

ABSTRACT
Human cytomegalovirus (HCMV) has a large (∼235 kb) genome with more than 200 predicted open reading frames that exploits numerous cellular factors to facilitate its replication. A key feature of HCMV-infected cells is the emergence of a distinctive membranous cytoplasmic compartment termed the virion assembly compartment (vAC). Here, we report that host protein WD repeat domain 11 (WDR11) plays a key role in vAC formation and virion morphogenesis. We found that WDR11 was upregulated at both mRNA and protein levels during HCMV infection. At the late stage of HCMV replication, WDR11 relocated to the vAC and colocalized with markers of the trans-Golgi network (TGN) and vAC. Depletion of WDR11 hindered HCMV-induced membrane reorganization of the Golgi and TGN, altered vAC formation, and impaired HCMV secondary envelopment and virion morphogenesis. Further, motifs critical for the localization of WDR11 in TGN were identified by alanine-scanning mutagenesis. Mutation of these motifs led to WDR11 mislocation outside the TGN and loss of vAC formation. Taken together, these data indicate that host protein WDR11 is required for efficient viral replication at the stage of virion assembly, possibly by facilitating the remodeling of the endomembrane system for vAC formation and virion morphogenesis.
IMPORTANCE During the late phase of human cytomegalovirus (HCMV) infection, the endomembrane system is dramatically reorganized, resulting in the formation of a unique structure termed the virion assembly compartment (vAC), which is critical for the assembly of infectious virions. The mechanism of HCMV-induced vAC formation is still not fully understood. In this report, we identified a host factor, WDR11, that plays an important role in vAC formation. Our findings argue that WDR11 contributes to the relocation of the Golgi and trans-Golgi network to the vAC, a membrane reorganization process that appears to be required for efficient virion maturation. The present work provides new insights into the vAC formation and HCMV virion morphogenesis and a potential novel target for antiviral treatment.
KEYWORDS: human cytomegalovirus, WDR11, virion assembly compartment, virion morphogenesis, virion structure
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous pathogen that persistently infects approximately 50 to 90% of adults globally (1). In China, the seroprevalence is as high as 93.7% (2). Following primary infection, the virus establishes latency and lifelong persistence in the host. Although 85 to 90% of primary HCMV infections in immunocompetent individuals are either asymptomatic or mildly symptomatic (3), latent HCMV can reactivate and cause severe diseases in immunocompromised or immunodeficient individuals, such as solid organ or bone marrow transplant recipients and AIDS patients (4–8). HCMV is also a leading cause of congenital infections worldwide that in some cases can result in severe, symptomatic congenital CMV infection with a mortality rate of 7 to 12% in the early neonatal period (3).
HCMV belongs to the family of herpesviridae. All herpesviruses share a similar virion structure in which a DNA-containing icosahedral capsid is embedded in tegument layers and surrounded by a lipid envelope (9). Viral assembly often takes place in specific intracellular compartments where viral components concentrate. Unlike other herpesviruses, HCMV causes a profound remodeling of the secretory and endocytic system (the Golgi, trans-Golgi network, and early endosomes) to form a unique juxtanuclear membrane structure termed the virion assembly compartment (vAC) (5, 10–15). The vAC is composed of nested cylindrical layers encompassing the microtubule organization center (MTOC). A recent study showed that HSV-1 could also form a perinuclear assembly compartment similar to the vAC; this occurred in primary mouse neurons and undifferentiated human and mouse cancerous neuronal cells but not in fibroblasts and epithelial cells (16).
vAC formation has been suggested to be essential for efficient HCMV virion assembly and maturation. However, the detailed mechanisms for vAC formation have not been completely elucidated. Multiple viral proteins have been identified as important factors for vAC biogenesis, including pUL48, pUL71, pUL94, pUL97, pUL103, pUL132, and pUL136 (14, 17–20). In addition, HCMV-encoded miRNAs UL112-1, US5-1, and US5-2 have been shown to regulate the reorganization of the host secretory pathway to facilitate vAC formation (21). How these viral factors interact with host factors to reorganize cellular organelles is still unknown. Several protein components of the cellular trafficking system, including RhoB, IFITMs, STX5, dynein, BiP, bicaudal D1, Rab11, FIP4, and WDR5, have been found inside vAC (15, 22–28), but the roles of these proteins in vAC formation remain incompletely characterized.
Previously, we showed that a WD repeat (WDR)-containing protein WDR5 accumulates in vAC and promotes secondary envelopment of HCMV capsids (29). The human genome encodes up to 610 WDR proteins that participate in a diverse array of cellular processes (30). Here, we report the identification of WDR11, another WDR family member, as a host protein that contributes to vAC formation and HCMV virion morphogenesis. Previously, WDR11 has been shown to localize in various cellular compartments, play a role in transcriptional regulation, and contribute to the trafficking of endocytosed cargo (31). More recently, WDR11 has been shown to participate in the replication of HSV-1 (32). Most relevant to our findings, WDR11 has recently been reported to function in tethering AP-1-coated clathrin-coated vesicles (CCVs) to the trans-Golgi network (TGN) through interactions with TBC1D23 and Golgin-245 (31).
In the present study, we found that HCMV infection upregulates WDR11 expression and induces WDR11 to relocate to the vAC. WDR11 knockout led to incomplete restructuring of cellular organelles, such as the trans-Golgi network (TGN) and Golgi, and consequently altered vAC formation with defective virion morphogenesis. We further identified two sorting signals in WDR11 that are critical for its associated TGN localization and demonstrated that WDR11 localization to the TGN is essential for TGN remodeling and vAC formation.
RESULTS
HCMV infection upregulates WDR11.
In order to investigate the role of WDR11 in HCMV infection, we first analyzed the effects of HCMV on WDR11 transcription and protein levels in human foreskin fibroblasts (HFFs). The mRNA levels of WDR11 were measured by quantitative reverse transcriptase PCR (qRT-PCR) (Fig. 1A). Compared with mock-infected cells, the mRNA level of WDR11 was significantly increased by HCMV infection. This transcriptional difference was observed at 12 hours postinfection (hpi) and continuously increased to more than 2.5-fold at 72 hpi. Similar increases in WDR11 protein levels were observed that maintained at a higher level during the entire HCMV infection, accompanying the accumulation of immediate-early (IE1 and IE2), early/late (pUL44), and late glycoprotein B (gB) viral proteins (Fig. 1B).
FIG 1.

Human cytomegalovirus (HCMV) infection induces WDR11. HFFs were mock-infected (M) or infected with HCMV Towne strain (V) at a multiplicity of infection (MOI) of 3 and then harvested at the times indicated. (A) WDR11 mRNA levels were determined by quantitative reverse transcriptase PCR (qRT-PCR) and normalized to levels in mock-infected cells at 6 hours postinfection (hpi). (B) Infected cell lysates were assayed for the indicated proteins by immunoblotting (IB). Protein levels determined by densitometry and normalized to mock-infected cells at 24 h are graphed below. (C) HFFs were mock-infected (M), HCMV-infected (V), or infected with UV-inactivated HCMV (UV) at an MOI of 3 in the presence of vehicle (dimethyl sulfoxide [DMSO]), ganciclovir (GCV), or phosphonoacetic acid (PAA). At 72 hpi, samples were collected and analyzed by IB for the indicated proteins. Protein levels determined by densitometry and normalized to mock-infected cells are graphed below. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) serves as a loading control. The data were collected from three independent experiments and analyzed by one-way analysis of variance (ANOVA); the results are presented as averages ± standard deviation (SD). **, P < 0.01; ***, P < 0.001; NS, not significant; IE, immediate-early; gB, glycoprotein B.
To determine whether viral gene expression is required for the enhanced WDR11 expression, HFFs were infected with UV-irradiated or live HCMV and in the presence of viral DNA polymerase inhibitors. As expected, UV-inactivated HCMV infection resulted in no de novo synthesis of viral proteins, exemplified by IE1/2, pUL44, and gB (Fig. 1C). Comparably, ganciclovir (GCV) or phosphonoacetic acid (PAA) treatment reduced IE2 and pUL44 protein levels and completely blocked gB expression but did not affect IE1 expression. In the presence of GCV or PAA, the WDR11 protein level did not increase and remained lower than untreated HCMV-infected cells, and similar level as in mock and UV controls (Fig. 1C). These data imply that upregulation of WDR11 requires viral DNA replication or expression of viral late proteins.
Depletion of WDR11 reduces viral yields.
We next employed short hairpin RNA (shRNA) gene silencing to investigate the role of WDR11 in HCMV replication. Lentiviruses expressing scrambled shRNAs (shScram) or two WDR11-specific shRNAs (shW1 and shW2) were transduced into HFFs, and the knockdown cell lines were referred to as shScram, shW1, and shW2 cells, respectively. The WDR11 protein levels were reduced by 53% in shW1 cells and 77% in shW2 cells (Fig. 2A). In addition, WDR11 knockdown in HFFs did not exhibit cytotoxicity up to 5 days after transduction (Fig. 2B).
FIG 2.

Knockdown of WDR11 results in reduced virion yield. (A) Efficiency of WDR11 knockdown. Human foreskin fibroblasts (HFFs) were transduced with lentiviruses expressing scrambled shRNA (shScram) or WDR11-specific shRNAs (shW1 and shW2), and WDR11 levels were examined by IB. The WDR11 protein levels relative to shScram are listed below each blot. (B) Cell viability. Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Data were collected at the indicated times from three independent experiments and analyzed by one-way ANOVA. The results are presented as means ± SD. NS, not significant. (C, D) Growth kinetics of virus. Control cells (shScram) and WDR11 knockdown cells (shW1 and shW2) were infected with HCMV at MOIs of 3 or 0.05, and supernatants collected at the indicated times postinfection were titrated by plaque assay. The results were obtained from three independent experiments, each conducted in triplicate and analyzed by one-way ANOVA. **, P < 0.01; ***, P < 0.001.
