

Abstract
This work describes the first preparation and application of primary trifluoroborate-iminiums (pTIMs) as a new, easily accessible and valuable class of organoboron derivatives. An array of structurally diverse pTIMs was prepared from potassium acyltrifluoroborates in excellent yields. Highly efficient and enantioselective [(R,R)-TethTsDpen-RuCl] complex-catalyzed hydrogenation of pTIMs provided direct access to chiral primary trifluoroborate-ammoniums (pTAMs). Moreover, facile synthesis of a series of structurally diverse chiral α-aminoboronic acids from chiral pTAMs was accomplished through novel, operationally simple and efficient conversion using hexamethyldisiloxane/aqueous HCl. Using no chromatography at any point, this work allowed easy access to chiral α-aminoboronic acids, as exemplified by the synthesis of optically pure anti-cancer drugs bortezomib and ixazomib.
Starting with potassium acyltrifluoroborates (KATs), N-unprotected chiral α-aminoboronic acids are prepared in three simple steps without chromatography. This facile methodology will tap the broad potential of these valuable compounds.
Introduction
Organoboron compounds1 play a pivotal role in synthetic organic chemistry, where they have enabled a myriad of useful transformations2–4 and have also been used in the field of materials science.5 The most advantageous application of organoboron compounds was achieved in medicinal chemistry6 where several boron-containing drugs reached the market in the last two decades. Among those the key role is attributed to proteasome-inhibiting7 anti-cancer drugs bortezomib8 and ixazomib.9 Furthermore, a structural analog delanzomib is in the development phase.10 These drugs possess a unique chemical structure in the sense that they all contain an α-aminoboronic acid moiety (Scheme 1). Beside their application in medicinal chemistry, α-aminoboronic acid derivatives found numerous applications in organic synthesis.11 However, preparation of chiral α-aminoboronates is more challenging compared to their natural counterparts and methods for their preparation are limited. A seminal work on the synthesis of chiral α-aminoboronates was conducted by Matteson, who developed a powerful homologation reaction which provided chiral (α-chloroalkyl)boronic esters that could be transformed into α-aminoboronic acids (Scheme 1).12 In 2008, Ellman reported a novel approach to obtain chiral α-aminoboronic esters using highly diastereoselective Cu-catalyzed addition of bis(pinacolato)diboron to N-tert-butanesulfinyl aldimines (Scheme 1).13 Next, we reported an alternative asymmetric hydrogenation approach to Matteson's methodology to prepare chiral (α-chloroalkyl)boronic- and α-aminoboronic esters (Scheme 1).14 Subsequently, novel methodologies for the synthesis of α-aminoboronates were reported by groups of Yudin,15 Sawamura,16 Morken,17 and others.11,18 In recent years, this field has continued to flourish with many exciting methods being developed.19–33 Among recent methodologies, discoveries of Yudin32 and Bode33 caught our attention. Namely, in these reports trifluoroborate-iminiums (TIMs) and MIDA-protected iminoboronates were prepared from acylboranes,34 and converted to racemic N-substituted α-aminoboronic acid derivatives with borohydride reagents (Scheme 1). This stimulated us to explore Ir-catalyzed hydrogenation of benzyl-protected TIMs (Bn-TIMs) to benzyl-protected trifluoroborate-ammoniums (Bn-TAMs). Although a series of racemic Bn-TAMs was prepared and their conversion to α-aminoboronic acids was demonstrated (Scheme 1), an asymmetric approach was not successful as a consequence of E/Z-isomerism that was observed in Bn-TIMs.35
Scheme 1. (A) Boron-containing proteasome inhibitors and key syntheses of α-aminoboronates. (B) Asymmetric hydrogenation of unprotected N–H imines. (C) This work.

Despite significant advances in the area, a need for new synthetic routes that would allow the preparation of chiral N-unprotected α-aminoboronic acids exists. Many of the existing methods are either racemic, allow limited scope of substrates, or do not enable the synthesis of N-unprotected derivatives.11,18–33,35
Based on our recent research,35 we envisioned that unsubstituted primary TIMs (pTIMs) could provide access to chiral α-aminoboronic acids using asymmetric hydrogenation. Moreover, the use of pTIMs would eliminate the N-deprotection step to obtain the free amino group, thereby shortening the overall synthetic sequence. However, the unprotected N–H imine functionality is considered challenging and thus underexplored in the area of asymmetric hydrogenation (Scheme 1),36–38 which suggests that asymmetric hydrogenation of pTIMs might be a demanding task. Herein, we present the results of our investigation that started with the preparation of a structurally diverse array of a novel subtype of TIM, i.e. pTIMs, which underwent Ru-catalyzed asymmetric hydrogenation to chiral pTAMs, followed by mild and efficient conversion of the trifluoroborate moiety to boronic acid functionality (Scheme 1).
Results and discussion
At the outset of our studies, we investigated the preparation of pTIMs. Although secondary and tertiary TIMs were prepared before by Bode33 and Bn-TIMs in our group,35 the formation of pTIMs was not an obvious task. We began our research using potassium acyltrifluoroborate (KAT) 1a as a model substrate for the formation of the corresponding pTIM 2a upon reaction with NH4Cl as a simple nitrogen precursor (Scheme S2†).
We initially screened various reaction conditions to establish the viability of the envisioned approach, using NH4Cl as an environmentally benign and non-toxic nitrogen source. Interestingly, no reaction took place between 1a and NH4Cl (2 eq.) in MeCN and DMF even at 60 °C in the presence of molecular sieves (3 Å) (Table S1, entries 1–3†), although these solvents enabled the preparation of secondary and tertiary TIMs.33,35 More encouraging results with 38% yield of 2a in 20 h (0.2 M concentration) were observed in MeOH at room temperature (Table S1, entry 4†). Interestingly, the reaction conducted at 0.05 M concentration afforded 62% conversion due to increased solubility of NH4Cl (Table S1, entry 5†). Increasing the temperature to 40 °C gave full conversion both at 0.2 M and 0.05 M concentration, albeit at the expense of 5–10% of unknown side product formation (Table S1, entries 6 and 7†) and a similar outcome was observed with 1 eq. NH4Cl (Table S1, entry 8†). By increasing the amount of NH4Cl to 5 eq., a clean reaction was obtained with 100% conversion in 4 h (Table S1, entry 9†). Finally, even without molecular sieves, a clean reaction was observed between 1a (0.08 M) and NH4Cl (5 eq.) in MeOH at 40 °C and afforded pTIM 2a after 4 h in 94% yield (Table S1, entry 10†). With optimal reaction conditions in hand, we converted a series of aromatic and aliphatic KATs 1a–2p to pTIMs 2a–2p. Overall, these reactions proceeded smoothly and provided aromatic and aliphatic pTIMs 2 in excellent 90–99% yields and high purity (Scheme 2).
Scheme 2. Synthesis of pTIMs 2 from KATs 1.

