

Abstract
Rationale
The cystic fibrosis (CF) modulator drug, elexacaftor/tezacaftor/ivacaftor (ETI), proved highly effective in controlled clinical trials for individuals with at least one F508del allele, which occurs in at least 85% of people with CF.
Objectives
PROMISE is a postapproval study to understand the broad effects of ETI through 30 months’ clinical use in a more diverse U.S. patient population with planned analyses after 6 months.
Methods
Prospective, observational study in 487 people with CF age 12 years or older with at least one F508del allele starting ETI for the first time. Assessments occurred before and 1, 3, and 6 months into ETI therapy. Outcomes included change in percent predicted FEV1 (ppFEV1), sweat chloride concentration, body mass index (BMI), and self-reported respiratory symptoms.
Measurements and Main Results
Average age was 25.1 years, and 44.1% entered the study using tezacaftor/ivacaftor or lumacaftor/ivacaftor, whereas 6.7% were using ivacaftor, consistent with F508del homozygosity and G551D allele, respectively. At 6 months into ETI therapy, ppFEV1 improved 9.76 percentage points (95% confidence interval [CI], 8.76 to 10.76) from baseline, cystic fibrosis questionnaire–revised respiratory domain score improved 20.4 points (95% CI, 18.3 to 22.5), and sweat chloride decreased −41.7 mmol/L (95% CI, −43.8 to −39.6). BMI also significantly increased. Changes were larger in those naive to modulators but substantial in all groups, including those treated with ivacaftor at baseline.
Conclusions
ETI by clinical prescription provided large improvements in lung function, respiratory symptoms, and BMI in a diverse population naive to modulator drug therapy, using existing two-drug combinations, or using ivacaftor alone. Each group also experienced significant reductions in sweat chloride concentration, which correlated with improved ppFEV1 in the overall study population.
Clinical trial registered with www.clinicaltrials.gov (NCT NCT04038047).
Keywords: cystic fibrosis, clinical trial, elexacaftor/tezacaftor/ivacaftor, PROMISE, modulators
At a Glance Commentary
Scientific Knowledge on the Subject
Elexaftor/tezacaftor/ivacaftor (ETI) is a modulator drug shown to provide substantial health benefits and improved cystic fibrosis transmembrane conductance regulator function in controlled clinical trials. This combination drug is effective in a large majority of people with cystic fibrosis and represents a milestone advance in managing this disease.
What This Study Adds to the Field
PROMISE is a postapproval, real-world, observational study to understand the effects of ETI in clinical use in the United States. We find similar substantial improvements across a range of clinical outcomes, which set a new benchmark for modulator drug therapy and highlight the anticipated positive impact of ETI in the management and care of this disease.
Cystic fibrosis (CF) is an autosomal recessive genetic disease resulting in life-shortening, multiorgan system dysfunction. Since the discovery of the CF transmembrane conductance regulator (CFTR) gene, a collective effort has been underway to correct the basic cellular defect. A major contribution has been the advent of CFTR modulators, small molecules that either correct protein misfolding and misprocessing or improve channel gating to enhance apical anion transport (e.g., chloride and bicarbonate) (1–3). By partially restoring channel function, CFTR modulators improve a range of clinical outcomes, but effects vary depending on the underlying CFTR mutations, modulator combination used, and individual clinical characteristics.
Until recently, the benchmark for highly effective CFTR modulator therapy was ivacaftor in people with CF (PwCF) who have G551D or other CFTR gating mutations (2). The biological and clinical effects of restoring CFTR function with ivacaftor have been substantial (4–9). Observational and patient registry studies, including the G551D Observational Trial (GOAL), demonstrated long-term clinical benefits and reduced mortality at the population level, but ivacaftor is highly effective as a monotherapy in less than 10% of all PwCF (4, 5, 10–13). Recently, the three-drug combination elexacaftor/tezacaftor/ivacaftor (ETI) was approved for individuals with at least one F508del allele. F508del is the most common CF mutation worldwide; thus, ETI has the potential to treat at least 85% of PwCF, underscoring its impact on CF care and prognosis. Randomized controlled trials of ETI demonstrated improvements in lung function, respiratory symptoms, risk of acute pulmonary exacerbations, and weight gain that met or exceeded those measured in the prior ivacaftor studies (14, 15), although its use in clinical practice and effects beyond endpoints necessary for clinical registration have not been widely reported.
Since regulatory approval in late 2019, ETI use has become widespread in the United States, raising intense interest in the clinical effects beyond outcomes measured in controlled trials. A deeper understanding of the biological and clinical impacts of CFTR correction in a broader array of patients will also support the goal of realizing highly effective CFTR-directed therapy for all PwCF.
Here we present results from the PROMISE study (NCT04038047); a CF Foundation-supported postapproval observational study of 487 participants age at least 12 years old initiating ETI therapy (16). Participants were assessed before starting ETI and are being followed for 30 months of drug use. The results presented are a preplanned analysis of the primary clinical outcomes after at least 6 months of ETI to determine its performance as a sustained clinical therapy and describe unique effects of ETI relevant to clinical care and the emerging CF landscape.
Methods
Study Design and Population
The PROMISE study is a prospective observational study that was described previously (16). Eligible participants were at least 12 years of age and had at least one copy of F508del and the intent to initiate ETI by the participant’s physician. Key exclusion criteria (see Table E1 in the online supplement) included use of ETI within 180 days of baseline, new chronic therapy initiation, or treatment for nontuberculous mycobacterial infection within 28 days of baseline, and initiation of acute antibiotics or systemic corticosteroids within 14 days of baseline. Participants enrolled and completed a baseline study visit before initiating ETI. Three subsequent visits occurred at 1, 3, and 6 months after initiating therapy. Additional 18- and 30-month study visits are planned. Site personnel were allowed to conduct the 6-month study visit outside the protocol-allowed window of 180 days after ETI initiation ± 14 days when necessary owing to the coronavirus disease (COVID-19) pandemic (17). Remote collection for certain procedures was implemented in response to the uncertain impact of the COVID-19 pandemic in an effort to mitigate against missing data, but the present analysis does not include any data collected at home for this purpose (e.g., home spirometry).
A core set of clinical assessments was conducted in all participants at each visit: spirometry, height, weight, and completion of the respiratory domain of the cystic fibrosis questionnaire-revised (CFQ-R RD) administered using electronic personal devices (18). Sweat chloride was collected at baseline and at 1- and 6-month visits. CFQ-R RDs were administered electronically within predefined windows when in-person visits were delayed because of the pandemic. Spirometry was performed according to the American Thoracic Society standards, and percent predicted FEV1 (ppFEV1) and FVC (ppFVC) were calculated using Global Lung Initiative Equations (19, 20). Body mass index (BMI) z-scores were calculated for participants younger than 18 years at baseline using the Center for Disease Control reference equations. Significant protocol adaptations were made in response to the COVID-19 pandemic (see the online supplement), but the impact on the core outcomes through 6 months was minor. Use of ETI was recorded at each visit by participant self-report.
PROMISE is organized into several organ system-based substudies (16). Outcomes from substudies will be reported separately. The target sample of at least 400 participants overall was chosen to allow for adequate enrollment into the various substudies without creating an impractically burdensome procedure load for each participant or study site. A sample of 400 participants provides more than 95% power to detect changes in ppFEV1 (estimated mean change, 6; SD, 10) and sweat chloride (estimated mean change, −50; SD, 22) as large as or larger (and with comparable variance) than those observed in the GOAL study of PwCF and the G551D mutation receiving ivacaftor alone (5). The study was registered at ClinicalTrials.gov (NCT04038047), and institutional review board approval was granted at participating sites.
Statistical Analysis
Clinical outcomes and change are presented using summary statistics and paired t tests with 95% confidence intervals (CI) at each time point. The primary outcomes are the change in sweat chloride and ppFEV1 at 6 and 30 months (30 months to be reported later). Sample size was determined based on the cumulative needs of the substudy efforts, as previously described, providing adequate power for the core clinical outcome measures reported here (16). In addition to the overall cohort, change statistics were calculated for strata defined by baseline modulator use before initiating ETI (none, ivacaftor monotherapy, or corrector–potentiator combination treatment [i.e., lumacaftor/ivacaftor or tezacaftor/ivacaftor]). P values for chronic medication use were generated using McNemar’s exact test. For exploratory univariate testing of effect modification by demographic characteristics, ANOVA tests determined whether there was a difference in the change from baseline to 6 months. False discovery rate in these exploratory tests was controlled using the Benjamini-Hochberg method, with a false discovery rate threshold of 5% within each outcome (21). Mean baseline values and change scores are revealed in each stratum if a significant association existed after correction. All analyses were performed with SAS version 9.4 and R version 4.1 (22).
