

ABSTRACT
All viruses must usurp host ribosomes for viral protein synthesis. Dicistroviruses utilize an intergenic region internal ribosome entry site (IGR IRES) to directly recruit ribosomes and mediate translation initiation from a non-AUG start codon. The IGR IRES adopts a three-pseudoknot structure that comprises a ribosome binding domain of pseudoknot II and III (PKII and PKIII), and a tRNA-like anticodon domain (PKI) connected via a short, one to three nucleotide hinge region. Recent cryo-EM structural analysis of the dicistrovirus Taura syndrome virus (TSV) IGR IRES bound to the ribosome suggests that the hinge region may facilitate translocation of the IRES from the ribosomal A to P site. In this study, we provide mechanistic and functional insights into the role of the hinge region in IGR IRES translation. Using the honeybee dicistrovirus, Israeli acute paralysis virus (IAPV), as a model, we demonstrate that mutations of the hinge region resulted in decreased IRES-dependent translation in vitro. Toeprinting primer extension analysis of mutant IRESs bound to purified ribosomes and in rabbit reticulocyte lysates showed defects in the initial ribosome positioning on the IRES. Finally, using a hybrid dicistrovirus clone, mutations in the hinge region of the IAPV IRES resulted in decreased viral yield. Our work reveals an unexpected role of the hinge region of the dicistrovirus IGR IRES coordinating the two independently folded domains of the IRES to properly position the ribosome to start translation.
IMPORTANCE Viruses must use the host cell machinery to direct viral protein expression for productive infection. One such mechanism is an internal ribosome entry site that can directly recruit host cell machinery. In this study, we have identified a novel sequence in an IRES that provides insight into the mechanism of viral gene expression. Specifically, this novel sequence promotes viral IRES activity by directly guiding the host cell machinery to start gene expression at a specific site.
KEYWORDS: ribosome, IRES, virus, translation, RNA, Israeli acute paralysis virus, RNA structure, RNA virus, infectious clones, internal ribosome entry site, translational control
INTRODUCTION
Viruses must use the host translational machinery to direct viral protein synthesis, and some have evolved noncanonical translational strategies to recruit the ribosome (1). One such mechanism is internal ribosome entry sites (IRESs). First identified in picornaviruses and common in the 5′ UTR of positive sense RNA viruses, IRESs are structured RNA elements that recruit the ribosome in a 5’cap-independent manner and generally require fewer translation initiation factors (2, 3). Extensive biochemical and reconstitution experiments have revealed that viral IRESs can be grouped into four main types based on sequence and structure conservation, and factor requirements (4). Type I and II IRESs are common among picornaviridae and require most canonical translation factors as well as IRES-trans-acting factors to recruit the 40S ribosome subunit. Type III IRESs, such as those from hepatitis C virus, foot-and-mouth disease virus, and classic swine fever virus, only require factors eIF3 and eIF2. Lastly, Type IV IRESs, found within the intergenic region (IGR) of the Dicistroviridae family, uses one of the most streamlined mechanisms recruiting the ribosome directly and initiates translation at a non-AUG codon. Studies on Type IV IRESs have provided a wealth of insights into viral translational mechanisms and fundamental ribosome function.
Dicistroviridae are positive single-stranded RNA viruses approximately 9 kb in length (5, 6). The viral RNA contains a viral genome-linked protein at the 5’end and a 3′ poly A tail (7). The genome contains two main open reading frames (ORFs), each driven by distinct IRESs. The ORFs are translated as a polyprotein and then processed by the viral 3C-like protease. Dicistroviridae mainly infect arthropods, with members including cricket paralysis virus (CrPV), the Panaeid shrimp infecting Taura syndrome virus (TSV), and several honey bee viruses including Israeli acute paralysis virus (IAPV), acute bee virus, and Kashmir bee virus.
The mechanism of IGR IRESs has been studied extensively through biochemical and structural studies, revealing an elaborate network of dynamic interactions between the IRES and the ribosome (8–13). Essential to this mechanism is the RNA structure of the IGR IRES. The IRES adopts two independently folded domains that mediate distinct functions to manipulate the ribosome. Pseudoknots II and III (PKII and PKIII) comprise one main domain that is responsible for 40S and 60S subunit binding, and PKI domain directs and positions the ribosome to start translation at the non-AUG codon. Moreover, unlike cap-dependent translation, which utilizes ∼12 core translation initiation factors, the IGR IRES does not require these factors for translation. Biochemical studies and multiple high resolution structural studies have provided insights into the mechanism of the IRES translocating through the ribosome. Initially, the IRES binds within the intersubunit space of the ribosome and spans all three ribosomal sites (9, 11, 13). The L1.1 domain of the IRES interacts with the L1 stalk of the 60S subunit, and the stem-loops IV (SLIV) and SLV interact with uS7 (9). Mutations in these domains disrupt 40S and 60S binding (14–16). The PKII and PKIII domain primarily occupies the ribosomal E and P sites and is connected via a hinge region to the PKI domain, which rests in the A-site. The conformation of PKI resembles a tRNA codon–anticodon interaction to set the translational reading frame (17, 18).
The translocation of the IRES through the ribosome is unprecedented as the IRES both acts as its own mRNA and occurs without peptide bond formation; thus, the event has been coined pseudotranslocation (19). Cryo-EM structures of the translocated IRES of the Taura syndrome virus (TSV) show that the IRES rearranges from an extended to bent to extended conformation in an inchworm-like fashion (11). During the initiation state, the IRES is extended where the front end, SLIV and SLV, interacts with S7, uS11, and eS25 within the head of the 40S, while the hind, PKI domain mimics an anticodon–codon interaction occupying the decoding center of the A site. During the first pseudotranslocation step, the PKI domain moves from the A– to the P– site and approaches the front end, shortening the distance between them and forming a bent inchworm-like conformation. The front end then departs its initial binding site, returning to an extended conformation. Recently, other translocated dicistrovirus IRES structures have been elucidated (9, 10, 20, 21). The honey bee dicistrovirus, Israeli acute paralysis virus (IAPV), IRES closely mimics a canonical P site tRNA in its posttranslocated state while its variable loop region (VLR) is in contact distance of the ribosomal E site (21). A double translocated IRES of the cricket paralysis virus (CrPV) also reveals a remarkable dynamic conformation whereby the PKI anticodon–codon mimic flips from the anticodon stem to the acceptor space in the ribosomal E site (12). In summary, the IGR IRES actively manipulates the ribosome to hijack specific steps of ribosome translocation.
The dicistrovirus IGR IRESs, to date, can be grouped into two main classes: Type I IRESs, such as those in CrPV and Drosophila C virus, and Type II exemplified by the IAPV and TSV. The main difference is that the Type II have an extra SLIII within PKI, where PKI and PKII/PKIII domains are functionally interchangeable (22–24). These findings suggest that both the PKI and PKII/PKIII domains work independently to direct distinct IRES-ribosomal functions. However, cross-communication between the two domains has been proposed, suggesting that there is coordination to drive IRES translation (14, 22, 25).
While much focus has been on helical stemmed structures of the IRES, unpaired sequences that connect between structured regions have major roles in IRES activity. For instance, the unpaired adenosines within PKII adopt an A-minor motif tertiary interaction to stabilize the PKII/PKIII globular domain (15, 26). Furthermore, the variable loop region within PKI promotes IRES translocation and ribosome positioning on the IRES (27, 28). Importantly, mutations in these regions have been shown to adversely affect productive virus infection (29). In both Type I and Type II IRESs, the PKI and PKII/III domains are typically linked by a short stretch of 1 to 3 nondiscriminate nucleotides, called the hinge domain (Fig. 1). Sequence analysis of the hinge domain does not show an obvious overall consensus identity or length; however, a subset contained either a UGC or UA hinge sequence. For example, Type II IGR IRESs typically contain two unpaired nucleotides; specifically, an UA hinge sequence is found in IAPV and other dicistroviridae honeybee viruses, whereas hinge sequences of CrPV and TSV are UGC and AA, respectively (23). Whether these sequences are important for IRES function has not yet been investigated. Interestingly, cryo-EM structures of the TSV IGR IRES translocated through the ribosome suggest that the hinge region interacts with uL5 and the C-terminal tail of eL42 to facilitate translocation (11). Using the IAPV IGR IRES as a model, we used extensive mutagenesis and biochemical and virological analysis to elucidate the role of the hinge region in IGR IRES:ribosome translation interaction, revealing that the hinge region is important for IRES translation and acts to coordinate PKII/PKIII and PKI domains for IRES translation initiation.
