

Abstract
Drug isomers may differ in their proarrhythmia risk. An interesting example is the drug sotalol, an antiarrhythmic drug comprising d- and l- enantiomers that both block the hERG cardiac potassium channel and confer differing degrees of proarrhythmic risk. We developed a multi-scale in silico pipeline focusing on hERG channel – drug interactions and used it to probe and predict the mechanisms of pro-arrhythmia risks of the two enantiomers of sotalol. Molecular dynamics (MD) simulations predicted comparable hERG channel binding affinities for d- and l-sotalol, which were validated with electrophysiology experiments. MD derived thermodynamic and kinetic parameters were used to build multi-scale functional computational models of cardiac electrophysiology at the cell and tissue scales. Functional models were used to predict inactivated state binding affinities to recapitulate electrocardiogram (ECG) QT interval prolongation observed in clinical data. Our study demonstrates how modeling and simulation can be applied to predict drug effects from the atom to the rhythm for dl-sotalol and also increased proarrhythmia proclivity of d- vs. l-sotalol when accounting for stereospecific beta-adrenergic receptor blocking.
Keywords: Ion channel, Molecular dynamics, Arrhythmia, Beta-blocker, Stereochemistry, Enantiomer
1. Introduction
Drug stereoisomers are increasingly viewed as potentially useful means to develop and test pharmacological agents [1,2] due to potentially more potent and/or selective action on their intended protein targets. Stereoisomerism also plays a crucial role in drug toxicity profiles with enantiomers possessing different, on- and off-target effects and capacities for metabolic degradation [3]. This phenomenon is well-described in the drug-induced cardiac toxicity [4] for commonly used drugs including an opioid methadone [5,6], an anti-malarial drug quinine and its diastereomer quinidine [7,8], the local anesthetics bupivacaine and ropivacaine [9–13] as well as for the calcium channel blocker verapamil used in the context of cancer chemotherapy [14,15].
A fascinating and puzzling example of isomerism to impact cardiac safety is the antiarrhythmic sulfonamide drug sotalol. The d-isomer was infamously shown in the SWORD (Survival With ORal D-sotalol) clinical trial [16] to increase mortality and risk of sudden cardiac death in patients, leading to its withdrawal [17]. The racemic mixture comprising d- and l-sotalol, however, is widely used as an effective antiarrhythmic – although not entirely without risk [18–29]. For instance, a prevalence of torsades de pointes (TdP) arrhythmia was estimated to be 0.1–7% depending on therapeutic doses, sex (significantly larger for women) and co-morbidities [28,30–35]. The apparent disparity in cardiac risks of the racemic mixture compared to a pure d-enantiomer could be attributed to differences in interactions between sotalol stereoisomers and cardiac K+ channel proteins, or from the higher efficacy of l-sotalol to act as a beta-blocker [36–40]. Here, we developed a multiscale approach combining cellular and tissue functional kinetic modeling with atomistic simulations and experiments to reveal the specific ion channel interactions with d- and l-sotalol and unveil elusive molecular mechanisms underlying stereospecificity of arrhythmia provoking drug-induced cardiotoxicity.
Cardiotoxicity in the form of abnormal heart rhythm is one of the most critical regulatory concerns for drugs and has resulted in withdrawal of a number of therapeutic agents. Drug-induced arrhythmia has been estimated to affect pharmaceuticals from multiple drug classes [41], with an estimated 3% of all prescription drugs worldwide harboring pro-arrhythmic side effects [42]. Indeed, cardiotoxicity is one of the leading causes of drug attrition [43], and accounts for 22–28% of US post-marketing drug withdrawal [44]. The problem of cardiotoxicity has even plagued drugs intended to treat arrhythmias like sotalol, encainide or flecainide [17,45]. Such deleterious drug side effects have been linked to blockade of the potassium current through the cardiac ion channel protein KV11.1, encoded by the human Ether-à-go-go-Related Gene (hERG) [46–48]. Block of cardiac hERG current (IKr), causes prolongation of the QT interval on the surface electrocardiogram (ECG), sometimes causing potentially deadly arrhythmias [48–50]. While not all hERG block and QT prolongation leads to increased risk of arrhythmia, there is no reliable method to distinguish unsafe hERG blockers from safe drugs [51,52], which hinders the pharmacological assessment of cardiotoxicity and may lead to attrition of safe and effective pharmaceuticals.
A recently proposed Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative is intended to address this need by improving predictions of drug pro-arrhythmia proclivities through the combination of multi-scale modeling and in vitro experimental assays [53,54]. However, the CiPA platform does not yet provide a ready-to-go recipe on how to predict drug arrhythmogenicity. The example of sotalol discussed in this study, where two stereoisomers have different pro-arrhythmia proclivities, is especially challenging. Atomistic molecular dynamics (MD) simulations of hERG – drug interactions, which we employed here, can be used to help identify stereoselective protein – drug interactions [55,56] and thus molecular determinants of drug-induced arrhythmogenesis. However, since cardiac arrhythmia is an emergent phenomenon, manifesting at the cardiac tissue and whole heart scales [57–60], we set out to create a link between atomistic-scale protein and drug structure-based MD simulations and functional kinetic simulations of cardiac cells and tissue. Recently, we developed such an integrative computational modeling approach that spans scales from the atom to the cardiac rhythm and were able to accurately predict pro-arrhythmia proclivities for dofetilide and moxifloxacin, hERG blockers with different cardiac safety profiles, and directly link them to clinical data [61].
Here, we set out to investigate and reveal the stereospecific molecular mechanisms of d- and l-sotalol causing ventricular arrhythmogenesis by applying a multi-scale modeling pipeline from the atomistic to the tissue scale. We have previously developed and validated atomistic force field parameters for cationic and neutral states of sotalol [62], and validated open conducting atomistic models of the hERG channel [61,63,64] based on a high-resolution cryo-EM structure (PDB ID: 5VA2) [65]. Sotalol is known to access the hERG channel pore via an open state, with subsequent hERG inactivation stabilizing the drug – channel interaction [66]. In this work, we used multi-microsecond long unbiased MD simulations with saturating sotalol concentrations to reveal key binding modalities for cationic and neutral d- and l-sotalol. Then, we used enhanced sampling MD simulations [61,63] to compute state-dependent affinities and “on”/”off” rates for open hERG channel – sotalol interactions, which we used as parameters for our functional kinetic model. We optimized the model for inactivated state block to reconcile and predict emergent pro-arrhythmia risks at cardiac cell and tissue scales [61]. To test model predictions, we obtained d- and l-sotalol through enantiomeric separation and conducted electrophysiological recordings of their hERG channel block. We found the affinities of both enantiomers to hERG channel to be comparable. Our MD simulation informed functional cardiac cell and tissue models predicted sotalol induced QT interval prolongation due to a combined effect of hERG channel block and stereoselective beta-adrenergic receptor (βAR) inhibition to be in good agreement with clinical data.
2. Methods
A brief overview is provided below. See Appendix A Supplementary Materials for a more detailed description.
2.1. Atomistic simulations
We used our atomistic structural model of open hERG channel [61], based on the cryo-EM structure (PDB ID: 5VA2) [65] and generated using Rosetta molecular modeling software [67–69] as well as all-atom force field parameters of cationic and neutral sotalol [62]. Standard CHARMM36 all-atom force fields for protein, lipid, ions [70–72] and TIP3P water model [73] were used as well. Our hERG channel model was embedded in a 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) bilayer and solvated by 0.15 M aqueous KCl solution and sotalol molecules using CHARMM-GUI [74] and equilibrated using a staged protocol by gradually reducing restraints [63] using NAMD program [75]. 8 μs long unbiased MD simulations with 23 sotalol molecules in aqueous solution corresponding to an initial 0.05 M concentration were run on Anton 2 [76]. Steered MD simulations with a sotalol molecule pulled from an intracellular aqueous into the hERG channel pore in five restrained 90 ns runs were used to seed umbrella sampling MD (US-MD) simulations [77]. Positional harmonic restraints for pore domain Cα and selectivity filter (SF) backbone non-H atoms to preserve an open hERG channel conformational state were reduced from 1.0 kcal·mol−1·Å−2 in steered MD runs to 0.2 kcal·mol−1·Å−2 during first 5 ns of US-MD runs and kept for the remainder of those and for the entire Hamiltonian replica exchange US-MD (HREUS-MD) [78] simulations. hERG channel residue sidechains and sotalol conformations were not restrained. US-MD simulations ran for 50 ns (including 10 ns equilibration) per each US-MD window, located at −50 ≤ z ≤ −5.5 Å in 0.5 Å intervals using 10 kcal·mol−1·Å−2 harmonic restraints. z = 0 is the center of mass of hERG SF Cα atoms. HREUS-MD simulations followed US-MD runs after 20 ns and continued for 60 ns per each window. Free energy profiles and diffusion coefficients were computed from those simulations and used to calculate dissociation constants (KD) as well as drug “on” (kon) and “off” (koff) rates [61] used as our functional model parameters.
2.2. Functional simulations
We used a previously published approach [61] to develop functional kinetic models of hERG channel – sotalol interactions based on the wild-type drug-free hERG Markov model [61]. MD-derived KD, kon and koff values were used to simulate open-state hERG channel block, whereas for the inactivated state block KDi was assumed to be 4.3-fold less in the model 1 based on ref. [79] and 63-fold less than KDo in the model 2 using a top-down optimization approach and ref. [80]. Rates for the inactivated state block were optimized using experimental dose response curves from this work and ref. [80] for models 1 and 2, respectively, and expression koff = kon · KD. These hERG – sotalol models were implemented into the O’Hara-Rudy human ventricular cardiac myocyte model [81] to simulate QT interval in pseudo-ECG recordings from a 1-dimensional strand of O’Hara-Rudy cells paced at 1000 ms basic cycle length (BCL) as was done previously [61]. Beta-adrenergic stimulation by isoproterenol (ISO) was implemented through setting protein kinase A (PKA) phosphorylation effect to maximum, and its inhibition by sotalol, i.e. beta-blockade, was modeled as 1/(1 + [sotalol]/Ki) where [sotalol] is drug’s plasma concentration and Ki is its inhibition constant, which is 38,000 ng/ml for d-sotalol and 650 ng/ml for l-sotalol [38]. Sotalol-induced QT prolongation was compared with clinical data at the matching BCL (RR interval) [82].
2.3. Experimental methods
Free sotalol base was prepared from sotalol hydrochloride salt through dissolving it in methanol and KOH solution, separation of supernatant, dissolving it in ethyl acetate, filtering and evaporating. High performance liquid chromatography (HPLC) resolution of the two sotalol enantiomers from free base sotalol was performed with an Agilent 1100 HPLC equipped with a Chiralpak IA 4.6 × 250 mm column, heated at 25 °C. An isocratic solvent system consisting of 20% mobile phase A (ethanol with 0.1% diethyl amine) and 80% mobile phase B (hexane with 0.1% diethyl amine) was used for 15 min for each run. Sotalol was detected by its absorption at 220 nm using an Agilent 1100 diode array detector (DAD). Under these conditions (S) sotalol enantiomer has tR = 7.99 min, while (R) sotalol enantiomer has tR = 11.88 min. Multiple runs using this method furnished about 25 mg of each enantiomer with almost 100% enantiomeric excess. The separated enantiomers were characterized by 1H NMR and proton decoupled 13C NMR (13C-NMR1H) spectra as well as X-ray crystallography using established methodology [83]. Please see details in the Appendix B Data Supplement and linked spectra files therein.
For electrophysiological recordings we used a human embryonic kidney (HEK) 293 cell line stably expressing hERG maintained in minimum essential medium supplemented with 10% fetal bovine serum and 400 μg/ml G418 as previously described [84]. HEK cells were continuously superfused with HEPES-buffered Tyrode solution. Membrane currents were recorded in the whole-cell configuration established using pipette suction [85]. Leak compensation was not used. Experiments were performed at room temperature (22–25 °C). The data were stored on a computer hard disk and analyzed using PatchMaster and Igor Pro 7 (WaveMetrics, Portland, OR). The IC50 for hERG block by sotalol were measured by fitting the data to Hill’s equation as follows:
where f(D) is the current ratio in the presence of the drug at a given concentration (D) and h is the Hill coefficient.
The data and datasets generated and analyzed in the current study can be accessed upon request sent to the corresponding author of the manuscript.
3. Results
3.1. hERG channel binding sites and pathways from sotalol flooding simulations
We previously developed atomistic force field parameters for cationic(+) and neutral(0) forms of sotalol and used MD simulations of their lipid membrane partitioning to validate those models showing water – membrane interface accumulation of sotalol(0) as well as significant free energy barrier and membrane perturbation for sotalol(+) [62]. This indicates ionization state-dependent energetics and kinetics of sotalol – lipid membrane interactions and may affect how the drug gains access to the hERG channel pore.
Here, we explicitly explored this using 8 μs long unbiased MD simulations of our open-state hERG channel model embedded in a POPC lipid bilayer and soaked by an aqueous solution with 0.15 M KCl and an initial 0.05 M concentration of sotalol in water, corresponding to 23 molecules in the simulation box. Utilizing these so-called drug flooding MD simulations, we were able to elucidate potential entry pathways and binding sites for d- and l- sotalol(+) and sotalol(0) interacting with the hERG channel. The distributions for cationic and neutral forms of each enantiomer were notably different (Fig. 1 and Appendix A Figs. S6–S7). d- and l-sotalol(0) (Fig. 1B, left panels) were found to embed into the membrane and interact with hERG channel, whereas d- and l-sotalol(+) (Fig. 1B, right panels) remain predominantly in aqueous solution, sampling much more of the solvent space over the duration of the simulation (see drug aqueous concentration time series and atomic density distribution plots, Appendix A Figs. S6 and S7).
Fig. 1.
