

Abstract
As one of the most common post-transcriptional modifications of mRNAs and noncoding RNAs, N6-methyladenosine (m6A) modification regulates almost every aspect of RNA metabolism. Evidence indicates that dysregulation of m6A modification and associated proteins contributes to glioblastoma (GBM) progression. However, the function of fat mass and obesity-associated protein (FTO), an m6A demethylase, has not been systematically and comprehensively explored in GBM. Here, we found that decreased FTO expression in clinical specimens correlated with higher glioma grades and poorer clinical outcomes. Functionally, FTO inhibited growth and invasion in GBM cells in vitro and in vivo. Mechanistically, FTO regulated the m6A modification of primary microRNA-10a (pri-miR-10a), which could be recognized by reader HNRNPA2B1, recruiting the microRNA microprocessor complex protein DGCR8 and mediating pri-miR-10a processing. Furthermore, the transcriptional activity of FTO was inhibited by the transcription factor SPI1, which could be specifically disrupted by the SPI1 inhibitor DB2313. Treatment with this inhibitor restored endogenous FTO expression and decreased GBM tumor burden, suggesting that FTO may serve as a novel prognostic indicator and therapeutic molecular target of GBM.
Keywords: glioblastoma, N6-methyladenosine, FTO, pri-miR-10a, SPI1, DB2313
Graphical abstract
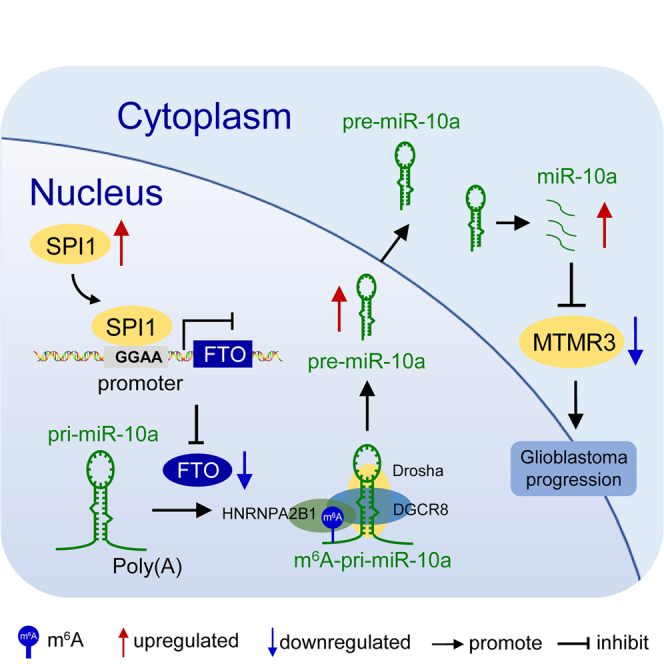
This comprehensive study showed that FTO modulates GBM tumorigenesis by regulating m6A modification of noncoding RNAs. FTO may serve as a novel prognostic indicator and therapeutic molecular target of GBM. Restoring the endogenous expression of FTO with the SPI1 inhibitor DB2313 may have therapeutic potential for GBM patients.
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor of the central nervous system and remains almost invariably fatal because of its high growth rate and aggressive nature.1,2 A previous study divided GBM into three subtypes, proneural (PN), classical (CL), and mesenchymal (MES), of which the MES subtype has the worst prognosis, exhibiting complicated inter- and intratumoral heterogeneity.3 Although recent efforts have shown no significant effective and sufficient therapeutic strategies for the treatment of patients with GBM,4 exploring other unidentified molecular modulators could contribute to our understanding of GBM malignancy and therapy resistance to provide new insights into possible treatments.
As one of the most common post-transcriptional modifications of mRNAs and noncoding RNAs, N6-methyladenosine (m6A) modification regulates almost every aspect of RNA metabolism, such as mRNA splicing, stability, translation, and microRNA (miRNA) maturation,5, 6, 7 and its function is determined mainly by m6A regulators called methyltransferase complexes (“writers”), demethylases (“erasers”), and RNA-binding proteins (“readers”), playing an important role in cellular proliferation, metabolism, and metastasis.8,9 Evidence is emerging that dysregulation of m6A modification and associated proteins contributes to the self-renewal, tumorigenesis, and radio-chemotherapy resistance of GBM stem cells.10, 11, 12, 13, 14 Fat mass and obesity-associated protein (FTO), a member of the Fe(II)- and α-ketoglutarate-dependent AlkB family, was reported to be the first identified m6A “eraser,”15 and plays various roles in tumorigenesis in some tumors by regulating m6A modification of mRNAs; inhibiting tumor progression in liver cancer,16 colorectal cancer,17 and ovarian cancer18; and promoting tumor progression in leukemia,19,20 presenting an opportunity for the development of effective targeted therapeutics. In a research letter, Tao et al.21 reported that FTO played a tumor-suppressive role in glioma by interacting with FOXO3a to enhance its nuclear translocation independent of its m6A demethylase activity. However, the specialized and systematic mechanisms of the m6A modification regulated by FTO remain largely unknown in GBM.
This study revealed that decreased expression of FTO in clinical specimens correlated with higher grades of gliomas and poorer clinical outcomes. We discovered that FTO inhibited GBM cell proliferation, migration, and invasion both in vitro and in vivo. Moreover, we found that FTO regulated m6A-mediated primary microRNA-10a (pri-miR-10a) processing, which could be recognized by the reader HNRNPA2B1, recruiting the core microprocessor complex DGCR8 and promoting the malignant progression of GBM. Furthermore, we discovered that the transcriptional activity of FTO could be inhibited by the transcription factor SPI1, and this effect could be specifically interrupted by the SPI1 inhibitor DB2313 in GBM cells. Treatment with this inhibitor restored the endogenous expression of FTO and decreased GBM tumor burden. Our findings revealed that the function of m6A modification regulated by FTO is important, and FTO may serve as a novel prognostic indicator and therapeutic molecular target of GBM. Thus, restoring the endogenous expression of FTO with the SPI1 inhibitor DB2313 may have therapeutic potential for GBM patients.
Results
Downregulated FTO expression correlated with poor prognosis of GBM patients
To explore FTO expression and its clinical significance in glioma patients, we analyzed FTO expression in The Cancer Genome Atlas (TCGA) low-grade glioma (LGG), Chinese Glioma Genome Atlas (CGGA), Repository for Molecular Brain Neoplasia Data (REMBRANDT), Gravendeel, Philips, and TCGA GBM microarray datasets. We discovered that FTO expression was lowest in GBM samples compared with low-grade glioma and normal brain tissue samples (Figure 1A). Kaplan-Meier survival analysis showed that glioma patients with low FTO expression had worse prognosis and shorter survival time than those with high FTO expression in all 6 datasets (Figure 1B). Additionally, FTO expression was lowest in the MES subtype of GBM, which is a more malignant GBM subtype, than in the PN and CL subtypes. Gene set enrichment analysis (GSEA) also showed that low FTO expression was strongly enriched in the MES-subtype gene set3 (Figure 1C). To explore the association between FTO expression and clinical characteristics (Table S1), we then compared FTO expression levels in 522 primary GBM patients from TCGA grouped by IDH1 status, CpG island methylated molecular phenotype (G-CIMP) status, MGMT promoter status, age, and sex. As shown in Figure S1A, the expression of FTO in IDH1 mutant samples was higher than that in IDH1 wild-type (WT) samples. Regarding G-CIMP status, FTO expression was higher in the G-CIMP group than in the non-G-CIMP group. There was no significant correlation between FTO expression and MGMT promoter methylation, age, or sex. Moreover, univariate Cox regression analysis of the overall survival of GBM patients in the TCGA GBM dataset showed that low FTO expression (hazard ratio [HR]: 0.863; p = 0.040) was an independent factor associated with a poor prognosis of GBM patients. In addition, in subsequent multivariate Cox regression analysis, FTO expression (HR: 0.839; p = 0.024) remained a statistically significant beneficial factor in GBM patients after adjustment for age, sex, molecular subtype, and G-CIMP status (Figures S1B and S1C; Table S2).
Figure 1.

Downregulated FTO expression correlated with poor prognosis of GBM patients
(A) Expression level of FTO in glioma patient tissues and normal brain tissues (NT) and (B) Kaplan-Meier survival curves of patients with high and low FTO expression in TCGA, CGGA, REMBRANDT, Gravendeel, and Philips datasets; log rank tests were used to identify the significance of the differences. (C) GSEA for mesenchymal signatures in FTO-high compared with FTO-low expression GBM samples. NES, normalized enrichment score; FDR, false discovery rate. (D) IHC analysis of FTO protein expression in normal tissue (NT) and glioma tissues with different WHO grades at 200× and 400× magnification. Scale bar, 50 μm. (E) Histogram representing statistical data of IHC. FTO-positive ratio was defined as the ratio of FTO-positive cell nucleus count to the total cell nucleus count in the same field (n = 3). (F) Western blot showing FTO protein expression in glioma tissue samples. GAPDH was used as control for normalization. (G) Protein expression levels of FTO in glioma stem cells (GSCs) of the proneural (PN) and mesenchymal (MES) subtypes. (H) Protein expression levels of FTO in normal human astrocytes (NHAs) and the glioma cell lines U87MG, U251, A172, LN229, U118MG, and P3. Comparisons between two independent samples and among multiple samples were performed using two-tailed t tests and one-way ANOVA, respectively. Error bars indicate at least three independent experiments, and data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Furthermore, immunohistochemistry (IHC) and western blot analysis of glioma clinical samples showed that FTO expression was lower in high-grade glioma (Figures 1D–1F), consistent with the results of the database analyses. We then tested the protein level in glioblastoma stem cells (GSCs) and discovered that FTO expression was lower in MES-subtype GSCs than in PN-subtype GSCs (Figure 1G). The protein level of FTO was lower in GBM cell lines than in normal human astrocyte (NHA) cells (Figure 1H). Pearson correlation analysis showed that FTO was negatively correlated with the proliferation marker (PCNA) and malignant invasion markers (vimentin and CD44) in all 5 glioma datasets (p < 0.0001; Figure S1D). Together, these results suggested that decreased FTO expression was associated with glioma malignancy and could be a potential prognostic marker in GBM patients.