Results
Study Population and Follow-Up
A total of 489 participants were enrolled at 56 U.S. CF Foundation Therapeutics Development Network sites between November 2019 and May 2020. Two participants were found to be ineligible after enrollment and were withdrawn, leaving 487 participants in the analysis population. Seven participants did not initiate ETI and withdrew before the 1-month visit (Figure E1). Participants were on average 25.1 years old (range, 11.6–64.6), 51% female at birth (n = 246), 94% Caucasian (n = 457), 6% Hispanic (n = 30), and 48% F508del homozygous. At baseline, participants were either not on a CFTR modulator (50.9%), on a two-drug CFTR modulator combination (lumacaftor/ivacaftor or tezacaftor/ivacaftor, 44.1%), or on ivacaftor alone (6.7%) (Table 1); prior modulator use corresponded with indication by underlying CFTR genotype.
Table 1.
Demographics and Baseline Characteristics
| Modulator Use at Baseline* |
||||
|---|---|---|---|---|
| Total (n = 487) |
None (n = 238) |
Iva (n = 34) |
Two-Drug (n = 215) |
|
| Age, yr | 25.1 (10.7) | 24.7 (10.8) | 27.4 (12.9) | 25.1 (10.1) |
| Sex at birth, female | 246 (50.5%) | 120 (50.4%) | 15 (44.1%) | 111 (51.6%) |
| Race | ||||
| White | 457 (93.8%) | 219 (92.0%) | 33 (97.1%) | 205 (95.3%) |
| Black or African American | 12 (2.5%) | 8 (3.4%) | 0 (0.0%) | 4 (1.9%) |
| Asian | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| American Indian or Alaska Native | 1 (0.2%) | 1 (0.4%) | 0 (0.0%) | 0 (0.0%) |
| Native Hawaiian or other Pacific Islander | 1 (0.2%) | 1 (0.4%) | 0 (0.0%) | 0 (0.0%) |
| More than one race | 13 (2.7%) | 6 (2.5%) | 1 (2.9%) | 6 (2.8%) |
| Other or unknown | 3 (0.6%) | 3 (1.3%) | 0 (0.0%) | 0 (0.0%) |
| Hispanic or Latino | 30 (6.2%) | 18 (7.6%) | 2 (5.9%) | 10 (4.7%) |
| ppFEV1 | 80.5 (22.7) | 80.2 (23.2) | 79.3 (20.8) | 81.1 (22.5) |
| ppFEV1 (by group) | ||||
| <65 | 130 (26.7%) | 64 (26.9%) | 10 (29.4%) | 56 (26.0%) |
| 65–90 | 161 (33.1%) | 76 (31.9%) | 9 (26.5%) | 76 (35.3%) |
| >90 | 196 (40.2%) | 98 (41.2%) | 15 (44.1%) | 83 (38.6%) |
| Weight, kg (age ⩾18) | 65.6 (13.6) | 64.6 (12.5) | 67.7 (11.5) | 66.4 (15.1) |
| Weight percentile (age <18) | 52.9 (29.2) | 50.8 (29.8) | 57.2 (27.8) | 54.6 (29.0) |
| BMI, kg/m2 (age ⩾18) | 23.1 (4.0) | 22.8 (3.9) | 23.5 (2.7) | 23.5 (4.3) |
| BMI Percentile (age <18) | 56.0 (26.5) | 53.8 (27.9) | 65.1 (24.5) | 57.1 (25.1) |
| Genotype group | ||||
| F508del homozygous | 236 (48.5%) | 27 (11.3%) | 0 (0.0%) | 209 (97.2%) |
| F508del heterozygous (MF†) | 195 (40.0%) | 192 (80.7%) | 0 (0.0%) | 3 (1.4%) |
| F508del heterozygous (G551D) | 35 (7.2%) | 2 (0.8%) | 33 (97.1%) | 0 (0.0%) |
| F508del heterozygous (other) | 21 (4.3%) | 17 (7.1%) | 1 (2.9%) | 3 (1.4%) |
Definition of abbreviations: BMI = body mass index; Iva = ivacaftor monotherapy; MF = minimal function; ppFEV1 = percent predicted FEV1; two-drug = tezacaftor/iva or lumacaftor/iva.
Most recent modulator used within 90 days of the baseline visit.
MF mutation defined by the VX-445-102 study eligibility list (15).
The COVID-19 pandemic impacted in-person research conduct, and therefore study visit completion was 86%, 40%, and 89% at 1, 3, and 6 months after starting ETI, respectively (17). More than 94% of 1-month visits were completed before March 11, 2020, when the World Health Organization declared a global pandemic and effects on in-person visits began. The 3-month study visits occurred during the greatest limitations to in-person research conduct. In response to these challenges, the allowable timeframe to complete the 6-month visit was extended. This greatly improved completion at this time point but extended median time from ETI initiation to 222 days (range, 142–416) (Figure E2). Mean changes in lung function and sweat chloride concentration were similar between the 1-month time point, largely completed before the pandemic, and the 3- and 6-month visits conducted during the pandemic (Table 2 and Table E4). When comparing changes from baseline in ppFEV1, sweat chloride, CFQ-R RD, and BMI between participants completing the 6-month study visit on time versus those for whom visits were delayed beyond the ±14-day window, no meaningful differences were observed. Of those completing a visit, the numbers reporting not using ETI at 1, 3, and 6 months were 4 (0.9%), 3 (1.5%), and 9 (2.1%), respectively.
Table 2.