FIG 1.

Mutational analysis of the hinge region of the IAPV IGR IRES. (A) Schematic of the secondary structure of the IRES within the intergenic region of the Israeli acute paralysis virus genome. Key stem-loops (SL) IV, V, and VI and pseudoknots (PK) I, II, and III are shown. The hinge mutations are highlighted to the right. U6562-G6618 wobble base pair (dashed line) mediates +1 frame translation ORFx. (B) Schematic of the bicistronic IRES reporter construct. The renilla luciferase reporter (RLuc) and firefly luciferase reporter (FLuc) are translated via scanning-dependent and IRES-dependent translation, respectively. Fluc is fused in the 0-reading frame in-frame with a short ORF2 sequence (sORF2) (71.1 kDa). Expression of the +1 frame ORFx produces an ∼11.1 kDa protein. (C) Translational activities of mutant IRESs. (Top) Bicistronic reporter constructs were linearized and incubated in Sf21 extracts for 120 min at 30°C in the presence of [35S]-methionine/cysteine. Reactions were analyzed by SDS-PAGE, followed by phosphorimager analysis. A representative gel is shown. (Bottom) Quantitations of radiolabeled protein products. The ratios of sORF2-FLuc (0 frame)/RLuc and ORFx (+1 frame)/RLuc normalized to wild-type 0 and +1 frame translation are shown. Averages are from at least three independent experiments ± SD are shown.
RESULTS
Mutational analysis of the hinge region of the IAPV IGR IRES.
To address whether the hinge region is important for dicistrovirus IGR IRES translation, we generated a series of mutations that varied in the length and identity of the UA hinge region sequence of the Type II IAPV IGR IRES (herein referred to as IRES), (Fig. 1A). We mutated the UA6547-8 sequence to other dicistrovirus hinge sequences such as AA (TSV) or UGC (CrPV) to determine whether there is specificity of these sequences between dicistrovirus IRESs. Translation of the bicistronic reporter constructs was tested in SF-21 insect extracts with [35S]-Met/Cys to monitor protein synthesis (30) (Fig. 1B). Here, the upstream cistron encoding renilla luciferase monitors scanning-dependent translation (∼35 kDa), and the downstream firefly luciferase cistron monitors IGR IRES translation (∼70 kDa). Since the IAPV IRES also directs +1 frame translation, this reporter construct also monitors +1 frame translation (∼11 kDa) for the protein referred to as ORFx in the same reaction (31) (Fig. 1B).
As shown previously(31), mutating the PKI base pair (ΔPKI; CC6615-6GG) abolished IAPV IRES activity, resulting in limited 0 and +1 frame translation compared to the wild-type IRES (Fig. 1C, lane 2). Mutating the wild-type UA hinge sequence to GC, UC, AU, or AA all decreased translation of both reading frames by 10% to 50% compared to wild-type IRES translation (Fig. 1C, lanes 3,4,5, and 8). Notably, the AA and UGC sequences, which are that of the TSV and CrPV IRES hinge regions, respectively, decreased translation in the 0 and +1 frames by ∼70%. (Fig. 1C, lanes 8 and 9). Interestingly, addition of an adenosine following the hinge sequence maintains 0 and +1 frame translation at ∼80–100% of the wild-type IRES. However, shortening the hinge region to a single uridine decreased translation by ∼80% (Fig. 1C, lane 7). Thus, these results demonstrate that not all dicistrovirus IGR IRES hinge regions function similarly in the IAPV IGR IRES and that the identity of the UA hinge sequence is important for IAPV IGR IRES activity.
Toeprinting analysis using purified salt-washed ribosomes.
As IRES translation involves translocation through the ribosome, we next examined whether the hinge mutations affected the position of the ribosome on the IRES. We tested this by toeprinting, a primer extension analysis on IRES:ribosome complexes. Briefly, reverse transcription will terminate upon encountering the leading edge of the ribosome, and the resultant cDNA (toeprint) can then be used to infer the specific nucleotides that occupy the ribosomal A and P sites. Ribosomal binding to the IGR IRES typically leads to a +14 nucleotide toeprint, placing the PKI domain in the A site, given that the first nucleotide of the A-site codon is +1 (17).
Toeprinting assays were performed on IRES-ribosome complexes using purified 40S and 60S ribosomal subunits from HeLa cells. As previously shown, ribosomes assembled on the wild-type IRES produced a dominant toeprint at A6628, termed Toeprint A, which is 14 nucleotides from the C (+1) of the CCU codon that occupies the ribosomal A site (17) (Fig. 1A, 2A). By contrast, disrupting the PKI base pairing by mutation of CC6615-6 to GG (ΔPKI), while maintaining the PII and PKIII that allows binding to the ribosome (15), abolished the +14 A6628 Toeprint A (Fig. 2B). This indicates that the ribosome is not positioned properly on the ΔPKI IRES, such that the tRNA anticodon–codon-like base pairing of PKI does not stably occupy the ribosomal A site (16). For each reaction, toeprints were quantified by radioactivity and normalized to the wild-type IRES (100%). Ribosomal binding to mutant IRESs containing hinge sequences GC, UC, AU, and U severely diminished the +14 toeprint signal to below 20% of the wild-type IRES, suggesting that the identity of the UA hinge sequence in the context of IAPV is important in ribosomal positioning (Fig. 2B). Mutant IRESs containing AA or UGC hinge sequences also decreased the toeprint signal, but was less inhibitory of the +14 toeprint binding at 31% and 42% of wild type, respectively (Fig. 2B). Interestingly, the UAA mutant retained a relatively high toeprint signal of 74%, compared to the wild type, suggesting that the additional adenosine nucleotide in the hinge does not affect the recruitment and positioning of the ribosome. These results indicate that the identity of the hinge region is important for proper ribosome positioning, as evidenced of a characteristic +14 Toeprint A in this toeprinting assay, which we interpret as a stable PKI domain docked in the decoding ribosomal A site primed for IRES translocation. Besides the A6628 (+14) toeprint A, two other prominent toeprints, B and C, are detected, which are located further upstream in the IRES at the base of SLIII and the PKIII stem-loop (Fig. 1A, 2A). Notably, Toeprints B and C intensities were detected in both reactions containing the IRES alone or the IRES:ribosome complex for all wild-type and mutant IRESs, indicating that RNA structures in the IRES block the reverse transcriptase. These results can infer that the overall structure of the IRESs is not dramatically affected by the mutations within the hinge.
FIG 2.
