

Abstract
Whole genome amplification (WGA) is a technology for non‐selective amplification of the whole genome sequence, first appearing in 1992. Its primary purpose is to amplify and reflect the whole genome of trace tissues and single cells without sequence bias and to provide sufficient DNA template for subsequent multigene and multilocus analysis, along with comprehensive genome research. WGA provides a method to obtain a large amount of genetic information from a small amount of DNA and provides a valuable tool for preserving limited samples in molecular biology. WGA technology is especially suitable for forensic identification and genetic disease research, along with new technologies such as next‐generation sequencing (NGS). In addition, WGA is also widely used in single‐cell sequencing. Due to the small amount of DNA in a single cell, it is often unable to meet the amount of samples needed for sequencing, so WGA is generally used to achieve the amplification of trace samples. This paper reviews WGA methods based on different principles, summarizes both amplification principle and amplification quality, and discusses the application prospects and challenges of WGA technology in molecular diagnosis and medicine.
Keywords: genomic DNA, isothermal amplification, molecular diagnosis, polymerase chain reaction, whole genome amplification
Whole genome amplification (WGA) is a technology for non‐selective amplification of the whole genome sequence. This paper reviews WGA methods based on different principles, summarizes both amplification principle and amplification quality, and discusses the application prospects and challenges of WGA technology in molecular diagnosis and medicine.
1. INTRODUCTION
In recent years, high‐throughput technologies such as DNA chips and NGS have greatly promoted the development and application of molecular diagnostic technology in various fields. However, the quantity and quality of nucleic acid samples are often important factors restricting the research. There are still many problems in obtaining high‐quality DNA samples as the first step of the work. WGA technology can effectively solve this problem by maximizing limited amounts of DNA as starting material while also representing the whole for high‐throughput analysis. 1 Requiring small amounts of starting material has allowed researchers to utilize WGA technology in single‐cell research. 2 , 3 Single‐cell WGA principle is to amplify and sequence the whole genome DNA of the single cell with high‐throughput sequencing. 4
WGA technology is divided into three categories: First is polymerase chain reaction (PCR) 5 such as primer extension preamplification PCR (PEP‐PCR), degenerate oligonucleotide primer PCR (DOP‐PCR), 6 tagged random primer PCR (T‐PCR), long and accurate PCR (LA‐PCR), ligation‐mediated PCR (LM‐PCR), interspersed repetitive sequence PCR (IRS‐PCR). The second category is isothermal amplification, 7 such as multiple displacement amplification (MDA), 8 multiple annealing and looping‐based amplification cycles (MALBAC), 9 and linear amplification via transposon insertion (LIANTI). 10 The final category is microfluidic amplification. 11 This paper will introduce these methods from the aspects of principle and application and summarize the advantages and disadvantages of each method. 12
2. PCR WGA TECHNOLOGY
2.1. PEP‐PCR
Among all the WGA methods, PEP‐PCR has been around the longest. 13 PEP uses random primers and genomic DNA (gDNA) to randomly amplify the whole gDNA. 14 The random primers are composed of 15 random nucleotides with 4 sequences. 15 , 16 During the amplification, the primers randomly anneal to a large number of gDNA sites starting at 37℃ and extend continuously to 55℃ to ensure that the random primers anneal to as many genomic sequences as possible. 17 After 50 cycles, around 75% of the gDNA is amplified. 18 Single‐cell PEP amplifies around 70% of the whole genome DNA 19 while using random primers and less intensive PCR cycle parameters, which may lead to uneven amplification results. 20
2.2. DOP‐PCR
DOP‐PCR is commonly used for samples with limited quantity to increase the amount of DNA yielded. 21 The schematic diagram of DOP‐PCR amplification is shown in Figure 1. DOP‐PCR uses non‐selective amplification to achieve whole genome sequencing. The principle of DOP‐PCR is similar to that of PEP, except that a fixed sequence with high frequency is added to the two ends of the six random primer sequences. The 3'‐terminal ATGTGG (A, adenine; T, thymine; G, guanine) plays a guiding role during annealing, while the six random sequences at the center play a role in strengthening the binding of primers and DNA, and the CCGACTCGAG (C, cytosine) sequence at the 5' end is used for terminal modification. DOP‐PCR consists of 50 cycles, with the annealing temperature of each cycle continuously rising, ensuring that different primers and as many genomic sequences as possible are annealed so that the whole genome can be amplified evenly in proportion. 22 , 23
FIGURE 1.
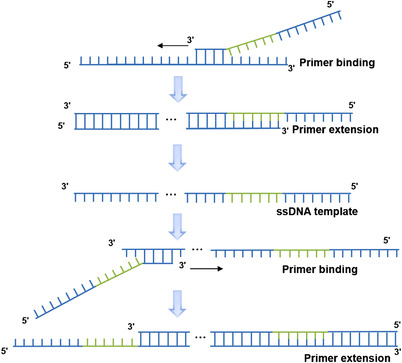
Polymerase chain reaction (PCR) reaction procedure of degenerate oligonucleotide primed PCR. The degenerate oligonucleotide primers are used in whole genome amplification. ssDNA template: single‐stranded DNA template (reproduced from Huang et al. 23 with permission from Annual Reviews)
Due to its exponential amplification, the gene coverage of DOP‐PCR technology is typically low. 24 Any slight differences in amplification factors between different sequences will be amplified exponentially, contributing to over‐amplified and under amplified areas in the genome, ultimately giving a low coverage rate. 25 This is due to the poor uniformity of amplification caused by exponential amplification, which worsens the coverage. However, this method can amplify an initial starting material of pg level, so when the initial amount of amplification template is very low, it can be used for pre‐amplification. Although DOP‐PCR lacks completeness in obtaining the whole genome, it can be applied to the measurement of copy number variation (CNV) in a large genome range (1 million base pairs). 26
There are multiple factors that can influence PCR amplification. The concentration of primers, template size, guanine‐cytosine (GC) content, and DNA secondary structure will affect the amplification efficiency of polymerase and any deficiencies can affect the whole genome coverage. 27 All of the above‐mentioned factors could lead to uneven amplification or non‐specific amplification and ultimately result in amplification bias. 28
2.3. T‐PCR
T‐PCR is a WGA technology, 29 which was first reported by Jeffreys in 1991. The 3' end of the primer is a random primer with 9–15 bases, and the 5' end is a fixed marker sequence with 17 bases. The reaction is divided into two steps: During the first step, 3' random primers are hybridized to the DNA template at 30–40°C and extended under the guidance of the Taq enzyme; in the second step, the newly synthesized DNA strand is used as a template with labeled base sequences at both ends; after 2–5 cycles, the unbound primers and primer dimers are removed by centrifugation. Finally, the PCR primers complementary to the marker sequence are added to amplify the products of the first step. Multiple replications can amplify the whole genome. 30
T‐PCR has the advantages of high amplification efficiency and strong specificity 29 but remains challenging to optimize the concentration of random primers. 31 Primer dimers that are unbound are washed away in the first step. 32 The initial template can be lost during the first washing step, and later, purification could lead to the loss of alleles. Thus, this is typically performed by a skilled technician.
2.4. LA‐PCR
There are two types of heat‐resistant DNA polymerase used in PCR technology 33 : The first does not contain 3'‐5' exonuclease activity, including Taq DNA polymerase, klentaq1 DNA polymerase, and pfu (exo‐) DNA polymerase; while the second contains 3'‐5' exonuclease activity, including pfu DNA polymerase, Vent DNA polymerase, and Deep Veep DNA polymerase, among others. The thermostable Taq DNA polymerase commonly used in PCR has a strong elongation ability, but it cannot correct the mismatched bases introduced in the process of amplification. The mismatched bases at the 3' end affect the smooth progress of the extension in the amplification process 34 so that the extension terminates ahead of time, thus limiting the length of the amplification fragment. Pfu DNA polymerase can replace Taq DNA polymerase (the fidelity of pfu DNA polymerase is 5–10 times higher than that of Taq DNA polymerase) while significantly reducing the production of mismatch bases, although studies have shown that a polymerase with exonuclease activity, 35 such as pfu, Vent and Deep Vent, cannot effectively amplify long fragments alone. This may be due to the enzyme degrading the primers during the long‐term amplification process due to the 3'‐5' exonuclease, which has high activity. Only the low level of 3'‐5' exonuclease activity can successfully amplify the long fragment. Based on this principle, a high level of thermostable DNA polymerase without exonuclease activity and a small amount of DNA polymerase with exonuclease activity were mixed, and the extension ability of the former and the correction ability of the latter were used to successfully amplify the long fragments of the two kinds of enzymes under appropriate proportions and reaction conditions. Presently, the mixed enzymes used in LA‐PCR include thermostable deoxyuridine 5'‐triphosphate pyrophosphatase (dUTPase) in addition to the above two enzymes, one enzyme does not contain 3'‐5' exonuclease activity and the other enzyme contains 3'‐5' exonuclease activity. 36 The deamination of deoxycytidine triphosphate (dCTP) will occur in the process of PCR and deoxyurodine triphosphate (dUTP). DNA polymerase with correction function irreversibly binds to deoxyuracil in vitro, which affects the efficiency of the enzyme and leads to the failure of LA‐PCR amplification. dUTPase will degrade dUTP, and effectively prevent dUTP from participating in the synthetic DNA during the PCR process. Hogrefe et al. found that heat‐resistant dUTPase can significantly improve the efficiency of pfu DNA polymerase amplification. 37 Hot start enzyme 38 has been widely used in LA‐PCR. 39 Hot start helps prevent the non‐specific amplification caused by the mismatch of primers or the formation of primer dimer in the PCR reaction, thus improving the amplification efficiency of target DNA fragments.
