

Abstract
Background and Objectives
This study aims to quantify microglial activation in individuals with Alzheimer disease (AD) using the 18-kDa translocator protein (TSPO) PET imaging in the hippocampus and precuneus, the 2 AD-vulnerable regions, and to evaluate the association of baseline neuroinflammation with amyloidosis, tau, and longitudinal cognitive decline.
Methods
Twenty-four participants from the Knight Alzheimer Disease Research Center (Knight ADRC) were enrolled and classified into stable cognitively normal, progressor, and symptomatic AD groups based on clinical dementia rating (CDR) at 2 or more clinical assessments. The baseline TSPO radiotracer [11C]PK11195 was used to image microglial activation. Baseline CSF concentrations of Aβ42, Aβ42/Aβ40 ratio, tau phosphorylated at position 181 (p-tau181), and total tau (t-tau) were measured. Clinical and cognitive decline were examined with longitudinal CDR and cognitive composite scores (Global and Knight ADRC-Preclinical Alzheimer Cognitive Composite [Knight ADRC-PACC] Score).
Results
Participants in the progressor and symptomatic AD groups had significantly elevated [11C]PK11195 standard uptake value ratios (SUVRs) in the hippocampus but not in the precuneus region. In the subcohort with CSF biomarkers (16 of the 24), significant negative correlations between CSF Aβ42 or Aβ42/Aβ40 and [11C]PK11195 SUVR were observed in the hippocampus and precuneus. No correlations were observed between [11C]PK11195 SUVR and CSF p-tau181 or t-tau at baseline in those regions. Higher baseline [11C]PK11195 SUVR averaged in the whole cortical regions predicted longitudinal decline on cognitive tests.
Discussion
Microglial activation is increased in individuals with brain amyloidosis and predicts worsening cognition in AD.
Classification of Evidence
This study provides Class II evidence that in patients with AD, higher baseline [11C]PK11195 SUVR averaged in the whole cortical regions was associated with longitudinal decline on cognitive tests.
The characteristic pathology of Alzheimer disease (AD) is the deposition of extracellular β-amyloid (Aβ) plaques and intracellular tau fibrils. Amyloid plaques and tau tangles are believed to initiate a cascade of pathology that includes neuroinflammation,1 synaptic dysfunction, and neuronal death, resulting in dementia. There is extensive literature documenting a microglial-mediated inflammatory response in AD.2 Multiple longitudinal studies demonstrate that inflammation and microglial activation begin early in AD, many years before dementia onset.3,4 It is also notable that microglia play a key role in neuroinflammatory response not only to amyloid deposition but also to tau accumulation in AD brains.5
Despite the association of neuroinflammation with AD, the exact role of inflammation in AD remains unclear. It has been suggested that in the early stages of AD, the initial microglial activation may serve a protective role trying to clear amyloid.6 In the later stages of AD, when clearance fails, microglia could play a detrimental role by producing proinflammatory cytokines, leading to progressive neurodegeneration.7 Furthermore, the relationship between neuroinflammation, amyloidosis, and cognitive decline is still unclear and warrants further investigation. The hippocampus and precuneus are involved in the earliest neuropathologic changes in AD and are sensitive to AD pathologies of amyloid and tau.8-11 Focusing analyses on those regions would shed light on the relationships among the neuroinflammation, amyloid, tau, and cognition.
After activation, microglia proliferate and express a series of genes for proinflammatory cytokines and certain receptors on their surface, including the 18-kDa translocator protein (TSPO). The advent of PET radioligands that bind the microglial protein, TSPO, including [11C]PK11195, allows assessment of activated microglia in many neurologic disorders, including AD.12 The aim of this study was to measure microglial activation with [11C]PK11195 PET imaging in cognitively normal and symptomatic AD participants and evaluate its association with CSF biomarkers of AD pathology and performance on cognitive tests. We hypothesized that microglial activation would be associated with amyloidosis and tau and would predict cognitive decline. These primary research questions have been addressed in this study.
