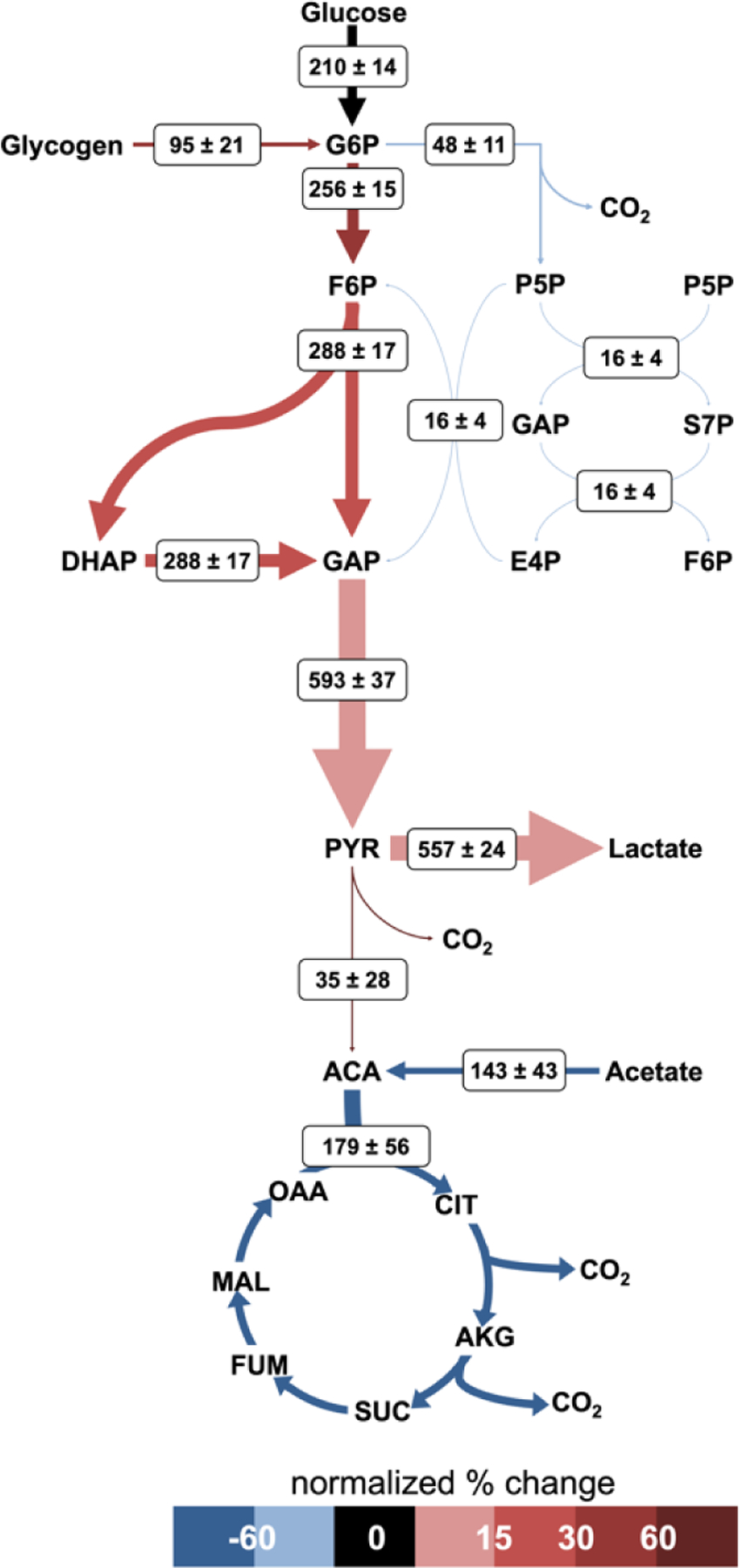
Net fluxes and arrow thicknesses are shown as previously described for Fig 6. Arrow color represents the percent change of the thrombin condition normalized flux from the resting condition normalized flux. Fluxes are normalized to the total glucose uptake rate. [please use color for figure in print]
DISCUSSION
The goal of this study was to quantitatively measure central metabolic fluxes in resting and thrombin activated human platelets using 13C-MFA. In order to use 13C-MFA for the first time on human platelets, we had to address the unique challenges posed by platelet physiology. Although previous studies have measured platelet metabolism through global and indirect methods, and even proposed predicted platelet flux maps (Jóhannsson et al., 2018; Paglia et al., 2014; Thomas et al., 2015), these techniques lack the reaction-level resolution and direct quantitative solutions provided by MFA. The approach taken here offers the primary advantage of removing inference in interpreting results: carbon flux measurements are quantitative, include upper and lower bounds, and measurements are not subject to confounding factors that are present in more global measurement types.