LA‐PCR technology overcomes the shortcomings of traditional PCR technology, such as short amplification fragment (often below 3 kb) and low fidelity. Its amplification length can exceed 10 times of traditional PCR technology and can survive longer reaction times. 40
2.5. LM‐PCR
LM‐PCR is a unilateral PCR technique 41 consisting of five steps. The first step uses chemical cleavage to cleave gDNA at specific sites 42 to produce 5'‐phosphate molecules. The second step uses the DNA fragments produced in the first step as a template and uses primer extension reactions guided by genome‐specific primers to produce flat‐end DNA molecules. During the third step, a common linker connects to the flat‐end DNA molecule produced above. The fourth step uses genome‐specific second primers and linker primers for PCR amplification of the ligated products. Finally, the labeled genome‐specific third primer was used to extend the PCR product, followed by visual electrophoresis and sequencing. The primers or probes can also be engineered to bind T7 RNA polymerase transcription to ligation as shown in Figure 2. Thus, a combined DNA strand consisting of two DNA probes 43 can be used as a transcription template reaction following the PCR process shown in Figure 2.
FIGURE 2.
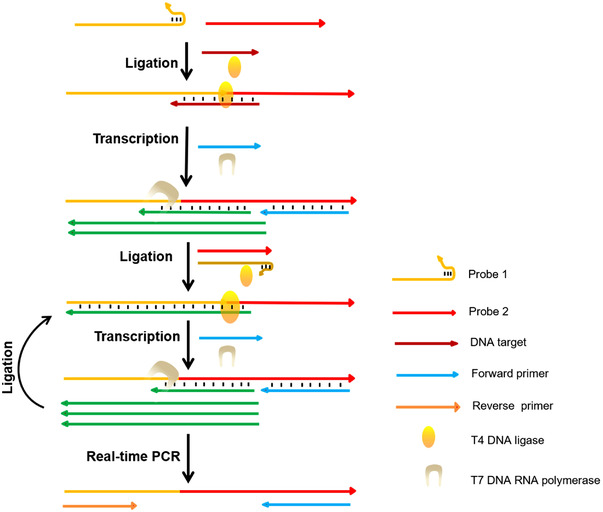
Short‐length DNA detection by using T7 RNA polymerase with LM‐PCR (reproduced from Yu et al. 44 with permission from American Chemical Society)
LM‐PCR generally adopts the method of three primers, and the connection between the three is that the extension of the second primer is located at the 3' end of the first primer, and so is the connection between the third primer and the second primer. This design greatly enhances the specificity of the reaction. 45
When using LM‐PCR for in vivo foot printing or sequencing, we are most concerned about the following three aspects: sensitivity, fidelity, and convenience of primer design. According to the principle of LM‐PCR, only when the third labeled primer competes effectively with the second primer in the PCR reaction, and annealing of PCR products template, the PCR product can be efficiently labeled by the labeled primer. When designing the third marker primers, a partial overlap should be left between the third primer and the second primer, and the melting temperature (Tm) value of the third primer should be higher than that of the second primer. This requirement greatly limits the design of PCR primers in the application of this technique, 46 especially when studying the interaction between DNA proteins rich in adenine‐thymine (AT) regulatory sequences and determining the nucleotide sequence.
2.6. IRS‐PCR
IRS is a widely distributed and highly conserved interval repeat sequence in gDNA. 47 The primer synthesized IRS‐PCR amplifies DNA between two IRS sequences. Alu sequence is a human unique IRS sequence, which contains about 900,000 copies in the human haploid genome, with an average interval of 4 kb. Due to the uneven distribution of Alu sequences in the human genome and different repetition times, the amplification efficiency of Alu‐PCR varies greatly in different regions of the genome, causing uneven amplification products, which limits its application.
3. ISOTHERMAL AMPLIFICATION TECHNOLOGY
3.1. MDA
MDA is a WGA technique developed in recent years. It is the most popular method of whole genome isothermal amplification at present. 48 MDA uses DNA polymerase with high extension activity and random primers resistant to exonuclease to amplify the genome under isothermal conditions. This method is based on chain substitution and was originally used to amplify huge circular DNA such as a plasmids or bacteriophage DNA. Now it is also used to amplify linear DNA. 49 As shown in Figure 3, it can uniformly amplify the whole genome with high fidelity, amplify 10 to 100 kb sized fragments, and provide a large number of uniform and complete whole genome sequences. 50 It can be amplified by random primers resistant to exonuclease activity and template annealing under the action of φ29 DNA polymerase with strong strand replacement activity and exonuclease activity. 51
FIGURE 3.
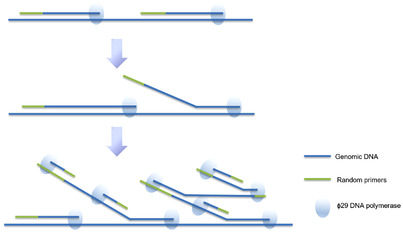
Multiple displacement amplification of genomic DNA. This reaction is an isothermal amplification reaction using random primers for φ29 DNA polymerase. (reproduced from Huang et al. 23 with permission from Annual Reviews)
Φ29DNA polymerase has a strong error correction function, and the amplification product has high fidelity, which ensures the consistent yield of different templates. 52 The longest amplification product is 100 kb, with an average of more than 10 kb. Using a 100 μl reaction system, the amplification product can be kept around 20–30 μg, which can be used for many aspects of DNA analysis. 53 MDA is simple to operate, reduces human error, avoids DNA loss, and is available in commercial kits. Although some studies have found that when used with low concentration DNA starting template, it has some shortcomings, such as poor repeatability, allele loss, and inconsistent location. 54
To verify the advantage of MDA in WGA, researchers have demonstrated the overall effectiveness of MDA in maintaining the quality of sequencing data and the abundance of species measurements in eight paired metagenomic samples and one titrated mixed control sample, 55 while providing higher high‐fidelity DNA yields and increasing the number of high fidelity DNA in low biomass samples. 56
Uneven amplification in MDA causes a lot of problems with biological applications of copy number estimation. 54 Under optimized conditions, related studies have proved that the whole reaction system can be physically separated into many microchambers or droplets using microfluidic devices, which can effectively reduce this deviation. 57 This improved MDA technique, also known as micro‐channel MDA (μCMDA), allows MDA reagents to be dispersed into one‐dimensional tubules. Due to the dual effects of soft segmentation of high molecular weight DNA molecules and limited diffusion of small particles, 58 μCMDA shows significant effectiveness in improving amplification uniformity. μCMDA can effectively improve our efficiency and sensitivity in detecting single nucleotide variants (SNVs). 59 Additionally, this simple method requires neither customized instruments nor complex operations and has gradually developed into an off‐the‐shelf technology in almost all biological laboratories. 60
It is also possible to sequence the DNA of a single cell by using expanded DNA as a template. MDA amplification can amplify a few micrograms of DNA in a bacterium to micrograms of DNA 61 that can be used for sequencing. It is currently recognized as the best single‐cell genome amplification technology. 60 As a great breakthrough in environmental research methods, MDA can be used to guide new research strategies in microbial genetics, ecology, and infectious diseases research. 62
Previously, newly discovered microbes can only be sequenced by growing enough strains to provide enough DNA templates. However, less than 1% of microbes have been successfully cultured. A large number of new unculturable organisms will appear in the natural environment, which will become a major obstacle to the traditional genome research methods. In this work, the emergence of single‐cell sequencing has opened up a new field in which DNA templates can be obtained directly from a single cell without the need for improved culture methods. 63 Using MDA, amplified DNA from single‐celled microorganisms can be sequenced. The high molecular weight DNA of micrograms can be amplified from several Feike (10–15 g) DNA in bacteria. The DNA amplified by high molecular weight DNA is suitable for preparing DNA libraries and Sanger sequencing. The DNA generated by MDA can also be directly used as a template for pyrophosphate sequencing. The linkage of gene sequences between individual cells is a powerful tool, which can be used to guide the construction of gene chips for multiple organisms obtained from extracted environmental DNA (macro genomic sequence) by shotgun sequencing. 49
The MDA method has a higher genome coverage than DOP‐PCR. 62 The high efficiency and high fidelity of φ29DNA polymerase allows MDA to possess significant advantages in the analysis of SNV and the construction of large fragment libraries. However, like DOP‐PCR, MDA is still an exponential amplification; thus, sequence preference of PCR response is still present. 64 However, unlike DOP‐PCR, this preference cannot be repeated, thus causing the MDA method to be unsuitable for CNV analysis.
