

Abstract
The antibiotic linezolid, the first clinically approved member of the oxazolidinone class, inhibits translation of bacterial ribosomes by binding to the peptidyl transferase center. Recent work has demonstrated that linezolid does not inhibit peptide bond formation at all sequences but rather acts in a context-specific manner, namely when alanine occupies the penultimate position of the nascent chain. However, the molecular basis for context-specificity has not been elucidated. Here we show that the second-generation oxazolidinone radezolid also induces stalling with a penultimate alanine, and we determine high-resolution cryo-EM structures of linezolid- and radezolid-stalled ribosome complexes to explain their mechanism of action. These structures reveal that the alanine side chain fits within a small hydrophobic crevice created by oxazolidinone, resulting in improved ribosome binding. Modification of the ribosome by the antibiotic resistance enzyme Cfr disrupts stalling due to repositioning of the modified nucleotide. Together, our findings provide molecular understanding for the context-specificity of oxazolidinones.
During translation, peptide bond formation occurs in the peptidyl transferase center (PTC) of the ribosome. The PTC is located within the large ribosomal subunit and catalyzes extension of the polypeptide chain through proper positioning of the peptidyl-tRNA in the P site and aminoacyl tRNA in the A site. Owing to its functional importance, the PTC of the bacterial ribosome is a common target for antibiotics that inhibit translation1,2.
The PTC antibiotic linezolid (LZD, Fig. 1a) was the first clinically approved member of the synthetic oxazolidinone class of antibiotics3. LZD is used to treat drug-resistant Gram-positive infections, including those caused by methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococci4. Initial crosslinking experiments identified the binding site for LZD within the PTC A site only when performed with actively translating ribosomes5,6, suggesting that other translation components may be involved in LZD binding. However, existing structures of LZD-bound ribosomes have been obtained with ribosomes devoid of charged transfer RNAs (tRNAs)7,8 or ribosomes containing a P site tRNA lacking a nascent chain9. Recent evidence obtained through ribosome profiling and single-molecule studies demonstrated that LZD does not indiscriminately inhibit the formation of every peptide bond, but rather interferes with translation at certain messenger RNA (mRNA) sites10,11. Robust inhibition of translation and ribosome stalling by LZD is strongly favored when the amino acid alanine occupies the penultimate, (−1), position within the nascent chain. Together, these results suggest that interactions between LZD and the nascent peptide may be important for stabilizing antibiotic binding to the ribosome. However, the exact nature of these interactions is yet to be elucidated.
Fig. 1 |. Radezolid induces ribosome stalling with alanine in the penultimate position.
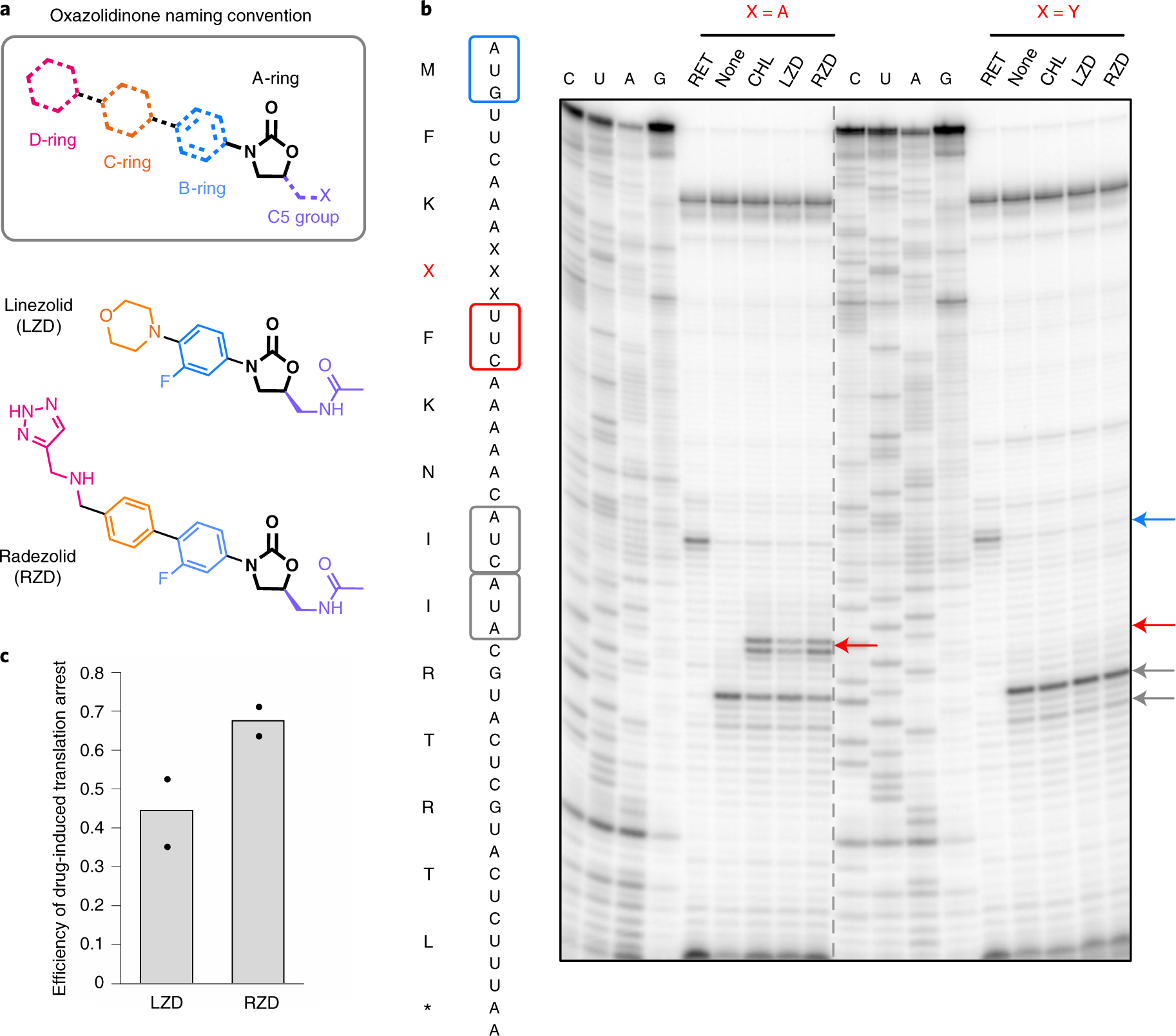
a, Molecular architecture of oxazolidinone antibiotics. The oxazolidinone (A-ring) portion of the molecule is conserved amongst the oxazolidinone class and is shown in black. Chemical moieties that vary among the oxazolidinone derivatives, the C5 moiety and the B, C and D rings are designated by their respective color. b, Toeprinting assays performed on model mRNAs encoding stalling and non-stalling peptides. The control antibiotic retapamulin (RET) was used to stall ribosomes at the start codon, indicated by the blue arrow51. ‘None’ designates reactions lacking ribosome-targeting antibiotics. The control antibiotic CHL was used to stall with alanine at the penultimate position10,11. The toeprint bands corresponding to prominent stall sites observed in reactions containing CHL, LZD or RZD when the fourth amino acid is an alanine, but not tyrosine, are indicated by the red arrow. Owing to the inclusion of the Ile-tRNA synthetase inhibitor mupirocin in all toeprinting reactions, any ribosomes not stalled at an upstream codon are trapped at the downstream Ile codons designated by gray arrows. All antibiotics were added to a final concentration of 50 μM. The presented gel is representative of two independent experiments. c, The extent of translation inhibition by LZD and RZD calculated as the ratio of intensities of the drug-specific toeprint bands (red arrow, b) to the sum of intensities of the drug-specific and trap codon bands (gray arrows, b). The bar graph shows the mean of two independent experiments, with individual data points indicated by black dots.
Despite the clinical success of LZD, emerging resistance mechanisms have threatened its clinical utility. Such mechanisms of resistance include alteration of ribosomal RNA (rRNA) and ribosomal proteins12,13 and ribosome protection through ABC-F proteins14. A prevalent form of transferable resistance to LZD is the rRNA-modifying enzyme Cfr, which methylates the ribosome within the PTC to perturb antibiotic binding15–17. Emerging resistance has sparked the development of second-generation oxazolidinone derivatives, such as radezolid (RZD), with improved potency18. RZD (Fig. 1a) is in clinical development for application to bacterial acne and community-acquired pneumonia19. Compared to LZD, RZD retains the aryl-oxazolidinone core (rings A and B) and C5 group, but has alterations to the C/D-ring system. Given that key chemical elements are conserved between LZD and RZD, it is plausible that RZD can also act as a context-specific inhibitor of translation. The importance of both conserved and distinct structural features of second-generation oxazolidinones to translation inhibition and context-specificity, if any, is yet to be determined.
In this work we show that RZD exhibits ribosome stalling behavior similar to that of LZD, arresting translating ribosomes when alanine occupies the penultimate position within the nascent peptide chain. Capitalizing on the stalling preference of these two antibiotics, we generated high-resolution cryo-EM structures of LZD- and RZD-stalled ribosome complexes. Direct comparison of the drug-stalled translation complexes with structures of vacant ribosomes bound to the same antibiotics enabled identification of molecular contacts that improve oxazolidinone binding to the ribosome. Specifically, we find that the penultimate alanine fits within a shallow hydrophobic pocket created by the oxazolidinone molecules, providing a structural rationale for context-specificity and highlighting the role of the growing peptide chain in antibiotic binding. Our analysis of RZD action on ribosomes modified by the oxazolidinone resistance enzyme Cfr and the structure of RZD bound to the Cfr-modified ribosome provides insights into how this second-generation oxazolidinone interacts with an LZD-resistant ribosome.
Results
LZD and RZD have similar ribosome stalling behavior.
Previous in vitro toeprinting experiments have demonstrated that LZD induces ribosome stalling on a model mRNA sequence when the amino acid alanine is located in the penultimate (−1) position of the nascent polypeptide chain10,11 (Fig. 1b). LZD-induced ribosome stalling is abolished when the penultimate alanine is replaced with tyrosine (Fig. 1b). To evaluate whether the second-generation oxazolidinone RZD shows similar stalling behavior, we carried out in vitro toeprinting analysis to monitor the position of stalled ribosomes on the previously described mRNAs encoding the peptide sequences MFKAFKNIIRTRTL and MFKYFKNIIRTRTL11.
Similarly to LZD, the presence of RZD permits the formation of the first peptide bond with both mRNA transcripts as templates (Fig. 1b). This result indicates that RZD is likely not a universal inhibitor of translation and also may not act as an initiation inhibitor as suggested for LZD by some earlier studies20. Although no inhibition of translation at the early mRNA codons was observed on the MFKYFK-encoding template, the presence of RZD or LZD led to selective stalling of the ribosome during translation of the MFKAFK-encoding template. The stalling occurred at the F5 codon when an alanine residue was in the penultimate position within the nascent peptide chain. These results indicate that RZD- or LZD-bound ribosomes are unable to catalyze peptide bond formation between F5 and K6 when the MFKAF nascent peptide occupies the exit tunnel. Comparison of the relative intensity of the stalled ribosome toeprint bands suggests that RZD is a stronger inducer of ribosome stalling than LZD (Fig. 1c). The observation that RZD is unable to stall the ribosome if the critical alanine residue is replaced with a tyrosine (MFKYF) is consistent with the specificity of action of LZD (Fig. 1b).
Formation of stalled ribosome complexes and cryo-electron microscopy analysis.
Guided by the in vitro stalling behavior, we designed a peptide to capture LZD- and RZD-stalled ribosome complexes (SRCs) for structural analysis, herein referred to as LZD-SRC and RZD-SRC, respectively. Stalled complexes were generated by conducting coupled in vitro transcription–translation reactions with Escherichia coli ribosomes in the presence of oxazolidinone antibiotics (Fig. 2a). To favor formation of the desired SRCs, we designed the template such that the open reading frame encoding the MFKAF stalling peptide was truncated after the F5 codon (Fig. 2a and Supplementary Fig. 1). The 70S SRCs were purified away from other components of the translation reaction by sucrose gradient fractionation and vitrified on carbon grids for cryo-EM analysis. Refinement and reconstruction were performed using the cisTEM software suite21 to obtain the 2.5 Å resolution structure of LZD-SRC and 2.5 Å resolution of RZD-SRC (Fig. 2b, Table 1 and Extended Data Fig. 1).
Fig. 2 |. Cryo-EM structures of LZD- and RZD-stalled ribosome complexes.
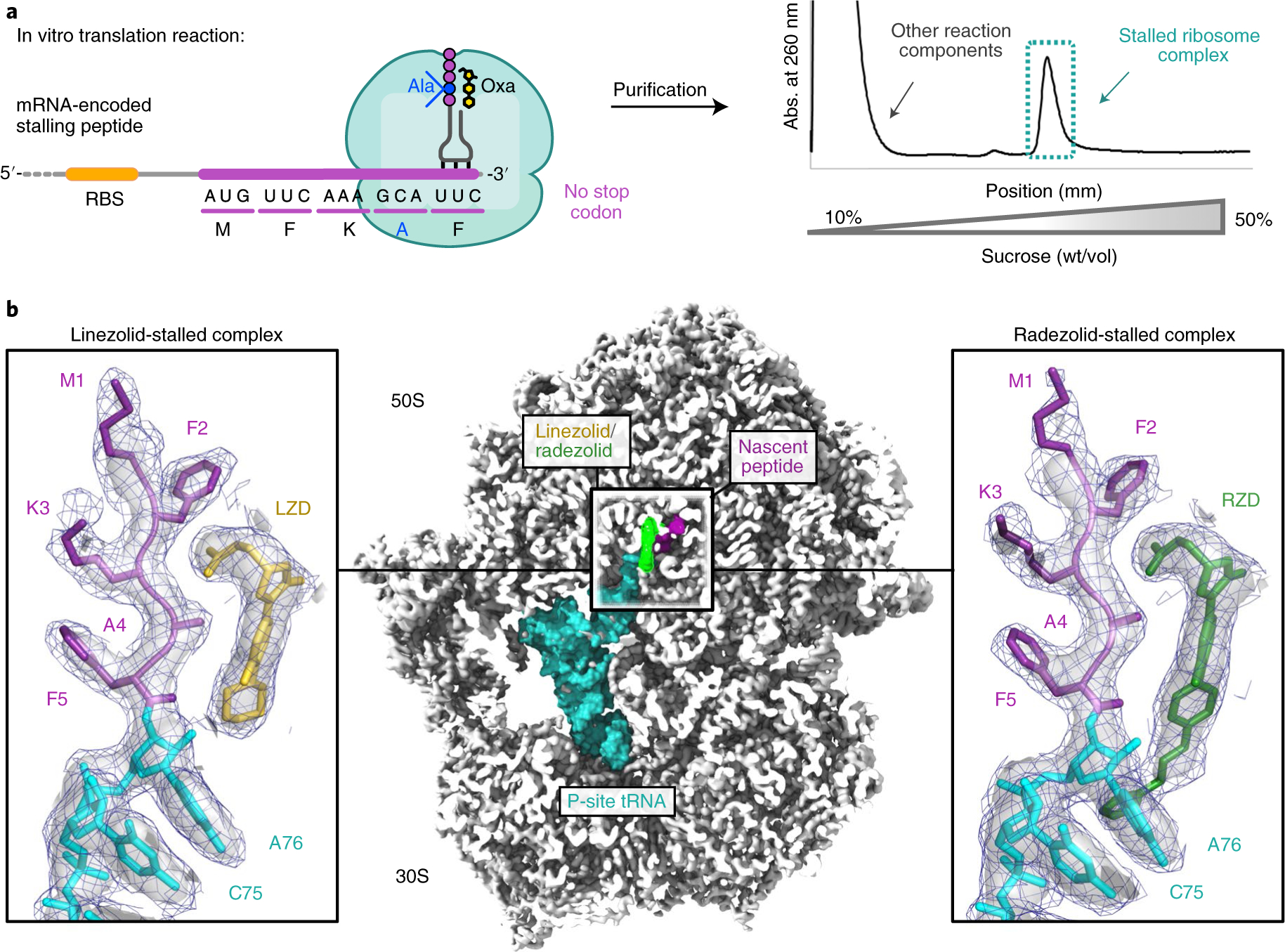
a, Stalled complexes were generated by performing coupled in vitro transcription–translation reactions in the presence of the oxazolidinone (Oxa) antibiotic LZD or RZD. Complexes were further purified by sucrose gradient fractionation. b, Cross-section of the cryo-EM density map of the stalled 70S ribosome in complex with peptidyl-tRNA and oxazolidinone. Insets: close-up views of LZD or RZD in complex with the MFKAF nascent peptide. Coulomb potential density is contoured at 4.0σ in the surface representation and 1.0σ in the mesh representation. The figure was prepared using unsharpened cryo-EM density maps.