Methods
Study Participants
Study participants were enrolled in ongoing studies at the Knight Alzheimer Disease Research Center (Knight ADRC) at Washington University School of Medicine (St. Louis, MO). The Human Research Protection Office at Washington University approved all studies, and written informed consent was obtained from all participants. Both cognitively normal and symptomatic AD participants with an age range from 55 to 90 years who underwent [11C] PK11195 PET scans were included in this study. The cognitively normal and the symptomatic AD participants were evaluated by the Clinical Core of the Knight ADRC. Detailed clinical assessments of the participants were performed in accordance with the Uniform Data Set protocol of the National Alzheimer's Coordinating Center.13 A clinical diagnosis of symptomatic AD, where appropriate, was made in accordance with criteria developed by working groups from the National Institute on Aging and the Alzheimer's Association.14 The severity of dementia was measured with the global clinical dementia rating (CDR),15 whereby CDR 0 is cognitively unimpaired, 0.5 is very mild dementia, 1 is mild dementia, 2 is moderate dementia, and 3 is severe dementia. The CDR sum of boxes (CDR-SB) was used as a more granular measure of clinical impairment.16 The Mini-Mental State Examination (MMSE) was also administered.17
Participants were categorized into 3 groups. Participants with normal cognition (CDR = 0) at the time of the PET scan who remained cognitively normal over 15 years were categorized as stable cognitively normal. Participants who were cognitively normal (CDR = 0) at the time of the PET scan and developed symptomatic AD (encompassing both mild cognitive impairment due to AD and AD dementia [CDR> 0]) over an average of 7 years of follow-up were categorized as progressors. Participants with symptomatic AD dementia at the time of PET scan (CDR > 0) were categorized as symptomatic AD.
Standard Protocol Approvals, Registrations, and Patient Consents
The local ethics committee of the Washington University School of Medicine approved the study, and all participants provided written informed consent before entering the study.
Cognitive Measures
As a measure of cognitive performance, we used the Knight ADRC Preclinical Alzheimer Cognitive Composite (Knight ADRC-PACC) score and a global composite score for each individual from the Knight ADRC cognitive battery. Consistent with previously published methods, the Knight ADRC-PACC score was calculated by averaging the z score across 5 cognitive tests measuring memory, attention, and processing speed.18 The cognitive tests were performed annually for each individual. Specifically, the included tests were MMSE, the Digit Symbol subtask of the Wechsler Adult Intelligence Scale, the animal naming test, the associate learning summary score from the Wechsler memory scale, and the free and cued selective reminding test. The global score was derived by averaging the z score for each individual across all common tasks collected by the neuropsychologic battery of the Knight ADRC. In addition to the tasks described earlier, the global score also included the Trail A and Trail B subtasks of the trail making test, the Boston naming test, the summary score of the crossing-off task, the mental control subtask of the Wechsler Memory Scale, the summary score for the S & P word fluency task, the digit span forward and backward subtasks of the revised Wechsler Memory Scale, and the information and block subtasks of the Weschler Adult Intelligence Scale. The resulting Knight ADRC-PACC and global scores were used as measures of specific and overall cognitive performance in subsequent analyses, respectively. The baseline cognitive scores used the cognitive assessment closest to the TSPO PET scan (all were within 1 year).
APOE Genotyping
DNA was extracted from peripheral blood samples by standard procedures. APOE genotyping was performed as previously described.19 Individuals carrying at least 1 APOE ε4 allele were classified as APOE ε4 positive (APOE ε4+).
CSF Collection and Analysis
CSF was collected under a standard protocol. Participants underwent a lumbar puncture at approximately 8 am after overnight fasting. Twenty to 30 mL of CSF was collected in a 50-mL polypropylene tube through gravity drip using an atraumatic Sprotte 22-gauge spinal needle. CSF was kept on ice and centrifuged (2,000 g, 10 minutes) within 2 hours of collection to pellet any cellular debris and then transferred to another 50 mL tube. CSF was aliquoted in 500-µL volumes into polypropylene tubes and stored at −80°C, as previously described.20
Before analysis, samples were brought to room temperature. Samples were vortexed and transferred to provided cuvettes for analysis. Concentrations of Aβ40, Aβ42, tau phosphorylated at position 181 (p-tau181), and total tau (t-tau) were measured by chemiluminescent enzyme immunoassay using a fully automated platform (LUMIPULSE G1200, Fujirebio, Malvern, PA) according to manufacturer's specifications. A single lot of reagents was used for all samples.
MRI Acquisition
Anatomic MRI was obtained on each participant with T1-weighted magnetization-prepared rapid acquisition gradient echo (MPRAGE) sequences using a Siemens Vision 1.5T scanner or a Siemens Avanto 1.5T scanner (Siemens, Erlangen, Germany). The acquisition parameters for MPRAGE were the following: TR, 9.7 ms; TE, 4.0 ms; inversion time, 20 ms; imaging resolution, 1 × 1 × 1.25 mm3. The T1-weighted MRI was used for FreeSurfer parcellation21 (freesurfer.net/).