Overexpression of FTO inhibited growth, migration, and invasion of GBM cells in vitro and in vivo
To evaluate the functional roles of FTO in GBM cells, three GBM cell lines, U87MG, A172, and U118MG, which have relatively low FTO expression compared with others, as previously detected (Figure 1H), were transduced with overexpression lentivirus and negative control lentivirus vectors. FTO overexpression efficiency was tested using western blotting and real-time qPCR (Figures 2A and S2A). The proliferation ability of GBM cells was decreased upon FTO overexpression, as determined by the CCK-8 and EdU assays (Figures 2B, S2B, and S2C). Transwell assays showed that overexpression of FTO inhibited both the migration and invasion abilities of GBM cells (Figure 2C). Consistent with the results of Transwell assays, the three-dimensional (3D) tumor spheroid invasion assay also showed that overexpression of FTO decreased the ability of GBM cells to invade adjacent areas (Figure 2D). Furthermore, overexpression of FTO decreased the protein expression levels of PCNA, CD44, vimentin, and other molecules closely related to tumor cell proliferation, invasion, and migration (Figure 2E). To determine whether FTO is important for GSC self-renewal, we performed neurosphere formation and extreme limiting dilution assays (ELDAs) in MES-subtype GSC267 cells, showing that overexpression of FTO significantly decreased the neurosphere formation frequency of GSC267 cells (Figures 2F and 2G). Moreover, western blot assays revealed that overexpression of FTO reduced the expression of CD44 and YKL40 (Figure 2H), the core markers of the MES subtype, confirming the inhibitory effect of FTO on GSC self-renewal and malignant phenotypic transformation. To clarify the function of FTO in GBM progression in vivo, FTO-overexpressing and negative control U87MG-luc cells were implanted into BALB/c mice in situ. Bioluminescent imaging displayed that FTO inhibited tumor growth in vivo (Figure 2I). H&E staining showed a smaller tumor volume and a rougher and more unclear border in the FTO overexpression group compared with the normal control (NC) group (Figure 2J). We showed that overexpression of FTO in GBM cells reduced the survival of tumor-bearing mice (Figure 2K).
Figure 2.

Overexpression of FTO inhibited growth, migration, and invasion of GBM cells in vitro and in vivo
(A) Expression levels of FTO in U87MG, A172, and U118MG cells after transduction with control lentivirus (ovNC) and FTO-overexpressing lentivirus (ovFTO). GAPDH was used as control. (B) The proliferation capacity of U87MG and A172 cells transduced with lentivirus was assessed using CCK-8 assay. (C) Transwell assays of U87MG and A172 cells. Representative images are shown. Scale bar, 50 μm. Histograms representing the number of migrating or invading cells. Data are presented as mean ± SD; n = 3. (D) Three-dimensional tumor spheroid invasion assay of U87MG cells transduced with lentivirus. Images at 0, 24, 48, and 72 h are shown; scale bar, 200 μm. (E) Protein levels of ZEB1, CD44, N-cadherin, PCNA, mmp2, and vimentin in U87MG, A172, and U118MG cells transduced with ovNC or ovFTO lentivirus. GAPDH was used as control. (F) Representative images of tumor sphere formation of GSC267 cells transduced with lentivirus and quantified by the diameter of spheres. Scale bar, 100 μm. (G) Extreme limiting dilution assay of GSC267 cells transduced with lentivirus. Data represent mean ± SD from three independent experiments. (H) Protein levels of FTO, CD44, and YKL40 in GSC267 cells transduced with lentivirus. β-Actin was used as control for normalization. (I) In vivo bioluminescent imaging analysis of tumor growth in xenograft nude mice at day 12 after implantation. (J) H&E staining of xenograft sections from FTO-overexpressing or negative control U87MG cell tissues on the day of sacrifice; scale bars, 1,000 μm (left) and 100 μm (right). (K) Survival analysis of nude mice orthotopically implanted with U87MG cells transduced with lentivirus overexpressing the control sequence or FTO (n = 5/group); log rank tests were used to identify the survival significance of the differences. Comparisons between two independent samples and among multiple samples were performed using two-tailed t tests and one-way ANOVA, respectively. Error bars indicate at least three independent experiments, and data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Knockdown of FTO promoted growth, migration, and invasion of GBM cells in vitro and in vivo
To further confirm the function of FTO in GBM cells, we silenced FTO in LN229, U251, and P3 GBM cells using two distinct short hairpin RNAs (shRNAs) targeting FTO (shFTO-1 and shFTO-2) (Figure 3A). FTO depletion in GBM cells markedly promoted the proliferation, invasion, and migration ability of GBM cells (Figures 3B–3D). Furthermore, western blot results showed that knockdown of FTO elevated the protein expression levels of CD44, ZEB1, N-cadherin, PCNA, vimentin, and mmp2 (Figure 3E). Moreover, knockdown of FTO significantly promoted the neurosphere formation frequency and malignant phenotypic transformation of GSCs 8–11 (Figures 3F–3H). In vivo, FTO-knockdown and negative control LN229-luc cells were implanted into BALB/c mice in situ. FTO-knockdown tumors displayed elevated tumor growth and invasiveness and prolonged the survival of tumor-bearing mice (Figures 3I–3K and S2D). Therefore, these results supported the findings that FTO inhibited GBM malignant progression in vitro and in vivo.
Figure 3.

Knockdown of FTO promoted growth, migration, and invasion of GBM cells in vitro and in vivo
(A) Expression levels of FTO in U251, LN229, and P3 cells transduced with control lentivirus (shNC) or FTO-knockdown lentivirus carrying two short hairpin RNA sequences (shFTO-1 and shFTO-2). GAPDH was used as control. (B) The proliferation capacity of LN229 and U251 cells transduced with lentivirus was assessed using a CCK-8 assay. (C) Transwell assays of LN229 and U251 cells. Representative images are shown. Scale bar, 50 μm. Histograms represent the number of migrating or invading cells. (D) Three-dimensional tumor spheroid invasion assay of LN229 cells transduced with lentivirus. Images at 0, 24, 48, and 72 h are shown; scale bar, 200 μm. (E) Protein levels of ZEB1, CD44, N-cadherin, mmp2, PCNA, and vimentin in P3, LN229, and U251 cells transduced with shNC, shFTO-1, or shFTO-2 lentivirus. GAPDH was used as control. (F) Neural sphere formation assay of GSCs 8–11 transduced with shNC, shFTO-1, or shFTO-2 lentivirus. (G) Extreme limiting dilution assay of GSCs 8–11 transduced with shNC, shFTO-1, or shFTO-2 lentivirus. (H) Protein levels of FTO and YKL40 in GSCs 8–11 transduced with shNC or shFTO lentivirus. β-Actin was used as control for normalization. (I) In vivo bioluminescent imaging analysis of tumor growth in xenograft nude mice at day 6. (J) H&E staining of xenograft sections from FTO-knockdown or negative control LN229 cell tissues on the day of sacrifice; scale bars, 1,000 μm (left) and 100 μm (right). (K) Survival analysis of nude mice orthotopically implanted with LN229 cells transduced with shNC or shFTO lentivirus (n = 5/group); log rank tests were used to identify the survival significance of the differences. Comparisons between two independent samples and among multiple samples were performed using two-tailed t tests and one-way ANOVA, respectively. Error bars indicate at least three independent experiments, and data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
FTO regulated the maturation of miR-10a in an m6A-dependent manner
Wei et al.22 found that FTO-mediated demethylation of m6A was prominent in the cell nucleus, and our IHC analysis also showed that FTO was localized primarily in the nucleus in GBM tissues (Figure 1D). We then examined the role of FTO in modulating m6A modification. Overexpression of FTO led to markedly decreased m6A levels in both U87MG and A172 GBM cells, while knockdown of FTO in U251 and LN229 GBM cells showed the opposite results (Figure 4A).
Figure 4.