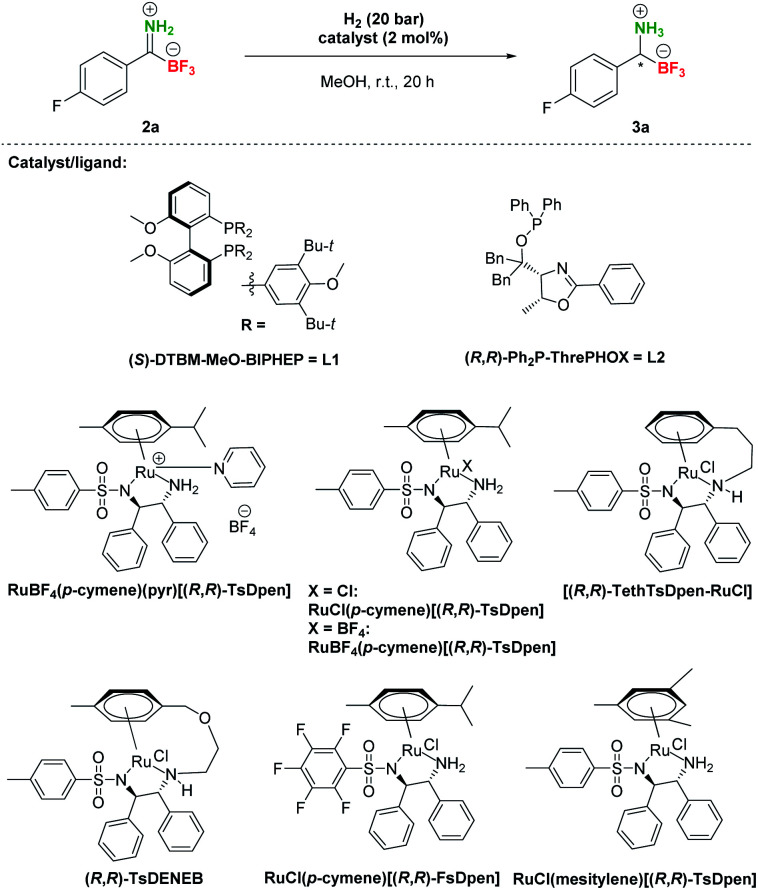
After the preparation of a series of pTIMs 2, we focused our attention on their asymmetric hydrogenation to chiral pTAMs 3 (Table 1). Initially, we decided to explore a diverse set of catalysts based on Ir, Ru and Rh metal precursors in combination with various P^P and P^N-ligands (Fig. S3†), as well as commercially available Noyori–Ikariya-type catalysts (Fig. S4†). The initial hydrogenation screening was done on substrate 2a at 20 bar H2 using Ir, Ru and Rh metal precursors (2 mol%) in combination with (S)-DTBM-MeO-BIPHEP (L1) and (R,R)-Ph2P-ThrePHOX (L2) ligands which provided good conversion in hydrogenation of Bn-TIMs to Bn-TAMs in the presence of t-BuOK base,35 albeit at moderate e.r.s. In contrast to Bn-TIMs, hydrolysis of 2a to 1a or decomposition of 2a was observed with selected catalytic systems when t-BuOK was used as an additive (Table 1, entries 1–4; Table S2, entries 1–6†) or no conversion of pTIM 2a to pTAM 3a was noticed in the absence of t-BuOK (Table 1, entries 5 and 6; Table S2, entries 7–10†). Encouraging first hit was obtained with the catalyst based on [Ru(p-cymene)Cl2]2 and (S)-DTBM-MeO-BIPHEP (L1) which enabled 23% conversion to 3a (racemic) in the absence of t-BuOK (Table 1, entry 7; Table S2, entry 11†). Furthermore, reaction using RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] provided 89% conversion to 3a and a promising e.r. of 79 : 21 when t-BuOK was not used (Table 1, entry 8; Table S2, entry 12†), which is in sharp contrast with a similar reaction using t-BuOK (Table 1, entry 4, Table S2; entry 6†) where only hydrolysis of 2a to 1a was observed. These results indicated that the base additive is detrimental to hydrogenation of pTIMs 2 and that only Ru-based catalysts are feasible for this reaction (Scheme 3). Currently, the price of Ru metal is more than seven times lower than that of Ir metal and thirtyfold lower compared to Rh metal.39 This implies that hydrogenation of pTIMs is not only a more atom economic approach, but also requires a less expensive metal catalyst than in the case of Bn-TIMs. For these reasons, we focused further on Ru-based catalysis.
Asymmetric hydrogenation of model pTIM 2aa.
| |||
|---|---|---|---|
| Entryb | Catalyst | 3a [%] | e.r. |
| 1c | [Ir(cod)]Cl/L1 | 0d | n.a. |
| 2c | [Ir(cod)(L2)]BArF | 0d | n.a. |
| 3c | RuCl[(p-cymene)]Cl/L1 | 0d | n.a. |
| 4c | RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] | 0d | n.a. |
| 5 | [Ir(cod)]Cl/L1 | 0e | n.a. |
| 6 | [Ir(cod)(L2)]BArF | 0e | n.a. |
| 7 | RuCl[(p-cymene)]Cl/L1 | 23 | 50 : 50 |
| 8 | RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] | 89 | 79 : 21 |
| 9 | RuCl(p-cymene)[(R,R)-TsDpen] | 81 | 89 : 11 |
| 10 | RuBF4(p-cymene)[(R,R)-TsDpen] | 100 | 89 : 11 |
| 11 | [(R,R)-TethTsDpen-RuCl] | 100 | 97 : 3 |
| 12 | (R,R)-TsDENEB | 88 | 92 : 8 |
| 13 | RuCl(p-cymene)[(R,R)-FsDpen] | 36 | 91 : 9 |
| 14 | RuCl(mesitylene)[(R,R)-TsDpen] | 40 | 88 : 12 |
| 15f | RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] | 100 | 77 : 23 |
| 16g | RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] | 100 | 80 : 20 |
| 17g | RuCl(p-cymene)[(R,R)-TsDpen] | 100 | 88 : 12 |
Cod = cyclooctadiene, BArF = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate.
Reactions were conducted by using 2a (0.1 mmol) and catalyst (2 mol%) in MeOH (0.05 M) with 20 bar H2 at room temperature for 20 h. The yields were determined by 1H-NMR and the e.r. (S/R) by chiral HPLC.
t-BuOK (1 eq.) as additive.
∼30% conversion to 1a observed.
No reaction observed.
With 50 bar H2.
Reaction time 44 h.
Scheme 3. Comparison of different reactivities of Bn-TIMs and pTIMs. Under the conditions that were successful in hydrogenation of the former, only partial hydrolysis to KAT was observed with model pTIM and an altogether different catalytic system was required. Catalyst and ligand structures are presented in Table 1.