3.2. MALBAC
MALBAC, shown in Figure 4, is the most advanced WGA technology at present. This technology can complete the high‐precision whole genome sequencing of a single cell. 65 During MALBAC, five rounds of MDA pre‐amplification are carried out to make the amplification products form closed ring molecules. Because the ring molecules cannot be further amplified, the amplification process becomes a linear amplification, and conventional PCR amplification is carried out. The template used at this time is more uniform, thus obtaining amplification products with less amplification offset and sufficient coverage. 23 Amplifiers complement each other and form a loop, preventing an exponential amplification of DNA, and reducing amplification bias so that 93% of the genomes in a single cell can be sequenced. This technique has high sensitivity, thus allowing a single cell and single chromosome or 0.5 pg gDNA to be amplified. The fragment length is mainly distributed among 0.8–10 kb, and the amplification uniformity is better than other WGA techniques, which is satisfied with a variety of downstream applications. 50
FIGURE 4.
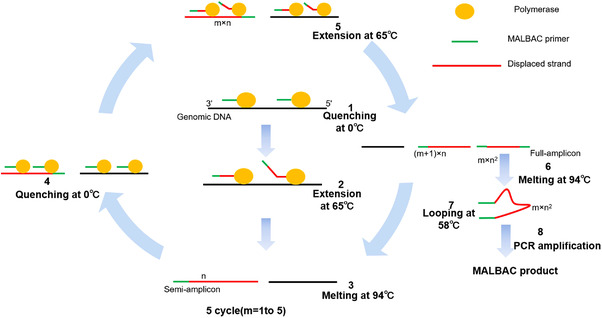
The schematic diagram of multiple annealing and looping‐based amplification cycles. m: the number of temperature cycles; n: the number of primers binding; (m + 1) × n: the number of semi‐amplicons appear at the mth cycle; m × n 2: the number of full amplicons produced in the mth cycle (reproduced from Huang et al. 23 with permission from Annual Reviews)
MALBAC makes it easier to detect smaller DNA sequence variations in a single cell, so genetic differences between individual cells can be found. Such differences can help explain the mechanism of cancer progression, the formation of germ cells, and even the differences of individual neurons. Although it still has a certain sequence preference, unlike MDA, MALBAC is repeatable between different cells, so CNV analysis can be carried out after standardizing the reference cells. 66 In addition to its repeatability, the knockout allele rate of MALBAC amplification is lower due to the homogeneity of its amplification. However, the fidelity of polymerase used in this method does not perform as well as φ29, 67 allowing the potential of more false positives in the detection of SNV. Also, low amplification regions on the genome are sometimes lost in the process of amplification due to its repeatable sequence preference. At present, MALBAC technology has been successfully applied to human single sperm, 68 blastocysts and polar bodies screened before implantation, 9 early developmental embryos, 69 tumor cells, 70 trace material evidence, and microorganisms at criminal investigation sites. 50
Under the same conditions, the genome coverage of MALBAC is higher and more uniform than MDA, 71 and at the same time, producing fewer false positives and maintaining higher accuracy. In addition, there is some difference between the two methods in variation detection. The identification of single nucleotide polymorphisms (SNPs) in sequenced samples revealed that more high‐quality SNPs were detected in MALBAC samples than in MDA samples, and the analysis showed that the MALBAC method had better performance in terms of uniformity and reproducibility of the whole genome amplification. 71
3.3. LIANTI
LIANTI is a new WGA method for single‐cell sequencing reported by Xie in Science 2017. 10 LIANTI can obtain adequate DNA through linear amplification, reducing errors caused by exponential amplification and non‐specific in MDA, MALBAC, and other methods. 72 The main differences between primary WGA methods are shown in Table 1. This amplification method is an improved single‐cell WGA technique based on the traditional methods, linearly amplified by the transposon. The transposon is a DNA that can change its position in the genome. It has a double‐stranded transposase‐binding site of 19 base pairs and a single‐stranded T7 promoter loop. The promoter is part of the DNA that starts the transcriptional process and is used to amplify downstream DNA and prepare DNA libraries. 73 First, Tn5 transposon was combined with LIANTI sequence to form Tn5 transposable complex with T7 promoter and then randomly inserted into gDNA. 74 After transposon, DNA was randomly fragmented. Subsequently, the T7 promoter performed in vitro transcription function to obtain a mass of linear amplified transcripts. After reverse transcription, substantial amplified products are obtained and DNA is ready for library preparation and sequencing as shown in Figure 5. There is no exponential amplification and non‐specific primers in the LIANTI process, which avoids the interference of PCR, dramatically reduces the amplification deviation and error, and improves the amplification stability. 75 The LIANTI is superior to the existing WGA methods. It can detect micro CNVs at kilobase resolution, which can be used to observe the random initiation of DNA replication from cell to cell. In addition, LIANTI is more accurate than MDA and MALBAC in detecting CNVs and SNVs. 76
TABLE 1.
Differences between whole genome amplification methods
| PEP‐PCR | DOP‐PCR | LM‐PCR | T‐PCR | MDA | MALBAC | LIANTI | |
|---|---|---|---|---|---|---|---|
|
Principle |
Completely random priming method |
Partial random priming method |
Adaptor ligation‐mediated PCR |
Two‐step PCR method using tagged random primer |
Multiple displacement amplification |
Multiple annealing and looping‐based amplification cycles | Using Tn5 transponson to achieve amplification without the non‐specific primers |
| Primer | Random primers containing 15 bases | Degenerate primers containing six random primers | Universal primer and an adaptor primer | Tagged random primers containing a 9 to 15 bp arbitrary 3' tail that can bind to any DNA sequence | Six random primers | Twenty‐seven universal primers and eight random primers | / |
| Enzyme | DNA polymerase | DNA polymerase | Taq DNA polymerase | Taq DNA polymerase | Phi29 DNA polymerase | Bst enzyme; Phi29 DNA polymerase | T7 RNA polymerase |
| Coverage | ∼40% | ∼50% | ∼96% | ∼37% | ∼70% | ∼90% | ∼97% |
| Uniformity | Low | Medium | Low | Medium | Low | High | High |
| Coefficient of variation | Medium | High | High | High | Medium | Medium | Low |
| GC preference | High | High | High | High | Medium | Low | Low |
| Advantages | The operation is simple, the quality of template DNA is low, the minimum starting template quantity is up to 5 pg | Simple to operate, minimum starting template up to 50 pg | The yield is high, the fragment is long, and the quality of template DNA is low | High amplification efficiency and product specificity | High yield, minimum initial amount up to 10 pg, good fidelity | Simple operation, high output, minimum starting template of several pocks, reliable and repeatable results | Small amplification deviation and high gene coverage |
| Disadvantages | Low output and poor fidelity | The amplification deviation is large when the initial template is very low | The operation is tedious, and the template DNA is easy to be lost by multi‐step operation | Low gene coverage | Large amplification deviation | It is more difficult to amplify when the initial template is very low, and it is easy to appear false positive | Prone to false positives |
| Application | LOH analysis, STR analysis, and so forth | FISH, SNP analysis, SSCP analysis, and so forth | CGH, SNP, STR analysis, Library establishment, Gene detection, and so forth | STR analysis, Forensic Medicine, DNA identification, and so forth | SNV detection, NGS, STR analysis, single‐cell sequencing, and so forth | Chromosome analysis, CNV detection, SNV detection, CGH, single‐cell sequencing, and so forth | CNV detection, SNV detection, single‐cell sequencing, haploid typing, and so forth |
| Reference | 86 | 9 , 87 , 88 | 44 , 89 , 90 , 91 | 44 | 17 , 28 | 4 , 83 , 92 | 93 , 94 |
Abbreviations: CGH, comparative genomic hybridization; DOP‐PCR, degenerate oligonucleotide primer PCR; FISH, fluorescence in situ hybridization; LIANTI, linear amplification via transposon insertion; LM‐PCR, ligation‐mediated PCR; LOH, loss of heterozygosity; MALBAC, multiple annealing and looping‐based amplification cycles; MDA, multiple displacement amplification; PCR, polymerase chain reaction; PEP‐PCR, primer extension preamplification PCR; SSCP, single‐strand conformation polymorphism; STR, short tandem repeat; T‐PCR, tagged random primer PCR; SNP, single nucleotide polymorphism; SNV, single nucleotide variant; CNV, copy number variation; NGS, next‐generation sequencing.
FIGURE 5.