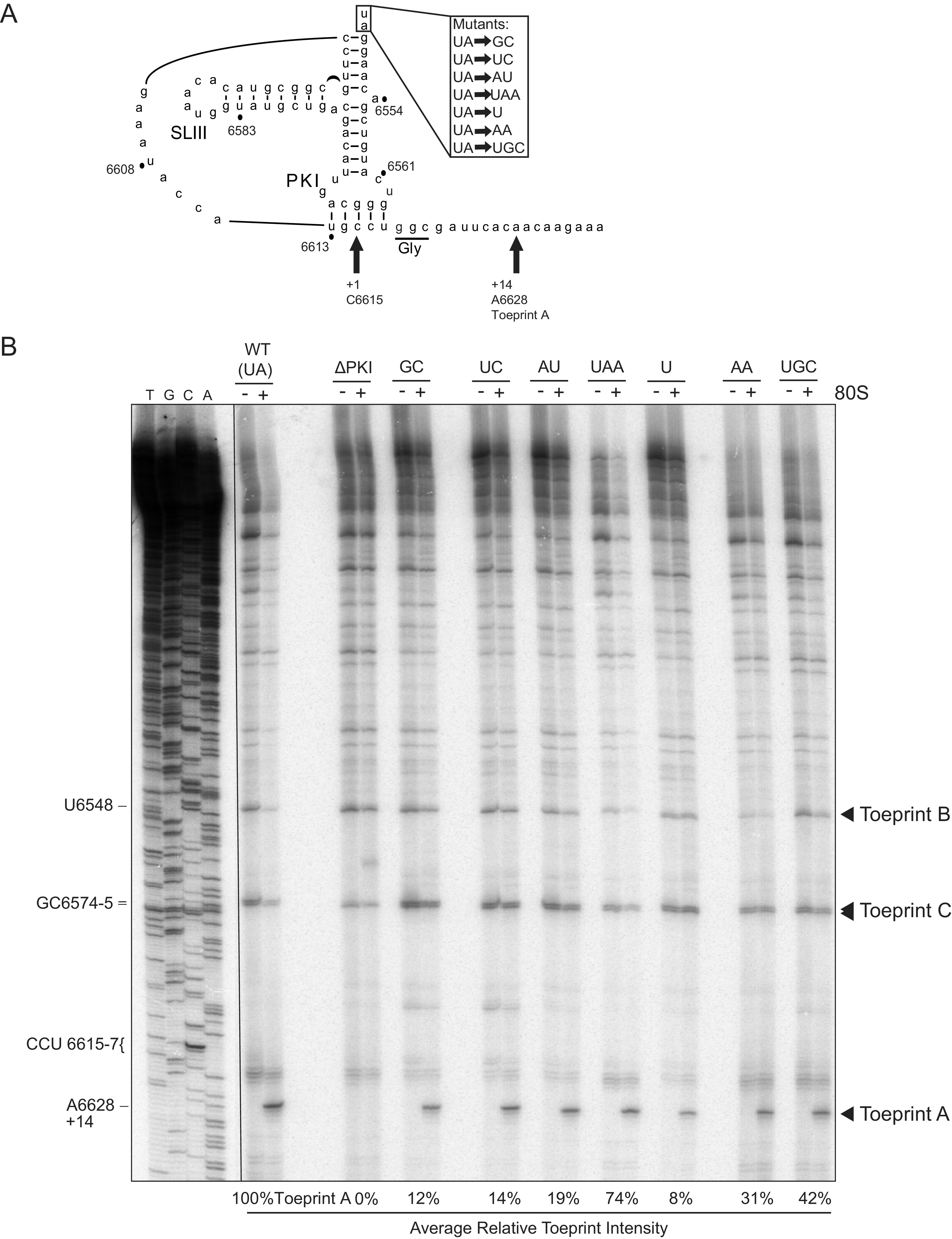
Toeprinting analysis of purified ribosomes bound to hinge mutant IRES. (A) Schematic of the PKI tRNA mimicry domain of the IAPV IGR IRES. Arrows denote the primary toeprint observed in (B). (B) In vitro transcribed bicistronic RNAs containing the wild-type or mutant IGR IRES were incubated with purified salt-washed human 80S ribosomes (100 nM) for 15 min at 30°C. Primer extension cDNA analysis was performed in the presence of α-[32P]-dATP, and the cDNA reaction products were resolved by denaturing polyacrylamide gel electrophoresis and visualized by autoradiography. (Left) Sequencing ladder for the wild-type IRES is shown with location of the major toeprints. Toeprint intensities were measured as a fraction of the radioactive counts for toeprint A6628 over the total radioactive counts within each lane normalized to that of the wild-type IGR IRES. Average is shown from two independent experiments.
To address whether the hinge mutants have an effect on ribosome recruitment, we assayed purified 80S binding on the IAPV IRES by filter binding (17). As previously indicated, U and UA hinge mutants inhibited both IRES translation and proper toeprinting at A6628 (+14), but it is unknown whether ribosomal recruitment to the IRES is a determining factor for this lapse in activity (Fig. 1 and 2). As shown previously, the wild-type IAPV IRES binds to 80S with a high affinity (KD ∼21 nM), whereas a triple pseudoknot IAPV IRES mutant, which disrupts all three major PKI basepairings, did not have appreciable 80S binding (Fig. 3) (25). Similar to the wild-type IAPV IRES, the U and UA hinge mutant IRESs bound to 80S ribosomes with a high affinity (KD ∼8-13 nM), indicating that the nucleotide identity of the hinge region is not necessary in ribosomal 80S recruitment, and strongly suggests that the overall structures within the IAPV IRES are intact.
FIG 3.

80S ribosome assembly on hinge mutant IAPV IGR IRESs. Wild type and the indicated mutant [32P]-IAPV IGR IRESs (0.5 nM) were incubated with increasing amounts of purified salt-washed 80S ribosomes, and the fractions bound were quantified by filter binding assay and phosphorimager analysis. The TM IAPV IRES denotes mutations that disrupt all three pseudoknots (25). Shown are averages ± standard deviation from at least three independent experiments.
Ribosome:IRES complexes at distinct translocation steps.
Previous cryo-EM structures revealed that the hinge region of the dicistrovirus TSV IGR IRES is in close proximity with ribosomal proteins uL5 and eL42, suggesting that these interactions may contribute to translocation of the PKI domain from the ribosomal A to P site (11). To examine the translocation of the IRES through the ribosome, we first established an in vitro translation system that exploits translation inhibitors to trap the IRES at distinct translocation steps, specifically the translocation of PKI from the A to P and from P to E sites. Cycloheximide (CHX), hygromycin B (HygB), and lactimidomycin D (LTM) are inhibitors that have been shown to block dicistrovirus IGR IRES translation, which we predict can trap the IAPV IRES PKI domain in each of the ribosomal A, P, and E sites, thereby providing a framework to monitor translocation in an in vitro translation assay (27, 32). In particular, CHX binds within the ribosomal E site occupied with a deacylated tRNA, thereby inhibiting translation elongation after two translocation events (32). LTM, like cycloheximide, binds the ribosomal E site but only if the E site is empty and can entrap initiating ribosomes (32). HygB binds near and stabilizes the A-site tRNA and disrupts translocation events by increasing the energy barrier for translocation, potentially generating a steric block to the movement of the tRNAs between the A and P sites in both the forward and reverse directions (33, 34).
We assessed the toeprints of ribosomes assembled on the IAPV IRES in rabbit reticulocyte lysates (RRL) in the absence or presence of CHX, HygB, or LTM. In the presence of LTM alone, a single toeprint at A6628 (+14) is detected, suggesting that LTM traps initiating ribosomes such that the PKI domain occupies the ribosomal A site (Fig. 4, lane 3). In the presence of CHX, ribosomes on the IAPV IRES translocate two elongation cycles to produce a prominent toeprint 6 nucleotides downstream (A6634 [+20] [Fig. 4, lane 1]) of the +14 A6628 Toeprint A, as shown previously (17). In the presence of HygB, a prominent toeprint is observed at A6631, three nucleotides downstream of the A6628 Toeprint A, which infers that the PKI domain has translocated a single step from the ribosomal A to P sites before HygB inhibition (Fig. 4, lane 2). This result also indicates that HygB cannot stabilize PKI in the A site. Despite the efficiency of the drugs, a minor A6628 (+14) toeprint was observed in extracts treated with CHX or HygB, which likely reflects a subset of ribosome:IRES complexes that did not initiate translocation. These results indicate that the use of these inhibitors can stabilize ribosome:IRES complexes within the first few steps of IRES-mediated pseudotranslocation.
FIG 4.
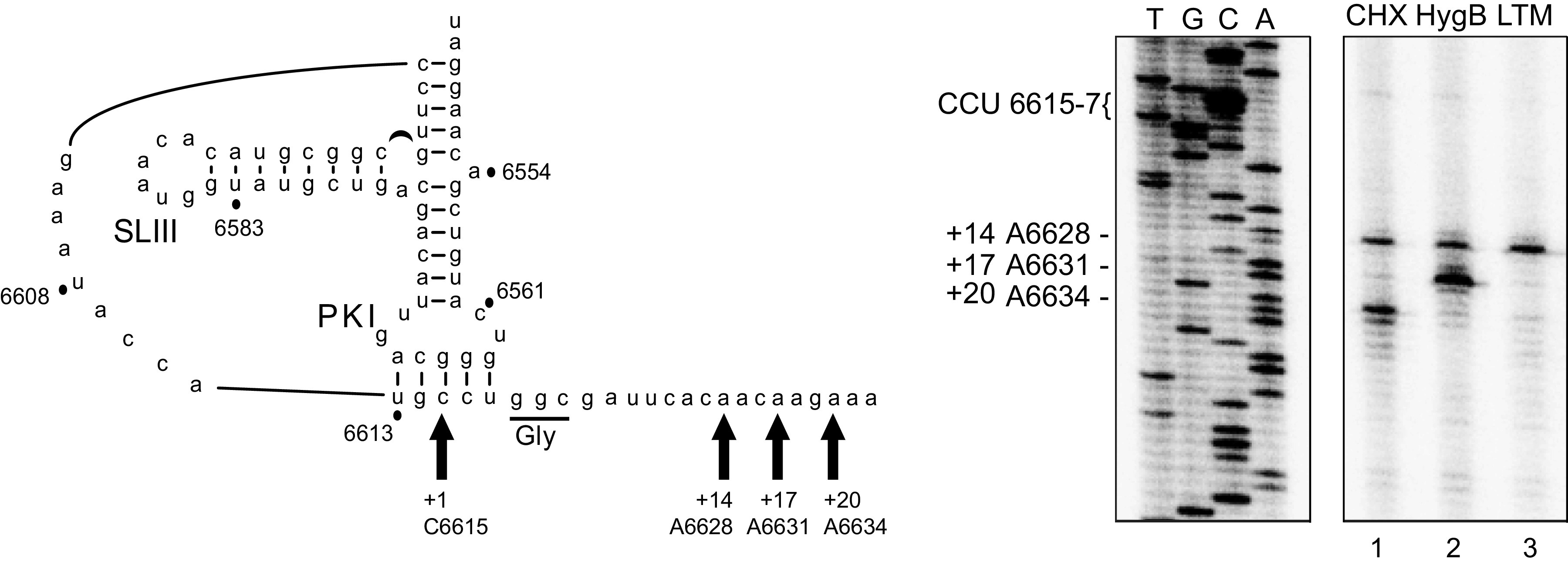
Toeprinting analysis of translocated ribosomes on the IAPV IGR IRES in RRL. (Left) Schematic of the PKI tRNA mimicry domain of the IAPV IGR IRES. Arrows denote the primary toeprints of primer extension reactions (right).