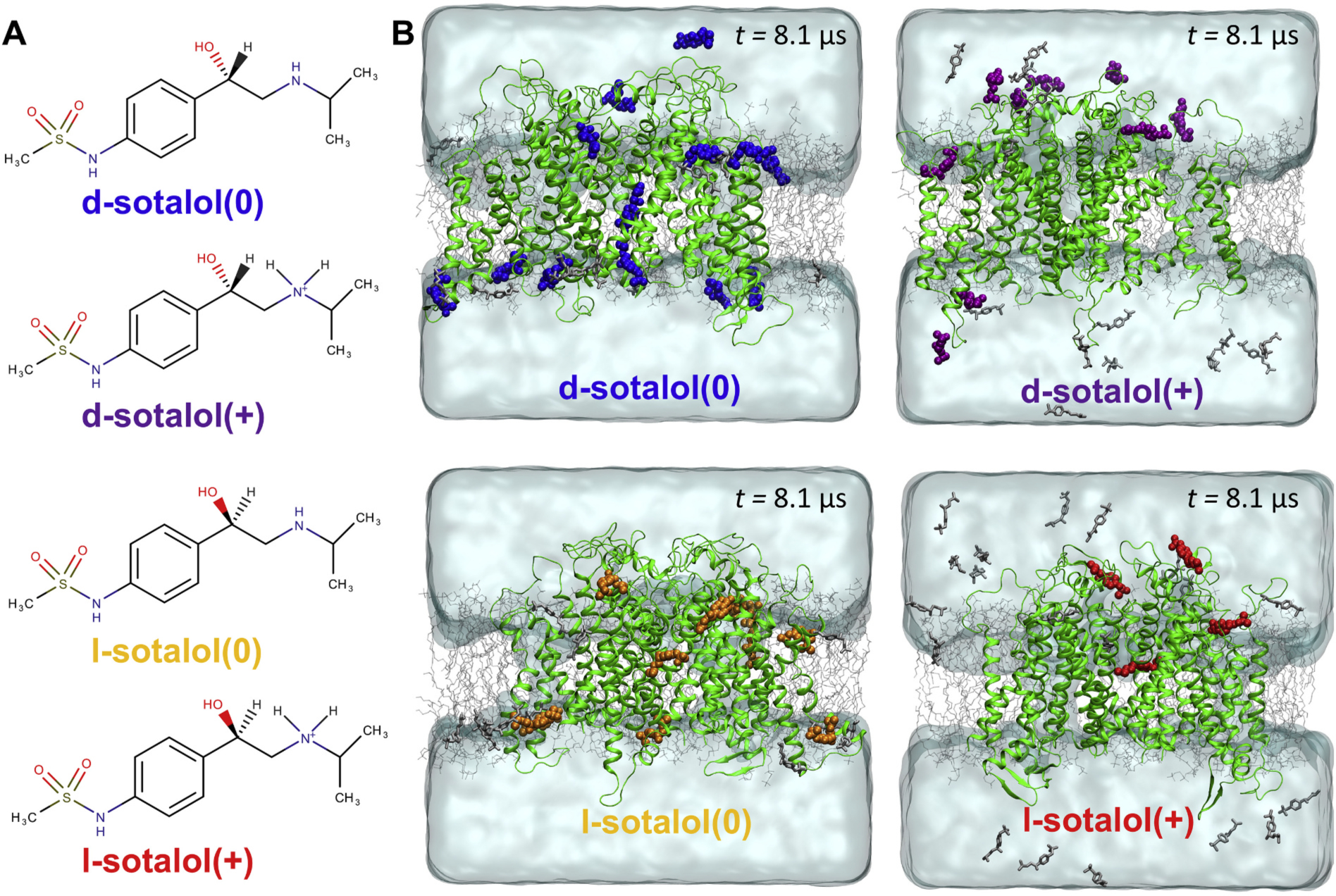
Distribution of cationic(+) or neutral(0) d- or l-sotalol around the hERG channel from multi-μs long unbiased MD simulations. (A) Chemical structures of neutral(0) and cationic(+) forms of d- and l-sotalol (B) Snapshots of the molecular systems consisting of the hERG channel embedded in the POPC bilayer, solvated with aqueous 150 mM KCl and initial 50 mM sotalol solution, at the end of 8.1 μs MD simulations. For sotalol molecules within 3.5 Å of hERG protein residues non‑hydrogen atoms are shown in the colored space filling representation, non-interacting sotalol molecules are shown as gray sticks. The hERG channel is shown as green ribbons, POPC lipid tails as thin gray sticks, water as aquamarine surface, K+ and Cl− ions are not shown for clarity. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Dominant binding poses of cationic and neutral d- and l-sotalol are shown in Fig. 2, whereas time series of drug binding z positions in the hERG pore are shown in Appendix A Fig. S8. Interestingly, one d-sotalol (0) or l-sotalol(+) molecule was observed to bind deep into the hERG pore, just below the SF region and interact with the canonical drug binding F656 and Y652 hERG residues [86–90] in the pore lining S6 helices (drug molecule M2 in Figs. 2A and D) for most of 8 μs long MD runs, while another drug molecule was observed to transiently bind below, interacting with F656 and/or Y652 from another domain as well as S660 and other residues at the bottom of S6 helices (M1 in Figs. 2A and D). Interestingly, for l-sotalol(0) and d-sotalol(+) systems, we observed only transient binding of one or two drug molecules at the bottom of hERG channel pore interacting with S660 and other S6 helix residues there (Figs. 2B and C and Appendix A Fig. S8B and C) Hence up to two sotalol molecules were able to bind to the hERG pore, in agreement with our electrophysiological data (as described below). Sotalol flooding MD simulations demonstrated that d- and l-sotalol are likely to enter the hERG channel pore through the aqueous intracellular gate and did not reveal any drug entry through lipid facing fenestrations as in the case of NaV channels [91–94] and also suggested for some hERG binding drugs such as ivabradine [95,96]. Thus, sotalol hERG pore binding pathways closely resemble those for dofetilide [61,63,95,96], which also has methanesulfonanilide moieties and fairly similar polarity [97].
Fig. 2.
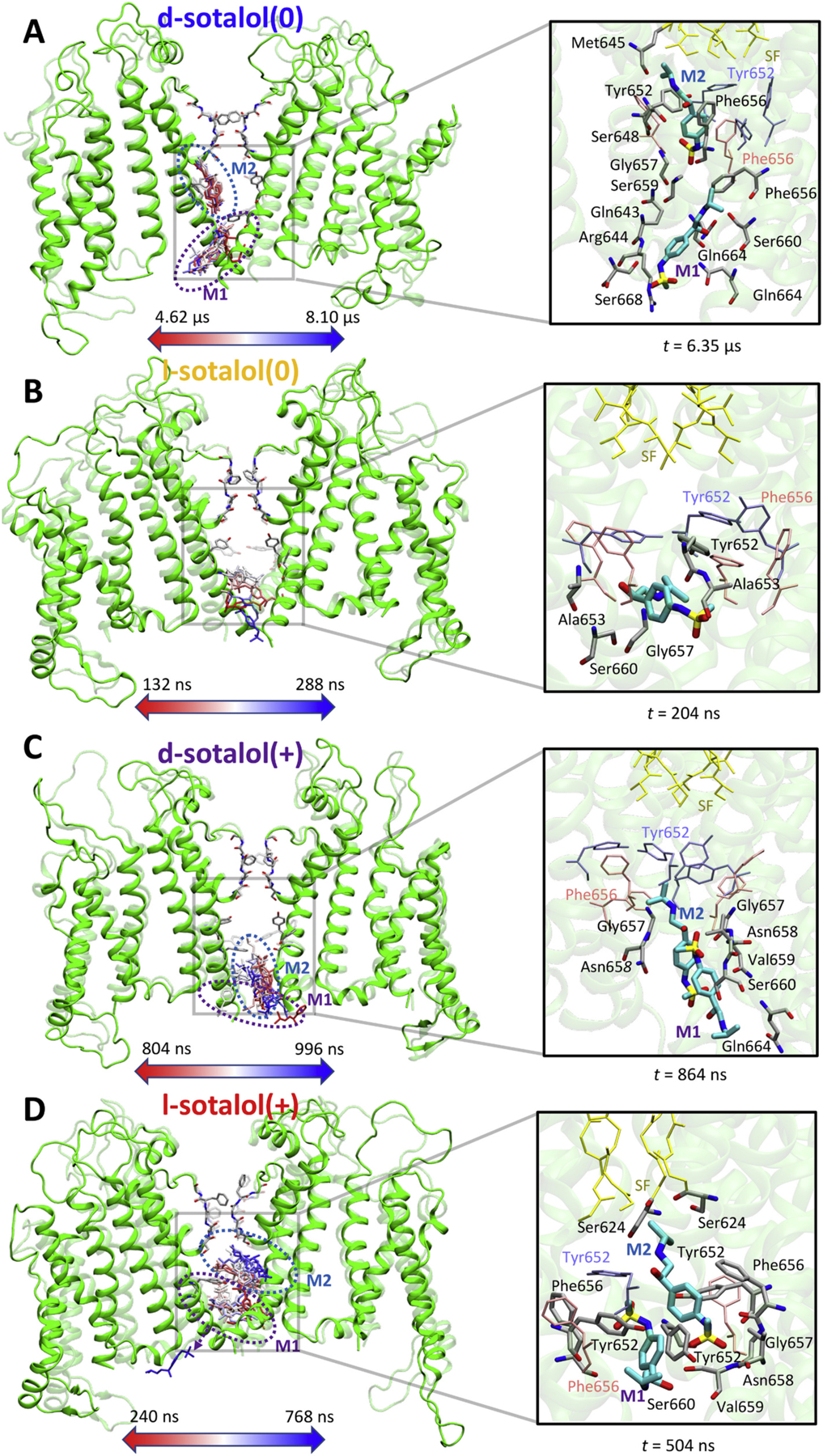
Binding sites of neutral(0) or cationic(+) d- or l-sotalol around the hERG channel from 8.1 μs long unbiased MD simulations. (A) d-sotalol(0); (B) l-sotalol(0); (C) d-sotalol(+); (D) l-sotalol(+). Left panels: Time-series rendering for binding of one or two sotalol molecules (labeled M1 and M2) within the hERG pore. Sotalol molecules in the frames are shown by colored sticks from the beginning (red) to the end (blue) of each representative binding event. The hERG channel is shown in the initial (transparent green ribbons) and the final (solid green ribbons) conformations. Canonical drug interacting residues Phe656 and Tyr652 as well as selectivity filter (SF) residues are shown as solid or transparent atom-colored ribbons (C – gray, O – red, N – blue). Right panels: Representative binding poses adopted by sotalol molecules (thick atom-colored sticks with C – cyan, S – yellow, others as above) in the hERG channel pore. Interacting hERG residues (within 3.5 Å of any non-H atoms of the drug) are shown as thick atom-colored sticks (C – gray, others are as above). Non-interacting hERG residues Phe656, Tyr652 as well as its SF residues are shown as thin pink, blue and yellow sticks. Hydrogen atoms are not shown for clarity. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
These qualitative observations do not exclude other possibilities since even very long simulation times of 8 μs might not be sufficient to fully sample other potential drug binding poses and pathways, which can be related to distortions of the hERG pore domain (Appendix A Fig. S9) including partial asymmetric pore closure as evidenced by decrease in interchain distances for S6 segment F656 and/or S660 residues (Appendix A Fig. S10) as well as SF distortions revealed by interchain distances of S624 to G628 SF residues (see Appendix A Fig. S11). These structural changes are similar to those observed for a non-conducting state of the hERG channel during a multi-microsecond long MD simulation under an applied transmembrane voltage [63] and may be indicative of a channel transition to a different conformational state, possibly inactivated. Moreover, sotalol contacts with hERG residues during drug flooding MD simulations were asymmetric and highly variable among MD runs (See Appendix A Figs. S12–S13) due to long-lasting interactions (Appendix A Fig. S14).
3.2. Energetics of sotalol – hERG channel binding from enhanced sampling simulations
Our multi-microsecond long drug flooding simulations indicated that enhanced sampling MD runs of a drug entry into the channel pore through an intracellular gate are needed to obtain quantitative information about hERG – sotalol interactions. Therefore, we applied umbrella sampling MD (US-MD) [77], and Hamiltonian replica exchange umbrella sampling MD (HREUS-MD) [78] techniques to compute drug binding affinities and rates. Weak harmonic restraints were applied to hERG pore domain backbone atoms to preserve open hERG conformational state.
First, we investigated binding poses of different sotalol forms, corresponding to free energy minima from US-MD runs as shown in Fig. 3. Both d- and l-sotalol occupy similar positions within the hERG pore (Fig. 3A) and interact with the canonical binding residues F656 and Y652 (see Fig. 3B). Sotalol molecules along with interacting protein side chains, which are not restrained during the simulations, adopt different orientations and conformations (see Fig. 3B and Appendix A Figs. S15–S20) to optimize those interactions. d- and l-sotalol(0) were bound slightly deeper into the pore than their cationic counterparts. Interestingly, all sotalol forms were found to bind just below a ring of Y652 residues, interacting with them, as long as other S6 segment residues from multiple chains (see Fig. 3B) in general agreement with previous studies [98,99]. Use of HREUS-MD methodology allowed us to facilitate sampling of multiple drug orientations and conformations in the hERG channel pore (Appendix A Figs. S16-S20).
Fig. 3.