Therefore, additional extensive catalyst screening was conducted using Ru-metal precursors in combination with various P^P and P^N-ligands and an extended list of Noyori–Ikariya-type catalysts40 (Table S3†). The study of ruthenium catalysts revealed that Ru–(p-cymene)–phosphine ligand complexes showed very low activity and no enantioselectivity in the hydrogenation of substrate 2a (Table S3, entries 2–14†). The RuOAc2–phosphine complexes showed limited enantioselectivity and also low conversions (Table S3, entries 15 and 16†), while the RuCl–diphosphine–diamine complexes performed slightly better in terms of enantioselectivity, but with similarly meagre conversions (Table S3, entries 17–24†). The dimethylamine adducts of RuCl–phosphine complexes were slightly more active but gave low enantioselectivity (Table S3, entries 25–27†). In contrast to these experiments, the best results for conversion of 2a to 3a were achieved with Noyori–Ikariya-type catalysts (Table 1). RuCl(p-cymene)[(R,R)-TsDpen] performed with slightly lower conversion compared to RuBF4(p-cymene)(pyr)[(R,R)-TsDpen], but gave higher e.r. (Table 1, entry 8 vs. entry 9). Furthermore, a full conversion was achieved with RuBF4(p-cymene)[(R,R)-TsDpen] along with a good e.r. (89 : 11, Table 1, entry 10). Gratifyingly, Wills's tethered catalyst [(R,R)-TethTsDpen-RuCl]41 provided full conversion and an excellent 97 : 3 e.r. (Table 1, entry 11). Catalysts (R,R)-TsDENEB®, RuCl(p-cymene)[(R,R)-FsDpen], and RuCl(mesitylene)[(R,R)-TsDpen] also afforded good enantioselectivities, albeit at incomplete conversions (Table 1, entries 12–14). Reactions with incomplete conversion could be completed by using higher hydrogen pressure (50 bar, Table 1, entry 15 vs. entry 8) with marginal loss of enantioselectivity, or by prolonging the reaction time to 44 h with retained e.r. (Table 1, entries 16 and 17 vs. entries 8 and 9).
Based on these screening results, [(R,R)-TethTsDpen-RuCl] was chosen as the optimal catalyst for the asymmetric reduction of TIMs 2 to TAMs 3. Next, the effect of the solvent was carefully examined (Table S4†), and MeOH was confirmed as the optimal choice. In addition, a mixture of MeOH/H2O (1–10% v/v) performed similarly at full conversion and nearly the same enantioselectivity, while the MeOH/H2O = 1 : 1 mixture or pure H2O promoted notable hydrolysis of 2a to 1a (Table S4, entries 1–9†). Furthermore, we established that concentration had no effect on conversion in the range of 0.025–0.1 M. Of note, the highest enantioselectivities were achieved at 0.05–0.10 M concentration (Table S4, entries 10–12†). Variations in [(R,R)-TethTsDpen-RuCl] loading were also examined (0.1–2.0 mol%) and while the reaction proceeded to completion also at 1 mol% catalyst (Table S4, entries 13–15†), 2 mol% catalyst loading was chosen in hydrogenation of other compounds to ensure full conversions across the whole substrate scope. Next, we proceeded with preparative hydrogenation of pTIMs 2a–p. Hydrogenation of aromatic derivatives 2a–h proceeded smoothly with full conversion at room temperature and 20 bar H2 with 2 mol% of [(R,R)-TethTsDpen-RuCl] in MeOH containing 2% water (v/v) giving the corresponding pTAMs 3a–i in excellent yields (88–95%) and high enantioselectivities with e.r.s ranging from 98 : 2 for 3h to 90 : 10 for 3i (Scheme 4). Interestingly, when aliphatic pTIM 2j was subjected to hydrogenation under the same conditions, low conversion (45%) was obtained (Table S5, entry 1†). Further optimization of the reaction conditions by increasing the catalyst loading to 4 mol% and prolongation of the reaction time to 72 hours provided 3j with a 94 : 6 e.r. at full conversion (Table S5, entry 4†). By applying these conditions, aliphatic pTIMs 2j–p were successfully hydrogenated and provided pTAMs 3j–p in 89–96% yields and excellent e.r.s that were in the range of 99 : 1 to 93 : 7, which is remarkable for such small compounds.36,37 Overall, aliphatic substrates gave slightly higher enantioselectivities compared to aromatic counterparts42 and provided the opposite enantiomer43 (Scheme 4).
Scheme 4. Scope of asymmetric hydrogenation of pTIMs 2.

In the final stage of our investigation, we studied the conversion of the trifluoroborate moiety in pTAMs 3 to the boronic acid functionality. Although hydrolysis of organic trifluoroborates to boronic acids in the presence of Cs2CO3 was extensively studied in connection with Suzuki–Miyaura coupling,44 general methods for the preparative hydrolysis of organic trifluoroborates to boronic acids are scarce and involve TMSCl/H2O,45 TMSCl/K2CO3/MeCN,46 silica gel/H2O,47 FeCl3/THF/H2O,48 alumina/H2O,49 and SiCl4/MeOH + HCl(aq.).33 This is associated with the fact that structurally diverse organic trifluoroborates exhibit notably different hydrolysis rates.50,51 As we have shown recently, only Bode's SiCl4 method33 enabled the conversion of pTAMs to the aminoboronic acids while other methods failed.35 However, huge excess (10 eq.) of difficult-to-handle SiCl4 was needed to transform pTAMs to aminoboronic acids.
Therefore, we decided to investigate other options for conversion of pTAMs 3 to α-aminoboronic acids 4 (Table S6†). Reactions using Cs2CO3 or silica gel/Cs2CO3 in MeOH or MeOH/H2O (95 : 5 v/v) gave no conversion of 3 to 4 (Table S6, entries 1–4†). The same was observed with TMSCl/Cs2CO3 in MeOH (Table S6, entry 5†). Surprisingly, TMSCl/Cs2CO3 in MeOH/H2O (95 : 5 v/v) provided 100% conversion of 3a, but the product 4a was contaminated with a side product (Table S6, entry 6†). Finally, replacement of the silicon source with hexamethyldisiloxane (HMDSO, O[Si(CH3)3]2) provided unexpected results (Table S6, entries 7–9†) and to our delight reaction of 3a with HMDSO (3 eq.) in the presence of aqueous HCl (4 eq.) in MeOH provided complete conversion to 4a (Table S6, entry 8†). It should be noted that HMDSO is non-toxic and has been studied as an MRI probe, where no toxic effects were observed when it was injected in mammals.52 Subsequent preparative transformation of pTAMs 3 to the corresponding α-aminoboronic acids 4 proceeded smoothly in excellent 94–100% yields (Scheme 5) without the loss of stereochemistry42 except for 3e where decomposition of the corresponding boronic acid to the benzyl amine was observed (Scheme S7 and Fig. S9†). Importantly, these highly polar and potentially labile compounds were isolated in pure form only by evaporation of volatiles.53
Scheme 5. Conversion of trifluoroborates 3 to boronic acids 4.