Table 1 |.
Cryo-EM data collection, refinement and validation statistics
| LZD-SRC (EMDB-24800, PDB 7S1G) | LZD-70S (EMDB-24801, PDB 7S1H) | RZD-SRC (EMDB-24802, PDB 7S1I) | RZD-70S (EMDB-24803, PDB 7S1J) | RZD-SRC* (EMDB-24804, PDB 7S1K) | |
|---|---|---|---|---|---|
| Data collection and processing | |||||
| Facility and electron | S2C2 | S2C2 | NCCAT | NCCAT | UCSF |
| microscope | Titan Krios | Titan Krios | Titan Krios | Titan Krios | Titan Krios |
| Camera | Gatan K3 | Gatan K3 | Gatan K2 Summit | Gatan K2 Summit | Gatan K3 |
| Magnification | 29,000 | 29,000 | 105,000 | 105,000 | 105,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 |
| Electron exposure (e−/Å2) | 67.8 | 67.8 | 52.8 | 52.8 | 69.0 |
| Defocus range (μm) | 0.3–0.8 | 0.3–0.8 | 0.5–1.5 | 0.5–1.5 | 0.3–0.8 |
| Pixel size (Å) | 0.8125 | 0.8125 | 0.8250 | 0.8250 | 0.8261 |
| Energy filter slit width (eV) | N/a | N/a | 20 | 20 | 20 |
| Symmetry imposed | C1 | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 332,502 | 441,587 | 292,882 | 179,529 | 369,975 |
| Final particle images (no.) | 176,779 | 360,239 | 240,596 | 146,245 | 276,799 |
| Map resolution (Å) | 2.48 | 2.35 | 2.48 | 2.47 | 2.42 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 2.2–4 | 2.2–4 | 2.2–4 | 2.2–4 | 2.2–4 |
| Refinement | |||||
| Initial model used (PDB code) | 6PJ6, 5JU8 | 6PJ6, 5JU8 | 6PJ6, 5JU8 | 6PJ6, 5JU8 | 6PJ6, 5JU8 |
| Model resolution (Å) | 2.48 | 2.35 | 2.48 | 2.47 | 2.42 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Model resolution range (Å) | 2.2–4 | 2.2–4 | 2.2–4 | 2.2–4 | 2.2–4 |
| Map sharpening B factor (Å2) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Model composition | |||||
| Total atoms | 240,937 | 238,267 | 240,938 | 238,218 | 240,967 |
| Hydrogens only | 96,780 | 95,859 | 96,769 | 95,822 | 96,780 |
| Nonhydrogen atoms | 144,157 | 142,408 | 144,169 | 142,396 | 144,187 |
| Protein residues | 5,661 | 5,656 | 5,661 | 5,656 | 5,661 |
| Nucleotides | 4,638 | 4,556 | 4,638 | 4,556 | 4,638 |
| Ligands | 199 | 209 | 212 | 202 | 210 |
| B factors (Å2) | |||||
| Protein | 88.86 | 98.22 | 92.45 | 99.33 | 108.17 |
| Nucleotide | 90.04 | 95.20 | 94.78 | 98.63 | 108.04 |
| Ligand | 65.50 | 68.61 | 69.48 | 75.30 | 70.36 |
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.002 | 0.002 | 0.002 | 0.002 | 0.003 |
| Bond angles (°) | 0.345 | 0.362 | 0.366 | 0.387 | 0.382 |
| Validation | |||||
| MolProbity score | 1.24 | 1.31 | 1.28 | 1.62 | 1.42 |
| Clashscore | 3.64 | 4.63 | 4.32 | 5.58 | 5.46 |
| Poor rotamers (%) | 0.80 | 0.99 | 0.95 | 1.77 | 0.89 |
| Ramachandran plot | |||||
| Favored (%) | 97.57 | 97.64 | 97.68 | 97.33 | 97.32 |
| Allowed (%) | 2.36 | 2.29 | 2.25 | 2.61 | 2.61 |
| Disallowed (%) | 0.07 | 0.07 | 0.07 | 0.05 | 0.07 |
| Nucleic acid geometry | |||||
| Probably wrong sugar puckers (%) | 0 | 0 | 0 | 0 | 0 |
| Bad backbone conformations (%) | 11 | 11 | 11 | 11 | 12 |
| EM ringer score | 3.21 | 3.34 | 3.68 | 2.53 | 2.97 |
The generated cryo-EM maps of LZD-SRC and RZD-SRC have well-defined densities for the oxazolidinone antibiotic and rRNA nucleotides, especially those within the PTC. Both cryo-EM maps also have distinct densities for the peptidyl-tRNA located in the P site. No A-site or E-site tRNAs are present. Modeling of the peptidyl-tRNA structure within the density maps unambiguously assigns the penultimate residue of the nascent peptide as alanine, as expected based on the mRNA template used in the experiment (Fig. 2). This assignment is further supported by cryo-EM densities corresponding to the mRNA:tRNA interaction because UUCmRNA:GAAtRNA-Phe offers a better fit than GCAmRNA:UGCtRNA-Ala (Extended Data Fig. 2).
To directly compare how the presence of the nascent peptide may influence the positioning of the antibiotic and/or conformation of the rRNA nucleotides, we also generated structures of vacant E. coli 70S ribosomes in complex with LZD or RZD alone, herein referred to as LZD-70S and RZD-70S, at 2.4 Å resolution and 2.5 Å resolution, respectively (Table 1 and Extended Data Fig. 1). Similar to the stalled complexes, antibiotic-only bound structures have unambiguous densities for the oxazolidinone antibiotic (Fig. 3a,b).
Fig. 3 |. Nascent peptide with penultimate alanine stabilizes oxazolidinone binding to the ribosome.
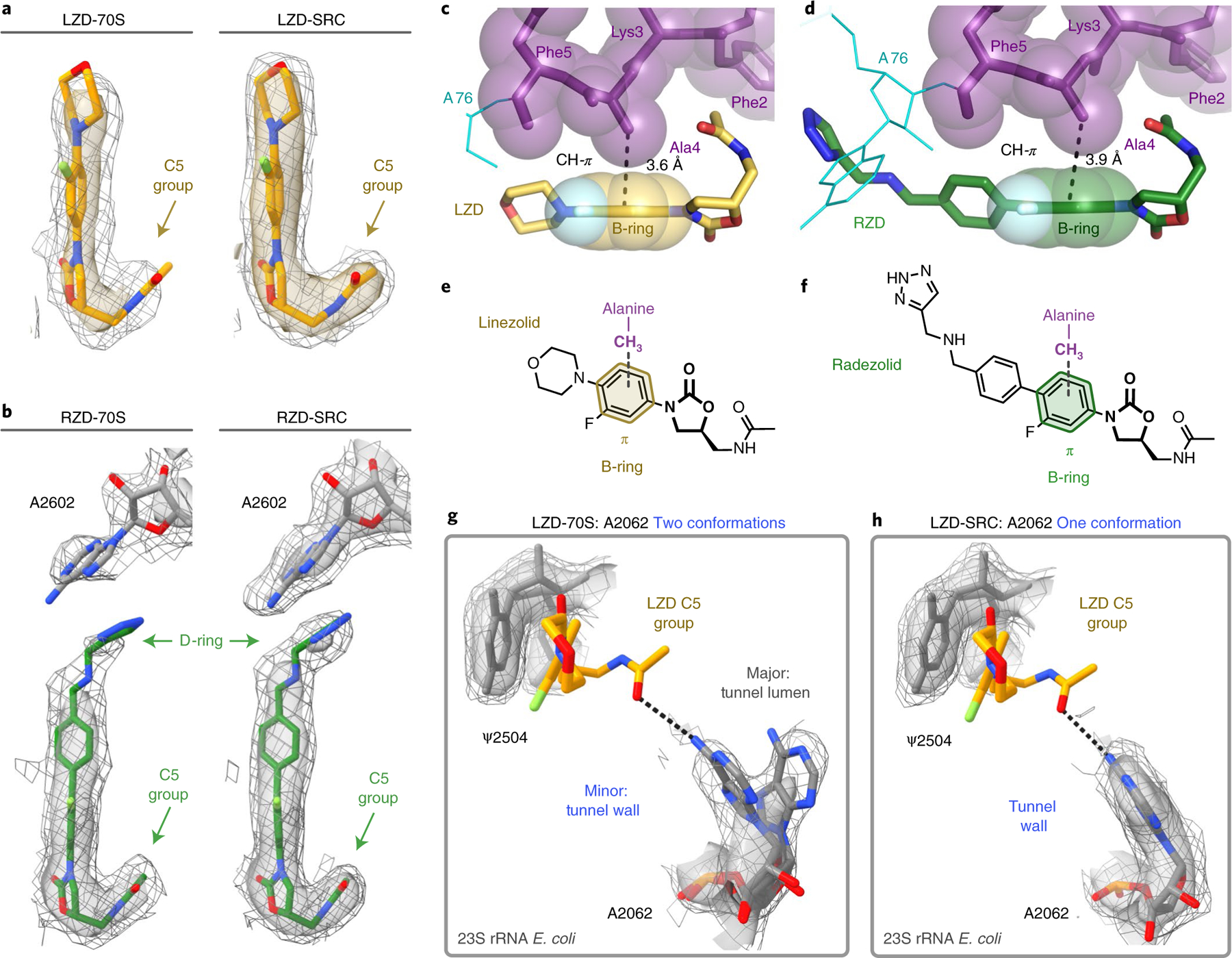
The Coulomb potential density is contoured at 4.0σ in the surface representation and 1.0σ in the mesh representation from unsharpened cryo-EM density maps. a, Comparison of LZD density in the LZD-only bound (LZD-70S) and the LZD-stalled complex (LZD-SRC). b, Comparison of RZD density in the RZD-only bound (RZD-70S) and the RZD-stalled complex (RZD-SRC). c, Close-up view of the CH–π interaction between the B-ring of LZD and the penultimate alanine (Ala4). d, Close-up view of the CH–π interaction between the RZD B-ring and the penultimate alanine. e,f, Schematics of the CH–π interaction involved in stabilizing antibiotic binding in LZD-stalled (e) and RZD-stalled (f) ribosome complexes. g, Density of the exit-tunnel rRNA nucleotide A2062 in the LZD-only bound structure, highlighting two conformations. h, Density of A2062 in the LZD-stalled structure, with only one observed conformation.
The penultimate alanine facilitates antibiotic binding.
The overall binding modes of LZD and RZD within the PTC in antibiotic-only bound and in SRCs are similar to those described previously7–9,22 (Extended Data Fig. 3). The binding modes of LZD and RZD are also similar to that of other oxazolidinone derivatives22–26, including contezolid and cadazolid, with the cadazolid structure also containing a P-site tRNA (Extended Data Fig. 3). The fluorophenyl moiety (B-ring) sits in the A-site cleft, a hydrophobic pocket formed by splayed out nitrogen bases of nucleotides C2452 and A2451 (Extended Data Fig. 3). The oxazolidinone ring (A-ring) of LZD and RZD is positioned in an offset π–π stacking interaction with ψ2504. Interestingly, we find that the carbonyl of the oxazolidinone ring does not interact with rRNA, but is instead chelated to a solvent molecule or ion (Extended Data Fig. 4a,b), corroborating previous structure–activity relationship studies that demonstrated the importance of an electron-pair donor for activity27.
The binding poses for both LZD and RZD are near identical in the antibiotic-only and stalled ribosome structures (Extended Data Fig. 4c,d). We do, however, observe improved density for both antibiotics in the SRC, suggesting that the presence of the stalling nascent peptide stabilizes the placement of LZD and RZD in the ribosome (Fig. 3a,b). Specifically, we observe improved density for the C5 acetamide group for both antibiotics, as well as enhanced density for the D-ring of RZD in the SRC structure. Of note, in previously published density maps of oxazolidinones bound to vacant ribosomes7–9,22, the acetamide is less well resolved and has been modeled in a variety of positions, probably due to its ability to sample multiple conformations (Extended Data Fig. 3). Our structures suggest that the nascent peptide present in the SRC stabilizes the C5 group, thereby providing a more biologically relevant view on drug placement.
The nascent protein chain and, more specifically, the penultimate Ala residue, contributes directly to the formation of the drug binding site (Figs. 2b and 3c,d). In contrast to the orientation of the flanking residues within the nascent peptide, the side chain of the penultimate alanine faces towards the oxazolidinone. In this orientation, the methyl group of alanine fits snugly within the hydrophobic crevice formed by the C5 group and A/B-ring system of the antibiotic. The crevice is deep enough to accommodate the small side chain of alanine, but is too shallow to fit bulkier side chains, including that of tyrosine, providing a rationale for the differential stalling preferences observed in our toeprinting experiments (Fig. 1b). In the stalled complex, the alanine’s side chain methyl group engages in a CH–π interaction with the aryl B-ring of the oxazolidinone (3.6 Å and 3.9 Å between the C atom and the plane of the B-ring for LZD-SRC and RZD-SRC, respectively; Fig. 3c–f). This interaction likely facilitates antibiotic binding to the A site of the PTC, resulting in enhanced occlusion of incoming aminoacyl tRNAs. Glycine, the smallest amino acid residue that lacks a side chain, would not clash with the ribosome-bound antibiotic when present in the penultimate position of the nascent peptide, yet it is not favored for drug-induced ribosome stalling10,11. The inability of glycine to form the alanine-specific CH-π interaction with the drug molecule likely explains this observation.
A similar CH-π interaction has recently been observed between the penultimate alanine and aryl ring of chloramphenicol (CHL), another PTC-targeting antibiotic that also exhibits context-specific inhibition of translation28. Superposition with the CHL complex reveals substantial overlap between the aryl groups of CHL and LZD and similar positioning with respect to the penultimate alanine of the nascent peptide, suggesting that these antibiotics likely exploit the same interaction to achieve context-specificity (Supplementary Fig. 2). In contrast to CHL, which also induces robust ribosome stalling when serine or threonine occupy the penultimate position, LZD exhibits a notably stronger preference for alanine10. Although in silico modeling of serine or threonine at the penultimate position in LZD-SRC reveals that serine can be accommodated, the methyl group of threonine generates a steric clash for all favored rotamers (Extended Data Fig. 5). Recent work hypothesized that CHL-induced stalling with serine in the penultimate position of the nascent peptide is stabilized by a hydrogen bond between the serine hydroxyl and Cl atom of CHL28. Because the C5 group of LZD does not have an analogous electron-pair donor, it is likely that the unsatisfied hydrogen-bond acceptor, rather than sterics, accounts for LZD’s strong preference for alanine over serine. Interestingly, these results suggest that previously developed LZD derivatives with C5 substitutions to more polar groups26, such as LZD-5, which contains a dichloroacetamide moiety similar to CHL, may exhibit expanded stalling profiles.
Additional interactions with rRNA nucleotides.