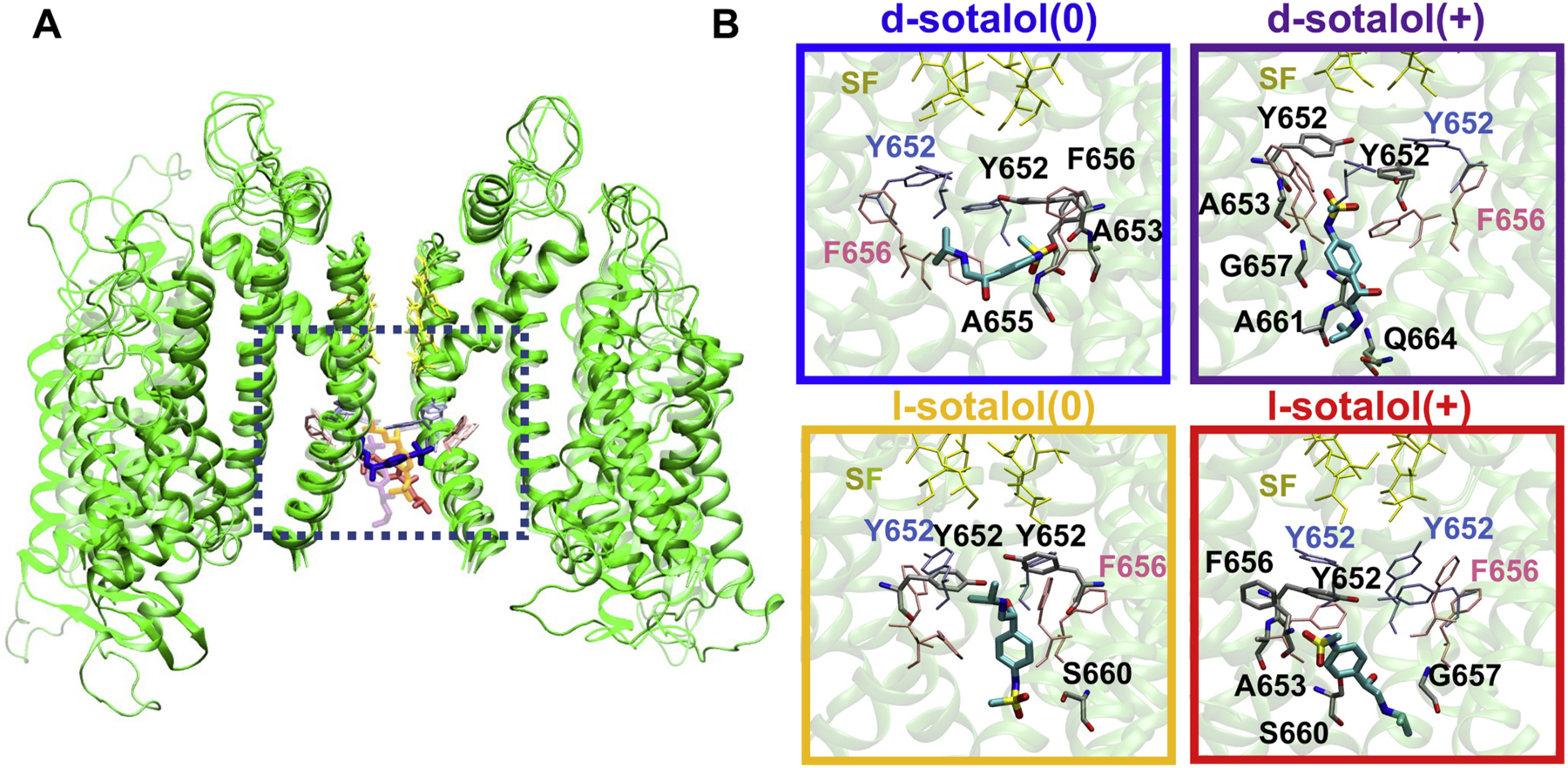
Representative binding poses of neutral(0) and cationic(+) d- and l-sotalol to open hERG channel obtained from US-MD simulations. (A) hERG channel – bound sotalol structures from four US-MD runs corresponding to free energy minima for each simulation are superimposed and represented by different shades. Two opposite chains of the open-state hERG channel structures are shown as green ribbons. Bound sotalol molecules are shown as thick colored sticks: d-sotalol(0) – blue, d-sotalol(+) – purple, l-sotalol(0) – orange, and l-sotalol(+) – red. hERG SF residues are shown as yellow thin sticks, and canonical binding residues F656 and Y652 as thin pink and ice-blue sticks. (B) Close-up views of sotalol hERG binding poses corresponding to a dotted black box location in panel A. Sotalol molecules are shown as thick atom-colored sticks (C – cyan, N – blue, O – red, S – yellow). hERG channel is shown by transparent green ribbons with residues within 3.5 Å of sotalol non‑hydrogen atoms shown by thin atom-colored sticks (C – gray, N – blue, O – red). Non-interacting SF, F656 and Y652 residues are shown by thin colored sticks as in panel A. Hydrogen atoms are not shown for clarity. Box border coloration in panel B corresponds to coloration of each isoform of d- and l-sotalol in panel A. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
US-MD and HREUS-MD simulations allowed us to estimate the free energies of drug binding, ΔGbind, and to compute dissociation constants (KD) [61,63] of cationic and neutral d- or l-sotalol binding to the hERG channel in the open state (see Table 1, Fig. 4 and Appendix A Figs. S21–S22), which shows good agreement between results obtained with US-MD and HREUS-MD methodologies. Our computed dissociation constants, KD, indicate one to two orders of magnitude stronger binding of neutral d- or l-sotalol to open hERG channel compared to their cationic counterparts (Table 1) in agreement with our previous results for dofetilide and moxifloxacin [61]. Combined KD values (taking into account neutral and cationic drug contributions at physiological pH = 7.4) of about 2 mM are very similar for d- vs. l-sotalol, and also for US-MD and HREUS-MD simulations (Table 1). They are also in excellent agreement with racemic sotalol IC50 values for a non-inactivated hERG S620T mutant [79].
Table 1.
Open hERG pore sotalol binding data from US-MS and HREUS-MD simulations. Molecular dynamics (MD) computed drug binding free energies (ΔGbind), dissociation constants (KD) and diffusion coefficients at the drug binding site (Dpore) as well as drug “on” (kon) and “off” (koff) rates used for parameterizing functional scale models. Data are means ± standard error of means (SEM) for 10-ns or 20-ns US-MD and HREUS-MD simulation blocks.
| ΔGbind (kcal mol−1) | KD (mM) | Dpore (10−6 cm2 s−1) | kon (μM−1 s−1) | koff (s−1) | ||
|---|---|---|---|---|---|---|
|
| ||||||
| d-Sotalol (0) | US-MD | −5.34 ± 0.74 | 0.17 ± 0.21 | 0.13 ± 0.02 | 7.4 · 102 | 1.3 · 105 |
| HREUS-MD | −5.03 ± 0.52 | 0.28 ± 0.24 | ||||
| d-Sotalol (+) | US-MD | −2.32 ± 0.07 | 23.3 ± 2.6 | 0.12 ± 0.02 | 3.6 · 102 | 8.3 · 106 |
| HREUS-MD | −2.44 ± 0.33 | 19 ± 10 | ||||
| d-Sotalol (pH = 7.4) | 2.3 (1.5) | |||||
| l-Sotalol (0) | US-MD | −4.57 ± 0.51 | 0.60 ± 0.49 | 0.16 ± 0.02 | 7.9 · 102 | 4.7 · 105 |
| HREUS-MD | −5.08 ± 0.24 | 0.26 ± 0.10 | ||||
| l-Sotalol (+) | US-MD | −3.60 ± 0.21 | 2.9 ± 1.0 | 0.17 ± 0.05 | 4.4 · 102 | 1.3 · 106 |
| HREUS-MD | −2.80 ± 0.08 | 10.5 ± 1.4 | ||||
| l-Sotalol (pH = 7.4) | 2.0 (2.0) | |||||
Fig. 4.
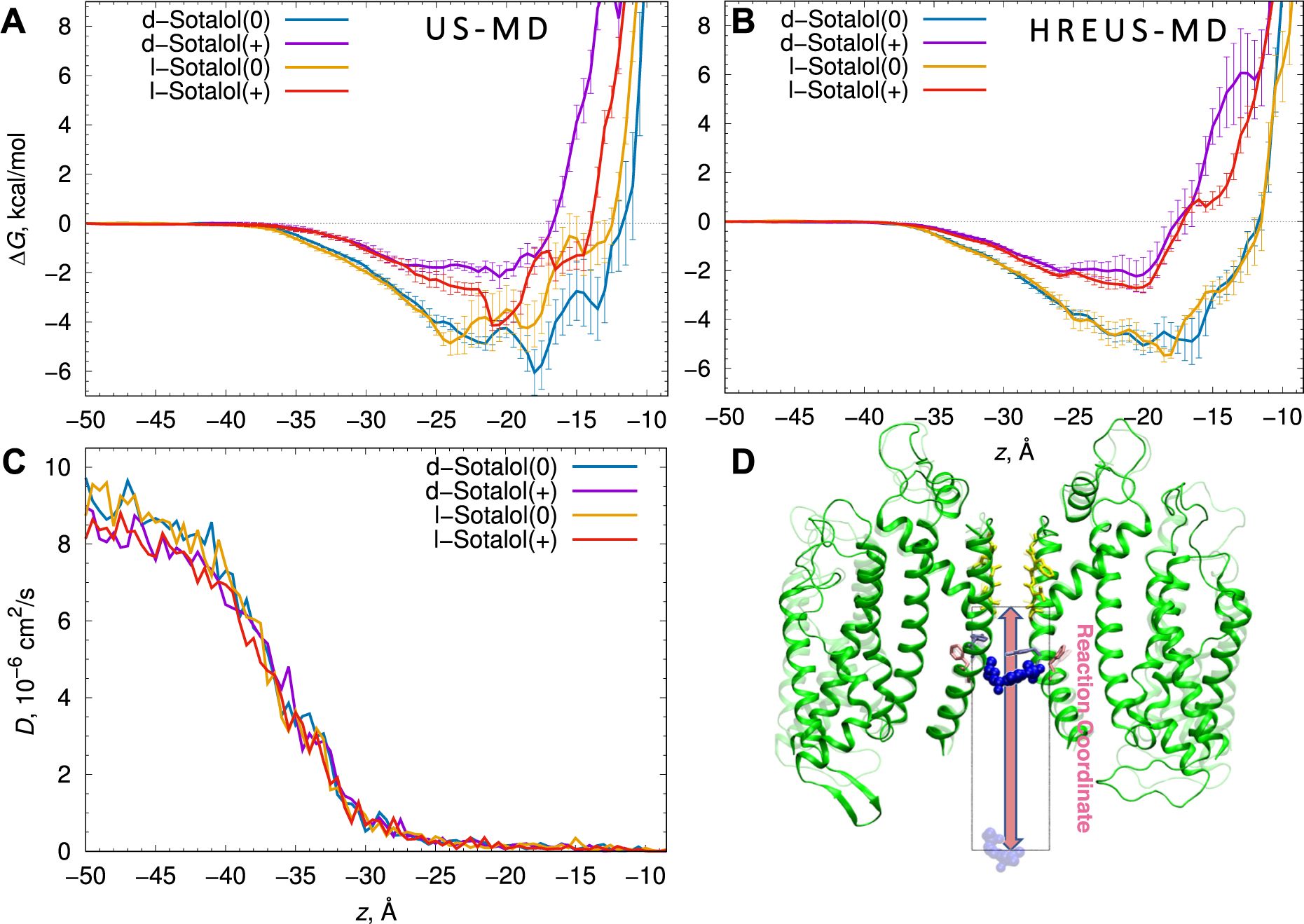
Thermodynamics and kinetics of d- and l-sotalol binding to the open hERG pore from US-MD simulations. Free energy, ΔG, profiles (A) and corresponding diffusion coefficient, D, profiles (C) computed from US-MD simulations, and (B) free energy, ΔG, profiles alternatively computed from HREUS-MD simulations for d-sotalol(0) (blue), d-sotalol(+) (purple), l-sotalol(0) orange, and l-sotalol(+) (red). Error bars are computed from block averaging and represent standard errors of mean. (D) Molecular snapshots of d-sotalol(0) simulated systems with two opposite chains of the open hERG channel shown as green ribbons, selectivity filter residues shown as yellow sticks, and sotalol molecule shown in blue space-filling representation at the pore binding site (opaque) or in the bulk aqueous solution (transparent). The reaction coordinate for these simulations is the z-coordinate with respect to the SF backbone Cα center of mass, leading from the intracellular bulk aqueous solution (z = −50 Å) to the bottom of the selectivity filter (z = −5.5 Å), shown as an arrow and a bounding box with a 20 Å width corresponding to a diameter of the flat-bottom restraint in the xy-plane. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
From hERG channel – sotalol US-MD simulations, we also computed position-dependent diffusion coefficient (D) profiles (see Fig. 4C), which are very similar for all the sotalol forms and indicate a steep ~70-fold drop in the drug diffusion as it enters the confinement of the hERG channel intra-cavitary space, as was also shown previously for dofetilide and moxifloxacin [61,63] as well as sotalol – lipid membrane partitioning [62]. Using ΔG and D profiles, we computed ingress or “on” rates, kon, which are very similar for d- and l-sotalol and somewhat smaller for cationic vs. neutral counterparts (see Table 1). Using computed KD and kon estimates we calculated their products corresponding to “off” or egress rates, koff, similar for d- and l- stereoisomers and 1–2 order of magnitude smaller for sotalol(0) compared to sotalol (+) due to stronger binding of the former (Table 1). kon and koff MD estimates will be used as parameters for functional kinetic models described in Section 3.4.
3.3. Experimental validations of stereospecificity in sotalol binding to hERG channel
Sotalol is commercially only available as a racemic mixture of its hydrochloride salt. We separated the racemic mixture into the S- and R-isomers known as d- and l-sotalol, respectively, using quantitative chiral HPLC technique to obtain multi-milligram quantities of both enantiomers (see Appendix A Fig. S1), which were tested using whole-cell patch-clamp measurements with hERG channels stably expressed in HEK-293 cells at room temperature with continuous superfusion of drug-containing external solution. We observed comparable hERG current inhibition by 0.3 mM d- or l-sotalol application, both at depolarized potentials and upon hyperpolarization, indicating classical drug-induced channel blockade (Fig. 5, top panels). Fitting of concentration response curves (Fig. 5, bottom panels) revealed IC50 values of 0.286 ± 0.007 mM and of 0.288 ± 0.010 mM for d- and l-sotalol, respectively. The Hill coefficients from these curves were 1.474 ± 0.051 for d-sotalol and 1.370 ± 0.010 for l-sotalol, respectively, suggesting cooperative interaction between more than one sotalol molecule to block hERG channel in agreement with flooding MD simulation data (Section 3.1 and Fig. 2). IC50 values also did not reveal any distinction in the ability of the two sotalol enantiomers to inhibit hERG current in good agreement with the results of enhanced sampling MD simulations reported above (Table 1). They are more favorable than MD Kd values of 1.5 to 2.3 mM, but the latter were computed for an open state hERG model, whereas in patch-clamp measurements using a voltage protocol in Fig. 5, the channel is mostly in the inactivated state, and sotalol binding is stabilized by channel inactivation [66]. Our experimental and MD data are in agreement with an electrophysiology study of hERG channel interaction with racemic sotalol, where the IC50 for wild-type hERG and S620T hERG mutant with abolished inactivation are 0.516 ± 0.036 mM and 2.22 ± 0.38 mM, respectively [79]. This suggests that our US-MD derived kinetic parameters (Table 1 and Section 3.2) are robust enough to be used for functional scale modeling.
Fig. 5.
