

Abstract
Transposable element (TE) mobilization is a significant source of genomic variation and has been associated with various human diseases. The exponential growth of population-scale whole-genome sequencing and rapid innovations in long-read sequencing technologies provide unprecedented opportunities to study TE insertions and their functional impact in human health and disease. Identifying TE insertions, however, is challenging due to the repetitive nature of the TE sequences. Here, we review computational approaches to detecting and genotyping TE insertions using short- and long-read sequencing and discuss the strengths and weaknesses of different approaches.
Keywords: long-read sequencing, mobile element, retrotransposition, structural variation
INTRODUCTION
Transposable elements (TEs) are ubiquitous in eukaryotes and constitute nearly half of the human genome (Lander et al., 2001). Most TE sequences are remnants of ancient proliferation, having lost their capacity to further mobilize; however, three TE families (L1, Alu, and SVA) can still mobilize by a copy-paste mechanism to transcribe themselves into RNA and insert cDNA reverse-transcribed from the TE RNA into a new genomic location. New copies of TEs are created in the human germline at an estimated rate of 1 out of every 20–200 live births (Feusier et al., 2019; Stewart et al., 2011); they have been found to account for ~40% of genomic structural variation in humans (Sudmant et al., 2015). In addition, more than a hundred TE insertions have been causally linked to Mendelian disorders and hereditary cancers (Hancks & Kazazian, 2016). A potential pathogenic role of TEs has also been reported in various common diseases, such as sporadic cancers and autoimmune, developmental, and neuropsychiatric disorders (Gardner et al., 2019; Hancks & Kazazian, 2016; Lee et al., 2012; Scott & Devine, 2017). Thus, the ability to detect TE insertions in genome sequencing data is important for determining whether TEs are associated with a genetic trait or disease.
TEs are large DNA elements (several hundreds of bases to several kilobases), and the human genome has numerous TE copies with similar, often nearly identical sequences. This makes it difficult to detect TE insertions from the current short-read (100–150 bp) sequencing data. Whereas conventional methods for structural variation (SV) detection mainly rely on uniquely mapped reads, short reads derived from inserted TE copies cannot uniquely map to the reference genome. Thus, new methods for TE insertion detection have been developed, with specialized handling of non-uniquely mapped reads derived from TEs. In general, there are two categories of TE insertion detection tools. Those in the first category detect insertions of known TE families, and thus are used for species with well-annotated TEs like humans, utilizing TE sequence libraries to which the reads not uniquely mapping to the reference genome are realigned. Those in the second category detect insertions of novel or unknown TE families without requiring TE sequence libraries, and thus can be used in species for which we have limited knowledge of active TEs.
Despite the common principles, the existing TE-calling methods differ in the types of insertions they are designed to detect (e.g., germline vs. somatic) and the applicable study design. One group of tools focuses on detecting germline insertions, which are non-reference TE insertions (i.e., absent in the reference genome) inherited from parents and thus present in every cell of the body (Fig. 1A). These tools typically analyze genome sequences from blood samples (Gardner et al., 2017; Thung et al., 2014; Wu et al., 2014; Zhuang, Wang, Theurkauf, & Weng, 2014; Bogaerts-Márquez et al., 2020). Tools in a second group analyze genome sequences from family members to identify de novo insertions, which are TE insertions present in individuals (often affected by diseases) but absent in their parents (Feusier et al., 2019; Gardner et al., 2019); Fig. 1B). Those in the last category were developed to identify somatic or mosaic TE insertions that occur in post-zygotic tissues, such as in cancer and non-cancerous brain tissues (Evrony et al., 2015; Lee et al., 2012; Tubio et al., 2014; also see Fig. 1C). These tools often analyze genome sequences from a pair of tissues—the tissue of interest and a blood or another control tissue—to ensure the somatic origin of detected insertions.
Figure 1.
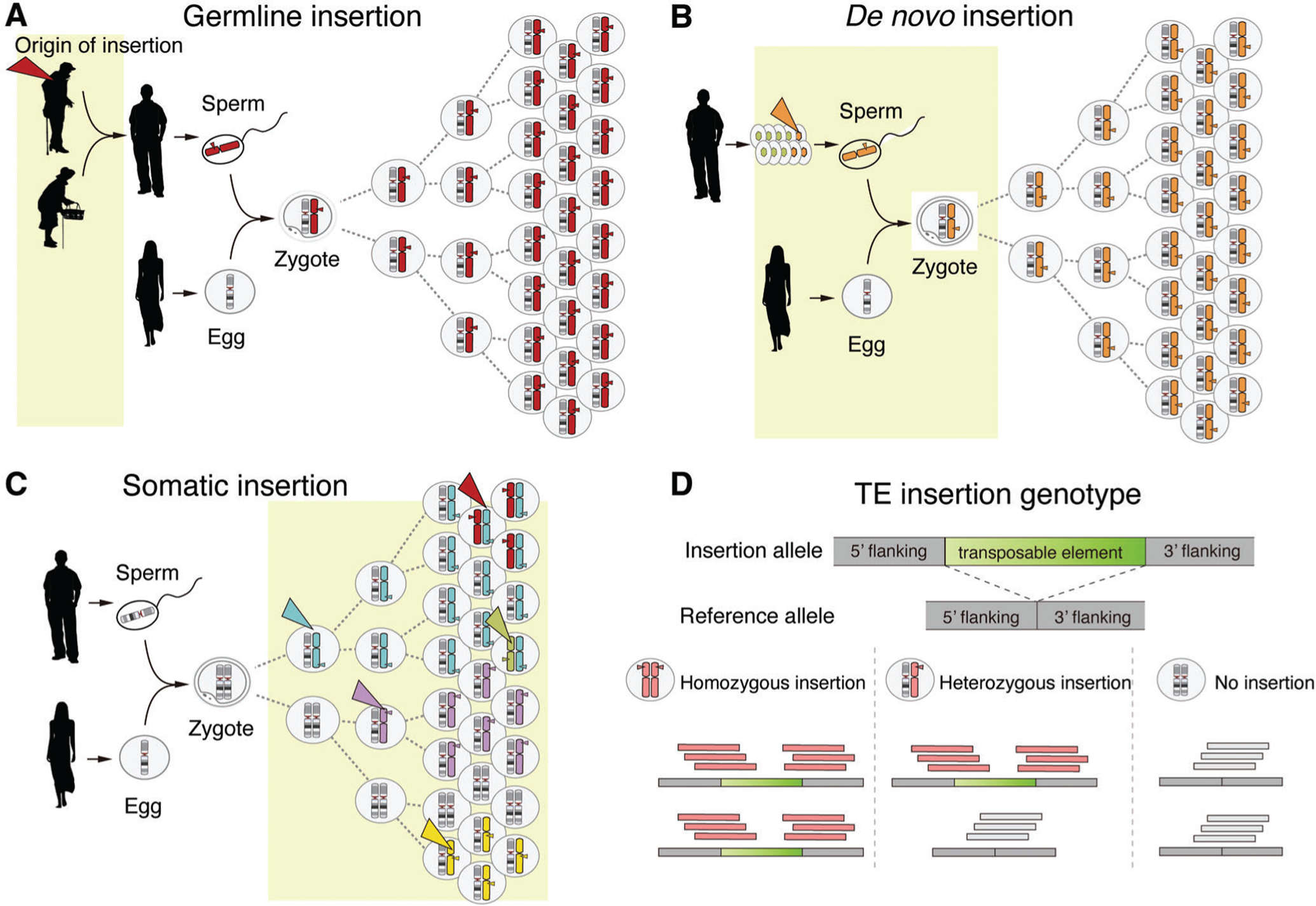
Different types of TE insertions and TE genotypes. (A-C) Three different types of TE insertions with light yellow boxes indicating the time frames for when each arises. The colored triangles point to the origin of chromosomes carrying insertions. (A) Germline TE insertions are inherited from parents, and thus are present in every cell of the body. (B) De novo TE insertions arise during gametogenesis of the parents or early embryogenesis of the child, and thus are not detected in blood samples of the parents. (C) Somatic insertions occur during development and aging and create genetic mosaicism in an individual. Depending on when and where insertions occur, they are detected in different tissues at different mosaic levels. (D) Every TE insertion in an individual genome has three genotypes: homozygous (1/1), heterozygous (0/1), or no insertion (0/0). A homozygous insertion produces TE-junction-spanning reads originating from two insertion alleles; a heterozygous insertion produces reads from both insertion and reference alleles. Chromosomes carrying a non-reference TE insertion and sequence reads derived from the chromosomes are marked in red.