Clinical Outcomes and Change from Baseline by Visit, Stratified by Baseline Modulator Use
| Baseline Modulator Use* |
||||||
|---|---|---|---|---|---|---|
| Outcome | Visit | All | None | Iva | Two-Drug | |
| ppFEV1 | Baseline | n | 487 | 238 | 34 | 215 |
| Mean ± SD | 80.5 ± 22.7 | 80.2 ± 23.2 | 79.3 ± 20.8 | 81.1 ± 22.5 | ||
| 1 mo | n [n chg.]† | 412 [412] | 198 [198] | 32 [32] | 182 [182] | |
| Mean ± SD | 88.5 ± 22.6 | 88.5 ± 22.7 | 84.6 ± 24.1 | 89.1 ± 22.2 | ||
| Change | +8.80 | +10.10 | +5.55 | +7.96 | ||
| CI (chg.) | (8.01 to 9.59) | (8.93 to 11.26) | (3.24 to 7.86) | (6.78 to 9.13) | ||
| 3 mo | n [n chg.]† | 190 [190] | 94 [94] | 11 [11] | 85 [85] | |
| Mean ± SD | 87.6 ± 23.1 | 88.4 ± 22.8 | 74.7 ± 23.9 | 88.3 ± 23.2 | ||
| Change | +10.31 | +12.57 | +3.94 | +8.63 | ||
| CI (chg.) | (8.99 to 11.63) | (10.55 to 14.59) | (1.24 to 6.65) | (6.85 to 10.40) | ||
| 6 mo | n [n chg.]† | 356 [356] | 166 [166] | 24 [24] | 166 [166] | |
| Mean ± SD | 90.9 ± 21.5 | 91.0 ± 20.8 | 87.5 ± 21.9 | 91.3 ± 22.2 | ||
| Change | +9.76 | +10.84 | +6.14 | +9.21 | ||
| CI (chg.) | (8.76 to 10.76) | (9.32 to 12.35) | (3.34 to 8.94) | (7.76 to 10.66) | ||
| ppFVC | Baseline | n | 487 | 238 | 34 | 215 |
| Mean ± SD | 91.9 ± 18.2 | 92.1 ± 18.4 | 93.2 ± 19.0 | 91.4 ± 17.8 | ||
| 1 mo | n [n chg.]† | 412 [412] | 198 [198] | 32 [32] | 182 [182] | |
| Mean ± SD | 96.9 ± 16.3 | 97.3 ± 15.8 | 94.6 ± 18.3 | 96.9 ± 16.5 | ||
| Change | +5.95 | +6.94 | +2.52 | +5.48 | ||
| CI (chg.) | (5.21 to 6.69) | (5.82 to 8.06) | (0.50 to 4.54) | (4.40 to 6.56) | ||
| 3 mo | n [n chg.]† | 190 [190] | 94 [94] | 11 [11] | 85 [85] | |
| Mean ± SD | 96.9 ± 17.1 | 97.9 ± 16.8 | 91.0 ± 18.3 | 96.6 ± 17.4 | ||
| Change | +7.50 | +9.22 | +3.48 | +6.12 | ||
| CI (chg.) | (6.34 to 8.66) | (7.44 to 10.99) | (0.87 to 6.09) | (4.51 to 7.74) | ||
| 6 mo | n [n chg.]† | 356 [356] | 166 [166] | 24 [24] | 166 [166] | |
| Mean ± SD | 98.8 ± 16.1 | 99.2 ± 15.7 | 97.7 ± 19.2 | 98.5 ± 16.1 | ||
| Change | +6.93 | +7.60 | +3.85 | +6.71 | ||
| CI (chg.) | (6.03 to 7.83) | (6.20 to 9.00) | (1.27 to 6.44) | (5.43 to 7.99) | ||
| Sweat chloride, mmol/L | Baseline | n | 462 | 221 | 33 | 208 |
| Mean ± SD | 88.0 ± 18.4 | 95.6 ± 12.8 | 52.6 ± 23.0 | 85.6 ± 15.2 | ||
| 1 mo | n [n chg.]† | 399 [387] | 187 [180] | 30 [29] | 182 [178] | |
| Mean ± SD | 49.1 ± 19.6 | 57.3 ± 17.9 | 25.3 ± 15.0 | 44.5 ± 17.2 | ||
| Change | −38.95 | −39.15 | −25.31 | −40.97 | ||
| CI (chg.) | (−40.60 to −37.29) | (−41.68 to −36.62) | (−31.43 to −19.19) | (−43.17 to −38.76) | ||
| 6 mo | n [n chg.]† | 383 [369] | 174 [165] | 31 [30] | 178 [174] | |
| Mean ± SD | 45.7 ± 21.2 | 53.5 ± 19.6 | 26.2 ± 20.4 | 41.5 ± 19.5 | ||
| Change | −41.70 | −43.15 | −23.92 | −43.40 | ||
| CI (chg.) | (−43.80 to −39.60) | (−46.21 to −40.09) | (−31.03 to −16.81) | (−46.38 to −40.41) | ||
| CFQ-R RD | Baseline | n | 410 | 205 | 23 | 182 |
| Mean ± SD | 70.3 ± 18.2 | 69.1 ± 18.5 | 71.0 ± 18.9 | 71.4 ± 17.9 | ||
| 1 mo | n [n chg.]† | 380 [342] | 187 [170] | 28 [22] | 165 [150] | |
| Mean ± SD | 82.7 ± 15.0 | 82.2 ± 14.8 | 83.3 ± 17.8 | 83.3 ± 14.7 | ||
| Change | +12.54 | +12.81 | +10.35 | +12.55 | ||
| CI (chg.) | (10.32 to 14.76) | (9.53 to 16.09) | (0.52 to 20.19) | (9.37 to 15.74) | ||
| 3 mo | n [n chg.]† | 352 [319] | 166 [154] | 27 [21] | 159 [144] | |
| Mean ± SD | 87.5 ± 13.5 | 87.2 ± 13.2 | 87.9 ± 15.2 | 87.7 ± 13.7 | ||
| Change | +16.95 | +17.97 | +14.81 | +16.16 | ||
| CI (chg.) | (14.76 to 19.13) | (15.00 to 20.95) | (5.75 to 23.87) | (12.69 to 19.63) | ||
| 6 mo | n [n chg.]† | 302 [273] | 144 [137] | 25 [18] | 133 [118] | |
| Mean ± SD | 90.5 ± 11.3 | 90.1 ± 12.2 | 91.8 ± 11.4 | 90.7 ± 10.4 | ||
| Change | +20.39 | +22.51 | +18.52 | +18.22 | ||
| CI (chg.) | (18.28 to 22.50) | (19.47 to 25.54) | (7.63 to 29.40) | (15.22 to 21.22) | ||
| BMI‡, adults, kg/m2 | Baseline | n | 326 | 157 | 24 | 145 |
| Mean ± SD | 23.1 ± 4.0 | 22.8 ± 3.9 | 23.5 ± 2.7 | 23.5 ± 4.3 | ||
| 1 mo | n vis. [n chg.] | 272 [272] | 128 [128] | 22 [22] | 122 [122] | |
| Mean ± SD | 23.7 ± 4.2 | 23.4 ± 4.2 | 23.8 ± 3.1 | 24.0 ± 4.3 | ||
| Change | +0.42 | +0.47 | +0.39 | +0.37 | ||
| CI (chg.) | (0.33 to 0.50) | (0.34 to 0.59) | (0.11 to 0.68) | (0.25 to 0.49) | ||
| 3 mo | n vis. [n chg.] | 136 [136] | 65 [65] | 10 [10] | 61 [61] | |
| Mean ± SD | 24.1 ± 3.8 | 23.9 ± 3.8 | 23.5 ± 2.7 | 24.4 ± 3.9 | ||
| Change | +0.85 | +1.08 | +0.86 | +0.62 | ||
| CI (chg.) | (0.68 to 1.03) | (0.85 to 1.30) | (-0.05 to 1.76) | (0.34 to 0.90) | ||
| 6 mo | n vis. [n chg.] | 268 [268] | 121 [121] | 22 [22] | 125 [125] | |
| Mean ± SD | 24.5 ± 4.6 | 24.2 ± 4.6 | 24.6 ± 3.2 | 24.8 ± 4.7 | ||
| Change | +1.24 | +1.31 | +1.28 | +1.17 | ||
| CI (chg.) | (1.05 to 1.44) | (1.01 to 1.61) | (0.47 to 2.09) | (0.89 to 1.44) | ||
| BMI, peds§ (z-score) |
Baseline | n | 159 | 80 | 10 | 69 |
| Mean ± SD | 0.2 ± 0.9 | 0.1 ± 0.9 | 0.5 ± 0.7 | 0.2 ± 0.8 | ||
| 1 mo | n [n chg.]† | 140 [140] | 70 [70] | 10 [10] | 60 [60] | |
| Mean ± SD | 0.3 ± 0.8 | 0.2 ± 0.9 | 0.5 ± 0.7 | 0.3 ± 0.8 | ||
| Change | +0.11 | +0.11 | +0.05 | +0.12 | ||
| CI (chg.) | (0.07 to 0.15) | (0.05 to 0.18) | (-0.07 to 0.18) | (0.08 to 0.16) | ||
| 3 mo | n [n chg.]† | 55 [55] | 28 [28] | 2 [2] | 25 [25] | |
| Mean ± SD | 0.3 ± 0.7 | 0.0 ± 0.8 | −0.5 ± 0.4 | 0.6 ± 0.5 | ||
| Change | +0.24 | +0.28 | +0.14 | +0.20 | ||
| CI (chg.) | (0.15 to 0.33) | (0.12 to 0.45) | (−1.92 to 2.19) | (0.11 to 0.29) | ||
| 6 mo | n [n chg.]† | 139 [139] | 67 [67] | 10 [10] | 62 [62] | |
| Mean ± SD | 0.5 ± 0.8 | 0.5 ± 0.8 | 0.7 ± 0.9 | 0.4 ± 0.8 | ||
| Change | +0.30 | +0.37 | +0.20 | +0.23 | ||
| CI (chg.) | (0.22 to 0.37) | (0.24 to 0.50) | (−0.06 to 0.45) | (0.14 to 0.31) | ||
Definition of abbreviations: BMI = body mass index; CFQ-R RD = Cystic Fibrosis Questionnaire–Revised, Respiratory Domain; chg. = change; CI = confidence interval; Iva = ivacaftor monotherapy; n vis. = number of participants attending the study visit; ppFEV1 = percent predicted FEV1; ppFVC = percent predicted FVC; two-drug = lumacaftor/iva or tezacaftor/iva.
Prior use of modulators defined as any use within 90 days of the baseline measurement.
[n chg.] is the number of participants contributing to the change estimate (having both baseline and follow-up measured).
