

Abstract
Rationale
Alveolar and endothelial injury may be differentially associated with coronavirus disease (COVID-19) severity over time.
Objectives
To describe alveolar and endothelial injury dynamics and associations with COVID-19 severity, cardiorenovascular injury, and outcomes.
Methods
This single-center observational study enrolled patients with COVID-19 requiring respiratory support at emergency department presentation. More than 40 markers of alveolar (including receptor for advanced glycation endproducts [RAGE]), endothelial (including angiopoietin-2), and cardiorenovascular injury (including renin, kidney injury molecule-1, and troponin-I) were serially compared between invasively and spontaneously ventilated patients using mixed-effects repeated-measures models. Ventilatory ratios were calculated for intubated patients. Associations of biomarkers with modified World Health Organization scale at Day 28 were determined with multivariable proportional-odds regression.
Measurements and Main Results
Of 225 patients, 74 (33%) received invasive ventilation at Day 0. RAGE was 1.80-fold higher in invasive ventilation patients at Day 0 (95% confidence interval [CI], 1.50–2.17) versus spontaneous ventilation, but decreased over time in all patients. Changes in alveolar markers did not correlate with changes in endothelial, cardiac, or renal injury markers. In contrast, endothelial markers were similar to lower at Day 0 for invasive ventilation versus spontaneous ventilation, but then increased over time only among intubated patients. In intubated patients, angiopoietin-2 was similar (fold difference, 1.02; 95% CI, 0.89–1.17) to nonintubated patients at Day 0 but 1.80-fold higher (95% CI, 1.56–2.06) at Day 3; cardiorenovascular injury markers showed similar patterns. Endothelial markers were not consistently associated with ventilatory ratios. Endothelial markers were more often significantly associated with 28-day outcomes than alveolar markers.
Conclusions
Alveolar injury markers increase early. Endothelial injury markers increase later and are associated with cardiorenovascular injury and 28-day outcome. Alveolar and endothelial injury likely contribute at different times to disease progression in severe COVID-19.
Keywords: COVID-19, respiratory distress syndrome, alveolar epithelial cells, endothelium, renin-angiotensin system
At a Glance Commentary
Scientific Knowledge on the Subject
Alveolar and endothelial injury have both been implicated in severe coronavirus disease (COVID-19) pneumonia and acute respiratory distress syndrome (ARDS). How these disease processes evolve over time is not well described.
What This Study Adds to the Field
This observational study of hypoxemic patients with COVID-19 found that alveolar injury markers peaked early, whereas endothelial injury markers increased later and were associated with renin–angiotensin system dysfunction, cardiorenovascular injury, and 28-day outcome. These results suggest alveolar and endothelial injury contribute at different times to disease progression in severe COVID-19.
Severe coronavirus disease (COVID-19) pneumonia frequently involves progression to hypoxemic respiratory failure and acute respiratory distress syndrome (ARDS). ARDS is a heterogenous syndrome (1, 2). In patients with a direct insult etiology (e.g., pneumonia, aspiration), ARDS is associated with higher plasma levels of pulmonary epithelial injury markers (3), whereas with indirect insults (e.g., sepsis, trauma, etc.), ARDS displays higher endothelial injury markers (3). Given that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes pneumonia, an alveolar injury–predominant phenotype might be expected, yet the high prevalence of venous thromboembolism and shunt physiology (4–7) and reported postmortem pulmonary endothelialitis (8) suggest significant endothelial dysfunction.
Whether epithelial and endothelial markers are indicative of clinical progression, extrapulmonary organ dysfunction, and/or patient outcomes in SARS-CoV-2 infection is unknown. Direct comparisons of epithelial and endothelial injury patterns are limited, and longitudinal analyses are lacking. Understanding their dynamics over time would facilitate biological interpretation of clinical trial results. We investigated the evolution of SARS-CoV-2 illness using serially measured pulmonary epithelial, endothelial, and organ injury markers in a cohort of prospectively enrolled patients with COVID-19 presenting with respiratory distress. We hypothesized these markers would display distinct associations with clinical variables and would vary over time in ways that reflect the dynamics of underlying disease biology.
Some of the results have been previously reported in the form of an abstract (9).
Methods
Overall Design and Aims
We performed a secondary analysis of a prospectively enrolled observational cohort of patients with severe COVID-19, with three objectives: 1) describe epithelial versus endothelial markers over time by level of respiratory support; 2) determine whether these markers correlate with markers of systemic inflammation, renin-angiotensin system (RAS) activation, and nonpulmonary organ dysfunction; and 3) determine whether epithelial and endothelial markers associate with 28-day patient outcomes. The study was designed after enrollment had completed but without consideration of the results of the primary cohort analysis.
Patients
Patients were enrolled in the emergency department of an urban academic hospital in Boston, Massachusetts during the peak of the initial COVID-19 surge (March 24 to April 30, 2020) as described (10). The institutional review board waived informed consent. Inclusion criteria were age ⩾ 18 years, clinical concern for COVID-19, and one or more of the following: 1) respirations ⩾ 22 breaths/min; 2) oxygen saturation as measured by pulse oximetry (SpO2) ⩽ 92% on room air; 3) supplemental oxygen requirement; or 4) positive-pressure ventilation. For this study, we excluded enrolled patients without COVID-19 subsequently confirmed by PCR or not requiring respiratory support, defined as supplemental oxygen or invasive mechanical ventilation. Per hospital policy during the study period, no patients received noninvasive mechanical ventilation because of aerosolization concerns.
Timeline and Clinical Data
Subjects contributed dedicated research blood samples with their initial clinical blood draw on Day 0 and, if still hospitalized, at Day 3 and Day 7. Clinical course was followed for 28 days after enrollment or until discharge, whichever occurred later. Recorded clinical data included demographics, comorbidities, home medications, presenting symptoms, serial vital signs, laboratory values, Sequential Organ Failure Assessment (SOFA) score, and World Health Organization (WHO) scale. Organ dysfunction criteria are described in the online supplement.
Patients were categorized by respiratory support level based on WHO scale. Among intubated patients, dead space ventilation was estimated by ventilatory ratio (11), calculated using the formula: ventilatory ratio = (e × PaCO2)/(predicted body weight × 100 × 37.5). Ventilatory ratios ⩾2.0 were considered high (11).
Biomarker Assay
Specimen processing, banking, and assay were as described (10). Plasma biomarkers were measured using the Olink proximity extension assay, an oligonucleotide-labeled antibody assay for high-specificity high-dimensional multiplex that allows measurement of low-abundance proteins. Because the signal is amplified by PCR, measurements are expressed in normalized protein expression units, reflecting relative abundance on a log2 scale, rather than absolute concentration. This assay allows within-analyte comparisons between different samples but not between-analyte comparisons. For example, angiopoietin-2 levels between two different patients can be compared, but angiopoietin-2 level cannot be directly compared with IL-8 level in the same patient. Analytical performance validation for each protein assay is available at https://www.olink.com/.
Biomarker Selection
We a priori selected markers from the Olink protein library based on prior literature (12) and hypothesized importance. Markers were selected without consideration of the results of subsequently published analyses of this cohort. To facilitate interrogation of alveolar, club cell, and endothelial injury; systemic inflammation; RAS activation; and nonpulmonary organ injury, the selected analytes included cell-type–specific markers (see Table E1 in the online supplement). To confirm specificity, biomarkers were cross-referenced against the Human Protein Atlas: Tissue and Blood Atlases (www.proteinatlas.org) (13).
Statistical Analysis
Detailed methods are in the online supplement. Briefly, to study how respiratory illness severity influenced each marker’s 7-day course, we constructed mixed-effects repeated measures models, adjusting for age, sex, and chronic heart, lung, and kidney disease. To account for within-subject correlation, models included a random effect for subject with study day treated categorically as a repeated measure. Class variables for respiratory support level and interaction terms for day and support level were included.
To ensure results were robust and consistent, each biomarker was analyzed with three different approaches to specifying the respiratory support variable. First, a binary variable was used for whether the patient was invasively ventilated or deceased at that time point versus not invasively ventilated and alive (strategy-1). Second, respiratory support was treated as a categorical variable with three levels: invasively ventilated or deceased, alive and requiring supplemental oxygen, or alive without respiratory support (strategy-2). Third, the respiratory status variable was kept fixed based on Day 0 status: invasively ventilated or requiring supplemental oxygen (strategy-3). As additional sensitivity analyses, we evaluated representative markers under complete-case analysis and when excluding patients who received steroids or tocilizumab.
C-statistics were calculated to measure marker discrimination for intubation or death at Days 0 and 3.