FTO regulated the maturation of miR-10a in an m6A-dependent manner
(A) Quantification assay m6A levels in U87MG, A172, LN229, and U251 cells transduced with ovNC, ovFTO, shNC, shFTO-1, and shFTO-2 lentiviruses. (B) Volcano plots showing the miRNA sequencing results of up- and downregulated miRNAs in GBM tissues compared with normal brain tissue (NT). (C) Venn plot displaying the significantly upregulated miRNAs (log FC > 4, padj < 0.05) and miRNAs affected by METTL3 and HNRNPA2B1 simultaneously. Mature miR-10a-5p and pri-miR-10a expression levels in (D) U87MG and A172 cells after transduction with ovNC and ovFTO lentiviruses or (E) LN229 and U251 cells after transduction with shNC, shFTO-1, and shFTO-2 lentiviruses. (F) Detection of pri-miR-10a m6A modification levels in control or FTO-overexpressing U87MG cells and in shNC, shFTO-1, or shFTO-2 LN229 cells, as detected by MeRIP-qPCR assays. (G) Mature miR-10a-5p and pri-miR-10a expression levels in U87MG and LN229 cells after transfection with small interfering RNA control (siNC) or small interfering RNA HNRNPA2B1 1 (siHNRNPA2B1-1) and 2 (siHNRNPA2B1-2). (H) Interaction between DGCR8 and HNRNPA2B1. U87MG cells were transduced with ovNC or ovFTO lentiviruses and used for co-immunoprecipitation with an anti-DGCR8 antibody. Western blots for HNRNPA2B1, FTO, and DGCR8 are shown. Quantification histogram represented relative HNRNPA2B1 enrichment level. (I) Co-immunoprecipitation by anti-DGCR8 antibody in LN229 cells transduced with shNC, shFTO-1, and shFTO-2 lentiviruses. Quantification histogram represented relative HNRNPA2B1 enrichment level. (J) Detection of the abundance of pri-miR-10a binding to HNRNPA2B1 in U87MG cells by immunoprecipitation with an anti-HNRNPA2B1 antibody followed by RIP-qPCR assays. (K) Schematic of dual luciferase plasmid design. (L) Dual luciferase assay of U87MG cells. Cells were co-transfected with the wild-type (WT) or mutant (Mut) reporter vectors and siNC or siHNRNPA2B1 as indicated. (M) Schematic of pri-miR-10a mutation design. Comparisons between two independent samples and among multiple samples were performed using two-tailed t tests and one-way ANOVA, respectively. Error bars indicate at least three independent experiments, and data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
To determine the precise mechanism by which FTO regulates GBM progression, we performed differential analysis of GBM samples from TCGA and CGGA datasets on the basis of the median FTO expression cut-off (Tables S3 and S4). Gene Ontology (GO) biological process (BP) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses showed that downregulated genes in the FTO high-expression group were significantly involved in cell proliferation (such as the cell cycle and cell death), cell migration (such as extracellular matrix organization, cell migration, and wound healing), immune response (such as myeloid cell activation involved in the immune response and leukocyte migration), and carcinogenic activation pathways (Figure S3A, S3B, S4A, and S4B). To further explore the biological behaviors among GBM samples with differential FTO expression, we used the gene set variation analysis (GSVA) algorithm to estimate oncogene pathway enrichment scores for individual samples (see Supplemental method). As shown in Figures S3C and S4C, with increasing FTO expression, the cancer-promoting signaling and immune infiltration pathways were significantly inhibited, accompanied by a decrease in the immune/stromal score and an increase in tumor purity, which have been identified as a common theme in the MES-subtype signature3 and correlated with increased intratumoral heterogeneity and treatment resistance. These results suggest that FTO could affect not only the biological function of tumor cells themselves but also the tumor microenvironment to further promote the malignant progression of GBM.
miRNAs are involved in multiple cellular processes and are essential for cell development, cell differentiation, and homeostasis, demonstrating the powerful regulatory role of these small RNAs, and aberrant miRNA expression has been associated with many different diseases, including tumor progression and immune disorder.23,24 Previously, Alarcón et al.25,26 found that m6A was enriched in primary miRNA transcripts (pri-miRNAs), a subset of which (55 miRNAs; Table S5) could be recognized by the m6A reader HNRNPA2B1 by recruiting the miRNA microprocessor complex protein DGCR8 and thus regulating miRNA maturation. Therefore, we wondered whether FTO could regulate pri-miRNA processing in a HNRNPA2B1-dependent manner. We then performed miRNA sequencing (miRNA-seq) and differential analysis to explore differentially regulated miRNAs in 11 GBM tissues compared with 8 normal brain tissues (Figure 4B; Table S6). We found that the expression of miR-10a and miR-445, simultaneously regulated by m6A and HNRNPA2B1, was significantly upregulated in GBM tissues (log2 fold change [FC] > 4, padj < 0.05; Figure 4C). In our miRNA sequencing results, the expression of both miR-10a-3p and miR-10a-5p was upregulated in GBM samples (Table S6). Our previous study determined that hypoxia promoted the release of miR-10a-5p into exosomes by GBM cells, stimulating the expansion and immunosuppressive function of myeloid-derived suppressor cells (MDSCs) in vivo and in vitro.27 It could also promote GBM progression by targeting myotubularin-related protein 3 (MTMR3),28 suggesting that miR-10a, whose biogenesis mechanism is not clear, is an important potential molecular target for GBM therapy. Thus, it would be of great significance to elucidate the mechanism of its maturation. Then, we tested whether FTO exerted its antitumoral effect by regulating the expression of miR-10a in GBM cells. First, we found that miR-10a-5p expression was significantly decreased in FTO-overexpressing cells and significantly upregulated in FTO-knockdown cells. Moreover, pri-miR-10a levels increased in FTO-overexpressing cells and decreased in FTO-knockdown cells (Figures 4D and 4E). The m6A RNA immunoprecipitation (MeRIP)-qPCR assay showed that the m6A modification level of pri-miR-10a was significantly decreased in FTO-overexpressing U87MG cells and increased in FTO-knockdown LN229 cells (Figure 4F). Next, to verify the function of HNRNPA2B1 in pri-miR-10a processing, we transfected small interfering RNA (siRNA) of HNRNPA2B1 into GBM cells. Real-time qPCR assays showed that knockdown of HNRNPA2B1 decreased miR-10a-5p expression and increased pri-miR-10a expression in GBM cells (Figure 4G). Next, a co-immunoprecipitation (co-IP) assay showed that overexpression of FTO decreased the interaction between DGCR8 and HNRNPA2B1 (Figure 4H). Knockdown of FTO enhanced the interaction of these two proteins (Figure 4I), but it was not interrupted by RNase (Figure S5), suggesting that the interaction is induced at the protein-protein level, consistent with a previous study.26 RNA immunoprecipitation (RIP)-qPCR assays also showed that HNRNPA2B1 could specifically bind to pri-miR-10a (Figure 4J). To further demonstrate the essential role of m6A in the regulation of pri-miR-10a processing, we predicted potential m6A modification sites on 100 sequences upstream of pre-miR-10a using the SRAMP (http://www.cuilab.cn/sramp) database.29 The results revealed only a potential m6A site, “GAA[m6A]CU,” with high confidence near the pre-miR-10a region (Figure S6). We then designed “GAACU” WT and “GATCU” Mut pri-miR-10a dual luciferase reporter plasmids30, 31, 32 (Figure 4K). Thus, mutant pri-miR-10a resists to the modification of m6A. The reporter plasmids were co-transfected into GBM cells with siNC or siHNRNPA2B1 as indicated. As expected, luciferase activity in WT pri-miR-10a transfected cells increased when HNRNPA2B1 was silenced, while knockdown of HNRNPA2B1 showed no effect on the expression of the mutant pri-miR-10a-fused reporter (Figure 4L), suggesting that the modulation of pri-miR-10a expression was under the control of HNRNPA2B1-associated m6A modification. Meanwhile, compared with the WT group, mutation on the m6A consensus sequences increased the expression of pri-miR-10a. In summary, pri-miR-10a processing is regulated by FTO-mediated m6A modification and recognized by HNRNPA2B1 through recruitment of the core microprocessor complex DGCR8 (Figure 4M).
The inhibitory effects of FTO on GBM progression are reversed by miR-10a
To confirm the role of miR-10a in GBM progression as a downstream target of FTO, we first analyzed the expression correlation of FTO and MTMR3, a target of miR-10a in GBM cells28, in the TCGA, CGGA, REMBRANDT, Gravendeel, and Philips databases. As shown in Figure 5A, FTO was significantly positively correlated with MTMR3. Then, we validated the expression of MTMR3 in GBM cells with FTO overexpression or knockdown. As shown in Figures 5B–5F, the mRNA and protein expression levels of MTMR3 were increased in FTO-overexpressing GBM cells but decreased in FTO-knockdown cells. The effect was reversed by miR-10a overexpression in FTO-overexpressing GBM cells and miR-10a inhibition in FTO-knockdown GBM cells. To further confirm that the observed FTO-mediated phenotypes were mediated by the dysregulation of miR-10a expression, we performed functional rescue assays. As shown by the CCK-8 and Transwell assays, FTO overexpression induced decreased proliferation, migration, and invasion capacity in U87MG cells, which could be reversed by miR-10a mimics transfection. Accordingly, the increased proliferation, migration, and invasion capacity in FTO-knockdown LN229 GBM cells could be rescued by suppressing miR-10a expression (Figures 5G–5I). The IHC assay also demonstrated that the MTMR3 protein level in FTO-overexpressing xenograft samples was higher than that in ovNC samples (Figure 5J). Furthermore, to confirm that the observed FTO-mediated phenotype is mediated by dysregulated expression of the target MTMR3 downstream of miR-10a, functional rescue assays were performed. As shown in Figures S7A–S7D, the decreased proliferation, migration, and invasion capacity in FTO-overexpressing GBM cells could be rescued by suppressing MTMR3 expression. In summary, these results confirmed that miR-10a is a downstream target of FTO and is involved in GBM progression.
Figure 5.

