

Abstract
Background and Objectives
Opsoclonus-myoclonus-ataxia syndrome (OMAS) is a rare disorder of the nervous system that classically presents with a combination of characteristic eye movement disorder and myoclonus, in addition to ataxia, irritability, and sleep disturbance. There is good evidence that OMAS is an immune-mediated condition that may be paraneoplastic in the context of neuroblastoma. This syndrome may be associated with long-term cognitive impairment, yet it remains unclear how this is influenced by disease course and treatment. Treatment is largely predicated on immune suppression, but there is limited evidence to indicate an optimal regimen.
Methods
Following an international multiprofessional workshop in 2004, a body of clinicians and scientists comprising the International OMS Study group continued to meet biennially in a joint professionals and family workshop focusing on pediatric OMAS. Seventeen years after publication of the first report, a writing group was convened to provide a clinical update on the definitions and clinical presentation of OMAS, biomarkers and the role of investigations in a child presenting with OMAS, treatment and management strategies including identification and support of long-term sequelae.
Results
The clinical criteria for diagnosis were reviewed, with a proposed approach to laboratory and radiologic investigation of a child presenting with possible OMAS. The evidence for an upfront vs escalating treatment regimen was reviewed, and a treatment algorithm proposed to recognize both these approaches. Importantly, recommendations on monitoring of immunotherapy response and longer-term follow-up based on an expert consensus are provided.
Discussion
OMAS is a rare neurologic condition that can be associated with poor cognitive outcomes. This report proposes an approach to investigation and treatment of children presenting with OMAS, based on expert international opinion recognizing the limited data available.
Opsoclonus-myoclonus-ataxia syndrome (OMAS) is a rare disorder of the nervous system with onset usually in the second year of life. This condition classically presents with a combination of characteristic eye movement disorder and myoclonus, in addition to ataxia, irritability, and sleep disturbance. In around 50% of children presenting with OMAS, there is an underlying neuroblastoma. There is good evidence that OMAS is an immune-mediated condition that may be paraneoplastic in the context of neuroblastoma.1 In 2004, following an international multiprofessional workshop, a report on immune pathogenesis, clinical features, acute and chronic neurologic manifestations, and current and therapeutic approaches to OMAS was published.2 This international body of clinicians and scientists continued to meet biennially in a joint professionals and family workshop focusing on many aspects of this neurologic syndrome.3
Although there have been significant advances, including earlier recognition and diagnosis of the clinical syndrome, many challenges remain. Moreover, treatment is largely predicated on immune suppression, although there is limited evidence to guide optimal treatment regimens. OMAS can be associated with long-term cognitive impairment, yet it remains unclear how this is influenced by disease course and treatment. Recent series, however, suggest that higher intensity of therapy may predict better outcomes.4,5 Following the series of biennial meetings and workshops of the International OMAS study group, a writing group was convened to provide a clinical update on the definitions and clinical presentation of pediatric OMAS, biomarkers and the role of investigations in a child presenting with OMAS, treatment and management strategies including identification and support of long-term sequelae (Methods attached as supplementary material, links.lww.com/NXI/A702).
Background, Definitions, and Clinical Presentation
Multiple acronyms have been used to describe the same phenomenon: opsoclonus-myoclonus-ataxia syndrome (OMAS), opsoclonus-myoclonus syndrome (OMS), opsoclonus-myoclonus-ataxia (OMA), and dancing eye syndrome. The incidence of OMAS has been reported for the United Kingdom in a prospective study as 0.18 cases per million of the population6 and in a retrospective study in Japan as 0.27 cases per million children.7 The mean age at presentation was 18 months in the prospective study, and the median age was 16.5 months in the retrospective study. In a large retrospective single-center study of 356 children, the mean age at onset in children was not significantly different between those with tumor (1.7 years ± 0.89 SD) and those without (2.1 years ± 1.4 SD), with the vast majority presenting before age 5 years.8 Those children presenting after age 24 months were found to be less likely to present with tumor than those younger than 24 months (<12 months, 58%, 12–24 months, 52%, >24 months, 38%, p = 0.03).9 No cases in this large published series presented after age 10.8
OMAS falls along the spectrum of acquired cerebellar ataxias of childhood, which may include syndromes presenting purely with ataxia, but is distinct for its paraneoplastic association with neuroblastoma (26%–44%)6,7,10 and the presence of opsoclonus and myoclonus. Opsoclonus (vimeo.com/208498940), due to saccadic bursts, produces chaotic eye movements, without an intersaccadic interval, unlike nystagmus that is rhythmic and has fast or slow waves depending on the etiology. Because children with OMAS often present with gait imbalance, initial diagnosis is often acute cerebellar ataxia, an infectious or postinfectious illness, which in some cases is associated with varicella zoster or Mycoplasma pneumoniae.9 The development of opsoclonus and/or myoclonus declares the OMAS diagnosis. Occasionally, eye movement abnormalities present first and can resemble the sensory nystagmus seen in patients with visual loss. In such cases, primary ophthalmologic disorders should be considered, especially in children with onset less than 3 months of age.
Kinsbourne's initial description of 6 children with OMAS was of a myoclonic encephalopathy,11 which would include the differential of pediatric neurodegenerative conditions with or without associated epilepsy, such as Batten disease. However, these conditions may be distinguished by the absence of the other features of OMAS, and an often prolonged time course in comparison to OMAS, where onset is noted to be acute or subacute in over half (57%) of patients.9
Although the majority of children who eventually receive a diagnosis of OMAS present to the clinician with opsoclonus, importantly, the first sign may be ataxia (staggering and falling)8 with later development of opsoclonus. Opsoclonus can be transient, difficult to see during examination and smartphone videos may be a useful adjunct to diagnosis. Opsoclonus may be elicited by saccades, and by a squeeze test in which the child forcefully closes the eyelids, which are held open by the examiner.