To determine the association of each marker at Days 0 and 3 with Day 28 clinical status, we used multivariable proportional-odds regression, with clinical status a three-level categorical variable: 1) died; 2) invasive ventilation; and 3) off invasive respiratory support. Covariates were age, sex, Day 0 SOFA, and chronic heart, lung, and kidney disease. Models assessing Day 3 biomarker levels’ association with clinical status included an adjustment for the Day 0 biomarker level. The proportional-odds assumption was assessed graphically in each case and found to be reasonable. We report models in ascending format, such that a higher odds ratio (OR) indicates higher probability of worse outcome at Day 28.
Analyses were done in SAS (SAS Institute). Figures were produced in SAS or Prism (GraphPad Inc.).
Results
Study Population and Clinical Course
Among 384 enrolled patients, 274 required respiratory support at Day 0. Forty-nine of these proved to not have COVID-19, yielding 225 for the current longitudinal analysis (Figure E1). At Day 0, 151 (67%) subjects received supplemental oxygen only, and 74 (33%) received invasive ventilation (Table 1). Attrition at Day 3 was minimal: 10 (4%) patients died and 10 (4%) were discharged (Figure 1A). By Day 28, 37 (16%) patients had died, 37 (16%) continued receiving invasive ventilation, and 148 (68%) had been discharged (Figure 1A). Severe hypoxemia was most prevalent on Day 0, whereas severe abnormalities of renal, coagulation, and circulatory function became more prevalent over time (Figure 1B–1F; missing data prevalence in Tables E2 and E3).
Table 1.
Cohort Characteristics at Day 0
| Variable | All Patients | Supplemental O2 | Invasive Ventilation |
|---|---|---|---|
| No. | 225 | 151 | 74 |
| Age, yr | 61 (18) | 60 (19) | 64 (16) |
| Female, n (%) | 103 (46) | 74 (49) | 29 (39) |
| Body mass index | 31.0 (7.6) | 31.3 (7.5) | 30.4 (8.0) |
| Comorbidities, n (%) | |||
| Heart failure | 27 (12) | 20 (13) | 7 (9) |
| Coronary disease | 21 (9) | 17 (11) | 4 (5) |
| Hypertension | 112 (50) | 74 (49) | 38 (51) |
| COPD without home O2 | 16 (7) | 14 (9) | 2 (3) |
| COPD with home O2 | 6 (3) | 4 (3) | 2 (3) |
| Current smoker | 63 (28) | 43 (19) | 20 (27) |
| Prehospital baseline creatinine, mg/dl | 1.2 (1.1) | 1.1 (0.5) | 1.6 (1.8) |
| Chronic kidney disease without hemodialysis | 31 (14) | 19 (13) | 12 (16) |
| End-stage renal disease | 3 (1) | 0 (0) | 3 (4) |
| Non–insulin-dependent diabetes | 56 (25) | 29 (19) | 27 (36) |
| Insulin-dependent diabetes | 16 (7) | 12 (8) | 4 (5) |
| Immunosuppression | 20 (9) | 10 (7) | 10 (14) |
| Active malignancy | 12 (5) | 7 (5) | 5 (7) |
| Home ACE inhibitor | 20 (9) | 11 (7) | 9 (12) |
| Presenting characteristics | |||
| Symptom duration, days | 7 (4–11) | 7 (4–11) | 7 (3–10) |
| Bilateral radiographic opacities, n (%) | 188 (84) | 122 (81) | 66 (89) |
| S/F | 210 (105) | 264 (87) | 101 (24) |
| P/F | — | (n = 0) | 194 (52) (n = 59) |
| Hypoxemia severity* | |||
| P:F > 300 or S:F > 315 | 54 (24) | 54 (36) | 0 (0) |
| P:F 200–300 or S:F 235–315 | 46 (20) | 46 (30) | 0 (0) |
| P:F < 200 or S:F < 235 | 125 (56) | 51 (34) | 74 (100) |
| ARDS on Day 0† | 145 (64) | 79 (52) | 66 (89) |
| Initial SOFA score | 4.4 (3.8) | 2.4 (2.6) | 8.5 (2.5) |
| Mean arterial pressure, mm Hg | 72 (13) | 77 (11) | 63 (11) |
| Lactate, mmol/L | 2.0 (1.3) | 1.7 (0.8) | 2.3 (1.6) |
| High-sensitivity troponin-T, ng/L | 11 (6–31) | 8 (6–24) | 17 (7–47) |
| Creatinine, mg/dl | 1.3 (1.3) | 1.1 (0.9) | 1.6 (1.8) |
| Fold-change in creatinine from prehospital baseline | 1.11 (0.7) | 1.06 (1.06) | 1.21 (0.87) |
| Bicarbonate | 23.1 (3.7) | 23.5 (3.7) | 22.2 (3.5) |
| C-reactive protein, mg/dl | 137.8 (69.1–189.3) | 104.2 (58.4–168.3 | 149.5 (124.7–225.0) |
| D-dimer, ng/ml | 1,201 (760–1,985) | 1,082 (710–1,784) | 1,533 (951–2,301) |
| Absolute lymphocyte count | 0.94 (0.62–1.27) | 1.01 (0.71–1.36) | 0.78 (0.49–1.06) |
| Absolute neutrophil count | 5.70 (4.22–7.96) | 5.27 (3.92–7.54) | 6.57 (4.93–8.23) |
| Platelet count | 209 (160–281) | 208 (160–280) | 209 (158–287) |
| In-hospital treatments | |||
| Steroids | 23 (10) | 14 (9) | 9 (12) |
| Remdesivir | 6 (3) | 4 (3) | 2 (3) |
| Tocilizumab | 16 (7) | 15 (10) | 1 (1) |
| Inhaled nitric oxide | 12 (5) | 4 (3) | 8 (11) |
Definition of abbreviations: ACE = angiotensin-converting enzyme; ARDS = acute respiratory distress syndrome; COPD = chronic obstructive pulmonary disease; S:F = SpO2/FiO2 ratio; SOFA = Sequential Organ Failure Assessment; SpO2 = oxygen saturation as measured by pulse oximetry; P:F = PaO2/FiO2 ratio.
Continuous variables presented as mean (SD) or as median (interquartile range).
Hypoxemia severity based hierarchically on P:F or, when P:F unavailable, the S:F.
Indicates ARDS by modified Berlin criteria (i.e., both bilateral radiographic opacities and either P:F < 300 or S:F < 315 believed unlikely to be in the setting of cardiac failure or volume overload). Hypoxemia despite > 5 cm H2O of positive end-expiratory pressure could not be assessed among patients who received only supplemental oxygen.
Figure 1.
