

Abstract
Rheumatoid arthritis (RA) is an inflammation-involved disorder and features the disruption of CD4+ T lymphocytes. Herein, we describe that microRNA-10b-5p (miR-10b) promotes RA progression by disrupting the balance between subsets of CD4+ T cells. MiR-10b-deficient mice protected against collagen antibody-induced arthritis (CAIA) model. RNA sequencing results indicated that disordered genes in miR-10b−/− CAIA model are closely associated with CD4+ T cells differentiation. Moreover, miR-10b mimics promoted Th1/Th17 and suppressed Th2/Treg cells differentiation, whereas miR-10b inhibitor induced contrary effects. In addition, GATA3 and PTEN was confirmed as two targets of miR-10b, and GATA3 siRNA could increase Th1 and reduce Th2 cells meanwhile PTEN siRNA could increase Th17 and decrease Treg cells. Furthermore, miR-10b inhibitor significantly ameliorated collagen-induced arthritis (CIA) development by attenuating the dysfunctional CD4+ T cell subpopulations. The present findings suggest that miR-10b could disrupt the balance of CD4+ T subsets, while suppressed miR-10b could attenuate the severity of experimental arthritis, which provided us a novel mechanistic and therapeutic insight into the RA.
Keywords: RA, miR-10b, Th1/2, Th17/Treg, GATA3, PTEN
Graphical abstract
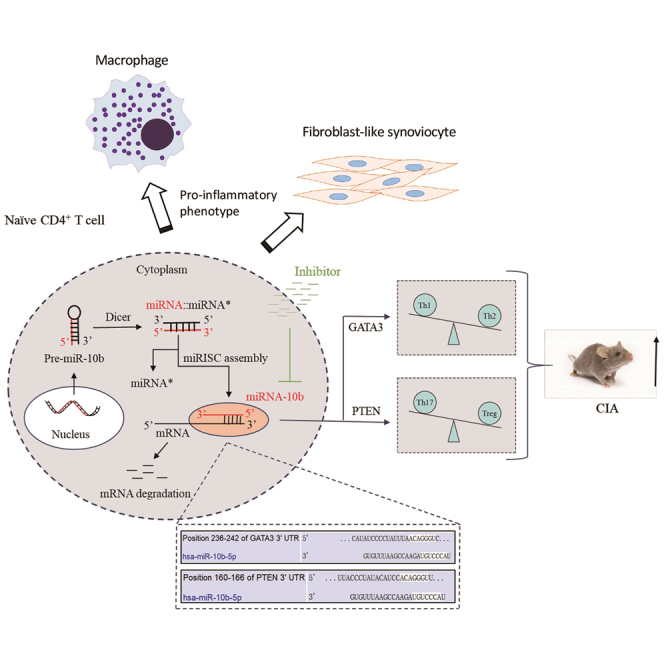
Rheumatoid arthritis (RA) is an inflammation-involved disorder and features the disruption of CD4+ T lymphocytes. Dr. Tu and his colleagues describe that microRNA-10b-5p promotes RA progression by disrupting the balance between subsets of CD4+ T cells via targeting GATA3 and PTEN, providing a novel mechanistic and therapeutic insight into the RA.
Introduction
Rheumatoid arthritis (RA) is a chronic and immune-mediated arthritis characterized by synovial inflammation, hyperplasia, angiogenesis, and progressive damage in the cartilage and bone, which affects approximately 1% of the population worldwide.1 Although the etiology of RA remains indefinable, both T lymphocytes (T cells) and cytokines are responsible for its development.2,3 The uncontrolled infiltration of activated and inflammatory T cells cooperates with other infiltrated and resident cells, such as B lymphocytes (B cells), producing pro-inflammatory mediators and/or through direct cell-to-cell interaction,4 ultimately resulting in persistent inflammation and cartilage damage.1
Abundant CD4+ T cells infiltrate the inflammatory synovium in patients with RA and participate in the initiation and perpetuation of RA.5,6 Stimulated by antigenic and cytokine signaling, naive CD4+ T cells become activated and differentiate into functionally distinct T helper (Th) cells, such as Th1, Th2, and Th17, and regulatory T (Treg) cells.7 Th1 and Th17 cells promote synovial inflammation and osteoclast formation, eventually leading to joint destruction,8 whereas Th2 and Treg cells ameliorate the progression of arthritis.9, 10, 11 However, the precise mechanisms resulting in CD4+ T cell differentiation into distinct phenotypes are various and need to be studied further to better understand the crucial role of these cells in RA.
MicroRNAs (miRNAs) negatively regulate target gene expression by promoting mRNA cleavage or inhibiting mRNA translation.12 Mounting evidence indicates that abnormal miRNAs might be involved in the immunological pathogenesis of autoimmune disorders by regulating T cells, including RA. For example, miR-21 was reported to be downregulated in peripheral blood mononuclear cells (PBMCs) and CD4+ T cells from RA patients and was verified to regulate Th17/Treg imbalances in RA patients by targeting STAT3.13 MiR-26a, miR-146a, miR-150, and other miRNAs are overexpressed in RA PBMCs and are significantly increased during Th17 differentiation.14 Increased miR-34a in RA PBMCs and CD4+ T cells impairs the Treg/Th17 balance by targeting Foxp3.15 However, the specific function and mechanisms of miRNAs in T cells of RA patients are elusive, given that previous studies either lack a knockout mouse model of arthritis or insufficient therapeutic evidence in vivo.
MicroRNA-10b-5p (miR-10b), one of the first identified anomalous miRNAs in cancer, has been extensively studied in various cancer types since its inaugural report.16,17 Recent data suggest that it participates in inflammation-associated diseases by regulating T cells.18,19 However, the function of miR-10b in RA, and especially in CD4+ T cells, has not been investigated previously. In this research, we described that miR-10b is overexpressed in RA patients, and that these upregulated levels might lead to the initiation and development of RA by disequilibrating Th cell subsets.
Results
MiR-10b is overexpressed and disrupts the subtypes of RA CD4+ T cells
To explore the effects of miR-10b in RA, we first examined expression levels in PBMCs of RA patients. The data showed that, compared with those of healthy donors, miR-10b was abundantly expressed in the total PBMCs and CD4+ T cells of RA patients (Figures 1A and 1B), while miR-10b did not significantly change in B cells or monocytes (Figure S1). As synovium hyperplasia is a main characteristic of RA patients, the miR-10b expression was assessed in the surgically removed RA synovium. Using fluorescence in situ hybridization (FISH) and qPCR, we demonstrated that miR-10b had higher levels in the RA synovium than in healthy donors (Figures 1C and 1D). miR-10b was predominantly distributed at sites of neovascularization of the RA synovium (Figure 1D), which was colocalized with CD4+ T cells in RA synovium (Figure S2), indicating that miR-10b might play a pathogenic role in T cells in RA synovium. In addition, to illustrate the function of upregulated miR-10b in RA, and especially in the differentiation of CD4+ T cells, CD4+ T cells were FACS-sorted from human PBMCs (Figure S3). The proportion of T helper cells was detected after transfecting CD4+ T cells with miR-10b mimic and corresponding mimic negative controls. Compared with that of the mimic NC, the overexpression miR-10b significantly elevated the percentage of Th1/Th17 cells but significantly decreased the percentage of Th2/Treg cells in human CD4+ T cells sorted from PBMCs (Figure 1E). In contrast, the silencing of miR-10b with its inhibitor obviously led to the opposite result in human CD4+ T cells sorted from PBMCs (Figure 1F). In addition, miR-10b promoted subset-specific cytokines, including TNF-α and IL-17, and transcriptional factors, such as T-bet and RORγt in human CD4+ T cells (Figure S4). On the contrary, miR-10b also repressed subset-specific cytokines, including IL-4 and IL-10, and subset-specific transcriptional factors, such as GATA3 and Foxp3, in human CD4+ T cells (Figure S4). Together, these data demonstrate that miR-10b is upregulated in RA and disrupts the proportion of different subtypes of CD4+ T cells.
Figure 1.

MiR-10b is overexpressed in RA CD4+ T cells and disrupts its balance
MiR-10b expression levels were determined in (A) PBMCs (n = 21), (B) in sorted CD4+ T (n = 12), and (C) synovium (n = 6) from heathy donors and RA patients by qRT-PCR. The graph displays the mean ± SD. Student’s t test; ∗∗p < 0.01, ∗∗∗p < 0.001. (D) RNA fluorescence in situ hybridization analysis of the expression and location of miR-10b in synovium of healthy donors and RA patients. Scale bar, 50 μm. (E and F) The percentage of Th cell subpopulations after transfection with miR-10b mimic and inhibitor in FACS-sorted CD4+ T cells from human PBMCs were analysis by flow cytometry. CD4+ T cells were treated with PMA, ionomycin, and BFA before flow cytometry analysis. Figures are presented as mean ± SD from at least five independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01.