Pregnant participants were excluded from analyses of BMI.
z-scores are based on Centers for Disease Control and Prevention percentiles.
Changes in Clinical Outcomes
Improvements from baseline to 6 months occurred in all outcome measures in the overall study population and in each subgroup defined by baseline modulator use before starting ETI (Figure 1 and Table 2). Lung function measured by ppFEV1 improved in the entire cohort by an average of 9.8 percentage points (95% CI, 8.8 to 10.8 points). The largest average changes in ppFEV1 were in those previously using no modulator (10.8; 95% CI, 9.3 to 12.4) or a two-drug combination (9.2; 95% CI, 7.8 to 10.7). The subgroup entering the study on ivacaftor had an average of 6.1 (95% CI, 3.3 to 8.9) increase in ppFEV1 at 6 months (Table 2). Similarly, average sweat chloride changes were largest among the two-drug (−43.4 mmol/L; 95% CI, −46.4 to −40.4) and no-modulator subgroups (−43.2; 95% CI, −46.2 to −40.1), but still substantially improved in the ivacaftor subgroup (−23.9; 95% CI, −31.0 to −16.8).
Figure 1.
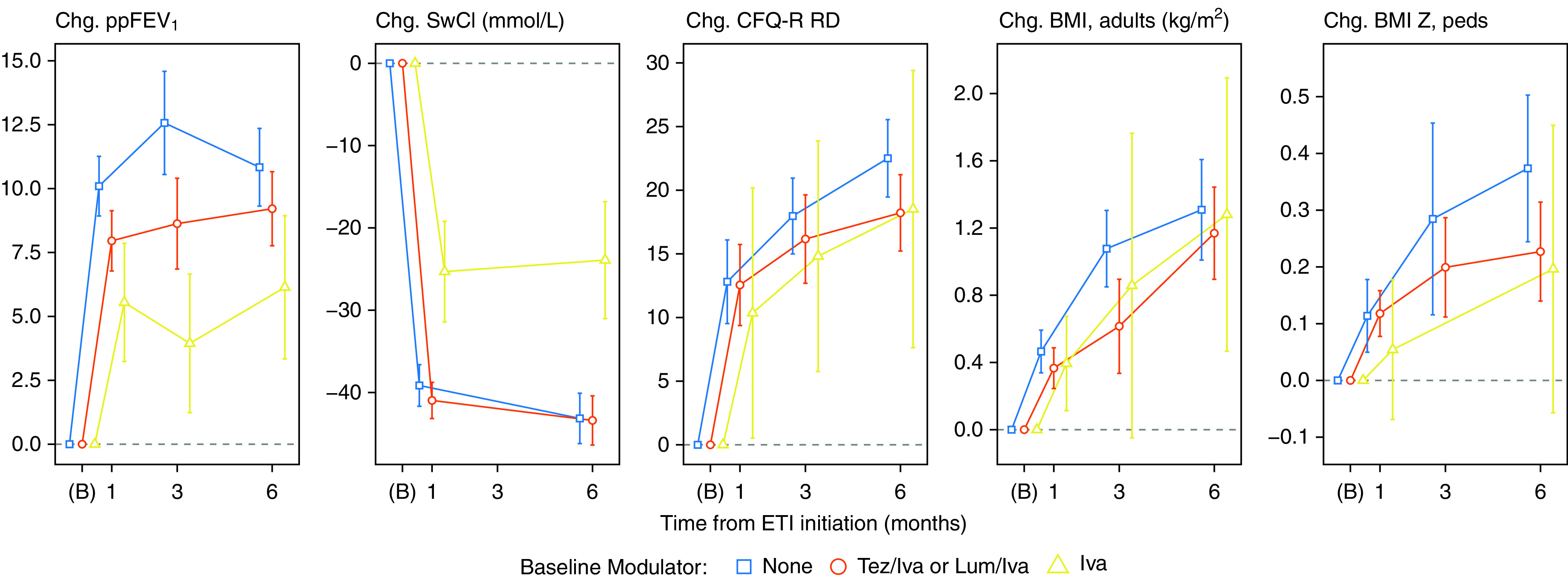
Changes from baseline with 95% confidence intervals, stratified by cystic fibrosis transmembrane conductance regulator modulator use at baseline (iva = ivacaftor monotherapy). Times of observation are pre-elaxaftor/tezacaftor/ivacaftor baseline (B) and planned visit times. Participants who were pregnant at a visit were excluded from analyses of body mass index (BMI). Confidence intervals with five or fewer participants are not shown, and low follow-up at 3 months requires additional caution in interpretation (see Table 2). CFQ-R RD = Cystic Fibrosis Questionnaire–Revised, Respiratory Domain; Chg. = change; ETI = elaxaftor/tezacaftor/ivacaftor; Lum = lumacaftor; ppFEV1 = percent predicted FEV1; SwCl = sweat chloride; Tez = tezacaftor.
Respiratory signs or symptoms of illness decreased from baseline to 6 months, with a mean 20.4-point (95% CI, 18.3 to 22.5 points) improvement in the CFQ-R RD score in the entire cohort. BMI also improved significantly, with a 6-month mean increase from baseline of 1.2 kg/m2 in adults and 0.3 z-score in adolescents. Improvements in BMI and CFQ-R RD were similar between baseline modulator subgroups (Table 2).
At the 6-month visit, the overall mean ppFEV1 improved to 90.9 in the study cohort with mean age of 25 years (Table 2). Mean sweat chloride was 45.7 mmol/L, mean CFQ-R RD was 90.5 of a maximum 100 points, and mean BMI was 24.5 kg/m2 (0.5 z-score in adolescents). The 6-month absolute values across these outcomes were similar among subgroups apart from sweat chloride, which was lower (23.9 mmol/L) in the subgroup previously treated with ivacaftor (i.e., those with F508del and a gating or residual function mutation) (Figure 2 and Table 2). Increasing numbers of participants reached the maximum possible score for CFQ-R RD. Among 302 subjects who completed the questionnaire at 6 months, 32% had the maximum possible CFQ-R RD score of 100, and an additional 50% had a score above 80. Large improvements in CFQ-R RD scores occurred even in those starting with relatively low lung function (<65 ppFEV1) at baseline (Figure 3).
Figure 2.
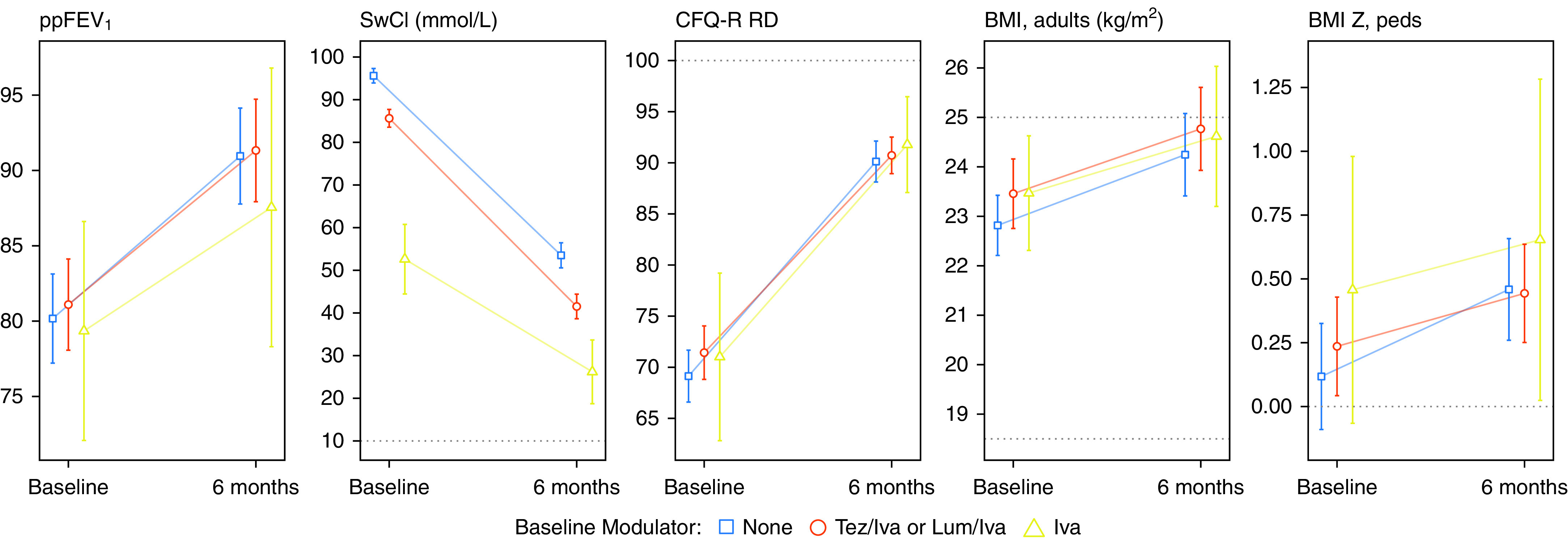
Cross-sectional estimates and 95% confidence intervals of outcomes, stratified by cystic fibrosis transmembrane conductance regulator modulator use at baseline (iva = ivacaftor monotherapy). Times shown are baseline and the 6-month post-elaxaftor/tezacaftor/ivacaftor visit. Participants who were pregnant at a visit (n = 2 at baseline; n = 7 at 6 mo) were excluded from analyses of body mass index (BMI). Dotted lines show limits of the instrument (sweat chloride [SwCl] and Cystic Fibrosis Questionnaire–Revised, Respiratory Domain [CFQ-R RD]) or thresholds (BMI 25 for overweight, BMI 18 for underweight, and BMI z-score 0 for median). Lum = lumacaftor; ppFEV1 = percent predicted FEV1; Tez = tezacaftor.