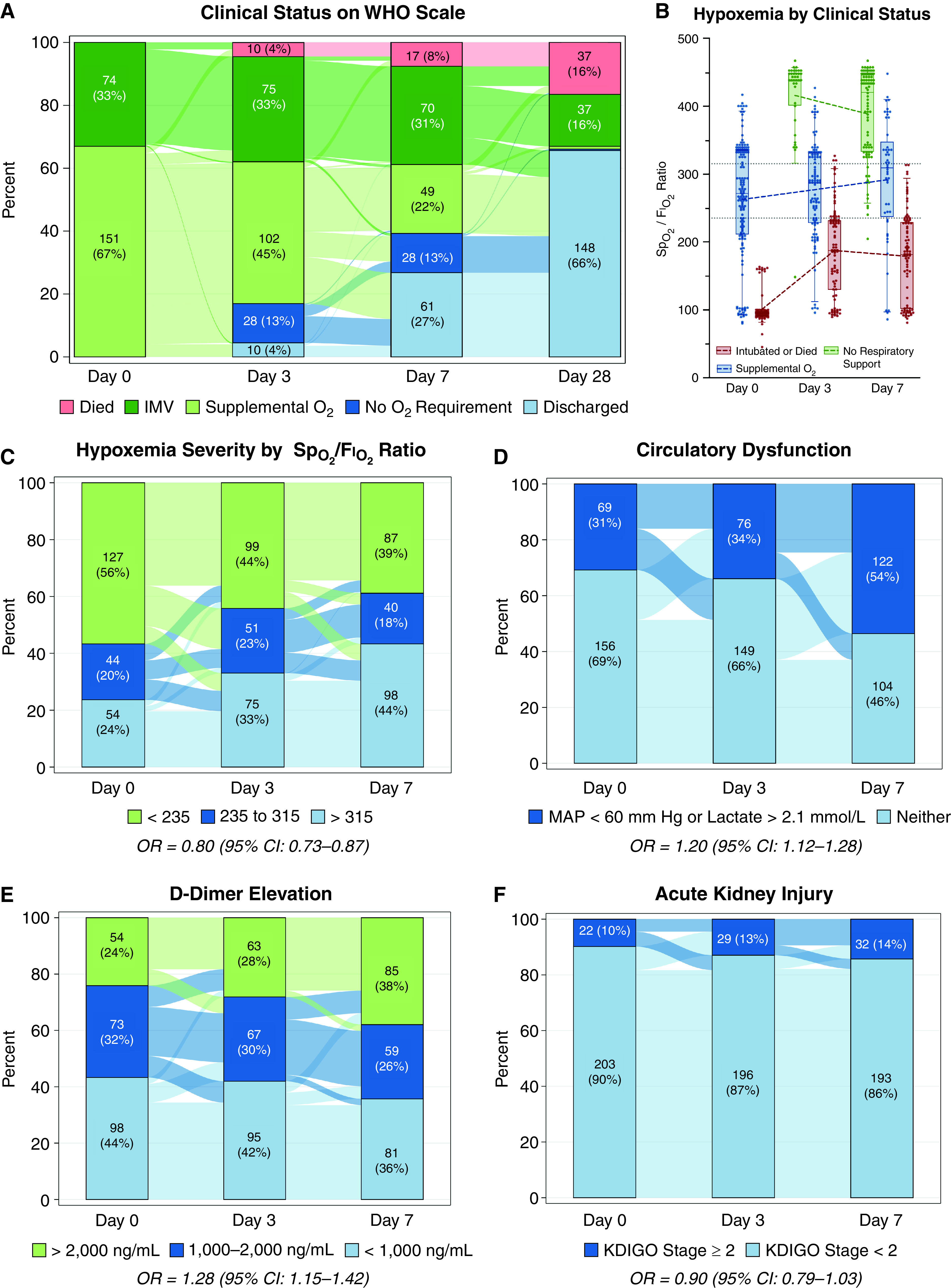
Longitudinal clinical status and organ dysfunctions. (A) Longitudinal distribution of patients by modified World Health Organization (WHO) scale. Lines reflect the temporal redistribution or maintenance of patients between WHO scale levels. (B) Oxygen saturation as measured by pulse oximetry (SpO2)/FiO2 ratio for each patient at Day 0, 3, and 7 by level of respiratory support. Box, 25th to 75th percentiles; whiskers, 5th to 95th percentiles; dots, individual patients. (C–F) Severity of hypoxemia, presence of circulatory dysfunction, degree of D-dimer elevation, and presence of stage 2 or higher acute kidney injury, respectively. Odds ratios (ORs) display the change in odds of most severe dysfunction per day in a simple mixed effects logistic model with subject as a random effect and day as a fixed effect. Plotted as in A. CI = confidence interval; IMV = invasive mechanical ventilation; KDIGO = Kidney Disease: Improving Global Outcomes; MAP = mean arterial pressure. Panel A adapted by permission from Reference 9.
Among patients intubated on Day 0, hypoxemia generally improved over time. Although ventilatory ratios also increased over time (Figure 2), suggesting increasing dead space ventilation, a minority of patients reached a high ventilatory ratio (⩾2.0), increasing from 7 (9%) on Day 0 to 23 (31%) by Day 7.
Figure 2.
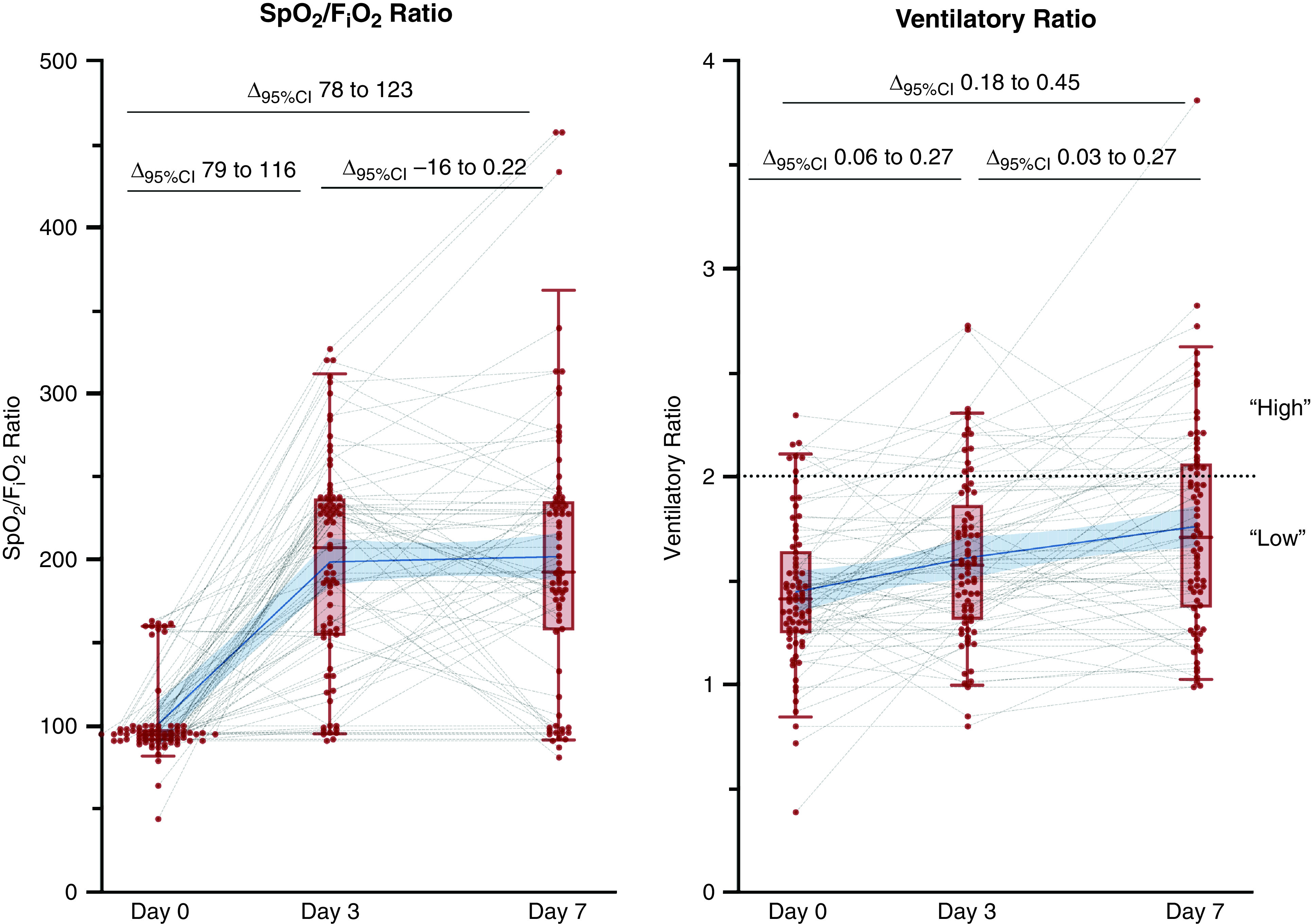
Oxygen saturation as measured by pulse oximetry (SpO2)/FiO2 and ventilatory ratios over time among patients who were invasively ventilated at Day 0. Gray dotted lines show individual patients’ trajectories. Red dots are individual patient data points. Δ95% indicates the 95% confidence interval (CI) for average slope between the two corresponding time points (Day 0, Day 3, or Day 7). Blue line and shaded area indicate mean slope between time points and 95% CI for the mean, respectively. The dotted horizontal line on the ventilatory ratio graph demarcates ventilatory ratios > 2.0, which are considered high.
Alveolar Injury Markers Peak Early and Decrease over Time
Day 0 alveolar injury markers were generally higher in patients requiring invasive ventilation than those requiring only supplemental oxygen (Figures 3, 4, E2, and E3). Although remaining higher among invasively ventilated patients, these markers decreased over time in both groups (Figure 4; modeling strategy-2), and the differences between the two became less pronounced (Figure 3; modeling strategy-1). This pattern was consistent across the alveolar injury markers receptor for advanced glycation endproducts (RAGE) and surfactant proteins A1 and A2, but not surfactant protein D or lysosome-associated membrane protein-3 (LAMP3). Alveolar markers had moderate to poor discrimination for invasive ventilation at Day 0 (Table E4). C-statistics ranged between a maximum of 0.76 (95% confidence interval [CI], 0.69–0.83) for RAGE to a minimum of 0.60 (95% CI, 0.51–0.68) for LAMP3. At Day 3, the C-statistic was ⩽0.72 for all alveolar injury markers.
Figure 3.