MiR-10b deficiency protects against CAIA in mice by unbalancing the polarization of CD4+ T cells
To further determine the pathogenic effect of miR-10b in RA, miR-10b knockout (miR-10b−/−) mice were used as experimental animals to induce the CAIA animal model (Figure 2A). Firstly, the immunological status of miR-10b−/− mice was examined. Compared with miR-10b+/+ mice, the percentage of T cells, B cells, and monocytes did not significantly change in miR-10b−/− mice at steady state (Figure S5). After induction of the CAIA model, moderate arthritis was observed on days 7–8, which peaked at approximately days 11–13 in wild-type (miR-10b+/+) mice. When endogenous miR-10b was absent (Figure S6), the paws and ankle swelling (Figure 2B), arthritis index (Figure 2C), and swollen joint count (Figure 2D) were obviously decreased compared with those in miR-10b+/− and miR-10b+/+ CAIA mice. Simultaneously, histopathology results revealed that miR-10b+/− and miR-10b+/+ CAIA mice showed more severe signs of inflammation, a massive pannus, narrowed joint spaces, infiltration of inflammatory cells, and damaged cartilage than miR-10b−/− mice (Figures 2E and 2F).
Figure 2.

MiR-10b deficiency protects against CAIA in mice by balancing CD4+ T cells
(A) The time indicates the of the CAIA induction model. Mouse monoclonal anti-collagen-II 5-clone antibody cocktail, combined with an immunogenic boost of LPS, rapidly induced arthritis in miR-10b+/+, miR-1b+/−, and miR-10b−/− mice. (B) Representative paw photographs of CAIA mice are shown. (C) The arthritis index and the swollen joints count (D) of CAIA mice were assessed every day after LPS stimulation. (E) Representative photomicrographs of joint histopathology by H&E (upper) and Safranin O-fast green staining (lower) are shown on day 16 from CAIA mice. Histopathological changes, including synovial hyperplasia (black arrowhead), pannus (red arrowhead), bone erosion (yellow arrowhead), and inflammatory cell infiltration (white arrowhead). (F) Pathological grading of joints was evaluated. Figures are presented as the mean and ± SD from three independent experiments with five mice per group. Two-way ANOVA followed by multiple comparison test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, no significance. (G and H) KEGG pathway analysis in the PBMCs and synovium from miR-10b+/+ and miR-10b−/− CAIA mice. (I and J) The percentage of Th cell subpopulations after transfected with miR-10b mimic and inhibitor in FACS-sorted CD4+ T cells from mouse spleens were analysis by flow cytometry. CD4+ T cells were treated with PMA, ionomycin, and BFA before flow cytometry analysis. Figures are presented as mean ± SD from at least four independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further identify the pathogenic mechanisms of miR-10b in RA, RNA sequencing was performed with miR-10b+/+ and miR-10b−/− CAIA mice. By analyzing all upregulated and downregulated genes in the PBMCs and synovium, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that these abnormal genes were mainly associated with Th1/Th2 and Th17 cell differentiation and inflammation-related signaling pathways in the PBMCs and synovium (Figures 2G and 2H). As expected, in FACS-sorted CD4+ T cells from mouse spleen (Figure S3), the miR-10b mimic significantly elevated the percentage of Th1/Th17 cells but significantly decreased the percentage of Th2/Treg cells (Figure 2I), and silencing of miR-10b with its inhibitor obviously led to the opposite results (Figure 2J). Correspondingly, subset-specific cytokines and the expression of transcription factors were also regulated by miR-10b in mouse CD4+ T cells (Figure S7). Collectively, these findings demonstrate that knockout of the miR-10b gene significantly ameliorates the development of experimental arthritis by unbalancing CD4+ T cells.
MiR-10b regulates CD4+ T cells priming by directly binding to GATA3 and PTEN
Next, open-source software (TargetScan, MiRDB, and miRWalk) were used for predicting the target genes of miR-10b that might be involved in T cell differentiation. GATA3 binding protein (GATA3), a transcription factor specific for Th2 cells, and phosphate and tensin homolog (PTEN), were predicted as presume targets of miR-10b (Figures 3A and 3C). To verify the predicted binding, dual-luciferase reporter assays were then performed. As displayed in Figures 3B and 3D, the miR-10b mimic+pmirGLO-GATA3-3′ UTR or the miR-10b mimic+pmirGLO-PTEN-3′ UTR markedly reduced the luciferase activity, whereas the mimic NC+pmirGLO mimic, mimic NC+pmirGLO-GATA3-3′ UTR, or mimic NC+pmirGLO-PTEN-3′UTR and miR-10b mimic+pmirGLO had no suppressive effect on luciferase activity. Moreover, miR-10b mimic reduced the protein expression of GATA3 (Figure 3E) and PTEN (Figure 3F). Besides, as shown in Figures 3G and 3H, GATA3 and PTEN were mainly expressed near the lymphatic vessels but were hardly expressed in other cells in the healthy donor synovium. However, unlike that in the healthy controls, the expression of GATA3 and PTEN in RA patients was repressed in RA synovium. More importantly, GATA3 silencing with siRNA (Figure S8), which successfully elevated Th1 but reduced Th2 cells (Figure 3I) while PTEN silencing with siRNA (Figure S9), increasing Th17 but reducing Treg cell percentages in human CD4+ T cells (Figure 3J). In addition, GATA3 and PTEN was restored in miR-10b−/− CAIA mice when compared with miR-10b+/+ mice (Figure S10). As expected, similar results were observed in mouse CD4+ T cells (Figures 3K and 3L) by using mouse siRNA of GATA3 and PTEN (Figures S11 and S12). Furthermore, GATA3 and PTEN siRNA sufficiently imitated the regulatory effect of miR-10b on CD4+ T cell differentiation and expression of cytokine and subset-specific transcription factors (Figure S13). These results suggest that miR-10b disrupt CD4+ T cell differentiation by directly binding the 3′ UTRs of GATA3 and PTEN.
Figure 3.

MiR-10b directly binds to the 3′UTR of GATA3 and regulates the differentiation of CD4+ T cells
(A and C) Schematic representation of the binding site for miR-10b in the GATA3 3′ UTR. (B and D) Luciferase activity in HEK-293T cells co-transfected with the pmirGLO-GATA3-3′ UTR or the pmirGLO-PTEN-3′ UTR sequence, and miR-10b mimic. The results represent the mean ± SD from three independent experiments. Student’s t test; ∗p < 0.05. (E and F) Relative protein expression levels of GATA3 and PTEN in Jurkat cells and U87MG cells, respectively, after transfection with the miR-10b mimic or negative control. The results are presented as the mean ± SD from six independent experiment, ∗p < 0.05. (G and H) The representative expression of GATA3 and PTEN in synovium from RA patients and healthy donors using immunohistochemical staining. (I and K) The percentage of Th1 and Th2 cells in FACS-sorted CD4+ T cells from PBMCs of healthy donors (30 donors) and mouse spleens (4 mice) after transfection with human GATA3 siRNA and mouse GATA3 siRNA were analyzed by flow cytometry. (J and L) The percentage of Th17 and Treg cells in FACS-sorted CD4+ T cells from PBMCs of healthy donors (30 donors) and mouse spleens (4 mice) after transfection with human PTEN siRNA and mouse PTEN siRNA were analyzed by flow cytometry. CD4+ T cells were treated with PMA, ionomycin, and BFA before flow cytometry analysis. Figures are presented as mean ± SD from at least three independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01.
Silencing miR-10b ameliorates inflammatory phenotypes in collagen-induced arthritis mice
To evaluate the potential of miR-10b as a therapeutic target of RA in vivo, an miR-10b inhibitor (antagomir-10b), along with the corresponding negative control (antagomir NC FAM), was injected into the DBA1/J mouse collagen-induced arthritis (CIA) model (Figure 4A). When the mice showed swelling, they were divided randomly into different groups and treated i.v. with antagomir NC FAM and antagomir-10b every 3 days from days 29 to 56, while the arthritis index (AI), swollen joint count (SJC), thickness of paws, and body weight changes were recorded. As displayed in Figures 4B and 4C, the initiation of macroscopic redness and swelling joints appeared on approximately day 29 after the primary injection (on day 0), and the SJC and AI of the mice gradually increased with the course of arthritis, reaching a peak at approximately day 38 and then gradually decreased. Compared with that in normal mice, CIA mice showed obviously swollen ankles and thick paws (Figure 4D). qPCR data showed that miR-10b was expressed at higher levels in the CIA group and the antagomir NC FAM group than normal group in PBMCs (Figure 4E) and synovium (Figure 4F) of mice. However, antagomir-10b decreased endogenous expression of miR-10b in CIA mice PBMCs and synovium (Figures 4E and 4F). Moreover, antagomir-10b significantly reduced the AI (Figure 4G) and SJC (Figure 4H) of CIA mice and decreased the thickness of the front (Figure 4I) and hind paws (Figure 4J). In addition, treatment with antagomir-10b increased the body weight on day 47 and restored the body weight on day 56 compared with that in the CIA group and the antagomir NC FAM group (Figure 4K). Collectively, antagomir-10b treatment could alleviate CIA progression in vivo.
Figure 4.