Having verified the use of these inhibitors, translocation was then assessed in hinge mutants IAPV IRESs. In the presence of LTM, as expected, the wild type but not the negative control, ΔPKI IRES, did not produce Toeprint A at A6628 (+14) (Fig. 5A, lanes 1 and 2). Mutating the hinge to GC, UC, AU, U, or AA reduced the A6628 (+14) toeprint by 60–80%, whereas UAA and UGC mutations had only moderate effects, decreasing the toeprint by 20–30% (Fig. 5A, lanes 3–9). In general, the trend of relative toeprint intensities of LTM-treated RRL is similar to that observed using purified ribosomes (Fig. 2 and 4A). For example, the UAA mutant IRESs produced the most intense A6628 toeprints compared to the other mutant IRESs in both LTM-treated RRL and purified ribosomes. Overall, these results support the conclusion that the hinge region of the IRES contributes to proper positioning of ribosomes on the IRES.
FIG 5.

Toeprinting analysis of IAPV IGR IRES in RRL in the presence of translation inhibitors. Bicistronic RNAs containing the wild-type or hinge mutant IGR IRESs were incubated in absence or presence of (A) lactimidomycin D (LTM 5 μM), (B) cycloheximide (CHX 17.8 μM), or hygromycin B (HygB 1 μM). Primer extension analysis was carried out in the presence of [32P]-dATP, and the reaction products were resolved by denaturing polyacrylamide gel electrophoresis and visualized by autoradiography. (Left) Sequencing ladder for the wild-type IRES is shown with location of the major toeprints. Primary detected toeprints are +14, +17, and +20 nucleotides downstream of the CCU 6615-7 in the ribosomal A-site, given that the first C is +1. A representative gel is shown with the indicated drug treatments. (Below) Quantitation of toeprints. Toeprint intensities were measured as a fraction of the radioactive counts for each toeprint over the total radioactive counts within each lane, normalized to that of the respective toeprints of the wild-type IGR IRES. Averages from at least three independent experiments ± SD are shown for the cycloheximide experiments, and averages from two independent experiments are shown for the hygromycin B experiment.
We next examined toeprints of ribosomes on the mutant IRESs in the presence of HygB or CHX (Fig. 5B). As mutations in the hinge region had a defect in initial ribosome positioning, it was not surprising to observe that translocated ribosomes on the IRES, A6631 (+17) and A6634 (+20) toeprints, were all decreased compared to that of the wild-type IRES. As observed, A6631 (+17) and A6634 (+20) toeprint signals from the mutant IRESs were greatly decreased, and followed the trend of the A6628 (+14) toeprint signal observed in both the LTM-treated RRL reaction and purified 80S:IRES reactions (Fig. 2 and 5B). Overall, these support that the identity of the IRES hinge region contributes to proper positioning of ribosomes on the IRES.
We next plotted the translational activity to the A6628 (+14) toeprint intensity of the wild-type and mutant hinge IAPV IRES in RRL (Fig. 6). In general, the toeprint intensities correlated with the translational activity (r = 0.84), indicating that the defect associated with the hinge mutation is at the level of IRES ribosome positioning. However, the correlation of translation and toeprint intensity did not fit with three mutant IRESs, UGC, GC, and UC, suggesting that these mutations may have other effects on IRES function, such as ribosome assembly and/or translocation.
FIG 6.
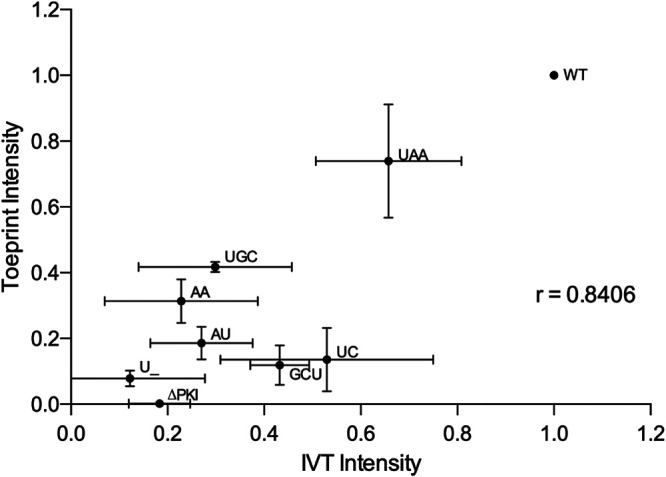
Correlation between the translation activities and toeprint intensities of hinge mutant IAPV IGR IRESs. The mean values of the translation activities and mean intensities of the toeprint AA6628 observed in LTM-treated RRL for each mutant IRES from Fig. 1C and 5A, respectively. Vertical and horizontal error bars represent standard deviations of toeprinting intensities (Fig. 4A) and relative IRES translational activities (Fig. 1C), respectively. Linear regression analysis was performed, and the Pearson correlation coefficient was calculated. P value is two-tailed with 95% confidence interval.
Pseudotranslocation of ribosomes on mutant hinge IRESs.
The inchworm-like movement of translocation of the IRES through the ribosome begins with the IRES in an extended form, which then compacts as PKI translocates from the ribosomal P to A sites per translocation cycle before going back to an extended conformation posttranslocation step (11). Within one of the intermediate steps of translocation, the hinge region of the IRES appears to interact with uL5 and eL42, possibly the hinge domain having a functional role in the initial IRES pseudotranslocation step (11). To address this, we examined whether the hinge mutations had a defect in the timing of translocated intermediates through the sequential addition of inhibitors. To monitor the first IRES-mediated translocation step (the PKI domain movement from the ribosomal A to P site), we incubated wild-type IAPV IRES in RRL in the presence of HygB, and then subsequently added LTM at times starting at 10 s after addition of HygB (Fig. 7). Over the course of the experiment, we predicted to observe an increase in the translocated ribosome-IRES complex toeprint A6631 (+17) and a decrease in the A6628 (+14) toeprint. Surprisingly, we did not observe a relative change in the A6628 (+14) and A6631 (+17) toeprints, which were consistently 90% and 10%, respectively, of the total toeprint signal intensities (Fig. 7). Similarly, addition of LTM and HygB at the beginning of the reaction resulted in a similar intensity of A6628 (+14) and A6631 (+17) toeprints (Fig. 7, lane 1). These results indicate that in the presence of LTM and HygB, the IRES translocated one cycle, leading to a stable ribosome:IRES complex in the ribosomal P site.
FIG 7.

Translocation of the IAPV IRES from the ribosomal A to P sites in the presence of HygB and LTM. Bicistronic RNAs containing the wild-type IGR IRES were incubated in the presence of the HygB (1 μM) and LTM (5 μM) in RRL for 15 min (lane 1). (Lanes 2–9) LTM was added at the indicated times after starting the reaction in the presence of HygB. Primer extension analysis was carried out in the presence of [32P]-dATP. Primer extension products were resolved by denaturing polyacrylamide gel electrophoresis and visualized by autoradiography. Quantitations of toeprints +17 and +14 are shown below at the indicated times of addition of LTM. Fraction of toeprints was calculated by the radioactive counts for toeprint +17 or +14 divided by the total counts of toeprints +14 and +17 which represent the binding and translocation of ribosomes on the IAPV IRES. Shown is a representative gel and quantitation from three independent experiments.
