

Abstract
Budigalimab, a novel anti–PD-1 monoclonal antibody, demonstrated efficacy and biomarker pharmacodynamics in patients with head and neck squamous cell carcinoma (HNSCC) or non–small cell lung cancer (NSCLC) consistent with those reported by other PD-1 inhibitors. Herein are presented additional outcomes of biomarker analyses from the phase 1 study of budigalimab monotherapy in patients with HNSCC and NSCLC (NCT03000257). PD-1 inhibitor naive patients with advanced HNSCC (n=41) or NSCLC (n=40) received budigalimab intravenously at 250 mg every 2 weeks (Q2W) or 500 mg Q4W until progression. Archival tumor specimens were evaluated by immunohistochemistry for CD8 and tumor PD-1 ligand 1 (PD-L1) expression, RNA, and whole-exome sequencing. Serum and whole blood samples were acquired at baseline and at select on-treatment time points. As of October 2019, best overall response of 15% in HNSCC and 18% in NSCLC was observed in all treated patients; both cohorts reported responses in PD-L1+ and PD-L1– tumors. Treatment with budigalimab was associated with increases in multiple soluble biomarkers including interferon gamma-induced chemokines. Expanded overall T-cell counts, total CD8 T-cell counts, and percentages of CD8+CD45RA–CD62L– effector memory T cells were observed at cycle 1, day 15 in responders. Univariate analysis demonstrated an association between prolonged progression-free survival and higher tumor mutational burden/neoantigen load, smaller tumor size, lower platelet-lymphocyte ratios, lower CCL23, lower colony-stimulating factor 1, and lower interleukin-6 levels at baseline. The biomarker analysis presented herein identified additional early pharmacodynamic biomarkers associated with anti–PD-1 activity and improved clinical responses to budigalimab in patients with advanced HNSCC and NSCLC.
Key Words: budigalimab, head and neck squamous cell carcinoma, non-small cell lung cancer, PD-1 inhibitor, pharmacodynamics
The programmed cell death protein 1 (PD-1) and ligand 1 (PD-L1) immune checkpoints are increasingly seen as important targets for anticancer therapy, due to the role of PD-1 in downregulating cell signaling pathways important for an effective immune response for many types of cancer. In patients with advanced solid tumors, including head and neck squamous cell carcinoma (HNSCC) and non–small cell lung cancer (NSCLC), treatment with anti–PD-1 blocking antibodies as immuno-oncology checkpoint inhibitors is an effective strategy associated with improved overall survival (OS) and reduction in risk of death.1 These drugs interfere with negative signaling through PD-1 expressed on activated T cells, which permits tumor cells (TCs) to evade immune surveillance and thereby stimulate antitumor T-cell activity.2 However, not all patients respond to therapy, and an improved understanding of PD-1–mediated checkpoint inhibition is needed to predict patient response and improve treatment outcomes. Subgroup analyses have shown OS benefits for anti–PD-1/PD-L1 antibodies in patients with PD-L1+tumors, those with a history of smoking, and when used as first-line therapy compared with later lines of treatment.1 In contrast, OS is not improved in patients aged 75 years and older, whereas treatment efficacy is not affected by age, performance status, tumor histology, or treatment type.
In the context of PD-1/PD-L1 blockade, identifying and validating predictors of treatment response has become a priority. Companion immunohistochemistry (IHC) assays have clinical utility in identifying patients with higher levels of TCs or PD-L1–expressing tumor-infiltrating immune cells that may have a greater chance of responding to PD-1/PD-L1 inhibitors.1,3 However, some PD-L1–patients are potential responders, and the response rate among patients with PD-L1–expressing TCs remains below 50%. In addition, IHC methods differ according to the anti–PD-1/PD-L1 antibody used for treatment.3 These challenges highlight the need for additional biomarkers that are prognostic for outcome and/or predictive of treatment response, and which reflect not only tumor microenvironment, but also peripheral blood cell composition. Some of these efforts have been successful in achieving this goal. For instance, in addition to PD-L1, several biomarkers have been identified as predictors of response to PD-1/PD-L1 blockade across multiple tumor types, such as melanoma and NSCLC.4 These include tumor mutational burden (TMB) and specific mutations identified using whole-exome and RNA sequencing approaches, as well as markers of immune cell activation in the periphery.5–8
Budigalimab (formerly ABBV-181), a novel humanized, recombinant immunoglobulin G1 monoclonal antibody targeting PD-1, is currently in development and has shown similar efficacy and biomarker pharmacodynamics to that reported for other PD-1 inhibitors.9 Budigalimab has been evaluated in a phase 1 study as monotherapy and in combination with other anticancer therapies in patients with advanced solid tumors (NCT03000257). The study included patients with HNSCC and NSCLC naive to anti–PD-1 treatment for whom chemotherapy had previously failed and who were followed for response according to Response Evaluation Criteria In Solid Tumors (RECIST) and modified immunotherapy (iRECIST) criteria.9 As previously published, budigalimab monotherapy in these cohorts demonstrated a response rate consistent with that achieved by other PD-1 inhibitors.9–13 Archival tumor biopsy samples were available for most patients; these can provide a cross-sectional profile of pre-existing antitumor immune responses, as well as intrinsic tumor biomarkers such as mutation status. Minimally invasive blood sampling for subsequent profiling of immune cells and secreted factors was collected for every patient at baseline and multiple assessments over time, as such samples can provide insight into immune fitness and drug pharmacodynamic effects.14 In addition, baseline blood sampling may have predictive value, and early time points in cycle (C)1 of anti–PD-1 therapy (24 h to 4 wk) may indicate a dynamic profile of tumor response to therapy.15,16
The present analysis used response data, archival tumor tissue, and peripheral blood samples from patients enrolled in the phase 1 study of budigalimab to characterize biomarkers associated with anti–PD-1 activity and response to identify patients with HNSCC and NSCLC who were more likely to respond to budigalimab. The analysis considered baseline predictors and early pharmacodynamic indicators of response to budigalimab.
METHODS
Study Design and Patients
The present biomarker analysis is based on a subset of patients included in a phase 1 dose-escalation and dose-expansion study undertaken to determine the recommended phase 2 dose and maximum tolerated dose of budigalimab, and to evaluate its safety, pharmacokinetic profile, and preliminary efficacy as monotherapy. The study was approved by the institutional review board at each participating site before initiation of any screening or study-specific procedures. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, as defined by the International Conference on Harmonisation. Written informed consent was obtained from each individual participating in the study.
Briefly, following the dose-finding part of the study, 41 patients with HNSCC (oral cavity, oropharynx, hypopharynx, or larynx) and 40 patients with NSCLC (squamous or nonsquamous, including 7 patients with epidermal growth factor receptor [EGFR] mutations or anaplastic lymphoma kinase [ALK] rearrangements) were enrolled across multiple sites into monotherapy expansion cohorts, between November 2017 and December 2018. Key inclusion criteria were previous failure of platinum-based therapy and naive to anti–PD-1 or anti–PD-L1 therapy. Budigalimab was administered by intravenous infusion at either 250 mg every 2 weeks (Q2W) or 500 mg Q4W, established as the recommended phase 2 dose. Patients were treated until disease progression (PD) per iRECIST10 or unacceptable toxicity. Efficacy endpoints included in this analysis were best overall response (BOR; complete response [CR], partial response [PR], or stable disease [SD]) and progression-free survival (PFS). Tumor assessments by radiographic imaging (contrast-enhanced computed tomography or magnetic resonance imaging) were performed at baseline and repeated every 2 treatment cycles for the first 12 months and every 3 cycles thereafter by the investigator per RECIST v1.1 and iRECIST. All treated patients were included in response calculations. Patients who dropped out before first radiographic clinical assessment were considered as having progressed. Additional details regarding study design and eligibility criteria are summarized in Italiano et al.9
Tissue Assessments
All patients consented to provide either archived formalin-fixed paraffin-embedded tumor tissue or a pretreatment fresh tumor biopsy for biomarker analysis. Sufficient tumor tissue for IHC analysis was obtained from 38 HNSCC and 33 NSCLC patients. Tumor tissue was analyzed for tumor and lymphocyte content by hematoxylin and eosin staining, PD-L1 expression (PD-L1+ [≥1%]) by the Dako 28-8 PharmDx IHC assay (Agilent, Santa Clara, CA), and for CD8+ T-cell infiltration by IHC using the Dako C8/144B clone (Agilent). In a subset of patients, RNA and DNA were extracted from tumor formalin-fixed paraffin-embedded samples using Qiagen Allprep (Germantown, MD), with RNA sequencing (RNAseq) libraries prepared using Sigma SeqPlex (Sigma-Aldrich Corp, St. Louis, MO) and paired-end 150-bp sequencing conducted using Illumina platforms (San Diego, CA). Whole-exome sequencing (WES) library preparation was performed using Agilent Clinical Research Exome (CRE) V2 baits (Agilent) and Illumina paired-end 150-bp sequencing, which targeted 200× coverage. Details of data processing and outlier identification of these data sets, including analysis methods for differential gene expression, somatic mutations, human leukocyte antigen (HLA) genotyping, and neoantigens are included in the Supplemental Digital Content Methods (Supplemental Digital Content 1, http://links.lww.com/JIT/A666). Following removal of poor-quality or outlier data, in the HNSCC cohort, 32 patient tumors generated RNA data and 22 generated DNA data; in the NSCLC cohort, 12 and 10 tumors, respectively, generated RNA and DNA data.