The inhibitory effects of FTO on GBM progression are reversed by miR-10a
(A) Analyses of the correlation between the expression of FTO and MTMR3, a miR-10a target, in five datasets. Statistical analysis was determined using Pearson correlation coefficient analyses. (B) MTMR3 mRNA expression in GBM cells transduced with FTO overexpression, knockdown, and the corresponding control lentiviruses. (C) Protein expression of MTMR3, CD44, PCNA, FTO, and GAPDH in U87MG cells co-transduced with ovNC or ovFTO lentivirus and mimics NC or miR-10a-5p mimics as indicated. Quantification histogram represents relative protein expression. (D) LN229 cells were co-transduced with shNC, shFTO-1, or shFTO-2 lentivirus and inhibitor NC or miR-10a-5p inhibitor as indicated. Quantification histogram represents relative protein expression. RNA expression of (E) miR-10a-5p and (F) MTMR3 in U87MG cells co-transduced with ovNC or ovFTO lentivirus and mimics NC or miR-10a-5p mimics as indicated. LN229 cells were co-transduced with shNC, shFTO-1, or shFTO-2 lentivirus and inhibitor NC or miR-10a-5p inhibitor as indicated. (G) CCK-8 assay of U87MG cells transfected with (left) mimics NC or miR-10a-5p mimics in the presence or absence of FTO overexpression of LN229 cells transfected with inhibitor NC or (right) miR-10a-5p inhibitor in the presence or absence of FTO knockdown. (H) Transwell assay of U87MG cells transfected with mimics NC or miR-10a-5p mimics in the presence or absence of FTO overexpression as indicated. Scale bar, 50 μm. (I) Transwell assay of LN229 cells transfected with inhibitor NC or miR-10a-5p inhibitor in the presence or absence of FTO knockdown. Scale bar, 50 μm. (J) IHC of MTMR3 expression in U87MG cells in xenograft sections of nude mice. Scale bar, 50 μm. Comparisons between two independent samples and among multiple samples were performed using two-tailed t tests and one-way ANOVA, respectively. Error bars indicate at least three independent experiments, and data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
SPI1 inhibited the transcriptional activity of FTO
To explore why FTO expression was downregulated in GBM cells, we predicted transcriptional regulators that might regulate FTO expression in brain tissues using the hTFtarget database (http://bioinfo.life.hust.edu.cn/hTFtarget/), and the transcription regulatory factors YY1, ZBTB33, and SPI1 had the three highest enrichment scores in brain tissues (Table S7). Of these, only the expression of SPI1 was significantly upregulated in GBM tissues, compared with normal brain and LGG samples in TCGA and CGGA microarray datasets (Figure S8A), and patients with high SPI1 expression had poorer prognosis than patients with low expression (Figure S8B). Moreover, univariate Cox regression analysis of the overall survival of GBM patients in the TCGA GBM dataset showed that high SPI1 expression (HR: 1.191; p = 0.011) was an independent risk factor associated with the prognosis of GBM patients. In addition, in subsequent multivariate Cox regression analysis, SPI1 expression (HR: 1.170; p = 0.032) remained a statistically significant risk factor in GBM patients after adjustment for age, sex, molecular subtype, and G-CIMP status (Figure S8C; Table S8). The binding site of SPI1 on the FTO promoter was predicted by the hTFtarget database to be Chr16,53703677,53,704,387, in which the only binding motif “GGAA” of SPI1, predicted by the JASPAR database (http://jaspar.genereg.net/matrix/MA0080.2/),33 which has been validated in numerous studies,34, 35, 36 was identified near the transcription start site (TSS) (Figure 6A).
Figure 6.

SPI1 inhibited the transcriptional activity of FTO
(A) Predicted SPI1 DNA-binding sequences in the FTO promoter region. (B) The correlation between FTO and SPI1 and the correlation between SPI1 and MTMR3 in TCGA and CGGA databases. Statistical analysis was determined using Pearson correlation coefficient analyses. (C) The relative mRNA expression of SPI1 (left) and FTO (right) in GBM cells transfected with siNC, siSPI1, or siSPI1-2. (D) The protein expression of SPI1, FTO, MTMR3, N-cad, CD44, PCNA, and vimentin in GBM cells transfected with siNC, siSPI1, or siSPI1-2. (E) ChIP-PCR of the FTO promoter region in GBM cells. (F) Relative FTO promoter luciferase activity in GBM cells transfected with siNC, siSPI1-1, or siSPI1-2. (G) Relative FTO promoter luciferase activity in the indicated GBM cells with or without the mutation of the SPI1 binding motif. (H) Transwell assays of U87MG and U118MG GBM cells transfected with siNC or siSPI1 in the presence or absence of FTO as indicated. Scale bar, 50 μm. (I) CCK-8 assays of U87MG and U118MG GBM cells transfected with siNC or siSPI1 in the presence or absence of FTO as indicated. Comparisons between two independent samples and among multiple samples were performed using two-tailed t tests and one-way ANOVA, respectively. Error bars indicate at least three independent experiments, and data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
We then performed differential analysis of GBM samples of TCGA and CGGA datasets on the basis of the median SPI1 expression cut-off (Tables S9 and S10). GO BP and KEGG enrichment analyses showed that upregulated genes in the SPI1 high-expression group were significantly involved in cell proliferation, cell migration, and immune response-related terms, similar to the downregulated genes in the FTO-high group (Figures S9A, S9B, S10A, and S10B). In addition, with increasing SPI1 expression, the cancer-promoting signaling and immune infiltration pathways were significantly enriched, accompanied by a decrease in immune/stromal score and an increase in tumor purity (Figures S9C and S10C), estimated via the GSVA algorithm, which was opposite to the FTO-regulated phenotype.
Furthermore, we found that FTO and MTMR3 expression was negatively correlated with SPI1 expression in TCGA and CGGA datasets (Figure 6B). We also confirmed that inhibition of SPI1 successfully upregulated FTO and MTMR expression and downregulated CD44, vimentin, and PCNA expression (Figures 6C and 6D). The chromatin immunoprecipitation (ChIP)-qPCR assay showed that the FTO promoter region was recognized by SPI1 (Figure 6E), and luciferase promoter assays demonstrated that SPI1 knockdown relieved the inhibition of FTO expression (Figure 6F). Furthermore, the SPI1 binding site mutation in the FTO promoter canceled this transcriptional regulation (Figure 6G). To further demonstrate that SPI1 inhibited the transcriptional activity of FTO, we conducted functional rescue assays. As CCK-8 and Transwell assays showed, SPI1 knockdown induced the inhibition of proliferation, migration, and invasion capacity in GBM cells, which could be reversed by suppression of FTO expression (Figures 6H and 6I).
The SPI1 inhibitor DB2313 blocked the transcriptional inhibition of FTO
As SPI1 knockdown significantly upregulated the expression of FTO and inhibited the malignant progression of GBM cells, we reasoned that targeting SPI1 may provide a novel therapy for GBM. DB2313, which harbors the amidine-benzimidazole-phenyl platform that provides excellent recognition for expanded AT sites, was selected as the SPI1 inhibitor.36 The binding model between SPI1 and DB2313 i shown in Figures 7A and 7B. Then, we tested the half maximal inhibitory concentration (IC50) of DB2313 in GBM cells in vitro (Figure S11A). According to the results, we chose 5 and 10 μg/mL in vitro and 10 mg/kg/day in vivo for subsequent experiments. We demonstrated that DB2313 restored FTO expression and inhibited PCNA, CD44, and MTMR expression in the U118 and U87MG cell lines (Figures 7C and S11B). After 10 μg/mL DB2313 treatment on day 0, U118MG and U87MG cell proliferation, invasion, and migration were inhibited compared with those after DMSO treatment in vitro, as shown by CCK-8 and Transwell assays (Figures 7D and 7E). The Molinspiration Cheminformatics database (http://www.molinspiration.com) predicted that DB2313 had a low possibility to penetrate the blood-brain barrier. Thus, we used subcutaneous xenograft model to evaluate the therapeutic efficacy of DB2313 in vivo. The results showed that DB2313 inhibited tumor burden significantly (Figures 7F and 7G). We also evaluated the tumor proliferation and invasion index in these solid tumors. The DB2313-treated tumors exhibited increased FTO expression and decreased Ki67 and CD44 expression compared with the control group (Figure 7H). In summary, SPI1 inhibited FTO expression at the transcriptional level. DB2313, which disrupts the interaction of SPI1 and FTO, may be a potential novel GBM therapeutic method.
Figure 7.

The SPI1 inhibitor DB2313 interrupted the transcriptional inhibition of FTO
(A) The two-dimensional (2D) binding mode of SPI1 and DB2313. (B) The binding model of DB2313 on molecular surface of SPI1. DB2313 is colored in cyan, and the molecular surface of SPI1 is colored in pale yellow (top); 3D binding mode of SPI1 and DB2313. DB2313 is colored in cyan, the surrounding residues in the binding pockets are colored in yellow, and the backbone of the receptor is depicted as white cartoon with transparency (below). (C) Protein expression of FTO, MTMR3, CD44, PCNA, and GAPDH in GBM cells treated with DMSO or DB2313. (D) CCK-8 assay of GBM cells treated with DMSO or DB2313 (10 μg/mL). (E) Transwell assay of GBM cells treated with DMSO or DB2313 as indicated. Scale bar, 50 μm. (F) DB2313 (10 mg/kg/day) or vehicle was intraperitoneally injected into mice (n = 5 per group) after subcutaneous implantation of U87MG cells. (G) Tumor volumes and weights were measured from sacrificed mice and are shown as box-and-whisker plots. (H) Represented images of immunofluorescence staining for FTO, Ki67, and CD44 in in xenograft sections from U87MG GBM cell subcutaneous xenograft mice treated with DB2313 or matched group. Scale bar, 50 μm. (I) Proposed model underlying the roles of FTO-mediated pri-miR-10a processing in GBM tumors. Comparisons between two independent samples and among multiple samples were performed using two-tailed t tests and one-way ANOVA, respectively. Error bars indicate at least three independent experiments, and data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Discussion
GBM, most of which is isocitrate dehydrogenase 1/2 (IDH1/2) wild-type, is the most aggressive primary nervous system cancer, and understanding its molecular pathogenesis is crucial to improving the efficacy of GBM diagnosis and treatment.37, 38, 39 Dysregulation of m6A-methylated mRNA can affect cancer initiation and progression.40,41 FTO, as a genuine m6A demethylase, has been suggested to play various functions in tumor progression.16,17,20 The specific functions and molecular regulatory mechanisms of FTO in GBM are still unclear. Our results revealed that m6A RNA modifications regulated by FTO play a central role in regulating GBM progression. First, we found that in glioma specimens, the degree of malignancy increased with the downregulation of FTO expression and confirmed that FTO significantly inhibited the proliferation, invasion and migration of GBM cells in vitro and in vivo. Second, knockdown of FTO significantly promoted the m6A modification of pri-miR-10a, which was then recognized by HNRNPA2B1 and further promoted the maturation of miRNA-10a by recruiting DGCR8, thus promoting the malignant progression of GBM cells by targeting the tumor suppressor protein MTMR3. Third, transcriptional activity of FTO could be inhibited by the transcription factor SPI1, and SPI1 knockdown or inhibition of transcriptional activity by DB2313 compound restored FTO expression and prevented GBM tumor progression (Figure 7I). To our knowledge, this is the first comprehensive study to show that FTO modulates GBM progression by regulating m6A modification of noncoding RNAs. Together, our findings demonstrate that FTO may serve as a novel prognostic indicator and therapeutic molecular target of GBM. Thus, restoring the endogenous expression of FTO with the SPI1 inhibitor DB2313 may have therapeutic potential for GBM patients.