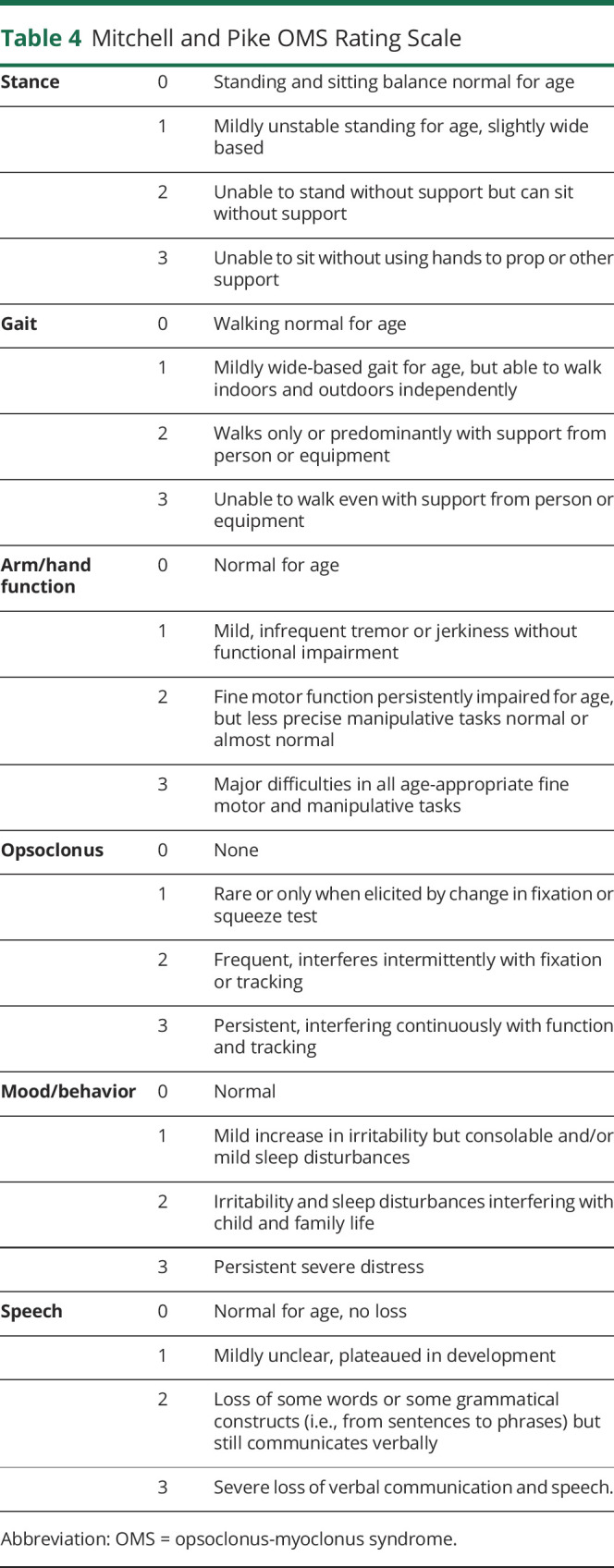
Behavioral abnormalities, including sleep disturbance, irritability, and inconsolable crying, are frequent (over half of patients).8 Many children lose previously acquired motor and language skills, with this regression related to age at presentation. The Mitchell-Pike OMS rating scale is the most frequently used disease-specific scale and involves the grading of 6 categories on an ordinal scale: stance, gait, arm/hand function, opsoclonus, mood/behavior, and speech, with higher scores representing more severe clinical presentations.12 Some studies, including the Children's Oncology Group (COG) study13 and EU study (NCT01868269), used only 5 of the 6 items, excluding speech and language, for a 15 point maximum. Initial presentation may be variable, with atypical presentation in 20% of children in a prospective UK study.6 Based on an expert panel recommendation, a diagnosis may be made with 3 of 4 features are present: (1) opsoclonus, (2) ataxia or myoclonus, (3) behavior change or sleep disturbance, and (4) neuroblastoma.2
Immunopathogenesis
The very specific association of peripheral neuroblastic tumors (neural antigens) and the relatively specific pathognomic clinical features make a specific adaptive response very likely in OMAS. Peripheral neuroblastic tumors are identified in 50% of children with OMAS,9 and some have suggested the presence of occult tumor in many more, with spontaneous regression in these cases.14 No clinical characteristics distinguish children with OMAS with and without occult tumor.8 Neuroblastoma (73%) is the most common tumor type, followed by ganglioneuroblastoma (22%) and ganglioneuroma (4%). The tumors are never in the brain: all are located along the sympathetic chain or in the adrenals, including the neck, thoracic, abdominal, or pelvic cavities.9 Notably, 93% of neuroblastoma is early stage (stage 1 or 2) in this population.9 Although rare, OMAS has been reported in adolescents with ovarian teratoma.15
A variety of infections have been reported in association with OMAS, including Mycoplasma, Streptococcus, Epstein-Barr virus, adenovirus, HIV, and dengue virus, among others,16 suggesting that no single etiologic infectious agent exists. Importantly, however, as an infectious prodrome may occur even in children with underlying tumor,9 all children presenting with OMAS should be screened for peripheral neuroblastic tumor.
The search for a specific and reproducible biomarker has to date been disappointing. Currently available biomarkers are available primarily on a research basis (Table 1) and do not discriminate paraneoplastic from idiopathic cases. Many nonspecific biomarkers have been described in OMAS. CSF in OMAS typically contains a small number of immune cells, but the cells can be immunophenotyped to determine whether there are aberrant or skewed immune cell populations. B-cell expansion in the CSF is reported in OMAS,17 correlating with severity and response to the B cell–depleting biologic therapy.18 Similarly, CSF oligoclonal bands have been found in 58% of untreated patients with OMAS, inferring intra-CNS production of antibodies.19
Table 1.
Biomarkers in OMAS and Neuroblastoma

CSF neopterin is elevated in the acute phase of OMAS, correlates with OMAS severity, and reduces rapidly with treatment,20 but is nonspecific. Likewise, cytokines and chemokines involved in B-cell (BAFF and CXCL13)21 and T-cell (CCL17, CCL19, CCL21, and CCL22),23,24 and microglial25 activation are elevated in CSF of OMAS but are also nonspecific. Finally, CSF elevation of neurofilament, a marker of axonal injury, is associated with OMAS outcome.26 Taken together, although these CSF biomarkers correlate with some degree with disease activity and therefore offer some hope for monitoring, in practice they lack sensitivity and specificity. Future studies to evaluate their utility in longitudinal assessments of response to therapy are needed.
The elevation of cytokines and chemokines involved in B-cell activation, alongside CSF oligoclonal bands, may argue for a role for a pathogenic humoral autoimmune process in OMAS,27 and discovery of a pathogenic autoantibody would advance the field substantially. This is supported by evidence of IgG binding to the cell surface of neurones in a minority of patients with OMAS.28 An alternate explanation is that the driving autoimmune process is T cell mediated, resulting in greater challenges from a discovery and biomarker perspective.
Investigating the Child Presenting With OMAS
Although a diagnosis may be made in the presence of 3 of 4 features: opsoclonus, ataxia or myoclonus, behavior change or sleep disturbance, and neuroblastoma,2 presentation may be atypical. In a prospective UK study, 3 of 15 children subsequently diagnosed with OMAS were initially misdiagnosed, 2 as acute cerebellar ataxia and 1 as Guillain-Barre syndrome.6 In the absence of a disease biomarker, investigations to exclude other neurologic and inflammatory conditions that may mimic some or all of the features seen in OMAS are performed. Notably, the presence of neuroblastoma associates strongly with OMAS; therefore, early investigation for neuroblastoma may allow timely diagnosis and immune treatment in those children with an atypical or incomplete presentation. Reexamining the child with an atypical presentation after 2 weeks is important in distinguishing acute cerebellar ataxia (in whom the clinical course is of improvement, without myoclonus or opsoclonus) from OMAS.29
CSF
Lumbar puncture, including cell count, protein, and glucose level, should be assessed at onset to exclude potential mimics of OMAS. As mild elevation of CSF WBC (>4/mm3) has been observed in 14% of untreated patients and oligoclonal bands positive in 58% of OMAS, with higher positivity in those over 2 years at symptom onset than those under 2 years, evaluation of serum and CSF oligoclonal bands should be performed.9 If possible, lymphocyte flow cytometry should be performed on CSF as the presence of relative expansion of B cells is a biomarker of active OMAS and correlates with response to B cell–depleting therapies.17
As noted above, many pathogens have been associated with the occurrence of the disease and differ according to patient geographical localization and history, and evidence of infection may be sought accordingly with optimized investigations (Table 2).