Figure 3.
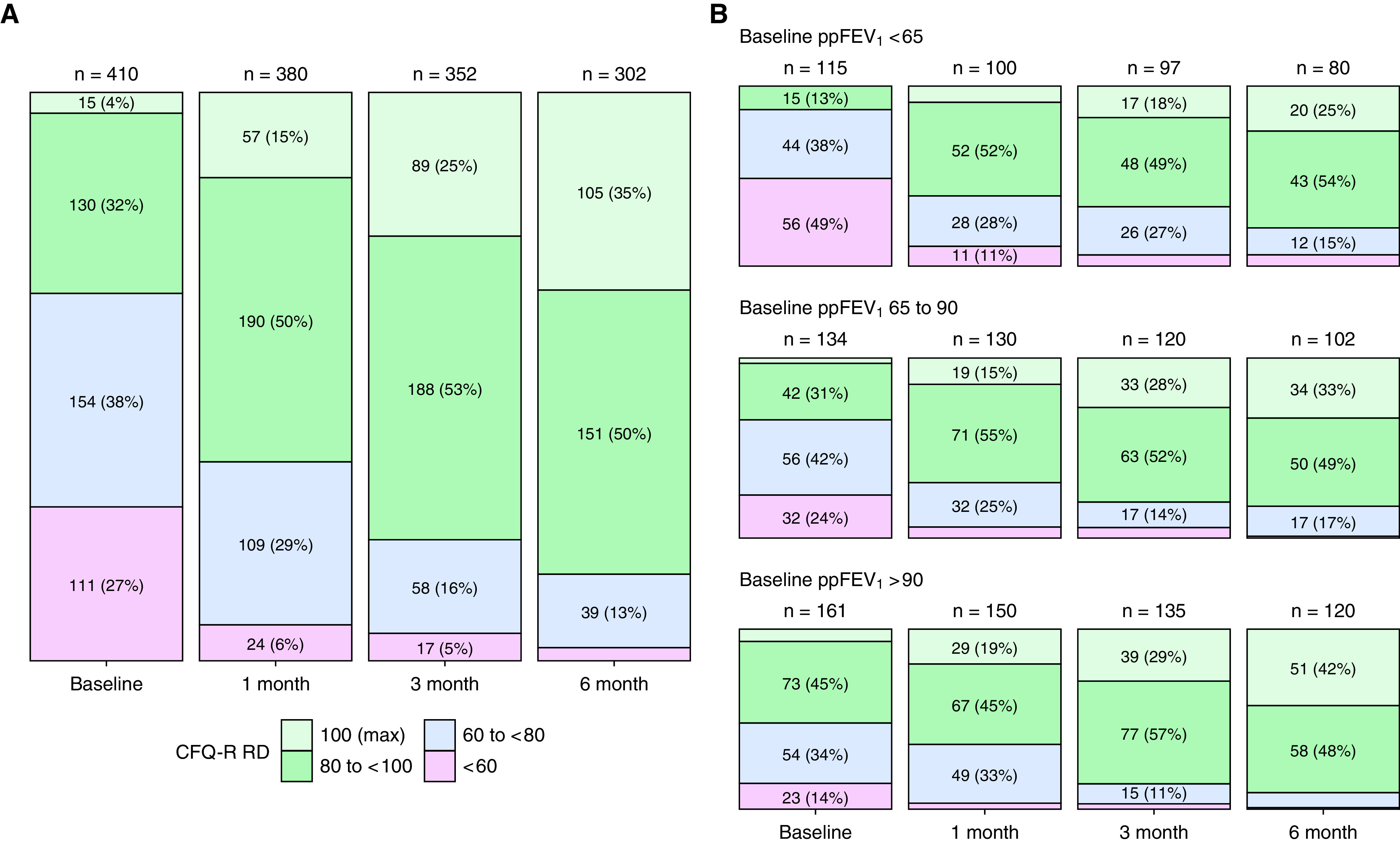
(A) Cystic Fibrosis Questionnaire–Revised, Respiratory Domain (CFQ-R RD) with participants grouped into categories at each visit. The top category (100, light green) represents the maximum achievable score, which a substantial number of participants obtained 6 months after elaxaftor/tezacaftor/ivacaftor initiation. Labels are omitted for categories with too few participants. (B) The same plot split into categories based on baseline percent predicted FEV1 (ppFEV1). The trend toward the maximum score was observed at each level of baseline lung function, with higher proportions in the high baseline lung function cohort reporting maximal CFQ-R RD at 6 months.
Participants were encouraged to continue baseline medications through at least the 6-month study visit to better understand the impacts of ETI. In addition to routine collection of concomitant medications, self-reported use of four common chronic therapies was specifically queried at each visit. At the 6-month visit, the proportion using dornase alfa decreased by 6%, hypertonic saline by 9.8%, azithromycin by 9.1%, and inhaled antibiotics by 34% relative to baseline (Table 3). Changes in medication use did not significantly differ between subgroups based on modulator use before ETI.
Table 3.
Use of Four Chronic Daily Medications Assessed at Each Visit by Self-Report
| Outcome | Visit | Using/Observed (%) | P Value |
|---|---|---|---|
| Inhaled antibiotics | Baseline | 248/486 (51.0) | — |
| 1 mo | 186/417 (44.6) | — | |
| 3 mo | 97/195 (49.7) | — | |
| 6 mo | 145/429 (33.8) | <0.005 | |
| Azithromycin | Baseline | 238/486 (49.0) | — |
| 1 mo | 206/417 (49.4) | — | |
| 3 mo | 94/195 (48.2%) | — | |
| 6 mo | 191/429 (44.5%) | 0.01 | |
| Hypertonic saline | Baseline | 368/486 (75.7%) | — |
| 1 mo | 308/417 (73.9%) | — | |
| 3 mo | 148/195 (75.9%) | — | |
| 6 mo | 293/429 (68.3%) | <0.005 | |
| Dornase alfa | Baseline | 424/486 (87.2%) | — |
| 1 mo | 365/417 (87.5%) | — | |
| 3 mo | 166/195 (85.1%) | — | |
| 6 mo | 350/429 (81.6%) | <0.005 |
Correlation between Change in Sweat Chloride and ppFEV1
Prior studies with CFTR modulators have only shown correlation between clinical outcome measures and CFTR function measured by sweat chloride if considering extended observation periods (i.e., >12 mo) or if combining multiple studies (4, 23). In our study, a correlation between changes in ppFEV1 and sweat chloride was observed over the first 6 months of ETI. In the entire study population, each additional 10-point decrease from baseline in sweat chloride was associated with an additional 0.89-point increase in ppFEV1 (Pearson r = −0.19; P < 0.005) (Figure 4).
Figure 4.