To demonstrate the significance of the presented methodology, we converted α-aminoboronic acid (R)-4m to bortezomib and ixazomib using a modified solid phase coupling approach (Scheme 6).54 Pure bortezomib and ixazomib were obtained in 44% and 50% yield respectively, which represents overall one of the shortest, simplest and most efficient approaches to these drugs (see Schemes S10–S15†).
Scheme 6. Conversion of (R)-4m to bortezomib and ixazomib.

Conclusions
In summary, we established a novel three-step synthetic strategy to a diverse array of chiral α-aminoboronic acids. The presented synthetic methodology is based on the conversion of KATs to their direct aza-analogs pTIMs 2, which are disclosed for the first time and could prove to be valuable organoboron reagents in their own right. Afterwards, highly stereoselective asymmetric hydrogenation of pTIMs 2 to chiral pTAMs 3 using the [(R,R)-TethTsDpen-RuCl] catalyst was successfully accomplished, followed by a novel and mild conversion of trifluoroborate to the boronic acid functionality in the presence of HMDSO and aqueous HCl. The represented transformations feature various aliphatic and aromatic substrates, proceed efficiently in high to excellent yields, are conducted in MeOH and water as sustainable solvents, and enable protecting-group free access to chiral α-aminoboronic acids. Our methodology towards these valuable building blocks will enable their broad deployment in access to libraries of novel proteasome inhibitors and beyond,11 as demonstrated by the synthesis of stereochemically pure drugs bortezomib and ixazomib.
Data availability
Data for this paper, including reactions screening and optimization experiments, detailed experimental procedures, overview of key synthetic procedures to bortezomib and characterization data, are provided in the ESI.†
Author contributions
A. Š. and Z. Č. conceived the study. A. Š. performed all experiments and collected the data. I. S. and Z. Č. guided the research. Z. Č. wrote the manuscript. A. Š. and I. S. performed editing of the manuscript. All authors discussed the results and contributed to the finalization of the paper.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the Slovenian Research Agency (research program P1-0208, P1-0230 and PhD grant to A. Š.). The authors acknowledge Lek Pharmaceuticals d.d. a Sandoz Company for support in chiral HPLC analysis, Mrs L. Kolenc and Dr N. Žigart for HPLC analyses and Mrs M. Frelih for HRMS analyses.
Electronic supplementary information (ESI) available: Preparation of pTIMs, enantioenriched pTAMs, and α-aminoboronic acids, details regarding reaction screening and optimization of reactions, determination of optical purities, and 1H, 13C, 11B, and 19F NMR spectra. See DOI: 10.1039/d1sc07065g
Notes and references
- For selected monographs on organoboron compounds and their chemistry, see: ; (a) Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. Hall, Wiley-VCH, Weinheim, 2011, vol. 1 and 2 [Google Scholar]; (b) Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer International Publishing, Cham, 2015 [Google Scholar]; (c) Boron-Based Compounds, ed. E. Hey-Hawkins and C. V. Teixidor, John Wiley & Sons, Ltd, Chichester, 2018 [Google Scholar]
- For selected and recent reviews on preparation of organoboron compounds and their reactivity, see: ; (a) Miyaura N. Suzuki A. Chem. Rev. 1995;95:2457–2483. doi: 10.1021/cr00039a007. [DOI] [Google Scholar]; (b) Mkhalid I. A. I. Barnard J. H. Marder T. B. Murphy J. M. Hartwig J. F. Chem. Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (c) Cid J. Gulyâs H. Carbó J. J. Fernândez E. Chem. Soc. Rev. 2012;41:3558–3570. doi: 10.1039/C2CS15291F. [DOI] [PubMed] [Google Scholar]; (d) Lennox A. J. J. Lloyd-Jones G. C. Chem. Soc. Rev. 2014;43:412–443. doi: 10.1039/C3CS60197H. [DOI] [PubMed] [Google Scholar]; (e) Ros A. Fernández R. Lassaletta J. M. Chem. Soc. Rev. 2014;43:3229–3243. doi: 10.1039/C3CS60418G. [DOI] [PubMed] [Google Scholar]; (f) Fyfe J. W. B. Watson A. J. B. Chem. 2017;3:31–55. doi: 10.1016/j.chempr.2017.05.008. [DOI] [Google Scholar]; (g) Hemming D. Fritzemeier R. Westcott S. A. Santos W. L. Steel P. G. Chem. Soc. Rev. 2018;47:7477–7494. doi: 10.1039/C7CS00816C. [DOI] [PubMed] [Google Scholar]; (h) Namirembe S. Morken J. P. Chem. Soc. Rev. 2019;48:3464–3474. doi: 10.1039/C9CS00180H. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Roscales S. Csáky A. G. Chem. Soc. Rev. 2020;49:5159–5177. doi: 10.1039/C9CS00735K. [DOI] [PubMed] [Google Scholar]; (j) Tian Y. M. Guo X. N. Braunschweig H. Radius U. Marder T. B. Chem. Rev. 2021;121:3561–3597. doi: 10.1021/acs.chemrev.0c01236. [DOI] [PubMed] [Google Scholar]; (k) Hu J. Ferger M. Shi Z. Marder T. B. Chem. Soc. Rev. 2021;50:13129–13188. doi: 10.1039/D0CS00843E. [DOI] [PubMed] [Google Scholar]; (l) Bose S. K. Mao L. Kuehn L. Radius U. Nekvinda J. Santos W. Westcott S. A. Steel P. G. Marder T. B. Chem. Rev. 2021;121:13238–13341. doi: 10.1021/acs.chemrev.1c00255. [DOI] [PubMed] [Google Scholar]