Diploid human genomes have three different genotypes for each germline TE insertion (Fig. 1D): homozygous (i.e., both chromosomes carry the insertion), heterozygous (i.e., insertion present in one chromosome), and reference (i.e., no insertion). TE genotype information is important for phasing—discerning which parental alleles carry TE insertions—and also for associating TE insertion polymorphisms with other types of variants such as single-nucleotide polymorphisms (SNPs) or expression quantitative trait loci (eTQLs).
Here we review the computational approaches and available tools to identify and genotype different types of TE insertions, mostly from whole-genome sequencing (WGS) data generated using short- and long-read platforms. In this review, we focus on methods for the human genome and recommend other reviews for tools for non-human species (Goerner-Potvin & Bourque, 2018).
TE CONSENSUS SEQUENCE AND REFERENCE ANNOTATION
Over evolutionary time, new TE families and subfamilies have emerged through different mechanisms, including the arms race between TEs and a host’s defense system that drives novel mutations in TEs (Jacobs et al., 2014; Kazazian, 2004). A TE phylogeny tree can be constructed by comparing TE sequences, with each clade in the tree defining a subfamily. A representative sequence for each subfamily, called the consensus sequence, is determined by multiple sequence alignments of individual TEs that belong to each clade. Repbase (Jurka et al., 2005), Dfam (Hubley et al., 2016), and UCSC Repeat Browser (Fernandes et al., 2020) provide well-curated libraries for consensus sequences of TE sub-families, including subfamilies of L1, Alu, and SVA. The most popular TE annotation tool is RepeatMasker (Smit, Hubley and Green, 2015), which annotates individual TE copies in the reference genome using BLAST to match the consensus sequence to the reference genome. The annotation includes information on the TE subfamily, the degree of sequence divergence, genomic coordinates, and the length of each TE copy in the reference genome. Most TE insertion calling methods use consensus sequences and reference annotations (Table 1).
Table 1.
Tools for TE Insertion Detection
| Sequence read type | Method | TE sequence library | TE-specific annotation |
|---|---|---|---|
| PE short reads | Tea (Lee et al., 2012) | Yes | Yes |
| PE short reads | MELT (Gardner et al., 2017) | Yes | Yes |
| PE short reads | Mobster (Thung et al., 2014) | Yes | Yes |
| PE short reads | RetroSeq (Keane et al., 2013) | Yes | Yes |
| PE short reads | TraFiC (Tubio et al., 2014) | Yes | Yes |
| PE short reads | TEMP (Zhuang et al., 2014) | Yes | Yes |
| PE short reads | T-lex3 (Bogaerts-Márquez et al., 2019) | Yes | Yes |
| PE short reads | Tangram (Wu et al., 2014) | Yes | Yes |
| PE short reads | ITIS (Jiang, Chen, Huang, Liu, & Verdier, 2015) | Yes | Yes |
| Long reads | Sniffles (Sedlazeck et al., 2018) | No | No |
| Long reads | SVIM (Heller & Vingron, 2019) | No | No |
| Long reads | pbsv (https://github.com/PacificBiosciences/pbsv) | No | No |
| Long reads | PBHoney (English, Salerno, & Reid, 2014) | No | No |
| Long reads | PALMER (Zhou et al., 2019) | No | Yes |
| Multiple platforms | xTea (https://github.com/parklab/xTea) | Yes | Yes |
| PE short reads | DD_DETECTION (Kroon et al., 2016) | No | Yes |
| PE short reads | TranSurVeyor (Rajaby & Sung, 2018) | No | Yes |
GENERAL STRATEGIES TO DETECT TE INSERTIONS FROM PAIRED-END SHORT READS
Most TE detection methods for paired-end short sequence reads use similar strategies to identify two types of read alignment patterns near the breakpoints of each insertion (Fig. 2; Table 1). First, the tools examine read pairs with one end mapping to the genomic region flanking a new TE copy and the mate read mapping to the TE sequence library. They typically have default TE sequence libraries consisting of TE consensus sequences. Some tools, such as Tea (Lee et al., 2012) and TraFiC (Tubio et al., 2014), also include sequences of individual TE copies from the reference genome; a subset of the tools, for example, MELT (Gardner et al., 2017) and xTea (https://github.com/parklab/xTea), can take customized TE libraries from users as input. Second, the tools identify “soft-clipped” (or “split”) reads that originate from the junction between a TE and the flanking region. When aligned to the reference genome, these junction-spanning reads show that only a part of the read maps to the flanking region and the remaining TE portion of the read is clipped.
Figure 2.