Resting washed human platelets experience balanced yet divided metabolism in which glucose consumption fully fuels lactate production from glycolysis and acetate uptake solely fuels the TCA cycle. Glycolysis and TCA flux are approximately equal, with TCA flux being >90% that of the glycolytic flux. The pentose phosphate pathway represents the smallest proportion of total flux, with 37% of carbon from G6P entering the pathway. Thrombin activated platelets experience global increases in intracellular pathway fluxes, fueled by a 3-fold increase in glucose uptake. INST-MFA flux results indicate that during thrombin activation, glucose uptake is supplemented by a 4-fold increase in glycogen depletion, consuming an excess of 0.044–0.134 (95% confidence LB-UB) μg glycogen/106 platelets versus the resting condition. This result is validated by experimental glycogen measurements, in which thrombin activated platelets had an average glycogen content 0.057±0.023 μg/106 platelets lower than resting platelets. While glucose and glycogen fluxes both increased, there was no change in acetate uptake upon activation. As a consequence, increases to oxidative mitochondrial metabolism require a metabolic shift, in which the previously unused pyruvate oxidation pathway is activated to redirect a portion of the glycolytic flux to the TCA cycle. Thrombin activated platelets also display increases in pentose phosphate pathway behavior, for a 1.5-fold change from the resting case; yet proportionally, thrombin treatment lowers pentose phosphate flux from 37% to 16% of the total flux through G6P.
INST-MFA calculated fluxes for resting platelets describe a central metabolism in which glucose is fully metabolized to lactate through glycolysis with approximately equal flux activity in the TCA cycle. In line with our results, many previous studies have observed stoichiometric conversion of glucose to lactate in resting platelets (Guppy et al., 1990; Murphy et al., 1992; Vasta et al., 1993). While originally thought to rely solely on oxidative phosphorylation, resting platelets are known to utilize both glycolysis and oxidative phosphorylation (Chacko et al., 2013; Nayak et al., 2018). Untargeted metabolomics of resting platelets suggest that platelet metabolism follows unique phenotypes related to storage length (Paglia et al., 2014); because our study was performed within hours post-collection from whole blood, the platelets measured here are expected to be comparable with the short-term storage phenotype associated with active glycolysis, active pentose phosphate pathway, and down-regulated TCA cycle (Paglia et al., 2014). Our results do support this summary, showing the greatest flux through glycolysis at 173 ± 37 nmol/1010 platelets/min, with 37% of G6P flux entering the pentose phosphate pathway, and the TCA flux approximately matching that of glycolysis at 157 ± 70 nmol/1010 platelets/min.
Platelet activation with thrombin has been reported to cause significant changes to platelet metabolism, particularly in regard to glucose uptake, extracellular acidification, and oxygen consumption (Aibibula et al., 2018; Nayak et al., 2018; Ravi et al., 2015). Agonist induced platelet activation initiates a step change in energy requirements, and as a result, activated platelets exhibit increased oxidative phosphorylation and glycolysis to accommodate increased ATP demands (Kholmukhamedov and Jobe, 2019). Our results show increased pentose phosphate pathway flux for activated platelets, likely a corollary to increased glycolysis (Kulkarni et al., 2019). During activation, 16% of G6P flux is sent through the pentose phosphate pathway, a value that is comparable to estimates from platelet storage literature (Baker et al., 2001; Jóhannsson et al., 2018; Murphy and Gardner, 1971). The pentose phosphate pathway is not suspected to play a major role in platelet energetics (Ravi et al., 2015) and indeed we see the smallest proportion of flux through this pathway and a relative decrease in pentose phosphate activity compared to the increase in glucose uptake. Previous studies have observed that activated platelets show increased glycolysis relative to mitochondrial phosphorylation (Kramer, 2014). Often this behavior of thrombin activated platelets is described as a “glycolytic phenotype” (Aibibula et al., 2018; Kholmukhamedov and Jobe, 2019). Our results support these prior studies, as we see the largest fold-changes from the resting to activated state occuring in glycolysis.
