

Abstract
The incorporation of CO2 into organometallic and organic molecules represents a sustainable way to prepare carboxylates. The mechanism of reductive carboxylation of alkyl halides has been proposed to proceed through the reduction of NiII to NiI by either Zn or Mn, followed by CO2 insertion into NiI-alkyl species. No experimental evidence has been previously established to support the two proposed steps. Demonstrated herein is that the direct reduction of (tBuXantphos)NiIIBr2 by Zn affords NiI species. (tBu-Xantphos)-NiI-Me and (tBu-Xantphos)NiI-Et complexes undergo fast insertion of CO2 at 22°C. The substantially faster rate, relative to that of NiII complexes, serves as the long-sought-after experimental support for the proposed mechanisms of Ni-catalyzed carboxylation reactions.
Keywords: carbon dioxide, nickel, reaction mechanisms, reduction, structure elucidation
Carbon dioxide (CO2) is a sustainable, inexpensive, and clean C1 source for chemical synthesis.[1] These advantages underlie recent advances in developing Ni-catalyzed reactions to incorporate CO2 into organic molecules.[2] Dong and co-workers reported the Ni-catalyzed carboxylation of organozinc reagents, giving rise to a variety of carboxylic acids.[3] Applying reductive conditions, the groups of Rovis,[4] Martin,[5] Tsuji,[6] and others[2] expanded the scope of carboxylation substrates to alkenes, aryl halides, and alkyl halides. The carboxylation of alkyl halides is proposed to initiate with the oxidative addition of Ni0 into an alkyl bromide, followed by the reduction of the NiII intermediate by either Mn or Zn to give a NiI-alkyl species (Scheme 1).[5a,b] Carboxylation of the NiI-alkyl intermediate gives rise to the carboxylate product. Computational studies on the reductive carboxylation of chlorobenzene suggest a similar mechanism, where a NiI-phenyl intermediate is responsible for carboxylation.[7] The reduction of NiII to NiI by either Mn or Zn and CO2 insertion into NiI-alkyl intermediates have not been established before. In light of the rapid growth of Ni-catalyzed carboxylation to harness CO2 as a chemical source, it is critical to obtain evidence for the proposed mechanism to inform future catalyst design.
Scheme 1.

Ni-catalyzed carboxylation of an alkyl bromide via a NiI-alkyl intermediate.[4]
The insertion of CO2 into organometallic molecules has been a long-time pursuit of fundamental studies.[8] While CO2 insertion into NiII-H complexes proved to be facile,[9] only three examples of CO2 insertion into Ni-carbyl complexes have been reported.[10] PCP-pincer-ligand-stabilized NiII-Me molecules undergo slow CO2 insertion at 150°C [Eq. (1)]. The
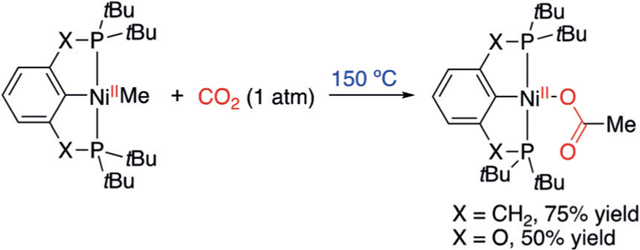 |
low reactivity contradicts the facile catalytic reactivity that has emerged in recent literature.[2] Computational studies predict that the more electron-rich NiI-carbyl complex undergoes faster CO2 insertion than NiII-carbyl complexes, as the carboxylation is nucleophilic in nature.[7] This hypothesis has found support in the recent observation of CO2 insertion into a NiI-aryl complex at room temperature.[10c] The Ni carboxylate product, however, was not characterized. The scarcity of examples of CO2 insertion into NiI-carbyl complexes is likely due to the difficulty of isolating well-defined NiI-carbyl and carboxylate molecules.[11,12] The previous NiI-alkyl[13] and NiIaryl[14] complexes have exceedingly bulky ligands to stabilize the open-shell molecules, and this is likely detrimental toward their reactivity with CO2.[14b,15] In light of the catalytic significance of NiI-carbyl complexes in mediating CO2 insertion and the fundamental interest in this bond-formation process, herein, we report the chemical reduction of NiII complexes to NiI and the first example of insertion of CO2 into NiI-alkyl complexes. This work serves as experimental support for the proposed mechanisms involving NiI species in Ni-catalyzed carboxylation reactions.