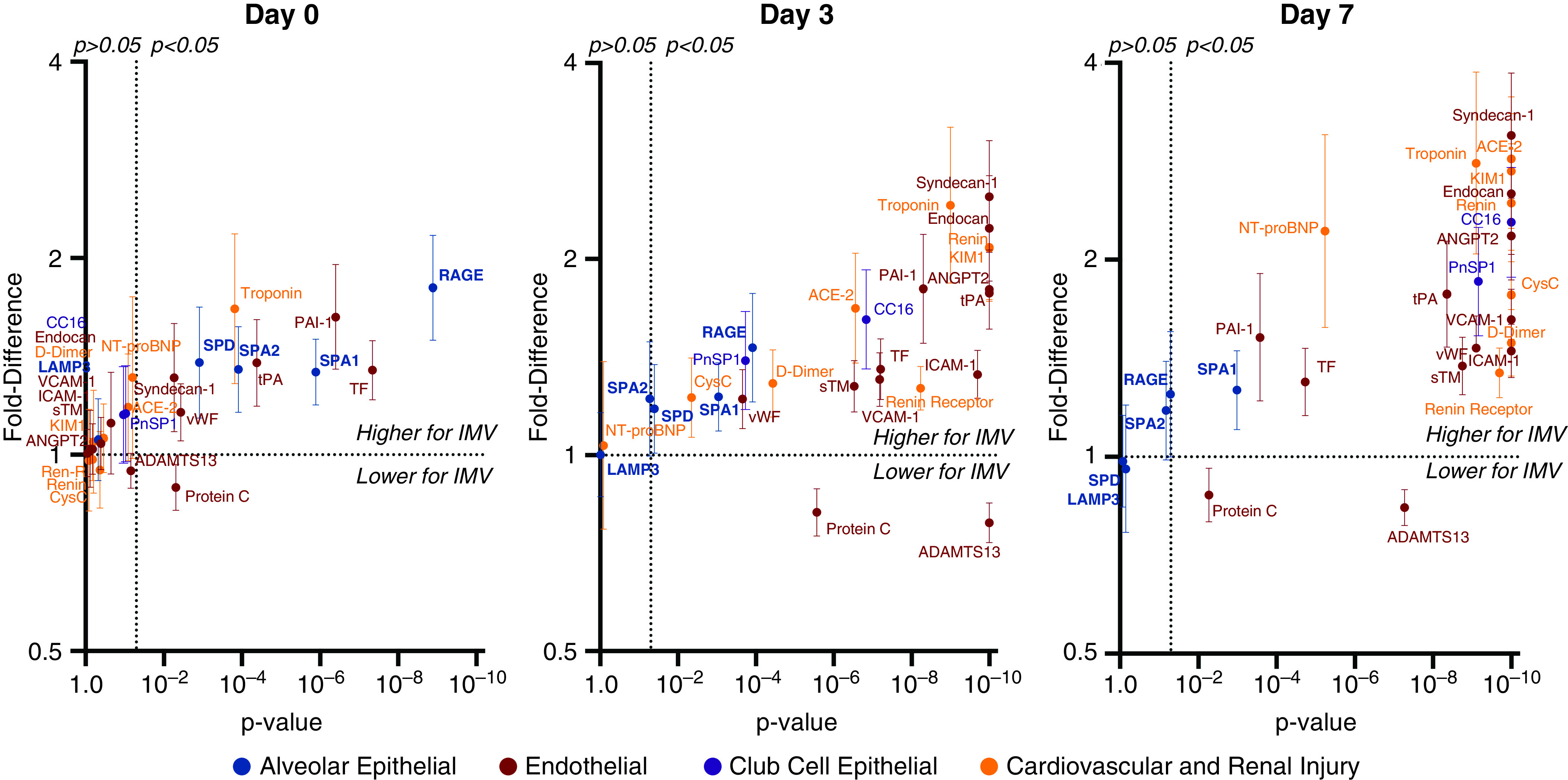
Differences in alveolar, endothelial, club cell, and cardiovascular injury markers among intubated versus nonintubated patients with coronavirus disease (COVID-19) at Day 0, 3, and 7. Multivariable estimates expressed as fold difference in biomarker levels between patients who were invasively ventilated or who died (n = 74) versus patients who were alive and ventilating spontaneously (n = 151), by study day. The y-axis shows the fold difference in biomarker level and the x-axis the P value. Error bars indicate the 95% confidence interval for the fold difference. The horizontal dotted lines indicate a fold difference of 1.0 (no difference). The vertical dotted lines indicate P = 0.05. IMV = invasive mechanical ventilation.
Figure 4.
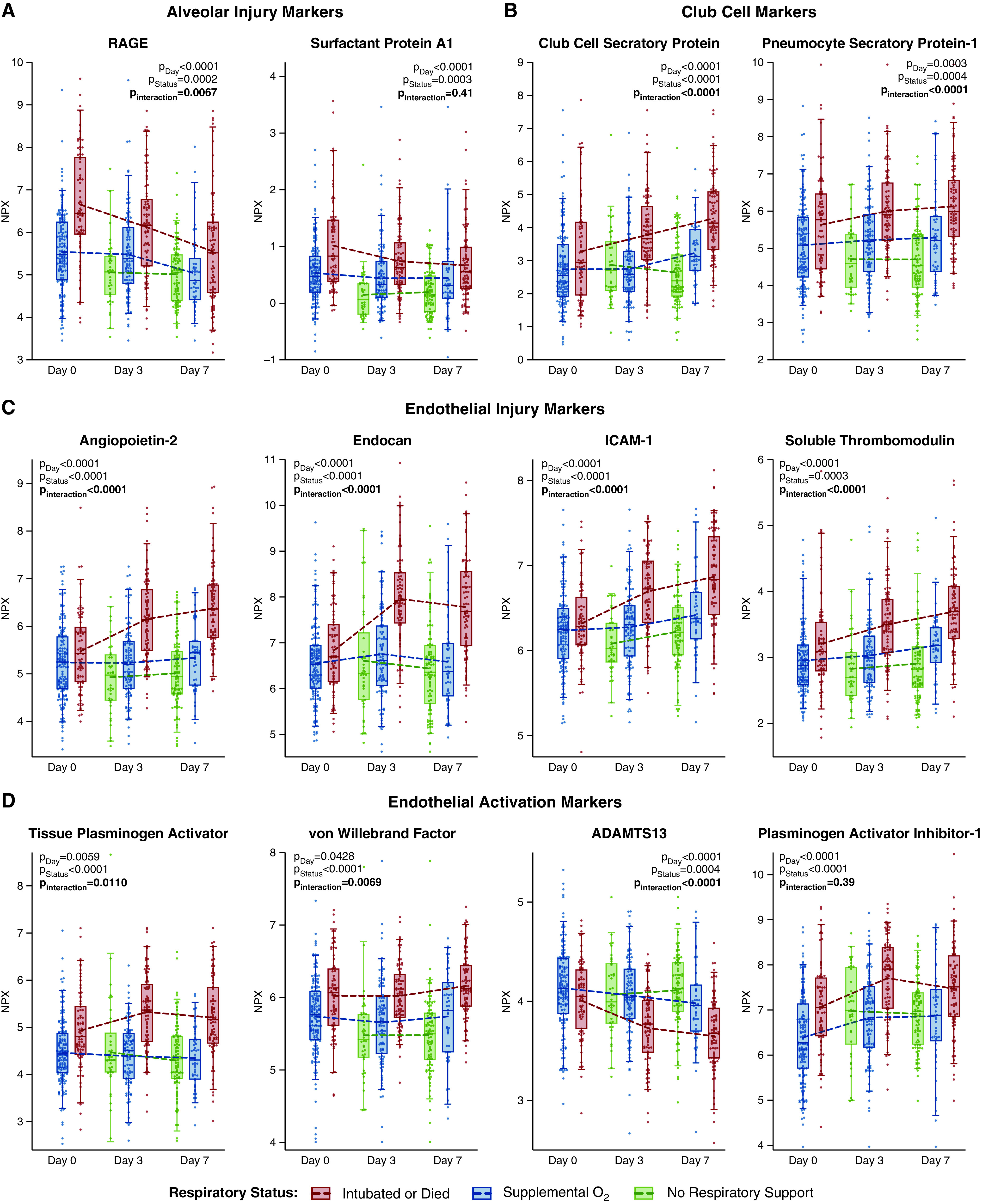
Plasma levels of alveolar injury, endothelial activation, and endothelial injury markers show distinct patterns over time. Representative markers of (A) alveolar, (B) club cell, and (C and D) endothelial injury and activation over the study period by level of respiratory support. P values indicate the statistical hypothesis tests from the multivariable mixed-effects repeated measures models for class differences by time in study (day), level of respiratory support (status), and whether respiratory support level is an effect modifier for time (interaction). Each dot represents an individual patient. Box, 25th to 75th percentiles; whiskers, 5th to 95th percentiles. Dashed lines connect the means at each time point. Normalized protein expression (NPX) units are on a log2 scale (i.e., a 1-unit increase corresponds to a doubling in level). ADAMTS13 = a disintegrin and metalloproteinase with thrombospondin motifs 13; ICAM-1 = intercellular adhesion molecule 1; RAGE = receptor for advanced glycation endproducts.
Results were similar across all three modeling strategies (Figures 3, 4, and E4; strategies 1, 2, and 3, respectively.).