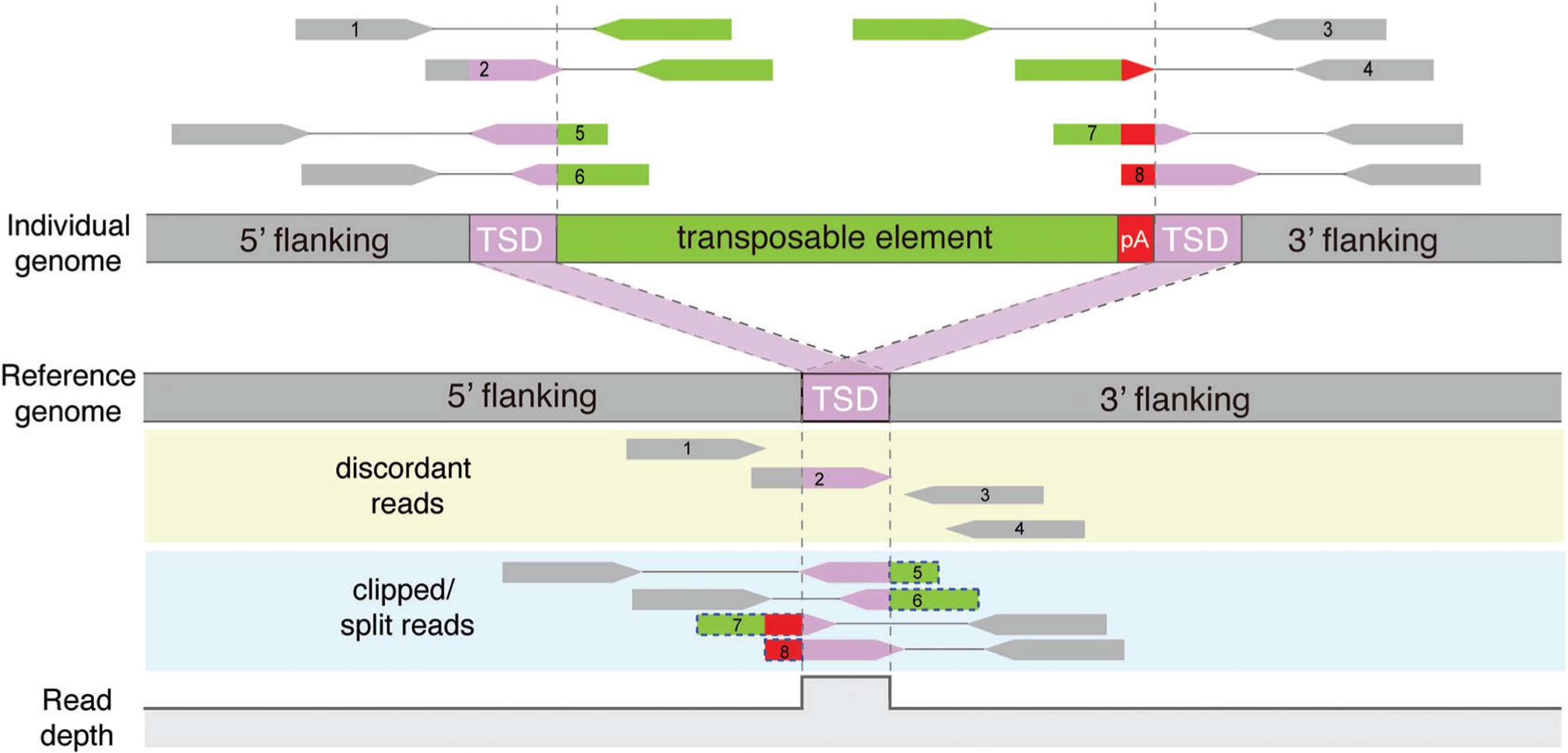
Identification of TE insertions from short-read sequencing data. Paired-end short reads from an individual with a TE insertion are aligned to the reference genome. A TE insertion is detected by identifying two types of read clusters near the insertion breakpoints: (i) discordant reads (reads 1–4) are uniquely aligned to flanking regions and have their mate-pair reads aligned to one of many reference TE copies remotely located from the breakpoints; and (ii) clipped reads or split reads (reads 5–8) span the insertion breakpoints, and thus have soft-clipped or split mapping to the reference (shown in dotted blue boxes). The change in read depth at a non-reference insertion site is shown at the bottom. Gray dashed lines indicate the boundary of TSDs.
From an input BAM or CRAM file including the reference read alignment, tools collect discordant read pairs where mate reads in a pair display alignment at an unexpected distance or orientation; some tools collect read pairs where one read is uniquely mapped while the mate read is ambiguously mapped to the reference genome. Tools identify candidate insertion sites by verifying whether uniquely mapped reads from the collected read pairs form clusters and whether their mate reads map to a TE copy. Tea, MELT, and several other tools align the reads to TE sequence libraries to verify whether the reads originated from a potentially new TE copy. Other tools, such as TEMP (Zhuang et al., 2014), simply verify whether the reported mapping position of the mate read is within a reference TE copy according to RepeatMasker annotation. Next, for each cluster of TE-derived discordant reads, clipped reads are examined by realigning the clipped sequence to TE sequence libraries to pinpoint insertion breakpoints.
For each insertion, several tools, such as Tea, TraFiC, MELT, Mobster (Thung et al., 2014), and xTea, also annotate TE-specific features (e.g., subfamily, insertion size, and orientation). Several features, such as the presence of target site duplication (TSD) and poly(A) tails, indicate the insertional mechanism. Given that all active human TEs are retrotransposons that mobilize via mRNA intermediates with poly(A) tails, a pileup of clipped reads with consecutive poly(A/T) bases on one side of a breakpoint is a strong indicator of target-primed reverse transcription (TPRT)−mediated retrotransposition rather than of other types of genomic rearrangement involving TE sequences. In addition, because the L1-encoding endonuclease responsible for retrotransposition of all active TE families cuts double-stranded DNA in a staggered manner with a ~15 bp overhang, the completion of retrotransposition leads to TSD, which is duplication of the short overhang sequence on both sides of the genomic flanking regions of the insertion (Gilbert, Lutz-Prigge, & Moran, 2002). The tools annotate and utilize these mechanistic signatures to filter false positive calls and improve detection specificity.
IDENTIFICATION OF DIFFERENT TYPES OF TE INSERTIONS
Germline and de novo TE insertions
Each tool is designed to analyze certain types of TE insertions. Specifically, tools such as MELT, RetroSeq (Keane, Wong, & Adams, 2013), Mobster, T-lex3 (Bogaerts-Márquez et al., 2020), and TEMP, were developed to identify germline insertions. Few of these tools provide a module to call de novo insertions; however, users can utilize the basic functions of the tools for de novo calling by considering the proband as a “case” and all other members in the family as “controls.” A straightforward approach adopted in multiple studies is to run the tool on each genome of both case and controls and then compare the outputs to filter out insertions shared in both case and controls. Due to the often lower sequencing depths of control samples, true TE insertions may fail to be detected in control genomes, resulting in false positive de novo calls in probands. One strategy to cope with this issue is to lower the stringency of calls for control samples. An extreme example of such filtering is to require control samples to have no split reads or discordant TE-mapping read pairs consistent with the insertion in the proband. One obvious caveat is the risk of missing true de novo events with such strict filtering. Most studies choose filtering schemes empirically, and so their performance may vary for different datasets. Of note is that the origin of some de novo insertions may be mosaic in their parents, i.e., present in the germ cell of a parent that generated the zygote and perhaps present in a low fraction of cells in the parent’s blood sample (Faulkner & Billon, 2018). These parental mosaic events are likely to be missed with stringent control filtering, and thus require a different approach to ensure their detection.
Somatic or mosaic TE insertions
Most tools for detecting somatic TE insertions, including Tea and TraFiC, were developed to study somatic insertions in cancer (Lee et al., 2012; Rodriguez-Martin et al., 2020; Tubio et al., 2014). These tools detect non-reference TE insertions from genome sequences of cancer and matched non-cancerous—mostly blood—samples from the same patients. They then filter out those events shared between the cancer and non-cancer samples, just as is done to detect de novo insertions. Detecting somatic insertions in cancer genomes is complicated by the abundant clipped and discordant reads that originate from various types of genomic rearrangement other than retrotransposition. Thus, the tools need effective filtering schemes to improve detection specificity, for example, using annotation of mechanistic signatures and code optimization to minimize run-time and resource requirements.
Somatic TE insertions also occur in non-cancer samples. Recent studies report post-zygotic somatic retrotransposition creating somatic mosaicism in the human brain and other tissues (Erwin et al., 2016; Evrony et al., 2015; Faulkner & Billon, 2018; Upton et al., 2015; Zhao et al., 2019). Identification of such somatic TE insertions in non-cancer samples is challenging due to the low fraction of cells carrying each insertion in a heterogeneous cell population. Thus, a single-cell approach has been used to identify somatic insertions in the human brain by leveraging the fact that somatic signals in a single cell have the same appearance as heterozygous germline insertions. scTea (single-cell Tea; Evrony et al., 2015) was developed to analyze MDA-amplified single-cell WGS data, and takes into account genome-amplification artifacts (e.g., chimeric reads, uneven genomic coverage, and allelic/locus dropout) to separate true insertion signals from noise. Several single-cell-targeted sequencing approaches including L1–IP (Evrony et al., 2012), RC–seq (Upton et al., 2015), and SLAV–seq (Erwin et al., 2016) have been used to profile somatic L1 insertions from a large number of brain cells. Different studies, however, have produced substantially different estimates of the somatic L1 insertion rate, necessitating rigorous bioinformatic analysis and experimental validation (Evrony, Lee, Park, & Walsh, 2016; Faulkner & Garcia-Perez, 2017).