- For selected reviews on specific classes of organoboron compounds and reagents, see: ; (a) Molander G. A. Ellis N. Acc. Chem. Res. 2007;40:275–286. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]; (b) Darses S. Genet J.-P. Chem. Rev. 2008;108:288–325. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]; (c) Neeve E. C. Geier S. J. Mkhalid I. A. I. Westcott S. A. Marder T. B. Chem. Rev. 2016;116:9091–9161. doi: 10.1021/acs.chemrev.6b00193. [DOI] [PubMed] [Google Scholar]; (d) Miralles N. Maza R. J. Fernández E. Adv. Synth. Catal. 2018;360:1306–1327. doi: 10.1002/adsc.201701390. [DOI] [Google Scholar]; (e) de Hiller N. J. do Amaral e Silva N. A. Tavares T. A. Faria R. X. Eberlin M. N. de Luna Martins D. Eur. J. Org. Chem. 2020;2020:4841–4877. doi: 10.1002/ejoc.202000396. [DOI] [Google Scholar]; (f) He J. Rauch F. Finze M. Marder T. B. Chem. Sci. 2021;12:128–147. doi: 10.1039/D0SC05676F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Li X. Hall D. G. Adv. Synth. Catal. 2021;363:2209–2223. doi: 10.1002/adsc.202000690. [DOI] [Google Scholar]; (h) Budiman Y. P. Westcott S. A. Radius U. Marder T. B. Adv. Synth. Catal. 2021;363:2224–2255. doi: 10.1002/adsc.202001291. [DOI] [Google Scholar]; (i) Maza R. J. Carbó J. J. Fernández E. Adv. Synth. Catal. 2021;363:2274–2289. doi: 10.1002/adsc.202100192. [DOI] [Google Scholar]; (j) Kamio S. Yoshida H. Adv. Synth. Catal. 2021;363:2310–2324. doi: 10.1002/adsc.202001460. [DOI] [Google Scholar]
- For selected reviews on boron compounds in activation and catalysis, see: ; (a) Hall D. G. Chem. Soc. Rev. 2019;48:3475–3496. doi: 10.1039/C9CS00191C. [DOI] [PubMed] [Google Scholar]; (b) Su Y. Kinjo R. Chem. Soc. Rev. 2019;48:3613–3659. doi: 10.1039/C9CS00072K. [DOI] [PubMed] [Google Scholar]; (c) Carden J. L. Dasgupta A. Melen R. L. Chem. Soc. Rev. 2020;49:1706–1725. doi: 10.1039/C9CS00769E. [DOI] [PubMed] [Google Scholar]; (d) Ma Y. Lou S. J. Hou Z. Chem. Soc. Rev. 2021;50:1945–1967. doi: 10.1039/D0CS00380H. [DOI] [PubMed] [Google Scholar]; (e) Feng J. J. Mao W. Zhang L. Oestreich M. Chem. Soc. Rev. 2021;50:2010–2073. doi: 10.1039/D0CS00965B. [DOI] [PubMed] [Google Scholar]
- For selected reviews on organoboron compounds in material science, see: ; (a) Alcaraz G. Grellier M. Sabo-Etienne S. Acc. Chem. Res. 2009;42:1640–1649. doi: 10.1021/ar900091a. [DOI] [PubMed] [Google Scholar]; (b) Galbraith E. James T. D. Chem. Soc. Rev. 2010;39:3831–3842. doi: 10.1039/B926165F. [DOI] [PubMed] [Google Scholar]; (c) Li D. Zhang H. Wang Y. Chem. Soc. Rev. 2013;42:8416–8433. doi: 10.1039/C3CS60170F. [DOI] [PubMed] [Google Scholar]; (d) Kumar R. Karkamkar A. Bowden M. Autrey T. Chem. Soc. Rev. 2019;48:5350–5380. doi: 10.1039/C9CS00442D. [DOI] [PubMed] [Google Scholar]; (e) Mellerup S. K. Wang S. Chem. Soc. Rev. 2019;48:3537–3549. doi: 10.1039/C9CS00153K. [DOI] [PubMed] [Google Scholar]; (f) Qin Y. Liu X. Jia P. P. Xu L. Yang H. B. Chem. Soc. Rev. 2020;49:5678–5703. doi: 10.1039/C9CS00797K. [DOI] [PubMed] [Google Scholar]; (g) Shi Z. Han X. Hu W. Bai H. Peng B. Ji L. Fan Q. Li L. Huang W. Chem. Soc. Rev. 2020;49:7533–7567. doi: 10.1039/D0CS00234H. [DOI] [PubMed] [Google Scholar]; (h) Bismillah A. N. Aprahamian I. Chem. Soc. Rev. 2021;50:5631–5649. doi: 10.1039/D1CS00122A. [DOI] [PubMed] [Google Scholar]; (i) Berger S. Marder T. B. Mater. Horiz. 2022;9:112–120. doi: 10.1039/D1MH00696G. [DOI] [PubMed] [Google Scholar]; (j) Ji L. Griesbeck S. Marder T. B. Chem. Sci. 2017;8:846–863. doi: 10.1039/C6SC04245G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Yan Y.-Q. Li Y.-B. Wang J.-W. Zhao C.-H. Chem.–Asian J. 2013;8:3164–3176. doi: 10.1002/asia.201300872. [DOI] [PubMed] [Google Scholar]; (l) Li S.-Y. Sun Z.-B. Zhao C.-H. Inorg. Chem. 2017;56:8705–8717. doi: 10.1021/acs.inorgchem.6b02847. [DOI] [PubMed] [Google Scholar]; (m) Miao J. Wang Y. Liu J. Wang L. Chem. Soc. Rev. 2022;51:153–187. doi: 10.1039/D1CS00974E. [DOI] [PubMed] [Google Scholar]; (n) Hirai M. Tanaka N. Sakai M. Yamaguchi S. Chem. Rev. 2019;119:8291–8331. doi: 10.1021/acs.chemrev.8b00637. [DOI] [PubMed] [Google Scholar]; (o) von Grotthuss E. John A. Kaese T. Wagner M. Asian J. Org. Chem. 2018;7:37–53. doi: 10.1002/ajoc.201700495. [DOI] [Google Scholar]; (p) Budy H. Gilmer J. Trageser T. Wagner M. Eur. J. Inorg. Chem. 2020;2020:4148–4162. doi: 10.1002/ejic.202000786. [DOI] [Google Scholar]; (q) Huang Z. Wang S. Dewhurst R. D. Ignat'ev N. V. Finze M. Braunschweig H. Angew. Chem., Int. Ed. 2020;59:8800–8816. doi: 10.1002/anie.201911108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Stępień M. Gońka E. Żyła M. Sprutta N. Chem. Rev. 2017;117:3479–3716. doi: 10.1021/acs.chemrev.6b00076. [DOI] [PubMed] [Google Scholar]