PET Acquisition and Processing
TSPO PET imaging was performed on a Siemens 962 HR + PET scanner using the radiotracer [11C]PK11195 [1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide]. A 6th-minute dynamic PET scan in 3-dimensional mode (septa retracted) was acquired (24 × 5-second frames; 9 × 20-second frames; 10 x 1-minute frames; 9 x 5-minute frames). PET scans were processed, as previously described, using the 10- to 60-minute motion-corrected frames for partial volume–corrected standard uptake value ratio (SUVR).22,23 The visual check on the registration between the [11C]PK11195 SUVR maps and MPRAGE images has been performed for each individual. Cerebellar cortex has the lowest density of microglia24 and was used as the reference region. Partial volume–corrected SUVRs23 were calculated for each [11C]PK11195 scan, using the 10- to 60-minute frames with the FreeSurfer regions and the calculated reference region of nonspecific binding in the cerebellar cortex. As the global index to reflect neuroinflammation, a mean cortical SUVR was calculated based on the cortical regions defined by FreeSurfer.
Regions of Interest
The hippocampus and precuneus are the 2 regions of interests selected based on previous work finding those 2 cerebral regions as sensitive to AD pathologies of amyloid and tau.9,23 Both regions are from FreeSurfer parcellation. [11C]PK11195 SUVRs were calculated for these regions and the whole cortex regions.
Statistical Analysis
Nonparametric Kruskal-Wallis and the χ2 tests were used for comparing continuous and categorical variables of the participants' demographics, respectively. One-way ANOVA was used to test the group differences in [11C]PK11195 binding, CSF biomarkers, and cognition. The Tukey honest significant differences were computed to perform multiple pairwise comparison between the mean values of groups. Spearman correlations were used to measure the strength of the associations between [11C]PK11195 SUVR and CSF biomarkers while adjusting for effects of age and sex.25 A random coefficient model26 was used to examine whether the baseline [11C]PK11195 SUVR was associated with the longitudinal cognitive change for all participants. This statistical method allows us to accommodate heterogeneous numbers of visits and intervals between visits for our included participants. The cognitive measure within 1 year of the [11C]PK11195 scan and all available follow-up cognitive measures for each individual were included in our statistical model. Age and sex were included in this model as the covariates. Of importance, time is considered a continuous variable (measured in years) in this model, representing the interval between the baseline cognitive assessment and each subsequent visit. Within this model, time is treated as both a fixed and random effect. The statistical model is the following:
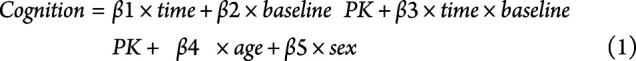 |
The estimate (β by type 3 sum of squares) and corresponding p values from the interaction term, time × baseline PK, were reported to investigate whether the baseline [11C]PK11195 SUVR predicts cognitive decline. The random coefficient model approach allows for different intercepts and slopes for each individual across variables of interest. This approach is particularly useful because it accommodates the heterogeneous number of visits and intervals between visits. All statistical analyses were performed using SAS, version 9.4 (SAS Institute Inc., Cary, NC) or R (R Core Team [2020]), and p < 0.05 was regarded as statistically significant after Bonferroni correction.
Data Availability
All data associated with this study are present in the article or the supplementary material. Deidentified data will be shared on reasonable request from a qualified investigator.
Results
Participant Demographics
Demographic data are summarized in Table 1. Individuals were categorized into the following groups: 9 stable cognitively normal (CDR = 0 at the PET scan and follow-up 13.2 ± 3.9 years later), 9 progressors (CDR = 0 at the PET scan and CDR > 0 at follow-up 7.4 ± 4.8 years later), and 6 symptomatic AD (3 with CDR = 0.5 and 3 with CDR = 1 at the PET scan). Individuals in the progressor and AD dementia groups were significantly older than individuals in the stable cognitively normal group (79.7 ± 6.2, 78.2 ± 7.5, and 67.9 ± 9.8 years [mean ± SD], respectively, p = 0.04). There were no significant group differences in sex, years of education, APOE ε4 status, or race. CDR and CDR-SB at baseline were higher (worse) in the symptomatic AD group compared with the progressor and stable cognitively normal groups (p < 0.001 and 0.004, respectively). However, there were no significant group differences in the MMSE, global composite scores, or Knight ADRC-PACC scores.
Table 1.