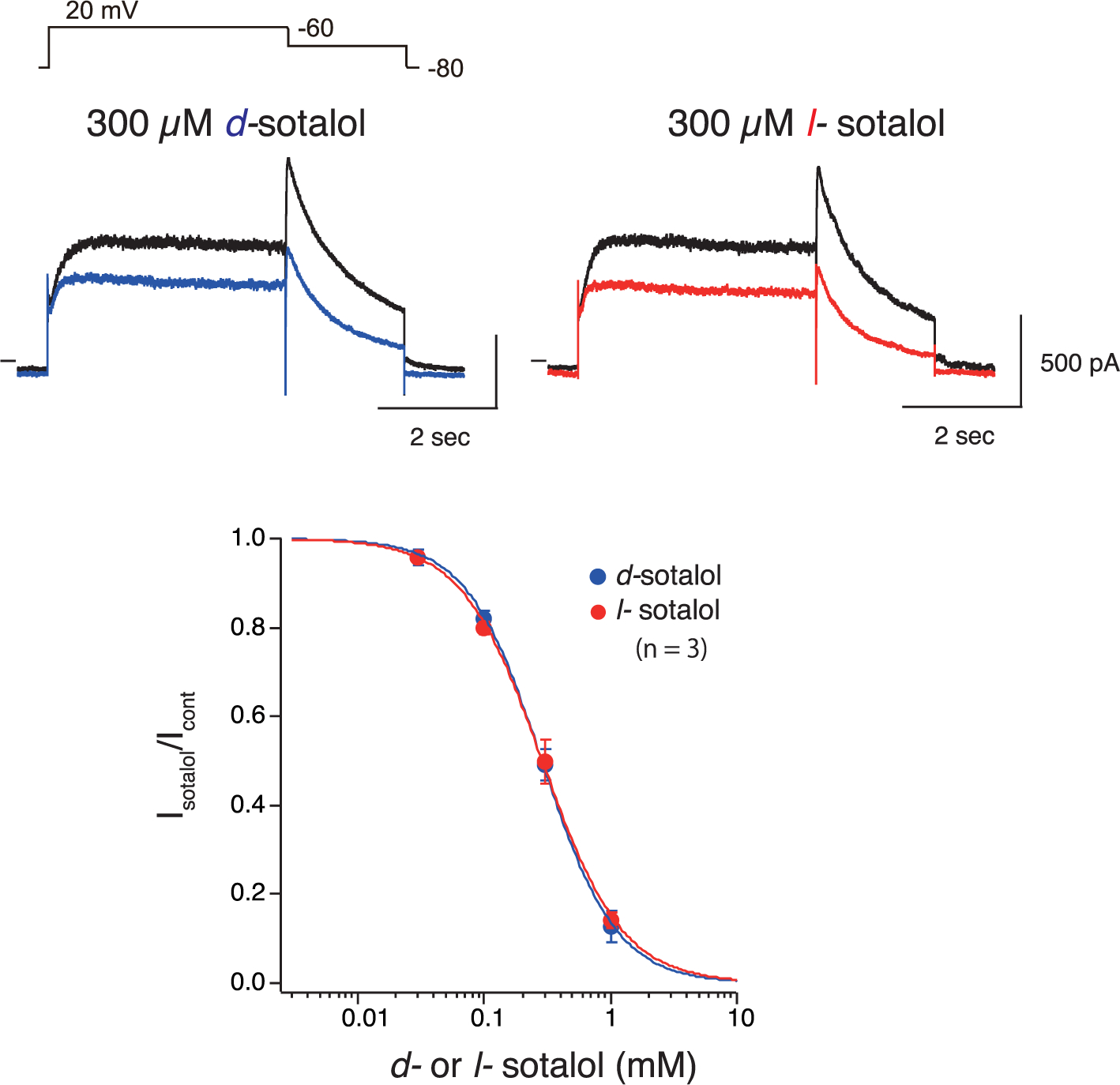
d- and l-Sotalol concentration-response data from patch clamp electrophysiology experiments. (Upper panels) Whole-cell currents from HEK-293 cells expressing hERG channels in response to voltage-clamp pulses from −80 mV to +20 mV and then to −60 mV to record pulse-current and tail currents, respectively. Black lines indicate the control currents and blue as well as red lines indicate the decreased currents in the presence of 300 μM of d- or l-sotalol, respectively. (Bottom panel) Concentration-response for block of hERG channels by d- (blue) or l-sotalol (red). Drug effects on tail current peak amplitude are plotted. Data are means ± standard error of means (SEM) from 3 cells each for d- or l-sotalol. Blue and red curves represent the fitted Hill equation.
3.4. Functional scale modeling of sotalol – hERG channel interactions and their effect on cardiac rhythm
To develop a functional kinetic model of sotalol – hERG channel interactions, we used the multiscale strategy we recently reported [61]. We used a drug-free hERG Markov model [61] and incorporated open and inactivated hERG channel – drug interactions via corresponding neutral and cationic drug “on” (kx_d and kxd) and “off” (rx_d and rxd) transition rates, where x = o or i for open and inactivated state, respectively (see Appendix A Fig. S2). We used US- MD computed drug “on” and “off” rates (Table 1) as our open-state hERG – sotalol(0) and sotalol(+) model estimates (ko_d, kod, ro_d and rod in Appendix A Fig. S2 and Tables S2–S3).
Since we lacked a structural atomistic model of inactivated hERG channel we relied on literature data for relative sotalol affinities for open versus inactivated states with experimental estimates indicating more potent inhibition for the latter [66,79]. We first used the reported 4.3-fold ratio of sotalol IC50 values for a non-inactivated S620T mutant to WT hERG channel from experiments in Chinese hamster ovary (CHO) cells at room temperature [79] as KDo / KDi in our Model 1 (solid light-blue and dotted black curves in Fig.6A) to optimize “on” rates, which provided excellent fit to experimental hERG inhibition data from this study (blue circles in Fig.6A). Notably, however, there is very little hERG inhibition for physiologically relevant drug concentrations from 1.4 to 14.7 μM [34,100] (black box in Fig. 6A): up to ~2% for this model.
Fig. 6.
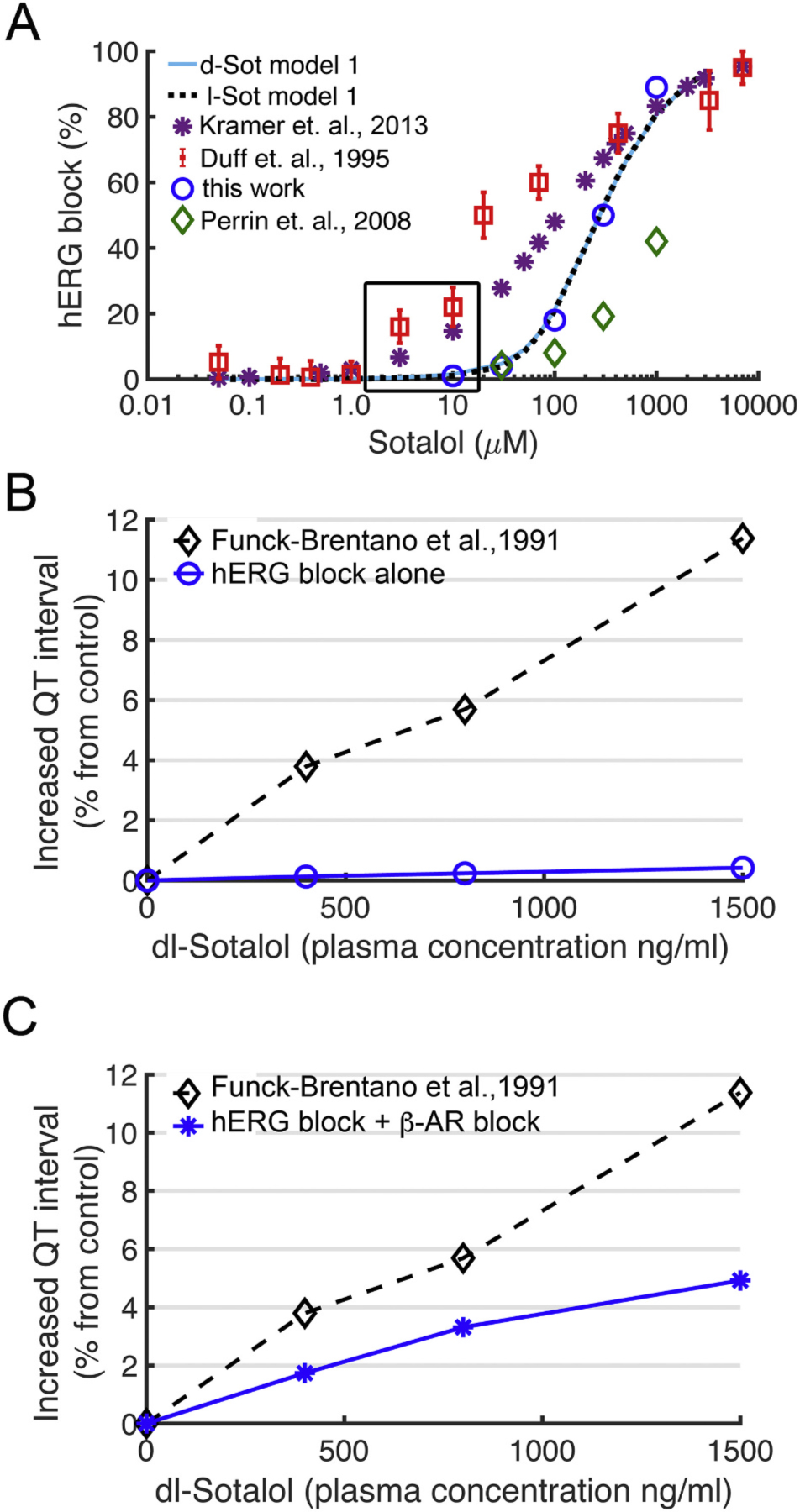
Concentration dependent block of hERG and QT prolongation by sotalol. (A) Experimentally measured dose dependent inhibition of hERG channel by sotalol (colored symbols) and model 1 optimization based on experiments by us and others from expressed channels in mammalian cell lines for d-sotalol (solid light-blue line) and for l-sotalol (dashed black line). Black box indicates therapeutic plasma concentrations. Experimental data are from: Kramer et. al, 2013 – ref. [117]; Duff et. al., 1995 – ref. [80]; Perrin et. al., 2008 – ref. [79]; and this work – see Fig. 5. (B) Concentration dependent increase in QT intervals by d,l-sotalol with hERG channel block alone (blue circles) compared to clinical data (black diamonds) from ref. [82]. (C) During sympathetic stimulation via concurrent ISO 1 μM application, simulations showed a concentration dependent increase in QT interval by d,l-sotalol dependent hERG block and βAR blockade (blue asterisks) compared to clinical data from ref. [82] (black diamonds). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To investigate the effect of the hERG channel – sotalol interactions on cardiac electrophysiology, we included hERG – sotalol model 1 (by swapping the drug-free IKr model) into the O’Hara-Rudy human ventricular cardiomyocyte model [81]. We simulated the effect of sotalol on the pseudo-ECGs computed from a 1D strand of O’Hara-Rudy ventricular myocytes and calculated the QT interval [61]. We observed negligible prolongation of the QT interval with model 1 when considering hERG channel block alone by the sotalol racemic mixture (d- and l-sotalol in a 1:1 ratio) as shown in Fig. 6B. In QT prolongation traces shown in Fig. 6C we simulated the sympathetic stimulation of 1D ventricular myocyte strand via the additive impact of 1 μM of potent beta-adrenergic agonist isoproterenol (ISO) and its stereoselective inhibition by sotalol [36–40]. l-Sotalol has a strong beta-blocking activity with Ki = 650 ng/ml vs. 38,000 ng/mol for d-sotalol [38]. Beta adrenergic stimulation did not notably increase the QT prolongation for model 1 (see Appendix A Fig. S4A&C for d- and l-sotalol, respectively). Simulating the stereospecific effect of beta-blockade in addition to hERG inhibition effect by racemic dl-sotalol, model 1 increases (due to l-sotalol, Appendix A Fig. S4C) but still significantly underestimated QT prolongation compared to clinically reported QT data [82]: compare blue and black curves in Fig. 6C.
When the functional model of hERG channel – sotalol interactions was optimized to data from expressed hERG channels in vitro to determine the relative strength of inactivated state block (as shown for model 1 above), we were unable to predict the clinical features of hERG channel block by sotalol. Therefore, we set out to make predictions about what relative affinities of sotalol to the open and inactivated state would allow reproduction of the clinical data. Because the open state affinity is determined by the MD simulations, we performed a “top down” model optimization to predict the inactivated state affinity of sotalol to hERG channel. In other words, we worked backward from the clinically reported effect of sotalol on the QT interval in humans to determine the inactivated state affinity since all the other parameters in the model are constrained by specific measurements or simulations.
The results of the top-down model optimization (model 2) are shown in Fig. 7. The model shown in Fig. 7A for d- and l-sotalol (solid red and dotted black curves, respectively) was identified as best able to reproduce the effect of sotalol on the QT interval. The model predicted that, consistent with some other hERG channel blockers such as dofetilide [79], the relative affinity of sotalol to the hERG inactivated state should be >50-fold the affinity to the hERG open state. We found that a 63-fold open to inactivated state affinity ratio yielded computed IC50 values of 20 μM during a voltage clamp protocol consistent with reference guinea pig ventricular myocyte hERG inhibition data [80] used for fitting this model. This functional kinetic hERG – sotalol interaction model 2 resulted in up to ~40% hERG inhibition for physiologically relevant d- and l-sotalol concentrations from 1.4 to 14.7 μM [34,100] as shown in Fig. 7A.
Fig. 7.