In LZD and RZD antibiotic-only 70S structures, the exit-tunnel nucleotide A2062 can adopt two distinct conformations (Fig. 3g). In its dominant conformation, the A2062 base projects into the lumen of the exit tunnel, whereas in the minor conformation, A2062 is rotated and lies flat against the tunnel wall. Strikingly, in the stalled ribosome complexes with the nascent protein chain occupying the tunnel, the A2062 base is found only in the rotated state juxtaposed against the wall of the tunnel (Figs. 3h and 4a). The rotated state of A2062 is stabilized by a hydrogen bond between the N1 atom of adenine and the stalling peptide backbone, as well as a non-canonical A:A base pair with m2A2503 (Fig. 4b). The non-canonical A:A base pair is also observed in ribosome structures containing the context-specific inhibitor CHL and a penultimate alanine/threonine-containing peptide28. Interestingly, the interaction between A2503 and A2602 was previously found to be essential for macrolide-induced stalling of bacterial ribosomes29–31 and is also a critical interaction for drug-like molecule PF846-mediated selective inhibition of translation termination in human ribosomes32. In this conformation, the exocyclic amine of A2062 is within hydrogen-bonding distance of the acetamide carbonyl of LZD/RZD (3.0 Å/3.3 Å), which likely explains why we observe improved density for the C5 group in the stalled complexes (Fig. 3a,b). This interaction has not been observed in existing structures of LZD/RZD-bound vacant ribosomes due to the alternate orientation of A2062. The peptide-induced interaction between A2062 and the oxazolidinone is distinct from that observed with CHL. In a structure of the ribosome–CHL complex that lacks the nascent peptide, A2062 is already in a rotated state to form a hydrogen bond with the antibiotic33.
Fig. 4 |. In the stalled complexes, rRNA nucleotides provide additional contacts with the oxazolidinone antibiotic.
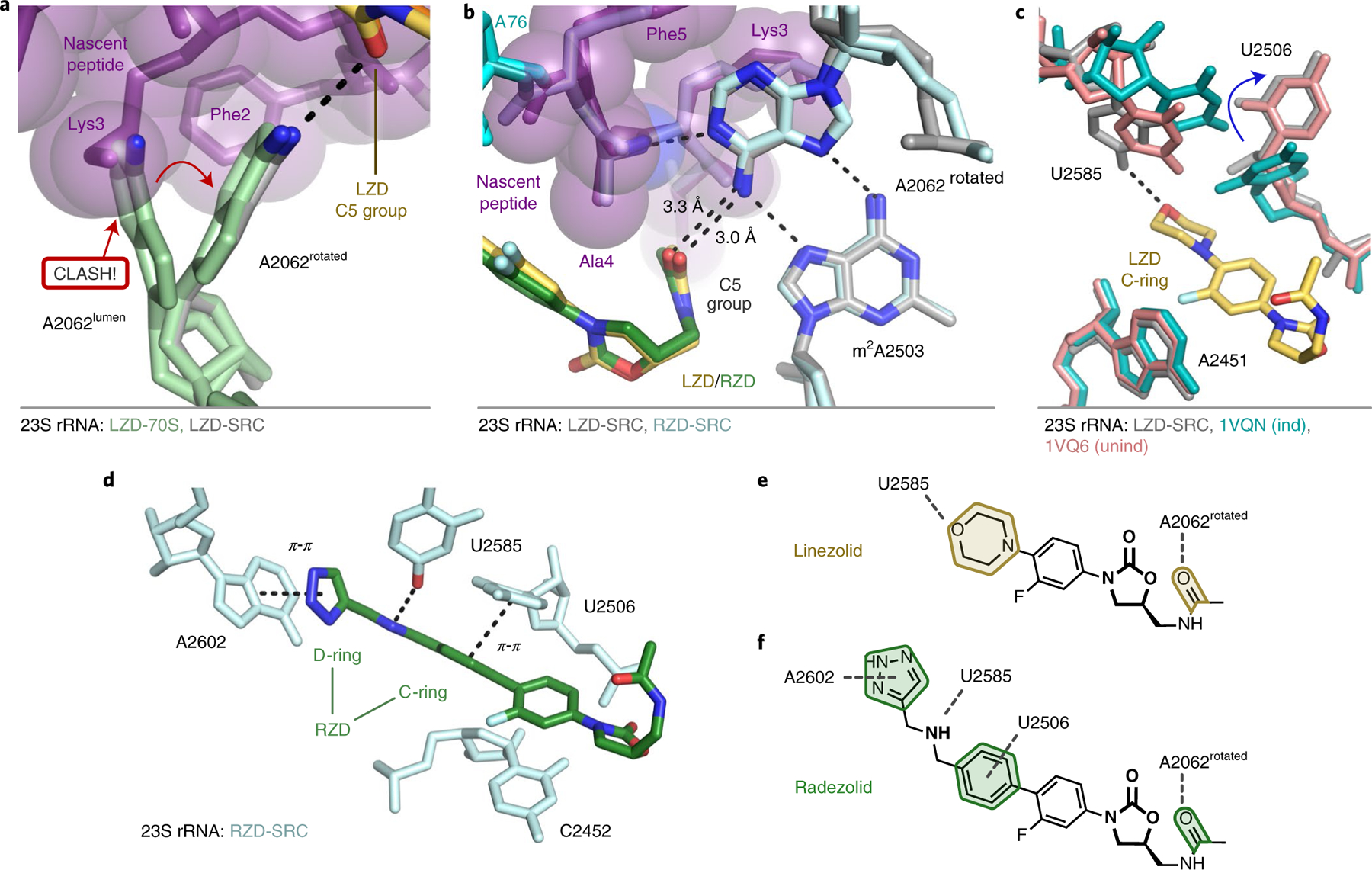
a, The rotated conformation of A2062 is adopted to accommodate the nascent chain (purple). b, In the rotated state, A2062 makes hydrogen-bond interactions with the nascent chain (purple), m2A2503 and the C5 group of LZD (yellow) and RZD (green). c, Additional interaction between U2585 and the morpholine ring of LZD (yellow). U2506 adopts a conformation consistent with the unaccommodated (unind, pink) state of the PTC34. The accommodated PTC state (ind, teal) is shown for comparison34. Labeling of 23S rRNA corresponds to E. coli numbering. d, Stabilized interactions between RZD and A2602, U2585 and U2506 observed in the stalled complex. e,f, Schematics of stabilized interactions for LZD (e) and RZD (f).
Compared to the oxazolidinone-only bound structures, we also observe stabilization of several dynamic PTC nucleotides in conformations that favor additional contacts with the antibiotics. We observe improved density for U2585, as the C4 enol tautomer could provide a hydrogen-bonding interaction with the O atom of the morpholine ring in LZD (Fig. 4c,e and Extended Data Fig. 6). Although U2506 does not make direct contact with LZD, we observe dramatically improved density for this nucleotide in a conformation analogous to an uninduced or nonproductive state of the PTC34 (Fig. 4c and Extended Data Fig. 6). In the RZD-stalled complex we observe improved densities for U2506, U2585 and A2602 (Extended Data Fig. 6). U2506 and A2602 provide π–π stacking interactions with the C- and D-ring, respectively, and U2585 engages in a hydrogen bond with the secondary amine of RZD (Figs. 3b and 4d,f). Interestingly, the D-ring interaction with A2602 was not observed in a previous ribosome structure with RZD22, but has been observed in structures with oxazolidinones containing other D-ring substituents23,24 (Extended Data Fig. 3e,f). The additional interaction with A2602 probably explains why RZD is a better inhibitor of translation than LZD, as the stabilized D-ring would account for improved drug binding and provide additional steric interference with incoming aminoacyl tRNAs (Extended Data Fig. 7). Together, these results suggest that, in addition to favorable interactions with the penultimate alanine, interactions between otherwise dynamic rRNA nucleotides and the oxazolidinone likely play a role in improving antibiotic binding to the ribosome to facilitate stalling.
Cfr methylation perturbs the stalled ribosome complex.
A prevalent resistance mechanism to LZD identified in multiple clinical isolates worldwide35–40 involves methylation of rRNA by the Cfr enzyme, which adds a methyl group at the C8 atom of A2503 (m8A2503) in 23S rRNA15,17,41–43. It has been speculated that the Cfr modification disrupts LZD binding to the ribosome by introducing a steric clash between the installed methyl mark and the C5 group of the antibiotic. Although this modification confers high levels of resistance to LZD, RZD retains modest efficacy against Cfr-positive strains44,45 (Extended Data Fig. 8a), likely due to retained interactions on the other side of the molecule involving the D-ring (Fig. 3b). However, this suggests that binding of RZD to Cfr-modified ribosomes would require structural changes, such as the C5 group of RZD adopting an alternate conformation to accommodate the C8 methyl group of m8A2503 or shifting of the modified nucleotide to open room for the C5 side chain. Given that positioning of both the C5 group and A2503 may influence context-specificity, we wanted to evaluate the ability of RZD to induce stalling of Cfr-modified ribosomes.
To perform in vitro assays, we expressed Cfr in E. coli and isolated ribosomes with near-complete methylation of m8A2503 as described previously43. As expected, RZD retains considerable activity against the m8A2503 ribosomes. In vitro translation of superfolder green fluorescent protein (sf-GFP) by Cfr-modified ribosomes was barely affected by LZD but could be inhibited by RZD, albeit with some loss in potency (half maximum inhibitory concentration (IC50) = 0.9 μM for wild-type and IC50 = 4.9 μM for Cfr-modified; Extended Data Fig. 8b). Toeprinting experiments showed that, as expected, the m8A2503 modification considerably reduced LZD-dependent ribosome arrest at the F5 codon of the MFKAF…-encoding ORF (Fig. 5a). Strikingly, RZD, although retaining its general translation inhibitory activity, was notably less efficient in arresting the m8A2503 ribosome at the F5 codon of the MFKAF template (Fig. 5a). Qualitatively similar results were observed when we performed toeprinting analysis using the sf-GFP coding sequence as a template. LZD and RZD induced stalling of unmodified ribosomes at several specific sites within the sf-GFP coding sequence. LZD-mediated stalling was essentially eliminated when translation was driven by Cfr-modified ribosomes. By contrast, RZD retained its ability to cause stalling of the Cfr-modified ribosomes, albeit with diminished efficiency (Extended Data Fig. 8c). These results suggest that RZD remains a good inhibitor of Cfr-modified ribosomes, because causing weak stalling at multiple sites within the mRNA ORFs would still be sufficient to inhibit expression of most cellular proteins. Together, these results suggest that the presence of the C8 methyl group at A2503 does not completely prevent binding of RZD, but partially diminishes its ability to arrest translation in the alanine-specific manner.
Fig. 5 |. The Cfr m8A2503 modification reduces RZD-dependent ribosome stalling.
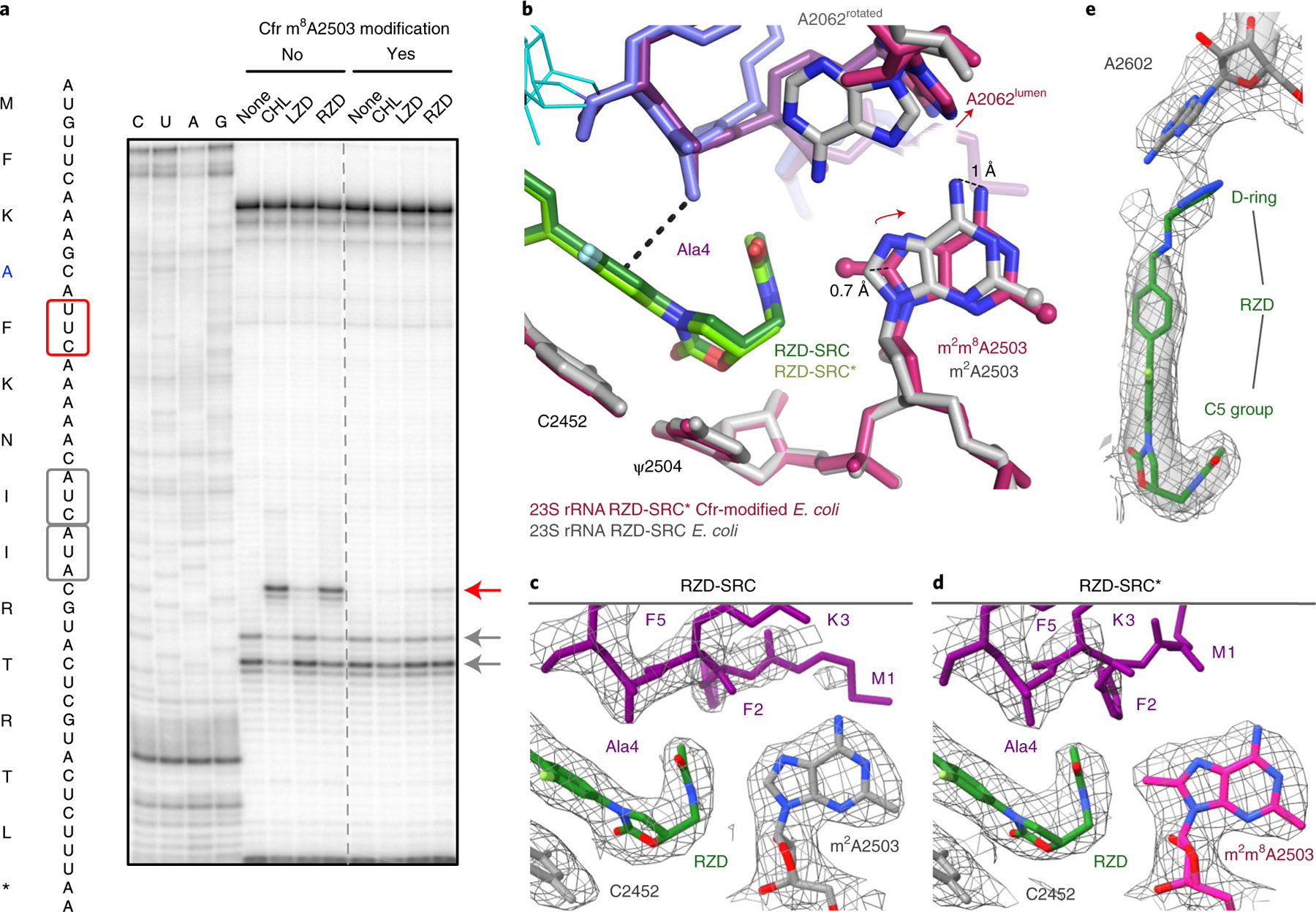
a, Toeprinting assays performed on the MFKAF stalling peptide sequence. Prominent stall sites observed in reactions containing CHL, LZD or RZD with WT ribosomes or Cfr-modified ribosomes, are indicated by the red arrow. ‘None’ designates reactions lacking ribosome-targeting antibiotics. Owing to the inclusion of the tRNA synthetase inhibitor mupirocin in all toeprinting reactions, any ribosomes not stalled at an upstream codon are forced to stall at the Ile codons designated by gray arrows. All antibiotics were added to a final concentration of 50 μM. Toeprinting experiments were performed at least twice with similar results. b, Structural rearrangements identified in the RZD-stalled complex with a Cfr-modified ribosome. The overlay was performed by alignment of 23S rRNA nucleotides 2000–3000. c,d, Coulomb potential density for A2503, RZD and the nascent chain contoured at 3.0σ for RZD stalled with the WT ribosome (RZD-SRC) (c) and RZD-stalled with the Cfr-modified ribosome (RZD-SRC*) (d). e, Retained interaction between the RZD D-ring and A2602 in the Cfr-modified ribosome. The Coulomb potential density is contoured at 4.0σ in the surface representation and 1.0σ in the mesh representation. Panels c–e were prepared using unsharpened cryo-EM density maps.