Correlation between the change in percent predicted FEV1 (ppFEV1) and the change in sweat chloride (mmol/L) from baseline to 6 months (visit 4). Pearson correlation coefficient is −0.19 (P < 0.005). The dark line shows the equivalent linear regression fit, indicating that each additional 10-point decrease in sweat chloride (SwCl) is associated with a mean 0.89 greater change in ppFEV1 from baseline to 6 months after elaxaftor/tezacaftor/ivacaftor initiation. This correlation remains significant if the influential points at (17, −31) and (−95, 35) are excluded, or if Spearman’s rank-based association statistic is substituted for the Pearson correlation statistic.
Outcomes and Baseline Demographics
Table E2 indicates effect modification of sex on sweat chloride change at 6 months, with larger improvements among females. As with previous work on CFTR modulators, weight and change in sweat chloride were marginally correlated (Figure E5) (24, 25). An exploratory analysis of post-ETI sweat chloride with sex, weight, and CFTR genotype as covariates showed that the association between sweat chloride and sex was significant even when adjusting for weight, but the association with weight was not significant after adjusting for sex (Figure E5). CFQ-R RD changes were, on average, larger among older and female participants who generally reported more symptoms of CF lung disease before starting ETI (Table E2). All groups subdivided by sex, age, or prior modulator use reported similar, near-maximum post-ETI scores (Figure 2 and Table E2). There were no identified interactions between ppFEV1 change and age, sex, ethnicity, or race. A stratification by baseline ppFEV1 (Table E5) shows significant improvements in each baseline lung function group. Owing to the study design, this breakdown cannot be used to demonstrate a ceiling effect (a reduction in change at high baseline levels) for ppFEV1.
CFTR Genotype Representation
Participants with one F508del allele had a wide variety of second CFTR mutations. Changes in ppFEV1 and sweat chloride are shown in Figure E3 for mutations other than G551D/F508del or F508del/F508del if present in at least six individuals in PROMISE. Mean sweat chloride values after starting ETI are also provided for groups containing data from at least three participants in Figure E4. PROMISE included 11 individuals with F508del plus one of the mutations added to the U.S. Food and Drug Administration (FDA)-approved label indication based largely on in vitro testing for improved CFTR function (26). These 11 individuals had a greater mean reduction in sweat chloride from baseline compared with a group identified as having F508del plus a mutation not expected to respond to ETI (i.e., a minimal function allele) (difference, 13.4 mmol/L; 95% CI, 2.4–24.3) (Table E3). Changes in ppFEV1 were not significantly different between these groups. Those with two minimal function mutations of particular interest owing to their prevalence and unique properties (W1282X, N1303K) were compared with the same comparator group (Table E3) as an exploratory analysis (27–29). No significant differences in change in sweat chloride or ppFEV1 were found, though available numbers of participants were small.
Discussion
This planned 6-month analysis of the PROMISE study enabled two important questions to be addressed: How does ETI drug therapy perform during clinical use, and what additional impacts of ETI can be identified that may be relevant to clinical care or future research priorities? We found clear evidence of substantial improvement in clinical outcomes among nearly 500 individuals despite a broad range of baseline demographic and other characteristics, including lung function, genotype, and prior CFTR modulator use (Table 2). The overall average improvement in ppFEV1 at 6 months was nearly 10%, the largest magnitude in a large observational study among PwCF. This occurred despite CFTR modulator use by the majority of participants at the time of enrollment (47% entered on two-drug combinations and 7% on ivacaftor) (Table 2). For comparison, the mean 6-month ppFEV1 improvement after starting ivacaftor to target the G551D mutation was 6.7% in the GOAL study, which enrolled a modulator-naive but similar population with respect to baseline age and lung function (5).
Lung function improved after initiating ETI therapy regardless of preexisting CFTR modulator use, which was closely associated with underlying CFTR genotype and generally consistent with drug indication (Table 1). Each subgroup, defined by modulator use before starting ETI (and associated genotype), had a baseline ppFEV1 between 79% and 81% (Table 2) and reached an average ppFEV1 near or above 90% predicted by 6 months (Figure 2). Even the substantially sized subset (n = 196) of those at or above 90% predicted at baseline (an exclusion criteria for the phase 3 trials) achieved a mean improvement of 6.5 ppFEV1 (Table E5). In this study, those below 65% predicted at baseline had a mean improvement of 12.2 ppFEV1 by 6 months into ETI therapy, and others have reported significant clinical improvement with ETI in people with even more advanced lung disease (30, 31). A high degree of CFTR functional improvement was achieved, as reflected by an overall mean reduction in sweat chloride of 45.7 mmol/L at 6 months, with all subgroups improving to a mean sweat chloride value below 60 mmol/L. Perhaps consequently, daily respiratory signs and symptoms significantly decreased with ETI therapy. At baseline, 36% of participants reported a CFQ-R RD score of at least 80 points, and 4% reported the maximum score of 100. After steady improvement, 85% had scores of at least 80, and 35% reported the maximum score of 100 by 6 months, suggesting many subjects resolved chronic respiratory symptoms. Given the substantial number of PwCF who reported a maximum score, it is possible that the CFQ-R RD may no longer be a dynamic endpoint for future studies enrolling people already treated with ETI. Substantial gains in CFQ-R RD were observed even when baseline ppFEV1 was less than 65% (Figure 3) (32). The mean overall improvement in this symptom score at 6 months was over 20 points compared with 7.4 points in GOAL, which had a similar open-label design that may bias self-reporting of respiratory symptoms (5, 18). For context, the minimal clinically important change in the CFQ-R RD has been identified as 4 points (18).
BMI also increased to an average of 24.5 kg/m2 among adults at 6 months and 65th percentile among adolescents and continued to increase even after changes in outcomes like sweat chloride and ppFEV1 had largely plateaued. These results suggest that goals for calorie supplementation may need to be reconsidered in many PwCF after starting ETI as chronic therapy (33). Future evaluation of BMI beyond 6 months of modulator therapy, body composition, and related endocrine and gastrointestinal outcomes planned within PROMISE will be of particular importance to better understand BMI trajectories, emergence of overweight/obesity, and the relationships of BMI with these additional outcomes.
Participants transitioning from ivacaftor to ETI, representing those with highly responsive CFTR gating mutations (97% had F508del/G551D), achieved the lowest sweat chloride concentration reported to date in response to modulator drug therapy. The group (n = 34) had a baseline mean sweat chloride on ivacaftor indicative of substantial drug response (52.6 mmol/L), but this value fell to 23.9 mmol/L at the 6-month visit after transitioning to ETI. This is below even the indeterminate range as a diagnostic test for CFTR dysfunction (34) and recapitulates recent clinical trial data in this population (35). Those entering the study on ivacaftor also experienced clinically meaningful and statistically significant improvements in ppFEV1, BMI, and respiratory symptoms when transitioning to ETI, supporting the benefit of drug-induced CFTR modulation even when CFTR function is already in the intermediate range.
In contrast to similar prior studies of CFTR modulator drugs, we observed a statistically significant correlation between change in sweat chloride and ppFEV1 (Figure 4) (5, 23, 36). We hypothesize that three factors contributed to this finding in the PROMISE study: 1) the large effect sizes caused by ETI; 2) the heterogeneity of the cohort by genotype and prior CFTR modulator use, increasing the spectrum of observed responses; and 3) the large sample size (37). This connection between restoration of CFTR function (i.e., change in sweat chloride) and clinical impact (i.e., improved FEV1, among other measures) supports the notion that sustained improvements in CFTR function with ongoing drug use will be associated with long-term clinical benefit, as observed with longer observation and natural history studies of ivacaftor therapy (4, 9, 38–40). Given the comparative 6-month findings of ETI in this study and ivacaftor in GOAL, we are particularly interested in monitoring this as the PROMISE study progresses. The strength of correlation between changes in sweat chloride and ppFEV1 in this study was modest, and additional work will be needed to better understand the use of sweat chloride as an indicator of clinical benefit, especially in individuals or small populations (41–43). It is important to recognize that small changes in sweat chloride did not preclude substantial improvement in clinical outcomes (Figure 4).