Demographic and Clinic Characteristics of the Participants

[11C] PK11195 PET SUVR Maps
Representative [11C]PK11195 PET SUVR images from 1 representative individual for each group are shown in Figure 1. Panel A is an individual from the stable cognitively normal group (mean cortical SUVR of 0.99), panel B is from the progressor group (SUVR 1.10), and panel C is from the symptomatic AD group (SUVR 1.18). The mean cortical SUVRs for the groups were as follows: stable cognitively normal, 1.10 ± 0.07 (mean ± SD); progressor, 1.11 ± 0.10; symptomatic AD, 1.14 ± 0.15. Although there were no significant group differences in mean cortical SUVR, there were significant regional elevations of [11C]PK11195 SUVR in the hippocampus (Figure 2A) in the progressor and symptomatic AD groups compared with those in the stable cognitively normal group. No group differences were found in the precuneus (Figure 2B).
Figure 1. Representative [11C]PK11195 PET SUVR Maps.

[11C]PK11195 PET SUVR maps of 3 representative cases from the stable cognitively normal (A), progressor (B), and symptomatic Alzheimer disease (C) groups.
Figure 2. [11C]PK11195 PET SUVR in the Hippocampus and Precuneus Regions.

Boxplots demonstrate the [11C]PK11195 PET SUVR in 3 groups in the hippocampus (A) and precuneus region (B). There is a significant increase in SUVR in the progressor and symptomatic Alzheimer disease groups compared with that in the stable cognitively normal group in the hippocampus but not in the precuneus region. The p values were produced from the Tukey honest significant differences test to demonstrate the between-group differences. Only the p values <0.05 are shown.
CSF Biomarkers
Of the 24 participants, 16 had available data on CSF Aβ42, Aβ42/Aβ40, p-tau181, and t-tau within 1 year of the PET scan. For CSF Aβ42, which is less accurate than other analytes in detecting brain amyloidosis, there were no significant group differences in CSF Aβ42 by omnibus testing (Figure 3A). Further data exploration revealed a trend toward lower CSF Aβ42 in the symptomatic AD group compared with the stable cognitively normal group (p = 0.06) (Figure 3A). Significant group differences were observed in CSF Aβ42/Aβ40, p-tau181, and t-tau (Figures 3, B–D). The ratio of CSF Aβ42/Aβ40, which is more sensitive to amyloidosis than CSF Aβ42,27 was lower in the symptomatic AD group when compared with the stable cognitively normal group (p = 0.009) (Figure 3B). CSF p-tau181 and t-tau were higher in the symptomatic AD group when compared with those in the stable cognitively normal group (Figures 3, C and D, p = 0.006 and p = 0.01, respectively).
Figure 3. CSF Measures of Amyloid and Tau.

Boxplots show CSF concentrations of Aβ42, Aβ42/Aβ40, p-tau181, and t-tau in the stable cognitively normal, progressor, and symptomatic AD groups. There were significant decreases in the CSF Aβ42 (A) and Aβ42/Aβ40 (B) in the Symptomatic AD group compared with the stable cognitively normal group. There were significant increases in CSF p-tau181 (C) and t-tau (D) in the Symptomatic AD group compared with those in the progressor and stable cognitively normal groups. The p values were produced from the Tukey honest significant differences test to demonstrate the between-group differences. Only the p values <0.05 are labeled here. AD = Alzheimer disease.
The relationship between CSF biomarkers and [11C]PK11195 SUVR was further evaluated in the entire subcohort with CSF biomarkers using Spearman correlations adjusting for age and sex. Significant negative correlations were found between CSF Aβ42 and [11C]PK11195 SUVR in the hippocampus and precuneus and the whole cortex regions as well (Figures 4, A, E, and I). Significant negative correlations between CSF Aβ42/Aβ40 and [11C]PK11195 SUVR were noted in the precuneus (Figure 4J). No significant correlations between CSF p-tau181 or t-tau and [11C]PK11195 SUVR were found. The Spearman rho (R) and p values are shown in Figure 4.
Figure 4. Correlations Between [11C]PK11195 SUVR and the CSF Biomarkers.

Scatterplots show the significant correlations between [11C]PK11195 SUVR and CSF levels of amyloidosis (A, E, I, and J). There were no significant correlations between [11C]PK11195 SUVR and CSF levels of p-tau181 (C, G, and K) and t-tau (D, H, and L). *, p < 0.05; adjusted by age and sex.