Blood Biomarker Assessments
All patients consented to provide blood for biomarker assessments. Standard laboratory tests for cell blood counts (lymphocytes, neutrophils, platelets, eosinophils) and soluble factors (lactate dehydrogenase, cholesterol) were conducted before first dose and at 2–4 weeks on study. Blood samples for exploratory biomarker assessment were collected before infusion (0 h, predose), 2 hours postinfusion, and on days (D) 2, 3, 8, and 15 of C1 and 3, and D1 and 15 of C2 and 4. Cellular biomarkers for PD-1 saturation, memory T-cell frequencies, and Ki67 proliferation were evaluated using validated flow cytometry assays (Covance Inc., USA) on freshly obtained anticoagulated blood. Information on the exact flow panels used and exemplary plots for the gating strategy are provided in the Supplemental Digital Content Methods (Supplemental Digital Content 1, http://links.lww.com/JIT/A666). Soluble biomarkers from cryopreserved serum taken at indicated treatment time points were evaluated using a pair of 92-plex proximity extension panels (Immuno-Oncology and Inflammation Panels, Olink Proteomics, Uppsala, Sweden) in terms of fold change (FC) from baseline. Additional assays applied to serum to quantify baseline and FC of soluble biomarkers included a Luminex panel tested at Myriad RBM (Austin, TX), including mitogen-inducible gene (MIG) and interferon-gamma (IFN-γ)–induced protein-10, and a Meso Scale Diagnostics panel (Rockville, MD) including IFN-γ, interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α). The numbers of patients with reportable data for each assessment are included in the relevant figures.
Statistical Analyses
Somatic TMB, representing the number of protein-coding somatic mutations/megabase, was calculated for each tumor WES sample by dividing the total number of these mutations by 38. Significant differences between CR/PR/iPR versus SD/PD for TMB and neoantigen load were assessed using the Wilcoxon rank-sum test. The association of clinical outcomes with baseline and FC measures of biomarkers at all available time points was assessed using response and other outcome data available at the data cutoff of October 31, 2019. Data for HNSCC and NSCLC indications were analyzed both individually and collectively for outcome associations with biomarkers. Patients from the NSCLC monotherapy arm included in the analysis were limited to those with wildtype EGFR and ALK tumor status. Univariate analyses used laboratory and biomarker data from flow cytometry, soluble biomarkers, and IHC (cleaned of duplicates and values less than the lower limit of detection), and were based on available samples from 41 patients with HNSCC and 33 patients with NSCLC (EGFR −, ALK-mutated patients excluded), including a total of 13 responders (CR/PR/iPR) and 27 patients with SD (Supplemental Digital Content Table 1, Supplemental Digital Content 1, http://links.lww.com/JIT/A666). An analysis of variance model was applied for associations with BOR, with a Cox proportional hazards model used for associations with PFS. Biomarkers with P-value <0.05 with either BOR or PFS are reported (Table 1).
TABLE 1.
Univariate Analysis of Tumor Biomarkers Associated With Response (CR, PR, or iPR) or With PFS
| Biomarker | Mean R | Mean NR | P for RNR | P for PFS | HR |
|---|---|---|---|---|---|
| Tumor baseline biomarkers | |||||
| Baseline tumor size | 54.923 | 73.117 | 0.058 | 0.004 | 1.501 |
| Tumor mutational burden (matched normal) | 5.254 | 2.624 | 0.010 | 0.122 | 0.769 |
| Neoantigen count | 182 | 76.148 | 0.003 | 0.040 | 0.716 |
| Gene.EDIL3 | 188.067 | 60.174 | 0.002 | 0.036 | 0.861 |
| Gene.LRRC32 | 217.232 | 88.698 | 0.009 | 0.163 | 0.870 |
| Gene.ITGA2B | 31.627 | 9.661 | 0.004 | 0.126 | 0.871 |
| Gene.MT1A | 11.378 | 1.387 | 0.030 | 0.466 | 0.904 |
| Gene.CDH2 | 115.086 | 14.568 | 0.014 | 0.338 | 0.928 |
| Gene. PCOLCE | 212.453 | 89.027 | 0.035 | 0.680 | 0.958 |
| Gene.IGFBP2 | 284.229 | 125.070 | 0.007 | 0.651 | 0.963 |
| Gene.CLU | 3076.621 | 672.702 | 0.031 | 0.946 | 0.994 |
| Gene.CXCL1 | 29.653 | 137.659 | 0.026 | 0.978 | 0.998 |
| Gene.PTGS2 | 171.929 | 629.011 | 0.037 | 0.825 | 1.016 |
| Gene.MEFV | 6.302 | 22.246 | 0.019 | 0.739 | 1.033 |
| Gene.IL1R2 | 26.998 | 220.249 | 0.018 | 0.569 | 1.034 |
| Gene.CCL20 | 4.011 | 37.988 | 0.027 | 0.507 | 1.039 |
| Gene.IL1B | 23.188 | 198.297 | 0.002 | 0.426 | 1.052 |
| Gene.DKK1 | 2.667 | 17.996 | 0.021 | 0.376 | 1.072 |
| Gene.MMP1 | 21.783 | 338.099 | 0.014 | 0.144 | 1.075 |
| Gene.CD207 | 3.115 | 23.415 | 0.006 | 0.184 | 1.094 |
| Gene.MX1 | 159.711 | 431.017 | 0.014 | 0.312 | 1.134 |
| Gene. TNFRSF13C | 34.072 | 76.320 | 0.030 | 0.082 | 1.138 |
| Gene. TNFSF10 | 620.231 | 2116.104 | 0.007 | 0.148 | 1.144 |
| Gene.ICOS | 5.041 | 24.668 | 0.017 | 0.082 | 1.158 |
| Gene. TNFAIP3 | 302.601 | 666.956 | 0.006 | 0.227 | 1.162 |
| Gene.SELL | 16.849 | 65.117 | 0.013 | 0.099 | 1.189 |
| Gene.CD6 | 21.499 | 81.263 | 0.024 | 0.019 | 1.223 |
| Gene.CCR7 | 16.864 | 97.291 | 0.005 | 0.023 | 1.228 |
| Gene.GATA3 | 15.335 | 52.612 | 0.013 | 0.013 | 1.258 |
| Gene.TYRO3 | 33.002 | 74.575 | 0.025 | 0.122 | 1.266 |
| Gene.TNFSF8 | 7.679 | 18.820 | 0.048 | 0.012 | 1.273 |
| Gene.IL21R | 18.981 | 44.443 | 0.046 | 0.019 | 1.278 |
| Gene.IRF7 | 35.858 | 76.732 | 0.006 | 0.043 | 1.282 |
| Gene.IFIT2 | 28.498 | 68.899 | 0.019 | 0.085 | 1.286 |
| Gene.FLT3LG | 7.952 | 22.423 | 0.014 | 0.003 | 1.354 |
| Gene.CD5 | 15.796 | 52.349 | 0.018 | 0.024 | 1.361 |
| Gene.RUNX3 | 111.279 | 257.059 | 0.000 | 0.003 | 1.557 |
Genes included are those immune-related genes (of 982 evaluated) with a P-value <0.05 for RNR and a >2-fold difference between responders and nonresponders, for which there was no significant indication interaction and the same direction of change in the combined HNSCC and NSCLC cohorts, excluding those with reported EGFR/ALK mutations. A total of 74 patients (41 HNSCC and 33 NSCLC) were included in the tumor size analyses, 34 patients (25 HNSCC and 9 NSCLC) were included in the exome analyses, and 45 patients (33 HNSCC and 12 NSCLC) were included in the gene expression analyses.
Italics values are significant by P<0.05.