This was the first opportunity to consider the effects of ETI in those with CFTR mutations that the FDA has subsequently approved for ETI use without an F508del allele based on in vitro testing (26). A group of 11 such individuals with no prior modulator exposure who had one of these mutations in combination with F508del had an average sweat chloride improvement of −53.0 mmol/L, 13.4 mmol/L larger than those with F508del and a mutation known to not respond to ETI (Table E3). These data support that ETI can improve CFTR function in individuals when targeting these mutations identified through in vitro testing. It is unknown whether ETI would be more or less effective in the absence of an F508del allele. We also acknowledge that the average changes in ppFEV1 were not statistically significantly greater than the F508del/minimal function group in this small, heterogeneous population.
Females experienced approximately 10 mmol/L greater reduction in sweat chloride compared with males, despite similar baseline values. An analysis of the subjects treated with ivacaftor in GOAL identified a similar association, in addition to noting an association between weight and change in sweat chloride (24). The association between sex and change in sweat chloride in our study remained even when adjusting for weight, and the biological basis for this observation is unclear (24, 25). In addition, 11 female participants were found to be pregnant during the study. Increased pregnancy rates associated with highly effective CFTR modulator use is an important consideration in the lives of PwCF, and we expect the need for further attention to reproductive and sexual health (44–46).
The safety of ETI was not assessed in this study, but we observed low self-reported discontinuation rates, consistent with clinical trials. Although participants were encouraged to maintain all other chronic medications through 6 months, significant reductions in use of supportive therapies, and in particular inhaled antibiotics, occurred by the 6-month visit (Table 3). Monitoring further changes in medication use and the association with clinical status will be important to understanding the future treatment landscape (47, 48). Changes in inhaled antibiotics may be particularly relevant, as ivacaftor use has been associated with reduced detection of Pseudomonas aeruginosa and some other pathogens in clinical respiratory cultures (4, 13, 39, 49). Microbiological changes are the focus of a substudy of PROMISE and are being carefully assessed through prospective, serial sputum collection.
In summary, the clinical results through 6 months of this prospective observational study find robust health benefits of ETI in real-world practice that are similar to those found in controlled clinical trials. Compared with prior similar studies in CF, these results set a new benchmark for highly effective modulator drug therapy (5). The full range of potential biological and clinical effects of ETI are important to understand; several organ-specific substudies focused on translational and clinical outcomes will follow. Similarly, a study in those aged 6–11 years is forthcoming concurrent with FDA label extension in younger patients. The measured health status of PROMISE participants at 6 months into ETI therapy demonstrates how daily morbidity has dramatically improved for those for whom ETI is indicated and available.
Certain limitations exist in these analyses, including the potential influence of the COVID-19 pandemic that contributed to a number of missing or delayed study visits at the 3- and 6-month time points (Figure E2). We compared the 6-month changes in outcomes between those completing the study visits by the end of the predefined window and those completing the visits late owing to the pandemic. We found no meaningful differences and overlapping 95% CIs for ppFEV1, sweat chloride, CFQ-R RD, and BMI (data not shown). It is fortunate that response to ETI occurs soon after starting drug therapy and that a large majority of the 1-month study visits were completed before pandemic-related restrictions began, though it is possible that our results were still affected. Completion of this study through at least 30 months of ETI will better characterize long-term outcomes and provide a robust understanding of the durability of these early health improvements—hopefully in a time with less need for social distancing. This study is collecting concomitant medications used, including antibiotics, but is not systematically capturing predefined pulmonary exacerbation events in real time. This decision, along with the relatively short timeframe reported in this first phase and potential impact of social distancing on risk of exacerbations, led us to postpone our analyses of antibiotic use until the end of the study. in addition, in the evaluation of mutations added to the United States Prescribing Information for approved drug use but not yet reported clinically, participants had at least one responsive F508del allele, which also contributed to their sweat chloride response. Despite these limitations, we are encouraged to find that ETI was broadly effective in individuals, including many who would not meet eligibility criteria for the randomized controlled trials that led to drug approval, and we hope that data from this study will support ongoing work to realize highly efficacious CFTR-directed therapy for all PwCF.
Acknowledgments
Acknowledgment
The authors thank the many people with cystic fibrosis and research teams across the United States who made this study possible.
Footnotes
A complete list of PROMISE Study Group members can be found in the online supplement.
Supported by the Cystic Fibrosis Foundation. Additional programmatic funding that supported this research was provided by the National Institute of Diabetes and Digestive and Kidney Diseases (P30DK072482 [S.M.R. and G.M.S.]) and NHLBI (P30DK089507 [D.P.N.], R35HL135816 [S.M.R.], UL1TR003096 [S.M.R.], UL1 TR002535 [S.D.S.], K24HL14166 [L.R.H.], and K08HL138153 [G.M.S.]). Funding agencies had no role in the design, management, data collection, analyses, or interpretation of the data or in the writing of the manuscript or the decision to submit for publication. The views expressed in this publication are those of the authors and not necessarily those of the Cystic Fibrosis Foundation or NIH.
Author Contributions: Study concept, design, and initiation: D.P.N., S.M.R., S.L.H., J.P.C., J.M.V.D., and M.H.K. Statistical analysis and advisement: A.C.P., S.L.H., and M.H.K. Analyses and interpretation of data: all authors. First drafting of manuscript: D.P.N., S.M.R., A.C.P., and S.L.H. Critical revisions and intellectual content: all authors. The PROMISE Study Group consists of key study team members at the central coordinating center and all lead investigators and research coordinators who are responsible for ethical board approval, participant recruitment, and conduct at local study sites.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202108-1986OC on November 16, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Bell SC, Mall MA, Gutierrez H, Macek M, Madge S, Davies JC, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med . 2020;8:65–124. doi: 10.1016/S2213-2600(19)30337-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mall MA, Mayer-Hamblett N, Rowe SM. Cystic fibrosis: emergence of highly effective targeted therapeutics and potential clinical implications. Am J Respir Crit Care Med . 2020;201:1193–1208. doi: 10.1164/rccm.201910-1943SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abou Alaiwa MH, Launspach JL, Grogan B, Carter S, Zabner J, Stoltz DA, et al. Ivacaftor-induced sweat chloride reductions correlate with increases in airway surface liquid pH in cystic fibrosis. JCI Insight . 2018;3:e121468. doi: 10.1172/jci.insight.121468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guimbellot JS, Baines A, Paynter A, Heltshe SL, VanDalfsen J, Jain M, et al. GOAL-e2 Investigators Long term clinical effectiveness of ivacaftor in people with the G551D CFTR mutation. J Cyst Fibros . 2021;20:213–219. doi: 10.1016/j.jcf.2020.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, et al. GOAL Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med . 2014;190:175–184. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. VX08-770-103 (ENVISION) Study Group Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med . 2013;187:1219–1225. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. VX08-770-102 Study Group A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med . 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med . 2017;377:2024–2035. doi: 10.1056/NEJMoa1709847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sawicki GS, McKone EF, Pasta DJ, Millar SJ, Wagener JS, Johnson CA, et al. Sustained benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am J Respir Crit Care Med . 2015;192:836–842. doi: 10.1164/rccm.201503-0578OC. [DOI] [PubMed] [Google Scholar]
- 10. Rosenfeld M, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. KLIMB study group An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2-5 years (KLIMB) J Cyst Fibros . 2019;18:838–843. doi: 10.1016/j.jcf.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ratjen F, Klingel M, Black P, Powers MR, Grasemann H, Solomon M, et al. Changes in lung clearance index in preschool-aged patients with cystic fibrosis treated with ivacaftor (GOAL): a clinical trial. Am J Respir Crit Care Med . 2018;198:526–528. doi: 10.1164/rccm.201802-0243LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hubert D, Dehillotte C, Munck A, David V, Baek J, Mely L, et al. Retrospective observational study of French patients with cystic fibrosis and a Gly551Asp-CFTR mutation after 1 and 2years of treatment with ivacaftor in a real-world setting. J Cyst Fibros . 2018;17:89–95. doi: 10.1016/j.jcf.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 13. Heltshe SL, Mayer-Hamblett N, Burns JL, Khan U, Baines A, Ramsey BW, et al. GOAL (the G551D Observation-AL) Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis . 2015;60:703–712. doi: 10.1093/cid/ciu944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. VX17-445-103 Trial Group Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet . 2019;394:1940–1948. doi: 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. VX17-445-102 Study Group Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med . 2019;381:1809–1819. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nichols DP, Donaldson SH, Frederick CA, Freedman SD, Gelfond D, Hoffman LR, et al. PROMISE: working with the CF community to understand emerging clinical and research needs for those treated with highly effective CFTR modulator therapy. J Cyst Fibros . 2021;20:205–212. doi: 10.1016/j.jcf.2021.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pearson K, Mayer-Hamblett N, Goss CH, Retsch-Bogart GZ, VanDalfsen JM, Burks P, et al. The impact of SARS-CoV-2 on the cystic fibrosis foundation therapeutics development network. J Cyst Fibros . 2021;20:195–197. doi: 10.1016/j.jcf.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest . 2009;135:1610–1618. doi: 10.1378/chest.08-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. ERS Global Lung Function Initiative Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J . 2012;40:1324–1343. doi: 10.1183/09031936.00080312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Graham BL, Steenbruggen I, Miller MR, Barjaktarevic IZ, Cooper BG, Hall GL, et al. Standardization of spirometry 2019 update. An Official American Thoracic Society and European Respiratory Society technical statement. Am J Respir Crit Care Med . 2019;200:e70–e88. doi: 10.1164/rccm.201908-1590ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B . 1995;57:289–300. [Google Scholar]
- 22.R Core Team 2021https://www.R-project.org/.