Table 2.
Recommendations for Initial Investigations of Children With OMAS

Neuroimaging
Initially, neuroimaging in children with acute or subacute onset of ataxia, regardless of whether they present with opsoclonus, is necessary to exclude focal lesions in the posterior fossa, including those due to tumor and other inflammatory or metabolic diseases. Neuroimaging of the brain is typically normal in the acute phase of OMAS, although cerebellar atrophy and cortical volume loss are occasionally seen longitudinally, and atrophy correlates with a higher OMAS score.30
Tumor Identification
High-resolution imaging, radioisotope imaging, and detection of excreted catecholamine metabolites are available methods in establishing the diagnosis of a peripheral neuroblastic tumor. Although no specific established hierarchy of investigation exists in OMAS, urine catecholamine metabolites, homovanillic acid, and vanillylmandelic acid (VMA/HVA) may be measured first but are very insensitive in OMAS. Most pediatric laboratories can do spot urine testing for VMA/HVA as a ratio to creatinine instead of 24-hour urine collections. As peripheral neuroblastic tumors associated with OMAS are generally small and thus minimally secreting or nonsecreting, the yield from this investigation is often disappointing, being as low as 24%,31 further confounded by challenges of collecting adequate urine samples in very young children. For these reasons, urine VMA screening alone is inadequate to exclude neuroblastoma.
Meta-iodo-benzyl-Guanidine (MIBG) scan scintigraphy is routinely used for screening and staging of neuroblastic tumors due to its high specificity.32 However, the false-negative rate of MIBG scintigraphy is around 8% and even higher in tumors with more differentiated pathology, and those are often associated with OMAS. In these cases, the false-negative rates can be as high as 24%.33 This is likely to be due to the nonsecreting nature and small size of low-grade neuroblastomas and ganglioneuroblastomas associated with OMAS.6 Fluorine 18 fluorodeoxyglucose PET/CT has been used in patients who have negative MIBG imaging as a less specific but more sensitive test.34 More evidence is needed to support the diagnostic accuracy of this test.34
In recent years, whole body imaging from the neck to pelvis, covering the wide range of possible tumor sites including the adrenals and the entire neural crest, has become the gold standard for detecting neuroblastic tumor in OMA. The majority of tumors, however, are either in the adrenals or in paraspinal regions of the chest and abdomen. Chest x ray and abdominal ultrasound alone are inadequate for detecting underlying neuroblastoma due to lower yields than other imaging modalities such as MRI, CT, and radioisotope imaging.6 Although there is no clear evidence of superiority of MRI over CT, the fact that CT needs to be performed both with and without IV contrast, for superior detection of small amounts of calcification seen in some tumors,35 and it involves a relatively high dose of radiation, has led to a shift toward the use of MRI. MRI avoids exposure to radiation, and its sensitivity reaches 100% in some studies,31 although general anesthesia is required in younger children. High resolution (4-mm cuts), imaging in multiple planes (axial, sagittal, and coronal), and diffusion weighted images are needed, together with interpretation by an experienced pediatric radiologist36 (Table 2).
Importantly, investigations to look for peripheral neuroblastic tumors may need to be repeated over time if no tumor is found at first attempt. The optimal timing and duration of repeat imaging has not been established, but repeat imaging at 6 months is practical and should detect the majority of tumors that may have been missed at first screening. Although in practice, MRI is frequently the initial investigation, complementary tests can be performed in the event of negative findings, in particular MIBG scintigraphy with single-photon emission computerized tomography or PET/CT.
Treatment of Associated Peripheral Neuroblastic Tumors
Peripheral neuroblastic tumors are the most frequent extracranial solid tumors in children,37 with an annual incidence of 8 cases/106 children younger than 15 years, per year. The median age at presentation is 22 months. Peripheral neuroblastic tumors are characterized by clinical and genetic heterogeneity. They represent a spectrum of disease, ranging from benign, well differentiated tumors (ganglioneuroma) to undifferentiated very aggressive malignancies (neuroblastoma).37
Although all types of peripheral neuroblastic tumor have been found in association with OMAS, most have been found to be differentiating neuroblastomas, with cases reported of intermixed ganglioneuroblastoma or even ganglioneuroma. Lymphocyte infiltration is abundant in neuroblastoma associated with OMAS, with follicular formation.38 The favorable prognosis of OMAS-associated peripheral neuroblastic tumor may suggest that this inflammatory infiltration is protective from a tumor perspective but creates the risk of a paraneoplastic process.
An increased frequency of segmental chromosomal abnormalities has been reported in tumor tissue compared with a control group of localized peripheral neuroblastic tumors not associated with OMAS.39 The genetic and molecular analysis of these tumors is complicated by the low percentage of tumor cells in the samples.
The risk stratification of peripheral neuroblastic tumors has evolved from anatomic and clinical40 (age, presence of metastases, and site of metastases) to genetic41 (N-myc-proto-oncogene amplification, 17q gain, 1p36 LOH, and 11q LOH) and molecular42 (tumor anaplastic lymphoma kinase [ALK] amplification or mutation burden, Ras pathway mutation burden, telomere maintenance mechanisms, and X-linked Alpha Thalassemia Mental Retardation [ATRX] mutation burden). In 2004, a landmark global collaboration developed a new International Neuroblastoma Risk Group classification system, using factors such as age at presentation, stage, pathology, MYCN amplification, 11q LOH, ploidy and stratifies patients in very low risk (overall survival [OS] > 85%), low risk (OS 75%–85%), intermediate risk (OS 50%–75%), and high risk (OS < 50%).
Accordingly, treatment has also evolved toward risk-adapted strategies. The majority of patients with peripheral neuroblastic tumor–associated OMAS have very low, low, or intermediate risk, nonmetastatic neuroblastoma. For low- and intermediate-risk patients, clinical trials have aimed at reduction of chemotherapy, without compromising the excellent outcome. In the COG P9641 trial, asymptomatic low-risk patients with neuroblastoma with or without OMAS achieved 5-year OS ≥ 95% with surgery alone.43 In the same study, chemotherapy was reserved for cases that were inoperable, symptomatic, or with progressive disease after surgery alone. Patients with stages 2a or 2b neuroblastoma received 2–8 cycles of chemotherapy (based on carboplatin, etoposide, cyclophosphamide, and doxorubicin), with 5-year OS > 85%. With a similar risk-adapted approach (4–8 cycles of chemotherapy based on carboplatin, etoposide, doxorubicin, and cyclophosphamide), patients with intermediate-risk neuroblastoma also achieve comparable 5-year OS above 90% in the A3961 trial.44 Similarly, in Europe, the Localized Neuroblastoma Europe Study 145 achieved OS > 90% in neuroblastoma stages 1 and 2 (non-MYCN-amplified) with surgery alone.