ALK indicates anaplastic lymphoma kinase; CR, complete response; EGFR, epidermal growth factor receptor; HNSCC, head and neck squamous cell carcinoma; HR, hazard ratio; iPR, immune partial response; NR, nonresponder; NSCLC, non–small cell lung cancer; PFS, progression-free survival; PR, partial response; R, responder; RNR, responder to nonresponder.
RESULTS
Patients
Patient demographics and baseline clinical characteristics are summarized in Supplemental Digital Content Table 1 (Supplemental Digital Content 1, http://links.lww.com/JIT/A666) and are reported in full elsewhere.9 Median age was 62 years for the 41 patients in the HNSCC cohort (range, 51–84), and 65 years for the 40 patients in the NSCLC cohort (range, 39–79). Mean measurable tumor burden at screening as measured by the sum of diameters was 66.2 mm (range, 17–173) for the HNSCC cohort and 75.8 mm (range, 10–246) for the NSCLC cohort. Patients had multiple prior lines of therapy before being dosed with budigalimab at either 250 mg Q2W or 500 mg Q4W. Disease response meeting RECIST or iRECIST definitions was observed in six of 41 patients with HNSCC for an ~15% response rate, and in 7 of 40 patients with NSCLC for an ~18% response rate.
Tumor-specific Biomarkers Associated With Clinical Response
As expected, clinical responses were observed in patients with both PD-L1+ (≥1% by Dako 28-8 PharmDx IHC assay) and PD-L1– tumors; tested PD-L1– tumors accounted for 2 clinical responses (CR/PR/iPR) in patients with HNSCC and 3 clinical responses in patients with NSCLC (Fig. 1A). In the HNSCC cohort, the use of more-stringent exploratory PD-L1 cutoffs did not alter the BOR rate. However, in the NSCLC cohort, using a ≥50% PD-L1 cutoff resulted in 2 of 8 (25%) patients with response (2 of 6 patients [33%] if patients with ALK/EGFR-mutated tumors were excluded from analysis), compared with 2 of 16 (12.5%) patients with ≥1% PD-L1+ tumors. The 28-8 PD-L1 assay assesses tumor PD-L1, but other PD-L1 assays may incorporate assessment of PD-L1 on infiltrating inflammatory cells. In an exploratory assessment, no association of inflammatory cell PD-L1 staining with clinical outcomes was observed with HNSCC, while low inflammatory cell PD-L1 staining (intensity <2) was associated with worse outcomes in NSCLC (Fig. 1B). IHC detection of CD8 expression had no clear value in stratifying responders and nonresponders in HNSCC. In the NSCLC cohort there was a trend to a higher response in patients with >15% CD8 T-cell infiltration (2 of 8 patients [25%]) compared with those with <15% CD8 T-cell infiltration (3 of 25 patients [12%]) (Fig. 1C).
FIGURE 1.
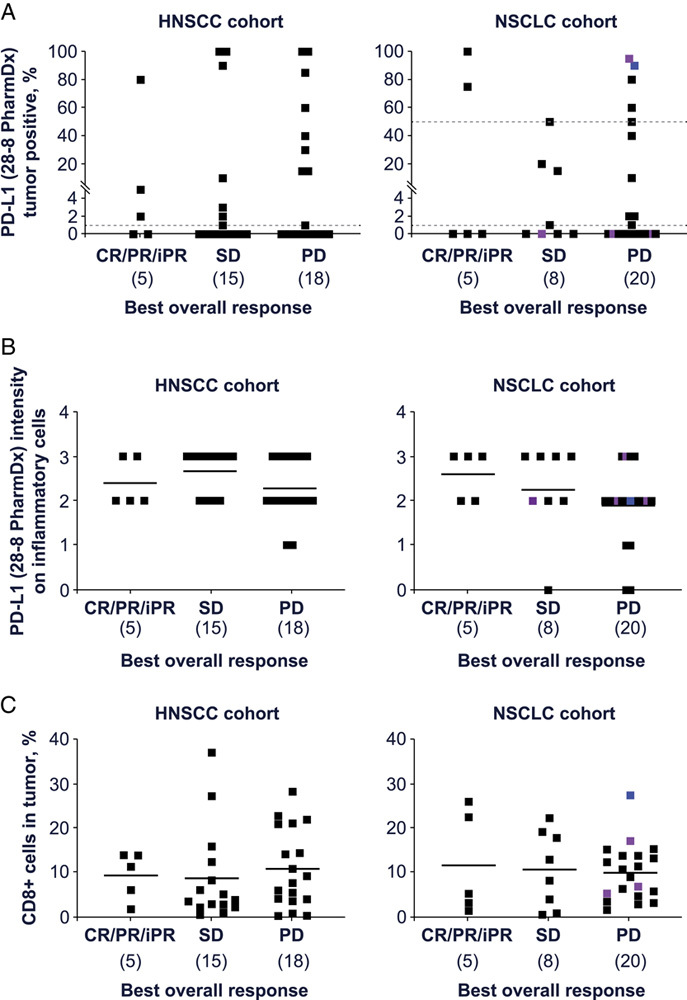
Immunohistochemical detection of PD-L1 percentage positive in tumor cells (A), PD-L1 intensity in inflammatory cells (B), and CD8 percentage positive (C) expression in archival tumor samples of patients with HNSCC and NSCLC treated with budigalimab monotherapy. A total of 71 patients (38 HNSCC and 33 NSCLC) were included in these analyses. Purple: EGFR mutated; blue: ALK rearrangement. + indicates positive; CR, complete response; HNSCC, head and neck squamous cell carcinoma; iPR, immune partial response; NSCLC, non–small cell lung cancer; PD, progressive disease; PD-L1, programmed cell death protein 1 ligand 1; PR, partial response; SD, stable disease.
Tumor exome data were available for 32 of 81 patients overall, 22 in the HNSCC cohort and 10 in the NSCLC cohort. Univariate analysis of baseline tumor data showed an association between response and TMB or predicted neoantigen load. Patients with higher TMB (cutoff optimized at 2.47 or 4.895) had a significantly higher likelihood of response and a trend to longer PFS (Table 1, Fig. 2A, B, Supplemental Digital Content Fig. 1A, Supplemental Digital Content 1, http://links.lww.com/JIT/A666). Correlation of TMB calculations using a matched normal versus without a matched normal is presented in Supplemental Digital Content Figure 1B (Supplemental Digital Content 1, http://links.lww.com/JIT/A666). Predicted neoantigen counts showed 94% correlation with TMB, and demonstrated significant association with both clinical response as well as PFS (Table 1, Supplemental Digital Content Figs. 1C–1E, Supplemental Digital Content 1, http://links.lww.com/JIT/A666). Homozygosity in at least 1 of 3 class-I HLAs (A, B, or C) was detected for 10/32 patients’ tumor WES samples. However, because this homozygosity was also detected in the germline for all 7 of the HLA-homozygous patients with matched normal blood samples, there is no evidence in this cohort supporting somatic loss of heterozygosity events for HLA class I molecules. The relationship between response and HLA homozygosity was not significant (data not shown).
FIGURE 2.
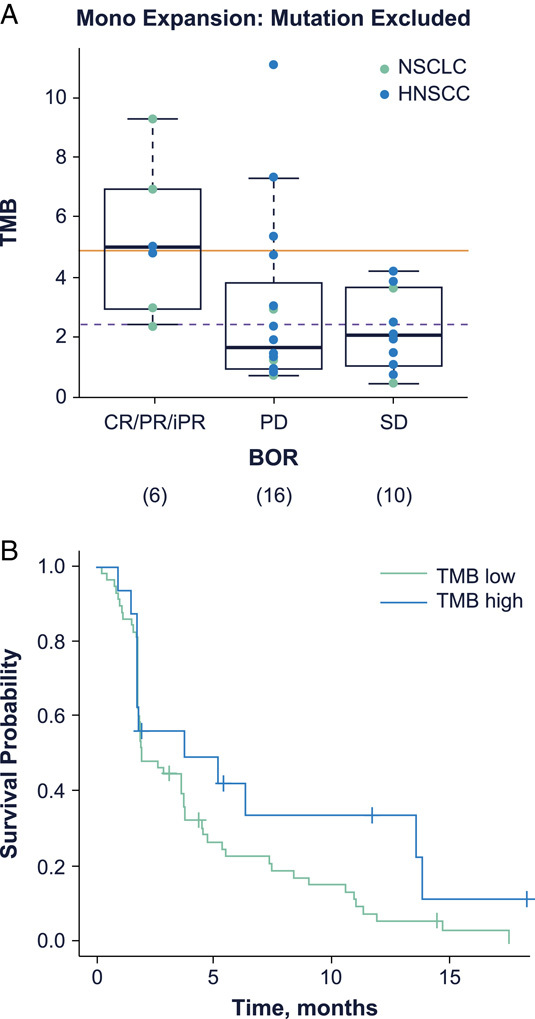
Exome sequencing showed a relationship between tumor mutational burden (TMB) and best overall response (A); weak association of TMB with PFS was also observed, with a hazard ratio of 0.77 and a P-value of 0.126 (B). The optimized (sensitivity plus specificity) cutoff of 4.895, shown by the solid line, and an optimized (maximal sensitivity) cutoff of 2.47, shown by the dotted line were determined by performance as shown in Supplemental Digital Content Figure 1 (Supplemental Digital Content 1, http://links.lww.com/JIT/A666). A total of 32 patients (22 HNSCC and 10 NSCLC) were included in these analyses. BOR indicates best overall response; CR, complete response; HNSCC, head and neck squamous cell carcinoma; iPR, immune partial response; mono, monotherapy; NSCLC, non–small cell lung cancer; PD, progressive disease; PFS, progression-free survival; PR, partial response; SD, stable disease.