[11C]PK11195 SUVR Predicts Cognitive Decline
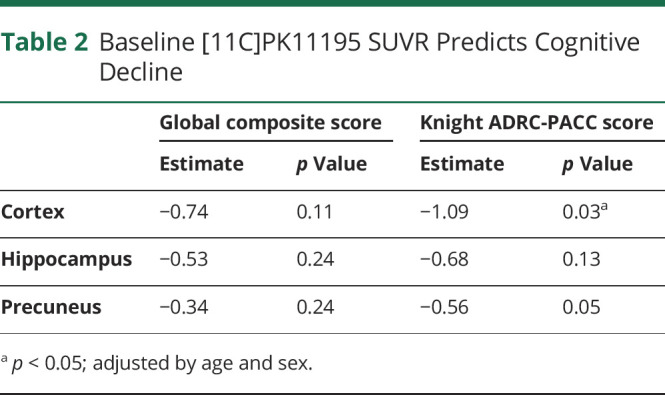
The ability of TSPO PET imaging to predict future cognitive decline was evaluated. The mean period of cognitive follow-up for the 3 groups (stable cognitively normal, progressor, symptomatic AD) were 13.2 ± 3.9, 7.4 ± 4.8, and 4.1 ± 2.9, respectively. We used the random coefficient model to investigate whether the baseline [11C]PK11195 SUVR predicts the cognitive decline. In this study, we report the estimate (β by type 3 sum of squares) and corresponding p values from the interaction term: time × baseline PK from Equation 1. We found that higher [11C]PK11195 SUVR in the whole cortex regions (p = 0.03) predicted the cognitive decline in Knight ADRC-PACC score, but not in global composite score (Table 2). [11C]PK11195 SUVR in the hippocampus and precuneus regions did not predict the decline of global and Knight ADRC-PACC scores with p > 0.05 (Table 2). The same statistical analysis was performed on 16 participants who have the CSF measures. We found that higher [11C]PK11195 SUVR in the whole cortical regions predicted the cognitive decline in the global composite score (p = 0.03) and Knight ADRC-PACC score (p = 0.01) (eTable 1 in the supplement, links.lww.com/NXI/A704). [11C]PK11195 SUVR in the precuneus but not in the hippocampus predicted cognitive decline in Knight ADRC-PACC score (p = 0.01) (eTable 1 in the supplement). When considering CSF Aβ42/Aβ40 and/or p-tau181 as covariates, [11C]PK11195 SUVR in the whole cortical regions still predicted the cognitive decline in global composite score and Knight ADRC-PACC score, and [11C]PK11195 SUVR in the precuneus predicted the cognitive decline in Knight ADRC-PACC score (eTables 2 and 3 in the supplement).
Table 2.
Baseline [11C]PK11195 SUVR Predicts Cognitive Decline
Classification of Evidence
This study provides Class II evidence that in patients with AD, higher baseline [11C]PK11195 SUVR averaged in the whole cortical regions was associated with longitudinal decline on cognitive tests.
Discussion
The goal of this study was to investigate the relationships between microglial activation measured by [11C]PK11195, CSF biomarkers of AD, and cognitive decline. We found that individuals categorized in the progressor and symptomatic AD groups had higher [11C]PK11195 SUVR in the hippocampus and precuneus regions compared with the stable cognitively normal group. Lower CSF Aβ42 and Aβ42/Aβ40, consistent with greater brain amyloidosis, was associated with higher [11C]PK11195 SUVR in the AD pathology–susceptible regionsthe hippocampus and precuneus. Of interest, no significant correlations CSF p-tau181 or t-tau were found with [11C]PK11195 SUVR in any region. Last, [11C]PK11195 SUVR in the overall cortical region predicted cognitive decline, as measured by the commonly used composite score, Knight ADRC-PACC score, in AD studies.
As one of the earliest developed PET TSPO radiotracers, [11C]PK11195 has been used to investigate the microglia activation in AD brains.28 The lack of group differences of the mean cortical SUVR may be attributed to the limited sample size. However, our region-based analysis found elevated [11C]PK11195 SUVR in the hippocampus of the progressor and symptomatic AD groups compared with the stable cognitively normal group. The hippocampus is an important subcortical region that is known to be severely and consistently affected by AD pathologic processes and shows a considerable shrinkage, distortion, and loss of neurons.29,30 Our previous work has demonstrated that the hippocampus volume demonstrated higher clinical diagnostic performance than the traditional volumetric measures for AD.8 The elevated [11C]PK11195 binding in the progressor group in this region suggests that microglial activation may be an early event in the pathogenesis of AD that precedes symptom onset.7,12,31
Although lower CSF Aβ42 is well-established as a biomarker of amyloidosis,32 the ratio of Aβ42 to Aβ40 (Aβ42/Aβ40) is superior to Aβ42 as a biomarker of brain amyloidosis.33,34 In this study, we investigated the relationship between microglial activation and amyloidosis measured by both CSF Aβ42 and Aβ42/Aβ40. We found both CSF levels of Aβ42 and Aβ42/Aβ40 were negatively correlated with [11C]PK11195 SUVR in the hippocampus and precuneus, suggesting that brain amyloidosis and microglial activation may be linked.