Endothelial Marker Dynamics Are Distinct from Alveolar Marker Dynamics
The dynamics of endothelial injury markers differed from those of alveolar markers (Figures 3, 4C, 4D, E3, and E4). At Day 0, endothelial injury markers were comparable for all respiratory support levels (Figures 3, 4, and E4). Unlike alveolar markers, after Day 0, endothelial markers increased (Figure 4), and by Day 3, most were significantly higher in invasively ventilated patients than nonintubated patients (Figures 3, 4, and E3). The endothelial markers tPA (tissue-type plasminogen activator), PAI-1 (plasminogen activator inhibitor 1), tissue factor, and protein C were exceptions, demonstrating significantly more anomalous levels in intubated patients at all times. ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin motifs 13), which is consumed during endothelial activation, showed an inverse pattern, where levels were significantly lower in invasively ventilated patients (Pinteraction < 0.0001). All three methods of specifying respiratory severity, complete-case analysis, and analysis excluding patients who received steroids or tocilizumab yielded similar results (Figures 3, 4, and E4–E7).
For nearly all endothelial injury markers, discrimination for invasive ventilation was initially poor but markedly increased over time (Table E4). Day 0 C-statistics were less than 0.60 for angiopoitein-2 (0.58; 95% CI, 0.50–0.66), endocan (0.59; 95% CI, 0.51–0.67), and ICAM-1 (intercellular adhesion molecule 1) (0.55; 95% CI, 0.47–0.63), whereas Day 3 C-statistics were 0.82 for angiopoietin-2 (95% CI, 0.76–0.88; Δc, 0.25), 0.78 for endocan (95% CI, 0.72–0.84; Δc, 0.19), 0.78 for ICAM-1 (95% CI, 0.71–0.84; Δc, 0.23) and greater than 0.70 for all others.
Among intubated patients, higher endothelial injury marker levels were not reliably associated with higher ventilatory ratios (Figure E8).
Club Cell Proteins Displayed Dynamics Similar to Those of Endothelial Markers
Club cell proteins followed the pattern of endothelial rather than alveolar markers (Figures 3, 4, E2, and E3). Club cell secretory protein (CC16) poorly discriminated respiratory severity at Day 0 (C-statistic, 0.59) but improved by Day 3 (C-statistic, 0.72; Δc, 0.13) (Table E4). Moreover, changes in club cell markers showed greater correlation with endothelial markers (Table E5).
COVID-19 versus Non–COVID-19
In multivariable regression, in invasively ventilated subjects, Day 0 alveolar injury markers were consistently higher in patients with COVID-19 than control subjects without COVID, while most endothelial injury markers were not (Figure E9). COVID-19–negative patients were not followed past Day 0.
RAS Activation and Cardiac and Renal Injury Markers Mirror the Endothelial Pattern
To explore whether endothelial abnormalities reflect transitions from predominantly pulmonary injury to dysregulated systemic responses, we examined markers of cardiac, renal, and vascular disturbances. Among these, markers of RAS activation, cardiac injury, and kidney injury showed dynamics similar to the endothelial pattern (Figures 3 and 5), with similarities between intubated and nonintubated patients at Day 0, yet at Days 3 and 7, significant and progressively higher levels in intubated versus nonintubated patients. Further suggestive of an increasingly systemic response, changes in cardiorenovascular injury markers were closely correlated with changes in endothelial, RAS activation, and club cell markers but not alveolar markers (Table E5 and E6).
Figure 5.

Plasma levels of renin–angiotensin system activation, cardiac injury, and renal injury markers over time show patterns similar to endothelial markers. Representative markers of (A) renin–angiotensin system activation, and (B and C) cardiorenal injury and dysfunction over the study period by level of respiratory support. P values indicate the results of the statistical hypothesis tests from the multivariable mixed-effects repeated measures models for class differences by time in study (day), level of respiratory support (status), and whether respiratory support level is an effect modifier for time (interaction). Each dot represents an individual patient. Box, 25th to 75th percentiles; whiskers, 5th to 95th percentiles. Dashed lines connect the means at each time point. Normalized protein expression (NPX) units are on a log2 scale (i.e., a 1-unit increase corresponds to a doubling in level). ACE-2 = angiotensin-converting enzyme-2; NT-proBNP = N-terminal pro-brain natriuretic peptide.
Some Inflammatory Markers Show Dynamics Similar to Endothelial Markers
Changes in soluble tumor necrosis factor receptor 1 (Pinteraction < 0.0001) and d-dimer (Pinteraction = 0.0001) levels mirrored the endothelial pattern and were more strongly correlated with cardiac and renal injury markers than IL-6, IL-8, or C-reactive protein. The latter, as described (14–17), were persistently higher among ventilated patients (Figures E10–E12 and Tables E7 and E8).
Patients with Delayed Respiratory Failure Had Late Increases in Endothelial Markers
Among patients on supplemental oxygen at Day 0, 17 (11%) were intubated or deceased by Day 7. Their biomarker course more closely resembled patients intubated on Day 0 than initially hypoxemic patients who were never intubated (Figure E13).
28-Day Outcomes Were More Closely Associated with Endothelial Markers than Alveolar Markers
In multivariable proportional-odds analysis, worse 28-day outcome was significantly associated with Day 0 values of protein C, but not the other 11 endothelial markers, and none of the 7 epithelial markers (Figure 6 and Table E9). However, Day 3 values were significantly associated with 28-day WHO scale for 11 of 12 (92%) endothelial versus 3 of 7 (43%) epithelial markers. Endothelial effect sizes at Day 3 were significantly larger than epithelial effect sizes (median adjusted OR, 3.49 [interquartile range (IQR), 2.41–5.46] versus 1.57 [IQR, 1.05–2.73]; P = 0.0297). Among endothelial markers, adjusted ORs were uniformly larger for Day 3 than Day 0 values (median adjusted OR, 3.49 vs. 1.26 [IQR, 1.22–1.40]; P = 0.0023).
Figure 6.
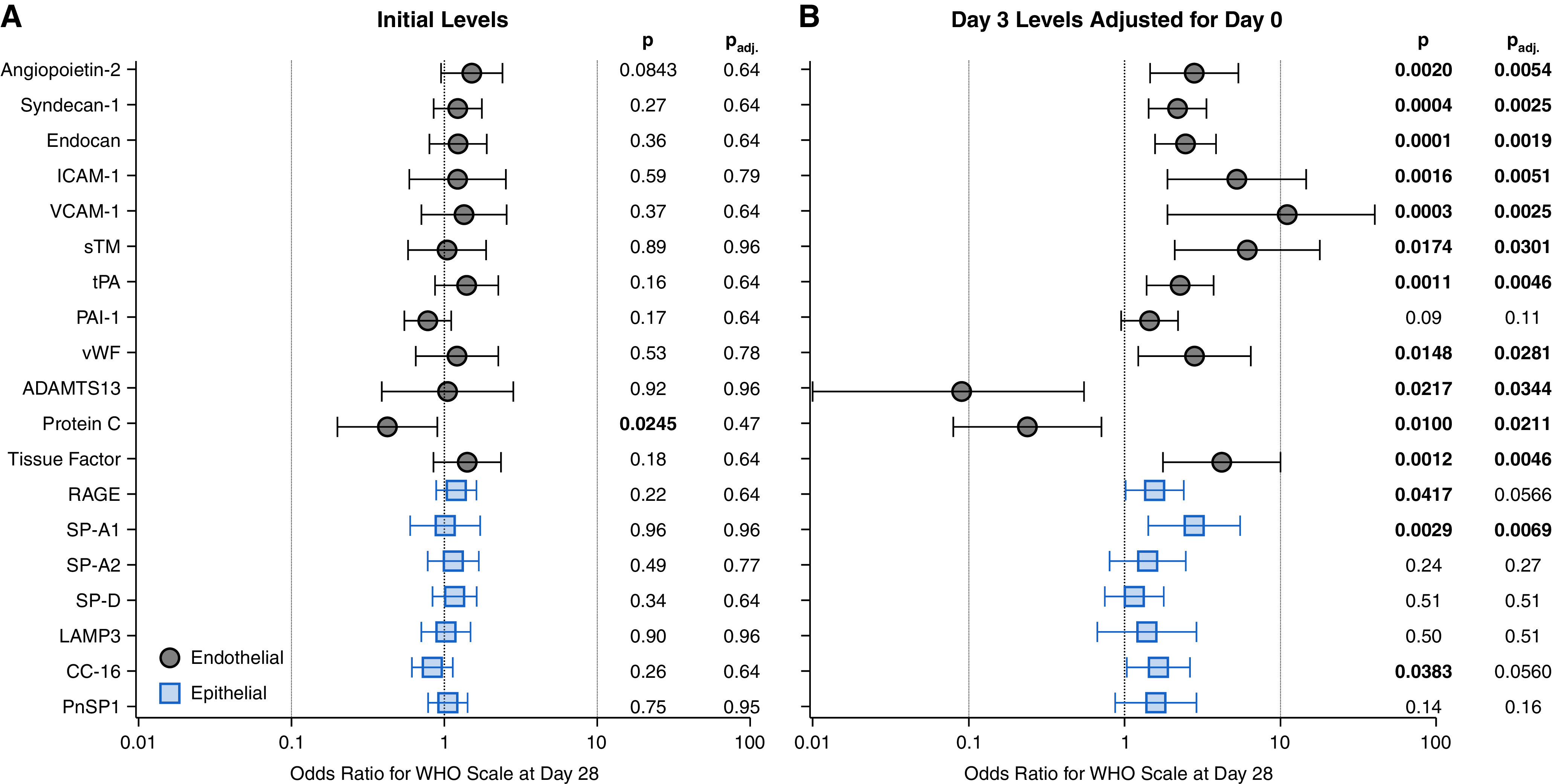
Association of initial levels and changes in level of plasma epithelial and endothelial markers with 28-day clinical outcome. Adjusted odds ratios of 12 endothelial and 7 pulmonary epithelial markers based on multivariable proportional odds models for modified World Health Organization (WHO) status at Day 28 for (A) Day 0 levels, and (B) changes in marker level from Day 0 to Day 3. Response levels were died (n = 37, 16%), invasive mechanical ventilation (n = 37, 16%), or off mechanical ventilation (n = 151, 67%; 148 [98%] of whom were discharged alive from the hospital at Day 28). The response is coded in ascending order such that a higher odds ratio indicates worse clinical status at Day 28. All models adjusted for age, sex, body mass index, initial Sequential Organ Failure Assessment (SOFA) score, heart failure, chronic kidney disease, and chronic obstructive pulmonary disease. Error bars, 95% confidence intervals. The right-hand columns display the model estimated P value (P) and the P value after a false-discovery rate correction (Padj). ADAMTS13 = a disintegrin and metalloproteinase with thrombospondin motifs 13; CC-16 = club cell secretory protein; ICAM-1 = intercellular adhesion molecule 1; LAMP3 = lysosome-associated membrane glycoprotein 3; PAI-1 = plasminogen activator inhibitor 1; PnSP1 = pneumocyte secretory protein 1; RAGE = receptor for advanced glycation endproducts; SP-A1 = pulmonary surfactant-associated protein A1; SP-A2 = pulmonary surfactant-associated protein A2; SP-D = pulmonary surfactant-associated protein D; sTM = soluble thrombomodulin; tPA = tissue-type plasminogen activator; VCAM-1 = vascular cell adhesion protein 1; vWF = von Willebrand factor.