In search of a molecular explanation for this result, we determined the structure of an RZD-stalled Cfr-modified ribosome containing the MFKAF-tRNA in the P site at 2.4 Å resolution (RZD-SRC*, Table 1 and Extended Data Fig. 1). Although the binding mode of the antibiotic is near identical to that observed in the stalled wild-type (WT) ribosome, m8A2503 is shifted away from RZD to accommodate the C5 group (Fig. 5b–d). A2503 positioning is also distinct from that observed in vacant Cfr-modified ribosomes, suggesting that repositioning is due to RZD accommodation and not due to C8 methylation per se (Extended Data Fig. 8d). Although the penultimate alanine is able to retain the CH-π interaction with RZD, the Cfr modification encroaches on the antibiotic binding pocket so that binding of RZD faces the increased energy barrier of partially displacing m8A2503 relative to its position observed in the unmodified SRC (Fig. 5b–d). The increased packing requirements of the alanine side chain, antibiotic and A2503 with the addition of the C8 methyl group (Extended Data Fig. 8e) likely restrict the conformational flexibility among these components to diminish formation of a stable stalled complex. Indeed, some destabilization of the SRC is revealed by the poor density for certain side chains of the nascent peptide (Extended Data Fig. 9).
Strikingly, we also find that A2062 adopts a single observable conformation within the lumen of the exit tunnel, which is now positioned too far away to engage in a hydrogen bond with RZD (Fig. 5b). This is distinct from the stalled WT complex, where A2062 is found in the rotated conformation against the tunnel wall, and also distinct from the RZD-only bound complex, in which both conformations are observed (Extended Data Fig. 10). Given that the non-canonical A:A base pair with A2503 is likely involved in stabilizing the rotated state of A2062, the shifted position of m8A2503 may contribute to reversion of A2062 to the lumen conformation. Interestingly, perturbation of the same A:A interaction has also been shown to disrupt macrolide-dependent ribosome stalling31.
Our structural findings also provide important insights into how RZD retains efficacy against LZD-resistant ribosomes. Although the Cfr modification of A2503 perturbs hydrogen bonding between the antibiotic and A2062, we note retention of other interactions. Although somewhat diminished, we observe densities for the D-ring and A2602 engaged in a π-π stacking interaction analogous to that observed in the WT, non-Cfr-modified complex (Fig. 5e). Because LZD does not contain the additional D-ring, its binding is more dramatically impacted by steric occlusion by m8A2503 and/or the lost interaction with A2062. Our results suggest that, in addition to shortening of the C5 group, which has been carried out with other oxazolidinones derivatives19,24,46, extension of the ring system on the opposite end of the molecule is an orthogonal, viable strategy for generating oxazolidinone antibiotics that overcome Cfr resistance. The combination of both approaches has been showcased in the development of tedizolid and cadazolid, both of which exhibit potent antimicrobial activity against Cfr-containing, LZD-resistant strains24,47.
Discussion
We identified two factors that contribute to the context-specificity of the oxazolidinones LZD and RZD. Although the antibiotic can still bind to the ribosome that lacks the nascent protein chain, as revealed by our structure as well as previously published structures of the vacant ribosome complexed with LZD and RZD, the presence of the nascent chain stabilizes binding of the drug by providing additional points of contact. Most importantly, the side chain of the penultimate alanine residue is intercalated into the complementarily shaped cavity formed by the drug molecule (Figs. 2b, 3c,d and 6a,b). Larger amino acids in the penultimate position of the nascent peptide would clash with the antibiotic, preventing its binding (Fig. 6a). In contrast, glycine cannot make a CH-π interaction with the drug, thereby making its binding less favorable (Fig. 6a). As a secondary effect of the alanine interaction, we also observe stabilization of dynamic nucleotides in conformations that provide additional contacts with the antibiotic (Fig. 4 and Extended Data Fig. 6). The culmination of these interactions leads to improved antibiotic binding to the ribosomal A site in the presence of nascent peptides containing a penultimate Ala, facilitating competition with incoming aminoacyl tRNAs and thus resulting in ribosome stalling (Fig. 6a,b). The mechanism by which the synthetically derived oxazolidinone antibiotics achieve context-specificity closely resembles that of chloramphenicol, a natural product. Chloramphenicol also capitalizes on interactions with the penultimate residue—namely alanine, threonine and presumably serine—to stabilize drug binding to the ribosome28. Despite their distinct origins, the shared mechanism by which these two antibiotic families accomplish translation inhibition highlights the importance of the interaction with the nascent chain for certain antibiotics that target the PTC.
Fig. 6 |. Model for oxazolidinone context-specific inhibition of translation.
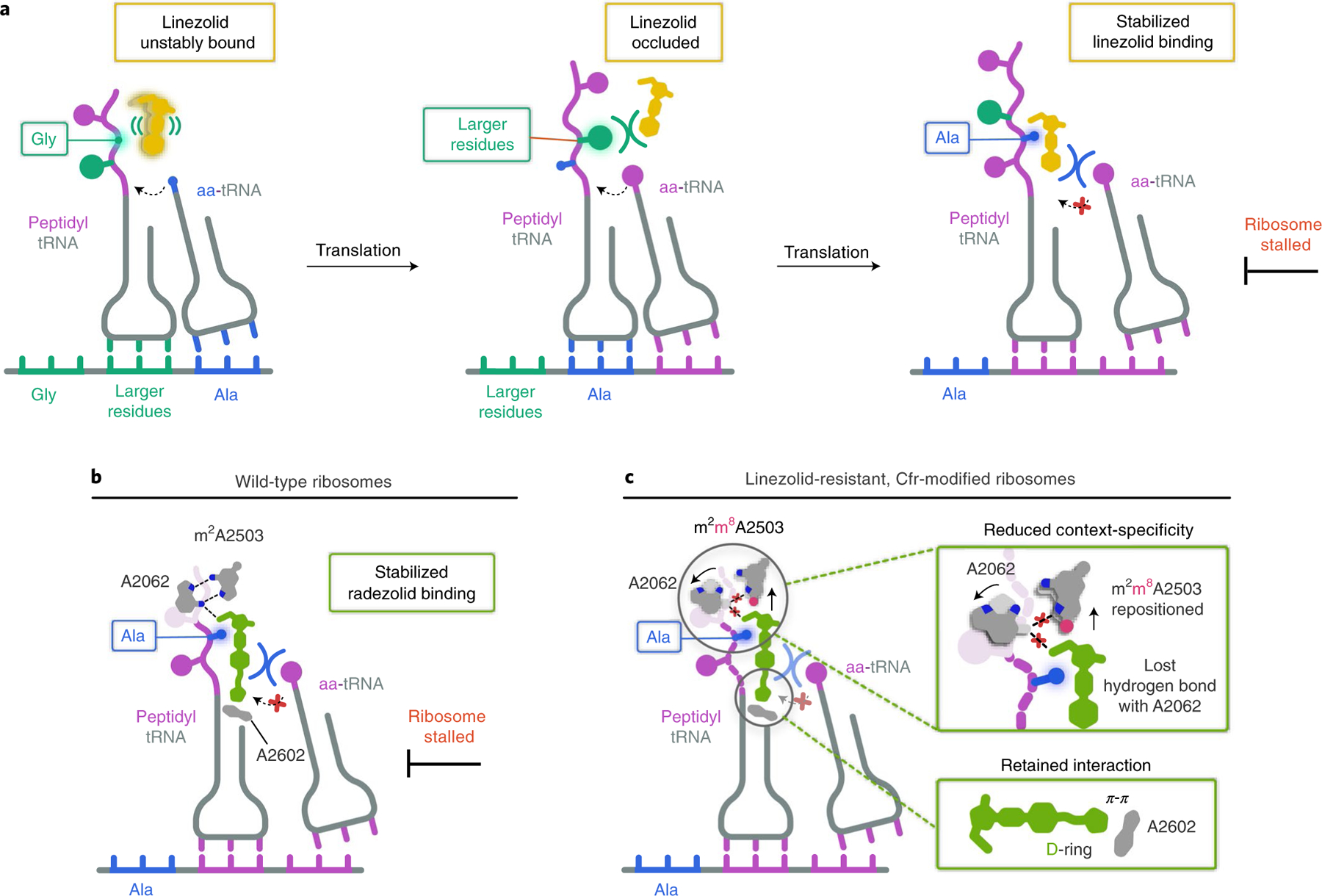
a, LZD (yellow) is either unstably bound or sterically occluded from binding to the ribosome when glycine (Gly) or larger residues occupy the penultimate position within the nascent chain. When alanine (Ala, blue) occupies the penultimate position, LZD becomes stably bound to the ribosomal PTC A site, enabling LZD to better compete away incoming aminoacyl tRNAs (aa-tRNAs) and resulting in ribosome stalling. b, RZD (green) also facilitates stalling of ribosomes with alanine in the penultimate position by enhancing RZD binding to the PTC A site. c, RZD context-specificity is diminished with Cfr-modified ribosomes due to repositioning of C8-methylated A2503 (pink circle) and the lost interaction with A2062. A retained interaction between the RZD D-ring and A2602 provides a rationale for why RZD can overcome Cfr-mediated resistance.
Our structural investigations into RZD binding to a Cfr-modified ribosome have revealed the importance of the D-ring in stabilizing ribosome engagement, providing insight into how RZD retains efficacy against Cfr-modified ribosomes (Fig. 6c). We find that, although RZD loses some potency, which is consistent with diminished stalling on alanine-containing peptides, RZD remains an overall effective inhibitor of m8A2503-containing ribosomes. Our structural findings suggest that the Cfr modification likely diminishes RZD stalling by forcing an energetically unfavorable repositioning of A2503, which also results in loss of an interaction between exit-tunnel nucleotide A2062 and the drug (Fig. 6c). In summary, our findings provide a unifying model for context-specific inhibition of translation by oxazolidinone antibiotics. Our observation that the antibiotic binding pocket is formed, in part, by the nascent peptide has revealed the ‘missing’ component of translation machinery involved in antibiotic binding from previous crosslinking experiments. The importance of Ala in the penultimate position of the nascent peptide for stabilizing the antibiotic provides the structural basis for context-specificity observed in ribosome profiling and single-molecule studies. Our structural insights into nascent peptide-specific inhibition of translation by oxazolidinones, and complementary work in related systems28,48–50, suggest prospects for the development of drugs that can modulate the activity of ribosomes in a protein-selective manner.
Methods
Generation of DNA templates for the PURExpress system.
DNA templates were prepared by performing polymerase chain reactions (PCRs) with AccuPrime Taq DNA polymerase (Thermo Fisher). The DNA templates used for toeprinting analysis were prepared by combining the following previously published primers11: 100 μM T7, 100 μM NV1, 10 μM SMFRET-Fwd, 10 μM SMFRET-Rev and either 10 μM SMFRET-Mid(FKAFK) (for the MFKAFK… template) or 10 μM SMFRET-Mid(FKYFK) (for the MFKYFK… template). The DNA templates used for the generation of stalled ribosome complexes were prepared by combining the following primers: 100 μM T7, 100 μM ORF_SD, 10 μM T7_MFKAF_Fwd and 10 μM SD_MFKAF_Rev. The primer sequences are listed in Supplementary Table 1. The PCR product was purified using a MiniElute PCR kit (Qiagen) following the manufacturer’s instructions, and quality was confirmed using an 8% Tris-borate-EDTA (Novagen) gel. The sequence architecture of the resulting DNA product is outlined in Supplementary Fig. 1.
Purification of 70S ribosomes.
Wild-type E. coli 70S ribosomes were purified from the MRE600 strain52. E. coli MRE600 were grown to mid-log phase in LB medium at 37 °C with shaking. Cfr-modified E. coli 70S ribosomes were purified from E. coli BW25113 expressing the evolved variant CfrV743. E. coli BW25113 transformed with pZA-encoded CfrV7 were grown to an optical density at 600nm (OD600) of ~0.7 in LB medium containing ampicillin (100 μg ml−1) and anhydrotetracycline (AHT) inducer (30 ngml−1) at 37 °C with shaking. Subsequent purification steps for WT and Cfr-modified ribosomes were identical. After lysis using a microfluidizer, clarified lysates were applied to a 32% wt/vol sucrose cushion. Tight-coupled 70S ribosomes were purified on a 15–30% wt/vol sucrose gradient. To eliminate sucrose, ribosomes were precipitated by the addition of PEG 20000 and subsequently resuspended in buffer containing 50mM Hepes-KOH (pH 7.5), 150mM KOAc, 20mM Mg(OAc)2, 7mM β-mercaptoethanol and 20Uml−1 SuperASE-In.
Determination of antibiotic resistance.
Antibiotic resistance was determined by broth microdilution following established protocols53. The E. coli strain BW25113 acrB::kan, in which efflux component AcrB is replaced with a kanamycin resistance cassette, was used for resistance testing of the oxazolidinones. In short, cultures of bacteria freshly transformed with empty pZA vector or pZA-encoded CfrV7 were grown at 37 °C with shaking for ~2.5 h. Cultures were then diluted to 106 cells and 50 μl of this dilution was dispensed into 96-well plates containing 50 μl of LB medium with the antibiotic of interest, ampicillin (100 μgml−1), kanamycin (50 μgml−1) and AHT (30 ngml−1). Resistance was evaluated using twofold serial dilution of the antibiotic with the following concentration ranges: LZD (1–256 μgml−1, Acros), RZD (0.5–128 μgml−1, Med Chem Express). The minimum inhibitory concentration (MIC) required to inhibit visible bacterial growth was determined after incubation at 37 °C with shaking for 18h. OD600 values were also recorded with a microtiter plate reader (SpectraMax M5, Molecular Devices).
In vitro assays.
Toeprinting assays were conducted as previously described11,49,54 using WT or Cfr-modified ribosomes purified as described above and previously55. Briefly, reactions were prepared using the PURExpress △ Ribosome Kit (New England Biolabs) in volumes of 5μl and were allowed to proceed for 15 min at 37 °C. Primer extension was initiated by the addition of AMV reverse transcriptase (New England Biolabs) and allowed to proceed for 10 min. All antibiotics were added to a final concentration of 50μM. Mupirocin is an Ile-tRNA synthetase inhibitor and was included in all toeprinting reactions so that any ribosomes not stalled at an upstream codon are forced to stall at the downstream hungry Ile codons.
The effect of antibiotics on protein expression was analyzed in a △ Ribosome PURExpress cell-free transcription–translation system (New England Biolabs). The plasmid pY7156 containing the sf-gfp gene under the control of T7 RNA polymerase promoter was used as a template. A typical 5-μl reaction, composed from the kit components as recommended by the manufacturer, contained 10pmol of WT or Cfr-modified ribosomes isolated as described above55. The reactions were prepared on ice and then placed in the wells of a 384-well black-wall/clear-bottom plate. The plate was incubated at 37 °C in a Tecan Infinite M200 Pro plate reader and the progression of the reaction was monitored by following sf-GFP fluorescence (λex = 488 nm; λem = 520 nm) over time.
Preparation of LZD- and RZD-only bound ribosomes for cryo-electron microscopy.
Purified 70S WT or Cfr-modified ribosomes were diluted to 1 pmol μl−1 in buffer containing 50mM Hepes-KOH (pH 7.5), 150mM KOAc, 20mM Mg(OAc)2, 7 mM β-mercaptoethanol and 20Uml−1 SuperASE-In. Diluted ribosomes (50 μl) were then incubated at 4 °C for 1.5 h after the addition of 3 μl of either 1mM LZD (Med Chem Express) or 1mM RZD (Med Chem Express), yielding a final antibiotic concentration of 60 μM (60× molar excess). Samples were then filtered for 5 min at 14,000g at 4 °C using a 0.22-μm low-binding Durapore PVDF filter (Millipore).
Preparation of stalled ribosome complexes for cryo-electron microscopy.