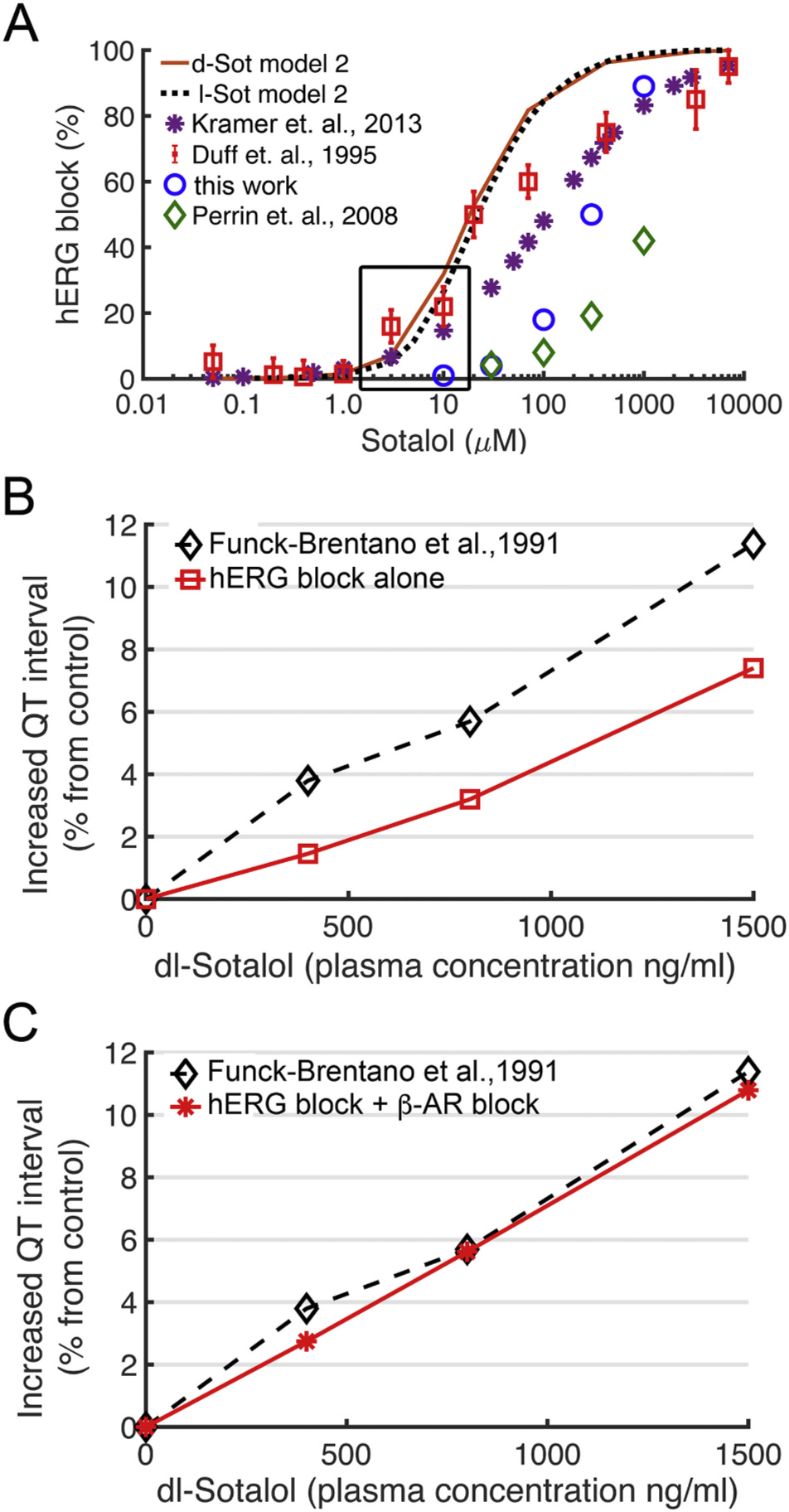
Validation of computational drug screening with human clinical data. (A) Experimentally measured dose dependent inhibition of hERG by sotalol (colored symbols) and model 2 optimization for d-sotalol (solid red line) and for l-sotalol (dashed black line) using data from ref. [80]. See Fig. 6 caption for sources of experimental data. (B) Concentration dependent increase in QT intervals by d,l-sotalol with hERG channel block alone (red circles) compared to clinical data (black diamonds) from ref. [82]. (C) During concurrent ISO 1 μM application to model sympathetic stimulation, model 2 simulations showed a concentration dependent increase in QT interval by d,l-sotalol dependent hERG block and βAR blockade (red asterisks), which compared well to clinical data from ref. [82] (black diamonds).
In Fig. 7B we showed that our predicted model 2 effects of dose-dependent racemic dl-sotalol induced hERG channel inhibition alone in a 1D strand of ventricular myocytes (red curve) underestimates clinical QT prolongation data [82] (black curve). However, after including the combined stereospecific beta-blocking effect of sotalol (dominated by l-sotalol as shown in Appendix A Fig. S4D) in addition to hERG inhibition effects by racemic dl-sotalol, we were able to reproduce clinical QT interval prolongation [82] by model 2 (compare black and red curves in Fig. 7C). Notably, using a 4.3 ratio for hERG open to inactivated state affinities as in model 1 resulted in worse fit to reference hERG inhibition data [80] and smaller QT prolongation compared to clinical data [82] as shown in Appendix A Fig. S5.
Thus, we observed that model 2 accurately predicts clinical QT prolongation by dl-sotalol if we take into account hERG block, beta-adrenergic stimulation and stereospecific beta-blockade. We tested a range of drug concentration effects as a function of time to predict arrhythmogenic impacts of d-, l- and dl-sotalol using model 2 as shown in Fig. 8. We observed that upon beta-adrenergic stimulation, hERG block alone (Fig. 8A–C, top rows) leads to appearance of early afterdepolarizations (EADs) on pseudo-ECGs (left panels for each condition) and variable effects on the cardiac rhythms in time (right panels for each condition indicate the peak positions of R and T waves). Upon beta-blockade pro-arrhythmia markers emerged for d-sotalol at all concentrations similar to the setting of hERG block alone (Fig. 8A, top two rows). For l-sotalol (Fig. 8B, middle rows) and the dl-sotalol racemic mixture (Fig. 8C, bottom rows), a different story emerged: In the setting of hERG block alone, arrhythmogenic patterns emerged for all drug concentrations tested, but when both hERG block and βAR blockade effects were simulated, the computed cardiac rhythms fully normalized. These tests were extended to a wide range of l-sotalol concentrations from 200 to 2400 ng/ml resulting in the same outcome (data not shown). These predictions suggest that a more potent beta-adrenergic blockade by l-sotalol may serve as the key plausible mechanism to reduced arrhythmogenicity with l-sotalol alone and for the racemic mixture.
Fig. 8.
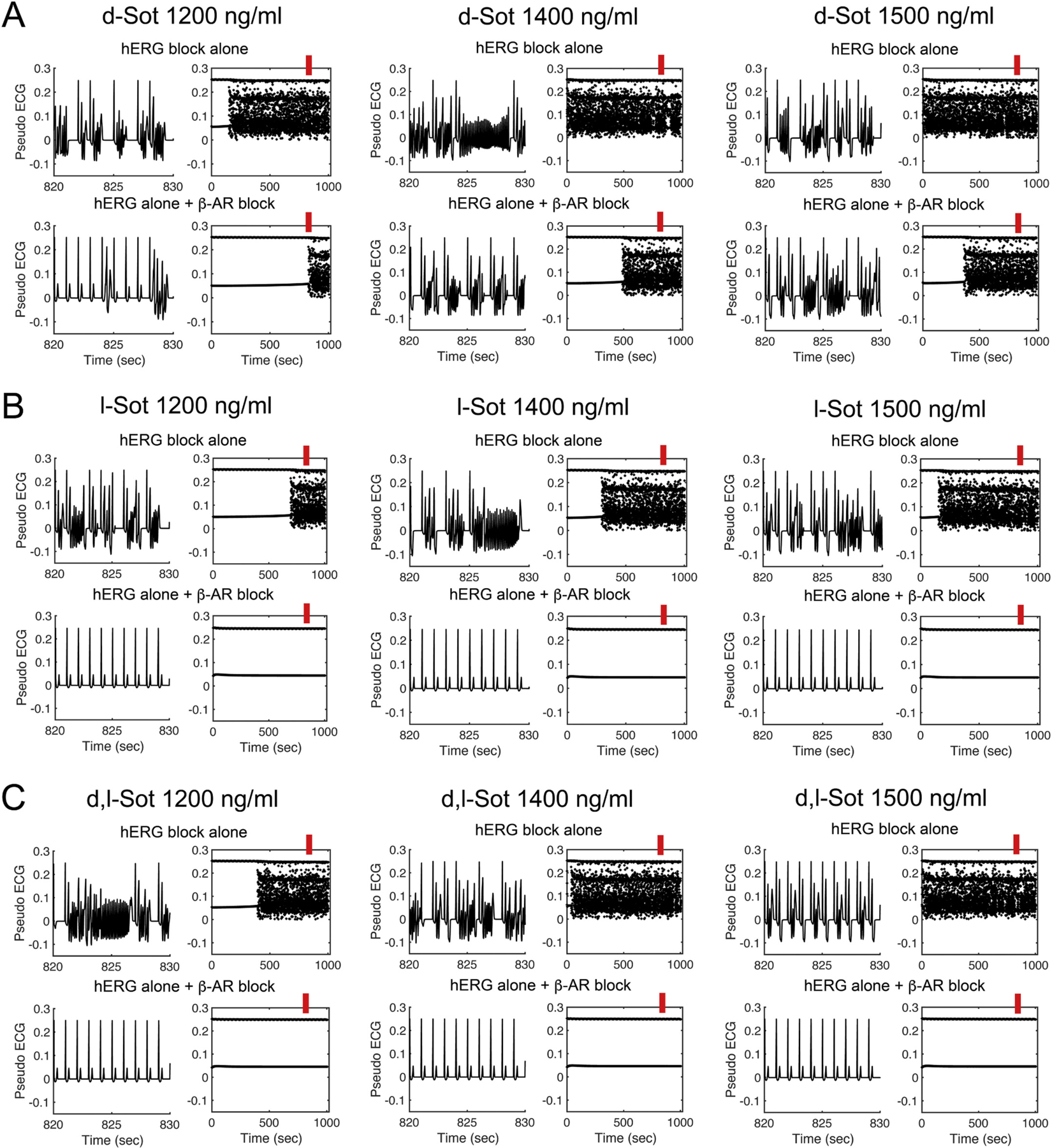
Model prediction of sotalol effects on hERG block and βAR blockade during ISO 1 μM application. The timecourse of pseudo ECGs of model 2 between 820 and 830 s (red bars) at three sotalol plasma concentrations are shown in the left columns of every panel. The peaks of R and T waves are plotted during a 1000 s long simulation timecourse as summary data shown in the right columns of every panel. (A) The predicted additive effect of d-sotalol to block hERG and βAR did not abolish EADs at any tested concentrations. (B) l-sotalol with both hERG and βAR blockade abolished EADs at 1200 ng/ml and higher concentrations. (C) Similar to l-sotalol, racemic d,l-sotalol 1200 ng/ml removed EADs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
4.1. A crucial link between sotalol stereochemistry and its arrhythmogenicity
Computational modeling and simulation approaches at various length and time scales have demonstrated usefulness to make predictions, suggest new experiments, and to reconcile seemingly disparate existing data [59,60,101,102]. In this study, multi-scale modeling and simulation approaches [57,61,103–105] allowed us to specifically ask questions about the importance of isomerism in determining fundamental mechanisms of arrhythmia proclivity induced by d- sotalol, l-sotalol and the dl-sotalol racemic mixture [36,39,40,106–109]. One benefit of computational modeling and simulation is the ease of performing component dissection to determine which attributes of a system yield emergent impacts [57,103]. Here, we show clearly in MD simulations, which were validated by experimental data, that d-sotalol and l-sotalol have similar affinity and interaction with the hERG channel (see Table 1 and Figs. 3–5), in contrast to observed or proposed stereospecific binding of some other hERG-blocking chiral drugs [4] such as methadone [6], quinidine vs. quinone [8], bupivacaine and ropivacaine [12,13]. Similar stereoisomer hERG channel affinities were previously reported for verapamil [110] and chromanol 293B [111]. Our findings are also in line with a previous experimental observation of similar effects of d-,l- and racemic dl-sotalol on action potential and time-dependent potassium current in isolated cardiac muscle and Purkinje fibers from animal models in the absence of catecholamines [36,112]. Therefore, we conclude that stereo-specificity of drug-induced cardiotoxicity attributed to d-sotalol is unlikely to result from differential binding affinities of sotalol stereoisomers to hERG channel. However, the comprehensive multiscale modeling approach [61] allowed us to go a step further and ask if the stronger beta blocking effect of l-sotalol [36–38,40,112–114] was necessary and sufficient to account for a reduction in pro-arrhythmic risk.
4.2. How to resolve disconnect between clinical and experimental data
One of the longstanding conundrums related to hERG block by sotalol is that relatively high concentration of drug required to block the channel when assessed in isolated cloned channels expressed in vitro compared to the relatively low plasma concentrations reported in humans that apparently result in substantial QT interval prolongation [34,82,115]. Human plasma concentrations from patients on sotalol are consistently reported in the 1.4–14.7 μM (500–4000 ng/ml) range [34,100]. Even smaller, 0.5 μM sotalol plasma concentration, results in a 50% likelihood of ≥10 ms increase in human QT interval [115]. This is in contrast to concentrations reported from in vitro measurements that suggest up to 100-fold higher sotalol concentrations required for substantial channel inhibition, although the range of experimentally reported IC50 values is variable [34,79,80,115–123].
We attempted to better understand this disconnect by building computational models that were optimized to experimental cell line data from our laboratory and were similar to previous studies (IC50 values of ~290 μM from this work, 586 ± 179 μM and 4.3-fold inactivated to open state inhibition from [79], 210–450 μM from [118], 103 μM from [115], etc.). Consistent with earlier reports [115,124,125], we also noted that models built on data from expressed channel – drug interactions [79] did not yield clinically observed prolongation of the QT interval [82]. However, when we performed a top-down model optimization, working backward from the clinically reported effects of sotalol on QT interval we found an IC50 value ~20 μM, which reflected ~63-fold higher affinity for inactive to open state. Interestingly, these values are similar to sotalol inhibition data of the reported low- and high-affinity dofetilide binding sites in guinea pig ventricular myocytes [80], although in the study reporting those binding assays, the authors interpret the low affinity site as non-specific binding, not necessarily related to hERG channel current inhibition [80,126]. Additional studies may be needed to provide further clarification.
There is no question that collecting data in the future from a physiologically relevant system with all of the interacting components like the guinea pig ventricular myocyte [80], is likely to yield more reliable data for model optimization. Nevertheless, we were able to use a top-down model approach to constrain model parameters and predict QT interval prolongation as a function of drug concentration that was an excellent agreement with clinically reported data [82] (see Fig. 7C). These results may suggest the critical importance of making drug affinity assessments in a physiologically relevant environment as the differences between the clinical dose and resulting effects cannot currently be reconciled with the dose response curves from expressed channels. Indeed, one possibility is that the induced pluripotent stem cell-derived cardiomyocyte (iPSC-CM) based drug screening technology may create new capacity and potential to do just that [127–129].
4.3. Limitations and future directions
In this study we were able to predict sotalol induced QT interval prolongation om the ECG and its stereospecific pro-arrhythmia proclivities with our functional hERG – sotalol models optimized using “on” and “off” drug rates for the open-state hERG channel. Since we lacked a structural atomistic hERG channel model in the inactivated state, we had to use a top-down approach to predict an optimal ratio of sotalol affinities to open and inactivated hERG channels, which turned out to correspond to a ratio of sotalol IC50 values for low and high affinity dofetilide binding sites in ventricular cardiomyocytes [80]. Yet, the low-affinity binding site in the Duff et al. study was interpreted as non-specific binding rather than hERG channel inhibition [80,126]. Such drug binding to other proteins or a lipid membrane itself will alter its effective intracellular concentration, availability for hERG channel inhibition and thus pro-arrhythmia proclivities, which can be investigated in future work. As an alternative, we also tested a ratio of sotalol IC50 values for wild-type and non-inactivated S620T mutant channels from cultured cells [79], with the latter agreeing well with our US-MD KD estimates. Functional models optimized using these data provided good fit to our experimental WT hERG inhibition dose response curves but failed to capture dose-dependent clinical QT prolongation [82] in line with previous reports suggesting a significant gaps between drug doses required to cause in vitro hERG channel inhibition and QT prolongation observed in vivo [115].