Bulky bidentate ligands with large bite angles can stabilize NiI complexes.[14b,15] We have recently demonstrated this concept in preparing (tBu-Xantphos)NiIAr complexes.[16] In an effort to prepare NiI-alkyl complexes, we treated (tBuXantphos)NiIIBr2 (1) with Zn in THF to generate a canary yellow solution (Scheme 2). X-ray crystallography established the product to be a 1:1 mixture of the ZnBr2-bound (tBuXantphos)NiIBr (2) and (tBu-Xantphos)NiIBr (3; see Figure S2 in the Supporting Information). The 1H NMR spectrum shows that no 3 is present in solution, indicating that the major species is 2. This single-electron reduction of NiII zinc supports a proposed intermediate step in catalytic carboxylation reactions (Scheme 1).[5a,b] Clean 3, without ZnBr2, can be obtained in high yields by the reduction of 1 with Cp2Co (Scheme 2). The EPR spectrum of 3 shows coupling to the bromide atom and both phosphines from the (tBu-Xantphos) ligand (see Figure S24).
Scheme 2.

Syntheses of (tBu-Xantphos)Ni-alkyl complexes and their reactivities toward CO2. THF=tetrahydrofuran.
Alkylation of 3 with MeLi and EtLi afforded (tBu-Xantphos)NiMe (4) and (tBu-Xantphos)NiEt (5), respectively, as dark brown solids (Scheme 2). The low yield of 5 is attributed to fast decomposition, likely by b-hydride elimination, followed by ejection of a hydrogen radical to form (tBu-Xantphos)Ni(N2).[16] Attempts to use a Ph-Xantphos ligand resulted in rapid decomposition. The crystal structure of 4 shows a slightly distorted trigonal-planar geometry, with the O-atom of tBu-Xantphos weakly interacting with Ni (Ni(1)–O(1)=2.6018(11) Å; Figure 1A). The Ni-C(1) bond length of 2.055(7) Å is longer than that of (terpy)NiMe (1.95(13) Å),[13a] and may be related to the redox activity of the terpy ligand. The structure of 5 is similar to that of 4, with a long β-H—Ni distance (3.18 c), suggesting the lack of a bagostic interaction (Figure 1B).[17] Addition of lithium acetylide reagents to 3 generated the NiI-acetylides 6 and 7. The crystal structure of 6 shows a Ni(1)-C(1)-C(2) angle of 174.53(14)°, consistent with an sp-hybridized C(1) (Figure 1C). The decreasing Ni(1)-C(1) bond lengths of 4, 5, 11,[16] and 6 reveal increasing bond strengths on the order of sp3 <sp2 <sp (Table 1). The spin-density plots of 4–6 obtained by DFT calculations reveal that the unpaired electron is primarily localized on the Ni center, with a small portion distributed to C(1) (Figure 1).[18] The spin-density is higher on C(1) for 4 and 5 than for 6 and 11, suggesting that the carbyl fragments on the former compounds have a more significant radical character. The EPR spectra of both 4 and 11 show strong coupling to the two phosphorus atoms of the (tBu- Xantphos) ligand and minimal coupling to the protons on the carbyl fragments (see Figures S25 and S26).
Figure 1.

X-ray structures[19] and spin-density plots of 4 (A), 5 (B), and 6 (C) at 50% probability thermal ellipsoids. Hydrogen atoms are omitted and tBu groups are truncated for clarity.
Table 1:
Bond parameters for 4, 5, 6, and 11.
| Bond length [Å] and angle [°] | 4 | 5 | 11 | 6 |
|---|---|---|---|---|
|
| ||||
| C(1) hybridization | sp3 | sp3 | sp2 | sp |
| Ni(1)-C(1) | 2.055(7) | 2.034(2) | 1.9795(14) | 1.9359(16) |
| Ni(1)···O(1) | 2.6018(11) | 2.6737(16) | 2.5184(10) | 2.5405(11) |
| Ni(1)-C(1)-C(2) | 118.07(18) | 174.53(14) | ||
Introducing 1 atm of CO2 to 4 immediately generated the NiI-carboxylate 9 as an orange compound [Eq. (2)]. The structure of 9 exhibits a trigonal-planar geometry with a secondary interaction between Ni and the Xantphos O(1) with a distance of 2.471 Å (Figure 2). The Ni-O(2) distance of 2.0145(13) Å is noticeably longer than that of the previously reported (PNP)NiII-formate (1.927(5) Å).[10a] The longer NiO(2) distance reflects a more electron-rich Ni center. The long distance of 2.9003(17) Å between Ni and O(3) reveals
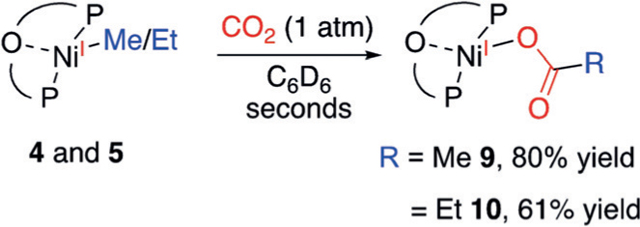 |
that the acetate is coordinated in an ɳ1 fashion. The EPR spectrum of 9 shows that the unpaired electron is coupled to phosphorus atoms of the (tBu-Xantphos) ligand and possibly to the protons on the acetate group (see Figure S26). The reaction of 5 with CO2 afforded 10 in 61% yield, as characterized by its analogous 1H NMR spectrum to that of 9. It is noteworthy that the carboxylation reactions are quite rapid, finishing in seconds at room temperature. This fast rate contrasts with those observed for (PCP)NiII-Me,[10] and is corroborated by previous DFT calculations.[7]
Figure 2.