As an alternative to single-cell sequencing with its high experimental costs and amplification bias, some recent studies have used a bulk sequencing approach with high sequencing coverage to detect low-level mosaic somatic insertions in the brain and other tissues. RetroSom is a machine learning–based tool that detects somatic L1 and Alu insertions from ultra-high-depth WGS, and its application to ~ 200× bulk WGS of sorted neurons and glia revealed two brain-specific somatic L1 insertions at ~1% mosaicism (Zhu et al., 2019). Furthermore, HAT-seq, a PCR-based targeted bulk sequencing approach, identified somatic L1 insertions with low–level mosaicism in neurons and non-brain tissues (Zhao et al., 2019). By employing a nucleotide–shifting design for semi-amplicon libraries, HAT-seq produces high-quality sequences that fully cover the 3′ insertion junction, which facilitate false-positive filtering based on both sequence and read-count features. The aforementioned studies based on single-cell WGS and bulk WGS or targeted capture approaches present mounting evidence of somatic retrotransposition in various human tissues creating inter-cellular genomic diversity within individuals. More studies are warranted to better understand the role of somatic TE activity in various tissues.
Non-canonical or complex TE insertions
Some TE insertions mobilize the flanking sequence of a source TE along with the TE sequence to a new insertion site when TE transcription uses an alternative upstream promoter or continues beyond the TE’s weak polyadenylation signal (Goodier, Ostertag, & Kazazian, 2000). This event is called transduction. ~30% of SVA insertions have 5′ or 3′ transduction (Damert et al., 2009), whereas ~15% of L1 insertions have 3′ transduction, with a few anecdotal examples of 5′ transduction in the human brain (Evrony et al., 2015; Sanchez-Luque et al., 2019). Reads originating from the transduced flanking sequences cannot be aligned to TE sequence libraries, which often results in a failure to call such insertions with transduction. To better detect TEs with transduction, MELT first runs canonical modules, and then for each candidate insertion it searches for potential transduction events. TraFiC detects somatic transduction events by determining whether the cluster of discordant read pairs points to the flanking region of either reference or polymorphic full-length TE copies. TraFiC also detects the so-called orphan transduction events where only flanking sequences are inserted without TE sequences due to early 3′ truncation upon retrotransposition. Similar to TraFiC, xTea also re-aligns the collected reads to flanking region sequences of both reference and polymorphic full-length copies to detect insertions with transduction, including orphan transductions.
Some TE insertions are also known to promote SVs upon retrotransposition in human cell lines (Gilbert et al., 2002) and in the human brain (Erwin et al., 2016). TraFiC-mem (Rodriguez-Martin et al., 2020) detects, in human tumors, different types of L1-associated SVs—deletion, duplication, inversion, and translocation—by examining the patterns of discordant read pairs that support an SV and an L1 insertion. For L1-promoted deletions, TraFiC-mem additionally utilizes read-depth-based copy number variation (CNV) calls and filters out candidates that are not supported by CNV calls. Requiring this additional CNV support improves specificity, but limited sensitivity and resolution of read-depth CNV calling, especially for small events, may lead to sensitivity loss for the detection of TE-mediated deletions. xTea provides an option to report the breakpoints of TE-mediated SVs without requiring CNV calls.
THE USE OF LONG READS FOR COMPREHENSIVE TE INSERTION DETECTION
Despite the improved performance of TE detection algorithms for short-read sequencing data, it is still difficult to detect certain subsets of TE insertions, including those that accompany complex genomic rearrangements or fall into repetitive genomic regions. Genomic regions with existing TE copies from the same TE subfamily, or centromeric or telomeric regions with many gaps, are particularly challenging for TE detection due to limited short read mappability (Bzikadze & Pevzner, 2019; Jain et al., 2018; Miga et al., 2019). Recent advances in long-read sequencing, notably PacBio and Oxford Nanopore (ONT) technologies, create ~10- to 15-Kbp-long reads. Data from these platforms allow one to construct the entire region that includes a new insertion and its flanking regions; thus markedly improving the identification of those challenging types of TE insertions.
Currently, there are only two tools specifically designed for TE calling using long-read sequencing: PALMER (Zhou et al., 2019) and xTea. PALMER detects L1 insertions from PacBio long reads, whereas xTea detects insertions of all TE families from long reads generated using PacBio and ONT, as well as the 10X barcode-linked read technology. Un-like the short reads, the long reads often encompass the inserted TEs, thus not necessitating the use of clipped or discordant reads and directly reporting the insertion sequences within each read alignment (i.e., in the CIGAR field of SAM/BAM/CRAM files). Some aligners, such as BLASR (Chaisson & Tesler, 2012) and BWA (Li & Durbin, 2009) rarely report insertions in the CIGAR field, but only report clipped reads. In contrast, recent aligners, such as NGLMR (Sedlazeck et al., 2018) and Minimap2 (Li, 2018) report insertions of small and intermediate size within the CIGAR field of each alignment. In general, the preferred approach would be to examine not only read clipping, but also internal insertion breakpoints and sequences reported in the CIGAR field.
Due to the high rate of sequencing errors in long-read sequencing, reads supporting TE insertions are clipped not at the exact insertion breakpoint but at highly variable positions. To define accurate breakpoints, different tools use different strategies (Fig. 3). xTea and Sniffles (Sedlazeck et al., 2018), a SV caller, use a similar approach, clustering breakpoints and filtering out clusters for which the standard deviation of breakpoint coordinates is larger than a pre-determined cutoff. By contrast, PALMER relies on the L1 annotation of the human reference genome rather than checking the clipped or internal CIGAR field. It first masks portions of long reads aligned to the reference L1 copies, and then searches the remaining portions of long reads against a hot full-length L1 sequence (GenBank: L19088) to identify reads with a putative L1 insertion. All supporting reads are then clustered and assembled into long contig sequences when reads are within 100 bp of each other. xTea and a few other SV callers, such as SMRT-SV (Huddleston et al., 2017), also provide this local assembly option that combines sequences from clipped reads and reads supporting internal insertions and compare the assembled contigs to the reference genome to define the insertion sequence. General, non-TE-specific SV callers, such as SMRT-SV, only report insertion sequences lacking TE-specific annotation.
Figure 3.