Rare cases of high-risk neuroblastoma and only anecdotal cases of MYCN-amplified tumors associated with OMAS have been reported. Molecularly targeted agents and immunotherapy are now being incorporated in frontline induction chemotherapy, with the intention to further intensify the initial treatment and prevent the development of resistance. Nevertheless, despite intensive multimodality treatment,46 these cases often have poor outcome (OS ∼ 40%).46
Follow-up of patients with neuroblastoma in remission after the end of treatment includes evaluation of urinary tumor markers (homovanillic and vanillylmandelic acid) if positive at presentation and surveillance by ultrasound or chest x rays. In rare cases, MRI is needed for surveillance of areas not adequately visualized with less invasive modalities. The role of MIBG scintigraphy in surveillance remains unclear, in particular in high-risk patients, whose prognosis after relapse is poor. Consideration should be given to the balance between risk of relapse and risk of exposure to radiation or repeated general anesthetics to minimize the long-term sequelae in survivors. Oncologic surveillance is recommended every 3 months in the first year after the end of treatment, every 4 months in the second year, and annually thereafter, until 5 years after the end of treatment. Tumor relapse occurs more frequently in the initial 2 years after the end of treatment, and few relapses occur greater than 5 years after the end of treatment. Tumor relapse is rare in the context of OMAS.47
Immune Treatment Strategies in OMAS
Although the optimal treatment for OMAS has not been established, the rarity of spontaneous remission, the high rate of chronic relapsing course, the risk of permanent neurologic sequelae, and evidence supporting an autoimmune cause have led to the widespread use of immunotherapy. Within this construct, there are 2 broad approaches. In many parts of the world including America and Australia, an upfront, aggressive approach using multiple agents is used, typically adrenocorticotropic hormone (ACTH) or corticosteroids, plasmapheresis, IV immunoglobulin (IVIg), and rituximab or cyclophosphamide.1,48 In a multinational European clinical trial (NCT01868269), now closed to recruitment, a stepwise approach was used starting with pulse oral dexamethasone (20 mg/m2/d for 3 consecutive days monthly) and reserving cyclophosphamide for patients who do not respond adequately by 3 months and rituximab for those who do not respond adequately to cyclophosphamide. In clinical practice, there has been a move to more rapid escalation than this protocol advocated and early introduction of rituximab before or in place of cyclophosphamide. With either approach, rapid diagnosis and early initiation of treatment is recommended with 1 study showing improved outcomes in patients treated within 2 months12
The upfront aggressive approach borrows from treatment paradigms in oncology, rheumatology, and neuroimmunology whereby the goal is to achieve remission quickly and then to maintain it. Considering the consistent paradigm change in neuroinflammation for earlier escalation to second-line therapy, usually within 4–6 weeks of initiation of 1st-line treatment,49 many would consider the timeline to escalation of the European trial protocol too lengthy. A revised algorithm is proposed (Figure 1) to outline the current treatment recommendations.
Figure 1. Immune Treatment Algorithm Whereby Both Treatment Upfront and Escalation Regimes Are Integrated.

Treatment generally persists at least for a full year. *Redosing of rituximab may be required if B-cell repopulation occurs quickly (<4 months) and/or clinical relapse. **Treatment may be extended if incomplete response or if there is clinical relapse and in this context is tailored to prior patient response and clinician experience.
Although some providers and studies have suggested that ACTH is superior to corticosteroids, little evidence supports this. However, the expert panel observes that some refractory patients may respond to ACTH and that patients who have not responded to dexamethasone alongside other first-line therapy should be considered for ACTH. Potential advantages of corticosteroids over ACTH include oral vs IM administration and lower cost. Two commonly used corticosteroid regimens include (1) IV pulse methylprednisolone (30 mg/kg/d for 3–5 days, maximum 1 g/d) followed by oral prednisone or prednisolone (starting dose 1–2 mg/kg/d) or (2) oral dexamethasone (20 mg/m2/d for 3 consecutive days as pulses 3–4 weekly), although a number of alternative steroid regimes are in use (Table 3). Daily oral steroids were used as part of standard therapy in a clinical trial (NCT00033293). IVIg is typically given as 2 g/kg over 2–5 days followed by 1–2 g/kg every 4 weeks for up to a year. In a clinical trial, children with OMAS associated with neuroblastoma were randomized to conventional treatment (prednisolone + cyclophosphamide) with (n = 26) or without (n = 27) concurrent IVIG. IVIG was associated with a higher treatment response rate (81% vs 41%) defined as a sustained improvement in OMS score (Table 4).13
Table 3.
Recommendations for the Dosing and Monitoring of Immunotherapeutic Agents in the Treatment of OMAS: Steroid Treatment and IVIg

Table 4.
Mitchell and Pike OMS Rating Scale
Rituximab is typically given in doses ranging from 375 mg/m2/dose for 2 to 4 weekly doses to 750 mg/m2/dose for 2 biweekly doses (Table 5). The potential effectiveness of rituximab in OMAS is supported by (1) an open-label study of 12 patients suggesting a reduction in relapse rate when combined with ACTH and IVIg,50 (2) higher IQ in a modern cohort (in which 11/15 received rituximab) compared with a historical cohort (in which 0/23 received rituximab),4 and (3) reduction in the duration of corticosteroid and IVIg treatment in those given rituximab.48 One study showed that 15 patients who received rituximab within 12 months of OMAS onset were more likely to achieve a good response (defined as modified Rankin score 0–2) compared with 16 patients who received it later.5 A retrospective comparison of children receiving multimodal treatment with dexamethasone, IVIG, and rituximab to those receiving steroids with or without IVIG found that multimodal treatment was associated with a more complete resolution of biomarkers of inflammation (B-cell frequencies, inflammatory markers, and oligoclonal bands).51 Children who are treated with rituximab should have lymphocyte subsets and immunoglobulin levels checked at baseline and serially. There are emerging data that younger age at rituximab initiation, neuroimmunologic diagnosis, and cumulative rituximab dose correlate with the risk of hypogammaglobulinemia,5 which should be carefully monitored.
Table 5.
Recommendations for the Dosing and Monitoring of Immunotherapeutic Agents in the Treatment of OMAS: Rituximab

Cyclophosphamide contributes both B-cell and T-cell suppression and is typically given IV as 750 mg/m2/dose monthly for 6 months (Table 6). Its potential benefit was first suggested by the finding that patients with OMAS who received tumor-directed chemotherapy (which commonly included cyclophosphamide) had better outcomes than those who did not.52 Although cyclophosphamide offers rapid recovery of immune function after cessation compared with rituximab, concerns around potential side effects, impact on fertility, and proposed B cell–mediated mechanism of OMAS have resulted in a tendency toward using rituximab in preference to cyclophosphamide.
Table 6.
Recommendations for the Dosing and Monitoring of Immunotherapeutic Agents in the Treatment of OMAS: Cyclophosphamide

Given the lack of definitive data on which treatments offer the best outcomes for OMAS, physicians must engage in a detailed discussion about potential risks and benefits of each treatment and single vs combined treatments. In general, the data suggest that multimodal immunotherapy, with concurrent initiation of rituximab, or rapid escalation of treatment may reduce relapses and improve long-term outcomes4 but may be associated with more serious potential side effects. Further studies are needed to guide patients, families, and providers in choosing treatments. Similarly, the optimal duration of immunotherapy in OMAS is not known. However, rapid tapers of steroids are often associated with OMAS relapses and should be avoided, and consideration given to the use of steroid sparing agents. The use of rituximab upfront may allow for more rapid tapering of corticosteroids and IVIg.48 A decision to initiate tapering of treatment should be strongly guided by the symptoms of OMAS, and reduction of therapy avoided while a patient remains symptomatic. Clinical symptoms of OMAS are suggestive of ongoing inflammation with potential for a poorer outcome.