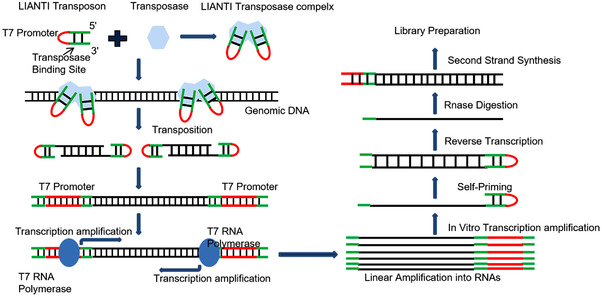
The schematic diagram shows the amplification principles and sequencing library preparation of the linear amplification via transposon insertion method (reproduced from Li et al. 74 with permission from MDPI)
However, LIANTI still has some limitations. It is highly dependent on Tn5 transposase and has not yet achieved large‐scale application. Moreover, its experimental operation is more tedious and lacks repeatability. 77 Therefore, the future development of LIANTI needs to be further explored.
4. MICROFLUIDICS‐BASED AMPLIFICATION TECHNOLOGY
In recent years, the progress of methodology has extensively promoted the development of molecular diagnosis technology. However, there are often many disadvantages in using traditional macro detection platforms for microscopic molecular level characterization and analysis, such as complex instruments, serious interference, complicated operation, easy cross‐contamination and poor stability. The timely emergence of microfluidic opens a new door for the research of molecular diagnosis. 78
Microfluidic technology refers to the science and technology of precise manipulation, analysis, and detection of a microfluidic or micro reaction system in the micron flow channel. Its core unit is the microfluidic chip. 79 The microfluidic chips can integrate complex fluid control systems and functional unit modules on a few square centimeters of a chip, with the benefits of automation, integration, miniaturization, and so on. 80 Therefore, it is called a micro‐complete analysis system or chip laboratory. The combination of microfluidic technology and micro reaction system has the advantages of small sample size, fast reaction speed, high sensitivity, and flexible manipulation. At the same time, the closed microfluidic chip can avoid cross‐contamination of the sample and reduce biohazard, which has a natural advantage in molecular diagnosis. 81 , 82 The method based on microfluidic technology provides an ideal platform for nucleic acid amplification. The nucleic acid amplification on the chip mainly depends on the rapid prototyping of polydimethylsiloxane or glass etching, which has the advantages of fast amplification speed, low detection limit, and high detection accuracy.
The amplification methods mentioned above, such as DOP‐PCR, MDA, and MALBAC, are all at the molecular level. The process of combining high fidelity DNA replication or linear amplification steps helps improve consistency, expand coverage, and reduce the error rate. Among these methods, MDA is easy to operate and provides higher fidelity and coverage. However, they all need to amplify the genome in vitro with DNA polymerase and then prepare a library for high‐throughput sequencing with a relatively short read length. The short sequence reads can affect the acquisition of gene information, especially for single‐cell research. Therefore, Xie et al. 4 reported the emulsion WGA technique in 2015, which combines emulsion, MDA, and microfluidic chip to accurately detect the genomic variation. This method showed significant improvement in the uniformity of amplification, the coverage of the genome, and the proportion of false positives. Later, Zhang made a further improvement based on this method. Namely, large fragments are evenly distributed into 24 independent reaction cells to amplify and prepare the library. 83 Single cells were lysed in a tube and then mixed with MDA reaction buffer. The emulsification used in the microfluidic cross‐connection device generates uniformly distributed droplets for DNA amplification, which maintains the high fidelity of MDA and improves the uniformity of amplification, which has attracted extensive attention in various single‐cell studies. 84 , 85
However, these microfluidic‐based methods still face various difficulties to meet the requirements of WGA. For example, these methods are both difficult to operate and to control droplets in parallel, limiting capturing the required droplets. In addition, a large number of monodisperse droplets are usually unstable, which may affect the uniform amplification of each droplet DNA fragment. Another disadvantage is that the droplet‐based method cannot be automated for WGA in integrated microfluidic chips. However, the market demand for molecular diagnosis and the continuous progress of microfluidic technology will promote the development of microfluidics in molecular diagnosis. With these advancements in microfluidics, advanced molecular diagnostic products based on microfluidics will appear centrally to provide better services for human health.
Based on PCR amplification, DOP‐PCR uses universal primers on both sides of the site to amplify the sample. 95 The detection accuracy of single‐cell CNV is high, but the coverage of calling SNV is low, creating higher false positive and negative rates. 96 The coverage of MDA has greatly improved, but the sensitivity in CNV determination is often low because its amplification gain varies along the genome and cannot be reproduced between different cells. 97 Through quasi‐linear amplification, MALBAC suppressed the random deviation of amplification, reduced the rate of allelic dropout, and produced a lower false negative of SNV detection. 68 Despite its shortcomings, MDA still provides genomic coverage equal to or higher than that of MALBAC in both individual diploid cells and aneuploid cells, such as mitotic 98 and cancer cells. 61 The main advantage of MDA is its low false‐positive rate of SNV detection, which is due to the use of pHi‐29, a highly progressive high‐fidelity polymerase. Microfluidic devices have been implemented for single‐cell WGA, 99 allowing contamination avoidance and parallel high‐throughput analysis of multiple single cells. The total reaction volume of the microfluidic device is very small (up to nanoliters or picoliters), which not only promotes the reaction efficiency but also greatly reduces the cost of the enzymes and reagents used. It is reported that in the WGA of a single bacterial cell, compared with the microliter device, the nanoliter volume of the microfluidic device improves the uniformity of amplification. 100
5. APPLICATION OF WGA TECHNOLOGY
WGA technology has a broad application prospect for applications with limited starting material such as forensics, paleontology, prenatal diagnosis for which only a single cell can be obtained from maternal peripheral blood and amniotic fluid, and tumor molecular pathology for genetic analysis of multiple loci.
5.1. Application of WGA in prenatal diagnosis
Prenatal diagnosis is used to advance the diagnosis of genetic diseases and congenital malformations before the fetus’ birth and to provide sufficient and reliable information for families at high risk of genetic diseases and other factors causing malformations. 101 To avoid the spread of pathogenic gene variation and detect genetic diseases as soon as possible, prenatal gene testing is provided. 102 Similarily to abnormal fetal development testing by ultrasound, genetic testing may provide an accurate diagnosis so that family members can make appropriate choices for abnormal fetuses during pregnancy and take positive and effective measures to correct and reduce the birth of defective babies. 103 However, in prenatal diagnosis, the concentration of DNA extracted is often too low due to oligohydramnios, few cells in amniotic fluid, or too many fetal fat components, which cannot be used for genetic detection. 104 At this time, it is necessary to improve the quality of DNA through WGA technology to meet the detection requirements and amplify the whole genome of the qualified DNA quality control. For example, myotonic dystrophy type 1 can be diagnosed prenatally by WGA, using triplet‐primed PCR to detect amplified DMPK alleles. Using single‐tube haplotype analysis, 12 closely linked microsatellite markers with high polymorphism can be analyzed, thus avoiding the transmission of the disease to the next generation. 105 WGA can also amplify the whole genome from a single cell, provide a new path for prenatal diagnosis, obtain a large number of DNA sequences without changing genetic information, and provide a more accurate diagnosis for clinical work. 106
5.2. Application of WGA in pre‐implantation genetic diagnosis (PGD)
PGD refers to the genetic analysis of embryos before implantation. 107 Individual blastomere cells, polar bodies, or several trophoblast cells are taken from embryos cultured by in vitro fertilization for genetic examination. 108 PGD analysis can prevent the occurrence of genetically abnormal pregnancy and advance prenatal diagnosis before the embryo is implanted into the endometrium, to avoid repeating abortion or induced labor damage to pregnant women. 109
At present, PCR is widely used in PGD and has sufficient detection sensitivity, but it is only suitable for specific sequence amplification and cannot meet the needs of multi‐site complex amplification. 110 The material source of single‐cell DNA detection is limited, 111 and the time requirement of the test is very high; if there are not enough samples, it is difficult to meet the needs of rapid detection, but WGA technology can meet the needs of this aspect. 112 Since the first appearance of PEP and DOP‐PCR in the early 1990s, WGA has made remarkable progress in the accuracy, efficiency, and reliability of single‐cell DNA amplification and has gradually developed into a powerful tool for single‐cell gene diagnosis.
Akash et al. 113 have verified a method to infer the whole genome sequence of embryonic inheritance for PGD. They combined haplotype analysis of parental genome sequencing with rapid embryo genotyping to predict the whole genome sequence of a fifth‐day human embryo from a couple at risk of alpha‐thalassemia. They found that genetic prediction had about 3 million paternal or maternal heterozygote sites with an accuracy of more than 99% and successfully staged and predicted the spread α1‐globin gene/α2‐globin gene (HBA1/HBA2) deletions from each parent. The experimental results show that pre‐implantation whole‐genome prediction is helpful to the comprehensive diagnosis of embryonic diseases with a known genetic basis.