X-ray structure[19] and spin-density plot of 9 at 50% probability thermal ellipsoids. Hydrogen atoms are omitted and tBu groups are truncated for clarity. Selected bond lengths [Å]: Ni(1)-O-(1)=2.0145(13), Ni(1)···O(2)=2.471, Ni(1)···O(3)=2.900.
The other (tBu-Xantphos)NiI molecules 6, 7, 8, and 11, however, did not react with CO2 over 24 hours under either 1 atm or 4 atm of CO2 (Scheme 3). Addition of Lewis acids, including LiCl, LiBF4, BPh3, and ZnCl2 did not promote CO2 insertion.[10c] When the less bulky (Ph-Xantphos)NiIPh is formed in situ from the reaction of (Ph-Xantphos)NiIIBrPh and Zn under one atmosphere of CO2, benzoic acid is obtained in 48% yield (see page S8 in the Supporting Information). This result suggests that the greater Ni–Csp2 bond strength, relative to the Ni–Csp3 bond, is not the factor that hinders carboxylation of 11. Instead, we attribute the contrasting reactivity of Ni-alkyls with Ni-aryls/alkynyls to their different mechanisms of insertion (Scheme 4). NiI-alkyl complexes could undergo insertion by nucleophilic attack of the alkyl groups to CO2, as proposed in previous computational studies (Scheme 4A).[10a] In contrast, sp and sp2 nucleophiles cannot access such a pathway, but can only undergo migratory insertion (Scheme 4B). The approach of CO2 to these (tBu-Xantphos)NiI complexes is inhibited by the bulky substituents on the ligand.[8c] The lack of reactivity of 8 with CO2 can be attributed to the lower nucleophilicity of phenoxides compared with carbanions.
Scheme 3.

NiI-carbyl and phenoxy complexes that are unreactive toward CO2.
Scheme 4.

Possible pathways for NiI-mediated CO2 insertion.
Finally, to verify the catalytic relevance of (Xantphos)Ni complexes, we tested the catalytic carboxylation of BnZnCl, PhZnCl, and n-BuZnCl, as well as the reductive carboxylation of benzyl bromide.[3] Either with no ligand or with tBu-Xantphos, no carboxylation reaction took place for zinc reagents, as the tBu-Xantphos ligand stabilizes NiI species and does not allow catalytic turnover. The use of Ph-Xantphos led to carboxylation of all three organozinc reagents under mild reaction conditions to afford the corresponding benzylacetic, benzoic, and valeric acids (Scheme 5A). (Ph-Xantphos)NiIIBr2 could be reduced in situ to NiI and, in fact, (PhXantphos)NiIBr renders a better yield for the carboxylation of n-BuZnCl. The catalytic reactivity of PhZnCl contrasts with the lack of stoichiometric reactivity of 11, but is consistent with the reactivity of in situ formed (Ph-Xantphos)NiIPh (see page S8), highlighting the access to the migratory insertion pathway that comes with a smaller steric profile (Scheme 4B). (Ph-Xantphos)NiIIBr2 also catalyzes reductive carboxylation of BnBr, albeit in low yield, but the bulkier (tBu-Xantphos)NiIBr does not effectively turn over the reaction (Scheme 5B).
Scheme 5.

(Xantphos)Ni-catalyzed carboxylation of organozinc reagents (A) and reductive carboxylation of BnBr (B). DMF=N,N’-dimethylformamide.