Anti-arthritis effects of miR-10b inhibitor on CIA
Mice immunized with CII/CFA were randomly divided into three groups and administered miR-10b antagomir or antagomir NC FAM by tail intravenous administration at the indicated doses every 3 days since the grouping. (A) The time indicates the of induction of the CIA model and administration of miR-10b antagomir or antagomir NC FAM. (B) The arthritis index and (C) the swollen joint count of individual CIA mice from day 29 to day 59 after the first immunization. (D) The representative photographs of paws and effects of miR-10b antagomir. The effects of miR-10b inhibitor on the expression of miR-10b in (E) PBMCs and (F) synovium of mice. (G) The arthritis index and the swollen joint count (H) were evaluated, as were the effects of miR-10b antagomir. The thickness of (I) front paws and (J) hind paws of mice were measured. (K) The bodyweight of CIA mice was restored by miR-10b antagomir. Data are presented as mean ± SD from three independent experiments with at least five mice pre group. Two-way ANOVA followed by multiple comparison test; ∗∗∗p < 0.001 versus normal group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus antagomir NC FAM; ns, no significance.
miR-10b knockdown ameliorates histopathological changes of CIA mice
The well-established CIA model is characterized by synovial hyperplasia, the infiltration of inflammatory cells, and a massive pannus, as well as bone and cartilage damage, for which the initiation, development, and pathology are similar to those of clinical RA patients.20 As demonstrated in Figure 5A, there was a monolayer or two-layer synovium without infiltrating inflammatory cells or damaged cartilage in normal mice. Synovial hyperplasia, infiltrated inflammatory cells, massive neovascularization or a pannus, and cartilage destruction were observed in CIA and antagomir NC FAM-treated mice, which were attenuated in miR-10b inhibitor-treated mice. Moreover, the miR-10b inhibitor decreased scoring of the inflammation, pannus, cell infiltration, and synovial hyperplasia, as well as bone erosion in CIA mice (Figure 5A). Bone erosion and cartilage damage are well-known manifestations of RA.21 Thus, we evaluated the effects of the miR-10b inhibitor on bone and cartilage damage in CIA mice by X-ray. Consistent with the histopathological results, the X-ray data showed that both CIA and antagomir NC FAM-treated mice showed accompanying osteoporosis, narrowed joint spaces, and bone damage on day 59 after the first injection, and that the miR-10b inhibitor mitigated these signs and improved the crooked and misshapen phalangeal joints (Figure 5B).
Figure 5.

Silencing miR-10b ameliorates histopathological changes of CIA mice
(A) Representative photographs of knee joint histopathological alterations are displayed: in normal mice, an upward arrow indicates the synovium, and a rightward arrow indicates the cartilage. In CIA mice, an upward arrow indicates the synovial hyperplasia, a rightward arrow indicates the cartilage damage, a leftward arrow indicates the pannus, and a diagonal-left arrow indicates the inflammatory cell infiltration. (B) Local pathological changes of ankle joints from normal, CIA, antagomir NC FAM, and miR-10b antagomir groups were detected by X-ray. (C) Typical UBM B-mode scans and color Doppler of arthritic knees and severity grading. (D) Representative photographs of immunohistochemical staining of CD4 from the joints in normal, CIA, antagomir NC FAM, and miR-10b antagomir mice. (E) Representative photographs of immunohistochemical staining of GATA3 and PTEN from the joints in normal, CIA, antagomir NC FAM, and miR-10b antagomir mice. Data are presented as mean ± SD from three independent experiments with at least five mice pre group. Two-way ANOVA followed by multiple comparison test; ∗∗∗p < 0.001 versus normal group; #p < 0.05, ##p < 0.01 versus antagomir NC FAM; ns, no significance.
In addition, an ultrasound biomicroscope (UBM) was used to evaluate the histopathological changes in CIA mice.22 Under B-mode, the bright tendon-tibia-femur triangle, homogeneous with sharp limits but without a hypoechogenic zone, was observed in normal mice. However, variation in the hypoechogenic zone and an enlarged distance between the skin and femur appeared in CIA and antagomir NC FAM-treated mice, whereas the miR-10b inhibitor reduced the hypoechogenic zone and decreased the UBM joint score (Figure 5C). Neovascularization, defined as the establishment of new capillaries from pre-existing vasculature,23 is a crucial and early event in the progression of RA as it contributes to the initiation and perpetuation of the disorder by facilitating the infiltration of inflammatory cells and cytokines into the synovium, resulting in synovial hyperplasia and bone damage.24,25 The use of power Doppler allowed investigation of vascularization and blood flow, which echoed inflammation severity within the synovium. As shown in Figure 5C, stronger signals were observed in CIA and antagomir NC FAM-treated mice, whereas fewer signals appeared in miR-10b inhibitor-treated mice, which was accompanied by decreased color Doppler scores. Besides, as displayed in Figure 5D, CD4+ T cells were widely stained in the CIA and antagomir NC FAM-treated mouse synovium, whereas the miR-10b inhibitor significantly decreased infiltration of CD4+ T cells in the corresponding synovium tissue. Finally, GATA3 and PTEN was expressed at basal levels in synovial tissue of the knee joint of CIA and antagomir NC FAM-treated mice, but restored in miR-10b inhibitor-treated mice (Figure 5E). In summary, these results together demonstrated that silencing miR-10b improves the articular histopathological changes associated with experimental arthritis.
Downregulation of miR-10b ameliorates the development of CIA by regulating abnormally activated CD4+ T cells
Next, we confirmed the histopathological changes in the spleen of CIA mice by hematoxylin and eosin (H&E) staining. As displayed in Figure 6A, increased GCs, lymphoid follicles, periarteriolar lymphoid sheaths (PLAs), marginal zones, and red pulp were observed in CIA and antagomir NC FAM-treated mice, whereas miR-10b silencing reduced these inflammatory characteristics in CIA spleens. Moreover, compared with those in normal mice, there were large numbers of CD4+ T cells in CIA and antagomir NC FAM-treated mouse spleens, whereas the miR-10b inhibitor reduced CD4+ T cells in CIA spleens (Figure 6B). Interestingly, splenomegaly was present in CIA mice, which partially reflected the inflammatory condition in the immune system. As shown in Figure 6C, the spleen index increased in CIA and antagomir NC FAM-treated mice compared with that in normal mice, whereas silencing miR-10b with its inhibitor could reduce the spleen index. We also observed a similar enlargement of the thymus in CIA and antagomir NC FAM-treated mice, which could be attenuated by the miR-10b inhibitor (Figure S14). These findings suggested that silencing miR-10b sufficiently decreased inflammation and improved the pathological changes in CIA mice.
Figure 6.

Silencing miR-10b ameliorates the development of CIA by regulating abnormally activated CD4+ T cells
(A) Representative photographs of spleen histopathology are shown: in normal mice, a rightward arrow indicates the white pulp, and a leftward arrow indicates the red pulp. In CIA mice, a leftward arrow indicates the red medullary congestion, a diagonal-left arrow indicates the lymphoid follicular hyperplasia, a diagonal-right arrow indicates the GC, and a rightward arrow indicates the PLA. (B) Immunohistochemical staining of CD4 from the spleens in normal, CIA, CIA + antagomir NC FAM, and CIA + miR-10b antagomir mice. (C) The effects of miR-10b on spleen index are shown based on the ratio of spleen weight to mouse bodyweight. (D) The proportion of CD4+ T cells in mice PBMCs and spleens (E) in normal, CIA, CIA + antagomir NC FAM, and CIA + miR-10b antagomir mice. CD4+ T cells were treated with PMA, ionomycin, and BFA before flow cytometry analysis. Figures represented the mean ± SD from three independent experiments with at least five mice pre group. Two-way ANOVA followed by multiple comparison test; ∗∗∗p < 0.001 versus normal group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus antagomir NC FAM; ns, no significance.
As mentioned, the miR-10b inhibitor reduced the proportion of Th1 and Th17 cells but increased the proportion of Th2 and Treg cells in vitro. To evaluate the effects of the miR-10b inhibitor on attenuating abnormally activated Th cells in CIA mice, Th1, Th2, and Th17, as well as Treg, were detected in mouse splenocytes and PBMCs. Compared with those in normal mice, higher proportions of Th1 and Th17 cells and lower proportions of Th2 and Treg cells were found in CIA and antagomir NC FAM-treated mice, whereas the miR-10b inhibitor reduced Th1 and Th17 cells and increased the percentage of Th2 and Treg cells in PBMCs (Figure 6D) and mouse splenocytes (Figure 6E), respectively. In addition, the expression of cytokines and subset-specific transcription factors in CD4+ T cells from CIA mice were also regulated by the treatment of miR-10b antagomir (Figure S15). These results indicated that silencing miR-10b attenuated the development of CIA mice by regulating abnormal CD4+ T cells in vivo, which could partially explain the protective effect of the miR-10b inhibitor on splenic and articular histopathological changes.
miR-10b-overexpressing CD4+ T cells promote inflammatory phenotypes of fibroblast-like synoviocytes and macrophages in vitro
In addition to CD4+ T cells, fibroblast-like synoviocytes (FLS)26 and macrophages27 also play an important role in the development of RA. The interaction between these cellular components further aggravates the inflammation in joint synovium. An in vitro co-culture system was used to assess whether miR-10b-overexpressing CD4+ T cells can indirectly promote pro-inflammatory phenotypes of primary FLS and macrophages (Figure 7A). Figures 7B–7F show that miR-10b-overexpressing CD4+ T cells promote the proliferation, migration, and release of inflammatory factors of FLS. In addition, the polarization patterns of peritoneal macrophages showed less inflammatory status in both miR-10b−/− CAIA and CIA mice treated with miR-10b antagomir (Figure S16). Co-culture experiments of CD4+ T cells and macrophages have suggested that miR-10b knockdown CD4+ T cells could repress M1 polarization and matrix metalloproteinases ([MMPs], MMP1 and MMP3) of macrophages (Figures 7G–7I). The expression of inflammatory cytokines and phagocytic ability were also repressed in macrophages that co-cultured with miR-10b knockdown CD4+ T cells (Figure S17). Taken together, we confirm that the pro-inflammatory effects of miR-10b-overexpressing CD4+ T cells may further expand via modulating FLS and macrophages, resulting in the exacerbation of RA disease (Figure 8).