The shScram, shW1, and shW2 cells were then infected with HCMV at MOIs of 0.05 or 3, and yields of infectious virus in the culture supernatants were determined to generate multi- or single-step growth curves. Virus yields were significantly decreased in WDR11-knockdown cells. In the cells infected at high MOI (3), yield reductions were 4.6- and 7.5-fold in shW1 and shW2 cells at 5 days postinfection (dpi), respectively (Fig. 2C). More dramatic reductions were observed in cells infected at an MOI of 0.05 with viral titers from shW1 and shW2 cells 12.8- and 21.5-fold lower, respectively, than those from shScram cells at 8 dpi (Fig. 2D). These results demonstrate that decreased expression of WDR11 is associated with a decrease in HCMV replication.
Depletion of WDR11 does not affect viral entry, viral genome replication, and viral protein expression.
To further characterize the functions of WDR11 during HCMV replication, we established WDR11 knockout (WDR11-KO) and WDR11 overexpressing (WDR11-OE) cell lines. WDR11-KO was generated by transducing hTERT-immortalized HEL cells (HELFs) with a CRISPR/Cas9-based lentivirus targeting WDR11 (33), and deletion of the WDR11 coding sequence was confirmed (Fig. 3A). WDR11-OE was made by transducing HELFs with a lentivirus expressing the WDR11 cDNA (NM_018117.12) (33). WDR11 overexpression and knockout were confirmed by immunoblotting (IB) (Fig. 3B).
FIG 3.

Effect of WDR11 overexpression (OE) and knockout (KO) on viral entry, genome replication, and viral gene expression. (A) Sequence of genomic target site of WDR11 knockout clone. DNA was extracted from CTL and WDR11-KO cells, PCR-amplified with WDR11-specific primers, and sequenced. The PAM motif is underlined, and the gRNA sequence is in red. (B) HELFs were transduced with lentiviral vectors that overexpress WDR11 (WDR11-OE) or knock out WDR11 (WDR11-KO) or relative empty vector (Vector and CTL), then exposed to HCMV at an MOI of 5 for 1 h at 4°C to allow virus attach, and then transferred to 37°C incubation for 1 h to allow virus entry. Finally, cells were washed by citric acid (pH 3) to remove noninternalized virus and harvested by trypsinization. WDR11 and cell-associated pp65 levels were determined by IB. The numbers below each blot indicate WDR11 or pp65 levels, determined by densitometry, normalized to GAPDH and to controls. (C) Viral entry determined by pp65 staining. Cells on coverslips were infected with HCMV at an MOI of 1 and fixed 6 hpi. HCMV pp65 protein (red) was detected by immunofluorescence assay (IFA), and the nuclei were counterstained with 4´,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 50 μm. Total cells and nuclear pp65-positive cells were counted in 20 random fields selected from each experiment. The data were analyzed by one-way ANOVA. Representative images (left panel) and quantifications (right panel) are shown. The results are medians ± SD. NS, not significant. (D) Cells were infected with HCMV at an MOI of 1 and HCMV genome copy numbers were determined by quantitative PCR (qPCR) and analyzed by one-way ANOVA. The results shown are means ± SD. (E) Viral protein level. Cells were infected with HCMV at an MOI of 1, and lysates were analyzed by IB to detect the indicated proteins. The results shown are representative of three independent experiments.
The HCMV life cycle includes viral entry, genome replication, gene expression, capsid assembly, envelopment, and release of mature virions (5). During HCMV entry and uncoating, the tegument protein pp65 rapidly translocates to the nucleus and thus is commonly used as an entry indicator (34–36). We found that similar amounts of pp65 proteins accumulated at 1 hpi in WDR11-OE and WDR11-KO cells compared with the controls (Fig. 3B). In addition, pp65 in the nucleus at 6 hpi was quantified by immunofluorescence. As shown in Fig. 3C, neither WDR11-OE nor WDR11-KO cells had a noticeable effect on the percentage of nuclear translocation of pp65 compared with their controls. These data indicate that WDR11 is not involved in HCMV entry.
We next examined viral genome replication and viral protein at different phases, including IE1/2, early protein pUL44 and late protein pp28, in these two cell lines. Neither WDR11 overexpression nor knockout had a noticeable impact on HCMV viral genome replication or expression of viral proteins throughout the infection. However, we observed slightly lower viral genome copy numbers in WDR11-KO cells compared with the control cells (Fig. 3D). Also, reduced pp28 (late protein) expression was noticed at 48 hpi in the KO cells, but it recovered at 96 hpi, suggesting that WDR11 may play a minor role in the initiation of pp28 expression (Fig. 3E). These data suggest that loss of WDR11 expression does not impact HCMV entry, genome replication, and expression of immediate-early and early proteins but could impact delay expression of true late proteins such as pp28.
Depletion of WDR11 impairs maturation of progeny virions.
The effects of WDR11 depletion on viral yield raised the possibility that WDR11 could contribute to HCMV assembly. Virion morphogenesis in WDR11-KO and control (CTL) cells were examined by transmission electron microscopy (TEM). Initially, capsid numbers were counted in CTL and WDR11-KO cells at 96 hpi. Representative micrographs showed that HCMV capsids were produced in both cell lines (Fig. 4A, panels a and b). The average numbers of capsids/cell were calculated from three independent experiments. The number of capsids per WDR11-KO cell (212.6) was 10% lower than capsids per CTL cell (235.6), but this difference was not statistically significant (Fig. 4A, panel c). In infected cell nuclei, genome packaging results in three morphologically distinct capsid types designated A, B, and C capsids. Quantification showed that B capsids accounted for more than 60% of the total capsids in both cell lines and that there was no difference in the proportions of A, B, or C capsids between WDR11-KO and CTL cells (Fig. 4A, panel d). These results demonstrated that WDR11 knockout does not affect viral capsid assembly or viral genome encapsulation.
FIG 4.

Analysis of infected cells by transmission electron microscopy (TEM). Cells infected with HCMV at an MOI of 1 were analyzed by TEM. (A) Sections prepared 96 hpi were imaged at 1,700× (panels a and b), and nuclear compartments containing capsids (indicated by white dashed boxes) were enlarged to 7,800× (panels a1 and b1). Total numbers of capsids located within 10 nuclei from each culture were quantitated (panel c). The numbers of A, B, or C capsids in 10 nuclei from each culture were determined based on capsid morphology, and the percentage of each capsid type was calculated (panel d). (B) Sections prepared 120 hpi were imaged at 1,700× (panels a and b), and cytoplasmic compartments containing capsids (indicated by white-dashed boxes) were enlarged to 7,800× (panels a1 to a3 and b1 to b3). The magnified images of the matured virions (panel a3, indicated with arrowhead) and unenveloped virions (panel b3, indicated with arrow) are presented in panels a4 and b4, respectively. Typical (panel a) and abnormal vAC (panel b) are delineated by dotted red lines. Total numbers of cytoplasmic capsids in 10 cells from each culture were quantitated (panel c). Medians for three independent experiments are indicated to the right of each plot, and the means of the three experiments are labeled above each group. The data were analyzed by Kruskal-Wallis test. NS, not significant; ***, P < 0.001. Total numbers of enveloped or nonenveloped capsids in 10 cells from each culture were quantitated percentages of each were calculated (d). The data were analyzed by chi-square test. n, total numbers of capsids analyzed; ***, P < 0.001.
We next assessed the impact of WDR11 depletion on virion assembly in the cytoplasm. After HCMV infection, a distinct cytoplasmic structure containing numerous viral particles was observed in CTL cells, whereas multiple smaller cytoplasmic collections of viral particles were observed in WDR11-KO cells (Fig. 4B, panels a and b). In addition, there were a larger number of enveloped virions (Fig. 4B, panel a4, indicated with arrowhead) in the cytoplasm of CTL cells (Fig. 4B, panel a) compared with WDR11-KO cells (Fig. 4B, panel b). The median ratio of cytoplasmic virions/cell (10.8) in WDR11-KO cells was about 4-fold lower than that (42.0) in CTL cells (Fig. 4B, panel c). Depletion of WDR11 also resulted in a distinct defect in virion morphogenesis. In WDR11-KO cells, more than half of the viral particles in the cytoplasm appeared to be nonenveloped (Fig. 4B, panel b4, indicated with arrow). Taken together, these results indicate that WDR11 contributes to cytoplasmic virion morphogenesis, and the envelopment step of virion morphogenesis was decreased in cells lacking WDR11, presumably accounting for the observed decrease in infectious virus yield.
TGN-located WDR11 is present in the vAC at the late stage of HCMV infection.