In summary, the direct reduction of (tBu-Xantphos)NiIIBr2 by Zn generates (tBu-Xantphos)NiIBr. (tBu-Xant-phos)NiI-Me and (tBu-Xantphos)NiI-Et complexes undergo fast insertion of CO2 at 228C, whereas (tBu-Xantphos)NiIphenyl, acetylide, and phenoxide complexes gave no insertion products. This observation represents the first characterization of CO2 insertion into NiI–alkyl bonds and verifies the nucleophilic addition mechanism proposed by previous DFT calculations. The fast reaction rate corroborates the facile catalytic reactions and the predicted importance of NiI intermediates relative to NiII in carboxylation. Characterization of these stoichiometric reactions provides evidence to support mechanistic proposals for Ni-catalyzed carboxylation reactions of organic and organometallic reagents.
Supplementary Material
Acknowledgements
J.D. thanks Paul Peterson and John Eng (Princeton University) for assistance with the EPR measurements and Qiao Lin for simulating the EPR spectra. This work was supported by the National Science Foundation under Award Number CHE-1654483. J.D. is supported by the Margaret and Herman Sokol Fellowship and the Ted Keusseff Fellowship. T.D. is a recipient of the Alfred P. Sloan Research Fellowship (FG-2018-10354) and the Camille-Dreyfus Teacher-Scholar Award (TC-19-019). T.D. acknowledges NSF (CHE-1827902) for funding to acquire an EPR spectrometer.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201906005.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Behr A, Angew. Chem. Int. Ed. Engl 1988, 27, 661–678; Angew. Chem. 1988, 100, 681–698; [Google Scholar]; b) Aresta M, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010; [Google Scholar]; c) Aresta M, Dibenedetto A, Quaranta E, Catal J. 2016, 343, 2–45. [Google Scholar]
- [2].a) For reviews, see: Sakakura T, Choi J-C, Yasuda H, Chem. Rev 2007, 107, 2365–2387; [DOI] [PubMed] [Google Scholar]; b) Correa A, Martín R, Angew. Chem. Int. Ed 2009, 48, 6201–6204; Angew. Chem. 2009, 121, 6317–6320; [DOI] [PubMed] [Google Scholar]; c) Martín R, Kleij AW, ChemSusChem 2011, 4, 1259–1263; [DOI] [PubMed] [Google Scholar]; d) Huang K, Sun C-L, Shi Z-J, Chem. Soc. Rev 2011, 40, 2435–2452; [DOI] [PubMed] [Google Scholar]; e) Cokoja M, Bruckmeier C, Rieger B, Herrmann WA, Kghn FE, Angew. Chem. Int. Ed 2011, 50, 8510–8537; Angew. Chem. 2011, 123, 8662–8690; [DOI] [PubMed] [Google Scholar]; f) Tsuji Y, Fujihara T, Chem. Commun 2012, 48, 9956–9964; [DOI] [PubMed] [Google Scholar]; g) Zhang L, Hou Z, Chem. Sci 2013, 4, 3395–3403; [Google Scholar]; h) Yeung CS, Dong VM, Top. Catal 2014, 57, 1342–1350; [Google Scholar]; i) Liu Q, Wu L, Jackstell R, Beller M, Nat. Commun 2015, 6; [DOI] [PubMed] [Google Scholar]; j) Yu B, He L-N, ChemSusChem 2015, 8, 52–62; [DOI] [PubMed] [Google Scholar]; k) Bçrjesson M, Moragas T, Gallego D, Martin R, ACS Catal. 2016, 6, 6739–6749; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Juliá-Hernández F, Gaydou M, Serrano E, van Gemmeren M, Martin R, Top. Curr. Chem 2016, 374, 1–38; [DOI] [PubMed] [Google Scholar]; m) Li Y, Cui X, Dong K, Junge K, Beller M, ACS Catal. 2016, 6, 1077–1086; [Google Scholar]; n) Fujihara T, Tsuji Y, J. Jpn. Pet. Inst 2016, 59, 84–92; [Google Scholar]; o) Zhang L, Hou Z, Curr. Opin. Green Sustainable Chem 2017, 3, 17–21; [Google Scholar]; p) Tortajada A, Juliá-Hernández F, Börjesson M, Moragas T, Martin R, Angew. Chem. Int. Ed 2018, 57, 15948–15982; Angew. Chem. 2018, 130, 16178–16214. [DOI] [PubMed] [Google Scholar]