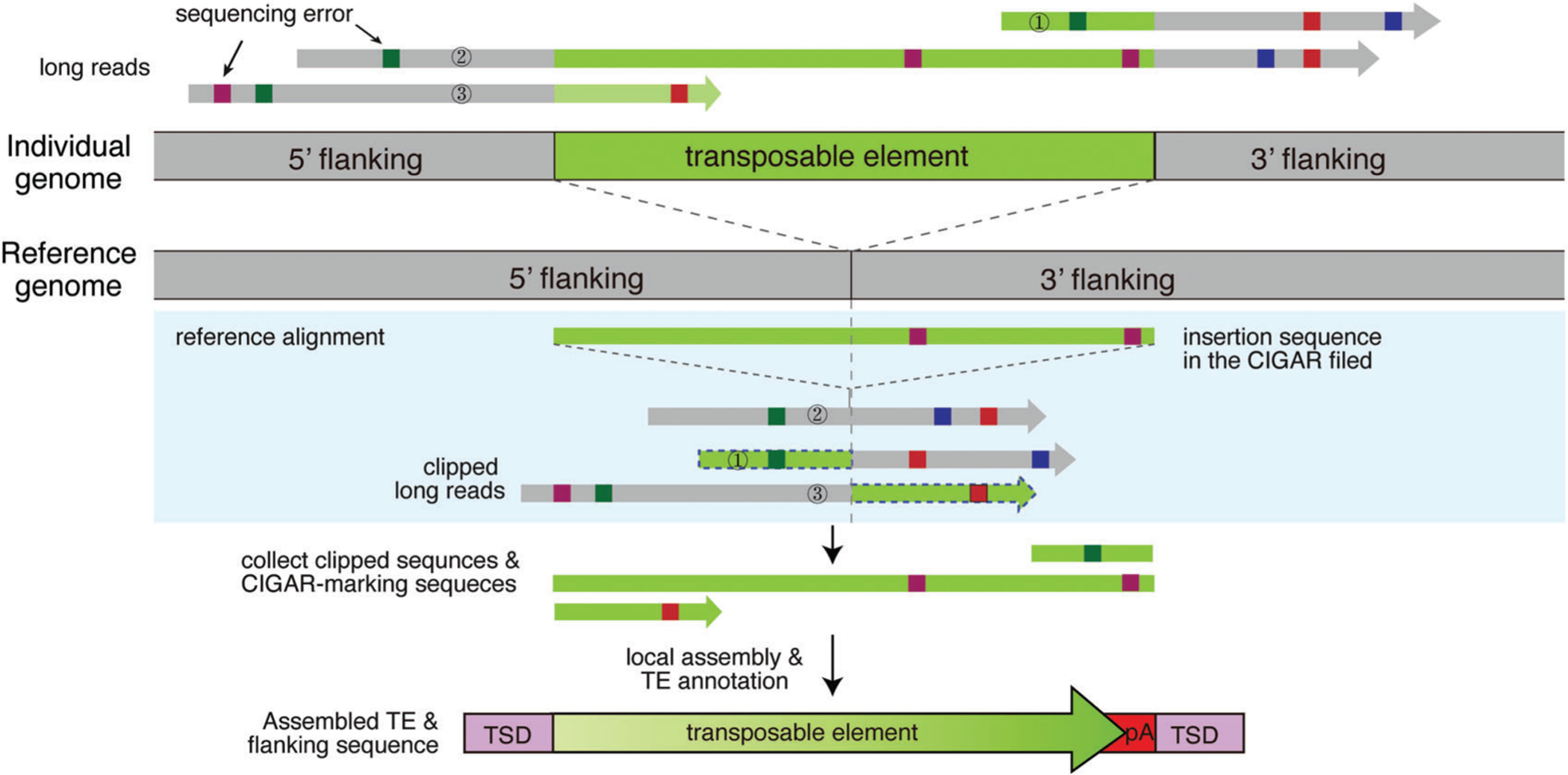
Identification of TE insertions from long-read sequencing data. Long reads from an individual with a TE insertion are aligned to the reference genome. First, insertion-supporting reads are collected: reads with a CIGAR field indicating internal insertions (read 2) and soft-clipped or split reads (read 1 and 3). Local assembly of these insertion-supporting reads is performed to create long contig sequences. The contigs are aligned to TE subfamily sequence libraries, and TE insertions are identified and annotated for multiple features, such as insertion size, TSD, and poly(A) tails.
TE INSERTION DETECTION FOR NOVEL TE FAMILIES
Some TE insertion callers do not rely on any reference TE annotation or any TE sequence library, and thus are well-suited to studying TEs in species in which there is limited information for active TEs. To our knowledge, only two tools—DD_DETECTION (Kroon et al., 2016) and TranSurVeyor (Rajaby & Sung, 2018)—support TE insertion calls for such de novo families. Similar to typical SV callers, DD_DETECTION (Kroon et al., 2016) collects discordant reads and clusters them according to their genomic coordinates. For each discordant read cluster, it examines clipped or split reads to define the breakpoints, with additional filtering to remove false positives, for example by checking the consistency of the two sides of discordant reads. TranSurVeyor (Rajaby & Sung, 2018) takes a similar approach but improves the performance by re-aligning one end of discordant reads originating from TEs and adopting an SNP-aware filter to remove incorrectly aligned reads in repeat regions. A major limitation of the existing de novo TE insertion callers is that they still only focus on detecting insertions, like other SV callers, but do not provide TE-specific annotation, such as TE family or target site duplication.
TE INSERTION GENOTYPING
Three tools—MELT, TypeTE (Goubert et al., 2020), and xTea—report the genotypes of germline TE insertions that are inferred from supporting read patterns. Notably, TypeTE provides genotypes of input TE insertions, but does not detect TE insertions. Since paternal and maternal chromosomes cannot be distinguished in general, the algorithms can only classify each insertion into one of three different genotype states (Fig. 1D; Table 2): neither paternal nor maternal alleles with the insertion (0/0 or homozygous reference), one parental allele with the insertion (0/1 or heterozygous), and both alleles with the insertion (1/1 or homozygous). TE genotyping is generally performed in two steps: feature extraction and statistical estimation of genotype.
Table 2.
Tools for TE Insertion Genotyping
| Read type | Method | Genotyping strategy | Graph alignment |
|---|---|---|---|
| Short reads | MELT (Gardner et al., 2017) | Maximum likelihood | No |
| Short reads | GHSTDEL (Chu et al., 2014) | Machine learning | No |
| Short reads | TypeTE (Goubert et al., 2020) | Maximum likelihood | No |
| Short reads | xTea (https://github.com/parklab/xTea) | Machine learning | Yes |
| Short reads | Paragraph (Chen et al., 2019) | Maximum likelihood | Yes |
| Short reads | GraphTyper2 (Eggertsson et al., 2019) | Maximum likelihood | Yes |
| Short reads | vg toolkit (Hickey et al., 2020) | Heuristic | Yes |
| Long reads | SVJedi (Lecompte et al., 2019) | Maximum likelihood | No |
| Long reads (phasing) | NanoSV (Cretu Stancu et al., 2017) | Heuristic | No |
Feature extraction for SV genotyping can be accomplished by aligning reads either to the linear reference genome or to genome graphs (Fig. 4A). All current TE-specific genotyping methods extract features using the linear reference genome. The tools utilize the number of discordant reads and the number of concordant reads across the breakpoints, and also the number of clipped or split reads at the breakpoints of each insertion to determine its genotype. xTea extracts additional features that support the reference allele, including the number of fully mapped reads at the breakpoints. These additional features can improve accuracy in TE genotyping.
Figure 4.