At the late stage of HCMV infection, the secretory and endocytic system (e.g., the Golgi, TGN, and early endosomes, etc.) reorganize to form vAC for efficient HCMV virion assembly and maturation (10). The reported localization of WDR11 in the TGN prompted us to examine its contribution to vAC formation. We first examined the subcellular distribution of WDR11 in mock- and HCMV-infected HELFs. Consistent with previous reports (31, 32), transiently transduced DsRed-WDR11 colocalized with TGN marker p230 in mock-infected HELFs. At the late stage of HCMV infection (72 hpi), both WDR11 and the TGN localized to a juxtanuclear aggregate and colocalized with pp28 within the interior of the vAC (Fig. 5A). These observations suggest that WDR11 relocalizes from or with the TGN to vAC. We further tested the effects of viral DNA polymerase inhibitors on WDR11 relocalization. As has been reported previously, both GCV and PAA prevented vAC formation (37). Accordingly, the HCMV-induced change of WDR11 localization to a typical vAC-like structure (large juxtanuclear aggregate) was not observed in the drug-treated cells (Fig. 5B). These results suggested that WDR11 relocation to the vAC formation requires early/late viral gene expression and/or de novo synthesis of viral proteins.
FIG 5.
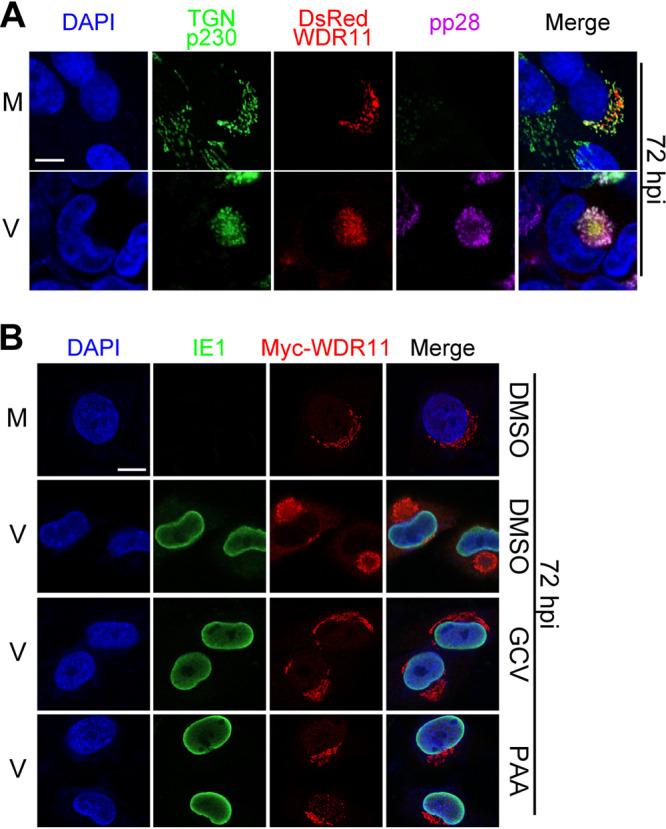
Subcellular localization of WDR11 redistribution to vAC. (A) Human telomerase (hTERT)-immortalized HEL cells (HELFs) were transfected with a plasmid expressing a DsRed-WDR11 fusion protein then mock-infected (M) or HCMV-infected (V) at an MOI of 1. At 72 hpi, the cells were fixed and costained with antibodies to viral protein pp28 (purple) and trans-Golgi network (TGN) marker p230 (green). Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm. (B) HELFs were transfected with a plasmid expressing a Myc-WDR11 fusion protein and infected as in panel A in the presence of dimethyl sulfoxide (DMSO), ganciclovir (GCV) (100 μg/ml), or phosphonoacetic acid (PAA) (100 μg/ml). At 72 hpi, the cells were fixed and costained with antibodies to Myc (red) and the HCMV IE1 protein (green). The nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
Mutation of sorting motifs in WDR11 eliminates its localization in the TGN.
We next asked whether the localization of WDR11 in the TGN is required for vAC formation. Protein trafficking to target sites like the TGN in the secretory pathway is usually mediated by the AP (adaptor protein) complexes and GGA (Golgi-localized, gamma-ear-containing, Arf-binding) proteins, which interact with sorting signals in the cytoplasmic tails of cargo proteins (38, 39). Two types of sorting signals have been described: a dileucine-based ([DE]XXXL[LI]) motif and a tyrosine-based (YXXØ) motif, where X is any amino acid, and Ø represents a bulky hydrophobic amino acid such as leucine, isoleucine, methionine, valine, or phenylalanine (39). Accordingly, we analyzed the amino acid sequence of WDR11 and predicted five [DE]XXXL[LI] signals (EIKNL886L887, DIKKL901L902 DQLLL1006L1007, EGVQL1069L1070, DTEKL1175L1176) and several tyrosine-based (YXXØ) motifs (Y140IVL, Y320NNI, Y405SPV, Y484QPL, Y582LAV, Y845LLV, Y967DVL, and Y1163GAF) (Fig. 6A).
FIG 6.

Alteration of WDR11 subcellular localization by mutagenesis of sorting motifs. (A) Illustration of WDR11 protein sequence showing putative sorting signals that were targeted by alanine substitutions in mutants indicated below. [DE]XXXL[LI] motifs are shown in green, tyrosine-based motifs (YXXØ) are blue, and amino acids substituted with alanines are red. (B) HEK293T cells were transfected with plasmids expressing wild-type WDR11 (WT) or the indicated WDR11 mutants and analyzed by immunoblotting (IB) 48 h posttransfection. (C) HELF cells were transfected as in panel B and costained by IF for TGN (green) and Myc (red). The nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
To assess the contribution of these sorting motifs to WDR11 intracellular distribution, we constructed a panel of expression plasmids that encoded mutations within these motifs by comprehensive alanine-scanning mutagenesis (Fig. 6A). The respective cassettes were inserted into pCDH-Myc-G418 vector for expressing recombinant WDR11 proteins containing a Myc epitope at the N terminus. The expression of each mutant was first confirmed by transient transfection of HEK293T cells (Fig. 6B). The same plasmids were transiently transfected into HELFs, and corresponding intracellular localizations of the WDR11 variants were assessed by immunofluorescence assay (IFA) using an anti-Myc antibody. Wild-type (WT) WDR11 colocalized almost exclusively with TGN marker p230 (Fig. 6C, panel a). Alanine substitutions in five putative N-terminal sorting motifs (Y140IVL, Y320NNI, Y405SPV, Y484QPL, and Y582LAV; designated N5) resulted in most of WDR11 being localized in the cytoplasm, but localization of WDR11 in the TGN was still observed (Fig. 6C, panel b). The intracellular localization of WDR11 mutant M3, which has alanine substitutions in three central motifs (Y845LLV, EIKNL886L887, and DIKKL901L902) was indistinguishable from that of wild-type WDR11 (Fig. 6C, panel c). In contrast to mutant N5, WDR11 mutants m967–1006 and C3 that contained substitutions in the centrally located Y967DVL and DQLLL1006L1007 motifs or terminal motifs EGVQL1069L1070, Y1163GAF, and DTEKL1175L1176, respectively, resulted in WDR11 localization outside the TGN (Fig. 6C, panels d and e). We further examined the contribution of individual sorting motifs of m967–1006 and mutations within the C3 to the intracellular distribution of WDR11. We found that single-motif mutations m967 (Y967DVL) and m1006 (DQLLL1006L1007) and double-motif mutation m1163–1175 (Y1163GAF and DTEKL1175L1176) had subtle effects on WDR11’s localization in the TGN (Fig. 6C, panels f to h), while the single-motif mutation m1069 (EGVQL1069L1070) profoundly altered WDR11 localization with almost no colocalization of WDR11 with the TGN maker p230 (Fig. 6C, panel i). Thus, we identified two critical sorting signals responsible for the localization of WDR11 in the TGN: single motif (EGVQL1069L1070/M1069) and double motifs (Y967DVL combined with DQLLL1006L1007/M967–1006).
Localization of WDR11 to the TGN is required for WDR11-mediated TGN restructuring and vAC formation during HCMV infection.
The m967–1006 and m1069 mutants enabled us to analyze the impact of WDR11 mislocalization on vAC formation and infectious virus yield. Since the KO cell line contained a puromycin-resistant gene, we generated pCDH-6×Myc-WDR11(WT)-G418 and relative mutant pCDH-6×Myc-WDR11(m967–1006/m1069)-G418 lentivirus and transduced the KO cells. Each cell line was selected with G418 sulfate (500 μg/ml; catalog no. A600958, Sangon Biotech), and the corresponding WDR11 expressions were confirmed by IB (Fig. 7A). Compared with the mock-infected controls, HCMV infection in CTL cells at 96 hpi exhibited altered TGN morphology with a juxtanuclear condensation in ring-like structures that also contained viral proteins gB and gM (Fig. 7B, panel a; Fig. 7C, panel c, indicated by arrowhead). However, the number of infected cells exhibiting typical vAC morphology was reduced during infection of WDR11-KO cells, and abnormal vAC appeared as multiple small perinuclear foci (Fig. 7B, panel b; Fig. 7C, panel d, arrow). Further quantitative analyses of vAC formation showed that the percentage of typical vAC decreased from 86.21% in CTL cells to 24.53% in WDR11-KO cells (Fig. 7D). This change in the morphology of the TGN and loss of typical vAC formation could be reversed by rescuing the KO cells with wild-type WDR11 (KO+WT), which increased typical vAC formation (Fig. 7B, panel c; Fig. 7C, panel e; Fig. 7D) (79.31%). However, the atypical vAC phenotype in WDR11-KO cells was only partially complemented when WDR11-KO cells were repaired with lentiviruses expressing the sorting motif mutants m967–1006 (Fig. 7B, panel d; Fig. 7C, panel f) (31.61%) or m1069 (Fig. 7B, panel e, and Fig. 7C, panel g) (30.17%). As expected, the virus yield in KO cells was 9.5-fold lower than in CTL cells at 96 hpi (MOI = 1), and reintroducing WT-WDR11 into KO cells rescued the yield of virus production to a level similar to that of CTL cells. Viral titers produced by KO+m967–1006 and KO+m1069 cells also failed to match those of KO+WT cells (P < 0.05). However, they were 2.85-and 1.85-fold higher than that of KO cells, respectively, although the difference was not statistically significant (Fig. 7E). These data suggest that authentic cellular trafficking of WDR11 is required for TGN remodeling, vAC morphogenesis, and wild-type levels of virus production.