Remission can be separately defined as remission on therapy, which always needs to be continuously reevaluated as treatment is weaned, and sustained remission off therapy. A clinical definition of remission used in the aforementioned European trial (NCT01868269) required disappearance of symptoms for 2 successive OMS scorings at minimum 4-week intervals as indicated by scores of zero in the categories of stance, gait, arm and hand function, and opsoclonus and a score 0 or 1 in the category of behavior (they did not include speech and language in definition of remission). In clinical practice, remission on therapy can be considered to be achieved when symptom free other than speech and behavioral scores of 1, for at least 2 months, with clinical judgment as to whether any residual behavioral symptoms are causally related to inflammation. Even where remission on therapy is achieved, immunotherapy should be continued for up to a year and reduced gradually.
To consider OMAS to be in sustained remission requires that symptoms have resolved and not recurred with intercurrent infection or stress, at a point where immunotherapies no longer have demonstrable biological effect. Specifically, if B-cell complement has not recovered after rituximab, remission is considered to be on therapy. However, at no point should sustained remission be considered equivalent to permanent remission, as relapses are possible even several years later and have been reported decades later53 with specific stresses such as pregnancy.54
Relapses reportedly occur in over half of cases in 1 series.10 Long-term sequelae including cognitive impairment and speech problems may be seen in up to 75% of patients but may potentially be mitigated by earlier and more aggressive immunotherapy.7,55 OMAS relapses or escalation of ongoing symptoms are typically associated with triggers including infections (typically common viral infections) and/or with reduction in immunotherapy.9 Patients with possible relapse should be evaluated promptly and assessed for treatable causes (i.e., intercurrent infection) and potentially reassessed for tumor depending on the timing and prior investigations.53 Temporary worsening of OMA symptoms during a febrile illness is not generally considered a relapse if it resolves promptly when febrile illness resolves. Conversely, OMA symptoms appearing during the recovery phase after an intercurrent illness are much more likely to represent a significant relapse, as they are occurring during a period of immune activation. Depending on the severity and timing of the relapse, increase in existing immunotherapy and/or escalation to more intensive immunotherapy should be strongly considered. For patients who relapse after previously receiving rituximab, B-cell number should be checked to see whether the relapse correlates with B-cell repopulation, in which case a repeat course can be considered. If B-cell (CD19) count is fully suppressed at the time of relapse, additional rituximab course is unlikely to be helpful and other modalities of treatment should be considered, such as switch from oral corticosteroids to ACTH. For patients who relapse after they are completely off immunotherapy, restarting at least a single modality of therapy is generally indicated, depending on prior response to therapies. Often, a course of pulse corticosteroids or a few months of pulse IVIG can reestablish remission, but some require a more aggressive approach to reinduce remission.
The Long-term Outcome and Management of OMAS
Long-term outcomes after childhood OMAS have largely been studied retrospectively. Patients usually recover relatively well, although incompletely in terms of motor function. Ataxia, apraxia, coordination deficits, and impaired fine motor skills or tremor have been described in 20%–60% in follow-up spanning 1–50 years4,12,31,56,57 but usually do not severely affect everyday function. In contrast, cognitive and behavioral deficits can severely affect function. Good cognitive outcome is reported in only 10%–50% of patients in older series. The range of cognitive and behavioural sequelae in children with OMAS includes intellectual disability, specific learning disabilities such as dyslexia, as well deficits in attention, expressive language, visuospatial function, and behavioral dysregulation, even in children with normal IQ.4,12,57,58
Cerebellar atrophy of the vermis and hemispheres has been described in patients who had follow-up imaging years after the acute phase, particularly in children who had severe presentations.12,30 In addition, a reduction of cerebral cortical thickness in visual and motor cortex areas has been reported on imaging 10–29 years after symptom onset in children.30 The correlation of these structural changes with cognitive and behavioral issues is not clear as variable results have been reported in small series, while cognitive testing in younger children is challenging.12,30 Further research in this area is needed.
Knowledge of risk factors associated with adverse cognitive outcome is limited. Delay in treatment,12 younger age at onset, and severe presenting symptoms have been reported to associate with poor outcome, but these findings are inconsistent across multiple retrospective studies. The presence or absence of neuroblastoma does not associate with neurologic outcome.12,56 Several retrospective studies have shown associations between a relapsing course and poor outcome.56,57 This has been confirmed by a recent prospective and retrospective analysis of a large cohort of patients in which a greater number of relapses was associated with a poorer cognitive outcome whereas clinical and demographic information at presentation did not.55 Furthermore, type of therapy may matter: 1 study has reported that more front-loaded immunosuppressive treatment associates with better cognitive outcomes.4 Finally, cognitive outcomes may evolve through time. One series of 3 patients described progressive cognitive dysfunction despite a stable clinical course following treatment.59
Given the significant symptoms and impact on development, treatment is targeted at inducing full remission, tolerating no symptoms, and acting promptly to avoid or mitigate relapses. Ongoing physical, occupational, and speech therapy should be recommended for all patients with OMAS, although many children will not tolerate therapies early in the course due to irritability. Often children may also require psychological support and behavioral therapy. As children with OMAS reach school age, neurocognitive testing should be performed, special services in the school are often necessary, and planning should begin early (Table 7).
Table 7.
Recommendations for the Monitoring of Children With OMAS

Restarting Immunizations in Patients With OMAS
Because of the age at onset, children with OMAS often receive treatment that interrupts the routine pediatric immunization schedule. Although most experts agree that immunizing patients with OMAS while on treatment should be avoided, there is no consensus on when to reimmunize or commence catch-up immunization in patients after treatment is complete. Several issues need to be considered: (1) the risk of reactivation of OMAS symptoms due to nonspecific immune stimulation by vaccination; (2) the risk of preventable illnesses; and (3) the ability of patients to mount an immune response after OMAS treatment.
Although there may be concerns regarding reactivation of OMAS symptoms with vaccination, in a series of 14 patients with neuroblastoma-associated OMAS, 6 had undergone revaccination a minimum of 2 years after treatment, none of whom had exacerbation of OMAS symptoms.47 Evidence for the risk of reactivation is extrapolated from large retrospective studies of patients with acquired demyelinating syndromes (ADS), yet several population-based studies suggest that the risk of an ADS within 3 years of a vaccination is not increased.60,61
Similarly, vaccine effectiveness while on immune treatment is generally extrapolated from the pediatric oncology and pediatric rheumatology experience, whereby live vaccinations are avoided and routine vaccination halted during active immunotherapy. Catch-up vaccination with or without booster doses depending on level of immune suppression are reinstituted following completion of immunotherapy,62 based on national vaccination schedules. The time lag to immune competency is often pragmatically considered from the immune biologic perspective (6 months) or longer based on physician or institutional preferences. Notably, such decisions should reflect critical outbreaks of life-threatening communicable disease whereby decision to immunize may be undertaken even if a suboptimal response may be expected. For infections where herd immunity cannot be relied on, such as seasonal influenza and SARS CoV2, immunization even while on immunotherapy should be strongly considered and potential risk and benefits discussed with families.