Figure 7.

miR-10b-overexpressing CD4+ T cells promote inflammatory phenotypes of FLS and macrophages
(A) The sorting strategy of FLS and macrophages from RA synovium tissues. (B–F) The co-culture system experiments showed that miR-10b-overexpressing CD4+ T cells promote the proliferation, migration, and the release of inflammatory factors of FLS. (G–I) Co-culture experiments of CD4+ T cells and macrophages suggest that miR-10b knockdown CD4+ T cells could repress M1 polarization and MMPs of macrophages. Figures are presented as mean ± SD from at least three independent experiments. Rank-sum test; ∗p < 0.01 and ∗∗p < 0.01 versus control group.
Figure 8.

Schema graph displaying miR-10b-regulated CD4+ T cell subsets in the development of the CIA model
MiR-10b negatively regulates GATA3 and PTEN expression by binding to their 3′ UTR in CD4+ T cells, leading to Th1/Th2 and Th17/Treg imbalance in the CIA model. The inhibitor of miR-10b repress miR-10b expression, balance Th1/Th2 and Th17/Treg subsets, and ultimately alleviate the development of the CIA model. In addition, miR-10b-overexpressing CD4+ T cells promote inflammatory phenotypes of primary macrophages and FLS from RA patients.
Discussion
RA, the most common chronic and systemic autoimmune disorder, is characterized by immune cell dysfunction. Although the precise pathogenic mechanism of RA remains unclear, infiltrated and hyper-activated CD4+ T cells undoubtedly contribute to RA progression.28 In general, Th1 cells were deemed to play a predominant role in RA development, as they participate in synovial inflammation by inducing a series of cytokines, including IFN-γ, TNF-α, and IL-1β, initiating RA development.29 Therefore, the neutralization of these inflammatory cytokines might be an effective treatment strategy for RA management. However, unexpectedly, neither IFN-γ nor IFN-γ receptor deficiency in experimental models, nor clinical monoclonal antibodies against IFN-γ, failed to work against pathogenic Th1 cells in RA.30, 31, 32 Similar to Th1 cells, Th17 cells participate not only in synovial inflammation and angiogenesis, but also in bone and cartilage destruction, contributing to RA development.33 Nevertheless, monoclonal antibodies against IL-17A34, 35, 36 and the IL-17A receptor37 have not shown successful therapeutic effects. These results suggest that RA is a systematic autoimmune disease that involves multiple cells and cytokines. Treatment with a single cytokine or receptor alone does not achieve sufficient therapeutic effects. As pro-inflammatory cytokines, IFN-γ and IL-17A, serve as “executors” in activating downstream effectors, leading to a vicious cycle of synovial inflammation and cartilage destruction, whereas the detrimental immune cells are the true “commanders.” Therefore, inhibiting the commanders by targeting crucial transcription factors and signaling molecules offers a novel promising therapeutic strategy for autoimmune diseases. For example, mice deficient in T-bet, the transcription factors of Th1 cells, show strongly decreased joint inflammation throughout the course of experimental arthritis.38 Furthermore, the pharmacological suppression of retinoid-related orphan nuclear receptor gamma t (RORγt), a master regulator of Th17 cells, sufficiently suppresses Th17 cell differentiation and the production of inflammatory cytokines in CIA mice.39,40 Moreover, tofacitinib, a small-molecular inhibitor of JAK, has been approved for the management of RA and was demonstrated to be more potent than or equally effective as biological disease modifying anti-rheumatic drugs for RA,41 partially by suppressing Th1 and Th17 subset differentiation simultaneously.42
Whereas Th1/Th17 cells aggravate the pathophysiology of RA, Th2/Treg cells protect against RA pathology. IL-4, an anti-inflammatory cytokine predominantly produced by Th2 cells, decreases pro-inflammatory cytokines and suppresses bone resorption in an ex vivo model of bone resorption in RA.43 In experimental arthritis, treatment with IL-4 sufficiently decreases the sensitivity and severity44 and protects against bone and cartilage damage.45 Treg cells also have a vital protective role in murine autoimmune arthritis; neutralization of Tregs aggravate CIA,10 whereas the adoptive transfer of Tregs successfully attenuates CIA.11 TNF-α inhibits the suppressive function of Tregs and partially decreases Foxp3, a specific transcription factor for Tregs, whereas neutralizing TNF-α with infliximab sufficiently elevates Foxp3 expression.46 In addition, treatment with IL-2 to induce Treg expression in vivo in patients with autoimmune diseases is a promising therapeutic method,47 and Treg differentiation and expression are dependent on IL2R signaling.48 In summary, it is undisputed that functional imbalances in Th1/Th2 and Th17/Treg subsets contribute to RA development. Thus, the simultaneous balancing of multiple commanders by regulating detrimental and protective Th subset differentiation provides a promising therapeutic strategy for the management of RA.
Here, we determined that miR-10b promotes Th1/Th17 cells but inhibits Th2/Treg cell differentiation by directly binding 3′ UTRs of GATA3 and PTEN. More significantly, miR-10b knockout or silencing with an inhibitor sufficiently protected against experimental arthritis. In this investigation, we report that miR-10b was overexpressed in RA patient PBMCs and CD4+ T cells, suggesting that miR-10b might be a pathogenic element in RA development. For exploring the specific effect of miR-10b in RA, we established a CAIA model using miR-10b+/+, miR-10b+/−, and miR-10b−/− mice. Compared with those in wild-type and heterozygote mice, miR-10b-deficient CAIA mice showed mild features of redness and swollen joints and ankles, a lower AI, and a decreased SJC. More importantly, miR-10b deficiency in CAIA mice decreased inflammation, cell infiltration, the pannus, bone erosion as well as synovial hyperplasia in knee joints. The knockout of miR-10b alleviated the severity of CAIA, including an attenuated AI and SJC and improved histopathological changes. To determine and illustrate the mechanisms underlying the effect of miR-10b in RA, RNA sequencing was performed using miR-10b-deficient and wild-type CAIA mice. KEGG pathway results indicated that abnormally expressed genes were closely related to CD4+ T cells differentiation, and inflammatory signaling pathways in the miR-10b-deficient CAIA model, suggesting that miR-10b might be an important factor in RA that functions by regulating CD4+ T cell differentiation. Therefore, we are constructing T cell-specific miR-10b knockout mice, hoping to further illustrate the specific role of miR-10b in T cells at different stages of arthritis development. However, we found that there is no significant change in immune cells from wild-type mice and miR-10b knockout mice. Only under collagen antibody-induced conditions, were wild-type and miR-10b knockout mice able to exert different reactions in the process of arthritis development, implying that miR-10b may not be a causative factor of RA.
Accumulating evidence has demonstrated that a single miRNA, such as miR-23b, could profoundly mediate immune responses, suggesting the prospect of therapeutic strategies for autoimmune diseases.49 miR-10b, mostly investigated in cancer, is not well known with respect to potential roles in autoimmune diseases.50 Recently, Chen et al.51 reported that miR-10b was upregulated in PBMC Th17 cells of ankylosing spondylitis patients. However, the function of miR-10b in RA, especially in CD4+ T cells, remains elusive. Our data indicated that miR-10b promoted Th1/Th17 cells, and suppressed Th2/Treg cell development, whereas the miR-10b inhibitor had the opposite effects. The findings of the current paper suggested that miR-10b is a potential diagnostic marker and therapeutic target of RA. We will continue to expand the sample size (assessing the correlation between miR-10b and different periods of RA patients) to evaluate the likelihood of miR-10b as a diagnostic marker for RA. In addition, we further investigate the effectiveness of miR-10b as a target for RA treatment. We structurally optimize the miR-10b inhibitor and systematically evaluate its adverse effects by using arthritis mice.
Intriguingly, transcription factors associated with Th cell differentiation were mutually exclusive and reciprocally regulated the differentiation of other T cell subpopulations via direct co-interactions and/or as an indirect consequence of cytokines secreted by other Th cell subpopulations. For example, T-bet could suppress the transcriptional activity of GATA3 by directly interacting with GATA3.52 On the contrary, GATA3 was reported to inhibit Th1 cell differentiation.53,54 Similarly, PTEN is a confirmed target gene of miR-10b,55 and is an important mediator of Th17 and Treg cells.56,57 In immune experimental arthritis, the intraarticular injection of PTEN was found to attenuate the severity of CIA in rats58 and p53-driven PTEN attenuates the signs of CIA in mice by acting on the dysregulated Th17/Treg balance.59 In this study, we demonstrated that GATA3 and PTEN might be capable of binding miR-10b and are accompanied by the regulation of Th1/Th2 and Th17/Treg differentiation. These results suggested that miR-10b might participate in RA development by regulating CD4+ T cell differentiation via GATA3 and PTEN.