Tumor RNAseq data were available for 44/81 patients, 32 in the HNSCC cohort and 12 in the non–EGFR/ALK-mutated NSCLC cohort. Of ∼40,000 genes with nonzero transcripts, significant differences (|FC| >1.5, P-value <0.05, false discovery rate <0.25) between responders and nonresponders were observed in HNSCC (1935 genes; Fig. 3A) and NSCLC (90 genes; Fig. 3B) as well as in the combined 44-patient cohort (794 genes; Fig. 3C). Using Ingenuity Pathway Analysis (Qiagen) we identified differential association of signaling pathways with response in HNSCC (Fig. 3D). Among other pathways, PXR signaling, apelin signaling, and fatty acid oxidation were positively associated with response, while amyloid processing, osteoarthritis, GP6 signaling, phosphatase and tensin homolog signaling, CDK5 signaling, and HOTAIR regulation were negatively associated with response in HNSCC. While tumors from responders generally did not display a greater IFN-γ gene expression signature than nonresponders (using the 20-gene signature known as the immunologic constant of rejection17), human papillomavirus (HPV)+ HNSCC did demonstrate a higher IFN-γ expression signature than HPV– HNSCC (Supplemental Digital Content Fig. 2, Supplemental Digital Content 1, http://links.lww.com/JIT/A666). There were too few differentially expressed genes in NSCLC for a strong pathway analysis. An additional univariate analysis focused on differentially expressed immune-related genes combining all patients regardless of indication. This analysis identified 34 genes with significant expression differences (|FC| >2, P-value <0.05). EDIL3 was higher in budigalimab responders and associated with both response and PFS (Figs. 3D–F, Table 1). Genes such as ITGA2B, CDH2, and CLU were also more strongly expressed in responders, while genes such as CXCL1, CCL20, CCR7, IL1B, and IL1R2 were more strongly expressed in nonresponders (Table 1). In addition, a smaller baseline tumor size (mean 54.9 mm in responders vs. mean 73.1 in nonresponders) was associated with better PFS (Table 1, Supplemental Digital Content Fig. 3, Supplemental Digital Content 1, http://links.lww.com/JIT/A666).
FIGURE 3.
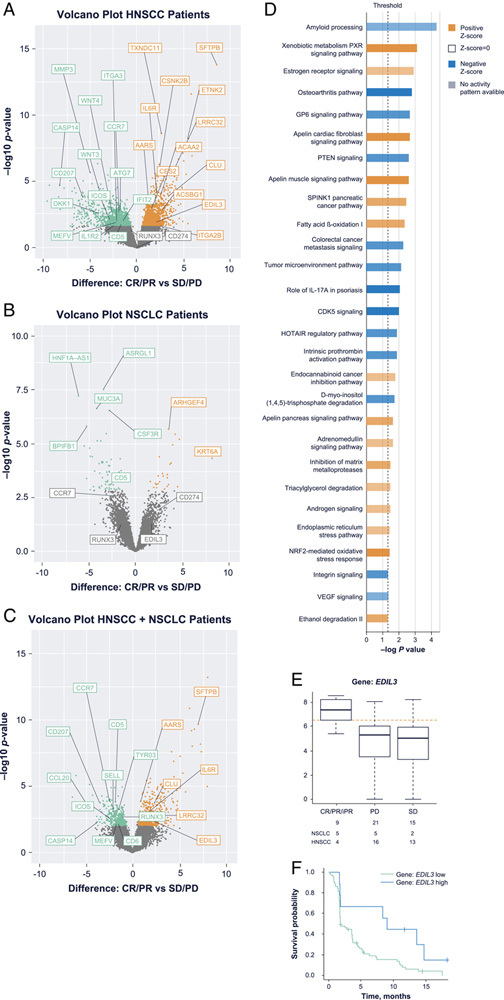
RNAseq analysis of the ∼40,000 transcripts that are nonzero in at least one sample detected multiple unique features with significant expression differences between responders and nonresponders in the indication-specific HNSCC, n=32 (A) and NSCLC, n=12 (B) data sets, as well as the combined (C) data set, n=44. Orange and green dots represent significantly differentially expressed transcripts (|FC| >2, P-value <0.05, FDR <0.25), with orange indicating higher expression in responders and green indicating lower expression in responders. Qiagen Ingenuity Pathway Analysis of differential gene expression by responders and nonresponders showed significantly upregulated pathways (orange) and downregulated pathways (blue) in responding patients with HNSCC (D). In the combined data set, EDIL3 was associated with both BOR (E) and PFS (F). AUC indicates area under the concentration-time curve; BOR, best overall response; CR, complete response; FC, fold change; FDR, false discovery rate; HNSCC, head and neck squamous cell carcinoma; HR, hazard ratio; iPR, immune partial response; NSCLC, non–small cell lung cancer; PD, progressive disease; PFS, progression-free survival; PR, partial response; RNAseq, RNA sequencing; SD, stable disease.
Blood Biomarkers of Immune Status at Enrollment Associated With Clinical Response
A wide range of baseline immune characteristics were observed in the later-line cancer patients enrolled in this study. Baseline peripheral biomarkers that significantly associated with better response and/or PFS included markers of better immune fitness, such as higher number or percentage of lymphocytes, higher T-cell counts (CD3), higher natural killer (NK) cell (CD3–/CD56+) counts, and higher %Ki67+ CD8 T cells (Table 2, Supplemental Digital Content Fig. 3 and 5, Supplemental Digital Content 1, http://links.lww.com/JIT/A666). Lymphocyte value percentages were significantly higher in responders (mean 24.15%) compared with nonresponders (mean 16.12%), which may reflect the below-reference-range median lymphocyte counts in patients with no clinical benefit (Table 2, Supplemental Digital Content Fig. 4, Supplemental Digital Content 1, http://links.lww.com/JIT/A666). Better response/PFS was also associated with a lower platelet-lymphocyte ratio, a lower percentage of naive CD8 T cells (detected both as CD45RA+CD62L+ and as CD28+CD95–), lower CCL23, lower colony-stimulating factor (CSF)-1, and lower IL-6 levels at baseline in both NSCLC and HNSCC (Table 2, Supplemental Digital Content Fig. 3, Supplemental Digital Content 1, http://links.lww.com/JIT/A666). In addition, responders in both indications had a higher baseline level of soluble PD-1 (PDCD1). Separation of patients into indication-specific cohorts identified additional baseline soluble biomarkers associated with better response and PFS in the NSCLC cohort, including higher levels of CAIX, CCL11, MCP-2, MCP-4, and soluble CD70 (Supplemental Digital Content Table 2, Supplemental Digital Content 1, http://links.lww.com/JIT/A666).
TABLE 2.