Following the same setup, we monitored the movement of the IRES from the ribosomal P to E site by adding HygB at various times after starting the reaction in the presence of CHX. In the presence of HygB alone, a prominent toeprint is observed at A6631 (+17), indicating that ribosome:IRES complexes are stable with the PKI domain in the ribosomal P site (Fig. 8, lane 2). Upon subsequent addition of CHX starting at 10 s after introduction of HygB, we observed a progressive loss of toeprint A6631 (+17) and an increase in A6634 (+20) toeprint, indicating that ribosomes translocating from the ribosomal P to E sites can be trapped kinetically in RRL. Using this framework, we examined whether mutations within the hinge region of the IRES could have a defect in this step of IRES translocation. We chose the AA mutant IAPV IRES to test as ribosomes assembled on this mutant IRES still produced a modest toeprint intensity in the presence of HygB and CHX (Fig. 8). Similar to the wild-type IRES, the AA mutant IRES showed a gradual decrease in toeprint intensity of A6631 (+17) and an increase in A6634 (+20) toeprint. However, the overall extent of the A6634 (+20) toeprint signal was impaired compared to the wild-type IRES (Fig. 4B). The wild-type and AA mutant IRESs mediate translocation of the IRES from the ribosomal P to E sites at relatively similar rates, thus suggesting that altering the hinge region from AU to AA does not impair this step of IRES pseudotranslocation.
FIG 8.
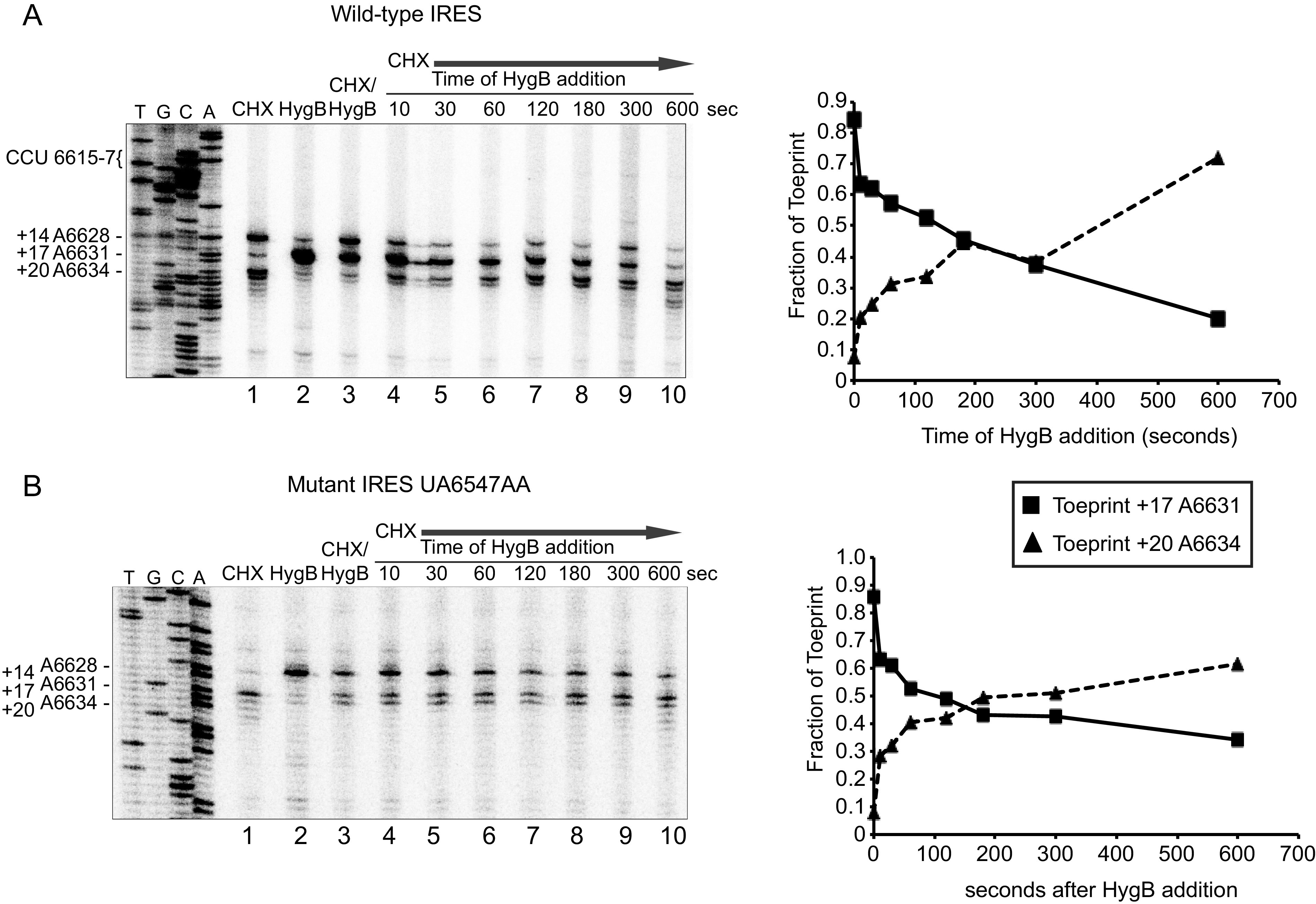
Translocation of wild-type and mutant IAPV IGR IRES from the ribosomal P to E sites in the presence of HygB and CHX. Bicistronic RNAs containing the (A) wild-type or (B) mutant (UA6547-8AA) IGR IRES were incubated in the presence of the indicated inhibitors HygB (1 μM) and CHX (17.8 μM) in RRL for 15 min (lanes 1–3). HygB was added at the indicated times after starting the reaction in the presence of CHX (lanes 4–10). Primer extension analysis was carried out in the presence of α-[32P]-dATP. Primer extension products were resolved by denaturing polyacrylamide gel electrophoresis and visualized by autoradiography. (below) Quantitations of toeprints +17 and +20 are shown below at the indicated times of addition of HygB. Fraction of toeprints was calculated by the radioactive counts for toeprint +17 or +20 divided by the total counts of both toeprints +17 and +20, which represent the binding and translocation of ribosomes on the IAPV IRES.
The hinge region of IAPV IGR IRES is important for virus infection.
We next investigated whether hinge mutations within the IAPV IRES plays a role in virus infection. Towards this, we introduced select IAPV IRES hinge mutations into a recently reported chimeric CrPV infectious clone whereby the CrPV IGR IRES is replaced by the IAPV IGR IRES (chimera CrPV/IAPV clone) (Fig. 9A) (25). We generated CrPV/IAPV chimeras containing the hinge AA and AU mutations, as these mutant IAPV IGR IRESs supported residual IRES translation and as such would be predicted to lead to some productive infection (∼40% of WT, Fig. 1B).
FIG 9.

Mutations of the IAPV hinge region disrupt virus translation in vitro and infection. (A) A hybrid CrPV/IAPV infectious clone was produced by replacing the CrPV IGR IRES in the CrPV infectious clone with the IAPV IGR IRES. (B) In vitro transcribed hybrid CrPV/IAPV containing the wild type (WT, hinge is UA), a mutant M2 (SLVI mutation) that disrupts IAPV IRES activity (25), a stop codon mutation in ORF1 (ORF1 Stop), or mutant hinge sequences AA or AU were incubated in an SF-21 insect cell lysate (2 h) in the presence of S35 labeled methionine/cysteine. Translation products were visualized by SDS-PAGE analysis and autoradiography. (C) Immunoblots of CrPV VP2 and tubulin or (D) viral titers of lysates of S2 cells transfected (48 h) with in vitro transcribed RNA (3 μg) of the indicated hybrid CrPV/IAPV RNAs. The ORF1 stop contains a stop codon within the N-terminus of ORF1, thus preventing ORF1 expression (25). Viral titers were calculated from endpoint dilution analysis. Shown are a representative immunoblot and viral titers from at least three independent experiments ± SD.
We first examined the effects of these hinge mutations in IRES translation of the CrPV structural proteins by incubating in vitro-transcribed chimera CrPV/IAPV RNAs containing wild-type or mutant IAPV IGR IRES in SF-21 insect cell lysates. As shown previously, the hybrid CrPV/IAPV genomic RNA supported translation of both viral nonstructural and structural proteins from ORF1 and ORF2 (25) (Fig. 9B, lane 1). Introducing a mutation that reduces IAPV IRES activity by disrupting stem-loop VI (SLVI) resulted in expression of unprocessed and processed ORF1 proteins (25, 35) (Fig. 9B, lane 2). Similar to that observed using bicistronic reporter RNAs, the mutant hinge CrPV/IAPV viral RNAs (AA and AU) led to reduced translation of processed structural proteins (VP2/VP3), indicating a defect in IAPV IRES function (Fig. 9B, lanes 3 and 4).