Although studies have demonstrated an association of neuroinflammation and CSF measures of tau,4 we did not find any significant correlations between [11C]PK11195 SUVR and CSF p-tau181 or t-tau. However, not reaching the significant level, the trends of positive associations between [11C]PK11195 SUVR and CSF measures of p-tau and tau in the whole cortex and precuneus regions have been shown in Figure 4, suggesting neuroinflammation could increase with the increase in tau pathology in AD. Notably, our participants exhibited very early AD. Accumulating evidence suggests that neuroinflammation may play a neuroprotective role in early AD, but this role may change and become pathologic in later stages of AD.7,35 Therefore, measures of neuroinflammation may vary in a nonlinear fashion across different disease stages, complicating studies. Large sample sizes and longitudinal evaluations are necessary to characterize changes in neuroinflammation over time.
Although several previous studies have found that TSPO PET uptake correlates inversely with cognitive measures,36,37 the relationship between [11C]PK11195 uptake and longitudinal cognitive decline has not been fully explored. Consistent with a recent study that demonstrated the ability of [11C]PK11195 to predict cognitive decline,38 we also found that [11C]PK11195 SUVRs in the whole cortical region predicted cognitive decline. Similar results were found on the subset participants who have CSF measures (eTable 1 in the supplement, links.lww.com/NXI/A704). A further analysis of whether microgliosis at baseline remains a significant predictor of subsequent cognitive decline after including amyloid and tau at baseline as independent predictors was performed. [11C]PK11195 SUVRs in the whole cortical region predicting cognitive decline still remained (eTables 2 and 3 in the supplement), suggesting microglial activation is a strong predictor of cognitive decline.
A linear regression model was also implemented to investigate the relationship between the baseline [11C]PK11195 binding and the rate of change in cognitive measures. The results were summarized in eTable 4 in the supplement, links.lww.com/NXI/A704. Significant negative correlations were found between the [11C]PK11195 binding and the rate of change in Global and Knight ADRC-PACC scores in the hippocampus region. The negative correlations between the [11C]PK11195 binding and the rate of change in cognitive measures in the whole cortex and precuneus regions were found but not reaching statistical significance. Those results also support our findings using the random coefficient model that the baseline [11C]PK11195 binding predicts cognitive decline in AD. The inverse correlation between [11C]PK11195 SUVR and the rate of change in cognitive composite score Knight ADRC-PACC found in this study suggests that a high level of microglial activation accelerates the cognition decline in AD. Although associations between amyloid deposition, tau load, and microglial activation have been established in both postmortem and in vivo neuroimaging studies,39,40 how those key AD pathologies independently or jointly promote the clinical progression of AD remains unclear. One recent study considered the interaction of those AD pathologies found the combined contribution of temporo-parietal tau pathology and anterior temporal neuroinflammation in predicting cognitive decline in patients with symptomatic AD.38 Further studies with larger sample size are desired along this direction.
There are several limitations in this study. First, the sample size is small. Studies with larger sample sizes are required to evaluate the interactions of AD pathologies in the prediction of cognitive decline. In addition, this study focused on the 2 earliest-affected AD pathology-sensitive regions—the hippocampus and precuneus. Further analysis on the whole brain will be necessary to deepen our understanding of the spatial relationships among these AD pathologies. Finally, CSF measures of amyloidosis and tauopathy do not provide information on the regional distribution of AD pathology. Future studies analyzing PET imaging of amyloid, tau, and microglial activation on the voxel level will enable the detailed investigation of the spatial relationships among amyloid, tau, and neuroinflammation in AD progression.
In summary, our results indicate a first-generation TSPO PET tracer developed to image microglial activation in AD. [11C]PK11195 demonstrates higher binding both in individuals with symptomatic AD and individuals who are cognitively normal at baseline and progress to symptomatic AD. We found that [11C]PK11195 binding correlated with global amyloidosis measured by CSF biomarkers in the hippocampus and precuneus, suggesting that microglial activation is associated with brain amyloidosis in early AD. Furthermore, regional [11C]PK11195 binding in the overall cortical region predicted cognitive decline in AD.
Acknowledgment
The authors thank all the research volunteers for their contributions.