Univariate Analysis of Peripheral Biomarkers Associated With Response (CR, PR, or iPR) or With PFS
| Biomarker | Mean R | Mean NR | P for RNR | P for PFS | HR |
|---|---|---|---|---|---|
| Peripheral baseline biomarkers | |||||
| PLR:screening | 6.198 | 7.986 | 0.211 | 0.024 | 1.415 |
| Lymphocytes (cells/μL):screening | 1303.462 | 987.109 | 0.161 | 0.015 | 0.605 |
| %Lymphocytes:C1D1 | 24.150 | 16.123 | 0.002 | 0.010 | 0.620 |
| CD3 (cells/μL):C1D1 | 1000.333 | 801.339 | 0.090 | 0.041 | 0.729 |
| Natural killer cells (cells/μL):C1D1 | 273.500 | 170.220 | 0.041 | 0.034 | 0.753 |
| %Ki67+(3+/4–/8+/45RA+):C1D1 | 1.522 | 0.960 | 0.085 | 0.048 | 0.780 |
| %CD28+/CD95–(CD3+/CD8+):C1D1 | 8.162 | 12.464 | 0.087 | 0.045 | 1.189 |
| %3+/4–/8+/45RA+/62L+:C1D1 | 3.209 | 5.348 | 0.019 | 0.205 | 1.285 |
| IL-6 (pg/mL):C1D1 | 2.176 | 5.007 | 0.035 | 0.011 | 1.335 |
| IL1ra (pg/mL):C1D1 | 170.615 | 247.656 | 0.050 | 0.146 | 1.375 |
| IO-CCL23 (NSCLC, NPX):C1D1 | 9.904 | 10.163 | 0.323 | 0.049 | 2.223 |
| IO-CCL23 (HNSCC, NPX):C1D1 | 11.614 | 12.251 | 0.037 | 0.013 | 2.000 |
| IO-CSF-1 (NSCLC, NPX):C1D1 | 10.410 | 10.575 | 0.143 | 0.012 | 12.375 |
| IO-CSF-1 (HNSCC, NPX):C1D1 | 9.146 | 9.364 | 0.033 | 0.168 | 3.748 |
| IO-PDCD1 (NSCLC, NPX):C1D1 | 4.885 | 4.478 | 0.045 | 0.002 | 0.209 |
| IO-PDCD1 (HNSCC, NPX):C1D1 | 4.640 | 4.265 | 0.160 | 0.036 | 0.504 |
| Peripheral pharmacodynamic biomarkers measured during budigalimab treatment at the cycle and day indicated | |||||
| FC:CD3/Lymphocytes:C1D2 | 0.947 | 0.961 | 0.565 | 0.024 | 0.008 |
| FC:IFN-γ:C1D2 | 3.397 | 1.996 | 0.065 | 0.043 | 0.754 |
| FC:IL8:C1D2 | 1.427 | 2.992 | 0.168 | 0.041 | 1.192 |
| FC:CXCL9:C1D2 | 1.950 | 1.645 | 0.224 | 0.049 | 0.601 |
| logFC:CCL23 (NSCLC):C1D2 | 1.361 | 1.179 | 0.143 | 0.003 | 0.143 |
| logFC:CCL23 (HNSCC):C1D2 | 1.318 | 1.112 | 0.155 | 0.038 | 0.414 |
| FC:%CD25+(CD3+/8+28–/95+):C1D3 | 0.608 | 0.875 | 0.057 | 0.012 | 1.829 |
| FC:%CD3+/CD4+:C1D8 | 0.930 | 0.984 | 0.210 | 0.016 | 6.869 |
| FC:%CD3+/CD8+:C1D8 | 0.941 | 1.029 | 0.120 | 0.044 | 6.745 |
| FC:%CD28+/CD95+(CD3+/CD8+):C1D8 | 0.911 | 0.959 | 0.350 | 0.049 | 9.734 |
| FC:event count lymphocytes:C1D15 | 2.468 | 1.003 | 0.122 | 0.043 | 0.636 |
| FC:event count CD3+:C1D15 | 2.539 | 1.007 | 0.104 | 0.026 | 0.614 |
| FC:event count CD3+/CD4–/CD8+:C1D15 | 2.799 | 0.996 | 0.087 | 0.021 | 0.601 |
| FC:event count CD3+/CD4+/CD8–:C1D15 | 2.210 | 1.011 | 0.119 | 0.042 | 0.650 |
| FC:%CD3+:C1D15 | 1.033 | 0.992 | 0.248 | 0.047 | 0.052 |
| FC:%3+/4–/8+/45RA–/62L–:C1D15* | 1.524 | 1.028 | 0.194 | 0.014 | 0.469 |
| FC:%CD3+/CD4–/CD8+:C1D15 | 1.051 | 0.993 | 0.158 | 0.017 | 0.053 |
| FC:%Ki67+(3+/4–/8+/45RA+):C1D15* | 1.198 | 2.617 | 0.061 | 0.047 | 1.269 |
| FC:eosinophils:C1D15 | 0.998 | 1.170 | 0.062 | 0.015 | 2.661 |
| FC:lactate dehydrogenase:C1D15 | 0.828 | 1.045 | 0.008 | 0.033 | 2.079 |
Peripheral biomarkers include those with a P-value <0.05 for RNR or PFS for which there was no significant indication interaction and the same direction of change in both HNSCC and NSCLC (patients with reported EGFR/ALK mutations excluded). Baseline measurement units are given in parentheses, with log2 normalized protein expression (NPX) given for proximity extension assay results. Pharmacodynamic measurements are given in fold change (FC) or logFC. A total of 74 patients (41 HNSCC and 33 NSCLC) were included in these analyses.
Thirty-nine patients included (21 HNSCC and 18 NSCLC).
Italics values are significant by P<0.05.
– indicates negative; +, positive; ALK, anaplastic lymphoma kinase; C, cycle; CR, complete response; D, day; EGFR, epidermal growth factor receptor; HNSCC, head and neck squamous cell carcinoma; HR, hazard ratio; iPR, immune partial response; NR, nonresponder; NSCLC, non-small cell lung cancer; PFS, progression-free survival; PLR, platelet-lymphocyte ratio; PR, partial response; R, responder; RNR, responder to nonresponder.
Dynamic Blood Biomarkers of Immune Response Associated With Clinical Response
Budigalimab treatment has been observed to modulate multiple peripheral pharmacodynamic biomarkers. As previously reported, complete (>95%) PD-1 receptor saturation on CD4 T central memory (CD28+CD95+) cells was rapidly detected in all budigalimab-treated patients.9 Induction of the proliferation marker Ki67 in CD4 and CD8 T cells suggested PD-1 receptor blockade led to the reinvigoration of a subset of experienced T cells, with similar peak induction at 2 weeks on treatment observed in both responders and nonresponders (Fig. 4A).
FIGURE 4.
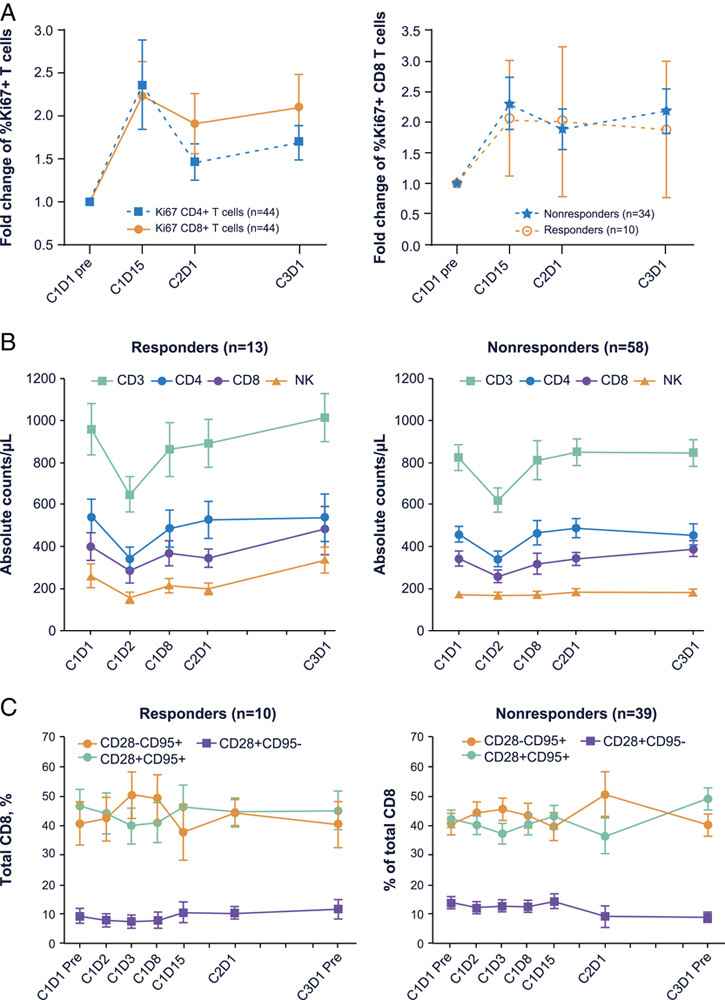
Immunophenotyping detected pharmacodynamic changes induced by budigalimab, including the percentages of Ki67+ CD4 and CD8 T cells [mean and SE shown from a total of 44 patients (24 HNSCC and 20 NSCLC) with baseline and postbaseline Ki67 assessments, with 9 responders] (A), circulating cell counts of CD3–, CD4–, and CD8+ T-cell counts (mean and SE shown from a total of 71 patients with baseline and postbaseline total T cells, helper T cells, cytotoxic T cells, B cells, and NK cells assessments, with 13 responders) (B), and percentages of CD8 T-cell subsets on the basis of CD28 and CD95 expression (mean and SE shown from a total of 49 patients with baseline and postbaseline memory subset assessments, with 10 responders) (C). – indicates negative; +, positive; C, cycle; D, day; HNSCC, head and neck squamous cell carcinoma; mem, memory; NK, natural killer; NSCLC, non–small cell lung cancer.