- For selected reviews on organoboron compounds in medicinal chemistry, see: ; (a) Baker S. J. Ding C. Z. Akama T. Zhang Y. K. Hernandez V. Xia Y. Future Med. Chem. 2009;1:1275–1288. doi: 10.4155/fmc.09.71. [DOI] [PubMed] [Google Scholar]; (b) Dembitsky V. M. Al Quntar A. A. A. Srebnik M. Chem. Rev. 2011;111:209–237. doi: 10.1021/cr100093b. [DOI] [PubMed] [Google Scholar]; (c) Baker S. J. Tomsho J. W. Benkovic S. J. Chem. Soc. Rev. 2011;40:4279–4285. doi: 10.1039/C0CS00131G. [DOI] [PubMed] [Google Scholar]; (d) Smoum R. Rubinstein A. Dembitsky V. M. Srebnik M. Chem. Rev. 2012;112:4156–4220. doi: 10.1021/cr608202m. [DOI] [PubMed] [Google Scholar]; (e) Adamczyk-Woźniak A. Borys K. M. Sporzyński A. Chem. Rev. 2015;115:5224–5247. doi: 10.1021/cr500642d. [DOI] [PubMed] [Google Scholar]; (f) Diaz D. B. Yudin A. K. Nat. Chem. 2017;9:731–742. doi: 10.1038/nchem.2814. [DOI] [PubMed] [Google Scholar]; (g) Fernandes G. F. S. Denny W. A. Dos Santos J. L. Eur. J. Med. Chem. 2019;179:791–804. doi: 10.1016/j.ejmech.2019.06.092. [DOI] [PubMed] [Google Scholar]; (h) Plescia J. Moitessier N. Eur. J. Med. Chem. 2020;195:112270. doi: 10.1016/j.ejmech.2020.112270. [DOI] [PubMed] [Google Scholar]; (i) Lence E. González-Bello C. Adv. Therap. 2021;4:2000246. doi: 10.1002/adtp.202000246. [DOI] [Google Scholar]; (j) Sangu K. G. Shinde A. U. Chopra S. Rode H. B. Future Med. Chem. 2021;13:229–232. doi: 10.4155/fmc-2020-0161. [DOI] [PubMed] [Google Scholar]
- For proteasome inhibitors, see: ; (a) Teicher B. A. Tomaszewski J. E. Biochem. Pharmacol. 2015;96:1–9. doi: 10.1016/j.bcp.2015.04.008. [DOI] [PubMed] [Google Scholar]; (b) E: Manasanach E. Orlowski R. Z. Nat. Rev. Clin. Oncol. 2017;14:417–433. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fricker L. D. Annu. Rev. Pharmacol. Toxicol. 2020;60:457–476. doi: 10.1146/annurev-pharmtox-010919-023603. [DOI] [PubMed] [Google Scholar]
- (a) Teicher B. A. Ara G. Herbst R. Palombella V. J. Adams J. Clin. Cancer Res. 1999;5:2638–2645. [PubMed] [Google Scholar]; (b) Orlowski R. Z. Stinchcombe T. E. Mitchell B. S. Shea T. C. Baldwin A. S. Stahl S. Adams J. Esseltine D.-L. Elliott P. J. Pien C. S. et al. . J. Clin. Oncol. 2002;20:4420–4427. doi: 10.1200/JCO.2002.01.133. [DOI] [PubMed] [Google Scholar]; (c) Richardson P. G. Sonneveld P. Schuster M. W. Irwin D. Stadtmauer E. A. Facon T. Harousseau J. L. Ben-Yehuda D. Lonial S. Goldschmidt H. et al. . N. Engl. J. Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- (a) Kupperman E. Lee E. C. Cao Y. Bannerman B. Fitzgerald M. Berger A. Yu J. Yang Y. Hales P. Bruzzese F. et al. . Cancer Res. 2010;70:1970–1980. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]; (b) Richardson P. G. Zweegman S. O'Donnell E. K. Laubach J. P. Raje N. Voorhees P. Ferrari R. H. Skacel T. Kumar S. K. Lonial S. Expert Opin. Pharmacother. 2018;19:1949–1968. doi: 10.1080/14656566.2018.1528229. [DOI] [PubMed] [Google Scholar]
- (a) Piva R. Ruggeri B. Williams M. Costa G. Tamagno I. Ferrero D. Giai V. Coscia M. Peola S. Massaia M. et al. . Blood. 2008;111:2765–2775. doi: 10.1182/blood-2007-07-100651. [DOI] [PubMed] [Google Scholar]; (b) Gallerani E. Zucchetti M. Brunelli D. Marangon E. Noberasco C. Hess D. Delmonte A. Martinelli G. Böhm S. Driessen C. et al. . Eur. J. Cancer. 2013;49:290–296. doi: 10.1016/j.ejca.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Ming W. Soor H. S. Liu X. Trofimova A. Yudin A. K. Marder T. B. Chem. Soc. Rev. 2021;50:12151–12188. doi: 10.1039/D1CS00423A. [DOI] [PubMed] [Google Scholar]
- (a) Matteson D. S. Chem. Rev. 1989;89:1535–1551. doi: 10.1021/cr00097a009. [DOI] [Google Scholar]; (b) Matteson D. S. Tetrahedron. 1989;45:1859–1885. doi: 10.1016/S0040-4020(01)80052-1. [DOI] [Google Scholar]; (c) Matteson D. S. Tetrahedron. 1998;54:10555–10607. doi: 10.1016/S0040-4020(98)00321-4. [DOI] [Google Scholar]; (d) Matteson D. S. J. Org. Chem. 2013;78:10009–10023. doi: 10.1021/jo4013942. [DOI] [PubMed] [Google Scholar]
- (a) Beenen M. A. An C. Ellman J. A. J. Am. Chem. Soc. 2008;130:6910–6911. doi: 10.1021/ja800829y. [DOI] [PubMed] [Google Scholar]; (b) Buesking A. W. Bacauanu V. Cai I. Ellman J. A. J. Org. Chem. 2014;79:3671–3677. doi: 10.1021/jo500300t. [DOI] [PubMed] [Google Scholar]
- (a) Gazić Smilović I. Casas-Arcé E. Roseblade S. J. Nettekoven U. Zanotti-Gerosa A. Kovačevič M. Časar Z. Angew. Chem., Int. Ed. 2012;51:1014–1018. doi: 10.1002/anie.201106262. [DOI] [PubMed] [Google Scholar]; (b) Gazić Smilović I. Časar Z. Chim. Oggi. 2013;31:20–25. [Google Scholar]
- (a) He Z. Zajdlik A. St Denis J. D. Assem N. Yudin A. K. J. Am. Chem. Soc. 2012;134:9926–9929. doi: 10.1021/ja304173d. [DOI] [PubMed] [Google Scholar]; (b) Zajdlik A. Wang Z. Hickey J. L. Aman A. Schimmer A. D. Yudin A. K. Angew. Chem., Int. Ed. 2013;52:8411–8415. doi: 10.1002/anie.201302818. [DOI] [PubMed] [Google Scholar]
- Kawamorita S. Miyazaki T. Iwai T. Ohmiya H. Sawamura M. J. Am. Chem. Soc. 2012;134:12924–12927. doi: 10.1021/ja305694r. [DOI] [PubMed] [Google Scholar]