Summary
There remains uncertainty as to the optimal management of children presenting with OMAS with or without neuroblastoma, and where data from well-controlled studies are lacking, the consensus of expert opinion has allowed several key points to be established:
Early diagnosis is important
Clinical severity scales are important to help evaluate and monitor OMAS (Tables 4 and 7)
Investigations are important, but biomarkers still do not have an established role in management (Tables 1 and 2)
Initiation of treatment to establish disease remission and prevent relapses is crucial (Tables 3, 5 and 6; Figure 1)
Children with OMAS must have long-term follow-up (Table 7)
Recommendations have been made to support clinicians in the investigation, treatment, and long-term support of children and families with OMAS. No one optimal treatment strategy has been established, but whether an upfront or escalating approach is taken, adherence to the key principles of rapidly establishing remission and preventing relapses is key, with informed decisions around the risk and benefit of more aggressive initial immune suppression. International collaboration will remain crucial in answering the ongoing uncertainty around this rare disease.
Acknowledgment
The authors thank the OMS Life Foundation and Dancing Eye Syndrome Support Trust for the enormous contributions to support clinicians, researchers, and families. They also thank Mike Michaelis for his continuous support of work done by the International Study Group, alongside all the remarkable efforts to foster greater knowledge of OMS. Here, the authors on behalf of the International OMAS community pay tribute to their friend, colleague, and physician extraordinaire, Dr. Michael Pranzatelli, who sadly passed away in October 2018. As will be evident from this review, he has been a pioneer, teacher, and champion for the many children and families who have bravely faced this condition.
Glossary
- ACTH
adrenocorticotropic hormone
- ADS
acquired demyelinating syndrome
- IVIg
IV immunoglobulin
- MIBG
meta-iodo-benzyl-guanidine scan
- OMA
opsoclonus-myoclonus-ataxia
- OMAS
opsoclonus-myoclonus-ataxia syndrome
- OMS
opsoclonus-myoclonus syndrome
- OS
overall survival
Appendix 1. Authors
Appendix 2. Coinvestigators

Contributor Information
Thomas Rossor, Email: trossor@nhs.net.
E. Ann Yeh, Email: ann.yeh@sickkids.ca.
Yasmin Khakoo, Email: khakooy@mskcc.org.
Paola Angelini, Email: paola.angelini@nhs.net.
Cheryl Hemingway, Email: cheryl.hemingway@gosh.nhs.uk.
Sarosh R. Irani, Email: sarosh.irani@ndcn.ox.ac.uk.
Gudrun Schleiermacher, Email: gudrun.schleiermacher@curie.fr.
Paramala Santosh, Email: paramala.1.santosh@kcl.ac.uk.
Tim Lotze, Email: tlotze@bcm.edu.
Russell C. Dale, Email: russell.dale@health.nsw.gov.au.
Kumaran Deiva, Email: kumaran.deiva@aphp.fr.
Barbara Hero, Email: barbara.hero@uk-koeln.de.
Andrea Klein, Email: andrea.klein@ukbb.ch.
Pedro de Alarcon, Email: pdealarc@uic.edu.
Mark P. Gorman, Email: mark.gorman@childrens.harvard.edu.
Wendy G. Mitchell, Email: wmitchell@chla.usc.edu.
Study Funding
No targeted funding reported.
Disclosure
T.E. Rossor reports no disclosures relevant to the manuscript. E.A. Yeh has received research funding from the NMSS, CMSC, CIHR, NIH, OIRM, SCN, CBMH Chase an Idea, SickKids Foundation, Rare Diseases Foundation, MS Scientific Foundation (Canada), McLaughlin Centre, and Mario Battaglia Foundation; investigator-initiated research funding from Biogen; scientific advisory—Biogen, Hoffmann-La Roche and Viela Bio; speaker honoraria—Saudi Epilepsy Society, NYU, MS-ATL; ACRS, and PRIME, all unrelated to this manuscript. Y. Khakoo receives support from the NCI Cancer Center Support Grant P30 CA008748. P. Angelini and C. Hemingway report no disclosures relevant to the manuscript. S. Irani has received grants from CSL Behring, UCB Pharma, and Ono Pharmaceutical and personal fees from UCB Pharma and ADC Therapeutics; is a coapplicant and receives royalties on patent application WO/210/046716; and is supported by the Wellcome Trust (104079/Z/14/Z), the UCB Oxford University Alliance, BMA Research Grants, Vera Down grant (2013) and Margaret Temple grant (2017), Epilepsy Research UK (P1201), the Fulbright UK-US Commission (MS Society research award), and the National Institute for Health Research Oxford Biomedical Research Centre. G. Schleiermacher, P. Santosh, and T. Lotze report no disclosures relevant to the manuscript. R. Dale is supported by NHMRC Investigator grant (Australia), Cerebral Palsy Alliance, and Petre Foundation. K. Deiva received speaker honoraria from Novartis, Biogen, Sanofi, and participation in scientific advisory boards of Novartis, Alexion, and Viela, all unrelated to this manuscript. B. Hero reports no disclosures relevant to the manuscript. A. Klein received speaker honoraria from Roche, Biogen, Santhera, and Sarepta and participated in scientific advisory boards of Novartis, Biogen, Santhera, Pfizer, Roche, and Sarepta, all unrelated to this manuscript. P. de Alarcon reports no disclosures relevant to the manuscript. M.P. Gorman has received research funding from Novartis, Biogen, Roche/Genentech, and Pfizer. W. Mitchell reports no disclosures relevant to the manuscript. M.J. Lim receives research grants from Action Medical Research, the DES society, the GOSH charity, the National Institute for Health Research, the MS Society, and the SPARKS charity; receives research support grants from the London Clinical Research Network and the Evelina Appeal; has received consultation fees from CSL Behring, Novartis and Octapharma; has received travel grants from Merck Serono; and was awarded educational grants to organize meetings by Novartis, Biogen Idec, Merck Serono, and Bayer. Go to Neurology.org/NN for full disclosures.