Since 2017, studies on the mechanism linking m6A methylation and oncogenesis in GBM cells have been continually reported and have drawn inconsistent conclusions. Some studies have shown that knockdown of METTL3 and METTL14, key components of the RNA methyltransferase complex, dramatically promote human GSC self-renewal and tumorigenesis, while inhibition of FTO suppresses tumor progression.42 In addition, inhibition of FTO by its specific inhibitor, MA2, significantly suppressed progression of GBM cells.43 On the other hand, several other independent research groups found that METTL3 could promote the malignant progression of GBM through different molecular mechanisms, such as by enhancing SOX2 mRNA stabilization,44 modulating nonsense-mediated mRNA decay (NMD) of splicing factors and alternative splicing isoform switches,45 and increasing the protein expression of ADAR1, a key adenosine-to-inosine (A-to-I) RNA editing factor.46 The controversial conclusions may be due to the samples used in these studies. These results indicate that the function of m6A modifications in GBM should be further investigated. Su et al.47 found that R-2-hydroxyglutarate (R-2HG), which accumulates to high levels in IDH1/2 mutant leukemia, inhibited FTO, in turn decreasing the stability of MYC/CEBPA transcripts in an m6A-dependent manner, leading to leukemia oncogenesis. In addition, the authors showed that R-2HG can inhibit the proliferation of glioma cell lines, suggesting that in low-grade IDH1/2 mutant gliomas, the levels of FTO, kept low by R-2HG, result in an overall high level of m6A methylation. These observations suggest that in low-grade gliomas with IDH1/2 mutations, FTO is kept low by R-2HG, leading to a significant increase in overall m6A RNA methylation and that among gliomas, the tumors with high FTO and low MYC levels should be the most sensitive to R-2HG therapy. Thus, a large number of mechanistic studies should be performed to confirm whether this is the case in glioma cells with that specific IDH mutation. In addition, another study found that the expression of FTO was significantly upregulated in IDH mutant glioma compared with IDH wild-type glioma by systematically analyzing the expression of thirteen widely reported m6A RNA regulators in 904 gliomas with RNA sequencing data from the Chinese Glioma Genome Atlas (n = 309) and The Cancer Genome Atlas (n = 595) datasets.48 Somatic mutations in the IDH1/2 gene occur in approximately 80% of grade II and III gliomas and secondary glioblastomas (sGBMs) and are very rare in grade IV GBM patients. In our study, we showed that FTO expression was negatively associated with World Health Organization (WHO) grades of glioma in multiple databases of glioma specimens and in our local tissues (Figure 1). Moreover, the expression of FTO was significantly upregulated in IDH-mutated glioma, and Cox analysis showed that FTO is a protective factor in GBM (Figure S1), consistent with previous reports.21,48 Using multiple GBM cell lines and GSCs, we also confirmed that FTO is a GBM tumor suppressor protein in vivo and in vitro.
Functional enrichment analysis of the sequencing data of tumor specimens from TCGA and CGGA showed that FTO not only regulates the biological functions of GBM cells themselves but also affects the tumor microenvironment (Figures S3 and S4). As important regulatory molecules of biological processes, miRNAs affect almost every aspect of tumors and the tumor microenvironment. During the initiation and development of tumors, miRNA expression profiles of cancer cells change significantly, in which the subsequent processing of pri-miRNA is tightly regulated.49 m6A has been reported to be enriched in pri-miRNA transcripts, marking them for recognition and processing by DGCR8.25 In a follow-up study, the same group also found that HNRNPA2B1 is a nuclear reader of the m6A mark, binds to m6A marks in a subset of pri-miRNA transcripts (including pri-miR-10a), interacts with DGCR8, and promotes pri-miRNA processing.26 Here, we explored the functions of FTO in miRNA maturation in GBM cells and discovered that FTO regulated the maturation of miR-10a in an m6A/HNRNPA2B1-dependent manner. It has been reported that miR-10a-5p promotes GBM progression by targeting MTMR3,28 which can interact with mTORC1 and suppress its activity.50 Therefore, our research demonstrated that FTO suppresses GBM progression through the miR-10a-5p/MTMR3 axis, inhibiting PI3k/akt/mTOR signaling and other carcinogenic pathways (Figures S3 and S4). In addition, miR-10a-5p plays an important role in the glioma tumor environment via exosome secretion, promoting MDSC immunosuppressive functions.27 Numerous studies have also shown that miR-10a can modulate metabolic reprogramming of other immune cells and inflammation,51, 52, 53, 54 suggesting the key role of miR-10a in tumor progression and immunoregulation. Thus, clarifying the mechanism of its biogenesis is of great importance in our search for effective treatments.
We used database predictions to identify transcription factors that lead to downregulation of FTO expression in GBM and found that SPI1 expression was significantly upregulated in GBM. Patients with high expression had a significantly poor prognosis and a negative correlation with FTO expression. Intriguingly, further functional enrichment analysis of the sequencing data of tumor specimens from TCGA and CGGA databases also showed that SPI1 not only regulates the biological functions of GBM cells themselves but also affects the tumor microenvironment, a function opposite to that of FTO (Figures S9 and S10). Further experiments proved that SPI1 is a transcriptional suppressor of FTO, the knockdown of which could reverse the expression of FTO and inhibit GBM progression. Therefore, blocking the inhibition of endogenous FTO expression by SPI1 may provide new clues and strategies for the treatment of patients with GBM. SPI1, also called PU.1/Sfpi1, has been reported to regulate tumorigenesis in various kinds of cancers.55,56 Specific transcriptional activation or repression function of SPI1 is various under different conditions. SPI1 can either activate or repress the transcription of genes by interacting with chromatin modifiers or cross-talk with other lineage-specific transcription factors.57 SPI1 was reported to bind the methyl CpG-binding protein 2 (MeCP2) to repress genes together with HDAC1-mSIN3a.58,59 Also, by forming complex with HDACs, SPI1 induces transcriptional repression in several gene promoters including the c-myc promoter.60 In leukemia differentiation, it was reported that SPI1 stimulates target gene transcription together with its coactivator c-Jun during myelopoiesis. Nevertheless, PU.1 blocks transcription by interacting with GATA-1 and inhibits histone H3K9 acetylation.61 On the basis of the characteristics of the expression level of SPI1 in sequencing data of tumor specimens from TCGA and CGGA databases and the results of our experiments, we found that SPI1 functions as a transcription suppressor to FTO, opening a new path for further understanding of the application of SPI1 inhibitor. DB2313, one of the heterocyclic diamidines, was demonstrated to inhibit PU.1 function by targeting the DNA minor groove.36,62 In this research, we found that DB2313 can disrupt the suppressing effect of SPI1 on FTO transcriptional activity and inhibit GBM cell proliferation, migration and invasion, indicating that DB2313 can be used as an anti-GBM tumor drug. However, one drawback of this compound is that it needs to be further optimized to better cross the blood-brain barrier. In recent years, pharmaceutical companies and some academic institutions have increased their research efforts to evaluate various non-invasive technologies for central nervous system treatment, including receptor-mediated transcytosis and the use of neurotropic viruses, nanoparticles, and exosomes, some of which have entered clinical trials63 and may provide us with effective drug delivery strategies in future work.
In summary, our work systematically revealed the tumor-suppressive properties of FTO in GBM. Moreover, FTO inhibited growth, migration and invasion of GBM cells in vitro and in vivo. We demonstrated that decreased FTO expression could induce the downregulation of MTMR3 expression by modulating the processing of pri-miR-10a in an m6A/HNRNPA2B1-dependent manner in GBM cells. Finally, FTO was suppressed by SPI1 at the transcriptional level, and this process could be interrupted by DB2313, which may have therapeutic potential for GBM patients. Thus, our findings indicate that FTO and SPI1 could serve as promising prognostic and treatment molecular targets for GBM patients.
Materials and methods
Patient specimens and public clinical datasets
Tissues of gliomas, including WHO grade II and III gliomas and GBM, were obtained from patients who were diagnosed with glioma and underwent surgery in the Department of Neurosurgery of Qilu Hospital at Shandong University. Normal brain tissue was obtained from patients who underwent craniocerebral decompression treatment for brain trauma. All participants provided written informed consent, and the research was approved by the Ethics Committee on Scientific Research of Shandong University Qilu Hospital (approval number KYLL-2018-324).
The TCGA HG-UG133A platform microarray data and corresponding clinicopathological parameters of GBM patients were obtained from https://xena.ucsc.edu/public. The CGGA GBM transcriptome data and corresponding clinicopathological parameters of GBM patients were obtained from the CGGA database (http://www.cgga.org.cn/). The RNA sequencing transcriptome data for FTO, SPI1, MTMR3, PCNA, vimentin, and CD44; the Kaplan-Meier survival analysis data for FTO and SPI1 in TCGA, CGGA, REMBRANDT, Gravendeel, and Philips datasets; and the correlation data for FTO and SPI1 were downloaded from the GlioVis database (http://gliovis.bioinfo.cnio.es/). The miRNA-seq sequencing data have been deposited in Genome Sequence Archive (GSA) under accession number CRA002339, to be released when the paper is published. The processed data and basic association analyses will be made available in supplementary data or from the corresponding author on reasonable request.