- [3].Yeung CS, Dong VM, J. Am. Chem. Soc 2008, 130, 7826–7827. [DOI] [PubMed] [Google Scholar]
- [4].Williams CM, Johnson JB, Rovis T, J. Am. Chem. Soc 2008, 130, 14936–14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Correa A, Martán R, J. Am. Chem. Soc 2009, 131, 15974– 15975; [DOI] [PubMed] [Google Scholar]; b) León T, Correa A, Martin R, J. Am. Chem. Soc 2013, 135, 1221–1224; [DOI] [PubMed] [Google Scholar]; c) Liu Y, Cornella J, Martin R, J. Am. Chem. Soc 2014, 136, 11212–11215; [DOI] [PubMed] [Google Scholar]; d) Moragas T, Cornella J, Martin R, J. Am. Chem. Soc 2014, 136, 17702–17705; [DOI] [PubMed] [Google Scholar]; e) Wang X, Nakajima M, Martin R, J. Am. Chem. Soc 2015, 137, 8924–8927. [DOI] [PubMed] [Google Scholar]
- [6].Fujihara T, Nogi K, Xu T, Terao J, Tsuji Y, J. Am. Chem. Soc 2012, 134, 9106–9109. [DOI] [PubMed] [Google Scholar]
- [7].Sayyed FB, Tsuji Y, Sakaki S, Chem. Commun 2013, 49, 10715–10717. [DOI] [PubMed] [Google Scholar]
- [8].a) Fan T, Chen X, Lin Z, Chem. Commun 2012, 48, 10808– 10828; [DOI] [PubMed] [Google Scholar]; b) Hazari N, Heimann JE, Inorg. Chem 2017, 56, 13655–13678; [DOI] [PubMed] [Google Scholar]; c) Obst M, Pavlovic L, Hopmann KH, J. Organomet. Chem 2018, 864, 115–127. [Google Scholar]
- [9].Darensbourg DJ, Darensbourg MY, Goh LY, Ludvig M, Wiegreffe P, J. Am. Chem. Soc 1987, 109, 7539–7540. [Google Scholar]
- [10].a) Schmeier TJ, Hazari N, Incarvito CD, Raskatov JA, Chem. Commun 2011, 47, 1824–1826; [DOI] [PubMed] [Google Scholar]; b) Jonasson KJ, Wendt OF, Chem. Eur. J 2014, 20, 11894–11902; [DOI] [PubMed] [Google Scholar]; c) Charboneau DJ, Brudvig GW, Hazari N, Lant HMC, Saydjari AK, ACS Catal. 2019, 9, 3228–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Horn B, Limberg C, Herwig C, Braun B, Chem. Commun 2013, 49, 10923–10925. [DOI] [PubMed] [Google Scholar]
- [12].a) Lin C-Y, Power PP, Chem. Soc. Rev 2017, 46, 5347–5399; [DOI] [PubMed] [Google Scholar]; b) Zimmermann P, Limberg C, J. Am. Chem. Soc 2017, 139, 4233–4242. [DOI] [PubMed] [Google Scholar]
- [13].a) Anderson TJ, Jones GD, Vicic DA, J. Am. Chem. Soc 2004, 126, 8100–8101; [DOI] [PubMed] [Google Scholar]; b) Laskowski CA, Bungum DJ, Baldwin SM, Del Ciello SA, Iluc VM, Hillhouse GL, J. Am. Chem. Soc 2013, 135, 18272–18275; [DOI] [PubMed] [Google Scholar]; c) Kuang Y, Anthony D, Katigbak J, Marrucci F, Humagain S, Diao T, Chem 2017, 3, 268–280; [Google Scholar]; d) Joannou MV, Bezdek MJ, Albahily K, Korobkov I, Chirik PJ, Organometallics 2018, 37, 3389–3393. [Google Scholar]
- [14].a) Schley ND, Fu GC, J. Am. Chem. Soc 2014, 136, 16588– 16593; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mohadjer Beromi M, Banerjee G, Brudvig GW, Hazari N, Mercado BQ, ACS Catal. 2018, 8, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kitiachvili KD, Mindiola DJ, Hillhouse GL, J. Am. Chem. Soc 2004, 126, 10554–10555. [DOI] [PubMed] [Google Scholar]
- [16].Diccianni JB, Katigbak J, Hu C, Diao T, J. Am. Chem. Soc 2019, 141, 1788–1796. [DOI] [PubMed] [Google Scholar]
- [17].Xu H, White PB, Hu C, Diao T, Angew. Chem. Int. Ed 2017, 56, 1535–1538; Angew. Chem. 2017, 129, 1557–1560. [DOI] [PubMed] [Google Scholar]
- [18].Neese F, Wiley Interdiscip. Rev.: Comput. Mol. Sci 2012, 2, 73–78. [Google Scholar]
- [19].CCDC 1869808, 1869810, 1869811, 1916123, 1916124, 1916125, 1916126, 19161237, and 1916128 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.References
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.