Using INST-MFA we now have a finer grain understanding of the platelet glycolytic phenotype, wherein a dual increase in glucose oxidation and fermentative glycolysis (sometimes referred to as aerobic glycolysis in other works to describe lactate production in the presence of oxygen) is observed following thrombin stimulation. The glycolytic flux culminates in a 3.4-fold increase to pyruvate compared to the resting platelet condition, 94% of which contributes to a 3.2-fold increase in lactate production with the balance fueling the previously unused pathway for pyruvate oxidation. We measured universal increases in absolute flux throughout all major pathways: glycolysis, pentose phosphate pathway, and the TCA cycle. However, we also observed a metabolic switch in which resting platelets perform no glucose oxidation, instead relying on acetate to feed TCA metabolism, and thrombin activated platelets incur large increases in glucose uptake in order to support increased fermentative glycolysis and glucose oxidation via the TCA cycle. For this reason, it is more appropriate to also describe resting platelets as displaying a glycolytic phenotype, because their utilization of glucose is 100% glycolytic and 0% oxidative, a state in which glucose is stoichiometrically converted to lactate.
Thrombin activated platelets display a glycolysis dominant phenotype in terms of absolute flux. However, from the perspective of glucose utilization, a portion of the glycolytic flux in thrombin activated platelets is oxidized in the TCA cycle, compared to none in the resting case, meaning that thrombin activated platelets trend toward a more oxidative phenotype than resting platelets. Acetate uptake remains unchanged between the two cases, allowing overall increases in TCA cycle flux as carbon from glucose and acetate uptakes are combined at the first step of the TCA cycle with acetyl-CoA.
In this study, we were also able to further define mitochondrial activity in terms of TCA cycle flux and quantify the magnitude between the two pathways. Resting platelets rely on glycolysis and mitochondrial TCA cycle approximately equally. Platelets activated with thrombin preferentially upregulate glycolysis, and begin utilizing glucose oxidation in the TCA cycle; both pathways increase but form a new balance in which flux through glycolysis exceeds that of the TCA cycle by approximately 3-fold.
Extracellular fluxes were measured from major carbon sources and sinks (glucose, glycogen, lactate, and acetate). CO2 is also a carbon sink, primarily from the TCA cycle, but there is also a non-trivial contribution to CO2 production from the oxidative pentose phosphate pathway. The flux maps shown in Figure 6 and Figure 7 are fully carbon balanced: extracellular glucose, extracellular acetate, and intracellular glucose from glycogen are the carbon sources. The fates of glucose and glycogen are lactate and CO2. Acetate is fully oxidized in the TCA cycle to CO2. Analogous studies in other cell types often have an additional carbon sink in the form of a biomass equation to account for the loss of carbon from central metabolism to support biosynthesis. A unified biomass equation was purposefully neglected on the basis that in vivo, platelets arise via differentiation from megakaryocytes and therefore do not divide. Additionally, precedent shows that neglecting the biomass equation is suitable during slow cell growth (Egnatchik et al., 2014): a lack of platelet division indicates that platelet growth rate is negligible. As a consequence of their origin, platelets lack a nucleus; however, they do contain mRNA and therefore can synthesize protein (Weyrich et al., 2009). The possibility of net flux leaving central metabolism to support protein synthesis was considered but was ultimately rejected. Again, there is precedent for neglecting the contribution of protein turnover, especially during INST-MFA when the labeling time of metabolic intermediates is expected to be significantly shorter than that of proteins (Schaub et al., 2008). However, to account for the possibility, alanine, aspartate, glutamate, and glutamine were included in the metabolic network. Flux results showed no net flux entering the amino acid pools, but their labeling profiles remained useful for flux calculation via exchange flux (see Supplemental Table 2).
A limitation of this study is the use of washed platelets. Cellular behavior is dependent on extracellular conditions, especially the availability of various carbon sources. For the purpose of isotope labeling, we choose to use washed platelets to avoid glucose carryover, which would require quantification of the dilution of the isotope tracer. However, we recognize that this deviates from the in vivo environment of circulating platelets. Further studies are needed to examine how platelet metabolism varies in more physiologic media like plasma or whole blood.