Cell culture
The glioma cell lines U87MG, U251, A172, LN229, and U118MG were obtained from the Type Culture Collection of the Chinese Academy of Sciences. All cells were cultured in DMEM (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin. All patient-derived GSC cell lines and neural progenitor cell (NPC) were kindly donated by Dr. Frederick F. Lang and Dr. Krishna P.L. Bhat (The University of Texas MD Anderson Cancer Center, Houston, TX).64, 65, 66 The P3 cell line isolated from human GBM tissue was kindly provided by Prof. Rolf Bjerkvig (University of Bergen). P3 cells and GSCs were cultured as previously described.67,68 All cells were maintained in a humidified incubator with 5% CO2 at 37°C. The cells were routinely tested for mycoplasma contamination.
RNA extraction and real-time quantitative PCR
Real-time qPCR was performed as previously described.69 In brief, total RNA was extracted from cells using TRIzol (Thermo Fisher Scientific) and reverse-transcribed using the ReverTra Ace qPCR RT kit (FSQ-101; Toyobo). Quantitative PCR was conducted using TB Green Premix Ex Taq (Takara) on a Roche Light Cycler 480 according to the protocol. Primers used in this study are listed in Table S10. The levels of miR-10a-5p and other RNAs were normalized to those of U6 and GAPDH respectively.
RNA interference transfection and lentivirus transduction
Human full-length FTO sequence and control sequence were cloned into the GV492 lentiviral vector to construct lentiviruses for stable overexpression (Genechem). Sequences of FTO knockdown (shFTO-1, shFTO-2), and the scramble control (shNC) were cloned into pLKD lentiviral vector to construct lentiviruses for FTO knockdown (Obio). GBM cells were infected with lentiviruses at 50% confluence after culture in 6-well plates. Stably transduced cells were selected using puromycin (4 μg/mL) for 2 weeks. miR-10a-5p mimics, inhibitor, and small interfering RNA targeting SPI1, MTMR3, and HNRNPA2B1 were synthesized by BioSune (Shanghai, China). Transfection was performed using the Lipofectamine 3000 kit (L3000015; Invitrogen) following the manufacturer’s protocol. All sequences are listed in Table S11.
Immunohistochemistry
Clinical tissue and nude mouse intracranial tumor implantation specimens were embedded in paraffin, and IHC assays were performed as previously described69 according to the manufacturer’s protocol for primary antibodies. The following primary antibodies were used in this research: anti-FTO (ab126605; Abcam), anti-CD44 (15675-1-AP; Proteintech), and anti-vimentin (5741; Cell Signaling Technology).
Western blot
Cell pellets were harvested and lysed using RIPA buffer containing 1% protease and phosphate inhibitor cocktail (P8340; Sigma-Aldrich). Western blotting was performed as previously described69 according to the manufacturer’s protocol with primary antibodies overnight and probing with secondary antibodies. Primary antibodies against the following antigens were used: ZEB1 (3396; Cell Signaling Technology; 1:1,000), PCNA (13110; Cell Signaling Technology; 1:1,000), DGCR8 (ab191875; Abcam; 1:1,000), FTO (ab126605; Abcam; 1:10,000), MTMR3 (21336-1-AP; Proteintech; 1:1,000), SPI1 (PU.1; 2266; Cell Signaling Technology; 1:1,000), β-actin (60008-1-Ig; Proteintech; 1:10,000), vimentin (5741; Cell Signaling Technology; 1:1,000), N-cadherin (13116; Cell Signaling Technology; 1:1,000), YKL40 (ab77528; Abcam; 1:1,000), CD44 (15675-1-AP; Proteintech; 1:1,000), MMP2 (40994; Cell Signaling Technology; 1:1,000), and GAPDH (ab8245; Abcam; 1:1,000).
Cell proliferation assays
We seeded 2000 GBM cells in 96-well plates and measured cell viability using the Cell Counting Kit-8 (CCK-8; CK04; Dojindo, Kumamoto, Japan) assay at 0, 24, 48, and 72 h according to the manufacturer’s protocol. EdU assays were performed using an EdU assay kit (C10310; RiboBio, Guangzhou, China) with GBM cells seeded in 24-well plates at 2 × 105 cells per well.
Transwell and 3D tumor spheroid invasion assays
Briefly, 2 × 104 GBM cells were used for the Transwell assay according to previously described methods.70 A total of 2 × 105 GBM cells were cultured in spheroid formation ECM for 72 h to generate tumor spheroids and used for the 3D tumor spheroid invasion assay with a 96-well 3D spheroid BME cell invasion assay kit (3500-096-K; Trevigen). Images were taken at 0, 24, 48, and 72 h using a Leica microscope.
Neurosphere formation assay and extreme limiting dilution assay
Glioma stem cells transduced by different lentiviruses were implanted into 6-well plates at a density of 1,000 cells/well. After 2 weeks culture, images were taken using a Leica microscope. The number of neurosphere in the field under the microscope was used for quantification. For extreme limiting dilution assay, glioma stem cells transduced by lentiviruses were implanted in 96-well plates at a density of 1, 2, 4, 8, 16, 32, 64, and 128 cells/well. After 1 week culture, the number of wells that developed neurosphere was recorded for analysis. Data were analyzed using ELDA software (http://bioinf.wehi.edu.au/software/elda/).71
Luciferase reporter assay
Pri-miR-10a sequence was cloned into a Pmir-GLO dual luciferase expression vector (GenePharma, Shanghai, China) containing Renilla luciferase (R-luc) and firefly luciferase (F-luc). Then, “GAACU” was replaced by “GATCU” to form Mut-pri-miR-10a reporter.30, 31, 32 GBM cells transfected with FTO promoter region reporter vectors WT pGL3-FTO and Mut pGL3-FTO (GenePharma) were further co-transfected with small interference sequences or negative control siRNA. The Dual Luciferase Reporter kit (Promega) was used to examine luciferase activity according to the manufacturer’s protocol. The ratios of firefly and Renilla luciferase activities were determined 48 h post-transfection.
Quantitative assay of relative m6A levels
We obtained total RNA of glioma cell lines transduced by lentiviruses using TRIzol and performed an RNA m6A quantitative assay using an EpiQuik m6A RNA Methylation Quantification Kit (colorimetric; P-9005-48; EpiGentek) according to the manufacturer’s instructions. The m6A level was tested first by OD450 and then compared with the standard curve.
M6A RNA immunoprecipitation
MeRIP was performed using a Magna RIP Kit (17-10499; Millipore) and anti-m6A antibody (202003; SYSY). In brief, cells transduced with lentivirus were harvested and washed with cold PBS. The cells were lysed with complete RIP lysis buffer on ice for 5 min. Lysates were stored at −80°C. Magnetic beads were prepared for immunoprecipitation, and antibodies were bound to the magnetic beads according to the manufacturer’s instructions. After the RIP lysate was thawed and centrifuged, 100 μL supernatant was removed and added to each bead-antibody complex. The complex was incubated overnight with rotation at 4°C. The complex was washed using a magnetic separator and RIP wash buffer. The RNA was purified using Proteinase K and phenol:chloroform:isoamyl alcohol according to the instructions. The purified RNAs were resuspended in 10–20 μL of RNase-free water for subsequent analysis. Real-time qPCR was used for RNA quantification. Ten percent of the lysis products were used for input and the rests were used for immunoprecipitation procedure. Percentage of input was determined by comparing relative expression level of pri-miR-10a in immunoprecipitation samples to 10 times of the expression level in input samples, respectively.
Co-immunoprecipitation
After transduction with lentiviruses to stably overexpress or knockdown FTO, GBM cells were used to perform co-IP using a Pierce Co-Immunoprecipitation Kit (26149; Thermo Fisher Scientific) according to the manufacturer’s protocol. In brief, immunoprecipitation of DGCR8 was performed using an anti-DGCR8 antibody overnight at 4°C. The next day, the complex was treated with RNase for 10 min at 37°C. Then, anti-HNRNPA2B1, anti-FTO, and anti-DGCR8 primary antibodies were used for western blot analysis.
Chromatin immunoprecipitation
ChIP assays were performed using a ChIP kit from Merck Millipore (17-295) according to the manufacturer’s instructions. DNA fragments at the promoter region co-immunoprecipitated with SPI1 were quantified using qPCR.
Animal studies
BALB/c male nude mice were 4 weeks old. GBM cells with stable overexpression or knockdown of FTO and ovNC or shNC were transduced with lentivirus expressing luciferase. The cells were intracranially injected at a density of 5 × 105/10 μL into every mouse to form an orthotopic xenograft model. Coordinates of injection were 1 mm anterior and 2.5 mm right to the bregma, at a depth of 3.5 mm (the right frontal lobes of the mouse). Every 6 days, bioluminescence imaging (IVIS Lumina Series III; PerkinElmer, Waltham, MA) was used to image the mouse. At 8 days, we randomly chose 5 mice from each group to euthanize them, and their brain tissues were fixed with paraformaldehyde for further study. Another 5 mice were used for survival time analysis. For DB2313 (563801; MedKoo) anti-tumor research, male nude mice were subcutaneously injected with 5 × 106 U87MG cells suspended in 0.1 mL PBS. After 7 days, mice were intraperitoneally injected with DB2313 at density of 10 mg/kg/day dissolved in PBS solvent containing 10% DMSO for 7 days. The other group treated with vehicle only was set as the control group. Two weeks after glioma cell injection, mice were euthanized for further research. All animal experiments were approved by the guidelines of the Institutional Animal Care and Use Committee of Qilu Hospital of Shandong University.