Stalled ribosome complexes containing the stalling peptide and corresponding oxazolidinone antibiotic were prepared by in vitro transcription–translation. Reactions (100-μl volume) were prepared using the PURExpress △ Ribosome Kit (New England Biolabs, E3313S) containing 0.8 U μl−1 of SuperASE-In, ~1,100ng of DNA template encoding the stalling peptide sequence (Supplementary Fig. 1), 5,000 pmol of LZD (Med Chem Express) or RZD (Med Chem Express) and 360 pmol of WT ribosomes or 250 pmol of Cfr-modified ribosomes. The reaction was halted by placing reactions on ice after incubation at 37 °C for 1h. The reaction was diluted to 190 μl by the addition of buffer C (50 mM Hepes-KOH pH 7.5, 150 mM KOAc, 20 mM Mg(OAc)2, 7 mM β-mercaptoethanol and 20 U ml−1 SuperASE-In) and purified by a 10–50% sucrose gradient also prepared in buffer C. Ultracentrifugation was performed using a SW Ti41 rotor (Beckman Coulter) at 60,000g for 16 h at 4 °C. Gradients were fractionated using a Bio-Comp fractionator in 20 fractions for which absorbance at 260 nm was continuously monitored. Fractions corresponding to stalled ribosome complexes were precipitated by the slow addition of PEG 20000 in buffer C at 4 °C to a final concentration of 8% wt/vol. Stalled complexes were isolated by centrifugation for 10 min at 17,500g at 4 °C. After removing the supernatant, samples were slowly resuspended in buffer C at 4 °C. Sample concentration was determined by a NanoDrop UV spectrophotometer (Thermo), where A260 = 1 corresponds to 24pmol of 70S ribosome. Purified stalled ribosome complexes (0.65 μM Cfr-modified stalled ribosome complex or 2.2 μM WT stalled ribosome complex) were then incubated with ~30× molar excess of RZD (20 μM for Cfr-modified stalled ribosome complexes and 60 μM for WT stalled ribosome complex) for 1 h at 4 °C. The final concentration of the antibiotic was ~4–60× over IC50 for translation (Extended Data Fig. 8b). Purified WT stalled ribosome complexes (0.418 μM) were incubated with >100× molar excess of LZD (60 μM) for 1h at 4 °C. The final concentration of antibiotic was ~3.5× over IC50 for translation (Extended Data Fig. 8b). Prior to freezing the grids, stalled ribosome complexes were filtered for 5 min at 14,000g at 4 °C using a 0.22-μm low-binding Durapore PVDF filter (Millipore).
Cryo-electron microscopy analysis.
The above-described samples were diluted in buffer C and deposited onto freshly glow-discharged (EMS-100 Glow Discharge System, Electron Microscopy Sciences, 30 s at 15 mA) copper Quantifoil (Quantifoil Micro Tools) grids with 2-nm-thick amorphous carbon on top. The grids were incubated for 30 s at 10 °C and 95% humidity, before blotting and vitrification by plunging into liquid ethane using a FEI Vitrobot Mark IV (Thermo Fisher). Ice thickness was controlled by varying the blot time, using Whatman #1 filter paper for blotting. The grids were screened for ice quality using an FEI Talos Arctica electron microscope (Thermo Fisher, 200 kV, at UCSF) before being transported via a dry shipper to other facilities or loaded into a UCSF FEI Titan Krios instrument (Thermo Fisher).
All datasets used for reconstruction were imaged on FEI Titan Krios microscopes (Thermo Fisher, 300 kV). The LZD-70S and LZD-SRC datasets were collected at the Stanford-SLAC CryoEM Center (S2C2) using SerialEM on a microscope equipped with a Gatan K3 direct electron detector (DED) but without an imaging filter. The RZD-70S and RZD-SRC datasets were collected at the National Center for CryoEM Access and Training (NCCAT) using Leginon/Appion on a microscope equipped with a Gatan K2 Summit DED and an imaging filter (20-eV slit). The RZD-SRC* dataset was collected at UCSF on a microscope equipped with a Gatan K3 DED and an imaging filter (20-eV slit). This dataset was collected on-axis, but all other datasets were collected using a nine-shot beam-image shift approach with coma compensation. All image stacks were collected in super-resolution mode. Pixel sizes, micrograph count, defocus values and exposures varied slightly between facilities and are reported in Table 1.
All image stacks were binned by a factor of two, motion-corrected and dose-weighted using UCSF MotionCor257. All reconstructions used dose-weighted micrographs. The initial contrast transfer function (CTF) parameters were determined using CTFFIND4 within the cisTEM (v1.0.0-beta)21 software suite. Micrographs with poor CTF fits or crystalline ice were excluded. Unsupervised particle picking using a soft-edged disk template was followed by two-dimensional classification in cisTEM. The initial and final particle counts are reported in Table 1. Only classes that clearly contained ice were omitted. An ab initio reconstruction was carried out in cisTEM on the RZD-SRC* dataset, which yielded a starting reference that was low-pass filtered and used as the initial reference for all five datasets. For SRC datasets, multi-class auto refinement in cisTEM was used to select for all particles that had tRNA present. These, and all non-SRC datasets, were then subjected to a two-class auto refinement in cisTEM to classify between ‘good’ particles and damaged ‘garbage’ particles and high-frequency noise. The good classes were carried forward into single class auto and manual refinement efforts, including per-particle CTF estimation. Care was taken not to increment the high-resolution cutoff in refinement to prevent overfitting. Unsharpened maps were used in model refinement and figure preparation. Pixel size was confirmed by comparison and cross-correlation between the resulting map and a crystallographically derived ribosome structure. 70S maps were used for model building and figure preparation. Map resolution values are reported as particle Fourier shell correlation (FSC) at 0.143.
Atomic model building and refinement.
Atomic models of 70S ribosome complexes with antibiotics and 70S stalled ribosome complexes were generated by rounds of model building in Coot58, molecular dynamics model fitting in ISOLDE59 and refinement in PHENIX60. An initial model for all ribosome complexes was obtained by combining (1) a model of the WT E. coli 50S subunit (PDB 6PJ6)52, (2) a model of the 30S subunit from the WT E. coli ErmBL-stalled ribosome structure (PDB 5JU8)61, (3) P-tRNA and mRNA extracted from the ErmBL-stalled ribosome structure (PDB 5JU8, mutated and remodeled in Coot58 to yield fully modified E. coli tRNAPhe and a short mRNA, respectively) and (4) the nascent peptide, which was modeled in Coot. Model refinement against the acquired LZD-SRC cryo-EM map was performed by multiple rounds of manual model building, molecular dynamics model fitting and restrained parameter refinement (rigid body fitting, real-space refinement, anisotropic displacement parameters (ADP) refinement and simulated annealing). Restraints for real-space refinement of modified nucleotides were generated using eLBOW62 within PHENIX52. For molecular dynamics, force fields for most modified nucleotides were available but inactive in standard releases of ISOLDE due to disfavored behavior under certain circumstances; these were toggled to active, and for nucleotides and the 4D4 amino acid without force fields available, the closest matching residue with force fields was substituted during molecular dynamics simulations as necessary. The antibiotics were held static for these simulations. The L3, L10 and L11 proteins were not modeled. Upon convergence of a satisfactory model in the LZD-SRC map, the same model was fit into the density in the other four maps, edited for chemical accuracy (removal of the peptidyl-tRNA and mRNA from non-SRC models, and removal of any ions not supported by the map), and then subjected to further molecular dynamics and refinement cycles until convergence. Overall, protein residues and rRNA nucleotides show well-refined geometrical parameters (Table 1). The figures were prepared using PyMOL Molecular Graphics System Version 2.4.1 (Schrödinger) or UCSF ChimeraX Version 1.2.563.
Extended Data
Extended Data Fig. 1 |. Processing workflow, classification tree, FSC curves, and local resolution estimation for each map.
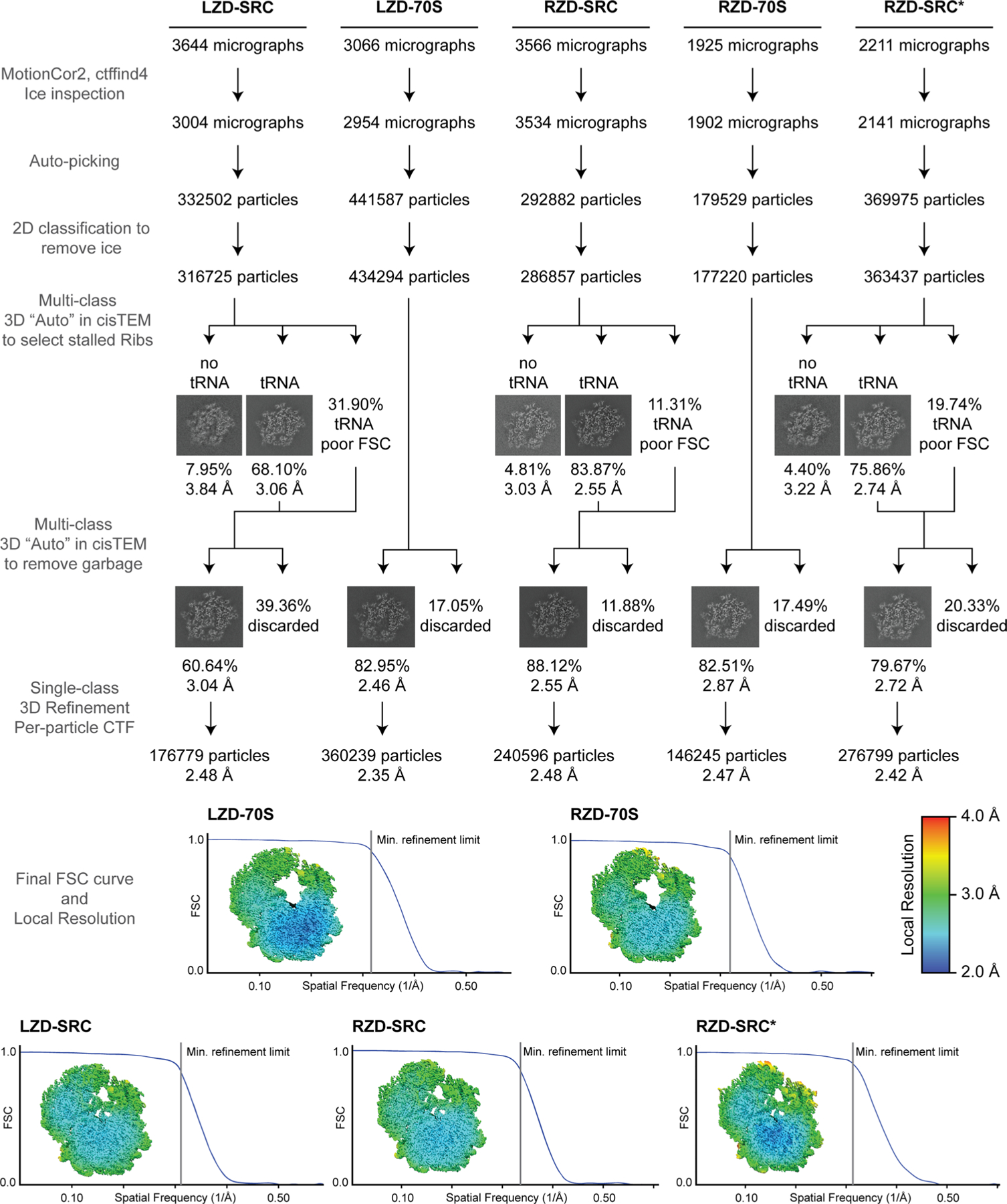
Micrographs were CTF corrected and curated for ice quality, followed by unsupervised particle picking. 2D classification was used to remove residual ice particles, with all others subjected to multi-class 3D classification and refinement approaches to remove particles without tRNA (for SRCs) and select for good particles. Final FSC curves are presented along with center-slab representation of local resolution. All steps were carried out within the cisTEM (v1.0.0-beta) framework with the exception of local resolution estimation, which used the Relion (v3.1.2) implementation.
Extended Data Fig. 2 |. Confirmation of oxazolidinone-induced ribosome stalling at the Phe5 codon.
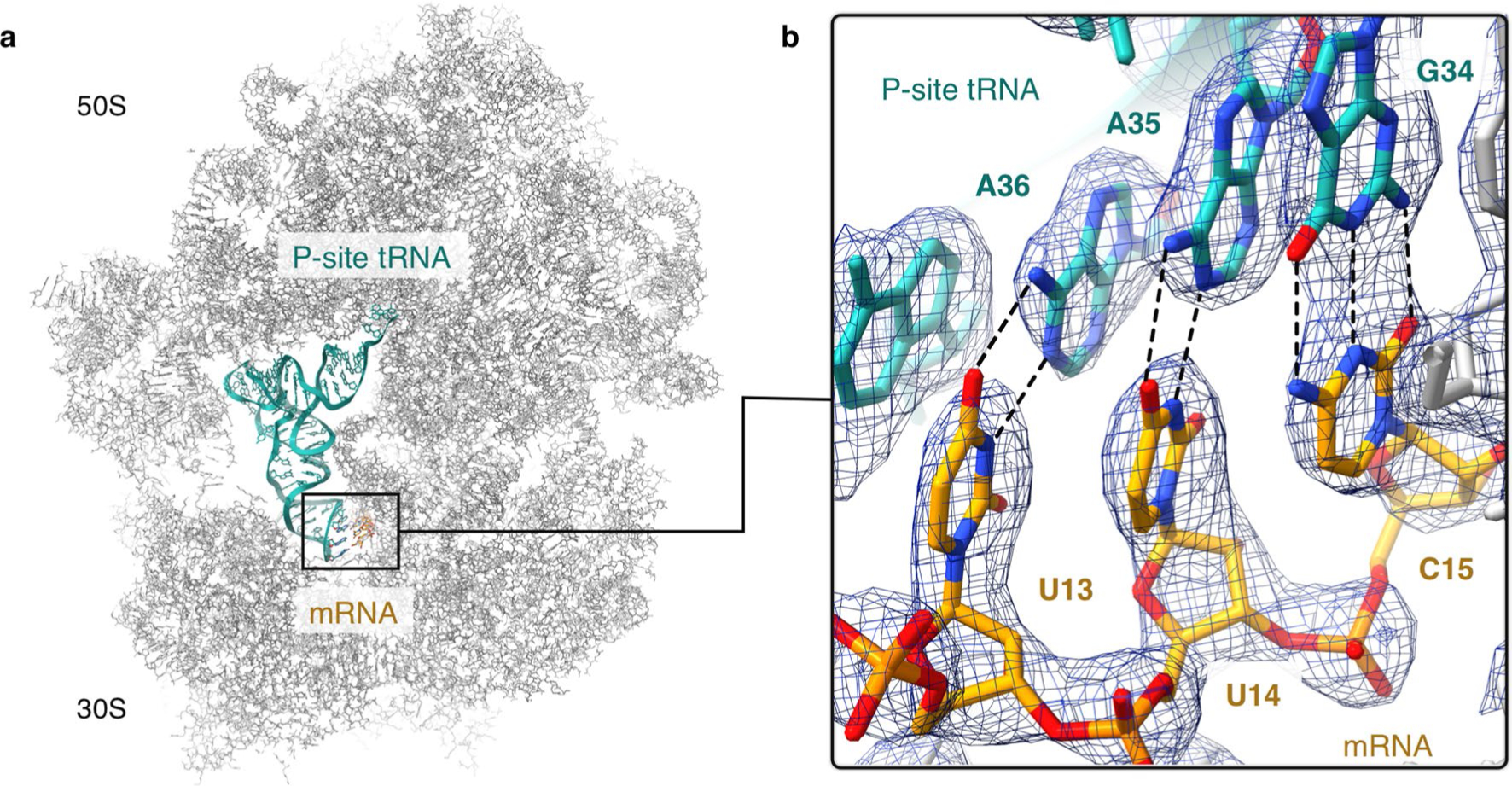
(a) Location of the codon-anticodon interaction within the linezolid-stalled 70S ribosome complex. (b) Density for the codon-anticodon interaction is best modeled as UUCmRNA:GAAtRNA-Phe rather than GCAmRNA:UGCtRNA-Ala which would correspond to stalling at the upstream Ala4 codon. The figure was generated from unsharpened maps and the density is contoured at 4σ.
Extended Data Fig. 3 |. Overview of the binding mode of linezolid and radezolid within the ribosomal PTC.