FIG 7.

WDR11 mutants with altered localization lower virus yields. (A) WDR11-KO cells (KO) were transduced with lentiviruses encoded wild-type WDR11 (WT) or mutants m967–1006 and m1069 and selected in G418 sulfate (500 μg/ml; catalog no. A600958, Sangon Biotech) for 1 week and generated KO+WT, KO+m967–1006, and KO+m1069 cells. Cell lysates were prepared and analyzed by IB to detect WDR11. (B and C) CTL, KO, KO+WT, KO+m967–1006, and KO+m1069 cells were infected with HCMV at an MOI = 1. At 96 hpi, cells were fixed and costained by IF for pp65 (red) and gM (green) in panel B or TGN (red), MCP (cyan), and gB (green) in panel C. The nuclei were counterstained with DAPI (blue). White arrowheads indicate typical vACs; white arrows indicate abnormal vACs. Scale bar, 10 μm. (D) Based on morphology of gB staining shown in panels B and C, vACs were classified as typical or abnormal, quantitated, and analyzed by chi-square test. n, number of infected cells used for quantitation; ***, P < 0.001. (E) Culture supernatants from CTL or WDR11-KO cells that were transfected and infected as in panels B and C were collected at 96 hpi and titrated by plaque assay. The results shown were from three independent experiments each conducted with triplicates. Significance was analyzed by one-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
WDR11 is required for reorganization of the Golgi in the vAC.
The failure of HCMV to induce TGN remodeling and formation of typical vAC in infected WDR11-KO cells prompted us to determine whether the reorganization of other cellular organelles in vAC formation was also impacted in the absence of WDR11. Thus, we compared the fractionation of cell organelles with previously described markers of the vAC markers in infected CTL and WDR11-KO cells. Subcellular fractionations were prepared by density gradient centrifugation with OptiPrep medium from both HCMV-infected WDR11-KO and CTL cells as described previously (40). The composition of each fraction was analyzed by organelle-specific markers including calnexin (CANX) for endoplasmic reticulum (ER), hepatocyte growth factor-regulated tyrosine kinase substrate (HGS) for the multivesicular body (MVB), early endosome antigen 1 (EEA1) for early endosome, and GM130 for Golgi. In addition, the presence of vAC was indicated by viral proteins gB and pp28, and viral genomes by viral DNA were quantified by qPCR. The most noticeable change of organelle distribution between mock- and HCMV-infected CTL cells is the shift of GM130-marked Golgi from high-density fractions 10 to 13 (Fig. 8A) to low-density fractions 8 and 9 (Fig. 8C). The coincidence of the peak distributions of the Golgi, vAC markers gB and pp28, and viral DNA in these fractions from infected CTL cells indicates that these fractions contain the essential components of vAC, and HCMV infection results in Golgi reorganization. This virus-induced Golgi reorganization to form the vAC is disrupted in WDR11-KO cells. In the absence of WDR11, GM130 entirely relocalized to fractions 12 and 13 at the bottom of the gradient (Fig. 8D) as it did in the mock-infected cells (Fig. 8A and B), while significant portions of gB, pp28, and viral DNA were also relocalized to these lower fractions (Fig. 8D). In contrast, WDR11-KO did not impact the distribution patterns of CANX and HGS. These findings suggest that WDR11 contributes to the process of Golgi reorganization to form vAC during HCMV infection. We also noticed that the distribution of EEA1 was not changed in the uninfected KO cells but slightly shifted to the low-density fractions in the infected KO cells. Thus, in the absence of WDR11, early endosomes may be utilized by the virus to form alternative viral assembly sites, which are probably the abnormal vACs we observed in the KO cells.
FIG 8.

Deletion of WDR11 alters Golgi recruitment to vAC. CTL and WDR11-KO cells were infected by mock (A, B) or by HCMV at an MOI of 1 (C, D). The cells were harvested at 96 hpi and fractionated as described under Material and Methods. Fractions were analyzed by IB for markers for early endosome (EE, early endosome antigen 1 [EEA1]), multivesicular body (MVB, hepatocyte growth factor-regulated tyrosine kinase substrate [HGS]), endoplasmic reticulum (ER, calnexin [CANX]), or Golgi (GM130), and viral vAC markers gB and pp28. In each fraction, protein levels were quantified by densitometry, and viral genome copies were determined by qPCR. All were expressed as percentages of the totals and plotted together versus fraction number.
WDR11 interacts with multiple viral proteins.
Because the impact of WDR11 on restructuring of cellular organelles and vAC formation depends on HCMV replication and/or de novo synthesis of viral proteins, an interaction between WDR11 and one or more viral proteins could be required for the effect of WDR11 on the endomembrane system (Fig. 4). To test this hypothesis, Myc-tagged WDR11 proteins were immunoprecipitated (IP) from cell lysates by anti-Myc antibody and subjected to SDS-PAGE to separate coimmunoprecipitated proteins. Coomassie staining identified at least five unique bands in virus-infected cells that were not present in mock-infected cells. These five bands were collected and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for protein identification. Multiple viral nonstructural proteins (pUL36, pUL44, pUL45, pUL54, pUL57, pUL87, pUL98, and pUL102) and viral structural proteins (pUL25, pUL48, pUL77, pUL88, pUL93, pp65, pp71, pp150, and MCP) were identified as WDR11-interacting viral proteins (Fig. 9A) (41).
FIG 9.

Identification of HCMV proteins that interact with WDR11. (A) HFFs were transduced with a Myc-WDR11-expressing lentivirus and mock-infected (M) or HCMV-infected (V) with an MOI of 1. At 72 hpi, cell lysates were immunoprecipitated (IP) with anti-Myc antibody followed by SDS-PAGE and Coomassie brilliant blue staining (left). Infected-cell-specific bands (arrows) in lane 6 were excised and analyzed by LC-MS/MS. The viral proteins identified and their corresponding spectral counts are shown (right panel). (B) HEK293T cells were cotransfected with plasmids expressing Myc-WDR11 and Flag-tagged viral proteins or empty vector. At 48 h, posttransfection lysates were immunoprecipitated using anti-Myc antibody, and both input and immunoprecipitated samples were immunoblotted and probed with antibodies to Myc, Flag, or GAPDH.
To confirm these WDR11-viral protein interactions, HEK293T cells were transfected with a plasmid expressing Myc-tagged WDR11 along with each of the Flag-tagged viral protein candidates or empty vector, followed by coimmunoprecipitation (co-IP) analysis. WDR11 was immunoprecipitated with an anti-Myc antibody, and the immunoprecipitates were probed for specific viral proteins by IB with an anti-Flag antibody. All Flag-tagged viral proteins except for pUL54, pUL77, and pUL98 were precipitated together with Myc-tagged WDR11 (Fig. 9B). These results demonstrate that WDR11 interacts with multiple viral proteins that may contribute to WDR11’s role in vAC formation.
DISCUSSION
Viral infections can lead to dynamic spatiotemporal reorganization of host cellular organelles, during which the viral components are localized to specific cellular compartments/organelles potentially to exploit host machinery for viral replication and virion assembly (42, 43). A distinct characteristic feature of HCMV infection is the formation of vAC, where tegumented capsids undergo final tegumentation and secondary envelopment during virion morphogenesis. A cluster of early/recycling endosomes and a Golgi-derived microtubule-organizing center reside in the center of vAC, surrounded by a cylindrical ring consisting of the Golgi and TGN (5, 10). In the present study, we identified WDR11 as an important host factor that contributes to Golgi/TGN restructuring for vAC formation and thereby promotes virion morphogenesis. A previous report revealed that HCMV-encoded microRNAs function to remodel organelles for vAC formation by downregulating key pathway members (i.e., CDC42, SNAP23, and Rab proteins) (21). Here, we found that WDR11 is upregulated in infected cells, consistent with a replication strategy in which HCMVs utilize the cellular machinery. Our findings are in agreement with previously reported studies using spatial proteomics to describe cellular responses to HCMV infection (44, 45). It seems that multiple strategies are employed coordinately by HCMV because disruption of a single strategy resulted in failed vAC formation.
In agreement with previous studies, our colocalization analysis showed that the TGN was reorganized as part of the morphogenesis of the vAC during HCMV infection (32). However, TGN reorganization and subsequent vAC formation were disrupted in WDR11 knockout cells, as evidenced by a significant loss of typical vAC in these cells. vAC formation could be restored by ectopic expression of wild-type WDR11 in these knockout cells. Nevertheless, the expression of WDR11 mutants without TGN sorting signals failed to rescue vAC formation, indicating that TGN localization of WDR11 is a prerequisite for its function in promoting TGN restructuring and vAC formation. Because the TGN and Golgi form closely intertwined layers in the vicinity of vAC, we further defined the colocalization of the Golgi and vAC by subcellular fractionation of the infected cells. Our findings showed that Golgi coexisted and comigrated with major viral vAC markers (gB and pp28) and viral DNA in the same fractions. In contrast, colocalization was altered in the absence of WDR11, providing additional evidence that WDR11 contributes to the formation of the vAC. Together, the evidence supports the contribution of WDR11 to the virus-induced Golgi and TNG restructuring process during vAC biogenesis.