In related studies, PCR and MDA+PCR were compared in the detection of PGD α‐thalassemia. The results showed that the total diagnosis rate of the MDA+PCR group (96.51%) was higher than that of the PCR group (86.16%). The rate of embryos available for transplantation in the MDA+PCR group (71.62%) was also significantly higher than that in the PCR group (62.23%), compared with the PCR group, the misdiagnosis rate was significantly lower. 114 The whole genome of single‐cell blastomere was amplified by MDA and analyzed by fluorescent PCR, which could effectively improve the accuracy of PGD in the diagnosis of Duchenne muscular dystrophy (DMD). This is also the first successful clinical application of MDA technology in the treatment of DMD in PGD. 115 An MDA‐PGD scheme for Marfan syndrome was designed by using MDA technology, which can be used for PCR analysis of five different sites of MDA products, and any known gene defects can be diagnosed with standard methods and conditions. 116 Lee et al. also evaluated the value of MDA in the diagnosis of fragile X syndrome PGD. 117 The fragile X mental retardation‐1 CGG repeat sequence, amelogenin, and two polymorphic markers were detected by fluorescent PCR, and 10 normal embryos, four mutant embryos, and six heterozygous carriers were diagnosed successfully, indicating that MDA and fluorescent PCR can be successfully used in the PGD of fragile X syndrome. This micro‐DNA amplification method can improve the sensitivity and reliability of PGD to complex monogenic diseases. PGD can also be used for monogenic disorders. The diagnostic efficiency can be improved by analyzing free gDNA in embryo culture fluid collected at the blastocyst stage. This requires a highly specific and efficient amplification method to provide sufficient samples for accurate diagnosis. WGA is considered a promising approach. 118
5.3. Application of WGA in tumor
Gene amplification in tumor cells is the main mechanism of oncogene activation. 119 Oncogene activation can cause tumor cells to escape growth inhibition and produce drug resistance. 120 As a genetic disease, gene diagnosis and analysis of cancer individuals are essential for the guidance and realization of individualized therapy but proves difficult to achieve the therapeutic purpose by amplifying fragments from sample sites, which may be due to the small number of DNA isolated. 121
Daisuke et al. used the MDA technique to amplify the whole genome DNA from plasma samples. They amplified two microsatellite markers, D1S243 and D19S246, thus showing microsatellite changes in patients with head and neck cancer. 122 DNA can also be extracted from a single lesion of prostatic intraepithelial neoplasia (HPIN) and prostate cancer (CaP) and can then be amplified by MDA for gDNA array comparative genomic hybridization (GaCGH). The combined detection of laser capture microdissection, MDA, and GaCGH is very suitable for identifying abnormal chromosome copy number abnormalities (CNAs) from small cell clusters. This detection can identify potential genomic markers and locate tumor suppressor genes or oncogenes that have not been previously reported in HPIN and CaP. 123 A microfluidic chip technology based on WGA technology has also been proposed, which can be used for whole genome sequencing of single‐cell circulating tumor cells (CTCs). 124 It can realize the whole process from blood filtration, enrichment, identification, and cleavage to WGA, providing a solid foundation for the whole genome sequencing of CTC and the precise treatment of cancer. 125 The progress of WGA technology in regards to gene amplification in tumors 126 will continue to play an important role in the diagnosis and treatment of cancers. 127 , 128
5.4. Application of WGA in forensic genetics laboratories
In recent years, the research on the amplification of trace DNA samples by WGA technology has become more and more indepth. 129 This technique can be used to amplify the whole genome of trace residual tissues, even single cells, and provide sufficient DNA templates for follow‐up multigene, multilocus analysis, and comprehensive genome research. 130 This allows testing of precious samples with small amounts of DNA or a large number of degraded DNA in medical biology. 131 WGA technology is particularly important to obtain a sufficient amount of DNA from a limited number of forensic samples for a large number of genetic analyses and tests.
B‐lymphoblastoid cells from female and nucleated cell samples from male venous blood have been genotyped at forensic sites with capillary electrophoresis and massively parallel sequencing (MPS) 132 platforms to assess their performance without WGA. The study found that in the absence of WGA, except for three and five samples of female cell samples with an accuracy of more than 25%, the rest had an accuracy of well under 10%. In male samples, the Y allele was promoted, occasionally leading to relatively high mean relative fluorescence units, while WGA combined with the MDA strategy performed relatively well on STR and SNP genotyping of low copy number samples on both CE and MPS, even for single‐cell samples. 133
The most common way to obtain DNA from forensic scenes is to extract DNA from evidence samples. A good DNA extraction method is the primary guarantee to obtain accurate genotyping results. 134 Different extraction methods will affect the accuracy of typing results. Ballantyne et al. 135 used the Chelex method to extract DNA. The amplification amount of Genome Plex WGA kit is more than that of the chemical extraction method. Erin et al. developed an improved PEP‐PCR. As a new WGA technique, it can effectively amplify DNA samples with low copy number (gDNA<100 pg) or environment‐induced degradation in evidence items, and can be used for subsequent microsatellite analysis. 136
Presently, WGA technology is only a technical method to improve the initial template DNA. To overcome the difficulty of forensic trace sample testing, it is necessary to combine WGA technology with other forensic DNA testing techniques to overcome the obstacles in forensic research. 137
6. CONCLUSION
In conclusion, WGA is a technology that can greatly maximize the original template DNA and provides a way to obtain a large amount of genetic information from trace gDNA. 138 In this paper, we reviewed multiple WGA methods along with their amplification principles, advantages, and disadvantages. WGA's disadvantages consist of amplification bias, 28 non‐specific amplification, 27 low sensitivity, poor repeatability, a high error rate of operation, and external contamination. 139 Researchers are continuously improving accuracy, coverage, convenience of amplification, and high sensitivity in trace samples. 140
At present, WGA has shown strong advantages in the microanalysis of sperm, fetal cells, lymphocytes, tissue sections, and other specimens. However, reducing or even avoiding the illusion of amplification in a small number of specimens will be an important topic in the future. 141 In addition, the application of DNA amplification in the body fluid of tumor patients, allows for a noninvasive diagnosis of tumor and amplification of processed DNA such as DNA methylation, reverse transcriptional cDNA and noninvasive prenatal diagnosis will be a promising development direction. WGA provides a powerful reference for the detection and diagnosis of human diseases.
AUTHORS' CONTRIBUTIONS
XYW was responsible for the manuscript. YPL played a guiding role in reference collection and manuscript revision. HNL and WJP were mainly responsible for the revision of English grammar and expression. JR, XMZ, YMT, ZC and YD checked different sections of the manuscript. HC, NYH and SL offered revision advice. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS APPROVAL
Not applicable.
ACKNOWLEDGMENTS
The authors appreciate the support of the National Key R&D Program of China (2021YFE0191400, 2021YFE0200400, 2019YFE0191000); the National Natural Science Foundation of China (No. 61971187, 61871180); Hunan Provincial Natural Science Foundation of China (2019JJ50122); Hunan Urgency Project (No. 2020SK3005) and Hunan Key R&D Projects (No. 2021SK2003).
Wang X, Liu Y, Liu H, et al. Recent advances and application of whole genome amplification in molecular diagnosis and medicine. MedComm. 2022;3:e116. 10.1002/mco2.116
Contributor Information
Hui Chen, Email: huier_88@vip.163.com.