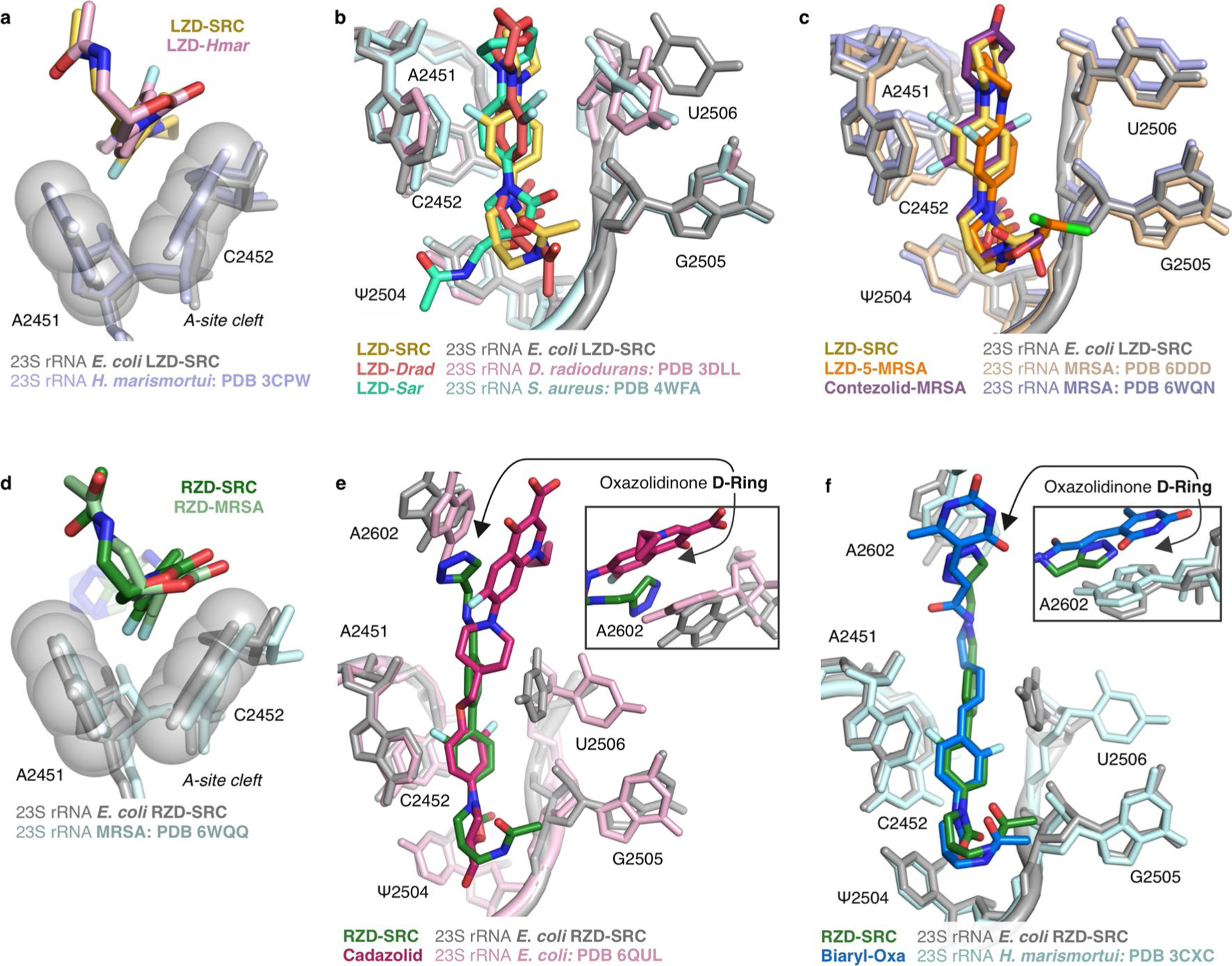
(a) Structural overlay of linezolid (LZD) bound to a H. marismortui ribosome (PDB: 3CPW) and linezolid-stalled E. coli ribosome complex (LZD-SRC), highlighting binding within the A-site cleft. (b) Overlay of E. coli LZD-SRC and LZD-only bound to a D. radiodurans (PDB: 3DLL) or S. aureus ribosome (PDB: 4WFA). (c) Overlay of E. coli LZD-SRC and MRSA ribosome bound to LZD analog (LZD-5, PDB: 6DDD) or contezolid (PDB: 6WQN). (d) Structural overlay of radezolid ( RZD)bound to a MRSA ribosome (PDB: 6WQQ) and radezolid-stalled E. coli ribosome complex (RZD-SRC), highlighting the A-site cleft. (e) Overlay of E. coli RZD-SRC and E. coli ribosome bound to cadazolid (PDB: 6QUL). (f) Overlay of E. coli RZD-SRC and H. marismortui ribosome bound to a biaryl-oxazolidinone (PDB: 3CXC). Insets for panels (e) and (f) highlight the p-stacking interaction between the oxazolidinone D-ring and A2602. All overlays in this figure were generated by alignment of 23S rRNA nucleotides. E. coli numbering is used for all figure panels.
Extended Data Fig. 4 |. Oxazolidinone binding modes in antibiotic-only and stalled ribosome complexes.
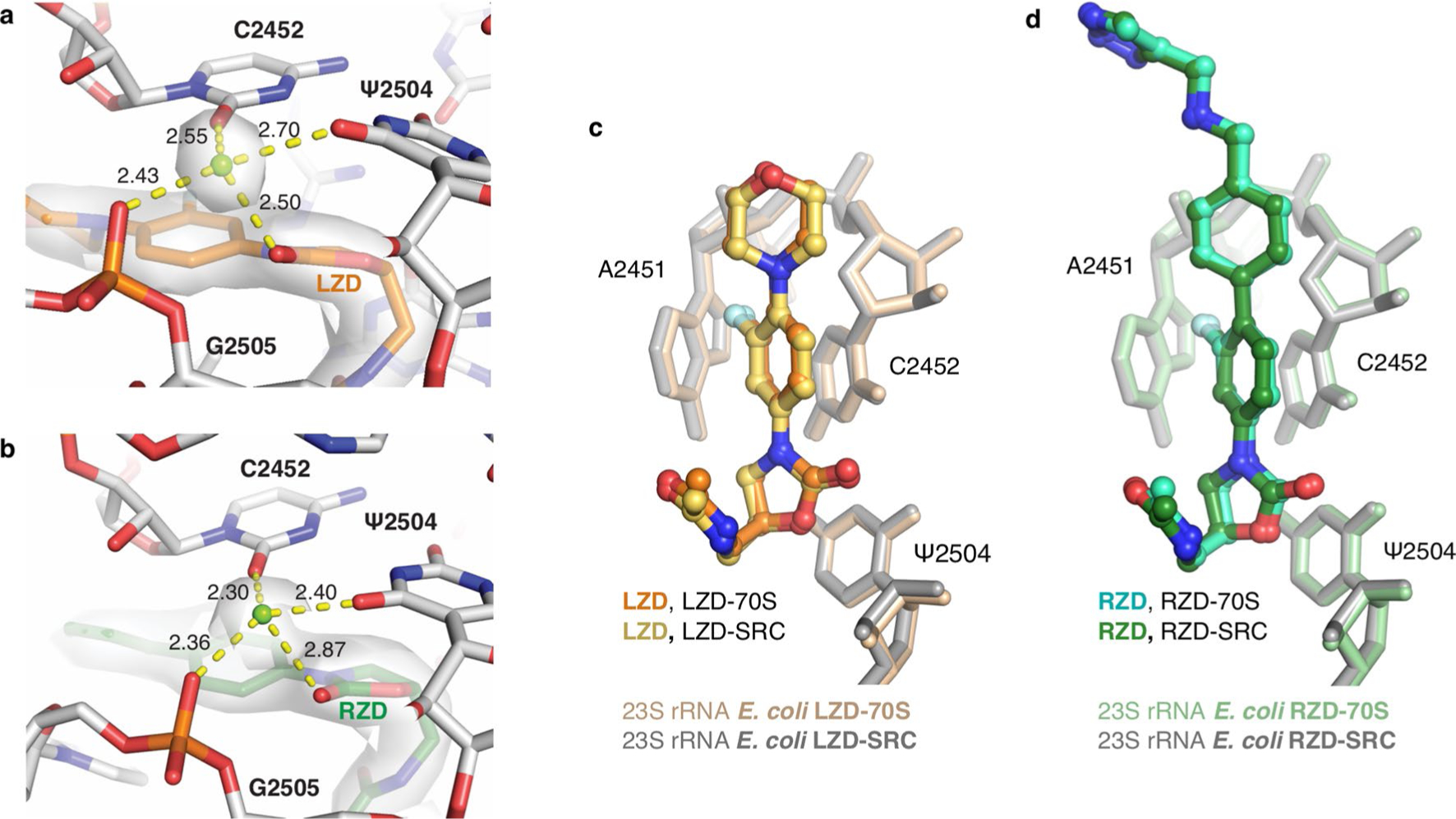
(a) Coordination of a solvent molecule or ion to (a) linezolid (LZD) and surrounding nucleic acids in the linezolid-stalled E. coli ribosome complex (LZD-SRC). Coulomb potential is shown 1.5 Å from the ion and LZD ligand, contoured at 5σ in the unsharpened map, and coordination distances are shown in Ångstroms. (b) Coordination of a solvent molecule or ion to radezolid (RZD) and surrounding nucleic acids in the radezolid-stalled E. coli ribosome complex (RZD-SRC). Coulomb potential is shown 1.5 Å from the ion and RZD ligand, contoured at 5s in the unsharpened map, and coordination distances are shown in Ångstroms. (c) Comparison of linezolid (LZD) binding modes in the antibiotic-only (LZD-70S, antibiotic in orange) versus LZD-stalled complex (LZD-SRC, antibiotic in yellow). (d) Comparison of radezolid (RZD) binding modes in the antibiotic-only (RZD-70S, antibiotic in teal) versus RZD-stalled complex (RZD-SRC, antibiotic in green). Structural overlays in this figure were performed by aligning the 23S rRNA chain.
Extended Data Fig. 5 |. In silico analysis of residues at the penultimate position.
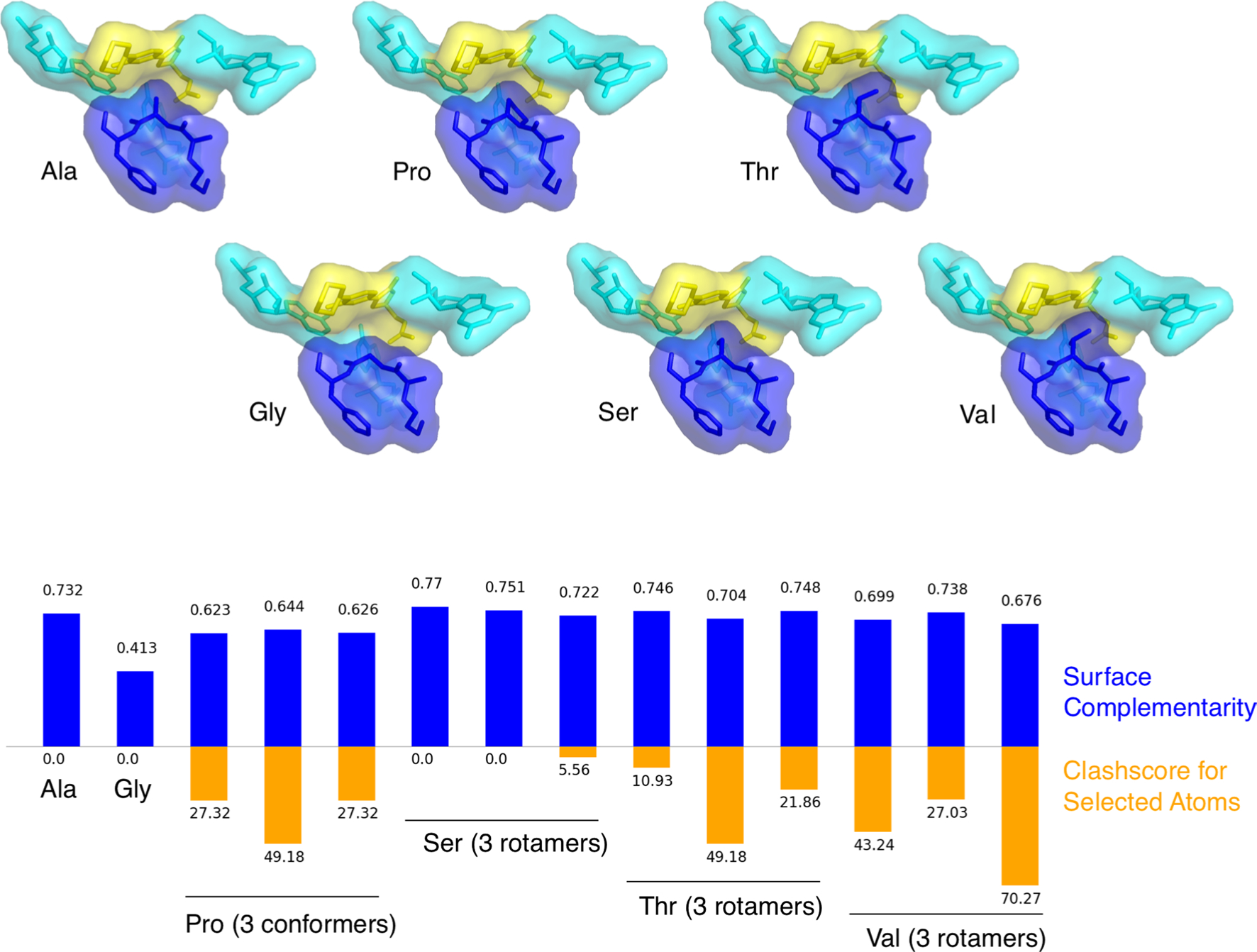
Substitution of glycine (Gly), proline (Pro), serine (Ser), threonine (Thr), or valine (Val) for alanine (Ala) at the penultimate position results in either reduced surface complementarity Sc as measured by the sc tool in CCP464 (blue bars) or increased clashscore (orange bars) in all cases except serine, which has two rotamers that slightly improve surface complementarity without clashing. Surface complementarity is calculated between two selections of atoms, the first of which is the sequence of three residues centered on the penultimate position, and the second of which is the linezolid ligand and the three nucleotides close enough to potentially interact either favorably or disfavorably with the peptide sequence in the first selection. Surface complementarity is calculated without hydrogens modeled due to limitations of the CCP4 implementation. Alanine and serine are most highly favored for ligand binding by these metrics.
Extended Data Fig. 6 |. Stabilization of PTC nucleotides in the oxazolidinone-stalled ribosome complexes.