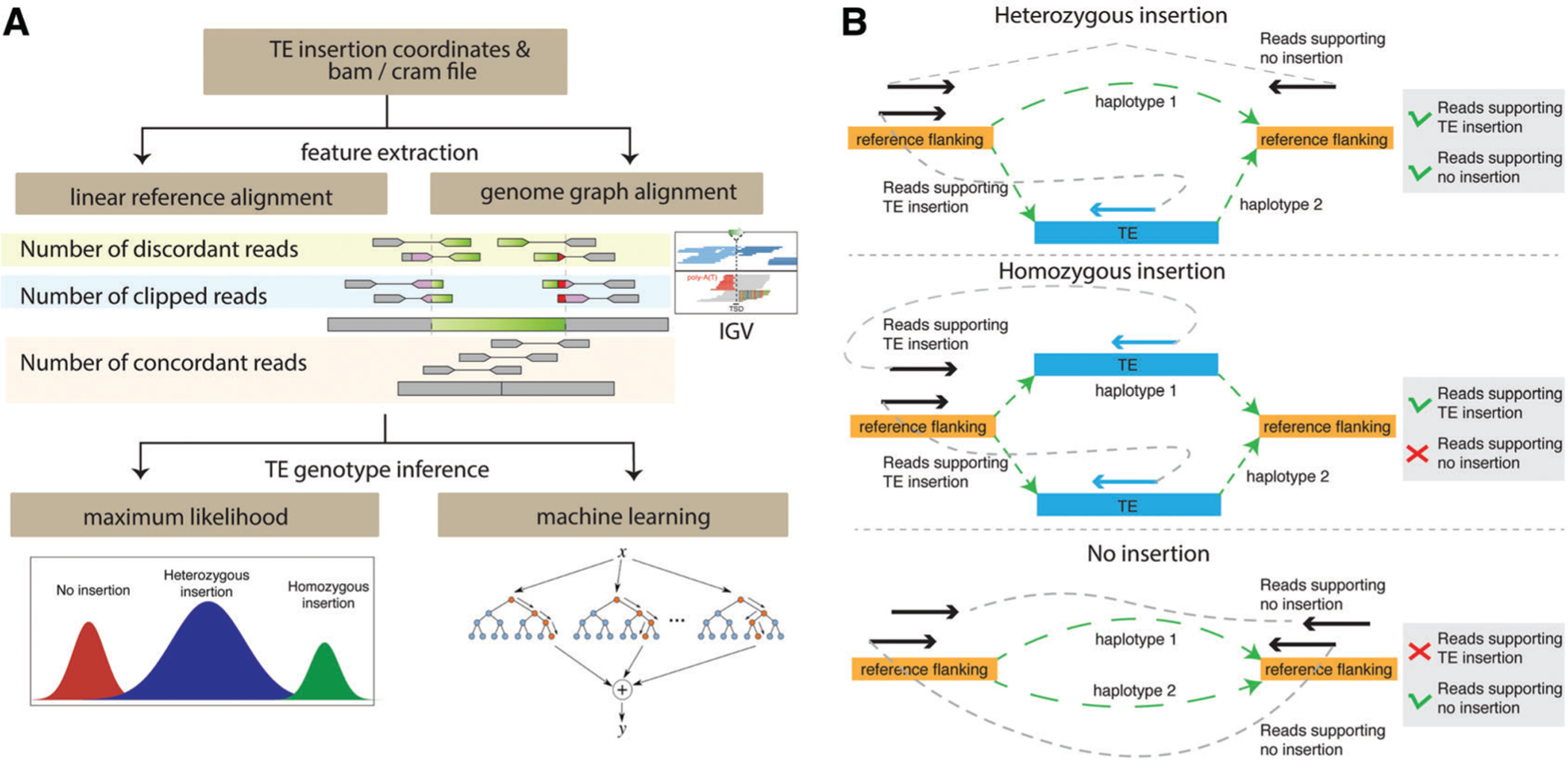
Overview of TE insertion genotyping. (A) In order to genotype TE insertions, informative features are extracted from read alignment to the linear reference genome or genome graphs. The IGV screenshot shows the local alignment patterns of a non-reference insertion with discordant and soft-clipped reads. Based on the extracted features, machine-learning or maximum-likelihood approaches are taken to estimate the genotype of each insertion. (B) Genome graph−based feature extraction is illustrated. Genome graphs represent two haplotypes of a heterozygous insertion (top), a homozygous insertion (middle), and no insertion (bottom). Using each genome graph, read pairs with one read aligned to the flanking region and the mate-pair read aligned to the TE insertion are counted as pairs supporting the presence of a TE insertion, while read pairs with both reads aligned to the reference flanking regions are counted as pairs supporting no TE insertion.
Although not specific to TE insertion detection, multiple SV genotyping tools use graph alignment to extract features to determine genotypes. Since SV genotyping tools can be applied to TE genotyping with additional TE annotation, we introduce some of the tools in this review to facilitate the development of better tools using this approach. Paragraph (Chen et al., 2019) and GraphTyper2 (Eggertsson et al., 2019) genotype SVs; vg toolkit (Hickey et al., 2020) genotypes SVs, SNVs, and indels; and SVJedi (Lecompte, Peterlongo, Lavenier, & Lemaitre, 2019) genotypes SVs for long-read sequencing data. These graph alignment tools construct a genome graph based on detected SVs and align reads to it (Fig. 4B). Genome graphs constructed for new local haplotypes consist of an SV sequence (e.g., an inserted TE sequence) connected to flanking sequences. Reads are then aligned to the graph. If a read originates from the TE insertion junction, it will fully map to the newly constructed haplotype; if a read originates from the reference homozygous allele, then it will fully align to the original reference genome. To estimate the genotype, reads fully aligned to the junction and reads aligned to the insertion are counted separately for each of the two haplotypes (with and without an insertion).
After feature extraction, genotype inference is performed using either the maximum-likelihood approach or a machine-learning approach. The maximum-likelihood approach assigns to each insertion the genotype with the highest probability of having the observed features. This approach has been widely adopted for SNP and indel genotyping (Li, 2011), and most tools, including MELT, TypeTE, Paragraph, GraphTyper2, and SVJedi, adopt this approach to genotype TE insertions and SVs. In contrast, xTea and GINDEL (Chu, Zhang, & Wu, 2014) take a machine-learning approach that views genotyping as a classification problem, with different class labels for each genotype. Different supervised machine-learning techniques are used with pre-labeled training data. For example, GINDEL uses a support vector machine and xTea uses random forest to train the model using simulation or curated training data. Rigorous performance evaluation of TE genotyping methods is warranted.
DISCUSSION
Several factors affect the sensitivity and specificity of TE insertion detection. In general, methods performing read realignment to TE sequence libraries show higher accuracy than methods that examine reference mapping and TE annotation without read realignment (Gardner et al., 2017). For example, SVA elements have internal Alu-like sequences, so some short reads from SVAs can be erroneously aligned to Alus in the reference genome. Thus, methods without read realignment may fail to correctly associate these SVA reads, and thus may fail to call some SVA insertions. In addition, most tools examine poly(A/T) tails and target site duplication, using the information to filter out false positives. However, a subset of germline TE insertions may have mutations in these features or have target site deletions rather than duplications. Thus, stringent filtering on these criteria may lead to loss of sensitivity. Long-read tools can detect insertions that are generally undetectable with short-read tools, especially in regions with low mappability; however, most long-read data have low sequencing depth due to high sequencing cost, sometimes leading to missed true insertions.
Precise genotyping and phasing of TE insertions is essential in genetic studies in order to associate TE insertions with other quantitative traits such as gene expression levels, or with SNPs from genome association studies (GWAS) of various genetic traits and diseases. Since GWAS SNPs are selected among tagging SNPs for which probes exist on SNP arrays, most of them are unlikely to represent true functional variants. To identify those more likely to be functional, TE phasing can be used to identify TE insertions with strong linkage to known GWAS variants (Payer et al., 2017). TE phasing is also important to identify a compound heterozygous TE insertion that causes a recessive genetic disease by confirming that the TE insertion inherited from one parent and the other pathogenic variant inherited from the other parent.
Currently, all TE-specific genotyping methods extract features for genotype estimation using read alignment to the linear reference genome. New TE genotyping methods need to be developed using genome graphs, as in recent SV genotyping approaches. To our knowledge, there is no existing tool for TE insertion phasing. With a large, population-scale dataset, SHAPEIT (Delaneau, Marchini, & Zagury, 2011), Eagle2 (Loh et al., 2016), and other algorithms can phase SNVs. However, since there are far fewer TE insertions than SNVs in each genome, these algorithms may not work for TE phasing. Heterozygous SNPs near TE insertions can be utilized to phase TEs, but only ~20% of TE insertions have such proximal heterozygous SNPs (Bohrson et al., 2019), making TE phasing with short reads challenging. Utilizing long reads is effective in constructing genomic haplotypes (Edge, Bafna, & Bansal, 2017; Martin et al., 2016; Mostovoy et al., 2016; Porubsky et al., 2019), and is likely to also be effective for TE insertion phasing.