Finally, with the goal of evaluating the therapeutic effects of miR-10b in experimental arthritis, we induced CIA in mice and treated them with an miR-10b inhibitor to silence miR-10b in vivo. The miR-10b inhibitor successfully ameliorated experimental arthritis severity in vivo. Compared with those in CIA and antagomir NC FAM-treated mice, the miR-10b inhibitor significantly decreased the AI, SJC, and thickness of paws and restored the body weight. Moreover, histopathological changes in the spleen and joint were attenuated in miR-10b inhibitor-treated CIA mice. In addition, the miR-10b inhibitor balanced the disruption of CD4+ T cells in CIA mice, which mechanistically illustrated the therapeutic effects of miR-10b in experimental arthritis.
There are several shortcomings in this paper that cannot be ignored. Firstly, it would be more convincing to establish an experimental arthritis model by using mice with miR-10b conditional knockout in CD4+ T cells. Secondly, the number of RA patients and healthy control included in this study is relatively limited. Finally, the safety and specificity of long-term in vivo usage of miR-10b inhibitor is an important issue for future evaluation. In response to these problems, we will further establish experimental arthritis model by using mice with miR-10b conditional knockout in CD4+ T cells. We also will continue to recruit more RA patients and normal donors. Moreover, the in vivo delivering system of miR-10b inhibitor and related issues about safety and specificity will be evaluated in the future.
Materials and methods
Specimens
The investigation approach was carried out based on the Declaration of Helsinki and authorized by the Ethics Committee of Anhui Medical University. RA peripheral blood (n = 21) and knee synovium (n = 21) were obtained from patients undergoing total knee replacement from Anhui Province Hospital, The First Affiliated Hospital of the University of Science and Technology of China. Blood samples from healthy controls were collected from the First Affiliated Hospital, Anhui Medical University. Written informed consent was obtained from each donor.
Mice
MiR-10b−/−, miR-10b+/−, and miR-10b+/+ littermate mice were raised in a specific pathogen-free (SPF) environment at Anhui Medical University. DBA/1 mice, 7-week-old males (18 ± 2 g; certificate no: 2017-0005), were purchased from Shanghai Slack Experimental Animal Limited Liability Company (Shanghai, China) and housed in the SPF environment. Wild-type C57BL/6J mice were purchased from the Experimental Animal Center of Anhui Medical University and raised in SPF conditions. All experiments were approved by the Ethical Review Committee of Animal Experiments of the Institute of Clinical Pharmacology, Anhui Medical University.
Cell culture
Human U87MG cells were maintained in Minimum Essential Medium (MEM), the human primary FLS and HEK 293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM), and human macrophages, Jurkat cells, mouse MPC-5 cells, and FACS-sorted CD4+ T cells were cultured in RPMI 1640, all the above cells were supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin. Cells were maintained at 37°C and in an atmosphere of 5% CO2. For the co-culture experiment, the proliferation of RA-FLS were tested by inoculating RA-FLS into the lower chamber of the transwell system (0.4 μm) and CD4+ T cells into the upper chamber for 3–6 days. The migration of RA-FLS was tested by inoculating RA-FLS into the upper chamber of the transwell system (8 μm) and CD4+ T cells into the lower chamber for 12 h. The polarization of macrophages was tested by inoculating macrophages into the lower chamber of the transwell system (0.4 μm) and CD4+ T cells into the upper chamber for 48 h.
Reagents and antibodies
Polymerase chain reaction (PCR) reagents included the following: 2× Taq PCR Master Mix (MT231) and DNA Marker (MD102) (Biomed, Beijing, China), agarose (1110GR100) (Biofrox, Berlin, Germany), Hairpin-itTM microRNA, U6 snRNA Normalization RT-PCR Quantitation Kit, and RNA FISH (Paraffin section) Kit (GenePharma, Shanghai, China). Reagents for inducing the CAIA and CIA model were as follows: 5-clone monoclonal antibody cocktail (53100) and Chick Type II Collagen (20011) (Chondrex, Washington, USA). Cell culture reagents included MEM (PM150410) (Procell, Wuhan, China), penicillin-streptomycin (P1400) (Solarbio, Beijing, China), DMEM (06-1055-57-1ACS), RPMI 1640 (01-100-1ACS), and FBS (04-001-1ACS) (Biological Industries, Beit Haemek, Israel). Further reagents included Lipofectamine RNAiMAX (13778150), the Cell Fixation/Permeabilization Kit (00-5123-43, 00-8333-56, 00-5223-56), Opti-MEM reduced serum medium (31985-070), TRIzol (15596026) and the Pierce BCA Protein Assay Kit (23227) (Thermo Fisher Scientific, MA, USA), 1× lymphocyte separation medium (7211011), and human lymphocyte separation medium (7111011) (Dakewe, Guangzhou, China), as well as protease inhibitor cocktail (CW2200) (CoWin Biosciences, Jiangsu, China) and chemiluminescence (ECL) reagent (WBKLS0100) (Millipore, MA, USA). Reagents and antibodies used for histopathological analysis were as follows: fast green stain (G1661) (Solarbio, Beijing, China), Safranin O (MB4620) (Meilunbio, Dalian, China), hematoxylin (C0107), eosin staining solution (C0109), and DAPI (C1005) (Beyotime, Shanghai, China), anti-GATA3 (ab106625) and anti-PTEN (ab31192) (Abcam, Cambridge, UK), GAPDH (60004-1-Ig) (Proteintech, Chicago, USA), (HRP)-conjugated goat anti-rabbit IgG (ZB-2301) and SPlink Detection Kits (SP-9000) (Zhongshan Jinqiao, Beijing, China), (HRP)-conjugated goat anti-mouse IgG (BA1050) (Boster Biological, Wuhan, China). The reagents and antibodies used to analyze human and mouse CD4+ T cell subsets were PMA (FMS-FZ207), ionomycin (FMS-FZ208), and BFA (FMS-FZ209) (Fcmacs Biotech, Nanjing, China), as well as anti-mouse CD4 (100509), anti-mouse IFN-γ (505808), anti-mouse IL-4 (504106), anti-mouse IL-17A (506904), anti-mouse CD25 (102012), anti-mouse/rat/human FOXP3 (320008), anti-human CD4 (344604), anti-human IFN-γ (502528), anti-human IL-4 (500714), anti-human IL-17A (512306), anti-human CD25 (302609), anti-human CD80 (305219), anti-human CD206 (321119), and anti-human FOXP3 (320108) (BioLegend, CA, USA).
CAIA mouse model
CAIA was induced in 24 mice (miR-10b−/−, miR-10b+/−, miR-10b+/+) as follows: on day 0, mice were intraperitoneally (i.p.) injected with 4 mg of 5-clone monoclonal antibody cocktail. On day 3, mice received 40 μg lipopolysaccharide (LPS) i.p. as described previously.60 On day 16, the mice were euthanized, and the knee joints were harvested for subsequent analysis.
CIA mouse model
Chick type II collagen was sufficiently dissolved in 5 mL of acetic acid (0.1 M) followed by emulsification with complete Freund’s adjuvant. On day 0, the mice were induced by intradermally injection with 0.15 mL CII emulsion. On day 21, the mice were re-stimulated by intradermally injecting them with 0.1 mL CII emulsion.61 On day 29, the success rate of the CIA model was 95%, and failed models were excluded. The mice were randomly divided into three groups: CIA model, CIA + antagomir NC FAM, and CIA + miR-10b antagomir. After the beginning of joint swelling at day 29, mice were treated i.v. with miR-10b antagomir and antagomir NC FAM every 3 days. The CIA model mice were treated with an equal volume of the vehicle. On day 59, the mice were euthanized, and the blood, spleen, and ankle joints were collected for subsequent studies.
Evaluation of arthritis
The progression of arthritis was monitored throughout the study. The severity of CAIA was assessed every day after LPS injection on day 3, and the severity of CIA was evaluated after the second sensitization on day 29. Mice were evaluated using the SJC and AI. The swollen joints included five phalanx joints and one ankle or wrist joint; thus, the maximum number of swollen joints for each mouse was 24. The AI was assessed in each paw with a scale of 0–4, where 0 was no macroscopic signs of inflammation, 1 was mild swelling or redness, 2 was moderate swelling of ankle or wrist joints, 3 was paws with severe swelling and redness, and 4 was paws with deformity or ankylosis. The maximum AI for each mouse was 16.
Mouse spleen and human PBMC CD4+ T lymphocyte isolation
Mouse spleens were collected and prepared in single-cell suspensions with 10 mL mouse 1× lymphocyte separation medium in a 100 × 20-mm dish. Lymphocytes were separated by density gradient centrifugation in a 15-mL tube. Whole blood (20 mL) from healthy donors was diluted 1:1 with PBS and layered onto human lymphocyte separation medium. Then, PBMCs were separated by density gradient centrifugation in a 50-mL tube. The lymphocytes and PBMCs were labeled with CD4 monoclonal antibodies. CD4+ T cells were FACS-sorted from lymphocytes and PBMCs using a FACSAria II cell sorter (BD Biosciences, USA) to a routine purity of 90%–95%.