RAS markers (renin, renin receptor, and angiotensin-converting enzyme-2 [ACE2]) displayed an endothelial-like association with 28-day outcome. Although Day 0 levels were not associated with 28-day outcome, worse Day 28 clinical status was significantly associated with Day 3 renin (OR, 1.76; 95% CI, 1.08–2.88; P = 0.0246), renin receptor (OR, 5.28; 95% CI, 1.64–17.04; P = 0.0054), and ACE2 (OR, 1.85; 95% CI, 1.19–2.88; P = 0.0065) (Table E10).
Discussion
In this cohort of 225 patients with COVID-19 requiring respiratory support at presentation, we observed distinct and consistent patterns in the dynamics of alveolar and endothelial injury markers: 1) elevations in alveolar injury markers diminish over time in both mechanically ventilated patients receiving lung-protective ventilation and spontaneously breathing patients; 2) elevations in endothelial markers are delayed, are limited to invasively ventilated patients, and correlate with nonpulmonary organ injury; and 3) among intubated patients, despite severe hypoxemia, ventilatory ratios are not prominently elevated early.
First, the finding that RAGE and surfactant proteins peak at Day 0 suggests that alveolar injury represents an early insult in COVID-induced respiratory failure. This is consistent with our prior proteomic analyses showing strong association of Day 0 RAGE levels with disease severity (10). Other cohorts also report higher RAGE levels in COVID-19 versus non-COVID ARDS (18, 19). The similar decrease in alveolar markers between intubated and nonintubated patients over time could indicate that lung-protective ventilation allows for alveolar recovery to a similar degree as spontaneous breathing. In the context of the controversy surrounding early intubation in COVID-19 and the opposing concerns of ventilator versus self-inflicted lung injuries, we note institutional practice during the study period was early intubation; noninvasive mechanical ventilation was prohibited. However, decreasing alveolar injury markers clearly do not equate with clinical recovery, as many patients required prolonged intubation despite downward trajectories in alveolar markers.
Second, the delayed peak in endothelial markers among intubated patients suggests that unlike alveolar injury, endothelial activation and injury are prominent features of later severe disease. Consistent with our findings, patients with COVID-19 display conspicuous pulmonary vasodilation (5), glycocalyx damage (20, 21), postmortem endothelialitis (8), and elevated von Willebrand factor with low levels of ADAMTS13 (22, 23). Comparisons of COVID-19 and non-COVID-19 ARDS report comparable to lower initial levels of endothelial injury markers, such as angiopoietin-2, in COVID-19 ARDS (18, 19).
The shift from alveolar- to endothelial-predominant injury may alternatively, or additionally, indicate that the “natural” course of COVID-19 respiratory failure involves a transition from a primarily lung-localized pathology to a systemic one. Herein, increased endothelial injury markers coincided with increased extrapulmonary organ injury and dysfunction and were more strongly associated with 28-day outcome than alveolar markers. In ARDS, trajectories of endothelial markers vary (24, 25), but higher levels consistently portend worse organ dysfunction and outcomes (25–29). In COVID-19, endothelial injury markers are associated with sustained elevations in viral mRNA (30). Therefore, the delayed increase in endothelial markers we observed might in part reflect increased systemic viral-induced injury.
Third, ventilatory ratios increased over time but were infrequently elevated and not consistently associated with endothelial injury. In contrast, SpO2/FiO2 was lowest at Day 0 and increased over time. A simple explanation might be imprecision in the ventilatory ratio as an estimate of dead space. However, intubated patients with COVID-19 undergoing right heart catheterization show increased cardiac output with elevated pulmonary capillary wedge pressure but atypically low pulmonary vascular resistance and comparable intrapulmonary shunt versus control subjects with non–COVID-19 ARDS (31). These observations suggest excess pulmonary vasodilation may contribute to severe COVID-19 hypoxemia, at least early on. This hypothesis is consistent with prominent early alveolar injury with minimal endothelial injury and does not necessarily invoke increased dead space ventilation. We note ventilatory ratios were infrequently elevated in our study but increased over time. This could simply reflect aggressive low-tidal-volume ventilation and permissive hypercapnia. Alternatively, it could suggest intrapulmonary thrombosis accrues over time.
Together, these results may explain the seemingly contradictory findings of the Accelerating COVID-19 Therapeutic Interventions and Vaccines 4 ACUTE (ACTIV-4A) trial. In ACTIV4A, full-dose heparin improved mortality in hospitalized nonintubated patients (32), but for intubated patients it was not beneficial and suggested harm (33). Heparin-treated critically ill patients showed no mortality benefit despite a nearly twofold reduction in major thrombotic events (5.7% vs. 10.3%) and similar frequency of (clinically detected) major bleeding (3.1% vs. 2.4%). Alveolar hemorrhage is common at autopsy in COVID-19 (7). Therapeutic anticoagulation likely exacerbates risk of alveolar hemorrhage in patients with severe alveolar injury. We found alveolar injury markers were highest on Day 0 and among intubated patients. The delayed endothelial injury occurring among intubated patients could reflect alveolar bleeding and vessel injury, rather than thrombosis alone, an interpretation supported by the lack of association between endothelial markers and ventilatory ratios. Both intubated and nonintubated patients in ACTIV4A were enrolled at presentation (our study’s Day 0). Therefore, our results suggest anticoagulation of intubated patients in ACTIV4A was initiated when alveolar bleeding risk was high, and thrombosis was not a significant contributor to hypoxemia. In contrast, at Day 0, nonintubated patients display less severe alveolar injury and similar levels of endothelial injury, such that these patients would be expected to have lower risk of alveolar bleeding and therefore be more likely to benefit from anticoagulation.
We included patients requiring both invasive and noninvasive respiratory support, reasoning their common insult (i.e., COVID-19 pneumonia) made them an ideal biological comparator. Extending validity to this approach, “inflammatory” ARDS phenotypes are validated in patients at risk of ARDS who do not yet meet Berlin criteria (25). Some patients on supplemental oxygen might have met Berlin ARDS criteria had they received positive end-expiratory pressure (PEEP); institutional policy prohibited noninvasive positive-pressure ventilation during the study period. In addition, not all patients had an arterial blood-gas on Day 0. For this reason, we used modified Berlin criteria (34) that did not consider PEEP and that used SpO2/FiO2 when PaO2 data were missing to designate ARDS.