Song Li, Email: sosong1980@gmail.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Jiang Z, Wang H, Michal JJ, et al. Genome wide sampling sequencing for SNP genotyping: Methods, challenges and future development. Int J Biol Sci. 2016;12(1):100‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blainey PC. The future is now: Single‐cell genomics of bacteria and archaea. FEMS Microbiol Rev. 2013;37(3):407‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hosokawa M, Nishikawa Y, Kogawa M, et al. Massively parallel whole genome amplification for single‐cell sequencing using droplet microfluidics. Sci Rep. 2017;7(1):5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fu Y, Li C, Lu S, et al. Uniform and accurate single‐cell sequencing based on emulsion whole‐genome amplification. Proc Natl Acad Sci U S A. 2015;112(38):11923‐11928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen N, Wang WM, Wang HL. An efficient full‐length cDNA amplification strategy based on bioinformatics technology and multiplexed PCR methods. Sci Rep. 2016;5:19420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheung VG, Nelson SF. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc Natl Acad Sci U S A. 1996;93(25):14676‐14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duan R, Lou X, Xia F. The development of nanostructure assisted isothermal amplification in biosensors. Chem Soc Rev. 2016;45(6):1738‐1749. [DOI] [PubMed] [Google Scholar]
- 8. Spits C, Le Caignec C, De Rycke M, et al. Whole‐genome multiple displacement amplification from single cells. Nat Protoc. 2006;1(4):1965‐1970. [DOI] [PubMed] [Google Scholar]
- 9. Hou Y, Fan W, Yan L, et al. Genome analyses of single human oocytes. Cell. 2013;155(7):1492‐1506. [DOI] [PubMed] [Google Scholar]
- 10. Chen C, Xing D, Tan L, et al. Single‐cell whole‐genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science. 2017;356(6334):189‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruan Q, Ruan W, Lin X, et al. Digital‐WGS: Automated, highly efficient whole‐genome sequencing of single cells by digital microfluidics. Sci Adv. 2020;6(50):eabd6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun G, Kaushal R, Pal P, et al. Whole‐genome amplification: relative efficiencies of the current methods. Leg Med (Tokyo). 2005;7(5):279‐286. [DOI] [PubMed] [Google Scholar]
- 13. Zhang L, Cui X, Schmitt K, et al. Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci U S A. 1992;89(13):5847‐5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Traeger‐Synodinos J. Pre‐implantation genetic diagnosis. Best Pract Res Clin Obstet Gynaecol. 2017;39:74‐88. [DOI] [PubMed] [Google Scholar]
- 15. Maciejewska A, Jakubowska J, Pawlowski R. Different whole‐genome amplification methods as a preamplification tool in Y‐chromosome Loci analysis. Am J Forensic Med Pathol. 2014;35(2):140‐144. [DOI] [PubMed] [Google Scholar]
- 16. Lasken RS, Egholm M. Whole genome amplification: abundant supplies of DNA from precious samples or clinical specimens. Trends Biotechnol. 2003;21(12):531‐535. [DOI] [PubMed] [Google Scholar]
- 17. Dean FB, Hosono S, Fang L, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99(8):5261‐5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moghaddaszadeh‐Ahrabi S, Farajnia S, Rahimi‐Mianji G, et al. A short and simple improved‐primer extension preamplification (I‐PEP) procedure for whole genome amplification (WGA) of bovine cells. Anim Biotechnol. 2012;23(1):24‐42. [DOI] [PubMed] [Google Scholar]
- 19. Sabina J, Leamon JH. Bias in whole genome amplification: Causes and considerations. Methods Mol Biol. 2015;1347:15‐41. [DOI] [PubMed] [Google Scholar]
- 20. Xu L, Bastus NC, Lu YJ. Sequencing a single circulating tumor cell for genomic assessment. In: Franco D, Francesco S, eds. Oncogenomics. Elsevier; 2019:219‐232. [Google Scholar]
- 21. Gawad C, Koh W, Quake SR. Single‐cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17(3):175‐188. [DOI] [PubMed] [Google Scholar]
- 22. Mendez P, Fang LT, Jablons DM, et al. Systematic comparison of two whole‐genome amplification methods for targeted next‐generation sequencing using frozen and FFPE normal and cancer tissues. Sci Rep. 2017;7(1):4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang L, Ma F, Chapman A, et al. Single‐cell whole‐genome amplification and sequencing: methodology and applications. Annu Rev Genomics Hum Genet. 2015;16:79‐102. [DOI] [PubMed] [Google Scholar]
- 24. Bonnette MD, Pavlova VR, Rodier DN, et al. dcDegenerate oligonucleotide primed‐PCR for multilocus, genome‐wide analysis from limited quantities of DNA. Diagn Mol Pathol. 2009;18(3):165‐175. [DOI] [PubMed] [Google Scholar]
- 25. Kwok PY. Making ‘random amplification’ predictable in whole genome analysis. Trends Biotechnol. 2002;20(10):411‐412. [DOI] [PubMed] [Google Scholar]
- 26. Baslan T, Kendall J, Ward B, et al. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res. 2015;25(5):714‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blagodatskikh KA, Kramarov VM, Barsova EV, et al. Improved DOP‐PCR (iDOP‐PCR): A robust and simple WGA method for efficient amplification of low copy number genomic DNA. PLoS One. 2017;12(9):e0184507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zong C, Lu S, Chapman AR, et al. Genome‐wide detection of single‐nucleotide and copy‐number variations of a single human cell. Science. 2012;338(6114):1622‐1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tarr AW, Boneham SP, Grabowska AM, et al. Tagged polymerase chain reaction subtractive hybridization for the enrichment of phage display random peptide libraries. Anal Biochem. 2005;339(1):61‐68. [DOI] [PubMed] [Google Scholar]
- 30. Vuong HG, Kondo T, Oishi N, et al. Genetic alterations of differentiated thyroid carcinoma in iodine‐rich and iodine‐deficient countries. Cancer Med. 2016;5(8):1883‐1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. White RB, Ziman MR. A comparative analysis of shotgun‐cloning and tagged‐random amplification‐cloning of chromatin immunoprecipitation‐isolated genome fragments. Biochem Biophys Res Commun. 2006;346(2):479‐483. [DOI] [PubMed] [Google Scholar]
- 32. Yang Z, Le JT, Hutter D, et al. Eliminating primer dimers and improving SNP detection using self‐avoiding molecular recognition systems. Biol Methods Protoc. 2020;5(1):bpaa004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Din RU, Khan MI, Jan A, et al. A novel approach for high‐level expression and purification of GST‐fused highly thermostable Taq DNA polymerase in Escherichia coli. Arch Microbiol. 2020:1‐10. [DOI] [PubMed] [Google Scholar]
- 34. Huang MM, Arnheim N, Goodman MF. Extension of base mispairs by Taq DNA polymerase: Implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 1992;20(17):4567‐4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sankar PS, Citartan M, Siti AA, et al. A simple method for in‐house Pfu DNA polymerase purification for high‐fidelity PCR amplification. Iran J Microbiol. 2019;11(2):181‐186. [PMC free article] [PubMed] [Google Scholar]
- 36. Alam MS, Moriyama H, Matsumoto M. Enzyme kinetics of dUTPase from the planarian Dugesia ryukyuensis. BMC Res Notes. 2019;12(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hogrefe HH, Hansen CJ, Scott BR, et al. Archaeal dUTPase enhances PCR amplifications with archaeal DNA polymerases by preventing dUTP incorporation. Proc Natl Acad Sci U S A. 2002;99(2):596‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Green MR, Sambrook J. Hot start polymerase chain reaction (PCR). Cold Spring Harb Protoc. 2018;2018(5):pdb.prot095125. [DOI] [PubMed] [Google Scholar]
- 39. Nishioka M, Mizuguchi H, Fujiwara S, et al. Long and accurate PCR with a mixture of KOD DNA polymerase and its exonuclease deficient mutant enzyme. J Biotechnol. 2001;88(2):141‐149. [DOI] [PubMed] [Google Scholar]
- 40. Green MR, Sambrook J. Long and accurate polymerase chain reaction (LA‐PCR). Cold Spring Harb Protoc. 2019;2019(3):pdb.prot095158. [DOI] [PubMed] [Google Scholar]
- 41. Carey MF, Peterson CL, Smale ST. In vivo DNase I, MNase, and restriction enzyme footprinting via ligation‐mediated polymerase chain reaction (LM‐PCR). Cold Spring Harb Protoc. 2009;2009(9):pdb.prot5277. [DOI] [PubMed] [Google Scholar]
- 42. Xu W, Xie Z, Tong C, et al. A rapid and sensitive method for kinetic study and activity assay of DNase I in vitro based on a GO‐quenched hairpin probe. Anal Bioanal Chem. 2016;408(14):3801‐3809. [DOI] [PubMed] [Google Scholar]
- 43. Tang X, Morris SL, Langone JJ, et al. Simple and effective method for generating single‐stranded DNA targets and probes. BioTechniques. 2006;40(6):759‐763. [DOI] [PubMed] [Google Scholar]
- 44. Yu CY, Yin BC, Wang S, et al. Improved ligation‐mediated PCR method coupled with T7 RNA polymerase for sensitive DNA detection. Anal Chem. 2014;86(15):7214‐7218. [DOI] [PubMed] [Google Scholar]
- 45. Kim TH, Dekker J. ChIP‐quantitative polymerase chain reaction (ChIP‐qPCR). Cold Spring Harb Protoc. 2018;2018(5):pdb.prot082644. [DOI] [PubMed] [Google Scholar]
- 46. Krawczyk B, Kur J, Stojowska‐Swedrzynska K, et al. Principles and applications of Ligation Mediated PCR methods for DNA‐based typing of microbial organisms. Acta Biochim Pol. 2016;63(1):39‐52. [DOI] [PubMed] [Google Scholar]
- 47. Hunter K. Application of interspersed repetitive sequence polymerase chain reaction for construction of yeast artificial chromosome contigs. Methods. 1997;13(4):327‐335. [DOI] [PubMed] [Google Scholar]