The availability of large WGS datasets from healthy and disease cohorts underscores the importance of scalable TE analysis methods. Such datasets are increasingly stored and shared through commercial cloud computing platforms, such as Amazon Web Services (AWS) and Google Cloud Platform (GCP), and the Broad Institute’s Terra (https://terra.bio/). Thus, TE insertion and genotyping methods need to be efficient in terms of both time and memory, and should provide platform-independent usability. For example, a Docker (https://docker.com) or Singularity-based container (https://singularity.lbl.gov/) can allow other researchers to easily use the tools on different computing platforms. Combined efforts in advancing sequencing technologies and TE analytical methods will enable us to scrutinize these previously underappreciated genetic elements in the diverse contexts of genomic studies and to understand the role of TEs in human health and disease.
ACKNOWLEDGEMENTS
E.A.L. was supported by grants from the NIA (K01AG051791), the Suh Kyungbae Foundation, the Charles H. Hood Foundation, and the Paul G. Allen Frontiers Group. B.Z. was supported by the Manton Center Pilot Project Award and Rare Disease Research Fellowship.
LITERATURE CITED
- Bogaerts-Márquez M, Barrón MG, Fiston-Lavier A-S, Vendrell-Mir P, Castanera R, Casacuberta JM, & González J (2020). T-lex3: An accurate tool to genotype and estimate population frequencies of transposable elements using the latest short-read whole genome sequencing data. Bioinformatics, 36(4), 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohrson CL, Barton AR, Lodato MA, Rodin RE, Luquette LJ, Viswanadham VV, … Park PJ (2019). Linked-read analysis identifies mutations in single-cell DNA-sequencing data. Nature Genetics, 51(4), 749–754. doi: 10.1038/s41588-019-0366-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzikadze AV, & Pevzner PA (2019). cen-troFlye: Assembling centromeres with long error-prone reads. bioRxiv, 772103. doi: 10.1101/772103. [DOI] [Google Scholar]
- Chaisson MJ, & Tesler G (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinformatics, 13, 238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Krusche P, Dolzhenko E, Sherman RM, Petrovski R, Schlesinger F, … Eberle MA (2019). Paragraph: A graph-based structural variant genotyper for short-read sequence data. Genome Biology, 20(1), 291. doi: 10.1186/s13059-019-1909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Zhang J, & Wu Y (2014). GINDEL: Accurate genotype calling of insertions and deletions from low coverage population sequence reads. PloS One, 9(11), e113324. doi: 10.1371/journal.pone.0113324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu Stancu M, Van Roosmalen MJ, Renkens I, Nieboer MM, Middelkamp S, De Ligt J, … Kloosterman WP (2017). Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications, 8(1), 1326. doi: 10.1038/s41467-017-01343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damert A, Raiz J, Horn AV, Lower J, Wang H, Xing J, … Schumann GG (2009). 5′-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Research, 19(11), 1992–2008. doi: 10.1101/gr.093435.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Marchini J, & Zagury J-F (2011). A linear complexity phasing method for thousands of genomes. Nature Methods, 9(2), 179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- Edge P, Bafna V, & Bansal V (2017). Hap-CUT2: Robust and accurate haplotype assembly for diverse sequencing technologies. Genome Research, 27(5), 801–812. doi: 10.1101/gr.213462.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggertsson HP, Kristmundsdottir S, Beyter D, Jonsson H, Skuladottir A, Hardarson MT, … Melsted P (2019). GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nature Communications, 10(1), 5402. doi: 10.1038/s41467-019-13341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, Salerno WJ, & Reid JG (2014). PBHoney: Identifying genomic variants via long-read discordance and interrupted mapping. BMC Bioinformatics, 15, 180. doi: 10.1186/1471-2105-15-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin JA, Paquola ACM, Singer T, Gal-lina I, Novotny M, Quayle C, … Gage FH (2016). L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nature Neuroscience, 19(12), 1583–1591. doi: 10.1038/nn.4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrony GD, Cai X, Lee E, Hills LB, El-hosary PC, Lehmann HS, … Walsh CA (2012). Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell, 151(3), 483–496. doi: 10.1016/j.cell.2012.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrony GD, Lee E, Mehta BK, Ben-jamini Y, Johnson RM, Cai X, … Walsh CA (2015). Cell lineage analysis in human brain using endogenous retroelements. Neuron, 85(1), 49–59. doi: 10.1016/j.neuron.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrony GD, Lee E, Park PJ, & Walsh CA (2016). Resolving rates of mutation in the brain using single-neuron genomics. eLife, 5, e12966. doi: 10.7554/eLife.12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner GJ, & Billon V (2018). L1 retrotransposition in the soma: A field jumping ahead. Mobile DNA, 9, 22. doi: 10.1186/s13100-018-0128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner GJ, & Garcia-Perez JL (2017). L1 mosaicism in mammals: Extent, effects, and evolution. Trends in Genetics, 33(11), 802–816. doi: 10.1016/j.tig.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Fernandes JD, Zamudio-Hurtado A, Claw-son H, Kent WJ, Haussler D, Salama SR, & Haeussler M (2020). The UCSC repeat browser allows discovery and visualization of evolutionary conflict across repeat families. Mobile DNA, 11, 13. doi: 10.1186/s13100-020-00208-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feusier J, Watkins WS, Thomas J, Farrell A, Witherspoon DJ, Baird L, … Jorde LB (2019). Pedigree-based estimation of human mobile element retrotransposition rates. Genome Research, 29(10), 1567–1577. doi: 10.1101/gr.247965.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EJ, Lam VK, Harris DN, Chuang NT, Scott EC, Pittard WS, … Devine SE (2017). The Mobile Element Locator Tool (MELT): Population-scale mobile element discovery and biology. Genome Research, 27(11), 1916–1929. doi: 10.1101/gr.218032.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EJ, Prigmore E, Gallone G, Danecek P, Samocha KE, Handsaker J, … Hurles ME (2019). Contribution of retrotransposition to developmental disorders. Nature Communications, 10(1), 4630. doi: 10.1038/s41467-019-12520-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Lutz-Prigge S, & Moran JV (2002). Genomic deletions created upon LINE-1 retrotransposition. Cell, 110(3), 315–325. doi: 10.1016/S0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Goerner-Potvin P, & Bourque G (2018). Computational tools to unmask transposable elements. Nature Reviews Genetics, 19(11), 688–704. doi: 10.1038/s41576-018-0050-x. [DOI] [PubMed] [Google Scholar]
- Goodier JL, Ostertag EM, & Kazazian HH Jr (2000). Transduction of 3′-flanking sequences is common in L1 retrotransposition. Human Molecular Genetics, 9(4), 653–657. doi: 10.1093/hmg/9.4.653. [DOI] [PubMed] [Google Scholar]