Immunofluorescence
For immunofluorescence study of tissues, sections were treated using deparaffinization and antigen retrieval. After blocking with serum of bovine or rabbit serum albumin for 30 min, the samples were incubated with primary FTO antibodies (abca dies [9449; Cell Signaling Technology; 1:500] and CD44 [15675-1-AP; Proteintech; 1:1,000]) for 2 h at room temperature or overnight at 4°C. The samples were then incubated with species-appropriate horseradish peroxidase (HRP)-labeled secondary antibodies for FTO and Ki67 primary antibodies, and immunofluorescent-labeled secondary antibodies for CD44 primary antibodies for 45 min at room temperature at a dilution of 1:500. DAPI (G1012; Servicebio) was used to mount coverslips. Then the slices were placed under the scanner (Pannoramic; MIDI:3Dhistech) to capture the images.
Statistical analysis
Data are presented as mean ± SD unless otherwise specified. All experiments related to cell culture were repeated at least three times. Comparisons between two independent samples and among multiple samples were performed using two-tailed t tests and one-way ANOVA, respectively. Survival data for mice and humans were analyzed by the Kaplan-Meier method. The Pearson correlation coefficient and R2 values were used to analyze the correlations between the expression of two genes in TCGA or other databases. p values < 0.05 were considered to indicate statistical significance. p values are indicated as follows: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Data analysis was carried out using GraphPad Prism version 8.0.2 for Windows. All data processing with R packages was performed using R Studio version 3.6.3 for Windows.
Data availability
The miRNA sequencing data have been deposited in GSA under accession number CRA002339. The processed data and basic association analyses will be made available in supplementary data or from the corresponding author on reasonable request.
Acknowledgments
GSC cell lines used in our study were kindly provided by Dr. Frederick F. Lang and Dr. Krishna P.L. Bhat. We thank the surgeons and patients who participated in these studies and the TCGA, CGGA, REMBRANDT, Gravendeel, and Phillips databases for providing large amounts of data. This work was financially supported by the National Natural Science Foundation of China (81902540 and 81702468) and the Natural Science Foundation of Shandong Province of China (ZR2019BH057 and ZR2020QH174).
Author contributions
S. Zhang and S. Zhao performed most experiments. Y.Q., B.L., Z.G., and Z.P. helped with the xenograft model. J.X., W.Q., Q.G., Y.F., Z.C., and S.W. helped with the in vitro study. R.Z. and C.J. performed bioinformatics analysis. S.N. and J.W. provided some reagents. H.X., H.W., and L.D. gave advice on study design. X.G., F.X., and G.L. contributed to funding acquisition, supervision, review of the manuscript, figures, and tables. All authors have read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.12.035.
Contributor Information
Rongrong Zhao, Email: qlyyzrr@email.sdu.edu.cn.
Gang Li, Email: dr.ligang@sdu.edu.cn.
Supplemental information
References
- 1.Jackson C.M., Choi J., Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat. Immunol. 2019;20:1100–1109. doi: 10.1038/s41590-019-0433-y. [DOI] [PubMed] [Google Scholar]
- 2.Bi J., Chowdhry S., Wu S., Zhang W., Masui K., Mischel P.S. Altered cellular metabolism in gliomas - an emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer. 2020;20:57–70. doi: 10.1038/s41568-019-0226-5. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q., Hu B., Hu X., Kim H., Squatrito M., Scarpace L., deCarvalho A., Lyu S., Li P., Li Y., et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32:42–56.e6. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan A., Ashley D., López G., Malinzak M., Friedman H., Khasraw M. Management of glioblastoma: state of the art and future directions. CA Cancer J. Clin. 2020;70:299–312. doi: 10.3322/caac.21613. [DOI] [PubMed] [Google Scholar]
- 5.Haussmann I., Bodi Z., Sanchez-Moran E., Mongan N., Archer N., Fray R., Soller M. mA potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–304. doi: 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- 6.Zaccara S., Ries R., Jaffrey S. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cel. Biol. 2019;20:608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 7.Frye M., Harada B., Behm M., He C. RNA modifications modulate gene expression during development. Science. 2018;361:1346–1349. doi: 10.1126/science.aau1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaccara S., Ries R.J., Jaffrey S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019;20:608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y., Hsu P.J., Chen Y.S., Yang Y.G. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616–624. doi: 10.1038/s41422-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui Q., Shi H., Ye P., Li L., Qu Q., Sun G., Sun G., Lu Z., Huang Y., Yang C.G., et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F., Yi Y., Miao Y., Long W., Long T., Chen S., Cheng W., Zou C., Zheng Y., Wu X., et al. N(6)-Methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 2019;79:5785–5798. doi: 10.1158/0008-5472.CAN-18-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S., Zhao B.S., Zhou A., Lin K., Zheng S., Lu Z., Chen Y., Sulman E.P., Xie K., Bögler O., et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Visvanathan A., Patil V., Abdulla S., Hoheisel J.D., Somasundaram K. N⁶-Methyladenosine landscape of glioma stem-like cells: METTL3 is essential for the expression of actively transcribed genes and sustenance of the oncogenic signaling. Genes (Basel) 2019;10:141. doi: 10.3390/genes10020141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Livneh I., Moshitch-Moshkovitz S., Amariglio N., Rechavi G., Dominissini D. The mA epitranscriptome: transcriptome plasticity in brain development and function. Nat. Rev. Neurosci. 2020;21:36–51. doi: 10.1038/s41583-019-0244-z. [DOI] [PubMed] [Google Scholar]
- 15.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y., et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X., Liu J., Xiao W., Zeng Q., Bo H., Zhu Y., Gong L., He D., Xing X., Li R., et al. SIRT1 regulates N -methyladenosine RNA modification in hepatocarcinogenesis by inducing RANBP2-dependent FTO SUMOylation. Hepatology. 2020;72:2029–2050. doi: 10.1002/hep.31222. [DOI] [PubMed] [Google Scholar]
- 17.Relier S., Ripoll J., Guillorit H., Amalric A., Achour C., Boissière F., Vialaret J., Attina A., Debart F., Choquet A., et al. FTO-mediated cytoplasmic mA demethylation adjusts stem-like properties in colorectal cancer cell. Nat. Commun. 2021;12:1716. doi: 10.1038/s41467-021-21758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang H., Wang Y., Kandpal M., Zhao G., Cardenas H., Ji Y., Chaparala A., Tanner E., Chen J., Davuluri R., et al. NFTO-dependent -methyladenosine modifications inhibit ovarian cancer stem cell self-renewal by blocking cAMP signaling. Cancer Res. 2020;80:3200–3214. doi: 10.1158/0008-5472.CAN-19-4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X.Y., Zhang J., Zhu J.S. The role of m(6)A RNA methylation in human cancer. Mol. Cancer. 2019;18:103. doi: 10.1186/s12943-019-1033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qing Y., Dong L., Gao L., Li C., Li Y., Han L., Prince E., Tan B., Deng X., Wetzel C., et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/mA/PFKP/LDHB axis. Mol. Cel. 2021;81:922–939.e9. doi: 10.1016/j.molcel.2020.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tao B., Huang X., Shi J., Liu J., Li S., Xu C., Zhong J., Wan L., Feng B., Li B. FTO interacts with FOXO3a to enhance its transcriptional activity and inhibits aggression in gliomas. Signal Transduct. Target. Ther. 2020;5:130. doi: 10.1038/s41392-020-00234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei J., Liu F., Lu Z., Fei Q., Ai Y., He P., Shi H., Cui X., Su R., Klungland A., et al. Differential mA, mA, and mA demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol. Cel. 2018;71:973–985.e5. doi: 10.1016/j.molcel.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodall G., Wickramasinghe V. RNA in cancer. Nat. Rev. Cancer. 2021;21:22–36. doi: 10.1038/s41568-020-00306-0. [DOI] [PubMed] [Google Scholar]
- 24.Mori M., Ludwig R., Garcia-Martin R., Brandão B., Kahn C. Extracellular miRNAs: from biomarkers to mediators of physiology and disease. Cell Metab. 2019;30:656–673. doi: 10.1016/j.cmet.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alarcón C., Lee H., Goodarzi H., Halberg N., Tavazoie S. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–485. doi: 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alarcón C., Goodarzi H., Lee H., Liu X., Tavazoie S., Tavazoie S. HNRNPA2B1 is a mediator of m(6)a-dependent nuclear RNA processing events. Cell. 2015;162:1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo X., Qiu W., Liu Q., Qian M., Wang S., Zhang Z., Gao X., Chen Z., Xue H., Li G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten pathways. Oncogene. 2018;37:4239–4259. doi: 10.1038/s41388-018-0261-9. [DOI] [PubMed] [Google Scholar]