- 23. Fidler MC, Beusmans J, Panorchan P, Van Goor F. Correlation of sweat chloride and percent predicted FEV1 in cystic fibrosis patients treated with ivacaftor. J Cyst Fibros . 2017;16:41–44. doi: 10.1016/j.jcf.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 24. Secunda KE, Guimbellot JS, Jovanovic B, Heltshe SL, Sagel SD, Rowe SM, et al. Females with cystic fibrosis demonstrate a differential response profile to ivacaftor compared with males. Am J Respir Crit Care Med . 2020;201:996–998. doi: 10.1164/rccm.201909-1845LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aalbers BL, Hofland RW, Bronsveld I, de Winter-de Groot KM, Arets HGM, de Kiviet AC, et al. Females with cystic fibrosis have a larger decrease in sweat chloride in response to lumacaftor/ivacaftor compared to males. J Cyst Fibros . 2021;20:e7–e11. doi: 10.1016/j.jcf.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Trikafta prescribing information 2021https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/212273s004lbl.pdf.
- 27. Mutyam V, Libby EF, Peng N, Hadjiliadis D, Bonk M, Solomon GM, et al. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros . 2017;16:24–29. doi: 10.1016/j.jcf.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rowe SM, Varga K, Rab A, Bebok Z, Byram K, Li Y, et al. Restoration of W1282X CFTR activity by enhanced expression. Am J Respir Cell Mol Biol . 2007;37:347–356. doi: 10.1165/rcmb.2006-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. DeStefano S, Gees M, Hwang TC. Physiological and pharmacological characterization of the N1303K mutant CFTR. J Cyst Fibros . 2018;17:573–581. doi: 10.1016/j.jcf.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O’Shea KM, O’Carroll OM, Carroll C, Grogan B, Connolly A, O’Shaughnessy L, et al. Efficacy of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease. Eur Respir J . 2021;57:2003079. doi: 10.1183/13993003.03079-2020. [DOI] [PubMed] [Google Scholar]
- 31. Burgel PR, Durieu I, Chiron R, Ramel S, Danner-Boucher I, Prevotat A, et al. French Cystic Fibrosis Reference Network Study Group Rapid improvement after starting elexacaftor-tezacaftor-ivacaftor in patients with cystic fibrosis and advanced pulmonary disease. Am J Respir Crit Care Med . 2021;204:64–73. doi: 10.1164/rccm.202011-4153OC. [DOI] [PubMed] [Google Scholar]
- 32. Davies J, Sheridan H, Bell N, Cunningham S, Davis SD, Elborn JS, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med . 2013;1:630–638. doi: 10.1016/S2213-2600(13)70182-6. [DOI] [PubMed] [Google Scholar]
- 33. Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H, Clinical Practice Guidelines on Growth and Nutrition Subcommittee Ad Hoc Working Group. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc . 2008;108:832–839. doi: 10.1016/j.jada.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 34. Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr . 2017;181S:S4–S15.e11. doi: 10.1016/j.jpeds.2016.09.064. [DOI] [PubMed] [Google Scholar]
- 35. Barry PJ, Mall MA, Álvarez A, Colombo C, de Winter-de Groot KM, Fajac I, et al. VX18-445-104 Study Group Triple therapy for cystic fibrosis Phe508del-gating and -residual function genotypes. N Engl J Med . 2021;385:815–825. doi: 10.1056/NEJMoa2100665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sagel SD, Khan U, Heltshe SL, Clancy JP, Borowitz D, Gelfond D, et al. Clinical effectiveness of lumacaftor/ivacaftor in patients with cystic fibrosis homozygous for F508del-CFTR: a clinical trial. Ann Am Thorac Soc . 2021;18:75–83. doi: 10.1513/AnnalsATS.202002-144OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Heltshe SL, Mayer-Hamblett N, Rowe SM. Understanding the relationship between sweat chloride and lung function in cystic fibrosis. Chest . 2013;144:1418. doi: 10.1378/chest.13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kawala CR, Ma X, Sykes J, Stanojevic S, Coriati A, Stephenson AL. Real-world use of ivacaftor in Canada: a retrospective analysis using the Canadian Cystic Fibrosis Registry. J Cyst Fibros . 2021;20:1040–1045. doi: 10.1016/j.jcf.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 39. Volkova N, Moy K, Evans J, Campbell D, Tian S, Simard C, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: data from national US and UK registries. J Cyst Fibros . 2020;19:68–79. doi: 10.1016/j.jcf.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 40. Kirwan L, Fletcher G, Harrington M, Jeleniewska P, Zhou S, Casserly B, et al. Longitudinal trends in real-world outcomes after initiation of ivacaftor. A cohort study from the Cystic Fibrosis Registry of Ireland. Ann Am Thorac Soc . 2019;16:209–216. doi: 10.1513/AnnalsATS.201802-149OC. [DOI] [PubMed] [Google Scholar]
- 41. Rowe SM. A simple test could extend cystic-fibrosis treatments to those left behind. Nature . 2020;583:S5. [Google Scholar]
- 42. Mayer-Hamblett N, van Koningsbruggen-Rietschel S, Nichols DP, VanDevanter DR, Davies JC, Lee T, et al. Building global development strategies for CF therapeutics during a transitional CFTR modulator era. J Cyst Fibros . 2020;19:677–687. doi: 10.1016/j.jcf.2020.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zemanick ET, Konstan MW, VanDevanter DR, Rowe SM, Clancy JP, Odem-Davis K, et al. Measuring the impact of CFTR modulation on sweat chloride in cystic fibrosis: rationale and design of the CHEC-SC study. J Cyst Fibros . 2021;20:965–971. doi: 10.1016/j.jcf.2021.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kazmerski TM, West NE, Jain R, Uluer A, Georgiopoulos AM, Aitken ML, et al. Family-building and parenting considerations for people with cystic fibrosis. Pediatr Pulmonol . 2021 doi: 10.1002/ppul.25620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. West NE, Kazmerski TM, Taylor-Cousar JL, Tangpricha V, Pearson K, Aitken ML, et al. Optimizing sexual and reproductive health across the lifespan in people with cystic fibrosis. Pediatr Pulmonol . 2021 doi: 10.1002/ppul.25703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jain R, Taylor-Cousar JL. Fertility, pregnancy and lactation considerations for women with CF in the CFTR modulator era. J Pers Med . 2021;11:418. doi: 10.3390/jpm11050418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mayer-Hamblett N, Nichols DP, Odem-Davis K, Riekert KA, Sawicki GS, Donaldson SH, et al. Evaluating the impact of stopping chronic therapies after modulator drug therapy in cystic fibrosis: the SIMPLIFY clinical trial study design. Ann Am Thorac Soc . 2021;18:1397–1405. doi: 10.1513/AnnalsATS.202010-1336SD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gifford AH, Mayer-Hamblett N, Pearson K, Nichols DP. Answering the call to address cystic fibrosis treatment burden in the era of highly effective CFTR modulator therapy. J Cyst Fibros . 2020;19:762–767. doi: 10.1016/j.jcf.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hisert KB, Heltshe SL, Pope C, Jorth P, Wu X, Edwards RM, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med . 2017;195:1617–1628. doi: 10.1164/rccm.201609-1954OC. [DOI] [PMC free article] [PubMed] [Google Scholar]