In the infected cell, the vAC is thought to be a site for the envelopment of subviral particles. Several studies have demonstrated that loss of vAC formation impairs infectious virion assembly (20, 22, 23). Consistent with these earlier findings, we observed that WDR11 deletion impaired virion maturation as more than 50% of virions in the cytoplasm of infected WDR11-KO cells were not enveloped compared to about 10% of cytoplasmic viral particles in control infected cells. These data suggest a critical role of WDR11 in HCMV secondary envelopment, but whether WDR11 is involved in this process directly or indirectly through Golgi/TGN reorganization needs further study.
The WD40-repeat proteins usually serve as a rigid scaffold for protein-protein interactions and facilitate the formation and/or stabilization of multiprotein complexes (46). WDR11 is a highly conserved WD40-repeat protein, and it has been proposed to promote the tethering of endosome-derived vesicles to the TGN. WDR11 binds to FAM91A1 and C17orf75 to form a trimeric complex in vesicles derived from endosomes (31). This WDR11 complex can recruit TBC1D23 as a bridge protein to dock itself to the TGN via TGN-associated protein golgin-245 (31, 47). Similarly, we speculate that HCMV viral proteins may interact with WDR11 and trigger the WDR11-mediated endocytic membrane remodeling process. Consistent with this hypothesis, our data show that WDR11’s relocation to vAC requires viral DNA/protein synthesis, indicating that newly synthesized viral proteins are needed for this process. Findings from our co-IP analyses identified multiple WDR11-interacting viral proteins, including MCP, pp65, pp71, pp150, pUL25, pUL48, pUL88, and pUL93. One of these proteins, pUL48, has been reported to be essential for HCMV vAC formation (14). What is more, pp65 (29), pp71 (48), pp150 (20), pUL25 (49), pUL48 (50), and pUL88 (51) have been shown to localize to the vAC. Further investigation of these WDR11-viral protein interactions will help dissect the detailed mechanism(s) of vAC formation.
Our lab previously revealed WDR5 as an import host regulator of HCMV virion assembly. Although in the same WDR family, the homology between WDR5 and WDR11 is only 19.38%. WDR11 is mainly distributed in the TGN, whereas WDR5 is originally located in the nucleus participating in HCMV nuclear egress. We did not observe the nuclear localization of WDR11, and the data do not support its participation in viral capsid capsulation as reported for WDR5. WDR5 is translocated to vAC to facilitate vAC formation at later time; similarly, the relocation of WDR11 to vAC was also observed in this report. A comparison of the interacting viral proteins of WDR5 and WDR11 identified that capsid MCP and tegument pp65 and pp150 are shared proteins, which likely contribute to relocalizations of the WDR proteins. WDR5 and WDR11 also have their specific interacting viral proteins, suggesting they may be recruited coordinately by the viral proteins to facilitate the virion assembly process.
WDR11 also plays a role in HSV-1 virion assembly. Unlike HCMV, HSV-1 induces host organelle rearrangement and forms multiple dispersed assembly compartments in epithelial and fibroblast cells, which causes WDR11 to disperse to the periphery of the cell and promotes HSV-1 assembly (32). Recently, it was found that HSV-1 could induce a unitary vAC-like structure in primary mouse neurons and undifferentiated human and mouse cancerous neuronal cells (16), but whether WDR11 participates in this process remains to be determined. It is noteworthy that WDR11 also interacts with HSV-1 viral proteins, including ICP0, VP16, and VP5. Thus, WDR11 might be a common host factor targeted by herpesviruses, and vAC formation may depend on the complex interplay between various viral and host factors.
MATERIALS AND METHODS
Ethics statement.
Human foreskin fibroblasts (HFFs) were isolated from neonatal human foreskins. The original source of the anonymized tissues was Zhongnan Hospital of Wuhan University (China). The cell isolation procedures and research plans were approved by the institutional review board (IRB) (approval no. WIVH10201202) according to the Guidelines for Biomedical Research Involving Human Subjects at Wuhan Institute of Virology, Chinese Academy of Sciences (34).
Cells and cell culture.
HFFs were isolated and maintained as described previously (34). HELFs are human embryonic lung fibroblasts that have been retrovirally transduced with human telomerase (hTERT) (29). HFFs, HELFs, and their derivatives were cultured in minimum essential medium (MEM, catalog no. 41500-034, Thermo Fisher) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (100 U/ml and 100 μg/ml, respectively; catalog no. 15140-122, Thermo Fisher). HEK293T were purchased from ATCC (CRL-11268) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; catalog no. 11995-123, Thermo Fisher) supplemented with 10% FBS and penicillin-streptomycin, as above. The cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Viruses and infections.
HCMV Towne strain (ATCC-VR 977) was propagated and titrated as described previously (29, 52). The cells were synchronized by serum starvation prior to infection and then incubated with viral inoculant for 2 h to allow for adsorption. The medium was then removed and replaced with fresh MEM.
Cell viability.
Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (catalog no. M2128, Merck) following the manufacturer’s instructions. Briefly, the cells were seeded at 5 × 103 cell/well in 96-well plates and incubated for 3, 4, 5, or 10 days. The medium was then replaced with 200 μl of fresh MEM without phenol red and containing 0.5% MTT. After incubation at 37°C for 4 h, the medium was removed, and 150 μl of dimethyl sulfoxide (DMSO) was added into each well. The plate was agitated, and the absorbance was determined at 570 nm with a reference wavelength of 630 nm using an Epoch microplate spectrophotometer (BioTek Instruments, USA).
Plasmid construction and alanine-scanning mutagenesis.
Primers used in this study are listed in Table 1. WDR11 cDNA (GenBank accession no. NM_018117.12) was derived by reverse transcription as described previously (29). The SV40 promoter and neomycin-resistance gene cassette were inserted in to BamHI- and SalI-digested pCDH-CMV-MCS-EF1α-Puro vector (catalog no. CD510B-1, System Biosciences) to replace the original EF-1α core promoter and puromycin-resistance gene cassette to generate pCDH-G418. Then, the 6×Myc tag was inserted in to NheI- and BamHI-digested pCDH-G418 to generate pCDH-6×Myc-G418. The pCDH-6×Myc-G418 vector was linearized by BamHI digestion, and pDsRed2-C1 vector was linearized by XhoI and BamHI digestion. The WDR11 cDNA was amplified by PCR and inserted into linearized vector by cloning recombination (ClonExpress II one-step cloning kit, catalog no. C112, Vazyme) and generated Myc-WDR11 and DsRed-WDR11. Open reading frames (ORFs) encoding UL25, UL32 (pp150), UL36, UL44, UL45, UL48, UL54, UL57, UL77, UL82 (pp71), UL83 (pp65), UL86 (MCP), UL87, UL88, UL93, UL98, and UL102 were PCR-amplified from Towne-BAC genome (53) and cloned into XhoI/BamHI-digested pHAGE-3×Flag by cloning recombination. Single-guide RNA (sgRNA) targeting exon 3 of WDR11 was designed using the online tool GPP sgRNA Designer (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). The sgRNA 5′-GAGCATGCCAAGCCTATCCA-3′ was cloned into the BbsI site of the lentiviral vector pLentiCRISPR-E (catalog no. 78852, Addgene). Double-stranded oligonucleotides corresponding to the targeting sequences were cloned into the pLKO.1 plasmid. The following sequences were targeted: WDR11-shRNA 1, 5′-GCAGTCGTATTCAGAGATAAA-3′; WDR11-shRNA 2, 5′-ACGTGTTCGAAGATCTTATAA-3′; and control Scramble-shRNA, 5′-TTCTCCGAACGTGTCACGT-3′. The alanine substitutions of WDR11 were introduced into pCDH-6×Myc-WDR11-G418 by fast multisite mutagenesis system (catalog no. FM201-01, TransGen Biotech).
TABLE 1.