The implications of the late increase in club cell markers are unclear. Injury of distal airways, the site of club cells, occurring after alveolar damage seems unlikely given that SARS-CoV-2 reaches alveoli via the airways. In vitro, club cells are particularly susceptible to SARS-CoV-2 infection, which induces inflammatory cytokine secretion (35). Although higher club cell markers were associated herein with greater disease severity, lower levels of club cell secretory protein have been previously associated with non-COVID-19 ARDS, albeit not with outcome (36, 37).
RAS activation markers displayed marked elevation, with dynamics similar to endothelial injury. They correlated closely with cardiac and renal injury, albeit only in intubated patients, consistent with trials showing RAS antagonism does not modify disease course in moderate and mild COVID-19 (38). Because our study did not measure angiotensin-II or ACE1, we cannot discriminate primary hyperreninemia (i.e., true RAS activation) from a compensatory response to a relative hypo-RAS state, as described in distributive shock (39). Suggesting the latter are high levels of plasma ACE2, which suppresses RAS and is catalytically active in COVID-19 (40, 41), and decreases in angiotensin-II levels over time in COVID-19 ARDS (42). The close correlations we observed between renin and markers of renal and cardiac injury further support attenuated RAS effects, as described in sepsis and distributive shock and after cardiac surgery (43–48). Alternatively, renin elevations could reflect responses to prolonged sedation-induced vasodilatation or hypovolemia following diuresis, representing the balance in ARDS management between protecting the lung and protecting the kidney. Regardless of mechanism, these findings indicate that renin’s utility as a marker of hypoperfusion (49) and prognosticator of cardiovascular and renal injury (39, 48) in severe COVID-19 and ARDS should be explored.
This work differs from the previously published analysis of this cohort (10) based on 1) prespecified biomarker selection rather than unbiased discovery analysis; 2) focus on patients presenting with hypoxemia rather than all comers; and 3) specific interrogation of lung epithelial, endothelial, and nonpulmonary organ injury markers. These tissue-specific injury markers may inform disease biology more than characterizations of nonspecific inflammatory markers alone.
Important limitations of this study include its single-center observational design, which facilitates thorough description but precludes causal evaluation of any markers under investigation. Specifically, we cannot definitively discern whether chronological differences between intubated and nonintubated patients result from COVID-19 severity or mechanical ventilation itself. In addition, because we enrolled subjects before the approval of remdesivir and dexamethasone treatment for COVID-19, these drugs were not administered to most subjects. Although this may limit extrapolation to current practice, it presents a valuable opportunity to observe biology unconfounded by the pleiotropic effects of steroids. Furthermore, although proximity extension assays increase assay sensitivity and enable detection of plasma proteins that cannot be detected by other methods, normalized protein expression units cannot be translated to ELISA-based readouts, which precluded applying validated models for ARDS phenotype identification (27). Multiple comparison adjustments were applied only to biomarker associations with patient outcomes. Finally, as with all biomarker studies, protein levels in plasma variably reflect those in tissue compartments.
Conclusions
Alveolar injury markers peak early in severe COVID-19 and decrease among both spontaneously breathing and invasively ventilated patients. Endothelial injury markers increase with delayed kinetics and are significantly associated with evidence of systemic inflammation, renin-angiotensin system activation, extrapulmonary organ injury, and 28-day outcome. In severe COVID-19, alveolar and endothelial injury likely contribute at different times to disease progression.
Acknowledgments
Acknowledgment
The authors thank Arthur, Sandra, and Sarah Irving for a gift that enabled this study and funded the David P. Ryan, M.D., Endowed Chair in Cancer Research (N.H.). They also thank Olink Proteomics Inc. for providing in-kind all proteomics assays presented in this work, without which our findings would not have been possible.
Footnotes
Supported in part by National Institutes of Health (NIH) grant U19 AI082630 (N.H.), an American Lung Association COVID-19 Action Initiative grant (M.B.G.), and grants from the Executive Committee on Research at MGH (M.B.G.) and the Chan-Zuckerberg Initiative (A.-C.V.). This work was also supported by the Harvard Catalyst/Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, NIH award UL1 TR 002541-01).
Author Contributions: Conceptualization: D.E.L., A.M., N.H., M.R.F., and M.B.G. Resources: M.R.F., N.H., M.B.G., and I.G. Methodology: D.E.L., A.M., N.H., M.R.F., M.B.G., M.S.-F., A.-C.V., B.A.P., I.G., and B.T.T. Investigation: All authors. Formal analysis: D.E.L. and A.M. Writing – original draft: D.E.L. Writing – review and editing: D.E.L., A.M., B.T.T., N.H., M.R.F., and M.B.G.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202106-1514OC on December 8, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers . 2019;5:18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilson JG, Calfee CS. ARDS Subphenotypes: understanding a Heterogeneous Syndrome. Crit Care . 2020;24:102. doi: 10.1186/s13054-020-2778-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Calfee CS, Janz DR, Bernard GR, May AK, Kangelaris KN, Matthay MA, et al. Distinct molecular phenotypes of direct vs indirect ARDS in single-center and multicenter studies. Chest . 2015;147:1539–1548. doi: 10.1378/chest.14-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iba T, Levy JH, Levi M, Connors JM, Thachil J. Coagulopathy of coronavirus disease 2019. Crit Care Med . 2020;48:1358–1364. doi: 10.1097/CCM.0000000000004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reynolds AS, Lee AG, Renz J, DeSantis K, Liang J, Powell CA, et al. Pulmonary vascular dilatation detected by automated transcranial Doppler in COVID-19 pneumonia. Am J Respir Crit Care Med . 2020;202:1037–1039. doi: 10.1164/rccm.202006-2219LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mirsadraee S, Gorog DA, Mahon CF, Rawal B, Semple TR, Nicol ED, et al. Prevalence of thrombotic complications in ICU-treated patients with coronavirus disease 2019 detected with systematic CT scanning. Crit Care Med . 2021;49:804–815. doi: 10.1097/CCM.0000000000004890. [DOI] [PubMed] [Google Scholar]
- 7. Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med . 2020;173:268–277. doi: 10.7326/M20-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med . 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leisman D, Mehta A. MGH COVID-19 Processing Team, Hacohen N, Filbin MR, Goldberg MB. Trajectories of pulmonary epithelial and endothelial injury markers in COVID-19 patients requiring respiratory support at presentation [abstract] Am J Respir Crit Care Med . 2021;203:A1061. [Google Scholar]
- 10. Filbin MR, Mehta A, Schneider AM, Kays KR, Guess JR, Gentili M, et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell Rep Med . 2021;2:100287. doi: 10.1016/j.xcrm.2021.100287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sinha P, Calfee CS, Beitler JR, Soni N, Ho K, Matthay MA, et al. Physiologic analysis and clinical performance of the ventilatory ratio in acute respiratory distress syndrome. Am J Respir Crit Care Med . 2019;199:333–341. doi: 10.1164/rccm.201804-0692OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ware LB, Calfee CS. Biomarkers of ARDS: what’s new? Intensive Care Med . 2016;42:797–799. doi: 10.1007/s00134-015-3973-0. [DOI] [PubMed] [Google Scholar]
- 13. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics: tissue-based map of the human proteome. Science . 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 14. Leisman DE, Ronner L, Pinotti R, Taylor MD, Sinha P, Calfee CS, et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir Med . 2020;8:1233–1244. doi: 10.1016/S2213-2600(20)30404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sinha P, Calfee CS, Cherian S, Brealey D, Cutler S, King C, et al. Prevalence of phenotypes of acute respiratory distress syndrome in critically ill patients with COVID-19: a prospective observational study. Lancet Respir Med . 2020;8:1209–1218. doi: 10.1016/S2213-2600(20)30366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. England JT, Abdulla A, Biggs CM, Lee AYY, Hay KA, Hoiland RL, et al. Weathering the COVID-19 storm: lessons from hematologic cytokine syndromes. Blood Rev . 2021;45:100707. doi: 10.1016/j.blre.2020.100707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arabi YM, Jawdat D, Hajeer AH, Sadat M, Jose J, Vishwakarma RK, et al. Inflammatory response and phenotyping in severe acute respiratory infection from the Middle East respiratory syndrome coronavirus and other etiologies. Crit Care Med . 2021;49:228–239. doi: 10.1097/CCM.0000000000004724. [DOI] [PubMed] [Google Scholar]