References
- 1.Gorman MP. Update on diagnosis, treatment, and prognosis in opsoclonus-myoclonus-ataxia syndrome. Curr Opin Pediatr 2010;22(6):745-750. [DOI] [PubMed] [Google Scholar]
- 2.Matthay KK, Blaes F, Hero B, et al. Opsoclonus myoclonus syndrome in neuroblastoma a report from a workshop on the dancing eyes syndrome at the advances in neuroblastoma meeting in Genoa, Italy, 2004. In: Cancer Letters. Vol 228. Ireland: Cancer Lett; 2005:275-282. doi: 10.1016/j.canlet.2005.01.051. [DOI] [PubMed]
- 3.Dancing eye syndrome/opsoclonus-myoclonus syndrome workshop report 2018. Accessed April 26, 2021, dancingeyes.org.uk/information-documents-and-reports/Published 2021
- 4.Mitchell WG, Wooten AA, O'Neil SH, Rodriguez JG, Cruz RE, Wittern R. Effect of increased immunosuppression on developmental outcome of opsoclonus myoclonus syndrome (OMS). J Child Neurol 2015;30(8):976-982. [DOI] [PubMed] [Google Scholar]
- 5.Dale RC, Brilot F, Duffy LV, et al. . Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology 2014;83(2):142-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ki Pang K, de Sousa C, Lang B, Pike MG. A prospective study of the presentation and management of dancing eye syndrome/opsoclonus-myoclonus syndrome in the United Kingdom. Eur J Paediatr Neurol 2010;14(2):156-161. [DOI] [PubMed] [Google Scholar]
- 7.Hasegawa S, Matsushige T, Kajimoto M, et al. . A nationwide survey of opsoclonus-myoclonus syndrome in Japanese children. Brain Dev 2015;37(7):656-660. [DOI] [PubMed] [Google Scholar]
- 8.Pranzatelli MR, Tate ED, McGee NR. Multifactorial analysis of opsoclonus-myoclonus syndrome etiology (“Tumor” vs. “No tumor”) in a cohort of 356 US children. Pediatr Blood Cancer 2018;65:e27097. [DOI] [PubMed] [Google Scholar]
- 9.Pranzatelli MR, Tate ED, McGee NR. Demographic, clinical, and immunologic features of 389 children with opsoclonus-myoclonus syndrome: a cross-sectional study. Front Neurol 2017;8(September):153-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tate ED, Allison TJ, Pranzatelli MR, Verhulst SJ. Neuroepidemiologic trends in 105 US cases of pediatric opsoclonus-myoclonus syndrome. J Pediatr Oncol Nurs 2005;22(1):8-19. [DOI] [PubMed] [Google Scholar]
- 11.Kinsbourne M. Myoclonic encephalopathy of infants. J Neurol Neurosurg Psychiatry 1962;25(3):271-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Grandis E, Parodi S, Conte M, et al. . Long-term follow-up of neuroblastoma-associated opsoclonus-myoclonus-ataxia syndrome. Neuropediatrics 2009;40(3):103-111. [DOI] [PubMed] [Google Scholar]
- 13.de Alarcon PA, Matthay KK, London WB, et al. . Intravenous immunoglobulin with prednisone and risk-adapted chemotherapy for children with opsoclonus myoclonus ataxia syndrome associated with neuroblastoma (ANBL00P3): a randomised, open-label, phase 3 trial. Lancet Child Adolesc Heal 2018;2(1):25-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto K, Ohta S, Ito E, et al. . Marginal decrease in mortality and marked increase in incidence as a result of neuroblastoma screening at 6 months of age: cohort study in seven prefectures in Japan. J Clin Oncol 2002;20(5):1209-1214. [DOI] [PubMed] [Google Scholar]
- 15.Armangue T, Titulaer MJ, Sabater L, et al. . A novel treatment-responsive encephalitis with frequent opsoclonus and teratoma. Ann Neurol 2014;75(3):435-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emamikhah M, Babadi M, Mehrabani M, et al. . Opsoclonus-myoclonus syndrome, a post-infectious neurologic complication of COVID-19: case series and review of literature. J Neurovirol 2021;27(1):26-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pranzatelli MR, Travelstead AL, Tate ED, Allison TJ, Verhulst SJ. CSF B-cell expansion in opsoclonus-myoclonus syndrome: a biomarker of disease activity. Mov Disord 2004;19(7):770-777. [DOI] [PubMed] [Google Scholar]
- 18.Pranzatelli MR, Tate ED, Travelstead AL, et al. . Rituximab (anti-CD20) adjunctive therapy for Opsoclonus-Myoclonus syndrome. J Pediatr Hematol Oncol 2006;28(9):585-593. [DOI] [PubMed] [Google Scholar]
- 19.Pranzatelli MR, McGee NR, Tate ED. Relation of intrathecal oligoclonal band production to inflammatory mediator and immunotherapy response in 208 children with OMS. J Neuroimmunol 2018;321:150-156. [DOI] [PubMed] [Google Scholar]
- 20.Pranzatelli MR, Hyland K, Tate ED, Arnold LA, Allison TJ, Soori GS. Evidence of cellular immune activation in children with opsoclonus-myoclonus: cerebrospinal fluid neopterin. J Child Neurol 2004;19(12):919-924. [DOI] [PubMed] [Google Scholar]
- 21.Pranzatelli MR, Tate ED, McGee NR, et al. . BAFF/APRIL system in pediatric OMS: relation to severity, neuroinflammation. Immunotherapy J Neuroinflammation 2013:10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pranzatelli MR, Tate ED, McGee NR, et al. . Key role of CXCL13/CXCR5 axis for cerebrospinal fluid B cell recruitment in pediatric OMS. J Neuroimmunol 2012;243(1-2):81-88. [DOI] [PubMed] [Google Scholar]
- 23.Pranzatelli MR, Tate ED, McGee NR, Colliver JA, Ransohoff RM. CCR4 agonists CCL22 and CCL17 are elevated in pediatric OMS sera: rapid and selective down-regulation of CCL22 by ACTH or corticosteroids. J Clin Immunol 2013;33(4):817-825. [DOI] [PubMed] [Google Scholar]
- 24.Pranzatelli MR, Tate ED, McGee NR, Ransohoff RM. CCR7 signaling in pediatric opsoclonus-myoclonus: upregulated serum CCL21 expression is steroid-responsive. Cytokine 2013;64(1):331-336. [DOI] [PubMed] [Google Scholar]
- 25.Pranzatelli MR, Tate ED, McGee NR. Microglial/macrophage markers CHI3L1, sCD14, and sCD163 in CSF and serum of pediatric inflammatory and non-inflammatory neurological disorders: a case-control study and reference ranges. J Neurol Sci 2017;381:285-290. [DOI] [PubMed] [Google Scholar]
- 26.Pranzatelli MR, Tate ED, McGee NR, Verhulst SJ. CSF neurofilament light chain is elevated in OMS (decreasing with immunotherapy) and other pediatric neuroinflammatory disorders. J Neuroimmunol 2014;266(1-2):75-81. [DOI] [PubMed] [Google Scholar]
- 27.Armangué T, Sabater L, Torres-Vega E, et al. . Clinical and immunological features of opsoclonus-myoclonus syndrome in the era of neuronal cell surface antibodies. JAMA Neurol 2016;73(4):417-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panzer JA, Anand R, Dalmau J, Lynch DR. Antibodies to dendritic neuronal surface antigens in opsoclonus myoclonus ataxia syndrome. J Neuroimmunol 2015;286:86-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poretti A, Benson J, Huisman TGM, Boltshauser E. Acute ataxia in children: approach to clinical presentation and role of additional investigations. Neuropediatrics 2013;44(3):127-141. [DOI] [PubMed] [Google Scholar]