Such discrepancies can be related to multiple factors not explicitly considered here. For instance, hERG channel isoforms, 1a and 1b, with the same transmembrane but different intracellular domain composition contribute to physiological IKr and have distinct gating kinetics [130], which may modulate drug effect on cardiac electrophysiology. Moreover, mutations in auxiliary hERG binding protein MiRP1 were found to affect drug-induced arrhythmogenicity also due to altered channel gating kinetics [52,131], although WT MiRP1 binding was shown not to affect sotalol hERG channel block [66].
Also, not only hERG channel – drug interaction, but also ventricular myocyte model may have an effect on predicted drug-induced QT prolongation and arrhythmogenicity as was tested and/or discussed previously [132–135]. Here and in previous studies [52,61,134,135] the O’Hara-Rudy (ORd) human ventricular cardiac myocyte model [81] was used providing good agreement with clinical QT prolongation data [82] for our myocyte-based model, whereas comparison with other widely used models such as Grandi – Bers [136], ToR-ORd (Tomek, Rodriguez following ORd) [137], and ten Tusscher – Panfilov [138] models will be performed in subsequent work. We recognize that all models have strengths and weaknesses and that models continue to undergo continuous development.
In this study, MD simulations used an available cryo-EM hERG channel structure [65], which we demonstrated to be in the open conducting state [61,63,64]. However, many hERG blocking drugs including sotalol are suggested to have a higher affinity for the inactivated channel state [48,66,79]. Structural information about potential hERG channel inactivated state just started to emerge from recent cryo-EM [139] and MD simulation [140] studies, but was not yet available for our present work. Interestingly, however, our multi-microsecond long unbiased drug flooding simulations resulted in a hERG channel structure with a distorted SF, pinched in the middle (in 3 out of 4 simulations) and significantly widened at the top, in line with some findings from those new studies [139,140]. A similar SF conformation was also shown to be non-conducting in our previous hERG channel simulations [63] and may potentially represent a transition to the inactivated state of the channel bearing some similarity to C-type inactivated KcsA [141,142] and Shaker family [143,144] channel structures, although there might be multiple structural mechanisms of K+ channel inactivation [144]. Such conformational transition may also explain stable sotalol binding poses, located deep inside the pore in some unrestrained drug flooding MD but not in any restrained US-MD runs, in line with previously suggested sotalol binding to open state and subsequent stabilization by hERG channel inactivation [66].
Finally, we found that beta-blocking properties of sotalol enantiomers are crucial for their pro-arrhythmia proclivities. Yet, in this study we did not explicitly study sotalol binding to beta-adrenergic receptors via MD simulations due to a complexity of this problem, requiring a separate study currently underway in our laboratory using recent human β1 adrenergic receptor (β1AR) structures [145] and state-specific functional models of sotalol – receptor interactions [146], We have also developed a functional model that explicitly couples the effects of the autonomic nervous system (ANS) stimulation on the sinoatrial node (SAN) and ventricular contractile cells and allows for coupled effects of changes in SAN pacing on ventricular myocyte electrophysiology (Clancy et al., manuscript in preparation). Those upcoming studies will allow for a comprehensive exploration of sotalol QT modulation effect at different BCL (RR interval) values [82].
5. Conclusions
Cardiotoxicity in the form of cardiac arrhythmia is a major reason for multiple drug withdrawals from the market. Such drug-induced arrhythmogenicity has been largely attributed to a blockade of hERG K+ channels, which repolarize cardiac myocytes. However, not all hERG-blocking drugs cause deadly arrhythmias. In the case of the anti-arrhythmic beta-blocker sotalol, a subtle change in drug chemistry due to stereoisomerism leads to different pro-arrhythmia outcomes. In this study, we investigated and predicted molecular determinants of stereospecific d- and l-sotalol pro-arrhythmia proclivities through a combination of atomistic molecular dynamics (MD) simulations with electrophysiological recordings as well as experiment- and MD-informed multi-scale modeling of cardiac electrophysiology at cell and tissue levels. MD simulations predicted that up to 2 sotalol molecules can bind in the hERG channel pore, demonstrated stronger binding of neutral drug forms as well as similar d- and l-sotalol binding poses and affinities for the open channel pore. These predictions are in good agreement with electrophysiology recordings of hERG channel inhibition in HEK cells by both sotalol enantiomers. MD simulation computed “on” and “off” rates for open hERG – sotalol interactions were used as parameters for functional kinetic models, which were optimized to reproduce hERG inhibition dose response curves using cultured cells from this work or cardiomyocyte data from a previous study. We also explored different estimates of sotalol affinities for open and inactivated hERG conformational states using a top-down approach. Optimized models were incorporated into standard ventricular cardiomyocyte cell and tissue simulations to predict dose-dependent d- and l-sotalol effect on heart rhythm. The model based on cardiomyocyte hERG inhibition data was able to predict clinically observed sotalol induced QT prolongation when taking into account both hERG channel and beta-adrenergic receptor inhibition, whereas the model based on a less potent sotalol effect on the hERG channel in cultured cells significantly underestimated clinical QT interval lengthening. The former model also correctly predicted higher pro-arrhythmia risk of d-sotalol compared to l-sotalol and racemic mixture attributing this to stereospecific beta-blocking properties, ameliorating the arrhythmogenic hERG channel inhibition effect.
Supplementary Material
Acknowledgments
The work in C.E.C., I.V., H.W., J.T.S., V.Y.Y., and S.Y.N. groups were supported by NIH NHLBI grants 5R01HL128537, 2R01HL128537, 5U01HL126273, NIH Common Fund 1OT2OD026580-01 and 3OT2OD026580-01 (C.E.C. and I.V.). Support was also provided by the following: American Heart Association Predoctoral Fellowship16PRE27260295 (K.R.D.) and Career Development Award 19CDA34770101 (I.V.), NIH NHLBI grants 1R01HL152681 (C.E.C. and I.V.) and R01HL128537 (S.Y.N.), Canadian Institutes of Health Research Project Program grant Funding Reference Number 156236 (S.Y.N.), Department of Physiology and Membrane Biology Research Partnership Fund (I.V. and C.E.C.), Extreme Science and Engineering Discovery Environment (XSEDE) Grant MCB170095 (I.V., C.E.C., K.R.D.), National Center for Supercomputing Applications (NCSA) Blue Waters Broadening Participation Allocation (C.E.C., I.V., K.R.D.), Texas Advanced Computing Center (TACC) Leadership Resource AllocationMCB20010 (I.V., C.E.C., K.R.D.), Oracle cloud for research allocation (I.V., C.E.C.), Pittsburgh Supercomputing Center (PSC) Anton 2 allocationsPSCA17085P, PSCA16108P, PSCA18077P, MCB160089P, and PSCA17021P (I.V., C.E.C., K.R.D., S.Y.N.). Anton 2 computer time was provided by the PSC through Grant R01GM116961 from the National Institutes of Health. The Anton 2 machine at PSC was generously made available by D.E. Shaw Research.
Abbreviations:
- βAR
beta adrenergic receptor
- BCL
basic cycle length
- CHARMM
chemistry at Harvard macromolecular mechanics
- CiPA
comprehensive in vitro proarrhythmia assay
- CryoEM
cryogenic electron microscopy
- EAD
early afterdepolarization
- ECG
electrocardiogram
- HREUS-MD
Hamiltonian replica exchange umbrella sampling molecular dynamics
- HEK
human embryonic kidney
- hERG
human Ether-à-go-go-related gene
- HPLC
high performance liquid chromatography
- IC50
the half maximal inhibitory concentration
- ISO
isoproterenol
- KV
voltage-gated potassium channel
- MD
molecular dynamics
- NAMD
nanoscale molecular dynamics
- NMR
nuclear magnetic resonance
- PD
pore domain
- PDB
protein data bank
- PKA
protein kinase A
- POPC
1-palmitoyl-2-oleoylphosphatidylcholine
- RMSD
root-mean-square deviation
- SEM
standard error of means
- SF
selectivity filter
- TdP
torsades de pointes
- US-MD
umbrella sampling molecular dynamics
- VMD
visual molecular dynamics
- VSD
voltage sensing domain
- WT
wild-type
Footnotes
Disclosures
None. The authors declare no competing interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2021.05.015.
References
- [1].Chhabra N, Aseri ML, Padmanabhan D, A review of drug isomerism and its significance, Int. J. Appl. Basic Med. Res. 3 (1) (2013) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gandhi K, Shah U, Patel S, Drug stereochemistry: a prodigy for pharmacology and drug development, Current Drug Discovery Technol. 17 (5) (2020) 565–573, 10.2174/1570163816666190502101803. [DOI] [PubMed] [Google Scholar]
- [3].Smith SW, Chiral toxicology: it’s the same thing… only different, Toxicol. Sci. 110 (1) (2009) 4–30. [DOI] [PubMed] [Google Scholar]
- [4].Sintra Grilo L, Carrupt P-A, Abriel H, Stereoselective inhibition of the hERG1 potassium channel, Front. Pharmacol. 1 (2010) 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Eap CB, Crettol S, Rougier JS, Schläpfer J, Sintra Grilo L, Déglon JJ, Besson J, Croquette-Krokar M, Carrupt PA, Abriel H, Stereoselective block of hERG channel by (S)-methadone and QT interval prolongation in CYP2B6 slow metabolizers, Clin. Pharmacol. Ther. 81 (5) (2007) 719–728. [DOI] [PubMed] [Google Scholar]
- [6].Ansermot N, Albayrak Ö, Schläpfer J, Crettol S, Croquette-Krokar M, Bourquin M, Déglon J-J, Faouzi M, Scherbaum N, Eap CB, Substitution of (R, S)-methadone by (R)-methadone: impact on QTc interval, Arch. Intern. Med. 170 (6) (2010) 529–536. [DOI] [PubMed] [Google Scholar]
- [7].White NJ, Cardiotoxicity of antimalarial drugs, Lancet Infect. Dis. 7 (8) (2007) 549–558. [DOI] [PubMed] [Google Scholar]
- [8].Yan M, Fan P, Shi Y, Feng L, Wang J, Zhan G, Li B, Stereoselective blockage of quinidine and quinine in the hERG channel and the effect of their rescue potency on drug-induced hERG trafficking defect, Int. J. Mol. Sci. 17 (10) (2016) 1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Calvey T, Isomerism and anaesthetic drugs, Acta Anaesthesiol. Scand. 39 (1995) 83–90. [DOI] [PubMed] [Google Scholar]
- [10].Graf BM, Abraham I, Eberbach N, Kunst G, Stowe DF, Martin E, Differences in cardiotoxicity of bupivacaine and ropivacaine are the result of physicochemical and stereoselective properties, Anesthesiol. J. Am. Soc. Anesthesiol. 96 (6) (2002) 1427–1434. [DOI] [PubMed] [Google Scholar]
- [11].Zapata-Sudo G, Trachez MM, Sudo RT, Nelson TE, Is comparative cardiotoxicity of S (−) and R(+) bupivacaine related to enantiomer-selective inhibition of L-type Ca2+ channels? Anesth. Analg. 92 (2) (2001) 496–501. [DOI] [PubMed] [Google Scholar]
- [12].González T, Arias C, Caballero R, Moreno I, Delpón E, Tamargo J, Valenzuela C, Effects of levobupivacaine, ropivacaine and bupivacaine on HERG channels: stereoselective bupivacaine block, Br. J. Pharmacol. 137 (8) (2002) 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Siebrands CC, Schmitt N, Friederich P, Local anesthetic interaction with human ether-a-go-go–related gene (HERG) ChannelsRole of aromatic amino acids Y652 and F656, Anesthesiol. J. Am. Soc. Anesthesiol. 103 (1) (2005) 102–112. [DOI] [PubMed] [Google Scholar]
- [14].Wainer I, Stereoisomers in clinical oncology: why it is important to know what the right and left hands are doing, Ann. Oncol. 4 (1993) S7–S13. [DOI] [PubMed] [Google Scholar]
- [15].Nguyen LA, He H, Pham-Huy C, Chiral drugs: an overview, Int. J. Biomed. Sci. 2 (2) (2006) 85. [PMC free article] [PubMed] [Google Scholar]
- [16].Waldo AL, Camm AJ, DeRuyter H, Friedman PL, MacNeil DJ, Pitt B, Pratt CM, Rodda BE, Schwartz PJ, Investigators S, Survival with oral d-sotalol in patients with left ventricular dysfunction after myocardial infarction: rationale, design, and methods (the SWORD trial), Am. J. Cardiol. 75 (15) (1995) 1023–1027. [DOI] [PubMed] [Google Scholar]
- [17].Waldo AL, Camm AJ, de Ruyter H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ, Veltri EP, Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol, Lancet 348 (9019) (1996) 7–12. [DOI] [PubMed] [Google Scholar]
- [18].Kirschenbaum HL, Rosenberg JM, Clinical experience with sotalol in the treatment of cardiac arrhythmias, Clin. Ther. 16 (3) (1994) 346–364. [PubMed] [Google Scholar]
- [19].Hohnloser SH, Woosley RL, Sotalol N Engl. J. Med. 331 (1) (1994) 31–38. [DOI] [PubMed] [Google Scholar]
- [20].Haverkamp W, Martinez-Rubio A, Hief C, Lammers A, Mühlenkamp S, Wichter T, Breithardt G, Borggrefe M, Efficacy and safety of d, l-sotalol in patients with ventricular tachycardia and in survivors of cardiac arrest, J. Am. Coll. Cardiol. 30 (2) (1997) 487–495. [DOI] [PubMed] [Google Scholar]
- [21].Kühlkamp V, Mermi J, Mewis C, Seipel L, Efficacy and proarrhythmia with the use of d, l-sotalol for sustained ventricular tachyarrhythmias, J. Cardiovasc. Pharmacol. 29 (3) (1997) 373–381. [DOI] [PubMed] [Google Scholar]
- [22].Kühlkamp V, Mewis C, Mermi J, Bosch RF, Seipel L, Suppression of sustained ventricular tachyarrhythmias: a comparison of d, l-sotalol with no antiarrhythmic drug treatment, J. Am. Coll. Cardiol. 33 (1) (1999) 46–52. [DOI] [PubMed] [Google Scholar]
- [23].Pacifico A, Hohnloser SH, Williams JH, Tao B, Saksena S, Henry PD, Prystowsky EN, Prevention of implantable-defibrillator shocks by treatment with sotalol, N. Engl. J. Med. 340 (24) (1999) 1855–1862. [DOI] [PubMed] [Google Scholar]