In summary, here we apply 13C-MFA for the first time in platelet cells to measure the central metabolism fluxes in resting and thrombin activated platelets and in doing so, lend additional quantitative context to the understanding of platelet energetics and activation. There is a lack of knowledge in the specific roles each metabolic pathway plays in platelets’ response to different agonists. Uncovering the relationships between platelet activation and metabolism may yield new drug targets, potentially for improved antiplatelet therapies. Additionally, very little is understood in how platelet metabolism and its response to agonists differs during cell stress (e.g. oxidative stress) and chronic inflammation (e.g. COVID-19, sepsis, etc.). The ability to characterize platelet metabolism has the potential to serve as a biomarker for platelet phenotype and provides a new tool for mechanistic study of platelet physiology. Our model has the potential to be expanded to include secondary pathways linked to platelet activation including fatty acid (Ravi et al., 2015; Slatter et al., 2016) and eicosanoid (Chen and Silverstein, 2018; Ju et al., 2012) metabolism. Such pursuits may require alternative techniques, for example, longer time scales to allow complete enrichment of more slowly labeled metabolites and/or alternate carbon labeling sources such as palmitate or arachidonic acid to successfully probe downstream pathways. Certain assumptions, such as metabolic steady state, may need to be reassessed before adapting to other conditions; if the metabolic steady state condition cannot be upheld, other specialized 13C-MFA techniques such as dynamic metabolic flux analysis (Antoniewicz, 2013) may be necessary.
Materials and Methods
A comprehensive list of abbreviations is available in Supplemental Table 3.
Ethics approval and consent to participate
The study received Institutional Review Board approval from the University of Colorado Anschutz Medical Campus.
Platelet collection and isolation
Blood samples were collected by venipuncture using a 21-gauge needle and collected into a syringe containing sodium citrate (3.8%). Anticoagulant citrate dextrose (ACD) solution was added to the whole blood in a 1:10 ratio, and the mixture was centrifuged at 200g to fractionate. Platelets were isolated and washed from the platelet rich plasma (PRP). In short, platelets were washed in modified Tyrode’s buffer with prostacyclin (PGI2) as an anticoagulant. Platelets were pelleted at 1000g for 10 minutes. After washing, isolated platelets were resuspended in Tyrode’s buffer, counted, and adjusted to a concentration of 8×108 cell/mL. Platelets were incubated at 37°C for 90 minutes prior to performing experiments.
Carbon labeling experiment
Prior to labeling, platelet suspensions were incubated with vehicle (ethanol) or thrombin (1 U/mL) for 15 minutes. Glucose and acetate tracers were administered in parallel labeling experiments and samples were taken at predetermined intervals over 60 minutes. For the glucose tracer experiment, 5 mM of 30% [1,2-13C2]glucose, 50% [U-13C6]glucose, and 20% unlabeled glucose was supplied. For the acetate tracer experiment, 20 mM of 25% [1-13C]acetate, 25% [2-13C]acetate, and 50% unlabeled acetate was used. 5 mM unlabeled glucose was added to the acetate labeling experiment and 20 mM unlabeled acetate was added to the glucose labeling experiment to maintain identical conditions between parallel labeling experiments. Samples were collected in triplicate at 0, 30 s, 5 min, 30 min, 60 min, and 180 min for glucose labeling and 0, 20 s, 2 min, 15 min, 45 min, and 180 min for acetate labeling. The 0 time-point samples were taken immediately prior to the addition of the label. Samples were quenched by rapidly plunging 0.25 mL portions into 1.25 mL of pre-chilled partially frozen normal saline and centrifuged to pellet at 16000g and 0°C. The supernatant was collected for extracellular metabolite analysis and the pellet was stored at −20°C prior to extraction.
Metabolite extraction
Intracellular metabolites were extracted using a method adapted from (Paglia et al., 2012). Platelet pellets were resuspended in 500 μL of 70% methanol and spiked with ribitol and PIPES as internal standards. Samples were frozen in liquid nitrogen, thawed on ice, and vortexed at 0°C for 5 minutes and 1000 rpm. This freeze/thaw/vortex cycle was repeated twice more, and samples were centrifuged at 16,000g for 5 minutes at −4°C in a Sorvall Legend Micro 17R microcentrifuge (Thermo Scientific). The supernatant (extract) was collected in a new tube and stored at −20°C. The extraction process was then repeated with 500 μL of 50% methanol and the second collected supernatant was added to the first. The extracted pellet was reserved at −20°C for glycogen analysis. The samples were then dried overnight under vacuum in a Savant SPD131DDA SpeedVac (Thermo Scientific). Dried extracts were resuspended in 150 μL Optima water and filtered with nylon filter tubes (Spin-X, Costar). Filters were rinsed with an additional 50 μL water for a total final extract volume of 200 μL for LC-MS/MS analysis.