Immunophenotyping also identified a transient drop in overall peripheral T-cell counts, observed at 24 hours after initial dosing in both responders and nonresponders, which recovered by 1–2 weeks in the treated population (Fig. 4B). Univariate analysis found associations with PFS, with responders demonstrating a larger drop in the CD3 population at 24 hours (0.947 in responders, 0.961 in nonresponders), and a more prolonged delay in the recovery of peripheral CD4 (0.93 for responders vs. 0.984 for nonresponders at 1 wk) and CD8 (0.941 for responders vs. 1.029 for nonresponders at 1 wk) T-cell populations (Table 2). This transient drop in peripheral T-cell memory subsets as defined by CD28/CD95 expression affected naive (CD28+CD95–) and central memory (CD28+CD95+) T-cell subsets more than effector memory (CD28–CD95+) T-cell subsets (Fig. 4C). By C1D15, an expansion of overall lymphocyte counts, CD4 and CD8 T-cell counts, and percentage of CD8+CD45RA–CD62L– effector memory T cells was observed in responders and associated with longer PFS (Table 2). In contrast, an expansion in eosinophil counts or in the percentage of Ki67+CD8+CD45RA+ naive T cells was associated with shorter PFS (Table 2).
We previously reported the induction of CXCL9 (MIG) and CXCL10 (IFN-γ–induced protein-10) by budigalimab.9 Here, we report additional soluble biomarker changes as a consequence of treatment with budigalimab and their associations with response. A broad evaluation of soluble biomarkers were investigated using a proximity extension assay, with relative changes in selected biomarkers correlating well with established detection methods for IFN-γ, IL-6, IL-8, CXCL9, and CXCL10. We detected a rapid significant induction of IFN-γ, IL-6, and CCL19 peaking at 24 hours along with rises in the IFN-γ–induced chemokines CXCL9, CXCL10, and CXCL11 and the soluble costimulatory molecules TNFRSF4 (OX40) and TNFRSF9 (4-1BB) that peaked at 4 week (Figs. 5A–C). No cytokine release syndrome (CRS) was reported in the phase 1 study, though increases in IL-6 and TNF-α at early time points were observed. Multiple novel biomarker movements were observed including early repression of soluble CD40 ligand (sCD40L) and the angiogenic factors EGF and ANGPT1 as well as later upregulation of CRTAM, GZMH, TNFRSF4, and TNFRSF9 (Supplemental Digital Content Figs. 5A, B, Supplemental Digital Content 1, http://links.lww.com/JIT/A666). In summary, novel induced biomarkers peaked as follows: CCL19 at C1D2; CXCL11, TNFRSF9, TNFRSF4, IL-12, MCP-3, GZMB, GZMH, CCL3, and CXCL13 at C2D1; and CRTAM, PTN, and PDCD1 at C4D1. Novel repressed biomarkers were detected as follows: EGF, ANGPT1, MCP-4, CD40L, and PDCD1 at C1D2. We also observed IL-10 upregulation at C1D2, and PDCD1 at C2D1. While a transient drop in PDCD1 was detected immediately after initial dosing at C1D2, by C2D1 posttreatment this biomarker was significantly upregulated.
FIGURE 5.
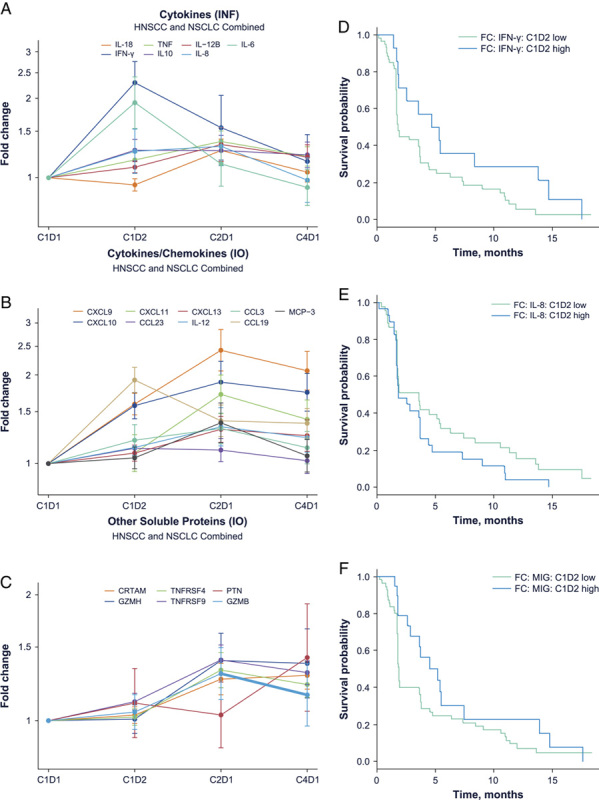
Budigalimab treatment induced soluble biomarkers of T-cell activity associated with differential response. Data shown are from a total of 65 patients with baseline and postbaseline assessments including 12 responders and 43 nonresponders. Longitudinal analysis of significantly upregulated biomarkers across all patients shows IFN-γ peak expression at the C1D2 time point (A), while CXCL9 (MIG), CXCL10, CXCL11, GZMH, TNFRSF4, and TNFRSF9 peak at the C2D1 time point (B, C). Significant PFS differences were observed between patients with the highest and lowest induction at C1D2 of IFN-γ (D), IL-8 (E), or CXCL9 (MIG) (F). C indicates cycle; D, day; FC, fold change; HSNCC, head and neck squamous cell carcinoma; IFN, interferon; IL, interleukin; INF, inflammatory; IO, immuno-oncologic; MIG, mitogen-inducible gene; NSCLC, non–small cell lung cancer; TNF, tumor necrosis factor.
Larger increases in IFN-γ and CXCL9 (MIG) at 24 hours were associated with better PFS, while increases in IL-8 at 24 hours were associated with worse PFS (Figs. 5D–F, Table 2). Increases in CCL23 were also associated with better PFS (Table 2). While multiple soluble biomarker movements were observed at time points later than C1D2, univariate analysis did not find these to be significantly correlated with response or PFS.
DISCUSSION
This biomarker analysis used archival tumor samples and pharmacodynamic blood sampling from patients with HNSCC and NSCLC enrolled in a phase 1 trial of budigalimab to confirm several previous findings relating to biomarkers of response to anti–PD-1 monoclonal antibody treatment. Consistent with other PD-1 inhibitors, budigalimab resulted in transient decreases in T-cell counts, enhanced T-cell proliferation, and increased CXCL9/CXCL10 levels, as previously reported for both pembrolizumab and nivolumab.18–20 In addition, we present several novel findings relating to the identification and expression of novel baseline and budigalimab treatment-modulated biomarkers that may associate with clinical outcomes. These biomarker findings confirm that budigalimab releases checkpoint inhibition of activated T cells and enhances T-cell activity.
Multiple reports have previously elucidated an association between tumor-specific biomarkers and clinical response to PD-1 blockade in advanced solid tumors such as HNSCC and NSCLC, including TMB, neoantigen burden, PD-L1 expression, CD8 infiltration, and IFN-γ–related mRNA profile.5,7,21–24 The present findings confirm earlier reports regarding higher TMB and neoantigen load as tumor biomarkers,22,23 with a higher likelihood of response associated with higher TMB, as well as higher PD-L1 expression predicting better response in NSCLC.21 Similar to previously published reports with other anti–PD-1 therapies, NSCLC patients with tumors that harbor EGFR-activating mutations and ALK rearrangements reported a lower response rate.25 Unsurprisingly, overall tumor size at enrollment was negatively correlated with response. However, in this study we were unable to confirm association of a 20-gene IFN-γ–related signature (the immune constant of rejection) with response, as has been reported using a 6-gene IFN-γ signature on NanoString data.5,23 Despite this limitation, our analysis revealed higher IFN-γ gene-related signatures in HPV+ patients with HNSCC, consistent with published findings.26 Archival samples may not be an ideal source of tumor tissue for analyses of immune infiltration, whether IHC or transcriptome based, as these may change over time with intervening treatments between biopsy and start of treatment. Consequently, a single IHC or RNAseq snapshot to profile a patient’s tumor biomarkers may have limited utility.