- 18. Spadaro S, Fogagnolo A, Campo G, Zucchetti O, Verri M, Ottaviani I, et al. Markers of endothelial and epithelial pulmonary injury in mechanically ventilated COVID-19 ICU patients. Crit Care . 2021;25:74. doi: 10.1186/s13054-021-03499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhatraju PK, Morrell ED, Zelnick L, Sathe NA, Chai XY, Sakr SS, et al. Comparison of host endothelial, epithelial and inflammatory response in ICU patients with and without COVID-19: a prospective observational cohort study. Crit Care . 2021;25:148. doi: 10.1186/s13054-021-03547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stahl K, Gronski PA, Kiyan Y, Seeliger B, Bertram A, Pape T, et al. Injury to the endothelial glycocalyx in critically ill patients with COVID-19. Am J Respir Crit Care Med . 2020;202:1178–1181. doi: 10.1164/rccm.202007-2676LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fraser DD, Patterson EK, Slessarev M, Gill SE, Martin C, Daley M, et al. Endothelial injury and glycocalyx degradation in critically ill coronavirus disease 2019 patients: implications for microvascular platelet aggregation. Crit Care Explor . 2020;2:e0194. doi: 10.1097/CCE.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delrue M, Siguret V, Neuwirth M, Joly B, Beranger N, Sène D, et al. von Willebrand factor/ADAMTS13 axis and venous thromboembolism in moderate-to-severe COVID-19 patients. Br J Haematol . 2021;192:1097–1100. doi: 10.1111/bjh.17216. [DOI] [PubMed] [Google Scholar]
- 23. Doevelaar AAN, Bachmann M, Hölzer B, Seibert FS, Rohn BJ, Bauer F, et al. von Willebrand factor multimer formation contributes to immunothrombosis in coronavirus disease 2019. Crit Care Med . 2021;49:e512–e520. doi: 10.1097/CCM.0000000000004918. [DOI] [PubMed] [Google Scholar]
- 24. Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA, NHLBI ARDS Network Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med . 2012;40:1731–1737. doi: 10.1097/CCM.0b013e3182451c87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kitsios GD, Yang L, Manatakis DV, Nouraie M, Evankovich J, Bain W, et al. Host-response subphenotypes offer prognostic enrichment in patients with or at risk for acute respiratory distress syndrome. Crit Care Med . 2019;47:1724–1734. doi: 10.1097/CCM.0000000000004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA, NHLBI ARDS Network Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med . 2014;2:611–620. doi: 10.1016/S2213-2600(14)70097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sinha P, Delucchi KL, McAuley DF, O’Kane CM, Matthay MA, Calfee CS. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: a secondary analysis of randomised controlled trials. Lancet Respir Med . 2020;8:247–257. doi: 10.1016/S2213-2600(19)30369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, et al. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med . 2018;6:691–698. doi: 10.1016/S2213-2600(18)30177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Short SAP, Gupta S, Brenner SK, Hayek SS, Srivastava A, Shaefi S. d-dimer and death in critically ill patients with coronavirus disease 2019. Crit Care Med . 2021;49:e500–e511. doi: 10.1097/CCM.0000000000004917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Schneider AM, Mehta A, Sade-Feldman M, Kays KR, Gentili M, et al. SARS-CoV-2 viremia is associated with distinct proteomic pathways and predicts COVID-19 outcomes J Clin Invest 2021131(13)e148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caravita S, Baratto C, Di Marco F, Calabrese A, Balestrieri G, Russo F, et al. Haemodynamic characteristics of COVID-19 patients with acute respiratory distress syndrome requiring mechanical ventilation: an invasive assessment using right heart catheterization. Eur J Heart Fail . 2020;22:2228–2237. doi: 10.1002/ejhf.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lawler PR, Goligher EC, Berger JS, Neal MD, McVerry BJ, Nicolau JC, et al. ATTACC Investigators ACTIV-4a Investigators; REMAP-CAP Investigators. Therapeutic anticoagulation with heparin in noncritically ill patients with Covid-19. N Engl J Med . 2021;385:790–802. doi: 10.1056/NEJMoa2105911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goligher EC, Bradbury CA, McVerry BJ, Lawler PR, Berger JS, Gong MN, et al. REMAP-CAP Investigators ACTIV-4a Investigators; ATTACC Investigators. Therapeutic anticoagulation with heparin in critically ill patients with Covid-19. N Engl J Med . 2021;385:777–789. doi: 10.1056/NEJMoa2103417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. ARDS Definition Task Force Acute respiratory distress syndrome: the Berlin Definition. JAMA . 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 35. Fiege JK, Thiede JM, Nanda HA, Matchett WE, Moore PJ, Montanari NR, et al. Single cell resolution of SARS-CoV-2 tropism, antiviral responses, and susceptibility to therapies in primary human airway epithelium. PLoS Pathog . 2021;17:e1009292. doi: 10.1371/journal.ppat.1009292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kropski JA, Fremont RD, Calfee CS, Ware LB. Clara cell protein (CC16), a marker of lung epithelial injury, is decreased in plasma and pulmonary edema fluid from patients with acute lung injury. Chest . 2009;135:1440–1447. doi: 10.1378/chest.08-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ware LB, Koyama T, Zhao Z, Janz DR, Wickersham N, Bernard GR, et al. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit Care . 2013;17:R253. doi: 10.1186/cc13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cohen JB, Hanff TC, William P, Sweitzer N, Rosado-Santander NR, Medina C, et al. Continuation versus discontinuation of renin-angiotensin system inhibitors in patients admitted to hospital with COVID-19: a prospective, randomised, open-label trial. Lancet Respir Med . 2021;9:275–284. doi: 10.1016/S2213-2600(20)30558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bellomo R, Forni LG, Busse LW, McCurdy MT, Ham KR, Boldt DW, et al. Renin and survival in patients given angiotensin II for catecholamine-resistant vasodilatory shock: a clinical trial. Am J Respir Crit Care Med . 2020;202:1253–1261. doi: 10.1164/rccm.201911-2172OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel SK, Juno JA, Lee WS, Wragg KM, Hogarth PM, Kent SJ, et al. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: implications for COVID-19 pathogenesis and consequences Eur Respir J 202157(5)2003730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kragstrup TW, Singh HS, Grundberg I, Nielsen AL, Rivellese F, Mehta A, et al. Plasma ACE2 predicts outcome of COVID-19 in hospitalized patients. PLoS One . 2021;16:e0252799. doi: 10.1371/journal.pone.0252799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ozkan S, Cakmak F, Konukoglu D, Biberoglu S, Ipekci A, Akdeniz YS, et al. Efficacy of serum angiotensin II levels in prognosis of patients with coronavirus disease 2019. Crit Care Med . 2021;49:e613–e623. doi: 10.1097/CCM.0000000000004967. [DOI] [PubMed] [Google Scholar]
- 43. du Cheyron D, Lesage A, Daubin C, Ramakers M, Charbonneau P. Hyperreninemic hypoaldosteronism: a possible etiological factor of septic shock-induced acute renal failure. Intensive Care Med . 2003;29:1703–1709. doi: 10.1007/s00134-003-1986-6. [DOI] [PubMed] [Google Scholar]
- 44. Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab . 1987;64:592–595. doi: 10.1210/jcem-64-3-592. [DOI] [PubMed] [Google Scholar]
- 45. Tumlin JA, Murugan R, Deane AM, Ostermann M, Busse LW, Ham KR, et al. Outcomes in patients with vasodilatory shock and renal replacement therapy treated with intravenous angiotensin II. Crit Care Med . 2018;46:949–957. doi: 10.1097/CCM.0000000000003092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leisman DE, Fernandes TD, Bijol V, Abraham MN, Lehman JR, Taylor MD, et al. Impaired angiotensin II type 1 receptor signaling contributes to sepsis-induced acute kidney injury. Kidney Int . 2021;99:148–160. doi: 10.1016/j.kint.2020.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leisman DE, Mastroianni F, Fisler G, Shah S, Hasan Z, Narasimhan M, et al. Physiologic response to angiotensin II treatment for coronavirus disease 2019-induced vasodilatory shock: a retrospective matched cohort study. Crit Care Explor . 2020;2:e0230. doi: 10.1097/CCE.0000000000000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Küllmar M, Saadat-Gilani K, Weiss R, Massoth C, Lagan A, Cortés MN, et al. Kinetic changes of plasma renin concentrations predict acute kidney injury in cardiac surgery patients. Am J Respir Crit Care Med . 2021;203:1119–1126. doi: 10.1164/rccm.202005-2050OC. [DOI] [PubMed] [Google Scholar]
- 49. Gleeson PJ, Crippa IA, Mongkolpun W, Cavicchi FZ, Van Meerhaeghe T, Brimioulle S, et al. Renin as a marker of tissue-perfusion and prognosis in critically ill patients. Crit Care Med . 2019;47:152–158. doi: 10.1097/CCM.0000000000003544. [DOI] [PubMed] [Google Scholar]