- 48. Picher AJ, Budeus B, Wafzig O, et al. TruePrime is a novel method for whole‐genome amplification from single cells based on TthPrimPol. Nat Commun. 2016;7:13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lasken RS. Genomic DNA amplification by the multiple displacement amplification (MDA) method. Biochem Soc Trans. 2009;37(Pt 2):450‐453. [DOI] [PubMed] [Google Scholar]
- 50. He F, Zhou W, Cai R, et al. Systematic assessment of the performance of whole‐genome amplification for SNP/CNV detection and beta‐thalassemia genotyping. J Hum Genet. 2018;63(4):407‐416. [DOI] [PubMed] [Google Scholar]
- 51. Liu N, Liu L, Pan X. Single‐cell analysis of the transcriptome and its application in the characterization of stem cells and early embryos. Cell Mol Life Sci. 2014;71(14):2707‐2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nelson JR. Random‐primed, Phi29 DNA polymerase‐based whole genome amplification. Curr Protoc Mol Biol. 2014;105(1):15.13.1‐15.13.16. [DOI] [PubMed] [Google Scholar]
- 53. Kamtekar S, Berman AJ, Wang J, et al. The phi29 DNA polymerase:protein‐primer structure suggests a model for the initiation to elongation transition. EMBO J. 2006;25(6):1335‐1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sidore AM, Lan F, Lim SW, et al. Enhanced sequencing coverage with digital droplet multiple displacement amplification. Nucleic Acids Res. 2015;44(7):e66‐e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hammond M, Homa F, Andersson‐Svahn H, et al. Picodroplet partitioned whole genome amplification of low biomass samples preserves genomic diversity for metagenomic analysis. Microbiome. 2016;4(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ahsanuddin S, Afshinnekoo E, Gandara J, et al. Assessment of REPLI‐g multiple displacement whole genome amplification (WGA) Techniques for metagenomic applications. J Biomol Tech. 2017;28(1):46‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li J, Lu N, Shi X, et al. 1D‐reactor decentralized MDA for uniform and accurate whole genome amplification. Anal Chem. 2017;89(19):10147‐10152. [DOI] [PubMed] [Google Scholar]
- 58. Li C, Yu Z, Fu Y, et al. Single‐cell‐based platform for copy number variation profiling through digital counting of amplified genomic DNA fragments. ACS Appl Mater Interfaces. 2017;9(16):13958‐13964. [DOI] [PubMed] [Google Scholar]
- 59. Li J, Lu N, Tao Y, et al. Accurate and sensitive single‐cell‐level detection of copy number variations by micro‐channel multiple displacement amplification (μcMDA). Nanoscale. 2018; 10(37):17933‐17941. [DOI] [PubMed] [Google Scholar]
- 60. Woyke T, Sczyrba A, Lee J, et al. Decontamination of MDA reagents for single cell whole genome amplification. PLoS One. 2011;6(10):e26161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu X, Hou Y, Yin X, et al. Single‐cell exome sequencing reveals single‐nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148(5):886‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lasken RS. Single‐cell genomic sequencing using multiple displacement amplification. Curr Opin Microbiol. 2007;10(5):510‐516. [DOI] [PubMed] [Google Scholar]
- 63. Peters BA, Kermani BG, Sparks AB, et al. Accurate whole‐genome sequencing and haplotyping from 10 to 20 human cells. Nature. 2012;487(7406):190‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gonzalez‐Pena V, Gawad C. Sequencing the genomes of single cells. Methods Mol Biol. 2019;1979:227‐234. [DOI] [PubMed] [Google Scholar]
- 65. Deleye L, De Coninck D, Christodoulou C, et al. Whole genome amplification with SurePlex results in better copy number alteration detection using sequencing data compared to the MALBAC method. Sci Rep. 2015;5:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sha Y, Sha Y, Ji Z, et al. Comprehensive genome profiling of single sperm cells by multiple annealing and looping‐based amplification cycles and next‐generation sequencing from carriers of robertsonian translocation. Ann Hum Genet. 2017;81(2):91‐97. [DOI] [PubMed] [Google Scholar]
- 67. Jiang Z, Zhang X, Deka R, et al. Genome amplification of single sperm using multiple displacement amplification. Nucleic Acids Res. 2005;33(10):e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lu S, Zong C, Fan W, et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole‐genome sequencing. Science. 2012;338(6114):1627‐1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Niu W, Wang L, Xu J, et al. Improved clinical outcomes of preimplantation genetic testing for aneuploidy using MALBAC‐NGS compared with MDA‐SNP array. BMC Pregnancy Childbirth. 2020;20(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lu S, Chang CJ, Guan Y, et al. Genomic analysis of circulating tumor cells at the single‐cell level. J Mol Diagn. 2020;22(6):770‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zheng D, Chen M, Song P, et al. Comparison of multiple displacement amplification (MDA) and multiple annealing and looping‐based amplification cycles (MALBAC) in single‐cell sequencing. PLoS One. 2014;9(12):e114520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yin Y, Jiang Y, Lam KG, et al. High‐throughput single‐cell sequencing with linear amplification. Mol Cell. 2019;76(4):676‐690. e610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Winterberg KM, Reznikoff WS. Screening transposon mutant libraries using full‐genome oligonucleotide microarrays. Methods Enzymol. 2007;421:110‐125. [DOI] [PubMed] [Google Scholar]
- 74. Li N, Jin K, Bai Y, et al. Tn5 transposase applied in genomics research. Int J Mol Sci. 2020;21(21):8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kang X, Liu A, Liu GE. Application of multi‐omics in single cells. Ann Biotechnol. 2018;2:1007. [Google Scholar]
- 76. Blanshard RC, Chen C, Xie XS, et al. Single cell genomics to study DNA and chromosome changes in human gametes and embryos. Methods Cell Biol. 2018;144:441‐457. [DOI] [PubMed] [Google Scholar]
- 77. Khoshkhoo S, Lal D, Walsh CA. Application of single cell genomics to focal epilepsies: A call to action. Brain Pathol. 2021;31(4):e12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li Z, Zhao J, Wu X, et al. A rapid microfluidic platform with real‐time fluorescence detection system for molecular diagnosis. Biotechnol Biotec EQ. 2019;33(1):223‐230. [Google Scholar]
- 79. Fang X. Microfluidic chip. In: Tang J, Pan S, eds. Clinical Molecular Diagnostics. Springer; 2021:357‐375. [Google Scholar]
- 80. Basha IHK, Ho ETW, Yousuff CM, et al. Towards multiplex molecular diagnosis—A review of microfluidic genomics technologies. Micromachines (Basel). 2017;8(9):266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yin J, Suo Y, Zou Z, et al. Integrated microfluidic systems with sample preparation and nucleic acid amplification. Lab Chip. 2019;19(17):2769‐2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Canals J, Franch N, Alonso O, et al. A point‐of‐care device for molecular diagnosis based on CMOS SPAD detectors with integrated microfluidics. Sensors(Basel). 2019;19(3):445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chu WK, Edge P, Lee HS, et al. Ultraaccurate genome sequencing and haplotyping of single human cells. Proc Natl Acad Sci U S A. 2017;114(47):12512‐12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Buenrostro JD, Wu B, Chang HY, et al. ATAC‐seq: a method for assaying chromatin accessibility genome‐wide. Curr Protoc Mol Biol. 2015;109(1):21.29.21‐21.29.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lan F, Demaree B, Ahmed N, et al. Single‐cell genome sequencing at ultra‐high‐throughput with microfluidic droplet barcoding. Nat Biotechnol. 2017;35(7):640‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tenaglia E, Imaizumi Y, Miyahara Y, et al. Isothermal multiple displacement amplification of DNA templates in minimally buffered conditions using phi29 polymerase. Chem Commun (Camb). 2018;54(17):2158‐2161. [DOI] [PubMed] [Google Scholar]
- 87. Feng X, Wang S, Duan X, et al. An improved PCR‐RFLP assay for the detection of a polymorphism rs2289487 of PLIN1 gene. J Clin Lab Anal. 2016;30(6):986‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Michaela A, Jan S. Degenerate oligonucleotide‐primed PCR. In: Bartlett JMS, Stirling D, eds, PCR Protocols. Methods in Molecular Biology. Humana Press; 2003:. 226‐229. [DOI] [PubMed] [Google Scholar]
- 89. Lo YT, Shaw PC. DNA barcoding in concentrated Chinese medicine granules using adaptor ligation‐mediated polymerase chain reaction. J Pharm Biomed Anal. 2018;149:512‐516. [DOI] [PubMed] [Google Scholar]
- 90. Wells WD, Shuang G, Wei S, et al. Correction to: An analytical pipeline for identifying and mapping the integration sites of HIV and other retroviruses. BMC Genomics. 2020;21(1):1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee WH, Wong CW, Leong WY, et al. LOMA: A fast method to generate efficient tagged‐random primers despite amplification bias of random PCR on pathogens. BMC Bioinformatics. 2008;9(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhou XX, Xu Y, Libo Z, et al. Comparison of multiple displacement amplification (MDA) and multiple annealing and looping‐based amplification cycles (MALBAC) in limited DNA sequencing based on tube and droplet. Micromachines (Basel). 2020;11(7):645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wu L, Jiang M, Wang Y, et al. scDPN for High‐throughput single‐cell CNV detection to uncover clonal evolution during HCC recurrence. Genomics Proteomics Bioinformatics. 2021;9:S1672‐0229(21)00151‐0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Xu J, Liao K, Yang X, et al. Using single‐cell sequencing technology to detect circulating tumor cells in solid tumors. Mol Cancer. 2021;20(1):1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Backx L, Thoelen R, Van Esch H, et al. Direct fluorescent labelling of clones by DOP‐PCR. Mol Cytogenet. 2008;1(1):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single‐cell sequencing. Nature. 2011;472(7341):90‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. De Bourcy CF, De Vlaminck I, Kanbar JN, et al. A quantitative comparison of single‐cell whole genome amplification methods. PLoS One. 2014;9(8):e105585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yu Z, Lu S, Huang Y. Microfluidic whole genome amplification device for single cell sequencing. Anal Chem. 2014;86(19):9386‐9390. [DOI] [PubMed] [Google Scholar]