- Goubert C, Thomas J, Payer LM, Kidd JM, Feusier J, Watkins WS, … Feschotte C (2020). TypeTE: A tool to genotype mobile element insertions from whole genome resequencing data. Nucleic Acids Research, 48(6), e36–e36. doi: 10.1093/nar/gkaa074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, & Kazazian HH Jr. (2016). Roles for retrotransposon insertions in human disease. Mobile DNA, 7, 9. doi: 10.1186/s13100-016-0065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller D, & Vingron M (2019). SVIM: Structural variant identification using mapped long reads. Bioinformatics, 35(17), 2907–2915. doi: 10.1093/bioinformatics/btz041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey G, Heller D, Monlong J, Sibbesen JA, Sirén J, Eizenga J, … Paten B (2020). Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biology, 21(1), 35. doi: 10.1186/s13059-020-1941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, … Wheeler TJ (2016). The Dfam database of repetitive DNA families. Nucleic Acids Research, 44(D1), D81–9. doi: 10.1093/nar/gkv1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J, Chaisson MJP, Steinberg KM, Warren W, Hoekzema K, Gordon D, … Eichler EE (2017). Discovery and genotyping of structural variation from long-read hap-loid genome sequence data. Genome Research, 27(5), 677–685. doi: 10.1101/gr.214007.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs FMJ, Greenberg D, Nguyen N, Haeussler M, Ewing AD, Katzman S, … Haussler D (2014). An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature, 516(7530), 242–245. doi: 10.1038/nature13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Olsen HE, Turner DJ, Stoddart D, Bulazel KV, Paten B, … Miga KH (2018). Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology, 36(4), 321–323. doi: 10.1038/nbt.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Chen C, Huang Z, Liu R, & Verdier J (2015). ITIS, a bioinformatics tool for accurate identification of transposon insertion sites using next-generation sequencing data. BMC Bioinformatics, 16, 72. doi: 10.1186/s12859-015-0507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, & Walichiewicz J (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research, 110(1–4), 462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kazazian HH Jr. (2004). Mobile elements: Drivers of genome evolution. Science, 303(5664), 1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Keane TM, Wong K, & Adams DJ (2013). RetroSeq: Transposable element discovery from next-generation sequencing data. Bioinformatics, 29(3), 389–390. doi: 10.1093/bioinformatics/bts697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon M, Lameijer EW, Lakenberg N, Hehir-Kwa JY, Thung DT, Slagboom PE, … Ye K (2016). Detecting dispersed duplications in high-throughput sequencing data using a database-free approach. Bioinformatics, 32(4), 505–510. doi: 10.1093/bioinformatics/btv621. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nus-baum C, Zody MC, Baldwin J, … International Human Genome Sequencing Consortium (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860–921. [DOI] [PubMed] [Google Scholar]
- Lecompte L, Peterlongo P, Lavenier D, & Lemaitre C (2019). SVJedi: Genotyping structural variations with long reads. bioRxiv, 849208. doi: 10.1101/849208. [DOI] [PubMed] [Google Scholar]
- Lee E, Iskow R, Yang L, Gokcumen O, Hase-ley P, Luquette LJ, … Park PJ (2012). Landscape of somatic retrotransposition in human cancers. Science, 337(6097), 967–971. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27(21), 2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2018). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics, 34(18), 3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, & Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25(14), 1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK, … Price AL (2016). Reference-based phasing using the Haplotype Reference Consortium panel. Nature Genetics, 48(11), 1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Patterson M, Garg S, Fischer SO, Pisanti N, Klau GW, … Marschall T (2016). WhatsHap: Fast and accurate read-based phasing. bioRxiv, 085050. doi: 10.1101/085050. [DOI] [Google Scholar]
- Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, … Phillippy AM (2019). Telomere-to-telomere assembly of a complete human X chromosome. bioRxiv, 735928. doi: 10.1101/735928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostovoy Y, Levy-Sakin M, Lam J, Lam ET, Hastie AR, Marks P, … Kwok P-Y (2016). A hybrid approach for de novo human genome sequence assembly and phasing. Nature Methods, 13(7), 587–590. doi: 10.1038/nmeth.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer LM, Steranka JP, Yang WR, Kryatova M, Medabalimi S, Ardeljan D, … Burns KH (2017). Structural variants caused by Alu insertions are associated with risks for many human diseases. Proceedings of the National Academy of Sciences of the United States of America, 114(20), E3984–E3992. doi: 10.1073/pnas.1704117114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Ebert P, Audano PA, Vollger MR, Harvey WT, Munson KM, … Marschall T (2019). A fully phased accurate assembly of an individual human genome. bioRxiv, 855049. doi: 10.1101/855049. [DOI] [Google Scholar]
- Rajaby R, & Sung W-K (2018). TranSurVeyor: An improved database-free algorithm for finding non-reference transpositions in high-throughput sequencing data. Nucleic Acids Research, 46(20), e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Martin B, Alvarez EG, Baez-Ortega A, Zamora J, Supek F, Demeulemeester J, … Tubio JMC (2020). Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nature Genetics, 52(3), 306–319. doi: 10.1038/s41588-019-0562-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Luque FJ, Kempen M-JHC, Gerdes P, Vargas-Landin DB, Richardson SR, Troskie R-L, … Faulkner GJ (2019). LINE-1 evasion of epigenetic repression in humans. Molecular Cell, 75(3), 590–604.e12. doi: 10.1016/j.molcel.2019.05.024. [DOI] [PubMed] [Google Scholar]
- Scott EC, & Devine SE (2017). The role of somatic L1 retrotransposition in human cancers. Viruses, 9(6), 131. doi: 10.3390/v9060131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, Von Haeseler A, & Schatz MC (2018). Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods, 15(6), 461–468. doi: 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, & Green P (2015) RepeatMasker Open-4.0 2013–2015
- Stewart C, Kural D, Strömberg MP, Walker JA, Konkel MK, Stütz AM, … Marth GT (2011). A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genetics, 7(8), e1002236. doi: 10.1371/journal.pgen.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, … Ko-rbel JO (2015). An integrated map of structural variation in 2,504 human genomes. Nature, 526(7571), 75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thung DT, De Ligt J, Vissers LEM, Stee-houwer M, Kroon M, De Vries P, … Hehir-Kwa JY (2014). Mobster: Accurate detection of mobile element insertions in next generation sequencing data. Genome Biology, 15(10), 488. doi: 10.1186/s13059-014-0488-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio JMC, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, … Campbell PJ (2014). Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science, 345(6196), 1251343. doi: 10.1126/science.1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton KR, Gerhardt DJ, Jesuadian JS, Richardson SR, Sánchez-Luque FJ, Bodea GO, … Faulkner GJ (2015). Ubiquitous L1 mosaicism in hippocampal neurons. Cell, 161(2), 228–239. doi: 10.1016/j.cell.2015.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lee W-P, Ward A, Walker JA, Konkel MK, Batzer MA, & Marth GT (2014). Tangram: A comprehensive toolbox for mobile element insertion detection. BMC Genomics, 15, 795. doi: 10.1186/1471-2164-15-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wu Q, Ye AY, Guo J, Zheng X, Yang X, … Huang AY (2019). Somatic LINE-1 retrotransposition in cortical neurons and non-brain tissues of Rett patients and healthy individuals. PLoS Genetics, 15(4), e1008043. doi: 10.1371/journal.pgen.1008043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Emery SB, Flasch DA, Wang Y, Kwan KY, Kidd JM, … Mills RE (2019). Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology. Nucleic Acids Research, 48(3), 1146–1163. doi: 10.1093/nar/gkz1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Wang J, Theurkauf W, & Weng Z (2014). TEMP: A computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Research, 42(11), 6826–6838. doi: 10.1093/nar/gku323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhou B, Pattni R, Gleason K, Tan C, Kalinowski A, … Urban AE (2019). Machine learning reveals bilateral distribution of somatic L1 insertions in human neurons and glia. bioRxiv, 660779. doi: 10.1101/660779. [DOI] [PMC free article] [PubMed] [Google Scholar]