Glossary
- AD
Alzheimer disease
- Aβ
β-amyloid
- CDR
clinical dementia rating
- CDR-SB
CDR Sum of Boxes
- Knight ADRC-PACC
Knight Alzheimer Disease Research Center Preclinical Alzheimer Composite Score
- MMSE
Mini-Mental State Examination
- MPRAGE
magnetization-prepared rapid acquisition gradient echo
- PET
positron emission tomography
- SB
sum of boxes
- SUVR
standard uptake value ratio
- TSPO
18 kDa translocator protein
Appendix. Authors

Footnotes
Class of Evidence: NPub.org/coe
Contributor Information
Qing Wang, Email: wangqing@wustl.edu.
Gengsheng Chen, Email: allisonchen@wustl.edu.
Suzanne E. Schindler, Email: schindler.s.e@wustl.edu.
Jon Christensen, Email: jonjaychristensen@gmail.com.
Nicole S. McKay, Email: n.mckay@wustl.edu.
Jingxia Liu, Email: esther@wustl.edu.
Sicheng Wang, Email: sicheng.wang@wustl.edu.
Zhexian Sun, Email: z.sun@wustl.edu.
Jason Hassenstab, Email: hassenstabj@wustl.edu.
Yi Su, Email: yi.su@bannerhealth.com.
Shaney Flores, Email: sflores@wustl.edu.
Russ Hornbeck, Email: russ@wustl.edu.
Lisa Cash, Email: lisacash@wustl.edu.
Carlos Cruchaga, Email: cruchagac@wustl.edu.
Anne M. Fagan, Email: fagana@wustl.edu.
Zhude Tu, Email: zhudetu@wustl.edu.
John C. Morris, Email: jcmorris@wustl.edu.
Mark A. Mintun, Email: mintun@lilly.com.
Tammie L.S. Benzinger, Email: benzingert@wustl.edu.
Study Funding
This study was supported partly by grants from the NIH, including the National Institutes on Aging (NIA) P01AG026276 (Antecedent Biomarkers of AD: the Adult Children Study, PI J.C. Morris); NIA P01AG003991 (Healthy Aging and Senile Dementia, PI J.C. Morris);NIA P30AG066444 (Alzheimer Disease Research Center, PI J.C. Morris); and NIA 1R01AG054567-01A1 (PIs T.L.S. Benzinger and Y. Wang). Q.W. is supported by NIA 1R03AG072375-01 (PI Q. Wang). S.E.S. is supported by K23AG053426 (PI S.E. Schindler). Additional support was generously provided by the Charles and Joanne Knight Alzheimer's Research Initiative and by the Fred Simmons and Olga Mohan Fund and the Paula and Rodger Riney Fund.
Disclosure
Go to Neurology.org/NN for full disclosures.
References
- 1.McGeer EG, McGeer PL. Inflammatory processes in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(5):741-749. [DOI] [PubMed] [Google Scholar]
- 2.Combs CK. Inflammation and microglia actions in Alzheimer's disease. J Neuroimmune Pharmacol. 2009;4(4):380-388. [DOI] [PubMed] [Google Scholar]
- 3.Brosseron F, Krauthausen M, Kummer M, Heneka MT. Body fluid cytokine levels in mild cognitive impairment and Alzheimer's disease: a comparative overview. Mol Neurobiol. 2014;50(2):534-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nordengen K, Kirsebom BE, Henjum K, et al. Glial activation and inflammation along the Alzheimer's disease continuum. J Neuroinflammation. 2019;16(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terada T, Yokokura M, Obi T, et al. In vivo direct relation of tau pathology with neuroinflammation in early Alzheimer's disease. J Neurol. 2019;266(9):2186-2196. [DOI] [PubMed] [Google Scholar]
- 6.Hamelin L, Lagarde J, Dorothée G, et al. Early and protective microglial activation in Alzheimer's disease: a prospective study using 18F-DPA-714 PET imaging. Brain. 2016;139(Pt 4):1252-1264. [DOI] [PubMed] [Google Scholar]
- 7.Fan Z, Brooks DJ, Okello A, Edison P. An early and late peak in microglial activation in Alzheimer's disease trajectory. Brain. 2017;140(3):792-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koenig LN, Day GS, Salter A, et al. Select Atrophied Regions in Alzheimer disease (SARA): an improved volumetric model for identifying Alzheimer disease dementia. Neuroimage Clin. 2020;26:102248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra S, Gordon BA, Su Y, et al. AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. Neuroimage. 2017;161:171-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Hoesen GW, Hyman BT. Hippocampal formation: anatomy and the patterns of pathology in Alzheimer's disease. Prog Brain Res. 1990;83:445-457. [DOI] [PubMed] [Google Scholar]
- 11.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446-452. [DOI] [PubMed] [Google Scholar]
- 12.Schuitemaker A, Kropholler MA, Boellaard R, et al. Microglial activation in Alzheimer's disease: an (R)-[(1)(1)C]PK11195 positron emission tomography study. Neurobiol Aging. 2013;34:128-136. [DOI] [PubMed] [Google Scholar]
- 13.Morris JC, Weintraub S, Chui HC, et al. The uniform data set (UDS): clinical and cognitive variables and descriptive data from Alzheimer disease centers. Alzheimer Dis Assoc Disord. 2006;20:210-216. [DOI] [PubMed] [Google Scholar]