Extracellular flux measurements
Glucose uptake, lactate excretion, and acetate uptake rates were determined by analyzing the change in the metabolites’ concentrations from the saved supernatants of the labeling experiment. The retained supernatant was thawed, mixed well, and a portion was removed for each assay. For glucose, the supernatant was analyzed using the Sigma starch assay kit (SA20). The lactate concentration was determined using the Megazyme L-lactic assay kit (K-LATE). Acetate was analyzed using the BioAssay EnzyChrom acetate assay kit. Concentration data over time was linearly fit using a least squares method, where the slope and its standard error were taken as extracellular flux. Glycogen from the reserved, extracted cell pellets was hydrolyzed to glucose using the method described by (Santos Rocha et al., 2014) and quantified on a Xylem YSI 2950D-3 Biochemistry Analyzer.
Analytical methods
Metabolite extracts were analyzed for targeted metabolites listed in Supplemental Table 4 using an LC-MS/MS method adapted from (Young et al., 2011). A Phenomenex 150 mm x 2 mm Synergi Hydro-RP column was used on an Agilent 1200 Series HPLC system coupled to an AB Sciex 5500 QTRAP system. LC was performed with an injection volume of 20 μL, using gradient elution of 10 mM tributylamine and 15 mM acetic acid (aqueous phase) with acetonitrile (organic phase) at a constant flow of 0.3 mL/min. The gradient profile of the organic phase is as follows: 0% B (0 min), 8% B (10 min), 16% B (15 min), 30% B (16.5 min), 30% B (19 min), 90% B (21.5 min), 90% B (26.5 min), 0% B (26.6 min), and 0% B (30.5 min). MS analysis was performed in negative mode using a multiple reaction monitoring (MRM) acquisition method. Data acquisition was performed on the ABSciex Analyst 1.7 software. Absolute quantification of intracellular metabolites was performed using the Analyst MultiQuant 3.0.3 Software. For isotope labeling profiles, MSConvert was used to convert LC-MS/MS data files (ABSciex .wiff and .wiff.scan) to an open-source format (Holman et al., 2014). A combination of the pyOpenMS (Rost et al., 2014) and SciPy (Virtanen et al., 2020) packages in Python, was used for the process of peak finding, window sizing, and noise assessment across multiple isotopes.
Isotopomer network and flux calculations
An isotopomer network model describing human platelet central metabolism was constructed primarily from the published, manually curated reaction network iAT-PLT-636 (Thomas et al., 2015). Carbon atom transitions were assigned with the assistance of the carbon fate maps from (Mu et al., 2007). Isotopic nonstationary metabolic flux analysis (INST-MFA) was performed in the Isotopomer Network Compartmental Analysis (INCA) software, along with the corresponding least-squares parameter regression and sensitivity analysis (Young, 2014). A list of the reactions and atom transitions for the model is listed in Supplemental Table 5.
Supplementary Material
Highlights.
Instationary isotope assisted metabolic flux analysis was applied for the first time to human platelets
Central carbon metabolic fluxes were determined for both resting and activated platelets
Resting platelets primarily convert glucose to lactate via glycolysis and acetate is oxidized in the TCA cycle
Activated platelets increase glucose consumption 3-fold and redistribute carbon dramatically
Acknowledgements.
Thank you to Allaura Cox from the Di Paola group for kindly sharing her platelet washing protocol.
Funding.
This project is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R61HL141794.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none.