Transfection
To assess the effects of miR-10b on CD4+ T cells, the FACS-sorted CD4+ T cells from human PBMCs or C57BL/6 mouse spleens were cultured in Opti-MEM reduced serum medium. The next day, the cells were transfected: miR-10b-5p mimic (20 μM), inhibitors (20 μM), negative controls (20 μM), and inhibitor NC (20 μM; GenePharma, China). Human GATA3 small-interfering RNA (siRNA) (100 μM), mouse GATA3 siRNA (100 μM), human PTEN siRNA (100 μM), and mouse PTEN siRAN (100 μM) (Cohesion Bioscience, UK) with Lipofectamine RNAiMAX based on the manufacturer's instructions. The cells were transfected at 5 × 105. The sequences for transfection are listed in Table S1. The transfection efficiency was evaluated using a 5-carboxyfluorescein-bound miRNA-negative control 4–6 h after transfection under a fluorescence microscope. The proportion of transfected cells with fluorescence was approximately 80%–90%.
PCR and qRT-PCR
Total DNA was obtained from the tail of mice, followed by amplification using a T100 Thermal Cycler (Bio-Rad, USA), and then separated by 2% agarose. To determine miR-10b levels, total RNA from PBMCs of RA patients and healthy donors to was extracted using TRIzol. The isolated RNA was used to synthesize cDNA using MMLV Reverse Transcriptase. The T100 Thermal Cycler and 7500 Real-Time PCR System (Thermo Fisher Scientific, USA) were used for reverse-transcription PCR (RT-PCR) and quantitative RT-PCR (qRT-PCR), respectively. The expression levels of miR-10b in RA patient PBMCs were calculated using the 2−ΔΔCt method, with U6 small RNA (U6 snRNA) as the endogenous standard. All experiments were performed following the manufacturer’s instructions for the Hairpin-itTM microRNA and U6 snRNA Normalization RT-PCR Quantitation Kit. The primer sequences used for PCR and qRT-PCR are shown in Table 1.
RNA sequencing
Joint tissues and PBMC samples from CAIA mice were analyzed by high-throughput sequencing by BGI (Shenzhen, China) to screen the abnormally and specifically expressed genes in miR-10b−/− CAIA mice. KEGG pathway analysis was carried out to estimate the function of these dysregulated genes in miR-10b KO CAIA mice. Specifically, total RNA from joint tissues and PBMC samples from CAIA mice was quantified using a NanoDrop2000 spectrophotometer (NanoDrop Technologies). Mice PBMCs and synovial tissues were collected. Two passing samples were included; the RNA integrity number values were all more than 7 and quality control (QC) (q30) were all more than 90%. PE150 paired-end sequencing and the MGI-2000 platform were used. For detection of whole mRNA expression, RNA sequencing was performed by BGI (China) according to standard procedure. In brief, the sequencing data were filtered with SOAPnuke (v.1.5.2) by: (1) removing reads containing sequencing adaptor; (2) removing reads whose low-quality base ratio (base quality less than or equal to 5) was more than 20%; and (3) removing reads whose unknown base (“N” base) ratio was more than 5%; subsequently, clean reads were obtained and stored in FASTQ format. The clean reads were mapped to the reference genome using HISAT2 (v.2.0.4). Bowtie2 (v.2.2.5) was applied to align the clean reads to the reference coding gene set; then the expression level of gene was calculated by RNA-Seq by Expectation-Maximization (RSEM) (v.1.2.12). The heatmap was drawn by pheatmap (v.1.0.8) according to the gene expression in different samples. Essentially, differential expression analysis was performed using the PossionDis9 with false discovery rate ≤ 0.001 and |log2ratio| ≥ 1. To gain insight into the change of phenotype, gene ontology (http://geneontology.org/) and KEGG (https://www.kegg.jp/) enrichment analysis of annotated different expressed genes was performed using Phyper (https://en.wikipedia.org/wiki/Hypergeometric_distribution) based on hypergeometric testing; the significant levels of terms and pathways were corrected by Q value with a rigorous threshold (Q ≤ 0.05) by Bonferroni.
Western blot
RIPA lysis buffer with protease and phosphatase inhibitors was used to isolate total cell proteins, whereas the BCA was used to measure the protein concentration. Total proteins (30 μg) were separated by 10% SDS-PAGE followed by transfer onto polyvinylidene fluoride membranes (IPVH00010, Millipore, USA). After blocking with 5% skim-milk, the membranes were then incubated with the anti-GATA3 and anti-PTEN primary antibodies followed by (HRP)-conjugated goat anti-rabbit IgG secondary antibody, and developed with a chemiluminescence (ECL) reagent after being washed. Finally, the gray values of the bands (versus GAPDH) were analyzed using ImageJ. Each experiment was independently repeated six times.
H&E and Safranin O-fast green analysis
The mice were euthanized on day 59 after the first immunization. The joints and spleens were immediately fixed in 4% paraformaldehyde followed by being embedded in paraffin. In addition, before paraffin embedding, the joint tissues were decalcified with formic acid (5%). Paraffin-embedded slides (5 μm) were stained with Safranin O-fast green and H&E and examined histologically. The severity of histopathological changes in the CIA joint and spleen were graded by two independent observers. For the joint, the synovial hyperplasia, bone erosion, inflammation, inflammatory cells infiltration, and pannus were scored from 0 to 4 (0 = no effect, 4 = severe effect). For the spleen, the germinal centers (GCs), cellularity of PLAs, lymphoid follicles, red pulp, and marginal zone were graded from 0 to 3 (0 = no effect, 3 = severe effect).62,63
Immunohistochemistry analysis
Synovium specimens from RA patients and healthy donors were fixed and paraffin embedded, as performed for mice, for subsequent immunohistochemistry and FISH. The slides were dewaxed and rehydrated routinely, followed by the quenching of endogenous peroxidase and antigen retrieval. Subsequently, the slides were blocked with goat serum and then incubated with rabbit anti-GATA3 and rabbit anti-PTEN polyclonal antibody at 4°C overnight, respectively. Next, the slides were incubated with goat anti-rabbit secondary antibody. After that, the slides were treated with 3,3′-diaminobenzidine and counterstained with hematoxylin, followed by sealing with neutral balsam.
FISH
The location of miR-10b in the synovium was determined by RNA FISH. The sections were dewaxed and rehydrated routinely, followed by denaturation and hybridization. For hybridization, the probes were resuspended with hybridization buffer and then denatured before being pipetted onto slides. Hybridization was performed at 37°C overnight, followed by DAPI staining. Finally, the slides were sealed with neutral balsam and observed under a fluorescence microscope. All experiments were performed using the manufacturer’s instructions for the RNA FISH (paraffin section) Kit.
Flow cytometry
To analyze expression in CD4+ T subsets, specifically Th1, Th2, Th17, and Treg, at the single-cell level, cells from human and mouse samples were stimulated with PMA, ionomycin, and BFA before examination. The cells were incubated with the surface markers (CD4), fixed, permeabilized, and then stained with intracellular cytokine monoclonal antibodies, including IFN-γ, IL-4, IL-17A, and Fxop3, respectively. Results were acquired using a FACS can cytometer (Beckman Coulter, USA) and analyzed with CytoExpert.
Dual-luciferase reporter assay
HEK293T cells grown in 24-well plates were transfected with 50 nM miR-10b mimic, 100 ng of pmirGLO vector (Promega, USA) tagged with GATA3 3′ UTR or PTEN 3′ UTR (containing the miR-10b-binding site), or empty pmirGLO plasmid using Lipofectamine 2000 (Invitrogen, USA). The firefly and Renilla luciferase activities in the cell lysates were assayed with a Dual-Luciferase Reporter Assay System (Promega) 48 h post-transfection.64
Macrophage phagocytosis
Macrophages were incubated with Dextran-FITC (Life Technology, USA) according to standard protocol. A flow cytometer (Beckman CytoFLEX) was used to detect phagocytosis.
Statistical analysis
GraphPad Prism v.8.2.1 (GraphPad, USA) was used for statistical analysis. Data in Figures are shown as the means ± standard deviations. One-way or two-way analysis of variance, Student’s t tests, and rank-sum test were used to determine the differences between experimental groups. p < 0.05 was considered significant.
Ethics approval and consent to participate
Ethics approval was granted by the Ethics Committee of Anhui Medical University. Animal experiments were performed according to the Anhui Medical University Animal Care and Use Guidelines. Moreover, the Ethics Committee approved the experimental protocols of Anhui Medical University. Written informed consent was obtained from all patients.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (31900616, 81673444, and 82003795), the Project of Improvement of Scientific Ability of Anhui Medical University (2020xkjT009), and Natural Science Foundation of Anhui Province for young scholars (1908085QH379).
Author contributions
J.T., D.H., H.J., X.T., and Z.X. performed the experiments. D.H. drafted the manuscript. X.W., W.H., T.L., and W.W. revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.12.022.