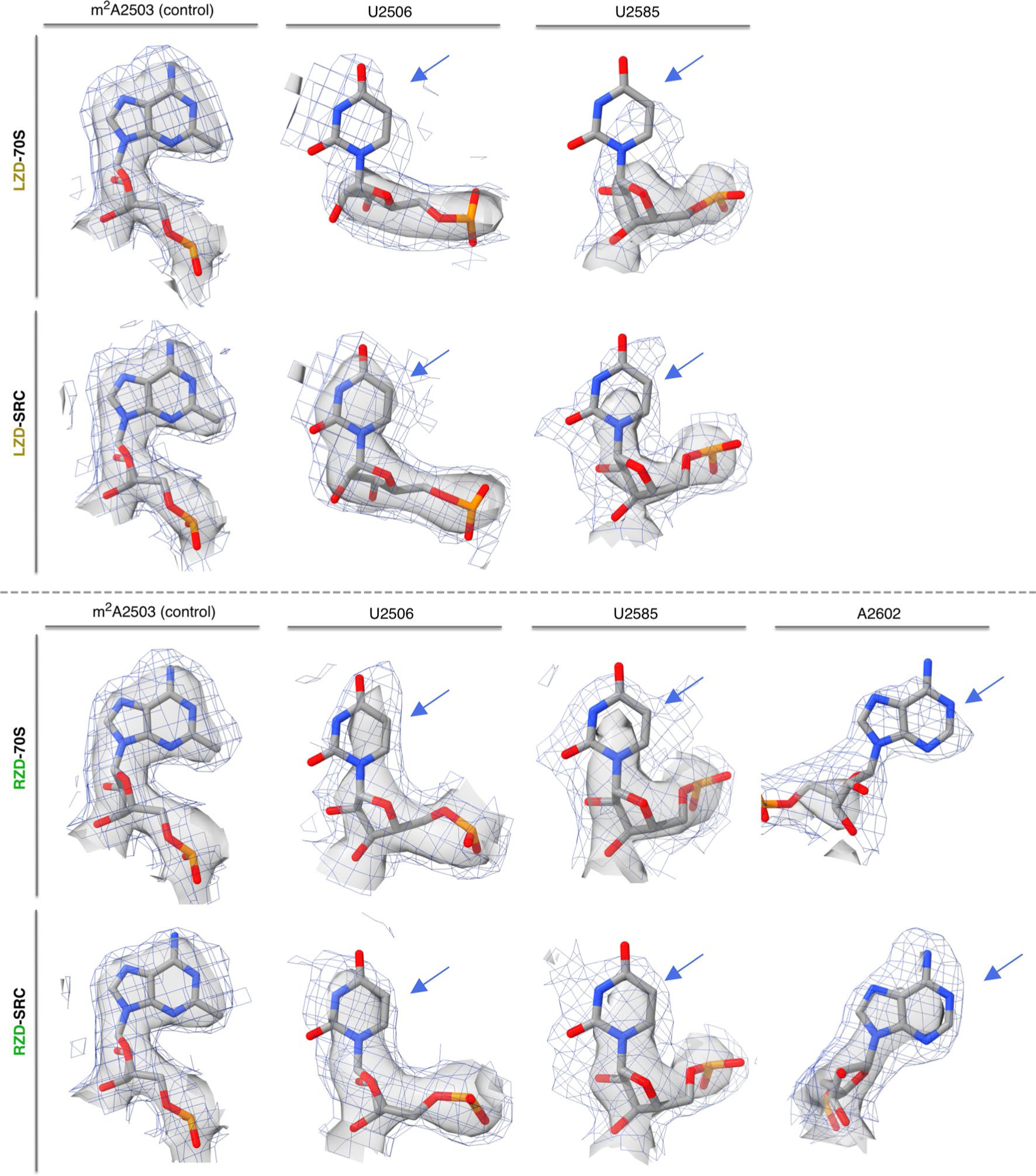
Direct comparison of nucleotide density in the linezolid-only bound (LZD-70S) and the linezolid-stalled complex (LZD-SRC), displaying m2A2503 as a control nucleotide and dynamic nucleotides U2506 and U2585. Direct comparison of nucleotide density in the radezolid-only bound (RZD-70S) and the radezolid-stalled complex (RZD-SRC), displaying A2503 and dynamic nucleotides U2506, U2585, A2602. Of note, U2506 is modeled in two conformations in LZD/RZD-70S; the relevant conformation for density comparison to SRCs is presented. Coulomb potential density is contoured at 4.0σ in surface representation and 1.0σ in mesh representation from unsharpened cryo-EM density maps. Blue arrows highlight the rRNA bases which exhibit the most prominent changes in density between the antibiotic-only and stalled complexes.
Extended Data Fig. 7 |. Steric occlusion of A-site tRNA binding by oxazolidinone antibiotics.
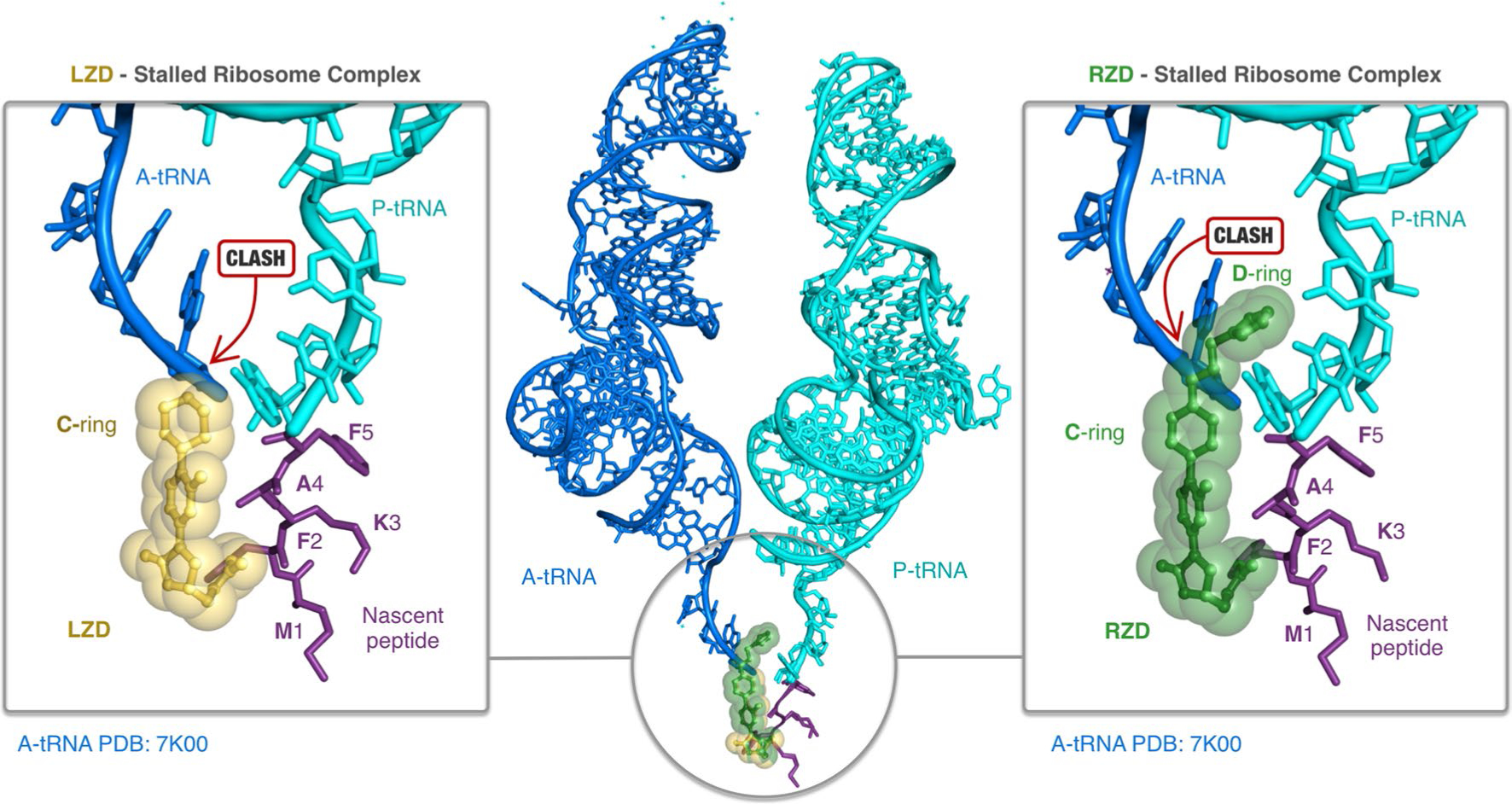
Linezolid (LZD, yellow) and radezolid (RZD, green) prevent binding of A-site tRNAs (blue, from PDB: 7K00). Prominent steric clashes between the A-tRNA and LZD C-ring and RZD C/D-ring are highlighted. Structural overlays were performed by alignment of 23S rRNA nucleotides.
Extended Data Fig. 8 |. LZD and RZD activity against Cfr-modified ribosomes.
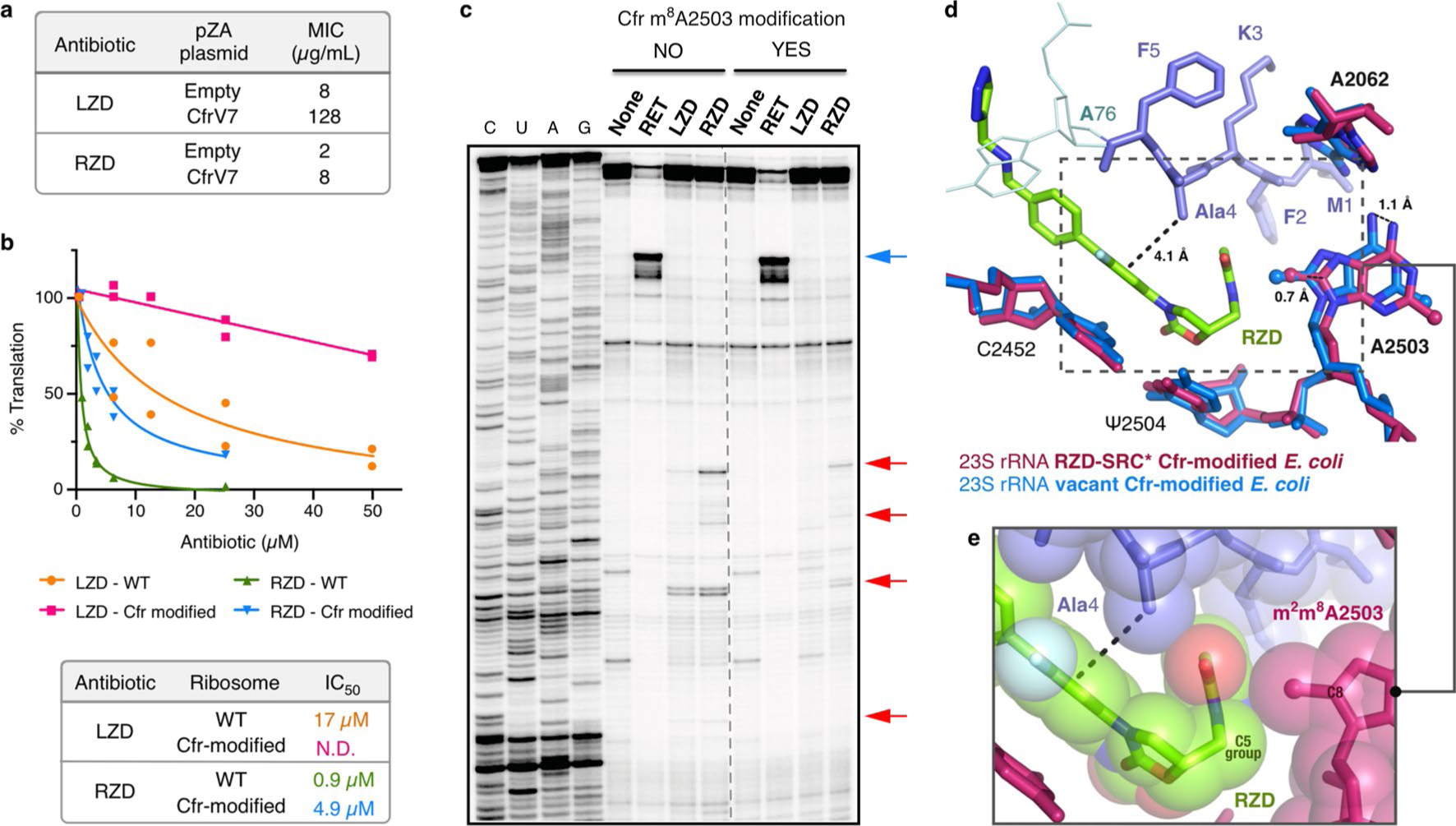
(a) Minimum inhibitory concentration of linezolid (LZD) and radezolid (RZD) required to inhibit growth of E. coli BW25113 acrB::kan transformed with either empty pZA plasmid or pZA encoding the evolved Cfr variant CfrV7, which achieves near-complete m8A2503 methylation43. MIC values were determined from two biological replicates. (b) In vitro activity of LZD and RZD against wildtype (WT) and Cfr-modified ribosomes determined by inhibition of sfGFP translation. Percent (%) translation calculated as the percentage of sfGFP translation at the tested antibiotic concentration compared to reactions containing no antibiotic determined from two independent experiments which are plotted as individual data points. N.D. indicates that the IC50 value was not determined. (c) Toeprinting analysis of LZD- and RZD-induced stalling of WT or Cfr-modified ribosomes within the 5’ region of the sfGFP ORF. Drug-specific toeprint bands are indicated by red arrows. ‘None’ designates reactions lacking ribosome-targeting antibiotics. The control antibiotic retapamulin (RET) was used to stall ribosomes at the start codon indicated by the blue arrow51. All antibiotics were added to a final concentration of 50 μM. Toeprinting experiments were performed at least twice with similar results. (d) Overlay of a vacant Cfr-modified E. coli ribosome43 with the RZD-stalled, Cfr-modified E. coli ribosome performed by alignment of 23S rRNA nucleotides 2000–3000. (e) Close-up view of the penultimate alanine, RZD, and C8-methylated A2503 in sphere representation.
Extended Data Fig. 9 |. Density for the nascent chain in WT and Cfr-modified RZD-stalled ribosome complexes.
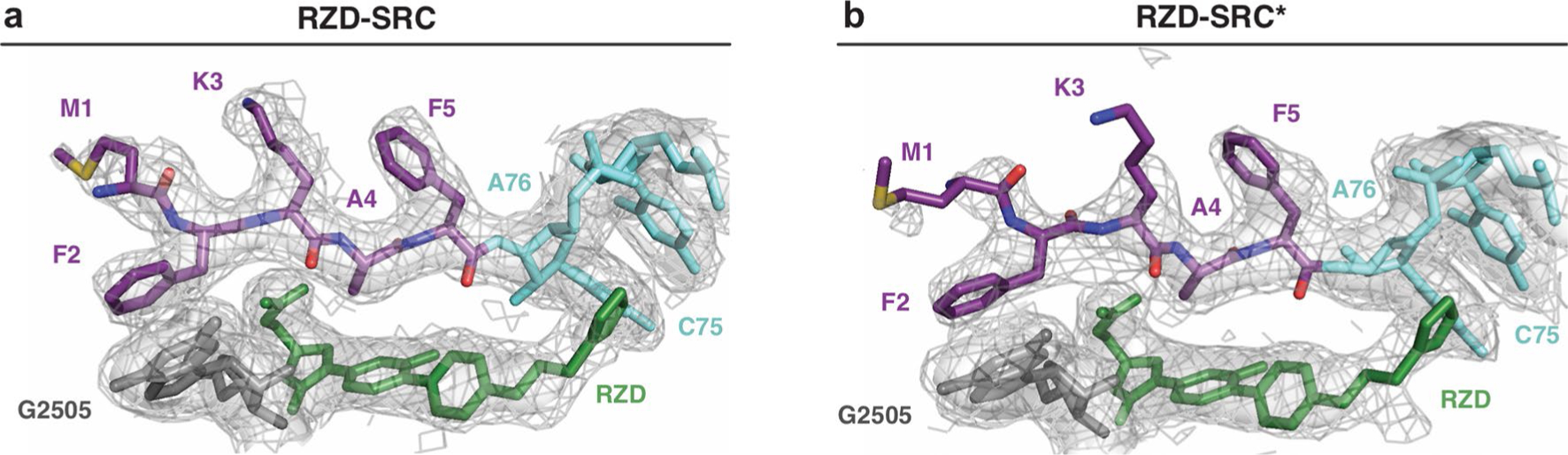
Density comparison for the alanine-containing MFKAF nascent peptide (purple) between RZD-stalled complexes with (a) WT and (b) Cfr-modified ribosome. RZD shown in green and G2505 shown in gray. Coulomb potential density is contoured at 4.0s in surface representation and 1.0s in mesh representation from unsharpened cryo-EM density maps and carved at 1.8 Ångstroms from the part of the model shown.
Extended Data Fig. 10 |. A2062 adopts a lumen conformation in the RZD-stalled complex with a Cfr-modified ribosome.