References
- Abonnenc M, Crettaz D, Marvin L, Grund B, Sonego G, Bardyn M, Tissot J-D, Prudent M, Rochat B, Lion N, 2016. Metabolomic profiling highlights oxidative damages in platelet concentrates treated for pathogen inactivation and shows protective role of urate. Metabolomics 12. [Google Scholar]
- Ahn WS, Antoniewicz MR, 2013. Parallel labeling experiments with [1,2-(13)C]glucose and [U-(13)C]glutamine provide new insights into CHO cell metabolism. Metab Eng 15, 34–47. [DOI] [PubMed] [Google Scholar]
- Aibibula M, Naseem KM, Sturmey RG, 2018. Glucose metabolism and metabolic flexibility in blood platelets. J Thromb Haemost 16, 2300–2314. [DOI] [PubMed] [Google Scholar]
- Antoniewicz MR, 2013. Dynamic metabolic flux analysis--tools for probing transient states of metabolic networks. Curr Opin Biotechnol 24, 973–8. [DOI] [PubMed] [Google Scholar]
- Antoniewicz MR, 2015. Methods and advances in metabolic flux analysis: a mini-review. J Ind Microbiol Biotechnol 42, 317–325. [DOI] [PubMed] [Google Scholar]
- Baker JM, Candy DJ, Hawker RJ, 2001. Influences of pH on human platelet metabolism. Platelets 12, 333–42. [DOI] [PubMed] [Google Scholar]
- Burkhart JM, Gambaryan S, Watson SP, Jurk K, Walter U, Sickmann A, Heemskerk JWM, Zahedi RP, 2014. What can proteomics tell us about platelets? Circ Res 114, 1204–1219. [DOI] [PubMed] [Google Scholar]
- Chacko BK, Kramer PA, Ravi S, Johnson MS, Hardy RW, Ballinger SW, Darley-Usmar VM, 2013. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest 93, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah YE, Young JD, 2018. Isotopically nonstationary metabolic flux analysis (INST-MFA): putting theory into practice. Curr Opin Biotechnol 54, 80–87. [DOI] [PubMed] [Google Scholar]
- Chen Y, Silverstein RL, 2018. Platelet metabolism meets thrombosis. Blood 132, 1089–1091. [DOI] [PubMed] [Google Scholar]
- Cohen P, Wittels B, 1970. Energy substrate metabolism in fresh and stored human platelets. J Clin Invest 49, 119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown SB, Antoniewicz MR, 2013a. Parallel labeling experiments and metabolic flux analysis: Past, present and future methodologies. Metab Eng 16, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown SB, Antoniewicz MR, 2013b. Publishing13C metabolic flux analysis studies: A review and future perspectives. Metab Eng 20, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Locasale JW, 2016. Understanding metabolism with flux analysis : From theory to application. Metab Eng 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davizon-Castillo P, McMahon B, Aguila S, Bark D, Ashworth K, Allawzi A, Campbell RA, Montenont E, Nemkov T, D’Alessandro A, Clendenen N, Shih L, Sanders NA, Higa K, Cox A, Padilla-Romo Z, Hernandez G, Wartchow E, Trahan GD, Nozik-Grayck E, Jones K, Pietras EM, DeGregori J, Rondina MT, Di Paola J, 2019. TNF-α-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 134, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dersch LM, Beckers V, Wittmann C, 2016. Green pathways: Metabolic network analysis of plant systems. Metab Eng 34, 1–24. [DOI] [PubMed] [Google Scholar]
- Egnatchik RA, Leamy AK, Noguchi Y, Shiota M, Young JD, 2014. Palmitate-induced activation of mitochondrial metabolism promotes oxidative stress and apoptosis in H4IIEC3 rat hepatocytes. Metabolism 63, 283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler TP, Campbell RA, Funari T, Dunne N, Balderas Angeles E, Middleton EA, Chaudhuri D, Weyrich AS, Abel ED, 2017. Deletion of GLUT1 and GLUT3 Reveals Multiple Roles for Glucose Metabolism in Platelet and Megakaryocyte Function. Cell Rep 20, 881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulliksson H, 1993. Storage of platelets in additive solutions: the effect of citrate and acetate in in vitro studies. Transfusion 33, 301–303. [DOI] [PubMed] [Google Scholar]
- Guppy M, Whisson ME, Sabaratnam R, Withers P, Brand K, 1990. Alternative fuels for platelet storage: a metabolic study. Vox Sang 59, 146–52. [DOI] [PubMed] [Google Scholar]