- [24].Anderson JL, Prystowsky EN, Sotalol: an important new antiarrhythmic, Am. Heart J. 137 (3) (1999) 388–409. [DOI] [PubMed] [Google Scholar]
- [25].Singh BN, Sotalol: current status and expanding indications, J. Cardiovasc. Pharmacol. Ther. 4 (1) (1999) 49–65. [DOI] [PubMed] [Google Scholar]
- [26].Highlights Of Prescribing Information: Sotalol hydrochloride injection for intravenous use U.S. Food and Drug Administration Silver Spring, MD, U.S.A, 2009. [Google Scholar]
- [27].Kerin NZ, Jacob S, The efficacy of sotalol in preventing postoperative atrial fibrillation: a meta-analysis, Am. J. Med. 124 (9) (2011) 875. e1–875. e9. [DOI] [PubMed] [Google Scholar]
- [28].Giardina E-G, Therapeutic Use and Major Side Effects of Sotalol, UpToDate, Inc, 2018. [Google Scholar]
- [29].Sotalol, Hydrochloride, Drugs.com, 2018. [Google Scholar]
- [30].Soyka LF, Wirtz C, Spangenberg RB, Clinical safety profile of sotalol in patients with arrhythmias, Am. J. Cardiol. 65 (2) (1990) 74–81. [DOI] [PubMed] [Google Scholar]
- [31].Falk RH, Proarrhythmia in patients treated for atrial fibrillation or flutter, Ann. Intern. Med. 117 (2) (1992) 141–150. [DOI] [PubMed] [Google Scholar]
- [32].MacNeil DJ, Davies RO, Deitchman D, Clinical safety profile of sotalol in the treatment of arrhythmias, Am. J. Cardiol. 72 (4) (1993) A44–A50. [DOI] [PubMed] [Google Scholar]
- [33].Lehmann MH, Hardy S, Archibald D, Quart B, MacNeil DJ, Sex difference in risk of torsade de pointes with d, l-sotalol, Circulation 94 (10) (1996) 2535–2541. [DOI] [PubMed] [Google Scholar]
- [34].Redfern W, Carlsson L, Davis A, Lynch W, MacKenzie I, Palethorpe S, Siegl P, Strang I, Sullivan A, Wallis R, Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development, Cardiovasc. Res. 58 (1) (2003) 32–45. [DOI] [PubMed] [Google Scholar]
- [35].Shantsila E, Watson T, Lip GY, Drug-induced QT-interval prolongation and proarrhythmic risk in the treatment of atrial arrhythmias, Europace 9 (suppl_4) (2007) iv37–iv44. [DOI] [PubMed] [Google Scholar]
- [36].Kato R, Ikeda N, Yabek SM, Kannan R, Singh BN, Electrophysiologic effects of the levo-and dextrorotatory isomers of sotalol in isolated cardiac muscle and their in vivo pharmacokinetics, J. Am. Coll. Cardiol. 7 (1) (1986) 116–125. [DOI] [PubMed] [Google Scholar]
- [37].Gomoll AW, Bartek MJ, Comparative β-blocking activities and electrophysiologic actions of racemic sotalol and its optical isomers in anesthetized dogs, Eur. J. Pharmacol. 132 (2–3) (1986) 123–135. [DOI] [PubMed] [Google Scholar]
- [38].Funck-Brentano C, Silberstein DJ, Roden DM, Wood AJ, Woosley RL, A mechanism of D-(+)-sotalol effects on heart rate not related to beta-adrenoceptor antagonism, Br. J. Clin. Pharmacol. 30 (2) (1990) 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Touboul P, Electrophysiologic properties of sotalol and d-sotalol. A current view, Eur. Heart J. 14 (suppl_H) (1993) 24–29. [DOI] [PubMed] [Google Scholar]
- [40].Funck-Brentano C, Pharmacokinetic and pharmacodynamic profiles of d-sotalol and d, l-sotalol, Eur. Heart J. 14 (suppl_H) (1993) 30–35. [DOI] [PubMed] [Google Scholar]
- [41].Behr ER, Roden D, Drug-induced arrhythmia: pharmacogenomic prescribing? Eur. Heart J. 34 (2) (2013) 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].De Ponti F, Poluzzi E, Montanaro N, QT-interval prolongation by non-cardiac drugs: lessons to be learned from recent experience, Eur. J. Clin. Pharmacol. 56 (1) (2000) 1–18. [DOI] [PubMed] [Google Scholar]
- [43].Ferri N, Siegl P, Corsini A, Herrmann J, Lerman A, Benghozi R, Drug attrition during pre-clinical and clinical development: understanding and managing drug-induced cardiotoxicity, Pharmacol. Ther. 138 (3) (2013) 470–484. [DOI] [PubMed] [Google Scholar]
- [44].Onakpoya IJ, Heneghan CJ, Aronson JK, Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature, BMC Med. 14 (1) (2016) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ruskin JN, The cardiac arrhythmia suppression trial (CAST), N. Engl. J. Med. 321 (6) (1989) 386–388. [DOI] [PubMed] [Google Scholar]
- [46].Sanguinetti MC, Jiang C, Curran ME, Keating MT, A mechanistic link between an inherited and an acquird cardiac arrthytmia: HERG encodes the IKr potassium channel, Cell 81 (2) (1995) 299–307. [DOI] [PubMed] [Google Scholar]
- [47].Trudeau MC, Warmke JW, Ganetzky B, Robertson GA, HERG, a human inward rectifier in the voltage-gated potassium channel family, Science 269 (5220) (1995) 92–95. [DOI] [PubMed] [Google Scholar]
- [48].Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP, hERG K(+) channels: structure, function, and clinical significance, Physiol. Rev. 92 (3) (2012) 1393–1478. [DOI] [PubMed] [Google Scholar]
- [49].Roden DM, Drug-induced prolongation of the QT interval, N. Engl. J. Med. 350 (10) (2004) 1013–1022. [DOI] [PubMed] [Google Scholar]
- [50].Sanguinetti MC, Tristani-Firouzi M, hERG potassium channels and cardiac arrhythmia, Nature 440 (7083) (2006) 463–469. [DOI] [PubMed] [Google Scholar]
- [51].Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N, Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the cardiac safety research consortium, Am. Heart J. 167 (3) (2014) 292–300. [DOI] [PubMed] [Google Scholar]
- [52].Romero L, Trenor B, Yang P-C, Saiz J, Clancy CE, In silico screening of the impact of hERG channel kinetic abnormalities on channel block and susceptibility to acquired long QT syndrome, J. Mol. Cell. Cardiol. 72 (2014) 126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Colatsky T, Fermini B, Gintant G, Pierson JB, Sager P, Sekino Y, Strauss DG, Stockbridge N, The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative - update on progress, J. Pharmacol. Toxicol. Methods 81 (2016) 15–20. [DOI] [PubMed] [Google Scholar]
- [54].Fermini B, Hancox JC, Abi-Gerges N, Bridgland-Taylor M, Chaudhary KW, Colatsky T, Correll K, Crumb W, Damiano B, Erdemli G, Gintant G, Imredy J, Koerner J, Kramer J, Levesque P, Li Z, Lindqvist A, Obejero-Paz CA, Rampe D, Sawada K, Strauss DG, Vandenberg JI, A new perspective in the field of cardiac safety testing through the comprehensive in vitro Proarrhythmia assay paradigm, J. Biomol. Screen. 21 (1) (2016) 1–11. [DOI] [PubMed] [Google Scholar]
- [55].Plazinska A, Plazinski W, Stereoselective binding of agonists to the β 2-adrenergic receptor: insights into molecular details and thermodynamics from molecular dynamics simulations, Mol. BioSyst. 13 (5) (2017) 910–920. [DOI] [PubMed] [Google Scholar]
- [56].Shen Q, Wang L, Zhou H, Yu L.-s., Zeng S, Stereoselective binding of chiral drugs to plasma proteins, Acta Pharmacol. Sin. 34 (8) (2013) 998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Moreno JD, Zhu ZI, Yang P-C, Bankston JR, Jeng M-T, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms, Sci. Transl. Med. 3 (98) (2011) (98ra83–98ra83). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Roberts BN, Yang P-C, Behrens SB, Moreno JD, Clancy CE, Computational approaches to understand cardiac electrophysiology and arrhythmias, Am. J. Phys. Heart Circ. Phys. 303 (7) (2012) H766–H783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang Y, Barocas VH, Berceli SA, Clancy CE, Eckmann DM, Garbey M, Kassab GS, Lochner DR, McCulloch AD, Tran-Son-Tay R, Multi-scale modeling of the cardiovascular system: disease development, progression, and clinical intervention, Ann. Biomed. Eng. 44 (9) (2016) 2642–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Clancy CE, An G, Cannon WR, Liu Y, May EE, Ortoleva P, Popel AS, Sluka JP, Su J, Vicini P, Multiscale modeling in the clinic: drug design and development, Ann. Biomed. Eng. 44 (9) (2016) 2591–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yang PC, DeMarco KR, Aghasafari P, Jeng MT, Dawson JR, Bekker S, Noskov S, Yarov-Yarovoy V, Vorobyov I, Clancy CE, A computational pipeline to predict Cardiotoxicity:from the atom to the rhythm, Circ. Res. 126 (2020) 947–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].DeMarco KR, Bekker S, Clancy CE, Noskov SY, Vorobyov I, Digging into lipid membrane permeation for cardiac Ion Channel blocker d-Sotalol with all-atom simulations, Front. Pharmacol. 9 (2018) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].DeMarco KR, Dawson JR, Yang P-C, Bekker S, Ngo VA, Noskov SY, Yarov-Yarovoy V, Clancy CE, Vorobyov I, Atomistic modeling towards predictive cardiotoxicity, BioRxiv (2019), 635441. [Google Scholar]
- [64].Miranda WE, DeMarco KR, Guo J, Duff HJ, Vorobyov I, Clancy CE, Noskov SY, Selectivity filter modalities and rapid inactivation of the hERG1 channel, Proc. Natl. Acad. Sci. 117 (6) (2020) 2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang W, MacKinnon R, Cryo-EM Structure of the Open Human Ether-a-go-go-Related K+ Channel hERG, Cell 169 (3) (2017) 422–430 (e10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Numaguchi H, Mullins FM, Johnson JP Jr., Johns DC, Po SS, Yang IC, Tomaselli GF, Balser JR, Probing the interaction between inactivation gating and Dd-sotalol block of HERG, Circ. Res. 87 (11) (2000) 1012–1018. [DOI] [PubMed] [Google Scholar]
- [67].Andre I, Bradley P, Wang C, Baker D, Prediction of the structure of symmetrical protein assemblies, Proc. Natl. Acad. Sci. U. S. A. 104 (45) (2007) 17656–17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Barth P, Schonbrun J, Baker D, Toward high-resolution prediction and design of transmembrane helical protein structures, Proc. Natl. Acad. Sci. U. S. A. 104 (40) (2007) 15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yarov-Yarovoy V, Schonbrun J, Baker D, Multipass membrane protein structure prediction using Rosetta, Proteins 62 (4) (2006) 1010–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Huang J, MacKerell AD Jr., CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data, J. Comput. Chem. 34 (25) (2013) 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, MacKerell AD Jr., Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles, J. Chem. Theory Comput. 8 (9) (2012) 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr., R.W. Pastor, Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types, J. Phys. Chem. B 114 (23) (2010) 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys. 79 (2) (1983) 926–935. [Google Scholar]
- [74].Jo S, Kim T, Iyer VG, Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM, J. Comput. Chem. 29 (11) (2008) 1859–1865. [DOI] [PubMed] [Google Scholar]
- [75].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K, Scalable molecular dynamics with NAMD, J. Comput. Chem. 26 (16) (2005) 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Shaw DE, Grossman JP, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, Forte A, Gagliardo J, Gill G, Greskamp B, Ho CR, Ierardi DJ, Iserovich L, Kuskin JS, Larson RH, Layman T, Lee LS, Lerer AK, Li C, Killebrew D, Mackenzie KM, Mok SYH, Moraes MA, Mueller R, Nociolo LJ, Peticolas JL, Quan T, Ramot D, Salmon JK, Scarpazza DP, Ben Schafer U, Siddique N, Snyder CW, Spengler J, Tang PTP, Theobald M, Toma H, Towles B, Vitale B, Wang SC, Young C, Anton 2: Raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer, Int Conf High Perfor (2014) 41–53. [Google Scholar]
- [77].Torrie GM, Valleau JP, Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling, J. Comput. Phys. 23 (2) (1977) 187–199. [Google Scholar]
- [78].Jiang W, Luo Y, Maragliano L, Roux B, Calculation of free energy landscape in multi-dimensions with Hamiltonian-exchange umbrella sampling on petascale supercomputer, J. Chem. Theory Comput. 8 (11) (2012) 4672–4680. [DOI] [PubMed] [Google Scholar]
- [79].Perrin MJ, Kuchel PW, Campbell TJ, Vandenberg JI, Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-a-go-go-related gene channels, Mol. Pharmacol. 74 (5) (2008) 1443–1452. [DOI] [PubMed] [Google Scholar]
- [80].Duff HJ, Feng ZP, Sheldon RS, High- and low-affinity sites for [3H]dofetilide binding to Guinea pig myocytes, Circ. Res. 77 (4) (1995) 718–725. [DOI] [PubMed] [Google Scholar]
- [81].O’Hara T, Virág L, Varró A, Rudy Y, Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation, PLoS Comput. Biol. 7 (5) (2011), e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Funck-Brentano C, Kibleur Y, Le Coz F, Poirier JM, Mallet A, Jaillon P, Rate dependence of sotalol-induced prolongation of ventricular repolarization during exercise in humans, Circulation 83 (2) (1991) 536–545. [DOI] [PubMed] [Google Scholar]
- [83].Sheldrick GM, A short history of SHELX, Acta Crystallographica, Section A 64 (2008) 112–121. [DOI] [PubMed] [Google Scholar]
- [84].Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT, Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature, Biophys. J. 74 (1) (1998) 230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hamill OP, Marty A, Neher E, Sakmann B, Sigworth F, Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches, Pflugers Arch. 391 (2) (1981) 85–100. [DOI] [PubMed] [Google Scholar]
- [86].Lees-Miller JP, Duan Y, Teng GQ, Duff HJ, Molecular determinant of high-affinity dofetilide binding toHERG1 expressed in Xenopus oocytes: involvement of S6 sites, Mol. Pharmacol. 57 (2) (2000) 367–374. [PubMed] [Google Scholar]
- [87].Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC, A structural basis for drug-induced long QT syndrome, Proc. Natl. Acad. Sci. U. S. A. 97 (22) (2000) 12329–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Perry M, Sanguinetti M, Mitcheson J, Symposium review: revealing the structural basis of action of hERG potassium channel activators and blockers, J. Physiol. 588 (17) (2010) 3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Butler A, Helliwell MV, Zhang Y, Hancox JC, Dempsey CE, An Update on the Structure of hERG, Frontiers in Pharmacology 10, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Dickson CJ, Velez-Vega C, Duca JS, Revealing molecular determinants of hERG blocker and activator binding, J. Chem. Inf. Model. 60 (1) (2019) 192–203. [DOI] [PubMed] [Google Scholar]