- Hong K. Morken J. P. J. Am. Chem. Soc. 2013;135:9252–9254. doi: 10.1021/ja402569j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For detailed recent reviews on the synthesis of α-aminoboronic acids, see: ; (a) Andrés P. Ballano G. Calaza M. I. Cativiela C. Chem. Soc. Rev. 2016;45:2291–2307. doi: 10.1039/C5CS00886G. [DOI] [PubMed] [Google Scholar]; (b) Šterman A. Sosič I. Gobec S. Časar Z. Org. Chem. Front. 2019;6:2991–2998. doi: 10.1039/C9QO00626E. [DOI] [Google Scholar]; (c) Thomas F. Pham T. L. Nachr. Chem. 2020;68:75–79. doi: 10.1002/nadc.20204096057. [DOI] [Google Scholar]; (d) Volochnyuk D. M. Gorlova A. O. Grygorenko O. O. Chem.–Eur. J. 2021;27:15277–15326. doi: 10.1002/chem.202102108. [DOI] [PubMed] [Google Scholar]
- Schwamb C. B. Fitzpatrick K. P. Brueckner A. C. Richardson H. C. Cheong P. H.-Y. Scheidt K. A. J. Am. Chem. Soc. 2018;140:10644–10648. doi: 10.1021/jacs.8b05045. [DOI] [PubMed] [Google Scholar]
- Bai X. Y. Zhao W. Sun X. Li B. J. J. Am. Chem. Soc. 2019;141:19870–19878. doi: 10.1021/jacs.9b10578. [DOI] [PubMed] [Google Scholar]
- Gao D. W. Gao Y. Shao H. Qiao T. Z. Wang X. Sanchez B. B. Chen J. S. Liu P. Engle K. M. Nat. Catal. 2020;3:23–29. doi: 10.1038/s41929-019-0384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W. Yang J. Hao F. ChemSusChem. 2020;13:121–125. doi: 10.1002/cssc.201902448. [DOI] [PubMed] [Google Scholar]
- Ming W. Liu X. Friedrich A. Krebs J. Marder T. B. Org. Lett. 2020;22:365–370. doi: 10.1021/acs.orglett.9b03773. [DOI] [PubMed] [Google Scholar]
- Reyes R. L. Sato M. Iwai T. Sawamura M. J. Am. Chem. Soc. 2020;142:589–597. doi: 10.1021/jacs.9b12013. [DOI] [PubMed] [Google Scholar]
- Ming W. Liu X. Friedrich A. Krebs J. Budiman Y. P. Huang M. Marder T. B. Green Chem. 2020;22:2184–2190. doi: 10.1039/D0GC00346H. [DOI] [Google Scholar]
- (a) Fan D. Zhang J. Hu Y. Zhang Z. Gridnev I. D. Zhang W. ACS Catal. 2020;10:3232–3240. doi: 10.1021/acscatal.9b04543. [DOI] [Google Scholar]; (b) Lou Y. Wang J. Gong G. Guan F. Lu J. Wen J. Zhang X. Chem. Sci. 2020;11:851–855. doi: 10.1039/C9SC04534A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Hall D. G. J. Am. Chem. Soc. 2020;142:9063–9069. doi: 10.1021/jacs.0c03207. [DOI] [PubMed] [Google Scholar]
- Guo L. Noble A. Aggarwal V. K. Angew. Chem., Int. Ed. 2021;60:212–216. doi: 10.1002/anie.202011739. [DOI] [PubMed] [Google Scholar]
- Sharma H. A. Essman J. Z. Jacobsen E. N. Science. 2021;374:752–757. doi: 10.1126/science.abm0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. Tan D. H. Fan W. X. Liu X. G. Wu J. Q. Huang Z. S. Li Q. Wang H. Angew. Chem., Int. Ed. 2021;60:3454–3458. doi: 10.1002/anie.202011872. [DOI] [PubMed] [Google Scholar]
- Kubota K. Miura D. Takeuchi T. Osaki S. Ito H. ACS Catal. 2021;11:6733–6740. doi: 10.1021/acscatal.1c01689. [DOI] [Google Scholar]
- Diaz D. B. Scully C. C. G. Liew S. K. Adachi S. Trinchera P. Denis J. D. St. Yudin A. K. Angew. Chem., Int. Ed. 2016;55:12659–12663. doi: 10.1002/anie.201605754. [DOI] [PubMed] [Google Scholar]
- Shiro T. Schuhmacher A. Jackl M. K. Bode J. W. Chem. Sci. 2018;9:5191–5196. doi: 10.1039/C8SC01486H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews on KATs, see:; (a) He Z. Zajdlik A. Yudin A. K. Acc. Chem. Res. 2014;47:1029–1040. doi: 10.1021/ar400210c. [DOI] [PubMed] [Google Scholar]; (b) Scharnagl F. K. Bose S. K. Marder T. B. Org. Biomol. Chem. 2017;15:1738–1752. doi: 10.1039/C6OB02425D. [DOI] [PubMed] [Google Scholar]; (c) Šterman A. Sosič I. Gobec S. Časar Z. ACS Omega. 2020;5:17868–17875. doi: 10.1021/acsomega.0c02391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu D. Taguchi J. Tanriver M. Bode J. W. Angew. Chem., Int. Ed. 2020;59:16847–16858. doi: 10.1002/anie.202005050. [DOI] [PubMed] [Google Scholar]; (e) Holownia A. . Apte C. N. Yudin A. Chem. Sci. 2021;12:5346–5360. doi: 10.1039/D1SC00077B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šterman A. Košmrlj J. Žigart N. Gobec S. Sosič I. Časar Z. Adv. Synth. Catal. 2021;363:2396–2402. doi: 10.1002/adsc.202001350. [DOI] [Google Scholar]
- (a) Hou G. Gosselin F. Li W. McWilliams J. C. Sun Y. Weisel M. O'Shea P. D. Chen C.-y. Davies I. W. Zhang X. J. Am. Chem. Soc. 2009;131:9882–9883. doi: 10.1021/ja903319r. [DOI] [PubMed] [Google Scholar]; (b) Hou G. Tao R. Sun Y. Zhang X. Gosselin F. J. Am. Chem. Soc. 2010;132:2124–2125. doi: 10.1021/ja909583s. [DOI] [PubMed] [Google Scholar]
- Zhao Q. Wen J. Tan R. Huang K. Metola P. Wang R. Anslyn E. V. Zhang X. Angew. Chem., Int. Ed. 2014;53:8467–8470. doi: 10.1002/anie.201404570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews on hydrogenation of imines, see:; (a) Fleury-Brégeot N. de la Fuente V. Castillón S. Claver C. ChemCatChem. 2010;2:1346–1371. doi: 10.1002/cctc.201000078. [DOI] [Google Scholar]; (b) Xie J.-H. Zhu S.-F. Zhou Q.-L. Chem. Rev. 2011;111:1713–1760. doi: 10.1021/cr100218m. [DOI] [PubMed] [Google Scholar]; (c) Yu Z. Jin W. Jiang Q. Angew. Chem., Int. Ed. 2012;51:6060–6072. doi: 10.1002/anie.201200963. [DOI] [PubMed] [Google Scholar]; (d) Seo C. S. G. Morris R. H. Organometallics. 2019;38:47–65. doi: 10.1021/acs.organomet.8b00774. [DOI] [Google Scholar]; (e) Barrios-Rivera J. Xu Y. Wills M. Vyas V. K. Org. Chem. Front. 2020;7:3312–3342. doi: 10.1039/D0QO00794C. [DOI] [Google Scholar]; (f) Morisaki K. Morimoto H. Ohshima T. ACS Catal. 2020;10:6924–6951. doi: 10.1021/acscatal.0c01212. [DOI] [Google Scholar]; (g) Peñafiel I., Mangas-Sánchez J. and Claver C., in Asymmetric Hydrogenation and Transfer Hydrogenation, ed. V. Ratovelomanana-Vidal and P. Phansavath, Wiley, 1st edn, 2021, pp. 281–305 [Google Scholar]