Supplemental information
References
- 1.McInnes I.B., Schett G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 2.Rao D.A., Gurish M.F., Marshall J.L., Slowikowski K., Fonseka C.Y., Liu Y., Donlin L.T., Henderson L.A., Wei K., Mizoguchi F., et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542:110–114. doi: 10.1038/nature20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kharlamova N., Jiang X., Sherina N., Potempa B., Israelsson L., Quirke A.M., Eriksson K., Yucel-Lindberg T., Venables P.J., Potempa J., et al. Antibodies to porphyromonas gingivalis indicate interaction between oral infection, smoking, and risk genes in rheumatoid arthritis etiology. Arthritis Rheumatol. 2016;68:604–613. doi: 10.1002/art.39491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beech J.T., Andreakos E., Ciesielski C.J., Green P., Foxwell B.M., Brennan F.M. T-cell contact-dependent regulation of CC and CXC chemokine production in monocytes through differential involvement of NFkappaB: implications for rheumatoid arthritis. Arthritis Res. Ther. 2006;8:R168. doi: 10.1186/ar2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nanki T., Lipsky P.E. Cytokine, activation marker, and chemokine receptor expression by individual CD4(+) memory T cells in rheumatoid arthritis synovium. Arthritis Res. 2000;2:415–423. doi: 10.1186/ar120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson A.E., Swan D.J., Wong O.Y., Buck M., Eltherington O., Harry R.A., Patterson A.M., Pratt A.G., Reynolds G., Doran J.P., et al. Tolerogenic dendritic cells generated with dexamethasone and vitamin D3 regulate rheumatoid arthritis CD4(+) T cells partly via transforming growth factor-beta1. Clin. Exp. Immunol. 2017;187:113–123. doi: 10.1111/cei.12870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crome S.Q., Wang A.Y., Levings M.K. Translational mini-review series on Th17 cells: function and regulation of human T helper 17 cells in health and disease. Clin. Exp. Immunol. 2010;159:109–119. doi: 10.1111/j.1365-2249.2009.04037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Annunziato F., Cosmi L., Liotta F., Maggi E., Romagnani S. Type 17 T helper cells-origins, features and possible roles in rheumatic disease. Nat. Rev. Rheumatol. 2009;5:325–331. doi: 10.1038/nrrheum.2009.80. [DOI] [PubMed] [Google Scholar]
- 9.van Hamburg J.P., Mus A.M., de Bruijn M.J., de Vogel L., Boon L., Cornelissen F., Asmawidjaja P., Hendriks R.W., Lubberts E. GATA-3 protects against severe joint inflammation and bone erosion and reduces differentiation of Th17 cells during experimental arthritis. Arthritis Rheum. 2009;60:750–759. doi: 10.1002/art.24329. [DOI] [PubMed] [Google Scholar]
- 10.Morgan M.E., Sutmuller R.P., Witteveen H.J., van Duivenvoorde L.M., Zanelli E., Melief C.J., Snijders A., Offringa R., de Vries R.R., Toes R.E. CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum. 2003;48:1452–1460. doi: 10.1002/art.11063. [DOI] [PubMed] [Google Scholar]
- 11.Morgan M.E., Flierman R., van Duivenvoorde L.M., Witteveen H.J., van Ewijk W., van Laar J.M., de Vries R.R., Toes R.E. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. 2005;52:2212–2221. doi: 10.1002/art.21195. [DOI] [PubMed] [Google Scholar]
- 12.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 13.Dong L., Wang X., Tan J., Li H., Qian W., Chen J., Chen Q., Wang J., Xu W., Tao C., et al. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J. Cell. Mol. Med. 2014;18:2213–2224. doi: 10.1111/jcmm.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niimoto T., Nakasa T., Ishikawa M., Okuhara A., Izumi B., Deie M., Suzuki O., Adachi N., Ochi M. MicroRNA-146a expresses in interleukin-17 producing T cells in rheumatoid arthritis patients. BMC Musculoskelet. Disord. 2010;11:209. doi: 10.1186/1471-2474-11-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie M., Wang J., Gong W., Xu H., Pan X., Chen Y., Ru S., Wang H., Chen X., Zhao Y., et al. NF-kappaB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J. Autoimmun. 2019;102:96–113. doi: 10.1016/j.jaut.2019.04.018. [DOI] [PubMed] [Google Scholar]
- 16.Jiajie T., Yanzhou Y., Hoi-Hung A.C., Zi-Jiang C., Wai-Yee C. Conserved miR-10 family represses proliferation and induces apoptosis in ovarian granulosa cells. Sci. Rep. 2017;7:41304. doi: 10.1038/srep41304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheedy P., Medarova Z. The fundamental role of miR-10b in metastatic cancer. Am. J. Cancer Res. 2018;8:1674–1688. [PMC free article] [PubMed] [Google Scholar]
- 18.Shams K., Kurowska-Stolarska M., Schutte F., Burden A.D., McKimmie C.S., Graham G.J. MicroRNA-146 and cell trauma down-regulate expression of the psoriasis-associated atypical chemokine receptor ACKR2. J. Biol. Chem. 2018;293:3003–3012. doi: 10.1074/jbc.M117.809780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen L., Al-Mossawi M.H., Ridley A., Sekine T., Hammitzsch A., de Wit J., Simone D., Shi H., Penkava F., Kurowska-Stolarska M., et al. miR-10b-5p is a novel Th17 regulator present in Th17 cells from ankylosing spondylitis. Ann. Rheum. Dis. 2017;76:620–625. doi: 10.1136/annrheumdis-2016-210175. [DOI] [PubMed] [Google Scholar]
- 20.Shu J.L., Zhang X.Z., Han L., Zhang F., Wu Y.J., Tang X.Y., Wang C., Tai Y., Wang Q.T., Chen J.Y., et al. Paeoniflorin-6'-O-benzene sulfonate alleviates collagen-induced arthritis in mice by downregulating BAFF-TRAF2-NF-kappaB signaling: comparison with biological agents. Acta Pharmacol. Sin. 2019;40:801–813. doi: 10.1038/s41401-018-0169-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi Y., Arron J.R., Townsend M.J. Promising bone-related therapeutic targets for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009;5:543–548. doi: 10.1038/nrrheum.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clavel G., Marchiol-Fournigault C., Renault G., Boissier M.C., Fradelizi D., Bessis N. Ultrasound and Doppler micro-imaging in a model of rheumatoid arthritis in mice. Ann. Rheum. Dis. 2008;67:1765–1772. doi: 10.1136/ard.2007.083915. [DOI] [PubMed] [Google Scholar]
- 23.Carmeliet P. Angiogenesis in health and disease. Nat. Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 24.Elshabrawy H.A., Chen Z., Volin M.V., Ravella S., Virupannavar S., Shahrara S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis. 2015;18:433–448. doi: 10.1007/s10456-015-9477-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clavel G., Valvason C., Yamaoka K., Lemeiter D., Laroche L., Boissier M.C., Bessis N. Relationship between angiogenesis and inflammation in experimental arthritis. Eur. Cytokine Netw. 2006;17:202–210. [PubMed] [Google Scholar]
- 26.Tu J., Hong W., Zhang P., Wang X., Korner H., Wei W. Ontology and function of fibroblast-like and macrophage-like synoviocytes: how do they talk to each other and can they be targeted for rheumatoid arthritis therapy? Front. Immunol. 2018;9:1467. doi: 10.3389/fimmu.2018.01467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tu J., Wang X., Gong X., Hong W., Han D., Fang Y., Guo Y., Wei W. Synovial macrophages in rheumatoid arthritis: the past, present, and future. Mediators Inflamm. 2020;2020:1583647. doi: 10.1155/2020/1583647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kotake S., Nanke Y., Mogi M., Kawamoto M., Furuya T., Yago T., Kobashigawa T., Togari A., Kamatani N. IFN-gamma-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. Eur. J. Immunol. 2005;35:3353–3363. doi: 10.1002/eji.200526141. [DOI] [PubMed] [Google Scholar]
- 29.Dayer J.M. Interleukin 1 or tumor necrosis factor-alpha: which is the real target in rheumatoid arthritis? J. Rheumatol. Suppl. 2002;65:10–15. [PubMed] [Google Scholar]
- 30.Sigidin Y.A., Loukina G.V., Skurkovich B., Skurkovich S. Randomized, double-blind trial of anti-interferon-gamma antibodies in rheumatoid arthritis. Scand. J. Rheumatol. 2001;30:203–207. doi: 10.1080/030097401316909530. [DOI] [PubMed] [Google Scholar]
- 31.Chu C.Q., Swart D., Alcorn D., Tocker J., Elkon K.B. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. 2007;56:1145–1151. doi: 10.1002/art.22453. [DOI] [PubMed] [Google Scholar]
- 32.Geboes L., De Klerck B., Van Balen M., Kelchtermans H., Mitera T., Boon L., De Wolf-Peeters C., Matthys P. Freund’s complete adjuvant induces arthritis in mice lacking a functional interferon-gamma receptor by triggering tumor necrosis factor alpha-driven osteoclastogenesis. Arthritis Rheum. 2007;56:2595–2607. doi: 10.1002/art.22791. [DOI] [PubMed] [Google Scholar]
- 33.Yang P., Qian F.Y., Zhang M.F., Xu A.L., Wang X., Jiang B.P., Zhou L.L. Th17 cell pathogenicity and plasticity in rheumatoid arthritis. J. Leukoc. Biol. 2019;106:1233–1240. doi: 10.1002/JLB.4RU0619-197R. [DOI] [PubMed] [Google Scholar]
- 34.Genovese M.C., Durez P., Richards H.B., Supronik J., Dokoupilova E., Mazurov V., Aelion J.A., Lee S.H., Codding C.E., Kellner H., et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann. Rheum. Dis. 2013;72:863–869. doi: 10.1136/annrheumdis-2012-201601. [DOI] [PubMed] [Google Scholar]