In our patient cohort we observed evidence that novel tumor-signaling pathways were associated with response to PD-1 blockade in HNSCC or NSCLC. In HNSCC, activation of fatty acid beta-oxidation pathways was positively correlated with response, while activation of osteoarthritis and IL-6 signaling pathways were negatively correlated with response. This corresponds with the negative association of high serum IL-6 with response. In addition, some genes showed association with response regardless of tumor indication. For instance, enhanced expression of EDIL3 (an integrin ligand) correlated with both better response and PFS, while enhanced expression of ITGA2B, CDH2, and CLU correlated with better response but not PFS. Expression of these cell adhesion factors may reflect a vascularized tumor stromal environment conducive to immune cell infiltration.27,28 In contrast, enhanced expression of chemokine receptors and factors such as CXCL1, CCL20, CCR7, IL1B, and IL1R2 are associated with nonresponse in these patients. These factors can be associated with tumor-associated macrophages, neutrophils, and regulatory T cells that promote tumor survival.29
Patients enrolled in phase 1 studies with multiple prior therapies may have suboptimal immune status. In this regard, systemic measures of immune status through routine laboratory tests (eg, neutrophils, NLR, cholesterol) together with custom lymphocyte profiling techniques can provide an indication of immune fitness. Higher NLR and platelet-lymphocyte ratio in patients has been associated with worse clinical outcomes.30,31 Other baseline peripheral biomarkers of immune status have been associated with better clinical response to PD-1 blockade, and include increased frequency of memory CD4+ T cells, decreased frequency of PD-L1+ NK cells and naive CD4+ T cells, and lower PD-1 expression on CD4+ T cells.6,8,16 The present findings confirm some of these earlier reports, with better immune fitness as indicated by greater lymphocyte, T-cell, and NK cell counts, as well as higher memory T cell counts associated with response to checkpoint inhibition with budigalimab in this study. Consistent with previous reports, baseline peripheral blood biomarkers associated with worse outcome to anti–PD-1 treatment were low lymphocyte numbers and high IL-6 levels, with the latter a marker of increased inflammation that is correlated with worse outcomes in HNSCC, NSCLC, and other solid tumors.32,33
In this study, we found additional serum cytokine levels that were associated with worse outcomes to anti–PD-1 treatment regardless of NSCLC or HNSCC indication. Higher CSF-1, IL1Rα, and CCL23 levels were noted in nonresponders. High CSF-1 could indicate more tumor-associated macrophage involvement, and interestingly, disruption of this pathway in combination with PD-1 pathway blockade in animal models improved tumor responses.34 In contrast, we observed a novel higher baseline PDCD1 level in responders from both indications, in HNSCC associated with PFS and in NSCLC associated with both BOR and PFS. PDCD1, the soluble form of PD-1, is also upregulated upon T-cell activation and can block PD-L1 interactions to promote antitumor immunity. Higher levels of this soluble form of PD-1 may indicate more engagement of the PD-1/PD-L1 pathway of tumor escape from immune control, hence the potential to benefit from PD-1 blockade.
We also observed differential baseline levels of serum proteins in NSCLC responders versus nonresponders, including increased levels of three chemokine ligands for CCR5: MCP-2 (aka CCL8), CCL11, and MCP-4 (aka CCL13) (Supplemental Digital Content Table 2, Supplemental Digital Content 1, http://links.lww.com/JIT/A666); CCR5 is highly expressed on T lymphocytes as well as macrophages, dendritic cells, and other cell types, and higher expression of these CCR5 ligands in responders may be reflective of an immune environment conducive to appropriate T-cell trafficking.
Following initiation of budigalimab treatment, changes in circulating T cells were rapidly detectable. We observed a transient drop in T-cell counts at 24 hours, which recovered by 1 week, as previously reported for other PD-1 inhibitors.18 This drop is potentially due to T-cell tissue redistribution, as following PD-1 blockade, novel T-cell clonotypes are reported to enter the tumor.35 It is interesting to note that different T-cell subtypes behaved differently in the first week, with circulating percentages of CD28–CD95+ effector memory cells expanding despite a contraction in circulating CD28+CD95+ central memory and CD28+CD95– naive cells. This is consistent with recently reported findings on early effector memory expansion in PD-1 inhibitor-treated patients.36 Invigoration of T cells was associated with a peak in T-cell proliferation, as measured by Ki67 at 2–4 weeks posttreatment, as reported for other PD-1 inhibitors,37,38 though this pharmacodynamic activity did not associate directly with clinical outcomes in our study.
The rapid, early peaking induction of IFN-γ observed together with minimal induction of IL-6 and TNF-α was consistent with the absence of CRS and low incidence of CRS issues in general with PD-1 inhibitors. CXCL9, CXCL10, and CXCL11 induction was observed in line with reports by other investigators.19–21 These are ligands for CXCR3 found highly expressed on TH1 CD4 and effector CD8 T cells and NK cells that play important roles in T-cell trafficking and function. In addition, we observed a rapid induction of CCL19, which can be produced by fibroblasts within the TME and enhance recruitment of CD8+ T cells to the tumor.39 In both HNSCC and NSCLC, elevated angiogenic factors have been reported as negative prognostic indicators.40,41 Our novel finding of rapid, early repression of factors involved in angiogenesis along with sCD40L repression could be a potential measure of tumor site activity, though these changes were not significantly associated with clinical outcomes. Increased levels of soluble TNFRSF4 (OX40), TNFRSF9 (4-1BB), GZMB, GZMH, and CRTAM observed in our study to reach peak sustained levels at C2D1 are consistent with enhancement of T-cell activity over time, as these are costimulatory and effector molecules upregulated by activated T cells and consistent with the observation that production of GZMA and sCD25 was associated with checkpoint blockade.19 The observed upregulation of PDCD1 at C2D1 in this study is consistent with early reports from PD-1 inhibitor-treated melanoma patients.42 This may suggest increased persistence of PD-1+ T cells in the presence of PD-1 blockade or reflect decreased metabolism of the soluble receptor when complexed with budigalimab.
This biomarker study also evaluated whether pharmacodynamic biomarkers of early immune activation associated with response. Early changes in soluble biomarkers (IFN-γ and CXCL9) were noted within 24 hours and correlated with improved PFS. In contrast, a strong increase in IL-8 at 24 hours correlated with worse PFS. This correlates with reports that melanoma patients responding to checkpoint inhibitors have increased serum CXCL9 and that high serum IL-8 is negatively associated with checkpoint inhibitor outcomes in NSCLC.43,44 Changes in lymphocyte populations and counts were observed within the first 2 weeks of treatment with budigalimab, with enhanced T-cell counts and particularly enhanced CD8 effector memory (CD45RA–CD62L–) at C1D15 associated with improved PFS. Others have shown an important role for the memory/effector T-cell population in response to cytotoxic T lymphocyte-associated protein 4 (CTLA-4) inhibitors and the NK cell subset in response to PD-1 inhibitors.45 In a study of patients treated with another PD-1 inhibitor, proliferative response of peripheral blood PD-1+/CD8+ T cells measured 7 days after treatment was identified as a potential biomarker for predicting response in patients with NSCLC.46 While a proliferative response was noted in CD8+ T cells in our study, it was not associated with clinical response.
In conclusion, elucidating the determinants of response to anti–PD-1 treatment in patients with advanced solid cancers such as HNSCC and NSCLC is key to optimal patient selection and appropriate therapeutic strategies to improve outcomes. In addition, studying predictive and PD biomarkers allows better understanding of the complex mechanisms involved with PD-1 inhibition. This biomarker analysis used archival tumor and longitudinal peripheral blood samples from patients enrolled in a phase 1 study of budigalimab to identify baseline and pharmacodynamic biomarkers associated with clinical outcomes. Patients in this study were heavily pretreated and numbers were small, but consistencies with previous reports of biomarker associations with clinical outcomes to PD-1 blockade therapy were observed. These observations highlight the usefulness of signals for peripheral blood as an emerging source of future predictive biomarkers of response to checkpoint inhibitors.
Novel biomarker associations observed in this study should be evaluated in other tumor indications with PD-1 inhibitor therapy to determine the breadth of applicability. Further evaluation of these biomarkers in the budigalimab clinical development program is ongoing.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.
ACKNOWLEDGMENTS
AbbVie and the authors thank the patients who participated in this clinical trial, the study coordinators, physicians, and support staff. They would also like to acknowledge Betty Wang for assistance with biomarker operations and setup, Merriam McClellan for assistance with flow assay validation and transfer, Kinjal Hew and Mary Saltarelli for assistance with vendor management, and Yan Sun for assistance with assay statistical validation. Medical writing support was provided by Mary L. Smith, PhD, CMPP, Aptitude Health, Atlanta, GA, funded by AbbVie.