- Holman JD, Tabb DL, Mallick P, 2014. Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Curr Protoc Bioinformatics 46, 13 24 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe EA, 1987. Cell biology of endothelial cells. Hum Pathol 18, 234–9. [DOI] [PubMed] [Google Scholar]
- Jóhannsson F, Guðmundsson S, Paglia G, Guðmundsson S, Palsson B, 2018. Systems analysis of metabolism in platelet concentrates during storage in platelet additive solution. Biochem J [DOI] [PubMed]
- Ju HK, Lee JG, Park MK, Park SJ, Lee CH, Park JH, Kwon SW, 2012. Metabolomic investigation of the anti-platelet aggregation activity of ginsenoside Rk(1) reveals attenuated 12-HETE production. J Proteome Res 11, 4939–46. [DOI] [PubMed] [Google Scholar]
- Kholmukhamedov A, Jobe S, 2019. Platelet respiration. Blood Adv 3, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PA, 2014. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic markers. Redox Biol 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni PP, Tiwari A, Singh N, Gautam D, Sonkar VK, Agarwal V, Dash D, 2019. Aerobic glycolysis fuels platelet activation: small-molecule modulators of platelet metabolism as anti-thrombotic agents. Haematologica 104, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepropre S, Kautbally S, Octave M, Ginion A, Onselaer MB, Steinberg GR, Kemp BE, Hego A, Wera O, Brouns S, Swieringa F, Giera M, Darley-Usmar VM, Ambroise J, Guigas B, Heemskerk J, Bertrand L, Oury C, Beauloye C, Horman S, 2018. AMPK-ACC signaling modulates platelet phospholipids and potentiates thrombus formation. Blood 132, 1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell RE, Aulak KS, Almoushref A, Melillo CA, Brauer BE, Newman JE, Tonelli AR, Dweik RA, 2020. Platelet glycolytic metabolism correlates with hemodynamic severity in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 318, L562–L569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Walther JL, Stephanopoulos G, 2009. Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J Biotechnol 144, 167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu F, Williams RF, Unkefer CJ, Unkefer PJ, Faeder JR, Hlavacek WS, 2007. Carbon-fate maps for metabolic reactions. Bioinformatics 23, 3193–9. [DOI] [PubMed] [Google Scholar]
- Mueller D, Heinzle E, 2013. Stable isotope-assisted metabolomics to detect metabolic flux changes in mammalian cell cultures. Curr Opin Biotechnol 24, 54–9. [DOI] [PubMed] [Google Scholar]
- Murphy S, 1995. The Oxidation of Exogenously Added Organic Anions by Platelets Facilitates Maintenance of pH During Their Storage for Transfusion at 22°C. Blood 85, 7. [PubMed] [Google Scholar]
- Murphy S, Gardner FH, 1971. Platelet storage at 22 degrees C; metabolic, morphologic, and functional studies. J Clin Invest 50, 370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S, Santiago M, Mark P-B, Eric N, 1992. Amino acid metabolism during platelet storage for transfusion. Br J Haematol 81, 6. [DOI] [PubMed] [Google Scholar]
- Nayak MK, Dhanesha N, Doddapattar P, Rodriguez O, Sonkar VK, Dayal S, Chauhan AK, 2018. Dichloroacetate, an inhibitor of pyruvate dehydrogenase kinases, inhibits platelet aggregation and arterial thrombosis. Blood Adv 2, 2029–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh K, Wahl A, Wiechert W, 2006. Computational tools for isotopically instationary 13C labeling experiments under metabolic steady state conditions. Metab Eng 8, 554–77. [DOI] [PubMed] [Google Scholar]
- Paglia G, Magnúsdóttir M, Thorlacius S, Sigurjónsson ÓE, Gudmundsson S, Palsson B, Thiele I, 2012. Intracellular metabolite profiling of platelets: Evaluation of extraction processes and chromatographic strategies. J Chromatogr B 898, 111–120. [DOI] [PubMed] [Google Scholar]
- Paglia G, Sigurjónsson ÓE, Rolfsson Ó, Hansen MB, Brynjólfsson S, Gudmundsson S, Palsson BO, 2015. Metabolomic analysis of platelets during storage: A comparison between apheresis- and buffy coat-derived platelet concentrates. Transfusion 55, 301–313. [DOI] [PubMed] [Google Scholar]