- 28.Yan Y., Yan H., Wang Q., Zhang L., Liu Y., Yu H. MicroRNA 10a induces glioma tumorigenesis by targeting myotubularin-related protein 3 and regulating the Wnt/β-catenin signaling pathway. FEBS J. 2019;286:2577–2592. doi: 10.1111/febs.14824. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91. doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin X., Chai G., Wu Y., Li J., Chen F., Liu J., Luo G., Tauler J., Du J., Lin S., et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 2019;10:2065. doi: 10.1038/s41467-019-09865-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Chen M., Wei L., Law C.T., Tsang F.H., Shen J., Cheng C.L., Tsang L.H., Ho D.W., Chiu D.K., Lee J.M., et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology (Baltimore, Md.) 2018;67:2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 32.Guo X., Li K., Jiang W., Hu Y., Xiao W., Huang Y., Feng Y., Pan Q., Wan R. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol. Cancer. 2020;19:91. doi: 10.1186/s12943-020-01158-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D., et al. Jaspar 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munde M., Wang S., Kumar A., Stephens C., Farahat A., Boykin D., Wilson W., Poon G. Structure-dependent inhibition of the ETS-family transcription factor PU.1 by novel heterocyclic diamidines. Nucleic Acids Res. 2014;42:1379–1390. doi: 10.1093/nar/gkt955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q., Yu J., Wang L., Tang Y., Zhou Q., Ji S., Wang Y., Santos L., Haeusler R., Que J., et al. Inhibition of PU.1 ameliorates metabolic dysfunction and non-alcoholic steatohepatitis. J. Hepatol. 2020;73:361–370. doi: 10.1016/j.jhep.2020.02.025. [DOI] [PubMed] [Google Scholar]
- 36.Antony-Debré I., Paul A., Leite J., Mitchell K., Kim H., Carvajal L., Todorova T., Huang K., Kumar A., Farahat A., et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J. Clin. Invest. 2017;127:4297–4313. doi: 10.1172/JCI92504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Rhun E., Preusser M., Roth P., Reardon D.A., van den Bent M., Wen P., Reifenberger G., Weller M. Molecular targeted therapy of glioblastoma. Cancer Treat Rev. 2019;80:101896. doi: 10.1016/j.ctrv.2019.101896. [DOI] [PubMed] [Google Scholar]
- 38.Gimple R.C., Bhargava S., Dixit D., Rich J.N. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019;33:591–609. doi: 10.1101/gad.324301.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang H., Weng H., Chen J. mA Modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37:270–288. doi: 10.1016/j.ccell.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vu L., Cheng Y., Kharas M. The biology of mA RNA methylation in normal and malignant hematopoiesis. Cancer Discov. 2019;9:25–33. doi: 10.1158/2159-8290.CD-18-0959. [DOI] [PubMed] [Google Scholar]
- 42.Cui Q., Shi H., Ye P., Li L., Qu Q., Sun G., Sun G., Lu Z., Huang Y., Yang C., et al. mA RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao L., Li X., Mu Z., Zhou J., Zhou P., Xie C., Jiang S. FTO inhibition enhances the antitumor effect of Temozolomide by targeting MYC-miR-155/23a cluster-MXI1 Feedback circuit in glioma. Cancer Res. 2020;80:3945–3958. doi: 10.1158/0008-5472.CAN-20-0132. [DOI] [PubMed] [Google Scholar]
- 44.Visvanathan A., Patil V., Arora A., Hegde A., Arivazhagan A., Santosh V., Somasundaram K. Essential role of METTL3-mediated mA modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–533. doi: 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- 45.Li F., Yi Y., Miao Y., Long W., Long T., Chen S., Cheng W., Zou C., Zheng Y., Wu X., et al. N-methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 2019;79:5785–5798. doi: 10.1158/0008-5472.CAN-18-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tassinari V., Cesarini V., Tomaselli S., Ianniello Z., Silvestris D., Ginistrelli L., Martini M., De Angelis B., De Luca G., Vitiani L., et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021;22:51. doi: 10.1186/s13059-021-02271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su R., Dong L., Li C., Nachtergaele S., Wunderlich M., Qing Y., Deng X., Wang Y., Weng X., Hu C., et al. R-2HG exhibits anti-tumor activity by targeting FTO/mA/MYC/CEBPA signaling. Cell. 2018;172:90–105.e123. doi: 10.1016/j.cell.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chai R., Wu F., Wang Q., Zhang S., Zhang K., Liu Y., Zhao Z., Jiang T., Wang Y., Kang C. mA RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gliomas. Aging. 2019;11:1204–1225. doi: 10.18632/aging.101829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Treiber T., Treiber N., Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cel. Biol. 2019;20:5–20. doi: 10.1038/s41580-018-0059-1. [DOI] [PubMed] [Google Scholar]
- 50.Hao F., Itoh T., Morita E., Shirahama-Noda K., Yoshimori T., Noda T. The PtdIns3-phosphatase MTMR3 interacts with mTORC1 and suppresses its activity. FEBS Lett. 2016;590:161–173. doi: 10.1002/1873-3468.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu W., He C., Liu C., Cao A., Xue X., Evans-Marin H., Sun M., Fang L., Yao S., Pinchuk I., et al. miR-10a inhibits dendritic cell activation and Th1/Th17 cell immune responses in IBD. Gut. 2015;64:1755–1764. doi: 10.1136/gutjnl-2014-307980. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi H., Kanno T., Nakayamada S., Hirahara K., Sciumè G., Muljo S., Kuchen S., Casellas R., Wei L., Kanno Y., et al. TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat. Immunol. 2012;13:587–595. doi: 10.1038/ni.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Z., Chen X., Zhang Z., Zhang X., Saunders L., Zhou Y., Ma P. Nanofibrous spongy microspheres to distinctly release miRNA and growth factors to enrich regulatory T cells and rescue periodontal bone Loss. ACS nano. 2018;12:9785–9799. doi: 10.1021/acsnano.7b08976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei Y., Corbalán-Campos J., Gurung R., Natarelli L., Zhu M., Exner N., Erhard F., Greulich F., Geißler C., Uhlenhaut N., et al. Dicer in macrophages prevents atherosclerosis by promoting mitochondrial oxidative metabolism. Circulation. 2018;138:2007–2020. doi: 10.1161/CIRCULATIONAHA.117.031589. [DOI] [PubMed] [Google Scholar]
- 55.Luo D., Ge W. MeCP2 promotes colorectal cancer metastasis by modulating ZEB1 transcription. Cancers (Basel) 2020;12:758. doi: 10.3390/cancers12030758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu G., Suo C., Yang Y., Shen S., Sun L., Li S.T., Zhou Y., Yang D., Wang Y., Cai Y., et al. MYC promotes cancer progression by modulating m(6) A modifications to suppress target gene translation. EMBO Rep. 2021;22:e51519. doi: 10.15252/embr.202051519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Riel B., Rosenbauer F. Epigenetic control of hematopoiesis: the PU.1 chromatin connection. Biol. Chem. 2014;395:1265–1274. doi: 10.1515/hsz-2014-0195. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki M., Yamada T., Kihara-Negishi F., Sakurai T., Hara E., Tenen D.G., Hozumi N., Oikawa T. Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene. 2006;25:2477–2488. doi: 10.1038/sj.onc.1209272. [DOI] [PubMed] [Google Scholar]
- 59.Kim M.Y., Yan B., Huang S., Qiu Y. Regulating the regulators: the role of histone deacetylase 1 (HDAC1) in erythropoiesis. Int. J. Mol. Sci. 2020;21:8460. doi: 10.3390/ijms21228460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kihara-Negishi F., Yamamoto H., Suzuki M., Yamada T., Sakurai T., Tamura T., Oikawa T. In vivo complex formation of PU.1 with HDAC1 associated with PU.1-mediated transcriptional repression. Oncogene. 2001;20:6039–6047. doi: 10.1038/sj.onc.1204756. [DOI] [PubMed] [Google Scholar]
- 61.Burda P., Laslo P., Stopka T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia. 2010;24:1249–1257. doi: 10.1038/leu.2010.104. [DOI] [PubMed] [Google Scholar]
- 62.Munde M., Wang S., Kumar A., Stephens C.E., Farahat A.A., Boykin D.W., Wilson W.D., Poon G.M. Structure-dependent inhibition of the ETS-family transcription factor PU.1 by novel heterocyclic diamidines. Nucleic Acids Res. 2014;42:1379–1390. doi: 10.1093/nar/gkt955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Terstappen G., Meyer A., Bell R., Zhang W. Strategies for delivering therapeutics across the blood-brain barrier. Nat. Rev. Drug Discov. 2021;20:362–383. doi: 10.1038/s41573-021-00139-y. [DOI] [PubMed] [Google Scholar]
- 64.Bhat K.P.L., Balasubramaniyan V., Vaillant B., Ezhilarasan R., Hummelink K., Hollingsworth F., Wani K., Heathcock L., James J.D., Goodman L.D., et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minata M., Audia A., Shi J., Lu S., Bernstock J., Pavlyukov M.S., Das A., Kim S.H., Shin Y.J., Lee Y., et al. Phenotypic plasticity of invasive edge glioma stem-like cells in response to ionizing radiation. Cell Rep. 2019;26:1893–1905.e7. doi: 10.1016/j.celrep.2019.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bhat K.P., Salazar K.L., Balasubramaniyan V., Wani K., Heathcock L., Hollingsworth F., James J.D., Gumin J., Diefes K.L., Kim S.H., et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011;25:2594–2609. doi: 10.1101/gad.176800.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Z., Wang H., Zhang Z., Xu J., Qi Y., Xue H., Gao Z., Zhao R., Wang S., Zhang S., et al. Cell surface GRP78 regulates BACE2 via lysosome-dependent manner to maintain mesenchymal phenotype of glioma stem cells. J. Exp. Clin. Cancer Res. 2021;40:20. doi: 10.1186/s13046-020-01807-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han M., Wang S., Fritah S., Wang X., Zhou W., Yang N., Ni S., Huang B., Chen A., Li G., et al. Interfering with long non-coding RNA MIR22HG processing inhibits glioblastoma progression through suppression of Wnt/β-catenin signalling. Brain. 2020;143:512–530. doi: 10.1093/brain/awz406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xue H., Yuan G., Guo X., Liu Q., Zhang J., Gao X., Guo X., Xu S., Li T., Shao Q., et al. A novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy. 2016;12:1129–1152. doi: 10.1080/15548627.2016.1178446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao X., Guo X., Xue H., Qiu W., Guo X., Zhang J., Qian M., Li T., Liu Q., Shen J., et al. lncTCF7 is a negative prognostic factor, and knockdown of lncTCF7 inhibits migration, proliferation and tumorigenicity in glioma. Sci. Rep. 2017;7:17456. doi: 10.1038/s41598-017-17340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu Y., Smyth G.K. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The miRNA sequencing data have been deposited in GSA under accession number CRA002339. The processed data and basic association analyses will be made available in supplementary data or from the corresponding author on reasonable request.