To determine if these hinge mutations affected viral infection, we transfected the in vitro-transcribed CrPV/IAPV chimeric RNA in S2 cells and monitored productive infection by immunoblotting for the CrPV structural protein, VP2. Detection of VP2 by immunoblotting is a general readout of productive virus infection (35). As shown previously, the hybrid CrPV/IAPV clone containing the wild-type IAPV IGR IRES but not a mutant clone containing a stop codon within ORF1 (ORF1 stop) led to VP2 expression (Fig. 9C) (35). The CrPV/IAPV clone containing the AA hinge mutant resulted in similar VP2 expression compared to the wild type. By contrast, VP2 was not detected in lysates transfected with the AU hinge mutant CrPV/IAPV chimeric RNA (Fig. 9C). To further validate these results, we monitored viral yield by endpoint dilution. Compared to the wild-type CrPV/IAPV clone, the AA mutant resulted in an approximately 50% decrease in viral yield, whereas the AU mutant did not lead to any virus production (Fig. 9D). These results demonstrate that the identity of the hinge region of the IAPV IGR IRES is necessary for productive virus infection.
Analysis of cryo-EM structures of IRES:ribosome complexes.
How do mutations in the hinge region disrupt proper ribosome positioning on the IAPV IRES? Given that hinge mutations primarily affect the initial ribosome positioning on the IAPV IRES, we reanalyzed the cryo-EM structures of ribosomes complexed with the IAPV and TSV IRESs, particularly focusing on the hinge region of the IRES and uL5/eL42 (11). From the three classes of 80S:IAPV IRES complexes identified (21), the UA6547-8 hinge region occupies the intersubunit space that is too distant to interact with ribosomal components that includes uL5/eL42 and adopts several conformations (Fig. 10A to C, Movie S1 in the supplemental material). Notably, UA6547-8 forms a network of potential hydrogen bond interactions with the preceding A6568, U6451, and G6604 and the 2'OH of the G6604 ribose within the VLR. U6547 is bent inwards like a wedge between A6546 and A6548. This structural analysis explains how mutations of either UA6546-7 disrupt key contacts that likely lead to structural alterations of the PKI domain. For example, mutation or deletion of the wedged U6547 to adenosine or altering A6548 to cytosine, all of which decrease IRES activity (Fig. 1), would alter the network of interactions and potentially disrupt how PKI docks in the ribosomal A site.
FIG 10.

Hinge residues U6547 and A6548 are important for pretranslocation mimicry by the IAPV IRES. Three conformational states of the pretranslocation IAPV IRES in complex with the 80S ribosome determined by cryo-EM are shown (A–C; PDB 6p5i, 6p5j, and 6p5k respectively) (21). These show small conformational changes in the hinge region and relative orientation with respect to uL5 (blue) and eL42 (orange), which do not form any direct contacts in the pretranslocation state (distance between uL5 Arg58 and U6547 indicated). Close analysis of the hinge region as shown in D (boxed in C) reveals that U6547 adopts a tight “wedge” structure to facilitate the turn in the RNA strand and subsequent positioning of the PKI domain. Both U6547 and A6548 form a specific network of potential hydrogen bond interactions with surrounding nucleotides. Also see Movie S1.
In the analysis of the 80S:TSV IRES complex intermediates that undergo the first eEF2-mediated pseudotranslocation step, it is clear as reported (11) that uL5/eL42 approach the hinge region (nucleotides 6886–6890), as the IRES mediates an inchworm-like bending state (Fig. S1, Movie S2). However, closer inspection showed that the upstream nucleotides, GA6686-7, interact with uL5/eL42, whereas the hinge region AA6889-90 is pointing away from uL5/eL42. A6889 (equivalent to U6547 of the IAPV IRES hinge domain) flips toward PKI before the IRES compresses into the inchworm-like bent state. When these findings are extrapolated to the IAPV IRES, these structural analyses explain why mutations within the hinge region UA6547-8 of the IAPV IRES do not affect translocation but rather play a role in a prior event in proper ribosome assembly on the IRES.
DISCUSSION
Dicistroviruses have evolved a streamlined mechanism to recruit the host ribosome for viral protein synthesis. Here, we show that the nucleotide identity and length of the hinge region that separates the IRES ribosome binding domain from the tRNA-like anticodon PKI domain are necessary for robust translation in an in vitro system. Moreover, we show that mutating the wild-type UA hinge sequence of the IAPV IGR IRES in a chimeric CrPV infectious clone prevents viral translation and replication in Drosophila S2 cells.
Cryo-EM structures of the TSV IGR IRES translocating through the ribosome reveal five distinct conformations (11), including a conformational intermediate that the hinge region of the IRES is in proximity with ribosomal protein uL5 and the C-terminus of eL42. We predicted that mutations in the hinge region may disrupt this interaction and prevent the first IRES pseudotranslocation step of PKI movement from ribosomal A to P site. We showed that mutation of the native IAPV UA hinge region decreases IRES translation in vitro (Fig. 1). Surprisingly, toeprinting analysis revealed a decrease in proper ribosome positioning (i.e., PKI stably in ribosomal A site) of initial ribosomes assembled on IRES hinge mutants, thus suggesting a defect in a step prior to translocation (Fig. 2 and 5). Furthermore, the hinge mutations do not affect 80S recruitment, indicating that the overall structure of the ribosome binding domain remains intact (Fig. 3). Importantly, the decrease in translational activity observed in most of the IRES hinge mutants can be attributed to a defect in initial ribosomal positioning on the IRES (Fig. 6).
The hinge region varies between 1 and 3 nucleotides in dicistrovirus IRESs. One potential hypothesis is that the hinge region length must match with the correct PKI of each IRES in order to adopt the proper space and alignment for PKI in the ribosomal A site. In support of this, our mutation analysis shortening or lengthening the hinge region decreases IRES translation (Fig. 1). However, IRES translation defects observed in hinge mutants that do not alter the hinge region length but instead alter the nucleotide identities hints at a more specific role of the hinge region. For example, changing the wild-type UA to AU or to AA disrupts IRES translation and the A6628 (+14) toeprint (Fig. 1, 2, and 4). Our structural analysis of the hinge region in IAPV IRES:ribosome complexes indicates that the UA6547-8 hinge region forms a network of interactions with surrounding nucleotides that undergo several conformations (Fig. 10). It is possible that the nucleotide identities of the hinge region may be specific for each IRES in order to adopt a distinct network of interactions with surrounding nucleotides.
In light of the new structural and biochemical insights into the hinge region of the IAPV IRES, mutations in UA6547-8 likely disrupt key interactions with surrounding bases including A6568, U6451, and G6604 and the 2′ hydroxyl of the G6604 ribose within the VLR that alter the conformational dynamics of the hinge region and the PKI domain, thus disrupting the initial steps of IRES translation. We propose that the identity of the hinge region allows for an optimal conformation(s) of the PKI domain such that it is stably docked within the decoding ribosomal A site for initiation of IRES translation. Specifically, the anticodon:codon mimic of the PKI domain is docked in the decoding A site with stabilizing interactions with the decoding bases 18S rRNA A1824 and A1825 and G626 (21). These stabilizing PKI interactions with the small ribosomal subunit lead to detection of the A6628 (+14) toeprint (i.e., proper ribosome positioning). On the other hand, mutations of the hinge region allow the PKI domain to be less conformationally constrained, leading the PKI domain to dynamically shift in and out of the decoding ribosomal A site in an undocked state or shift back and forth between the A and P sites (i.e., backtranslocate), whereby both scenarios would lead to decreased A6628 (+14) toeprints (Fig. 2 and 4). Investigations using higher resolution detection methods are needed in order to fully capture the effects of the hinge mutants on the PKI domain dynamics within the ribosome.
Proper ribosome positioning on IAPV IGR IRES is affected by the identity and function of various structures of the IRES, including the VLR and L1.1 domains, as well as SLVI of the IAPV IRES (14, 25, 28). In particular, previous studies inducing mutations in the L1.1 region within the PKII and PKIII domains suggest relatively long-range communication to the PKI domain for coordination to promote IRES function and ribosomal positioning. In sum, multiple domains within the IRES likely coordinate how the PKI anticodon–codon mimic is stably docked within the decoding A site.