- 100. Liu W, Zhang H, Hu D, et al. The performance of MALBAC and MDA methods in the identification of concurrent mutations and aneuploidy screening to diagnose beta‐thalassaemia disorders at the single‐and multiple‐cell levels. J Clin Lab Anal. 2018;32(2):e22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Levy B, Stosic M. Traditional prenatal diagnosis: Past to present. In: Levy, B , eds. Prenatal Diagnosis, Methods Mol Biol. Humana Press; 2019:3‐22. [DOI] [PubMed] [Google Scholar]
- 102. Torres‐Canchala L, Castaño D, Silva N, et al. Prenatal diagnosis of pfeiffer syndrome patient with FGFR2 C.940‐1G>C variant: A case report. Appl Clin Genet. 2020;13:147‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Evangelidou P, Sismani C, Ioannides M, et al. Clinical application of whole‐genome array CGH during prenatal diagnosis: Study of 25 selected pregnancies with abnormal ultrasound findings or apparently balanced structural aberrations. Mol Cytogenet. 2010;3(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ou J, Yang C, Cui X, et al. Successful pregnancy after prenatal diagnosis by NGS for a carrier of complex chromosome rearrangements. Reprod Biol Endocrinol. 2020;18(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lian M, Lee CG, Chong SS. Robust preimplantation genetic testing strategy for myotonic dystrophy type 1 by bidirectional triplet‐primed polymerase chain reaction combined with multi‐microsatellite haplotyping following whole‐genome amplification. Front Genet. 2019;10:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Peng W, Takabayashi H, Ikawa K. Whole genome amplification from single cells in preimplantation genetic diagnosis and prenatal diagnosis. Eur J Obstet Gynecol Reprod Biol. 2007;131(1):13‐20. [DOI] [PubMed] [Google Scholar]
- 107. Harper JC, Bui TH. Pre‐implantation genetic diagnosis. Best Pract Res Clin Obstet Gynaecol. 2002;16(5):659‐670. [DOI] [PubMed] [Google Scholar]
- 108. Sermon K, Lissens W, Joris H, et al. Adaptation of the primer extension preamplification (PEP) reaction for preimplantation diagnosis: single blastomere analysis using short PEP protocols. Mol Hum Reprod. 1996;2(3):209‐212. [DOI] [PubMed] [Google Scholar]
- 109. Sermon K, Van Steirteghem A, Liebaers I. Preimplantation genetic diagnosis. Lancet. 2004;363(9421):1633‐1641. [DOI] [PubMed] [Google Scholar]
- 110. Kovačević N. Magnetic beads based nucleic acid purification for molecular biology applications. In: Micic M, eds. Sample Preparation Techniques for Soil, Plant, and Animal Samples. Springer Protocols Handbooks; 2016:53‐67. [Google Scholar]
- 111. Gao FF, Chen L, Bo SP, et al. ChromInst: A single cell sequencing technique to accomplish pre‐implantation comprehensive chromosomal screening overnight. PloS One. 2021;16(5):e0251971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mallory XF, Edrisi M, Navin N, et al. Methods for copy number aberration detection from single‐cell DNA‐sequencing data. Genome Biol. 2020;21(1):1‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kumar A, Ryan A, Kitzman JO, et al. Whole genome prediction for preimplantation genetic diagnosis. Genome Med. 2015;7(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fu Y, Shen X, Zhou C. Multiple displacement amplification as the first step could increase diagnosis efficiency of preimplantation genetic diagnosis for α‐thalassemia. Fertil Steril. 2018;110(4):e417. [Google Scholar]
- 115. Ogilvie CM. Preimplantation genetic diagnosis. Lancet. 2003;362(9379):250. [DOI] [PubMed] [Google Scholar]
- 116. Lledo B, Ten J, Galan F, et al. Preimplantation genetic diagnosis of Marfan syndrome using multiple displacement amplification. Fertil Steril. 2006;86(4):949‐955. [DOI] [PubMed] [Google Scholar]
- 117. Lee HS, Kim MJ, Lim CK, et al. Methodology multiple displacement amplification for preimplantation genetic diagnosis of fragile X syndrome. Genet Mol Res. 2011;10(4):2851‐2859. [DOI] [PubMed] [Google Scholar]
- 118. El Tokhy O, Salman M, El‐Toukhy TJO, et al. Preimplantation genetic diagnosis. Obstet Gynaecol Reprod Med. 2021;31(5):157‐161. [Google Scholar]
- 119. Shi R, Tang YQ, Miao H. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm (2020). 2020;1(1):47‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rangel‐Pozzo A, Liu S, Wajnberg G, et al. Genomic analysis of localized high‐risk prostate cancer circulating tumor cells at the single‐cell level. Cells. 2020;9(8):1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Navin NE. Cancer genomics: One cell at a time. Genome Biol. 2014;15(8):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nakamoto D, Yamamoto N, Takagi R, et al. Detection of tumor DNA in plasma using whole genome amplification. Bull Tokyo Dent Coll. 2006;47(3):125‐131. [DOI] [PubMed] [Google Scholar]
- 123. Hughes S, Yoshimoto M, Beheshti B, et al. The use of whole genome amplification to study chromosomal changes in prostate cancer: insights into genome‐wide signature of preneoplasia associated with cancer progression. BMC Genomics. 2006;7(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Joaquin M, Marco G, Nava RD, et al. The promise of circulating tumor cell analysis in cancer management. Genome Bio. 2014;15(8):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Li R, Jia F, Zhang W, et al. Device for whole genome sequencing single circulating tumor cells from whole blood. Lab Chip. 2019;19(19):3168‐3178. [DOI] [PubMed] [Google Scholar]
- 126. Gajduskova P, Snijders AM, Kwek S, et al. Genome position and gene amplification. Genome Bio. 2007;8(6):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bagci O, Kurtgöz S. Amplification of cellular oncogenes in solid tumors. N Am J Med Sci. 2015;7(8):341‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Xu Y, Su GH, Ma D, et al. Technological advances in cancer immunity: From immunogenomics to single‐cell analysis and artificial intelligence. Signal Transduct Target Ther. 2021;6(1):1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Van Oorschot RAH, Szkuta B, Meakin GE, et al. DNA transfer in forensic science: A review. Forensic Sci Int Genet. 2019;38:140‐166. [DOI] [PubMed] [Google Scholar]
- 130. Hosono S, Faruqi AF, Dean FB, et al. Unbiased whole‐genome amplification directly from clinical samples. Genome Res. 2003;13(5):954‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Balogh MK, Børsting C, Sánchez Diz P, et al. Application of whole genome amplification for forensic analysis. International Congress Series. 2006;1288:725‐727. [Google Scholar]
- 132. Rope AF, Wang K, Evjenth R, et al. Massively parallel sequencing identifies a previously unrecognized X‐linked disorder resulting in lethality in male infants owing to amino‐terminal acetyltransferase deficiency. Genome Biol. 2011;12(S1):1‐27. [Google Scholar]
- 133. Chen M, Zhang J, Zhao J, et al. Comparison of CE‐and MPS‐based analyses of forensic markers in a single cell after whole genome amplification. Forensic Sci Int Genet. 2020;45:102211. [DOI] [PubMed] [Google Scholar]
- 134. Hawkins TL, Detter JC, Richardson PM. Whole genome amplification‐applications and advances. Curr Opin Biotechnol. 2002;13(1):65‐67. [DOI] [PubMed] [Google Scholar]
- 135. Ballantyne KN, Van Oorschot RAH, Mitchell RJ. Comparison of two whole genome amplification methods for STR genotyping of LCN and degraded DNA samples. Forensic Sci Int. 2007;166(1):35‐41. [DOI] [PubMed] [Google Scholar]
- 136. Hanson EK, Ballantyne J. Whole genome amplification strategy for forensic genetic analysis using single or few cell equivalents of genomic DNA. Anal Biochem. 2005;346(2):246‐257. [DOI] [PubMed] [Google Scholar]
- 137. Vidaki A, Kayser M. From forensic epigenetics to forensic epigenomics: Broadening DNA investigative intelligence. Genome Biol. 2017;18(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Naiyun L, Yi Q, Zheyun X, et al. Recent advances and application in whole‐genome multiple displacement amplification. Quantitative Biology. 2020;8(4):1‐16. [Google Scholar]
- 139. Nadal‐Ribelles M, Islam S, Wei W, et al. Sensitive high‐throughput single‐cell RNA‐seq reveals within‐clonal transcript correlations in yeast populations. Nat Microbiol. 2019;4(4):683‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Martelotto LG, Baslan T, Kendall J, et al. Whole‐genome single‐cell copy number profiling from formalin‐fixed paraffin‐embedded samples. Nat Med. 2017;23(3):376‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Gewaily MS, Noreldin AE, Dawood MA, et al. The distribution profile of glycoconjugates in the testis of brown‐banded bamboo shark (Chiloscyllium punctatum) by using lectin histochemistry. Microsc Microanal. 2021;27(5):1161‐1173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.