- [91].Raju S, Barber AF, LeBard DN, Klein ML, Carnevale V, Exploring volatile general anesthetic binding to a closed membrane-bound bacterial voltage-gated sodium channel via computation, PLoS Comput. Biol. 9 (6) (2013), e1003090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Boiteux C, Vorobyov I, French RJ, French C, Yarov-Yarovoy V, Allen TW, Local anesthetic and antiepileptic drug access and binding to a bacterial voltage-gated sodium channel, Proc. Natl. Acad. Sci. U. S. A. 111 (36) (2014) 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Martin LJ, Corry B, Locating the route of entry and binding sites of benzocaine and phenytoin in a bacterial voltage gated sodium channel, PLoS Comput. Biol. 10 (7) (2014), e1003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Nguyen PT, DeMarco KR, Vorobyov I, Clancy CE, Yarov-Yarovoy V, Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel, Proc. Natl. Acad. Sci. 116 (8) (2019) 2945–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Perissinotti L, Guo J, Kudaibergenova M, Lees-Miller J, Ol’khovich M, Sharapova A, Perlovich GL, Muruve DA, Gerull B, Noskov SY, The pore-lipid interface: role of amino-acid determinants of lipophilic access by ivabradine to the hERG1 pore domain, Mol. Pharmacol. 96 (2) (2019) 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kudaibergenova M, Guo J, Khan HM, Zahid F, Lees-Miller J, Noskov SY, Duff HJ, Allosteric coupling between drug binding and the aromatic cassette in the pore domain of the hERG1 channel: implications for a state-dependent blockade, Front. Pharmacol. 11 (2020) 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Yu Z, AP IJ, Heitman LH, Kv 11.1 (hERG)-induced cardiotoxicity: a molecular insight from a binding kinetics study of prototypical Kv 11.1 (hERG) inhibitors, Br. J. Pharmacol. 172 (3) (2015) 940–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zhang YH, Dempsey CE, Hancox JC, The basis for low-affinity herg potassium channel block by sotalol, J. Pharmacol. Pharmacother. 8 (3) (2017) 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Negami T, Araki M, Okuno Y, Terada T, Calculation of absolute binding free energies between the hERG channel and structurally diverse drugs, Sci. Rep. 9 (1) (2019) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Schulz M, Schmoldt A, Therapeutic and toxic blood concentrations of more than 800 drugs and other xenobiotics, Die Pharmazie An Int. J. Pharm. Sci. 58 (7) (2003) 447–474. [PubMed] [Google Scholar]
- [101].Qu Z, Garfinkel A, Weiss JN, Nivala M, Multi-scale modeling in biology: how to bridge the gaps between scales? Prog. Biophys. Mol. Biol. 107 (1) (2011) 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Walpole J, Papin JA, Peirce SM, Multiscale computational models of complex biological systems, Annu. Rev. Biomed. Eng. 15 (2013) 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Yarov-Yarovoy V, Allen TW, Clancy CE, Computational models for predictive cardiac ion channel pharmacology, Drug Discov. Today Dis. Model. 14 (2014) 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Yang PC, Moreno JD, Miyake CY, Vaughn-Behrens SB, Jeng MT, Grandi E, Wehrens XH, Noskov SY, Clancy CE, In silico prediction of drug therapy in catecholaminergic polymorphic ventricular tachycardia, J. Physiol. 594 (3) (2016) 567–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Yang PC, Perissinotti LL, Lopez-Redondo F, Wang Y, DeMarco KR, Jeng MT, Vorobyov I, Harvey RD, Kurokawa J, Noskov SY, Clancy CE, A multiscale computational modelling approach predicts mechanisms of female sex risk in the setting of arousal-induced arrhythmias, J. Physiol. 595 (14) (2017) 4695–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yabek SM, Kato R, Ikeda N, Singh BN, Cellular electrophysiologic responses of isolated neonatal and adult cardiac fibers to d-sotalol, J. Am. Coll. Cardiol. 11 (5) (1988) 1094–1099. [DOI] [PubMed] [Google Scholar]
- [107].Advani SV, Singh BN, Pharmacodynamic, pharmacokinetic and antiarrhythmic properties of d-sotalol, the dextro-isomer of sotalol, Drugs 49 (5) (1995) 664–679. [DOI] [PubMed] [Google Scholar]
- [108].Singh BN, Electrophysiologic basis for the antiarrhythmic actions of sotalol and comparison with other agents, Am. J. Cardiol. 72 (4) (1993) A8–A18. [DOI] [PubMed] [Google Scholar]
- [109].Hsieh Y-C, Horng T-L, Lin S-F, Lin T-C, Ting C-T, Wu T-J-D, l-Sotalol at therapeutic concentrations facilitates the occurrence of long-lasting non-stationary reentry during ventricular fibrillation in isolated rabbit hearts, Circulation J. 73 (1) (2009) 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Waldegger S, Niemeyer G, Mörike K, Wagner C, Suessbrich H, Busch A, Lang F, Eichelbaum M, Effect of verapamil enantiomers and metabolites on cardiac K+ channels expressed in Xenopus oocytes, Cell. Physiol. Biochem. 9 (2) (1999) 81–89. [DOI] [PubMed] [Google Scholar]
- [111].Yang IC-H, Scherz MW, Bahinski A, Bennett PB, Murray KT, Stereoselective interactions of the enantiomers of chromanol 293B with human voltage-gated potassium channels, J. Pharmacol. Exp. Ther. 294 (3) (2000) 955–962. [PubMed] [Google Scholar]
- [112].Carmeliet E, Electrophysiologic and voltage clamp analysis of the effects of sotalol on isolated cardiac muscle and Purkinje fibers, J. Pharmacol. Exp. Ther. 232 (3) (1985) 817–825. [PubMed] [Google Scholar]
- [113].Reid J, Duker G, Almgren O, Nerme V, (+)-Sotalol causes significant occupation of β-adrenoceptors at concentrations that prolong cardiac repolarization, in: Naunyn-Schmiedeberg’s Archives of Pharmacology 341(3), 1990, pp. 215–220. [DOI] [PubMed] [Google Scholar]
- [114].Groh WJ, Gibson KJ, McAnulty JH, Maylie JG, β-Adrenergic blocking property of dl-sotalol maintains class III efficacy in guinea pig ventricular muscle after isoproterenol, Circulation 91 (2) (1995) 262–264. [DOI] [PubMed] [Google Scholar]
- [115].Dubois V, Casarotto E, Danhof M, Della Pasqua O, Pharmacokinetic–pharmacodynamic modelling of drug-induced QTc interval prolongation in man: prediction from in vitro human ether-à-go-go-related gene binding and functional inhibition assays and conscious dog studies, Br. J. Pharmacol. 173 (19) (2016) 2819–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Mo Z-L, Faxel T, Yang Y-S, Gallavan R, Messing D, Bahinski A, Effect of compound plate composition on measurement of hERG current IC50 using PatchXpress, J. Pharmacol. Toxicol. Methods 60 (1) (2009) 39–44. [DOI] [PubMed] [Google Scholar]
- [117].Kramer J, Obejero-Paz CA, Myatt G, Kuryshev YA, Bruening-Wright A, Verducci JS, Brown AM, MICE models: superior to the HERG model in predicting torsade de pointes, Sci. Rep. 3 (1) (2013) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Ridder BJ, Leishman DJ, Bridgland-Taylor M, Samieegohar M, Han X, Wu WW, Randolph A, Tran P, Sheng J, Danker T, A systematic strategy for estimating hERG block potency and its implications in a new cardiac safety paradigm, Toxicol. Appl. Pharmacol. (2020) 114961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Crumb WJ Jr., Vicente J, Johannesen L, Strauss DG, An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel, J. Pharmacol. Toxicol. Methods 81 (2016) 251–262. [DOI] [PubMed] [Google Scholar]
- [120].Abi-Gerges N, Holkham H, Jones E, Pollard C, Valentin JP, Robertson G, hERG subunit composition determines differential drug sensitivity, Br. J. Pharmacol. 164 (2b) (2011) 419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Männikkö R, Overend G, Perrey C, Gavaghan C, Valentin JP, Morten J, Armstrong M, Pollard C, Pharmacological and electrophysiological characterization of nine, single nucleotide polymorphisms of the hERG-encoded potassium channel, Br. J. Pharmacol. 159 (1) (2010) 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kirsch GE, Trepakova ES, Brimecombe JC, Sidach SS, Erickson HD, Kochan MC, Shyjka LM, Lacerda AE, Brown AM, Variability in the measurement of hERG potassium channel inhibition: effects of temperature and stimulus pattern, J. Pharmacol. Toxicol. Methods 50 (2) (2004) 93–101. [DOI] [PubMed] [Google Scholar]
- [123].Orvos P, Kohajda Z, Szlovák J, Gazdag P, Árpádffy-Lovas T, Tóth D, Geramipour A, Tálosi L, Jost N, Varró A, Evaluation of possible Proarrhythmic potency: comparison of the effect of Dofetilide Cisapride, Sotalol, Terfenadine, and Verapamil on hERG and native I Kr currents and on cardiac action potential, Toxicol. Sci. 168 (2) (2019) 365–380. [DOI] [PubMed] [Google Scholar]
- [124].Davie C, Pierre-Valentin J, Pollard C, Standen N, Mitcheson J, Alexander P, Thong B, Comparative pharmacology of Guinea pig cardiac myocyte and cloned hERG (IKr) channel, J. Cardiovasc. Electrophysiol. 15 (11) (2004) 1302–1309. [DOI] [PubMed] [Google Scholar]
- [125].Zhou Q, Zygmunt AC, Cordeiro JM, Siso-Nadal F, Miller RE, Buzzard GT, Fox JJ, Identification of I Kr kinetics and drug binding in native myocytes, Ann. Biomed. Eng. 37 (7) (2009) 1294–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Fiset C, Feng Z-P, Wang L, Sheldon RS, Duff HJ, [3H] Dofetilide binding: biological models that manifest solely the high or the low affinity binding site, J. Mol. Cell. Cardiol. 28 (5) (1996) 1085–1096. [DOI] [PubMed] [Google Scholar]
- [127].Sube R, Ertel EA, Cardiomyocytes derived from human induced pluripotent stem cells: An in-vitro model to predict cardiac effects of drugs, J. Biomed. Sci. Eng. 10 (11) (2017) 527. [Google Scholar]
- [128].Sala L, Bellin M, Mummery CL, Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: has the time come? Br. J. Pharmacol. 174 (21) (2017) 3749–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Doss MX, Sachinidis A, Current challenges of iPSC-based disease modeling and therapeutic implications, Cells 8 (5) (2019) 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Perissinotti LL, De Biase PM, Guo J, Yang P-C, Lee MC, Clancy CE, Duff HJ, Noskov SY, Determinants of isoform-specific gating kinetics of hERG1 channel: combined experimental and simulation study, Front. Physiol. 9 (2018) 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA, MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia, Cell 97 (2) (1999) 175–187. [DOI] [PubMed] [Google Scholar]
- [132].Mirams GR, Davies MR, Brough SJ, Bridgland-Taylor MH, Cui Y, Gavaghan DJ, Abi-Gerges N, Prediction of thorough QT study results using action potential simulations based on ion channel screens, J. Pharmacol. Toxicol. Methods 70 (3) (2014) 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Lancaster MC, Sobie E, Improved prediction of drug-induced Torsades de pointes through simulations of dynamics and machine learning algorithms, Clin. Pharmacol. Therapeutics 100 (4) (2016) 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Britton OJ, Abi-Gerges N, Page G, Ghetti A, Miller PE, Rodriguez B, Quantitative comparison of effects of dofetilide, sotalol, quinidine, and verapamil between human ex vivo trabeculae and in silico ventricular models incorporating inter-individual action potential variability, Front. Physiol. 8 (2017) 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Dutta S, Chang KC, Beattie KA, Sheng J, Tran PN, Wu WW, Wu M, Strauss DG, Colatsky T, Li Z, Optimization of an in silico cardiac cell model for proarrhythmia risk assessment, Front. Physiol. 8 (2017) 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Grandi E, Pasqualini FS, Bers DM, A novel computational model of the human ventricular action potential and Ca transient, J. Mol. Cell. Cardiol. 48 (1) (2010) 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Tomek J, Bueno-Orovio A, Passini E, Zhou X, Minchole A, Britton O, Bartolucci C, Severi S, Shrier A, Virag L, Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block, Elife 8 (2019), e48890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Ten Tusscher KH, Panfilov AV, Cell model for efficient simulation of wave propagation in human ventricular tissue under normal and pathological conditions, Phys. Med. Biol. 51 (23) (2006) 6141. [DOI] [PubMed] [Google Scholar]
- [139].Asai T, Adachi N, Moriya T, Oki H, Maru T, Kawasaki M, Suzuki K, Chen S, Ishii R, Yonemori K, Cryo-EM structure of K+-bound hERG channel complexed with the blocker astemizole, Structure 29 (3) (2021) 203–212 (e4). [DOI] [PubMed] [Google Scholar]
- [140].Li J, Shen R, Reddy B, Perozo E, Roux B, Mechanism of C-type inactivation in the hERG potassium channel, Sci. Adv. 7 (5) (2021), eabd6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Cuello LG, Jogini V, Cortes DM, Perozo E, Structural mechanism of C-type inactivation in K+ channels, Nature 466 (7303) (2010) 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Cuello LG, Cortes DM, Perozo E, The gating cycle of a K+ channel at atomic resolution, Elife 6 (2017), e28032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Pau V, Zhou Y, Ramu Y, Xu Y, Lu Z, Crystal structure of an inactivated mutant mammalian voltage-gated K+ channel, Nat. Struct. Mol. Biol. 24 (10) (2017) 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Matthies D, Bae C, Toombes GE, Fox T, Bartesaghi A, Subramaniam S, Swartz KJ, Single-particle cryo-EM structure of a voltage-activated potassium channel in lipid nanodiscs, Elife 7 (2018), e37558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Xu X, Kaindl J, Clark MJ, Hübner H, Hirata K, Sunahara RK, Gmeiner P, Kobilka BK, Liu X, Binding pathway determines norepinephrine selectivity for the human β 1 AR over β 2 AR, Cell Res. (2020) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Amanfu RK, Saucerman JJ, Modeling the effects of β1-adrenergic receptor blockers and polymorphisms on cardiac myocyte Ca2+ handling, Mol. Pharmacol. 86 (2) (2014) 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.