Translation inhibitors were also explored in this study, showing that CHX and HygB were able to trap stable IRES:ribosome complexes during pseudotranslocation. Previous studies showed that LTM block dicistrovirus IGR IRES translation by inhibiting eEF2-mediated translocation (32). Specifically, LTM arrests translation at the first IRES pseudotranslocation step without affecting tRNA binding or peptide formation by trapping PKI in the A site and only binds to the E site if it is empty. Interestingly, our toeprinting results with successive addition of LTM in the presence of HygB (Fig. 7) indicated that this combination was not productive to arrest pseudotranslocation. It is possible that in the toeprinting reactions, LTM may not be bound stably to the ribosome to trap initial ribosomal assembly prior to pseudotranslocation, or that LTM is ineffective in binding to the ribosome in the presence of HygB. Therefore, the use of toeprinting analysis is likely not sensitive enough to monitor and delineate the kinetic effects of LTM on IRES translocation. Furthermore, it is probable the time of addition of or action of antibiotics in this system cannot resolve the hinge-induced effects on translocation.
Comprehensive mutagenesis on the IGR IRES has allowed for systematic delineation of specific functions within the IRES. Along with structural studies, molecular and biochemical studies have converged on a unifying model for IGR IRES translation (9, 11). Even though the hinge region is not conserved at the nucleotide level and initially appeared to be innocuous, our study further highlights the importance of likely all regions of the viral IRES in manipulating the ribosome. Given the limited coding capacity and limited genome size of dicistroviridae, and viruses more generally, our findings further extend the idea that the virus has evolved to maximize the nucleotide space for optimal viral translation and replication.
MATERIALS AND METHODS
DNA reporter constructs.
The bicistronic luciferase chimeric reporter constructs containing the IAPV IGR IRES have been previously described (31). Construction of the chimera CrPV/IAPV infectious clone has been previously described (35). IRES mutations were generated using PCR-based mutagenesis. All constructs and clones were sequence verified. CrPV (ORF1-stop) contains a UAA stop codon (C775T, G778T; numbering of nucleotides is based on CrPV-3 infectious clone KP974707).
In vitro transcription and translation.
Bicistronic DNA constructs containing the wild-type or mutant IRESs were linearized using XbaI. RNAs were transcribed in vitro using T7 RNA polymerase and purified using a RNeasy kit (Qiagen). RNA purity and integrity were confirmed by denaturing formaldehyde agarose gel electrophoresis. For in vitro translation assays, 3 μg of purified RNA was first prefolded at 65°C and then slow cooled to room temperature in buffer E (20 mM Tris hydrochloride [pH 7.5], 100 mM potassium acetate, 2.5 mM magnesium acetate, 0.25 mM spermidine, and 2 mM dithiothreitol) and then incubated in Spodoptera frugiperda (SF-21) extract (Promega). Reactions were analyzed by SDS-PAGE analyzed by PhosphorImager analysis (Typhoon, Amersham). Luciferase protein levels were analyzed by [35S]-methionine incorporation.
Purification of 40S and 60s ribosomal subunits.
We purified 40S and 60S ribosomal subunits from HeLa cell pellets (National Cell Culture Center) as previously described (14). Briefly, HeLa cells were lysed in Triton-X lysis buffer (15 mM Tris-HCl (pH 7.5), 300 nM NaCl, 1% (vol/vol) Trito X-100, 6 mM MgCl2, 1 mg/mL heparin), and the supernatant was subjected to brief centrifugation to remove cellular debris. The supernatant was applied to a 30% (wt/wt) sucrose cushion containing 0.5M KCl and centrifuged at 100,00 g to pellet ribosomes. Ribosomes were resuspended in buffer B (20 mM Tris-HCl [pH 7.5], 6 mM magnesium acetate, 150 mM potassium acetate, 6.8% (wt/wt) sucrose, 2 mM dithiothreitol) and subsequently treated with puromycin (2.3 mM final concentration) to dissociate ribosomes from the mRNAs. Potassium chloride was to obtain a final concentration of 0.5 M. The dissociated ribosomes were resolved on a 10–30% (wt/wt) sucrose gradient, and the corresponding fractions containing 40S and 60S subunits were collected, pooled, and concentrated in buffer C (20 mM Tris-HCl [pH 7.5], 0.2 mM EDTA, 10 mM KCl, 1 mM, MgCl2, 6.8% [wt/wt] sucrose) using Amicon Ultra spin concentration (Millipore). The concentrations of the ribosomal subunits were determined by spectrophotometry using the conversions 1 A260 = 50 nM and 1 A260 = 25 nM for the 40S and 60S subunits, respectively.
Toeprinting/primer extension analysis.
Toeprinting analysis of ribosomal complexes in RRL was performed as previously described (30). of Bicistronic IGR IRES RNAs (400 ng) were annealed to PrEJ 761 (5′-CATGGGGGTATCGATCCTATTTGGAG-3′) in 40 mM Tris (pH 7.5) and 0.2 mM EDTA by slow cooling from 65°C to 30°C. Annealed RNAs were incubated in RRL that was preincubated with 20 μM amino acid mix, 8 units of Ribolock (Fermentas), and 154 nM final concentration of potassium acetate (pH 7.5). Drug concentrations that were used: 17.8 μM cycloheximide; 1 μM hygromycin B; 5 μM of lactimidomycin. Reactions were incubated at 30°C for 15 min. Ribosome positioning was determined by primer extension using 5 units of AMV reverse transcriptase (Promega) in the presence of 415 μM each of dTTP, dGTP, dCTP, and 83 μM dATP, 0.33 μL of α-[32P] dATP (3.33 μM, 3000 Ci/mmol), and 6.7 mM MgOAc. Toeprinting and primer extension analysis using purified 80S ribosomes were performed in a similar manner using 150 ng of bicistronic RNA and 100 nM and 150 nM final concentrations of 40S and 60S ribosome subunits, respectively, at 30°C. Following reverse transcription for 1 h at 30°C, the samples were extracted by phenol chloroform (twice) and chloroform (once) and ethanol-precipitated. The cDNA was analyzed under denaturing conditions on 6% (wt/vol) polyacrylamide/8M urea gels, which were subsequently dried and subjected to PhosphorImager analysis.
Ribosome binding.
One picomole of 5′-end-labeled IRES RNA was incubated with increasing amounts of purified 40S, a 1.5-fold excess of 60S, and 25 ng/μL of noncompetitor RNA in buffer E (20 mM Tris hydrochloride [pH 7.5], 100 mM potassium chloride, 2.5 mM magnesium chloride, 0.25 mM spermidine, 2 mM dithiothreitol). The reaction mixtures were incubated for 20 min at room temperature, after which they were applied to a double membrane of nitrocellulose and nylon using a Bio-Dot filtration apparatus (Bio-Rad). The membranes were subsequently dried and subjected to autoradiography.
In vitro transcription, RNA transfection, and viral titers.
Plasmids containing the CrPV and chimeric CrPV/IAPV infectious clone were linearized with Ecl136II as described (35). RNA was transcribed using a T7 RNA polymerase reaction and then purified with an RNeasy kit (Qiagen). The integrity of the RNA was confirmed on a 1% denaturing formaldehyde agarose gel.
Transfection of in vitro synthesized RNA (3 μg) into 2.5 × 106 S2 cells was performed using Lipofectamine 3000 (Invitrogen) as per the manufacturer’s instructions. Viral titers were performed as described (35).
Western blots.
Equal amounts of S2 protein lysates were resolved on a 12% SDS-PAGE gel and then transferred onto a polyvinylidene difluoride Immobilon-FL membrane (Millipore). Membranes were blocked for 1 h at room temperature in 5% skim milk in TBST (50 mM Tris, 150 mM NaCl, 1% Tween 20, pH 7.4). Membranes were then incubated with the CrPV ORF2 (raised against CrPV VP2) rabbit polyclonal antibody (1:10000, Genscript) and mouse antitubulin antibody (1:1000; Santa Cruz). Membranes were washed with TBST three times. Membranes were then incubated with IRDye 680CW goat antimouse (1:10000; Li-Cor Biosciences) or IRDye 800CW goat antirabbit (1:20000; Li-Cor Bioscience) for 1 h at room temperature. Detection was performed on an Odyssey imager (Li-Cor Biosciences).
ACKNOWLEDGMENTS
We thank Jan and Allan labs for critical analysis and reading of the manuscript. We acknowledge that Che-Min Lee generated a subset of initial hinge mutant reporter constructs. This work was supported by CIHR grants to E.J. (PJT-148761), to D.W.A. (20R76320), and to N.C.J.S.
Footnotes
Supplemental material is available online only.