- Prices of metals are reported as of January 18th, 2022 in US dollars per troy ounce. Rh: 16 500 $, Ir: 4000 $, Ru: 550 $. Source: Johnson Matthey, Price Charts, http://www.platinum.matthey.com/prices/price-charts, accessed January 2022
- (a) Hashiguchi S. Fujii A. Takehara J. Ikariya T. Noyori R. J. Am. Chem. Soc. 1995;117:7562–7563. doi: 10.1021/ja00133a037. [DOI] [Google Scholar]; (b) Fujii A. Hashiguchi S. Uematsu N. Ikariya T. Noyori R. J. Am. Chem. Soc. 1996;118:2521–2522. doi: 10.1021/ja954126l. [DOI] [Google Scholar]
- (a) Hannedouche J. Clarkson G. J. Wills M. J. Am. Chem. Soc. 2004;126:986–987. doi: 10.1021/ja0392768. [DOI] [PubMed] [Google Scholar]; (b) Hayes A. M. Morris D. J. Clarkson G. J. Wills M. J. Am. Chem. Soc. 2005;127:7318–7319. doi: 10.1021/ja051486s. [DOI] [PubMed] [Google Scholar]; (c) Nedden H. G. Zanotti-Gerosa A. Wills M. Chem. Rec. 2016;16:2623–2643. doi: 10.1002/tcr.201600084. [DOI] [PubMed] [Google Scholar]; (d) Wills M. Top. Curr. Chem. 2016;374:14. doi: 10.1007/s41061-016-0013-7. [DOI] [PubMed] [Google Scholar]
- Stereochemical purity of aliphatic TAMs 3k–p was determined by their conversion to aliphatic α-aminoboronic acids 4k–p followed by the formation of Mosher amides and determination of e.r. with 19F-NMR: ; Allen D. A. Tomaso A. E. Priest O. P. Hindson D. F. Hurlburt J. L. J. Chem. Educ. 2008;85:698–700. doi: 10.1021/ed085p698. [DOI] [Google Scholar]; . For the α-aminoboronic acids 4 containing aromatic substituents the enantiomeric purity was also verified with Mosher amides of derivatives 4a and 4j which had comparable e.r. to the corresponding pTAM 3a and 3j (see the ESI†).
- With [(R,R)-TethTsDpen-RuCl], aliphatic pTIMs 2j–p provide (S)-pTAMs 3j–p as unambiguously confirmed by the synthesis of (S,S)-bortezomib from 4m and determination of optical rotation for 3j–p. Based on the opposite direction of optical rotation, aromatic pTIMs 2a–i provide (R)-pTAMs 3a–i. The correct diastereoisomer of bortezomib was synthesized using [(S,S)-TethTsDpen-RuCl] in conversion of 2m to (R)-3m.
- Lennox A. J. J., Organotrifluoroborate Preparation, Coupling and Hydrolysis, Springer, Heidelberg, 2013 [Google Scholar]
- (a) Yuen A. K. L. Hutton C. A. Tetrahedron Lett. 2005;46:7899–7903. doi: 10.1016/j.tetlet.2005.09.101. [DOI] [Google Scholar]; (b) Inglis S. R. Woon E. C. Y. Thompson A. L. Schofield C. J. J. Org. Chem. 2010;75:468–471. doi: 10.1021/jo901930v. [DOI] [PubMed] [Google Scholar]
- Churches Q. I. Hooper J. F. Hutton C. A. J. Org. Chem. 2015;80:5428–5435. doi: 10.1021/acs.joc.5b00182. [DOI] [PubMed] [Google Scholar]
- Molander G. A. Cavalcanti L. N. Canturk B. Pan P.-S. Kennedy L. E. J. Org. Chem. 2009;74:7364–7369. doi: 10.1021/jo901441u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins D. W. Yao M.-L. Yong L. Kabalka G. W. Tetrahedron Lett. 2011;52:6534–6536. doi: 10.1016/j.tetlet.2011.09.140. [DOI] [Google Scholar]
- Kabalka G. W. Coltuclu V. Tetrahedron Lett. 2009;50:6271–6272. doi: 10.1016/j.tetlet.2009.09.008. [DOI] [Google Scholar]
- Lennox A. J. J. Lloyd-Jones G. C. J. Am. Chem. Soc. 2012;134:7431–7441. doi: 10.1021/ja300236k. [DOI] [PubMed] [Google Scholar]
- Omari I. Yunker L. P. E. Penafiel J. Gitaari D. San Roman A. McIndoe J. S. Chem.–Eur. J. 2021;27:3812–3816. doi: 10.1002/chem.202004726. [DOI] [PubMed] [Google Scholar]
- (a) Kodibagkar V. D. Cui W. Merritt M. E. Mason R. P. Magn. Reson. Med. 2006;55:743–748. doi: 10.1002/mrm.20826. [DOI] [PubMed] [Google Scholar]; (b) Gulaka P. K. Rastogi U. McKay M. A. Wang X. Mason R. P. Kodibagkar V. D. NMR Biomed. 2011;24:1226–1234. doi: 10.1002/nbm.1678. [DOI] [PubMed] [Google Scholar]
- Hinkes S. P. A. Klein C. D. P. Org. Lett. 2019;21:3048–3052. doi: 10.1021/acs.orglett.9b00584. [DOI] [PubMed] [Google Scholar]
- For the synthesis of bortezomib and ixazomib we used a modified solid phase procedure of Klein et al. and Stivala et al., where Fmoc-protection/deprotection of α-aminoboronic acids was omitted from the synthesis and thus the overall two step shorter solid phase synthetic sequence was obtained and provided even cleaner products without chromatography (see the ESI†): ; (a) Behnam M. A. M. Sundermann T. R. Klein C. D. Org. Lett. 2016;18:2016–2019. doi: 10.1021/acs.orglett.6b00625. [DOI] [PubMed] [Google Scholar]; (b) Daniels B. E. Stivala C. E. RSC Adv. 2018;8:3343–3347. doi: 10.1039/C7RA13479G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hinkes S. P. A. Kämmerer S. Klein C. D. P. Chem. Sci. 2020;11:9898–9903. doi: 10.1039/D0SC03999C. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for this paper, including reactions screening and optimization experiments, detailed experimental procedures, overview of key synthetic procedures to bortezomib and characterization data, are provided in the ESI.†