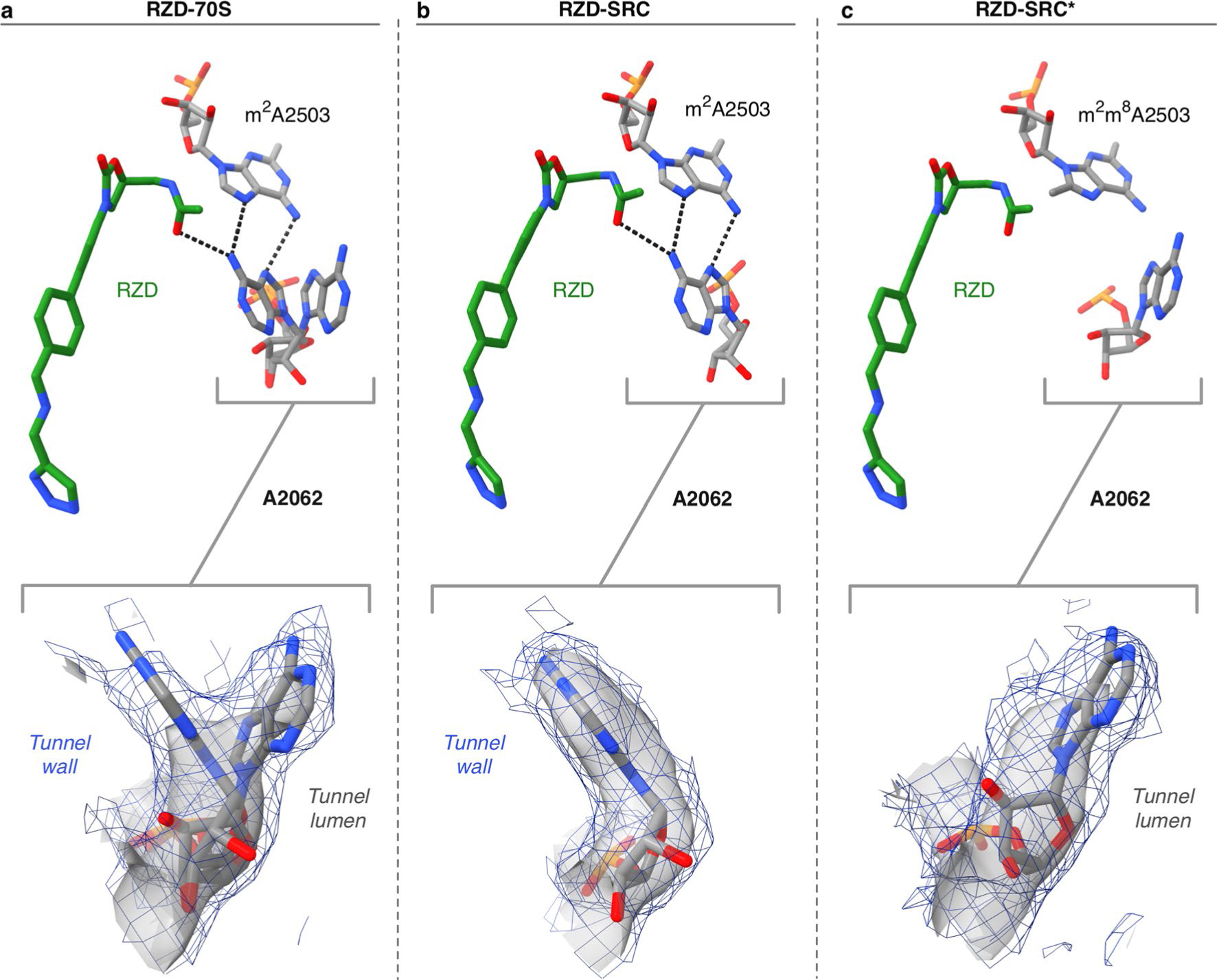
(a) A2062 adopts both the lumen and the rotated, tunnel wall conformation in the RZD-only bound ribosome. In the rotated wall conformation, A2062 engages in H-bonding interactions with A2503 and RZD. (b) A2062 is only observed in the rotated conformation to H-bond with A2503 and RZD in the WT stalled complex. (c) A2062 is observed in the lumen conformation in the RZD-stalled complex with a Cfr-modified ribosome. Coulomb potential density is contoured at 4.0s in surface representation and 1.0s in mesh representation from unsharpened cryo-EM density maps.
Supplementary Material
Acknowledgements
We thank D. Bulkley and G. Gilbert for technical support at the UCSF Center for Advanced CryoEM, which is supported by the National Institutes of Health (S10OD020054 and S10OD021741) and the Howard Hughes Medical Institute (HHMI). Some of this work was performed at the Stanford-SLAC Cryo-EM Center (S2C2), which is supported by the National Institutes of Health Common Fund Transformative High-Resolution Cryo-Electron Microscopy program (U24 GM129541). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank E. Eng, E. Kopylov and the rest of the staff for technical support at the National Center for CryoEM Access and Training (NCCAT) and the Simons Electron Microscopy Center located at the New York Structural Biology Center, which is supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129539) and by grants from the Simons Foundation (SF349247) and New York State. We acknowledge support from NIAID (R01AI137270 to D.G.F. and F32AI148120 to D.J.L.), a W.M. Keck Foundation Medical Research Grant (to J.S.F. and D.G.F.), a Sanghvi-Agarwal Innovation Award (J.S.F.), UCSF PBBR and Bowes Biomedical Investigator Program awards (D.G.F.), R01 AI 125518 (A.S.M. and N.V.-L.) and R35 GM 127124 (A.S.M.), NSF GRFP (1650113 to K.T.), the UCSF Discovery Fellowship (K.T.) and NIH F32-GM133129 (I.D.Y.).
Footnotes
Competing interests
The authors declare no competing interests.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41594-022-00723-9.
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this Article.
Extended data is available for this paper at https://doi.org/10.1038/s41594-022-00723-9.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41594-022-00723-9.
Peer review information Nature Structural & Molecular Biology thanks Daniel Wilson, Marina Rodnina and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Sara Osman was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
Atomic coordinates for all presented structures have been deposited in the Protein Data Bank and EMDB under the following accession numbers: LZD-SRC, PDB ID 7S1G, EMDB-24800; LZD-70S, PDB ID 7S1H, EMDB-24801; RZD-SRC, PDB ID 7S1I, EMDB-24802; RZD-70S, PDB ID 7S1J, EMDB-24803; RZD-SRC*, PDB ID 7S1K, EMDB-24804. The following datasets were used to generate the presented structures: WT E. coli 50S ribosomal subunit, PDB 6PJ6; ErmBL-stalled ribosome complex, PDB 5JU8. Source data are provided with this paper.
References
- 1.Wilson DN Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 12, 35–48 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Polacek N & Mankin AS The ribosomal peptidyl transferase center: structure, function, evolution, inhibition. Crit. Rev. Biochem. Mol. Biol. 40, 285–311 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Stevens DL, Dotter B & Madaras-Kelly K A review of linezolid: the first oxazolidinone antibiotic. Expert Rev. Anti. Infect. Ther. 2, 51–59(2004). [DOI] [PubMed] [Google Scholar]
- 4.Birmingham MC et al. Linezolid for the treatment of multidrug-resistant, gram-positive infections: experience from a compassionate-use program. Clin. Infect. Dis. 36, 159–168 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Colca JR et al. Cross-linking in the living cell locates the site of action of oxazolidinone antibiotics. J. Biol. Chem. 278, 21972–21979 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Leach KL et al. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Mol. Cell 26, 393–402(2007). [DOI] [PubMed] [Google Scholar]
- 7.Eyal Z et al. Structural insights into species-specific features of the ribosome from the pathogen Staphylococcus aureus. Proc. Natl Acad. Sci. USA 112, E5805–E5814 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson DN et al. The oxazolidinone antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc. Natl Acad. Sci. USA 105, 13339–13344 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ippolito JA et al. Crystal structure of the oxazolidinone antibiotic linezolid bound to the 50S ribosomal subunit. J. Med. Chem. 51, 3353–3356 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Marks J et al. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl Acad. Sci. USA 113, 12150–12155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi J et al. Dynamics of the context-specific translation arrest by chloramphenicol and linezolid. Nat. Chem. Biol. 16, 310–317 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long KS & Vester B Resistance to linezolid caused by modifications at its binding site on the ribosome. Antimicrob. Agents Chemother. 56, 603–612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belousoff MJ et al. Structural basis for linezolid binding site rearrangement in the Staphylococcus aureus ribosome. MBio 8, e00395–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson DN, Hauryliuk V, Atkinson GC & O’Neill AJ Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 18, 637–648 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Kehrenberg C, Schwarz S, Jacobsen L, Hansen LH & Vester B A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: methylation of 23S ribosomal RNA at A2503. Mol. Microbiol. 57, 1064–1073 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Long KS, Poehlsgaard J, Kehrenberg C, Schwarz S & Vester B The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins and streptogramin A antibiotics. Antimicrob. Agents Chemother. 50, 2500–2505 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giessing AMB et al. Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA 15, 327–336 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandit N, Singla RK & Shrivastava B Current updates on oxazolidinone and its significance. Int. J. Med. Chem. 2012, 159285 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw KJ & Barbachyn MR The oxazolidinones: past, present and future. Ann. N. Y. Acad. Sci. 1241, 48–70 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Swaney SM, Aoki H, Ganoza MC & Shinabarger DL The oxazolidinone linezolid inhibits initiation of protein synthesis in bacteria. Antimicrob. Agents Chemother. 42, 3251–3255 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant T, Rohou A & Grigorieff N cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright A et al. Characterization of the core ribosomal binding region for the oxazolidone family of antibiotics using cryo-EM. ACS Pharmacol. Transl. Sci. 3, 425–432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J et al. Design at the atomic level: design of biaryloxazolidinones as potent orally active antibiotics. Bioorg. Med. Chem. Lett. 18, 6175–6178 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Scaiola A et al. Structural basis of translation inhibition by cadazolid, a novel quinoxolidinone antibiotic. Sci. Rep. 9, 5634 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang K et al. Structural insights into species-specific features of the ribosome from the human pathogen Mycobacterium tuberculosis. Nucleic Acids Res. 45, 10884–10894 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belousoff MJ et al. CryoEM-guided development of antibiotics for drug-resistant bacteria. ChemMedChem 14, 527–531 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Renslo AR, Luehr GW & Gordeev MF Recent developments in the identification of novel oxazolidinone antibacterial agents. Bioorg. Med. Chem. 14, 4227–4240 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Syroegin EA et al. Structural basis for the context-specific action of the classic peptidyl transferase inhibitor chloramphenicol. Nat. Struct. Mol. Biol. 10.1038/s41594-022-00720-y (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vázquez-Laslop N, Ramu H, Klepacki D, Kannan K & Mankin AS The key function of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J. 29, 3108–3117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arenz S et al. Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol. Cell 56, 446–452 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koch M, Willi J, Pradère U, Hall J & Polacek N Critical 23S rRNA interactions for macrolide-dependent ribosome stalling on the ErmCL nascent peptide chain. Nucleic Acids Res. 45, 6717–6728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Chang ST-L, Ward FR & Cate JHD Selective inhibition of human translation termination by a drug-like compound. Nat. Commun. 11, 4941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svetlov MS et al. High-resolution crystal structures of ribosome-bound chloramphenicol and erythromycin provide the ultimate basis for their competition. RNA 25, 600–606 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmeing TM, Huang KS, Strobel SA & Steitz TA An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature 438, 520–524 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Toh S-M et al. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol. Microbiol. 64, 1506–1514 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonilla H et al. Multicity outbreak of linezolid-resistant Staphylococcus epidermidis associated with clonal spread of a cfr-containing strain. Clin. Infect. Dis. 51, 796–800 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Morales G et al. Resistance to linezolid is mediated by the cfr gene in the first report of an outbreak of linezolid-resistant Staphylococcus aureus. Clin. Infect. Dis. 50, 821–825 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Baos E, Candel FJ, Merino P, Pena I & Picazo JJ Characterization and monitoring of linezolid-resistant clinical isolates of Staphylococcus epidermidis in an intensive care unit 4 years after an outbreak of infection by cfr-mediated linezolid-resistant Staphylococcus aureus. Diagn. Microbiol. Infect. Dis. 76, 325–329 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Locke JB et al. Linezolid-resistant Staphylococcus aureus strain 1128105, the first known clinical isolate possessing the cfr multidrug resistance gene. Antimicrob. Agents Chemother. 58, 6592–6598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dortet L et al. Long-lasting successful dissemination of resistance to oxazolidinones in MDR Staphylococcus epidermidis clinical isolates in a tertiary care hospital in France. J. Antimicrob. Chemother. 73, 41–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan F et al. RlmN and Cfr are radical SAM enzymes involved in methylation of ribosomal RNA. J. Am. Chem. Soc. 132, 3953–3964 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan F & Fujimori DG RNA methylation by radical SAM enzymes RlmN and Cfr proceeds via methylene transfer and hydride shift. Proc. Natl Acad. Sci. USA 108, 3930–3934 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsai K et al. Directed evolution of the rRNA methylating enzyme Cfr reveals molecular basis of antibiotic resistance. Elife 11, e70017 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawrence L, Danese P, DeVito J, Franceschi F & Sutcliffe J In vitro activities of the Rx-01 oxazolidinones against hospital and community pathogens. Antimicrob. Agents Chemother. 52, 1653–1662 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Locke JB et al. Structure–activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations. Antimicrob. Agents Chemother. 54, 5337–5343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw KJ et al. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob. Agents Chemother. 52, 4442–4447 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhanel GG et al. Tedizolid: a novel oxazolidinone with potent activity against multidrug-resistant gram-positive pathogens. Drugs 75, 253–270 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Svetlov MS et al. Context-specific action of macrolide antibiotics on the eukaryotic ribosome. Nat. Commun. 12, 2803 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vazquez-Laslop N, Thum C & Mankin AS Molecular mechanism of drug-dependent ribosome stalling. Mol. Cell 30, 190–202 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Li W et al. Structural basis for selective stalling of human ribosome nascent chain complexes by a drug-like molecule. Nat. Struct. Mol. Biol. 26, 501–509 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meydan S et al. Retapamulin-assisted ribosome profiling reveals the alternative bacterial proteome. Mol. Cell 74, 481–493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stojkovic V et al. Assessment of the nucleotide modifications in the high-resolution cryo-electron microscopy structure of the Escherichia coli 50S subunit. Nucleic Acids Res. 48, 2723–2732 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wiegand I, Hilpert K & Hancock REW Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Orelle C et al. Identifying the targets of aminoacyl-tRNA synthetase inhibitors by primer extension inhibition. Nucleic Acids Res. 41, e144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohashi H, Kanamori T, Osada E, Akbar BK & Ueda T Peptide screening using PURE ribosome display. Methods Mol. Biol. 805, 251–259 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Bundy BC & Swartz JR Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein– protein click conjugation. Bioconjug. Chem. 21, 255–263 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Croll TI ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 74, 519–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arenz S et al. A combined cryo-EM and molecular dynamics approach reveals the mechanism of ErmBL-mediated translation arrest. Nat. Commun. 7,12026(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moriarty NW, Grosse-Kunstleve RW & Adams PD Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pettersen EF et al. UCSF ChimeraX: structure visualization for researchers, educators and developers. Protein Sci. 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr 50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates for all presented structures have been deposited in the Protein Data Bank and EMDB under the following accession numbers: LZD-SRC, PDB ID 7S1G, EMDB-24800; LZD-70S, PDB ID 7S1H, EMDB-24801; RZD-SRC, PDB ID 7S1I, EMDB-24802; RZD-70S, PDB ID 7S1J, EMDB-24803; RZD-SRC*, PDB ID 7S1K, EMDB-24804. The following datasets were used to generate the presented structures: WT E. coli 50S ribosomal subunit, PDB 6PJ6; ErmBL-stalled ribosome complex, PDB 5JU8. Source data are provided with this paper.