Contributor Information
Eric Jan, Email: ej@mail.ubc.ca.
Anne E. Simon, University of Maryland, College Park
REFERENCES
- 1.Jan E, Mohr I, Walsh D. 2016. A cap-to-tail guide to mRNA translation strategies in virus-infected cells. Annu Rev Virol 3:283–307. 10.1146/annurev-virology-100114-055014. [DOI] [PubMed] [Google Scholar]
- 2.Jang SK, Krausslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E. 1988. A segment of the 5' nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol 62:2636–2643. 10.1128/JVI.62.8.2636-2643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pelletier J, Sonenberg N. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334:320–325. 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- 4.Mailliot J, Martin F. 2018. Viral internal ribosomal entry sites: four classes for one goal. Wiley Interdiscip Rev RNA 9:e1458. 10.1002/wrna.1458. [DOI] [PubMed] [Google Scholar]
- 5.Bonning BC, Miller WA. 2010. Dicistroviruses. Annu Rev Entomol 55:129–150. 10.1146/annurev-ento-112408-085457. [DOI] [PubMed] [Google Scholar]
- 6.Warsaba R, Sadasivan J, Jan E. 2020. Dicistrovirus-host molecular interactions. Curr Issues Mol Biol 34:83–112. 10.21775/cimb.034.083. [DOI] [PubMed] [Google Scholar]
- 7.King LA, Moore NF. 1988. Evidence for the presence of a genome-linked protein in two insect picornaviruses, cricket paralysis and Drosophila C viruses. FEMS Microbiology Lett 50:41–44. 10.1111/j.1574-6968.1988.tb02908.x. [DOI] [Google Scholar]
- 8.Johnson AG, Grosely R, Petrov AN, Puglisi JD. 2017. Dynamics of IRES-mediated translation. Philos Trans R Soc Lond B Biol Sci 372:20160177. 10.1098/rstb.2016.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez IS, Bai XC, Murshudov G, Scheres SH, Ramakrishnan V. 2014. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell 157:823–831. 10.1016/j.cell.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muhs M, Hilal T, Mielke T, Skabkin MA, Sanbonmatsu KY, Pestova TV, Spahn CM. 2015. Cryo-EM of ribosomal 80S complexes with termination factors reveals the translocated cricket paralysis virus IRES. Mol Cell 57:422–432. 10.1016/j.molcel.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abeyrathne PD, Koh CS, Grant T, Grigorieff N, Korostelev AA. 2016. Ensemble cryo-EM uncovers inchworm-like translocation of a viral IRES through the ribosome. Elife 5:e14874. 10.7554/eLife.14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pisareva VP, Pisarev AV, Fernández IS. 2018. Dual tRNA mimicry in the cricket paralysis virus IRES uncovers an unexpected similarity with the Hepatitis C virus IRES. Elife 7:e34062. 10.7554/eLife.34062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh CS, Brilot AF, Grigorieff N, Korostelev AA. 2014. Taura syndrome virus IRES initiates translation by binding its tRNA-mRNA-like structural element in the ribosomal decoding center. Proc Natl Acad Sci USA 111:9139–9144. 10.1073/pnas.1406335111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang CJ, Lo MC, Jan E. 2009. Conserved element of the dicistrovirus IGR IRES that mimics an E-site tRNA/ribosome interaction mediates multiple functions. J Mol Biol 387:42–58. 10.1016/j.jmb.2009.01.042. [DOI] [PubMed] [Google Scholar]
- 15.Pfingsten JS, Costantino DA, Kieft JS. 2006. Structural basis for ribosome recruitment and manipulation by a viral IRES RNA. Science 314:1450–1454. 10.1126/science.1133281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jan E, Sarnow P. 2002. Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J Mol Biol 324:889–902. 10.1016/s0022-2836(02)01099-9. [DOI] [PubMed] [Google Scholar]
- 17.Au HH, Cornilescu G, Mouzakis KD, Ren Q, Burke JE, Lee S, Butcher SE, Jan E. 2015. Global shape mimicry of tRNA within a viral internal ribosome entry site mediates translational reading frame selection. Proc Natl Acad Sci USA 112:E6446–E6455. 10.1073/pnas.1512088112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costantino DA, Pfingsten JS, Rambo RP, Kieft JS. 2008. tRNA-mRNA mimicry drives translation initiation from a viral IRES. Nat Struct Mol Biol 15:57–64. 10.1038/nsmb1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson JE, Pestova TV, Hellen CU, Sarnow P. 2000. Initiation of protein synthesis from the A site of the ribosome. Cell 102:511–520. 10.1016/s0092-8674(00)00055-6. [DOI] [PubMed] [Google Scholar]
- 20.Murray J, Savva CG, Shin BS, Dever TE, Ramakrishnan V, Fernández IS. 2016. Structural characterization of ribosome recruitment and translocation by type IV IRES. Elife 5:e13567. 10.7554/eLife.13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acosta-Reyes F, Neupane R, Frank J, Fernández IS. 2019. The Israeli acute paralysis virus IRES captures host ribosomes by mimicking a ribosomal state with hybrid tRNAs. EMBO J 38:e102226. 10.15252/embj.2019102226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jang CJ, Jan E. 2010. Modular domains of the Dicistroviridae intergenic internal ribosome entry site. RNA 16:1182–1195. 10.1261/rna.2044610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakashima N, Uchiumi T. 2009. Functional analysis of structural motifs in dicistroviruses. Virus Res 139:137–147. 10.1016/j.virusres.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Hertz MI, Thompson SR. 2011. In vivo functional analysis of the Dicistroviridae intergenic region internal ribosome entry sites. Nucleic Acids Res 39:7276–7288. 10.1093/nar/gkr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Au HHT, Elspass VM, Jan E. 2018. Functional insights into the adjacent stem-loop in honey bee dicistroviruses that promotes internal ribosome entry site-mediated translation and viral infection. J Virol 92:e01725-17. 10.1128/JVI.01725-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costantino D, Kieft JS. 2005. A preformed compact ribosome-binding domain in the cricket paralysis-like virus IRES RNAs. RNA 11:332–343. 10.1261/rna.7184705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruehle MD, Zhang H, Sheridan RM, Mitra S, Chen Y, Gonzalez RL, Cooperman BS, Kieft JS. 2015. A dynamic RNA loop in an IRES affects multiple steps of elongation factor-mediated translation initiation. Elife 4:e08146. 10.7554/eLife.08146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Au HH, Jan E. 2012. Insights into factorless translational initiation by the tRNA-like pseudoknot domain of a viral IRES. PLoS One 7:e51477. 10.1371/journal.pone.0051477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kerr CH, Ma ZW, Jang CJ, Thompson SR, Jan E. 2016. Molecular analysis of the factorless internal ribosome entry site in cricket paralysis virus infection. Sci Rep 6:37319. 10.1038/srep37319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang QS, Au HH, Jan E. 2013. Methods for studying IRES-mediated translation of positive-strand RNA viruses. Methods 59:167–179. 10.1016/j.ymeth.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 31.Ren Q, Wang QS, Firth AE, Chan MM, Gouw JW, Guarna MM, Foster LJ, Atkins JF, Jan E. 2012. Alternative reading frame selection mediated by a tRNA-like domain of an internal ribosome entry site. Proc Natl Acad Sci USA 109:E630–E639. 10.1073/pnas.1111303109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6:209–217. 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holtkamp W, Cunha CE, Peske F, Konevega AL, Wintermeyer W, Rodnina MV. 2014. GTP hydrolysis by EF-G synchronizes tRNA movement on small and large ribosomal subunits. EMBO J 33:1073–1085. 10.1002/embj.201387465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borovinskaya MA, Shoji S, Fredrick K, Cate JH. 2008. Structural basis for hygromycin B inhibition of protein biosynthesis. RNA 14:1590–1599. 10.1261/rna.1076908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kerr CH, Wang QS, Keatings K, Khong A, Allan D, Yip CK, Foster LJ, Jan E. 2015. The 5' untranslated region of a novel infectious molecular clone of the dicistrovirus cricket paralysis virus modulates infection. J Virol 89:5919–5934. 10.1128/JVI.00463-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1<br>. Download jvi.01330-21-s0001.mov, MOV file, 18.3 MB (18.8MB, mov)
Movie S2<br>. Download jvi.01330-21-s0002.mov, MOV file, 11.2 MB (11.5MB, mov)
Fig. S1 and Supplemental Movie Legends<br>. Download jvi.01330-21-s0003.pdf, PDF file, 3.7 MB (3.8MB, pdf)