Constructs and primers used in this study
| Plasmids | Forward/reverse | Primers |
|---|---|---|
| Myc-WDR11 | Forward | 5′-GGCGCGGCAGCCGCTTCATTGCCCTACACAGTGAACTTCAAGG-3′ |
| Reverse | 5′-CCAGAGGTTGATTGTCGACTCAGAAGAACTCGTCAAGAAGGCG-3′ | |
| DsRed-WDR11 | Forward | 5′-CACCACCTGTTCCTGAGATTTGCCCTACACAGTGAACTTC-3′ |
| Reverse | 5′-CAGTTATCTAGATCCGGTTCACTCTTCAATGGGTTCTTCC-3′ | |
| Flag-pUL25 | Forward | 5′-GGCGCGGCAGCCGCTTCATCGTCGCGGCGTCGCAGCTCGTCAC-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTATCAGCAACAGTATTCCCCGCTGTC-3′ | |
| Flag-pp150 (UL32) | Forward | 5′-GGCGCGGCAGCCGCTTCAAGTTTGCAGTTTATCGGTCTACAGCG-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTATTCCTCCGTGTTCTTAATCTTC-3′ | |
| Flag-pUL36 | Forward | 5′-GGCGCGGCAGCCGCTTCAGACGACCTACGGGACACGTTGATG-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTATCAGTTGTTCATGTAGGCGTG-3′ | |
| Flag-pUL44 | Forward | 5′-GGCGCGGCAGCCGCTTCAGATCGCAAGACGCGCCTCTCGGAGCC-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTAGCCGCACTTTTGCTTCTTGGTG-3′ | |
| Flag-pUL45 | Forward | 5′-GGCGCGGCAGCCGCTTCAAGTTTGCAGTTTATCGGTCTACAGCG-3′ |
| Reverse | 5′-GAGGTTGATTAGGATCTATCGATCTATTCCTCCGTGTTCTTAATC-3′ | |
| Flag-pUL48 | Forward | 5′-GGCGCGGCAGCCGCTTCAAAAGTCACGCAGGCCAGCTGCCACC-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTACAAAAGATAGAGAAACCGCATGTG-3′ | |
| Flag-pUL54 | Forward | 5′-GGCGCGGCAGCCGCTTCATTTTTCAACCCGTATCTGAGCGGCG-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTAACAGCATTCGTGCGCCTTGACAC-3′ | |
| Flag-pUL57 | Forward | 5′-GGCGCGGCAGCCGCTTCAAGCCACGAGGAACTAACCGCGCTAG-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTACAACCGGCTGCGTTTGGCCGGC-3′ | |
| Flag-pUL77 | Forward | 5′-GGCGCGGCAGCCGCTTCAAGTCTGTTGCACACCTTTTGGCGGC-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTATTACAACACCGCCACGCTCGGAAG-3′ | |
| Flag-pp71 (UL82) | Forward | 5′-GGCGCGGCAGCCGCTTCATCTCAGGCATCGTCCTCGCCCGGTG-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTACTAGATGCGGGGTCGACTGCGTG-3′ | |
| Flag-pp65 (UL83) | Forward | 5′-GGCGCGGCAGCCGCTTCAGAGTCGCGCGGTCGCCGTTGTC-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTATCAACCTCGGTGCTTTTTGG-3′ | |
| Flag-MCP (UL86) | Forward | 5′-GGCGCGGCAGCCGCTTCAGAGAACTGGTCGGCGCTCGAGCTC-3′ |
| Reverse | 5′-GAGGTTGATTAGGATCTATCGATTCACGAGTTAAATAACATGG-3′ | |
| Flag-pUL87 | Forward | 5′-GGCGCGGCAGCCGCTTCAGCCGGCGCTGCGCCGCGCCGCCTCGGC-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTATCATCGTGATGCAAACCGCGCTC-3′ | |
| Flag-pUL88 | Forward | 5′-GGCGCGGCAGCCGCTTCAATGGAAGCCGCGGCCGCTGCCGCCGCG-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTACTAGGCACGCAGCAGAGCCACCAGG-3′ | |
| Flag-pUL93 | Forward | 5′-GGCGCGGCAGCCGCTTCAGAAACGCACCTGTATTCGGATCTGGCG-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTACTAAAGATCGTCGAACGGCAAGCG-3′ | |
| Flag-pUL98 | Forward | 5′-GGCGCGGCAGCCGCTTCATGGGGCGTCTCGAGTTTGGACTACGAC-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTATCAGGGGCTCACCGGGCGTGGTAC-3′ | |
| Flag-pUL102 | Forward | 5′-GGCGCGGCAGCCGCTTCAACCGCTCAGCCGCCGCTGCAC-3′ |
| Reverse | 5′-AGAGAGCGCACGAGTTCTTAAGCGTTGAGCCGGAAAAAC-3′ | |
| pCDH-G418 | Forward | 5′-GGCGCGGCAGCCGCTTCAGGATCCCTGAGGCGGAAAGAACCAGCTG-3′ |
| Reverse | 5′-CCAGAGGTTGATTGTCGACTCAGAAGAACTCGTCAAGAAGGCG-3′ | |
| pCDH-G418-6Myc | Forward | 5′-TAGAAGATTCTAGAGCTAGCCGATTTAAAGCTATGGAGCAAAA-3′ |
| Reverse | 5′-TCTTTCCGCCTCAGGGATCCTGAAGCGGCTGCCGCGCCCAAG-3′ |
Construction of WDR11 knockdown and knockout cells.
HFFs transduced with pLKO.1-WDR11 shRNA (shW1 and shW2) and scrambled shRNA (shScram) were selected by puromycin (1 μg/ml; catalog no. P8833, Merck) for 3 days in 10-cm dishes. Survivor cells had been propagated in the presence of puromycin until the experiment was conducted. WDR11-KO and CTL (empty vector) HELF cells were generated by transduction of lentiviruses based on pLentiCRISPR-E expressing a combination of guide RNAs (gRNAs) and Cas9. Clones were selected by puromycin (8 μg/ml) for 3 days. Survivors were harvested, and clonal lines were isolated by flow cytometry sorting and then subcultured in 96-well plates with the medium containing puromycin (8 μg/ml) for 2 weeks. Finally, the cells were transferred to 6-well plates for further subculture followed by IB to validate WDR11 knockout.
Transient transfection.
About 1.5 × 106 HEK293T cells were seeded into 100-mm dishes. The next day the medium was changed 2 h prior to transfection via Ca2(PO4)2 precipitation with 10 μg of Myc-WDR11 along with 10 μg of plasmids encoding Flag-tagged viral proteins or empty vector as described previously (34). 2 × 106 HFFs were seeded onto coverslips in 12-well plates. The medium was changed at 2 h prior to transfection the next day. Plasmids (1.2 μg for each well) expressing Myc-tagged WDR11 were transfected into HELFs using Lipofectamine 2000 reagent (catalog no. 11668-019; Thermo) according to the manufacturer’s instructions.
qRT-PCR.
HFFs were infected at an MOI of 3 and harvested at the indicated times postinfection. A total of 1 × 106 cells were used for total RNA extraction using RNAiso Plus reagent (catalog no. 9109, TaKaRa), followed by treatment with 10 U of DNase I (catalog no. 2270A, TaKaRa) to remove residual DNA. One μg RNA of each sample was reverse transcribed with a RevertAid H Minus first-strand cDNA synthesis kit (catalog no. K1631, Fermentas) with random primers. Then, qPCR was performed on a real-time thermocycler (Bio-Rad; Connect) using SYBR green PCR master mix (catalog no. 4309155, Applied Biosystems) in 20-μl reactions for 40 PCR cycles as described previously (34). The PCR primers for WDR11 were 5′-AGCAGGAGTAGCTCAGTGTGA-3′ and 5′-ATCGCGGGAAGCATCTTGATT’. The PCR primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were 5′-GAGTCAACGGATTTGGTCGT-3′ and 5′-GACAAGCTTCCCGTTCTCAG-3′.
Immunoblotting (IB).
Cells were harvested in cell lysis buffer (catalog no. P0013, Beyotime) containing protease inhibitor cocktail (catalog no. 04693159001, Roche) and homogenized by ultrasonication. Protein concentrations of lysates were determined by Bradford assay (catalog no. 500-0205, Bio-Rad). After boiling with loading buffer, cell lysates containing equal protein amounts were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (catalog no. ISEQ00010, Millipore). The membranes were sequentially probed with primary antibodies and appropriate peroxidase-conjugated secondary antibodies. All antibodies used for IB are listed in Table 2. The blots were developed using the SuperSignal West Femto Chemiluminescent Substrate (catalog no. 34095, Thermo Fisher). Signals were detected using a FluorChem HD2 System (Alpha Innotech) and quantified with ImageJ software (National Institutes of Health).
TABLE 2.
Primary and secondary antibodies applied in immunofluorescence assay (IFA) and immunoblotting (IB)a
| Antibody | Host/isotype | Source/catalog no. |
|---|---|---|
| Primary antibody | ||
| Epitope tag | ||
| Flag | Rabbit polyclonal | Proteintech/20543-1-AP |
| Myc | Rabbit polyclonal | Proteintech/16286-1-AP |
| Myc | Mouse monoclonal/IgG2a | Abcam/ab56 |
| Cellular | ||
| WDR11 (130 kDa) | Rabbit polyclonal | Abcam/ab93871 |
| GAPDH (36 kDa) | Rabbit polyclonal | Proteintech/10494-1-AP |
| p230 trans-Golgi (230 kDa) | Mouse monoclonal/IgG1 | BD Pharmingen/611280 |
| CANX (90 kDa) | Rabbit polyclonal | Proteintech/10427-2-AP |
| HGS (110 kDa) | Rabbit polyclonal | Proteintech/10390-1-AP |
| EEA1 (170 kDa) | Rabbit polyclonal | Proteintech/28347-1-AP |
| GM130 (130 kDa) | Rabbit polyclonal | Proteintech/11308-1-AP |
| HCMV | ||
| IE1/2 (UL123/122) (72/86 kDa) | Mouse monoclonal/IgG1 | Virusys/P1215 |
| pUL44 (ICP36) (46 kDa) | Mouse monoclonal/IgG1 | Virusys/P1202-1 |
| pp65 (UL83) (65 kDa) | Mouse monoclonal/IgG3 | (20) |
| gM (UL100) (42 kDa) | Mouse monoclonal/IgG1 | (20) |
| gB (UL55) (52 and 105 kDa) | Mouse monoclonal/IgG1 | Virusys/P1201 |
| pp28 (UL99) (28 kDa) | Mouse monoclonal/IgG2a | Virusys/CA004-1 |
| MCP (150kDa) | Mouse monoclonal/IgG2a | (20) |
| Secondary antibody | ||
| Peroxidase-anti-mouse IgG | Goat anti-mouse | Jackson ImmunoResearch Laboratories/115-035-003 |
| Peroxidase-anti-rabbit IgG | Goat anti-rabbit | Jackson ImmunoResearch Laboratories/111-035-003 |
| TRITC-anti-mouse-IgG3 | Goat anti-mouse | Southern Biotech/1100-03 |
| TRITC-anti-mouse-IgG2b | Goat anti-mouse | Southern Biotech/1090-03 |
| AF488-anti-mouse-IgG1 | Goat anti-mouse | Thermo Fisher/A-21121 |
| AF488-anti-mouse-IgG2b | Goat anti-mouse | Thermo Fisher/A-21141 |
| AF488-anti-mouse-IgG2a | Goat anti-mouse | Thermo Fisher/A-21131 |
| AF568-anti-mouse-IgG1 | Goat anti-mouse | Thermo Fisher/A-21124 |
| AF647-anti-mouse-IgG2a | Goat anti-mouse | Thermo Fisher/A-21241 |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CANX, calnexin; HGS, hepatocyte growth factor-regulated tyrosine kinase substrate; EEA, endosome antigen; IE, immediate-early; TRITC, tetramethyl rhodamine isothiocyanate.