- Paglia G, Sigurjõnsson ÓE, Rolfsson Ó, Valgeirsdottir S, Hansen MB, Brynjõlfsson S, Gudmundsson S, Palsson BO, 2014. Comprehensive metabolomic study of platelets reveals the expression of discrete metabolic phenotypes during storage. Transfusion 54, 2911–2923. [DOI] [PubMed] [Google Scholar]
- Quach ME, Chen W, Li R, 2018. Mechanisms of platelet clearance and translation to improve platelet storage. Blood 131, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi S, Chacko B, Sawada H, Kramer PA, Johnson MS, Benavides GA, O’Donnell V, Marques MB, Darley-Usmar VM, 2015. Metabolic plasticity in resting and thrombin activated platelets. PLoS One 10, e0123597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost HL, Schmitt U, Aebersold R, Malmstrom L, 2014. pyOpenMS: a Python-based interface to the OpenMS mass-spectrometry algorithm library. Proteomics 14, 74–7. [DOI] [PubMed] [Google Scholar]
- Sake CL, Metcalf AJ, Boyle NR, 2019. The challenge and potential of photosynthesis: unique considerations for metabolic flux measurements in photosynthetic microorganisms. Biotechnol Lett 41, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos Rocha D, de Souza SK, Hugo Onsten TG, Martins da Silva RS, Emilio Frizzo M, 2014. A Simple Method to Quantify Glycogen from Human Platelets. Journal of Cytology & Histology 05. [Google Scholar]
- Schaub J, Mauch K, Reuss M, 2008. Metabolic flux analysis in Escherichia coli by integrating isotopic dynamic and isotopic stationary 13C labeling data. Biotechnol Bioeng 99, 1170–85. [DOI] [PubMed] [Google Scholar]
- Schwechheimer SK, Becker J, Wittmann C, 2018. Towards better understanding of industrial cell factories: novel approaches for (13)C metabolic flux analysis in complex nutrient environments. Curr Opin Biotechnol 54, 128–137. [DOI] [PubMed] [Google Scholar]
- Semple JW, Freedman J, 2010. Platelets and innate immunity. Cell Mol Life Sci 67, 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatter DA, Aldrovandi M, O’Connor A, Allen SM, Brasher CJ, Murphy RC, Mecklemann S, Ravi S, Darley-Usmar V, O’Donnell VB, 2016. Mapping the Human Platelet Lipidome Reveals Cytosolic Phospholipase A2 as a Regulator of Mitochondrial Bioenergetics during Activation. Cell Metab 23, 930–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowton AP, Millington-Burgess SL, Murray AJ, Harper MT, 2018. Rapid kinetics of changes in oxygen consumption rate in thrombin-stimulated platelets measured by high-resolution respirometry. Biochem Biophys Res Commun 503, 2721–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Rahmanian S, Bordbar A, Palsson B, Jamshidi N, 2015. Network reconstruction of platelet metabolism identifies metabolic signature for aspirin resistance. Sci Rep 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meijden PEJ, Heemskerk JWM, 2019. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol 16, 166–179. [DOI] [PubMed] [Google Scholar]
- Vasta V, Meacci E, Farnararo M, Bruni P, 1993. Glutamine utilization in resting and stimulated platelets. J Biochem 114, 163–6. [DOI] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, van der Walt SJ, Brett M, Wilson J, Millman KJ, Mayorov N, Nelson ARJ, Jones E, Kern R, Larson E, Carey CJ, Polat I, Feng Y, Moore EW, VanderPlas J, Laxalde D, Perktold J, Cimrman R, Henriksen I, Quintero EA, Harris CR, Archibald AM, Ribeiro AH, Pedregosa F, van Mulbregt P, SciPy C, 2020. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich AS, Schwertz H, Kraiss LW, Zimmerman GA, 2009. Protein synthesis by platelets: historical and new perspectives. J Thromb Haemost 7, 241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechert W, 2001. 13C metabolic flux analysis. Metab Eng 3, 195–206. [DOI] [PubMed] [Google Scholar]
- Wiechert W, Noh K, 2005. From stationary to instationary metabolic flux analysis. Adv Biochem Eng Biotechnol 92, 145–72. [DOI] [PubMed] [Google Scholar]
- Young JD, 2014. INCA: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30, 1333–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD, Shastri AA, Stephanopoulos G, Morgan JA, 2011. Mapping photoautotrophic metabolism with isotopically nonstationary 13C flux analysis. Metab Eng 13, 656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.