- 14.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412-2414. [DOI] [PubMed] [Google Scholar]
- 16.Berg L, Miller JP, Storandt M, et al. Mild senile dementia of the Alzheimer type: 2. Longitudinal assessment. Ann Neurol. 1988;23(5):477-484. [DOI] [PubMed] [Google Scholar]
- 17.Folstein MF, Folstein SE, McHugh PR. Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189-198. [DOI] [PubMed] [Google Scholar]
- 18.Donohue MC, Sperling RA, Salmon DP, et al. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol. 2014;71(8):961-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talbot C, Lendon C, Craddock N, Shears S, Morris JC, Goate A. Protection against Alzheimer's disease with apoE epsilon 2. Lancet. 1994;343(8910):1432-1433. [DOI] [PubMed] [Google Scholar]
- 20.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59(3):512-519. [DOI] [PubMed] [Google Scholar]
- 21.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33(3):341-355. [DOI] [PubMed] [Google Scholar]
- 22.Su Y, D'Angelo GM, Vlassenko AG, et al. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One. 2013;8(11):e73377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su Y, Blazey TM, Snyder AZ, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage. 2015;107:55-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001;101(3):249-255. [DOI] [PubMed] [Google Scholar]
- 25.Kim S. ppcor: an R package for a fast calculation to semi-partial correlation coefficients. Commun Stat Appl Methods. 2015;22(6):665-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Longford NT. Random Coefficient Models. In: Arminger G, Clogg CC, Sobel ME, eds. Handbook of Statistical Modeling for the Social and Behavioral Sciences. Springer; 1995. [Google Scholar]
- 27.Schindler SE, Gray JD, Gordon BA, et al. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement. 2018;14(11):1460-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ching AS, Kuhnast B, Damont A, Roeda D, Tavitian B, Dollé F. Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imaging. 2012;3(1):111-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabuncu MR, Desikan RS, Sepulcre J, et al. The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol. 2011;68(8):1040-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jack CR Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55:484-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461-467. [DOI] [PubMed] [Google Scholar]
- 32.Fagan AM, Perrin RJ. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer's disease. Biomark Med. 2012;6(4):455-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schindler SE, Bollinger JG, Ovod V, et al. High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647-e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baldeiras I, Santana I, Leitao MJ, et al. Addition of the Abeta42/40 ratio to the cerebrospinal fluid biomarker profile increases the predictive value for underlying Alzheimer's disease dementia in mild cognitive impairment. Alzheimers Res Ther. 2018;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ismail R, Parbo P, Madsen LS, et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer's disease: a longitudinal PET study. J Neuroinflammation. 2020;17:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edison P, Archer HA, Gerhard A, et al. Microglia, amyloid, and cognition in Alzheimer's disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412-419. [DOI] [PubMed] [Google Scholar]
- 37.Yokokura M, Mori N, Yagi S, et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2011;38(2):343-351. [DOI] [PubMed] [Google Scholar]
- 38.Malpetti M, Kievit RA, Passamonti L, et al. Microglial activation and tau burden predict cognitive decline in Alzheimer's disease. Brain. 2020;143(5):1588-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayes A, Thaker U, Iwatsubo T, Pickering-Brown SM, Mann DM. Pathological relationships between microglial cell activity and tau and amyloid beta protein in patients with Alzheimer's disease. Neurosci Lett. 2002;331(3):171-174. [DOI] [PubMed] [Google Scholar]
- 40.Dani M, Wood M, Mizoguchi R, et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer's disease. Brain. 2018;141(9):2740-2754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data associated with this study are present in the article or the supplementary material. Deidentified data will be shared on reasonable request from a qualified investigator.