Immunoprecipitation (IP).
HEK293T cells cotransfected with plasmids expressing Myc-WDR11 and Flag-tagged viral proteins or empty vector and were harvested at 48 h posttransfection. The cells were lysed in IP lysis buffer (catalog no. P0013, Beyotime) for 1 h at 4°C and then centrifuged at 12,000 × g for 5 min to remove cell debris. IP was performed by incubation of the resulting lysates overnight at 4°C with mouse monoclonal anti-Myc antibody (catalog no. 60003-2-Ig, Proteintech). Protein A+G-agarose beads (catalog no. P2012, Beyotime) were added and incubated for 3 h at 4°C and then washed five times with lysis buffer. Loading buffer was added, and samples were boiled for 5 min before separation by SDS-PAGE followed by IB for detection of Myc, Flag, and GAPDH.
Immunofluorescent assay (IFA).
HELFs or HFFs were seeded onto coverslips and after attachment infected with HCMV at an MOI of 3. The coverslips were collected at the indicated times postinfection and fixed with 4% paraformaldehyde. Target proteins were detected by incubation with primary antibodies and appropriate secondary antibodies, as listed in Table 1, and as described previously (29). The nuclei were counterstained with 4´,6-diamidino-2-phenylindole (DAPI) (catalog no. D9542, Merck). The images were obtained using a UltraVIEW VoX (PerkinElmer) spinning disk laser confocal scanning microscope. The 3D z-axis image stacks were acquired with 0.2-μm spacing, and 3D modeling was performed using Volocity 5.5 software (PerkinElmer). The images were median-filtered to reduce noise, and contrast was enhanced to improve resolution.
Quantitation of viral genome copy number by qPCR.
DNA from each fraction was analyzed by qPCR to quantitate viral genome copy number using HCMV UL83 primers as described previously (34). Means and standard deviations (SDs) from at least three independent experiments were calculated.
Subcellular fractionation.
CTL and WDR11-KO cells were infected at an MOI of 1. The cells were collected at 96 hpi and resuspended in phosphate-buffered saline (PBS) containing 0.25 M sucrose supplemented with protease inhibitor cocktail (catalog no. 5892970001, Roche). The cells were then lysed with 200 passages in a type B Dounce homogenizer on ice and centrifuged at 2,500 × g for 10 min at 4°C to remove nuclei and cell debris. Supernatants were layered onto iodixanol gradients prepared by sequentially layering 800-μl volumes of 30, 20, and 10% iodixanol (catalog no. D1556, Merck) in PBS (vol/vol) and then centrifuged at 125,000 × g for 16 h at 4°C using an SW 55Ti rotor and an Optima MAX-XP ultracentrifuge. Fractions were collected from top to bottom of the gradient and analyzed by immunoblot.
Transmission electron microscopy (TEM).
Control HELFs (CTL) and the WDR11 knockout HELFs (WDR11-KO) were infected with HCMV at an MOI of 0.5 and harvested at 120 hpi. The cells were fixed with 2.5% (wt/vol) glutaraldehyde for 1 h at room temperature and then treated with 1% osmium tetroxide and dehydrated through a graded series of ethanol concentrations (from 30 to 100%) and embedded using an Embed 812 kit (Electron Microscopy Sciences, Fort Washington, PA). Ultrathin sections (60 to 80 nm) of embedded specimens were prepared and deposited onto Formvar-coated copper grids (200-mesh), stained with 2% (wt/vol) phosphotungstic acid (PTA, pH 6.8), and observed under a Tecnai transmission electron microscope (FEI) operated at 200 kV as described previously (29).
LC-MS/MS analysis.
LC-MS/MS analysis was performed on a Q Exactive mass spectrometer (Thermo Scientific) coupled to Easy nLC (Proxeon Biosystems) for 60 min in positive ion mode. The data were acquired using a data-dependent top 10 method dynamically choosing the most abundant precursor ions from the survey scan (300 to 1,800 m/z) for highly conserved domain (HCD) fragmentation. Automatic gain control (AGC) target was set to 3e6, and maximum inject time was set to 10 ms. Dynamic exclusion duration was 40.0 s. Survey scans were acquired at a resolution of 70,000 at m/z 200, resolution for HCD spectra was set to 17,500 at m/z 200, and isolation width was 2 m/z. Normalized collision energy was 30 eV, and the underfill ratio was defined as 0.1%. The instrument was run with the peptide recognition mode enabled.
Statistical analysis.
The data were analyzed by chi-square or one-way analysis of variance (ANOVA), as appropriate, using SPSS software (version 18.0; SPSS). The results were shown as means ± SD from three independent experiments. A value of P < 0.05 was considered significant. Statistical tests for TEM data were conducted using the Kruskall-Wallis test. Post hoc Dunn’s multiple-comparison tests were conducted if the overall Kruskal-Wallis test was statistically significant, and P < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Anna Du and Pei Zhang of the Core Facility, Wuhan Institute of Virology, Chinese Academy of Sciences (CAS), for technical support of electron microscopy. We thank Juan Min of the Core Facility, Wuhan Institute of Virology, CAS, for technical support of flow cytometry sorting.
This work was supported by grants 81620108021, 81427801, 81871660, 31900137, and 31900138 from the National Natural Science Foundation of China and grants 2019M652846 and 2019M662851 from the China Postdoctoral Science Foundation.
Contributor Information
Sitang Gong, Email: sitangg@126.com.
Min-Hua Luo, Email: luomh@wh.iov.cn.
Lori Frappier, University of Toronto.
REFERENCES
- 1.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. 2002. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62:3347–3350. [PubMed] [Google Scholar]
- 2.Wen L, Qiu Y, Cheng S, Jiang X, Ma YP, Fang W, Wang W, Cui J, Ruan Q, Zhao F, Hu F, Luo MH. 2018. Serologic and viral genome prevalence of HSV, EBV, and HCMV among healthy adults in Wuhan, China. J Med Virol 90:571–581. 10.1002/jmv.24989. [DOI] [PubMed] [Google Scholar]
- 3.Lazzarotto T, Guerra B, Gabrielli L, Lanari M, Landini MP. 2011. Update on the prevention, diagnosis and management of cytomegalovirus infection during pregnancy. Clin Microbiol Infect 17:1285–1293. 10.1111/j.1469-0691.2011.03564.x. [DOI] [PubMed] [Google Scholar]
- 4.Jean Beltran PM, Cristea IM. 2014. The life cycle and pathogenesis of human cytomegalovirus infection: lessons from proteomics. Expert Rev Proteomics 11:697–711. 10.1586/14789450.2014.971116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eddleston M, Peacock S, Juniper M, Warrell D. 1997. Severe cytomegalovirus infection in immunocompetent patients. Clin Infect Dis 24:52–56. 10.1093/clinids/24.1.52. [DOI] [PubMed] [Google Scholar]
- 7.Ho M. 1990. Epidemiology of cytomegalovirus infections. Rev of Infectious Diseases 12:S701–S710. 10.1093/clinids/12.Supplement_7.S701. [DOI] [PubMed] [Google Scholar]
- 8.Söderberg-Nauclér C, Fish KN, Nelson JA. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126. 10.1016/s0092-8674(01)80014-3. [DOI] [PubMed] [Google Scholar]
- 9.Bhella D, Rixon FJ, Dargan DJ. 2000. Cryomicroscopy of human cytomegalovirus virions reveals more densely packed genomic DNA than in herpes simplex virus type 1. J Mol Biol 295:155–161. 10.1006/jmbi.1999.3344. [DOI] [PubMed] [Google Scholar]
- 10.Alwine J. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog 8:e1002878. 10.1371/journal.ppat.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol 74:975–986. 10.1128/jvi.74.2.975-986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seo JY, Britt WJ. 2007. Cytoplasmic envelopment of human cytomegalovirus requires the postlocalization function of tegument protein pp28 within the assembly compartment. J Virol 81:6536–6547. 10.1128/JVI.02852-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das S, Ortiz DA, Gurczynski SJ, Khan F, Pellett PE. 2014. Identification of human cytomegalovirus genes important for biogenesis of the cytoplasmic virion assembly complex. J Virol 88:9086–9099. 10.1128/JVI.01141-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goulidaki N, Alarifi S, Alkahtani SH, Al-Qahtani A, Spandidos DA, Stournaras C, Sourvinos G. 2015. RhoB is a component of the human cytomegalovirus assembly complex and is required for efficient viral production. Cell Cycle 14:2748–2763. 10.1080/15384101.2015.1066535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White S, Kawano H, Harata NC, Roller RJ. 2020. Herpes simplex virus organizes cytoplasmic membranes to form a viral assembly center in neuronal cells. J Virol 94:e00900-20. 10.1128/JVI.00900-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bughio F, Umashankar M, Wilson J, Goodrum F. 2015. Human cytomegalovirus UL135 and UL136 genes are required for postentry tropism in endothelial cells. J Virol 89:6536–6550. 10.1128/JVI.00284-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dietz AN, Villinger C, Becker S, Frick M, von Einem J. 2018. A tyrosine-based trafficking motif of the tegument protein pUL71 is crucial for human cytomegalovirus secondary envelopment. J Virol 92:e00907-17. 