- 30.Anand G, Bridge H, Rackstraw P, et al. . Cerebellar and cortical abnormalities in paediatric opsoclonus-myoclonus syndrome. Dev Med Child Neurol 2015;57(3):265-272. [DOI] [PubMed] [Google Scholar]
- 31.Brunklaus A, Pohl K, Zuberi SM, De Sousa C. Investigating neuroblastoma in childhood opsoclonus-myoclonus syndrome. Arch Dis Child 2012;97(5):461-463. [DOI] [PubMed] [Google Scholar]
- 32.Pfluger T, Schmied C, Porn U, et al. . Integrated imaging using MRI and 123I metaiodobenzylguanidine scintigraphy to improve sensitivity and specificity in the diagnosis of pediatric neuroblastoma. Am J Roentgenol 2003;181(4):1115-1124. [DOI] [PubMed] [Google Scholar]
- 33.Biasotti S, Garaventa A, Villavecchia GP, Cabria M, Nantron M, De Bernardi B. False-negative metaiodobenzylguanidine scintigraphy at diagnosis of neuroblastoma. Med Pediatr Oncol 2000;35(2):153-155. [DOI] [PubMed] [Google Scholar]
- 34.Bleeker G, Tytgat GAM, Adam JA, et al. . 123I-MIBG scintigraphy and 18F-FDG-PET imaging for diagnosing neuroblastoma. Cochrane Database Syst Rev 2015;2015(9):CD009263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donaldson JS, Gilsanz V, Miller JH. CT scanning in patients with opsomyoclonus: importance of nonenhanced scan. Am J Roentgenol 1986;146(4):781-784. [DOI] [PubMed] [Google Scholar]
- 36.Slovis TL, Meza MP, Cushing B, et al. . Thoracic neuroblastoma: what is the best imaging modality for evaluating extent of disease?. Pediatr Radiol 1997;27(3):273-275. [DOI] [PubMed] [Google Scholar]
- 37.Matthay KK, Maris JM, Schleiermacher G, et al. . Neuroblastoma. Nat Rev Dis Prim 2016:2:16078. [DOI] [PubMed] [Google Scholar]
- 38.Gambini C, Conte M, Bernini G, et al. . Neuroblastic tumors associated with opsoclonus-myoclonus syndrome: histological, immunohistochemical and molecular features of 15 Italian cases. Virchows Arch 2003;442(6):555-562. [DOI] [PubMed] [Google Scholar]
- 39.Hero B, Clement N, Øra I, et al. . Genomic profiles of neuroblastoma associated with opsoclonus myoclonus syndrome. J Pediatr Hematol Oncol 2018;40(2):93-98. [DOI] [PubMed] [Google Scholar]
- 40.Evans AE, D'Angio GJ, Randolph J. A proposed staging for children with neuroblastoma. Children's cancer study group A. Cancer 1971;27(2):374-378. [DOI] [PubMed] [Google Scholar]
- 41.Ikegaki N, Shimada H. Subgrouping of unfavorable histology neuroblastomas with immunohistochemistry toward precision prognosis and therapy stratification. JCO Precis Oncol 2019;3(3):1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ackermann S, Cartolano M, Hero B, et al. . A mechanistic classification of clinical phenotypes in neuroblastoma. Science. 2018;362(6419):1165-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strother DR, London WB, Lou SM, et al. . Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 2012;30(15):1842-1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker DL, Schmidt ML, Cohn SL, et al. . Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 2010;363(14):1313-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Bernardi B, Mosseri V, Rubie H, et al. . Treatment of localised resectable neuroblastoma. Results of the LNESG1 study by the SIOP Europe Neuroblastoma Group. Br J Cancer 2008;99(7):1027-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ladenstein R, Pötschger U, Valteau-Couanet D, et al. . Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol 2018;19(12):1617-1629. [DOI] [PubMed] [Google Scholar]
- 47.Patel A, Fischer C, Lin YC, et al. . Treatment and revaccination of children with paraneoplastic opsoclonus-myoclonus-ataxia syndrome and neuroblastoma: the Memorial Sloan Kettering experience. Pediatr Blood Cancer 2020:67(8):e28319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilbur C, Yea C, Licht C, Irwin MS, Yeh EA. An upfront immunomodulatory therapy protocol for pediatric opsoclonus-myoclonus syndrome. Pediatr Blood Cancer 2019;66(8):e27776. [DOI] [PubMed] [Google Scholar]
- 49.Dale RC, Gorman MP, Lim M. Autoimmune encephalitis in children: clinical phenomenology, therapeutics, and emerging challenges. Curr Opin Neurol 2017;30(3):334-344. [DOI] [PubMed] [Google Scholar]
- 50.Pranzatelli MR, Tate ED, Swan JA, et al. . B cell depletion therapy for new-onset opsoclonus-myoclonus. Mov Disord 2010;25(2):238-242. [DOI] [PubMed] [Google Scholar]
- 51.Pranzatelli MR, Tate ED. Dexamethasone, intravenous immunoglobulin, and rituximab combination immunotherapy for pediatric opsoclonus-myoclonus syndrome. Pediatr Neurol 2017;73:48-56. [DOI] [PubMed] [Google Scholar]
- 52.Russo C, Cohn SL, Petruzzi MJ, De Alarcon PA. Long-term neurologic outcome in children with opsoclonus-myoclonus associated with neuroblastoma: a report from the Pediatric Oncology Group. Med Pediatr Oncol 1997;28(4):284-288. [DOI] [PubMed] [Google Scholar]
- 53.Pranzatelli MR, Tate ED. Trends and tenets in relapsing and progressive opsoclonus-myoclonus syndrome. Brain Dev 2016;38(5):439-448. [DOI] [PubMed] [Google Scholar]
- 54.Adhikari S, Thuringer A, Maali L, Jassam Y. Opsoclonus myoclonus syndrome in a postpartum period. Mult Scler Relat Disord 2021;50:102862. [DOI] [PubMed] [Google Scholar]
- 55.Sheridan A, Kapur K, Pinard F, et al. . IQ predictors in pediatric opsoclonus myoclonus syndrome: a large international cohort study. Dev Med Child Neurol 2020;62(12):1444-1449. [DOI] [PubMed] [Google Scholar]
- 56.Brunklaus A, Pohl K, Zuberi SM, De Sousa C. Outcome and prognostic features in opsoclonus-myoclonus syndrome from infancy to adult life. Pediatrics 2011;128(2):e388-e394. [DOI] [PubMed] [Google Scholar]
- 57.Mitchell WG, Brumm VL, Azen CG, Patterson KE, Aller SK, Rodriguez J. Longitudinal neurodevelopmental evaluation of children with opsoclonus-ataxia. Pediatrics 2005;116(4):901-907. [DOI] [PubMed] [Google Scholar]
- 58.Mitchell WG, Davalos-Gonzalez Y, Brumm VL, et al. . Opsoclonus-ataxia caused by childhood neuroblastoma: developmental and neurologic sequelae. Pediatrics 2002;109(1):86-98. [PubMed] [Google Scholar]
- 59.Goh EL, Scarff K, Satariano S, Lim M, Anand G. Evolving cognitive dysfunction in children with neurologically stable opsoclonus–myoclonus syndrome. Children 2020;7(9):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langer-Gould A, Qian L, Tartof SY, et al. . Vaccines and the risk of multiple sclerosis and other central nervous system demyelinating diseases. JAMA Neurol 2014;71(12):1506-1513. [DOI] [PubMed] [Google Scholar]
- 61.Karussis D, Petrou P. The spectrum of post-vaccination inflammatory CNS demyelinating syndromes. Autoimmun Rev 2014;13(3):215-224. [DOI] [PubMed] [Google Scholar]
- 62.van Tilburg CM, Sanders EAM, Rovers MM, Wolfs TFW, Bierings MB. Loss of antibodies and response to (re-)vaccination in children after treatment for acute lymphocytic leukemia: a systematic review. Leukemia 2006;20(10):1717-1722. [DOI] [PubMed] [Google Scholar]