- 35.Blanco F.J., Moricke R., Dokoupilova E., Codding C., Neal J., Andersson M., Rohrer S., Richards H. Secukinumab in active rheumatoid arthritis: a phase III randomized, double-blind, active comparator- and placebo-controlled study. Arthritis Rheumatol. 2017;69:1144–1153. doi: 10.1002/art.40070. [DOI] [PubMed] [Google Scholar]
- 36.Genovese M.C., Greenwald M., Cho C.S., Berman A., Jin L., Cameron G.S., Benichou O., Xie L., Braun D., Berclaz P.Y., et al. A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2014;66:1693–1704. doi: 10.1002/art.38617. [DOI] [PubMed] [Google Scholar]
- 37.Nirula A., Nilsen J., Klekotka P., Kricorian G., Erondu N., Towne J.E., Russell C.B., Martin D.A., Budelsky A.L. Effect of IL-17 receptor A blockade with brodalumab in inflammatory diseases. Rheumatology. 2016;55:ii43–ii55. doi: 10.1093/rheumatology/kew346. [DOI] [PubMed] [Google Scholar]
- 38.Wang J.S., Fathman J.W., Lugo-Villarino G., Scimone L., von Andrian U., Dorfman D.M., Glimcher L.H. Transcription factor T-bet regulates inflammatory arthritis through its function in dendritic cells. J. Clin. Invest. 2006;116:414–421. doi: 10.1172/JCI26631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue X.H., Soroosh P., De Leon-Tabaldo A., Luna-Roman R., Sablad M., Rozenkrants N., Yu J.X., Castro G., Banie H., Fung-Leung W.P., et al. Pharmacologic modulation of ROR gamma t translates to efficacy in preclinical and translational models of psoriasis and inflammatory arthritis. Sci. Rep. 2016;6:37977. doi: 10.1038/srep37977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang M.R., Lyda B., Kamenecka T.M., Griffin P.R. Pharmacologic repression of retinoic acid receptor-related orphan nuclear receptor gamma is therapeutic in the collagen-induced arthritis experimental model. Arthritis Rheumatol. 2014;66:579–588. doi: 10.1002/art.38272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kremer J.M., Bloom B.J., Breedveld F.C., Coombs J.H., Fletcher M.P., Gruben D., Krishnaswami S., Burgos-Vargas R., Wilkinson B., Zerbini C.A.F., et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo (vol 60, pg 1895, 2009) Arthritis Rheum. 2012;64:1487. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 42.Maeshima K., Yamaoka K., Kubo S., Nakano K., Iwata S., Saito K., Ohishi M., Miyahara H., Tanaka S., Ishii K., et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-gamma and interleukin-17 production by human CD4+ T cells. Arthritis Rheum. 2012;64:1790–1798. doi: 10.1002/art.34329. [DOI] [PubMed] [Google Scholar]
- 43.Miossec P., Chomarat P., Dechanet J., Moreau J.F., Roux J.P., Delmas P., Banchereau J. Interleukin-4 inhibits bone resorption through an effect on osteoclasts and proinflammatory cytokines in an ex vivo model of bone resorption in rheumatoid arthritis. Arthritis Rheum. 1994;37:1715–1722. doi: 10.1002/art.1780371202. [DOI] [PubMed] [Google Scholar]
- 44.Horsfall A.C., Butler D.M., Marinova L., Warden P.J., Williams R.O., Maini R.N., Feldmann M. Suppression of collagen-induced arthritis by continuous administration of IL-4. J. Immunol. 1997;159:5687–5696. [PubMed] [Google Scholar]
- 45.Joosten L.A., Lubberts E., Helsen M.M., Saxne T., Coenen-de Roo C.J., Heinegard D., van den Berg W.B. Protection against cartilage and bone destruction by systemic interleukin-4 treatment in established murine type II collagen-induced arthritis. Arthritis Res. 1999;1:81–91. doi: 10.1186/ar14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valencia X., Stephens G., Goldbach-Mansky R., Wilson M., Shevach E.M., Lipsky P.E. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–261. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klatzmann D., Abbas A.K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol. 2015;15:283–294. doi: 10.1038/nri3823. [DOI] [PubMed] [Google Scholar]
- 48.Yu A., Zhu L., Altman N.H., Malek T.R. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30:204–217. doi: 10.1016/j.immuni.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu S., Pan W., Song X., Liu Y., Shao X., Tang Y., Liang D., He D., Wang H., Liu W., et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-alpha. Nat. Med. 2012;18:1077–1086. doi: 10.1038/nm.2815. [DOI] [PubMed] [Google Scholar]
- 50.Ma L., Teruya-Feldstein J., Weinberg R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer (vol 449, pg 682, 2007) Nature. 2008;455:256. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 51.Chen L., Al-Mossawi M.H., Ridley A., Sekine T., Hammitzsch A., de Wit J., Simone D., Shi H., Penkava F., Kurowska-Stolarska M., et al. miR-10b-5p is a novel Th17 regulator present in Th17 cells from ankylosing spondylitis. Ann. Rheum. Dis. 2017;76:620–624. doi: 10.1136/annrheumdis-2016-210175. [DOI] [PubMed] [Google Scholar]
- 52.Hwang E.S., Szabo S.J., Schwartzberg P.L., Glimcher L.H. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science. 2005;307:430–433. doi: 10.1126/science.1103336. [DOI] [PubMed] [Google Scholar]
- 53.Nawijn M.C., Dingjan G.M., Ferreira R., Lambrecht B.N., Karis A., Grosveld F., Savelkoul H., Hendriks R.W. Enforced expression of GATA-3 in transgenic mice inhibits Th1 differentiation and induces the formation of a T1/ST2-expressing Th2-committed T cell compartment in vivo. J. Immunol. 2001;167:724–732. doi: 10.4049/jimmunol.167.2.724. [DOI] [PubMed] [Google Scholar]
- 54.Usui T., Nishikomori R., Kitani A., Strober W. GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL-12Rbeta2 chain or T-bet. Immunity. 2003;18:415–428. doi: 10.1016/s1074-7613(03)00057-8. [DOI] [PubMed] [Google Scholar]
- 55.Bahena-Ocampo I., Espinosa M., Ceballos-Cancino G., Lizarraga F., Campos-Arroyo D., Schwarz A., Maldonado V., Melendez-Zajgla J., Garcia-Lopez P. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016;17:648–658. doi: 10.15252/embr.201540678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Essig K., Hu D., Guimaraes J.C., Alterauge D., Edelmann S., Raj T., Kranich J., Behrens G., Heiseke A., Floess S., et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr cells. Immunity. 2017;47:1067–1082 e1012. doi: 10.1016/j.immuni.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 57.Artham S., Verma A., Alwhaibi A., Adil M.S., Manicassamy S., Munn D.H., Somanath P.R. Delayed Akt suppression in the lipopolysaccharide-induced acute lung injury promotes resolution that is associated with enhanced effector regulatory T cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020;318:L750–L761. doi: 10.1152/ajplung.00251.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang C.R., Shiau A.L., Chen S.Y., Lin L.L., Tai M.H., Shieh G.S., Lin P.R., Yo Y.T., Lee C.H., Kuo S.M., et al. Amelioration of collagen-induced arthritis in rats by adenovirus-mediated PTEN gene transfer. Arthritis Rheum. 2008;58:1650–1656. doi: 10.1002/art.23517. [DOI] [PubMed] [Google Scholar]
- 59.Lee S.H., Park J.S., Byun J.K., Jhun J., Jung K., Seo H.B., Moon Y.M., Kim H.Y., Park S.H., Cho M.L. PTEN ameliorates autoimmune arthritis through down-regulating STAT3 activation with reciprocal balance of Th17 and Tregs. Sci. Rep. 2016;6:34617. doi: 10.1038/srep34617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hutamekalin P., Saito T., Yamaki K., Mizutani N., Brand D.D., Waritani T., Terato K., Yoshino S. Collagen antibody-induced arthritis in mice: development of a new arthritogenic 5-clone cocktail of monoclonal anti-type II collagen antibodies. J. Immunol. Methods. 2009;343:49–55. doi: 10.1016/j.jim.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 61.Chen J., Wang Y., Wu H., Yan S., Chang Y., Wei W. A modified compound from paeoniflorin, CP-25, suppressed immune responses and synovium inflammation in collagen-induced arthritis mice. Front. Pharmacol. 2018;9:563. doi: 10.3389/fphar.2018.00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang L., Li P., Song S., Liu Y., Wang Q., Chang Y., Wu Y., Chen J., Zhao W., Zhang Y., et al. Comparative efficacy of TACI-Ig with TNF-alpha inhibitor and methotrexate in DBA/1 mice with collagen-induced arthritis. Eur. J. Pharmacol. 2013;708:113–123. doi: 10.1016/j.ejphar.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 63.Chang Y., Wu Y., Wang D., Wei W., Qin Q., Xie G., Zhang L., Yan S., Chen J., Wang Q., et al. Therapeutic effects of TACI-Ig on rats with adjuvant-induced arthritis via attenuating inflammatory responses. Rheumatology. 2011;50:862–870. doi: 10.1093/rheumatology/keq404. [DOI] [PubMed] [Google Scholar]
- 64.Tu J., Tian G., Cheung H.H., Wei W., Lee T.L. Gas5 is an essential lncRNA regulator for self-renewal and pluripotency of mouse embryonic stem cells and induced pluripotent stem cells. Stem Cell Res. Ther. 2018;9:71. doi: 10.1186/s13287-018-0813-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.