10.1128/JVI.00907-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azzeh M, Honigman A, Taraboulos A, Rouvinski A, Wolf DG. 2006. Structural changes in human cytomegalovirus cytoplasmic assembly sites in the absence of UL97 kinase activity. Virology 354:69–79. 10.1016/j.virol.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 20.Wu H, Kropff B, Mach M, Britt WJ. 2020. Human cytomegalovirus envelope protein gpUL132 regulates infectious virus production through formation of the viral assembly compartment. mBio 11:e02044-20. 10.1128/mBio.02044-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M, Landais I, Jeng S, McWeeney S, Britt W, Nelson JA. 2014. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 15:363–373. 10.1016/j.chom.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie M, Xuan B, Shan J, Pan D, Sun Y, Shan Z, Zhang J, Yu D, Li B, Qian Z. 2015. Human cytomegalovirus exploits interferon-induced transmembrane proteins to facilitate morphogenesis of the virion assembly compartment. J Virol 89:3049–3061. 10.1128/JVI.03416-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cruz L, Streck NT, Ferguson K, Desai T, Desai D, Amin S, Buchkovich NJ. 2017. Potent inhibition of human cytomegalovirus by modulation of cellular SNARE syntaxin 5. J Virol 91:e01637-16. 10.1128/JVI.01637-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchkovich NJ, Maguire TG, Alwine JC. 2010. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J Virol 84:7005–7017. 10.1128/JVI.00719-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J Virol 83:11421–11428. 10.1128/JVI.00762-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Indran SV, Ballestas ME, Britt WJ. 2010. Bicaudal D1-dependent trafficking of human cytomegalovirus tegument protein pp150 in virus-infected cells. J Virol 84:3162–3177. 10.1128/JVI.01776-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krzyzaniak MA, Mach M, Britt WJ. 2009. HCMV-encoded glycoprotein M (UL100) interacts with Rab11 effector protein FIP4. Traffic 10:1439–1457. 10.1111/j.1600-0854.2009.00967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang B, Yao Y, Wu H, Yang H, Ma XH, Li D, Wang XZ, Huang SN, Jiang X, Cheng S, Sun JY, Huang ZL, Zhao C, McVoy MA, Ahn JH, Zeng WB, Britt WJ, Gong S, Luo MH. 2021. Localization of the WD repeat-containing protein 5 to the virion assembly compartment facilitates human cytomegalovirus assembly. J Virol 95:e02101-20. 10.1128/JVI.02101-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang B, Liu XJ, Yao Y, Jiang X, Wang XZ, Yang H, Sun JY, Miao Y, Wang W, Huang ZL, Wang Y, Tang Q, Rayner S, Britt WJ, McVoy MA, Luo MH, Zhao F. 2018. WDR5 facilitates human cytomegalovirus replication by promoting capsid nuclear egress. J Virol 92:e00207-18. 10.1128/JVI.00207-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Hu XJ, Zou XD, Wu XH, Ye ZQ, Wu YD. 2015. WDSPdb: a database for WD40-repeat proteins. Nucleic Acids Res 43:D339–D344. 10.1093/nar/gku1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navarro Negredo P, Edgar JR, Manna PT, Antrobus R, Robinson MS. 2018. The WDR11 complex facilitates the tethering of AP-1-derived vesicles. Nat Commun 9:596. 10.1038/s41467-018-02919-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor KE, Mossman KL, Sandri-Goldin RM. 2015. Cellular protein WDR11 interacts with specific herpes simplex virus proteins at the trans-Golgi network to promote virus replication. J Virol 89:9841–9852. 10.1128/JVI.01705-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu XJ, Yang B, Huang SN, Wu CC, Li XJ, Cheng S, Jiang X, Hu F, Ming YZ, Nevels M, Britt WJ, Rayner S, Tang Q, Zeng WB, Zhao F, Luo MH. 2017. Human cytomegalovirus IE1 downregulates Hes1 in neural progenitor cells as a potential E3 ubiquitin ligase. PLoS Pathog 13:e1006542. 10.1371/journal.ppat.1006542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu Y-R, Liu X-J, Li X-J, Shen Z-z, Yang B, Wu C-C, Li J-F, Miao L-F, Ye H-Q, Qiao G-H, Rayner S, Chavanas S, Davrinche C, Britt WJ, Tang Q, McVoy M, Mocarski E, Luo M-H. 2015. MicroRNA miR-21 attenuates human cytomegalovirus replication in neural cells by targeting Cdc25a. J Virol 89:1070–1082. 10.1128/JVI.01740-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Viswanathan K, Smith MS, Malouli D, Mansouri M, Nelson JA, Fruh K. 2011. BST2/tetherin enhances entry of human cytomegalovirus. PLoS Pathog 7:e1002332. 10.1371/journal.ppat.1002332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Microbiol Mol Biol Rev 72:249–265. 10.1128/MMBR.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo JY, Britt WJ. 2006. Sequence requirements for localization of human cytomegalovirus tegument protein pp28 to the virus assembly compartment and for assembly of infectious virus. J Virol 80:5611–5626. 10.1128/JVI.02630-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kropff B, Koedel Y, Britt W, Mach M. 2010. Optimal replication of human cytomegalovirus correlates with endocytosis of glycoprotein gpUL132. J Virol 84:7039–7052. 10.1128/JVI.01644-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park SY, Guo X. 2014. Adaptor protein complexes and intracellular transport. Biosci Rep 34:e00123. 10.1042/BSR20140069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inoue J, Ninomiya M, Umetsu T, Nakamura T, Kogure T, Kakazu E, Iwata T, Takai S, Sano A, Fukuda M, Watashi K, Isogawa M, Tanaka Y, Shimosegawa T, McNiven MA, Masamune A. 2019. Small interfering RNA screening for the small GTPase Rab proteins identifies Rab5B as a major regulator of hepatitis B virus production. J Virol 93:e00621-19. 10.1128/JVI.00621-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, 2nd, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. 2005. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell 97:147–172. 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glingston RS, Deb R, Kumar S, Nagotu S. 2019. Organelle dynamics and viral infections: at cross roads. Microbes Infect 21:20–32. 10.1016/j.micinf.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jean Beltran PM, Mathias RA, Cristea IM. 2016. A portrait of the human organelle proteome in space and time during cytomegalovirus infection. Cell Syst 3:361–373.e6. 10.1016/j.cels.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, Lehner PJ, Gygi SP. 2014. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157:1460–1472. 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abraham E, Pantoja M, Kucherenko MM, Yatsenko AS, Shcherbata HR, Fischer KA, Maksymiv DV, Chernyk YI, Ruohola-Baker H. 2008. Genetic modifier screens reveal new components that interact with the Drosophila dystroglycan-dystrophin complex. PLoS One 3:e2418. 10.1371/journal.pone.0002418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin JJH, Gillingham AK, Begum F, Chadwick J, Munro S. 2017. TBC1D23 is a bridging factor for endosomal vesicle capture by golgins at the trans-Golgi. Nat Cell Biol 19:1424–1432. 10.1038/ncb3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hensel GM, Meyer HH, Buchmann I, Pommerehne D, Schmolke S, Plachter B, Radsak K, Kern HF. 1996. Intracellular localization and expression of the human cytomegalovirus matrix phosphoprotein pp71 (ppUL82): evidence for its translocation into the nucleus. J Gen Virol 77:3087–3097. 10.1099/0022-1317-77-12-3087. [DOI] [PubMed] [Google Scholar]
- 49.Battista MC, Bergamini G, Boccuni MC, Campanini F, Ripalti A, Landini MP. 1999. Expression and characterization of a novel structural protein of human cytomegalovirus, pUL25. J Virol 73:3800–3809. 10.1128/JVI.73.5.3800-3809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim Y-E, Oh SE, Kwon KM, Lee CH, Ahn J-H, Sandri-Goldin RM. 2016. Involvement of the N-terminal deubiquitinating protease domain of human cytomegalovirus UL48 tegument protein in autoubiquitination, virion stability, and virus entry. J Virol 90:3229–3242. 10.1128/JVI.02766-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar R, Cruz L, Sandhu PK, Buchkovich NJ. 2020. UL88 mediates the incorporation of a subset of proteins into the virion tegument. J Virol 94:e00474-20. 10.1128/JVI.00474-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan X, Li X-J, Liu X-J, Yuan H, Li J-F, Duan Y-L, Ye H-Q, Fu Y-R, Qiao G-H, Wu C-C, Yang B, Tian X-H, Hu K-H, Miao L-F, Chen X-L, Zheng J, Rayner S, Schwartz PH, Britt WJ, Xu J, Luo M-H. 2013. Later passages of neural progenitor cells from neonatal brain are more permissive for human cytomegalovirus infection. J Virol 87:10968–10979. 10.1128/JVI.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marchini A, Liu H, Zhu H. 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol 75:1870–1878. 10.1128/JVI.75.4.1870-1878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]