Conflicts of Interest/Financial Disclosures
AbbVie Inc. provided financial support for the study (NCT03000257) and participated in the design, study conduct, as well as the writing, review, and approval of the manuscript. Data analysis and interpretation was conducted by employees of AbbVie Inc.
S.L.L., C.Z., C.G., T.T., D.L.M., S.E., Y.F., J.S., R.T.M., C.T., and D.A.: AbbVie employees and may own stock. G.V.: former employee of AbbVie and may own stock.
Footnotes
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Contributor Information
Stacie L. Lambert, Email: stacie.lambert@abbvie.com.
Chun Zhang, Email: chun.zhang@abbvie.com.
Claire Guo, Email: claire.guo@abbvie.com.
Tolga Turan, Email: tolga.turan@abbvie.com.
David L. Masica, Email: david.masica@abbvie.com.
Stefan Englert, Email: stefan.englert@abbvie.com.
Yuni Fang, Email: yuni.fang@AbbVie.com.
James Sheridan, Email: james.sheridan@AbbVie.com.
Robert Tyler McLaughlin, Email: robert.mclaughlin@abbvie.com.
Catherine Tribouley, Email: catherine.tribouley@abbvie.com.
Greg Vosganian, Email: gregvosganian@gmail.com.
Daniel Afar, Email: daniel.afar@abbvie.com.
REFERENCES
- 1.Weng YM, Peng M, Hu MX, et al. Clinical and molecular characteristics associated with the efficacy of PD-1/PD-L1 inhibitors for solid tumors: a meta-analysis. Onco Targets Ther. 2018;11:7529–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prasad V, Kaestner V. Nivolumab and pembrolizumab: monoclonal antibodies against programmed cell death-1 (PD-1) that are interchangeable. Semin Oncol. 2017;44:132–135. [DOI] [PubMed] [Google Scholar]
- 3.Ancevski Hunter K, Socinski MA, Villaruz LC. PD-L1 testing in guiding patient selection for PD-1/PD-L1 inhibitor therapy in lung cancer. Mol Diagn Ther. 2018;22:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voong KR, Feliciano J, Becker D, et al. Beyond PD-L1 testing—emerging biomarkers for immunotherapy in non-small cell lung cancer. Ann Transl Med. 2017;5:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prat A, Navarro A, Paré L, et al. Immune-related gene expression profiling after PD-1 blockade in non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res. 2017;77:3540–3550. [DOI] [PubMed] [Google Scholar]
- 7.Yarchoan M, Albacker LA, Hopkins AC, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight. 2019;4:e126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng H, Liu X, Zhang J, et al. Expression of PD-1 on CD4+ T cells in peripheral blood associates with poor clinical outcome in non-small cell lung cancer. Oncotarget. 2016;7:56233–56240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Italiano A, Cassier PA, Lin CC, et al. First-in-human phase 1 study of budigalimab, an anti-PD-1 inhibitor, in patients with non-small cell lung cancer and head and neck squamous cell carcinoma. Cancer Immunol Immunother. 2021. Available at: https://link.springer.com/article/10.1007%2Fs00262-021-02973-w#citeas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143–e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. [DOI] [PubMed] [Google Scholar]
- 12.Gettinger SN, Horn L, Gandhi L, et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2015;33:2004–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–965. [DOI] [PubMed] [Google Scholar]
- 14.Vaidyanathan R, Soon RH, Zhang P, et al. Cancer diagnosis: from tumor to liquid biopsy and beyond. Lab Chip. 2018;19:11–34. [DOI] [PubMed] [Google Scholar]
- 15.Sanmamed MF, Perez-Gracia JL, Schalper KA, et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann Oncol. 2017;28:1988–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Juliá EP, Mandó P, Rizzo MM, et al. Peripheral changes in immune cell populations and soluble mediators after anti-PD-1 therapy in non-small cell lung cancer and renal cell carcinoma patients. Cancer Immunol Immunother. 2019;68:1585–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendrickx W, Simeone I, Anjum S, et al. Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis. Oncoimmunology. 2017;6:e1253654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das R, Verma R, Sznol M, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194:950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choueiri TK, Fishman MN, Escudier B, et al. Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma. Clin Cancer Res. 2016;22:5461–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362:eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu-Lieskovan S, Lisberg A, Zaretsky JM, et al. Tumor characteristics associated with benefit from pembrolizumab in advanced non-small cell lung cancer. Clin Cancer Res. 2019;25:5061–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berghoff AS, Bellosillo B, Caux C, et al. Immune checkpoint inhibitor treatment in patients with oncogene-addicted non-small cell lung cancer (NSCLC): summary of a multidisciplinary round-table discussion. ESMO Open. 2019;4:e000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gameiro SF, Ghasemi F, Barrett JW, et al. Treatment-naïve HPV+ head and neck cancers display a T-cell-inflamed phenotype distinct from their HPV- counterparts that has implications for immunotherapy. Oncoimmunology. 2018;7:e1498439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harjunpää H, Asens ML, Guenther C, et al. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front Immunol. 2019;10:1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeong D, Ban S, Oh S, et al. Prognostic significance of EDIL3 expression and correlation with mesenchymal phenotype and microvessel density in lung adenocarcinoma. Sci Rep. 2017;7:8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Argyle D, Kitamura T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front Immunol. 2018;9:2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diem S, Schmid S, Krapf M, et al. Neutrophil-to-lymphocyte ratio (NLR) and platelet-to-lymphocyte ratio (PLR) as prognostic markers in patients with non-small cell lung cancer (NSCLC) treated with nivolumab. Lung Cancer. 2017;111:176–181. [DOI] [PubMed] [Google Scholar]
- 31.Song X, Chen D, Yuan M, et al. Total lymphocyte count, neutrophil-lymphocyte ratio, and platelet-lymphocyte ratio as prognostic factors in advanced non-small cell lung cancer with chemoradiotherapy. Cancer Manag Res. 2018;10:6677–6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choudhary MM, France TJ, Teknos TN, et al. Interleukin-6 role in head and neck squamous cell carcinoma progression. World J Otorhinolaryngol Head Neck Surg. 2016;2:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silva EM, Mariano VS, Pastrez PRA, et al. High systemic IL-6 is associated with worse prognosis in patients with non-small cell lung cancer. PLoS One. 2017;12:e0181125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y, Yang J, Xu D, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitizes hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68:1653–1666. [DOI] [PubMed] [Google Scholar]
- 35.Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25:1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valpione S, Galvani E, Tweedy J, et al. Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer. 2020;1:210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang AC, Postow MA, Orlowski RJ, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamphorst AO, Pillai RN, Yang S, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017;114:4993–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng HW, Onder L, Cupovic J, et al. CCL19-producing fibroblastic stromal cells restrain lung carcinoma growth by promoting local antitumor T-cell responses. J Allergy Clin Immunol. 2018;142:1257–1271. [DOI] [PubMed] [Google Scholar]
- 40.Bremnes RM, Camps C, Sirera R. Angiogenesis in non-small cell lung cancer: the prognostic impact of neoangiogenesis and the cytokines VEGF and bFGF in tumours and blood. Lung Cancer. 2006;51:143–158. [DOI] [PubMed] [Google Scholar]
- 41.Khademi B, Soleimanpour M, Ghaderi A, et al. Prognostic and predictive value of serum vascular endothelial growth factor (VEGF) in squamous cell carcinoma of the head and neck. Oral Maxillofac Surg. 2014;18:187–196. [DOI] [PubMed] [Google Scholar]
- 42.Mehta A, Rucevic M, Sprecher E, et al. The use of plasma proteomic markers to understand the biology of immunotherapy response. J Clin Oncol. 2020;38(15 suppl):517s. [Google Scholar]
- 43.Chow MT, Ozga AJ, Servis RL, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. 2019;50:1498–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oyanagi J, Koh Y, Sato K, et al. Predictive value of serum protein levels in patients with advanced non-small cell lung cancer treated with nivolumab. Lung Cancer. 2019;132:107–113. [DOI] [PubMed] [Google Scholar]
- 45.Subrahmanyam PB, Dong Z, Gusenleitner D, et al. Distinct predictive biomarker candidates for response to anti-CTLA-4 and anti-PD-1 immunotherapy in melanoma patients. J Immunother Cancer. 2018;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim KH, Cho J, Ku BM, et al. The first-week proliferative response of peripheral blood PD-1+CD8+ T cells predicts the response to anti-PD-1 therapy in solid tumors. Clin Cancer Res. 2019;25:2144–2154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.