

Abstract
Increased local blood flow in response to neural activity is critical for brain function and the basis for functional imaging. Takano et al. now show that in vivo, astrocytes are central in translating neural activity into vasodilation via a mechanism involving COX1 metabolites.
Interruptions of blood flow as short as a few seconds can lead to unconsciousness, and interruptions of only a few minutes cause irrevocable damage, because of the loss of energy (oxygen and glucose) supplied by the blood. To circumvent these catastrophic effects, the brain is equipped with an array of auto-regulatory mechanisms that strive to maintain cerebral perfusion in the face of changing systemic blood pressure1. However, because neural activity consumes energy substrates, maintaining an adequate energy supply also requires that blood flow is increased to regions of the brain that are active. The brain’s ability to increase blood flow to active regions, termed functional hyperemia, was first described more than a century ago1, but interest in the mechanisms underlying this phenomenon has intensified in recent years both because it is the basis of functional brain imaging with MRI (fMRI) and because it is likely to be intimately involved in a variety of brain pathologies such as stroke2 and Alzheimer disease1. Although a variety of potential mechanisms have been considered3, recent attention has focused on astrocytes as a primary intermediary between neuronal activity and increased blood flow. However, attempts to elucidate the role of astrocytes with in vitro preparations have yielded conflicting results. In this issue, Takano et al.4 describe an elegant new approach to study functional hyperemia in vivo and show conclusively that the activation of astrocytes induces local vasodilation and increased blood flow and that the signaling pathway includes COX1 metabolites.
By sending specialized processes to both the vasculature5 and synaptic contacts6, individual astrocytes provide a clear anatomical link between neurons and arterioles (Fig. 1), and astrocytes sense and respond to synaptically released glutamate7,8. Notably, activation of astrocyte glutamate receptors raises cytoplasmic calcium in waves that spread along astrocytic processes (Fig. 1). Furthermore, synaptically released glutamate is removed primarily by uptake on astrocytic glutamate transporters, and glutamate uptake into astrocytes is thought to stimulate astrocytes to take up glucose from local blood vessels as a source of energy9,10. However, though astrocytes seem very qualified for the job, it has been difficult to directly test the role of astrocytes in functional hyperemia8,11. Teasing out the mechanisms underlying functional hyperemia is complicated because it is difficult to monitor activity in all the relevant elements (neurons, astrocytes, arterioles, pericytes and endothelial cells) while selectively manipulating specific elements. Recent efforts to address the role of astrocytes with in vitro preparations have yielded conflicting results, with one study indicating that Ca2+ signals in astrocytes induce vasodilation8 but another study finding that Ca2+ signals in astrocytes cause vasoconstriction11. This disagreement probably stems from differences in the pre-existing state of the vasculature in the respective in vitro preparations12 and highlights the importance of maintaining physiological vascular tone for studies of functional hyperemia.
Figure 1.
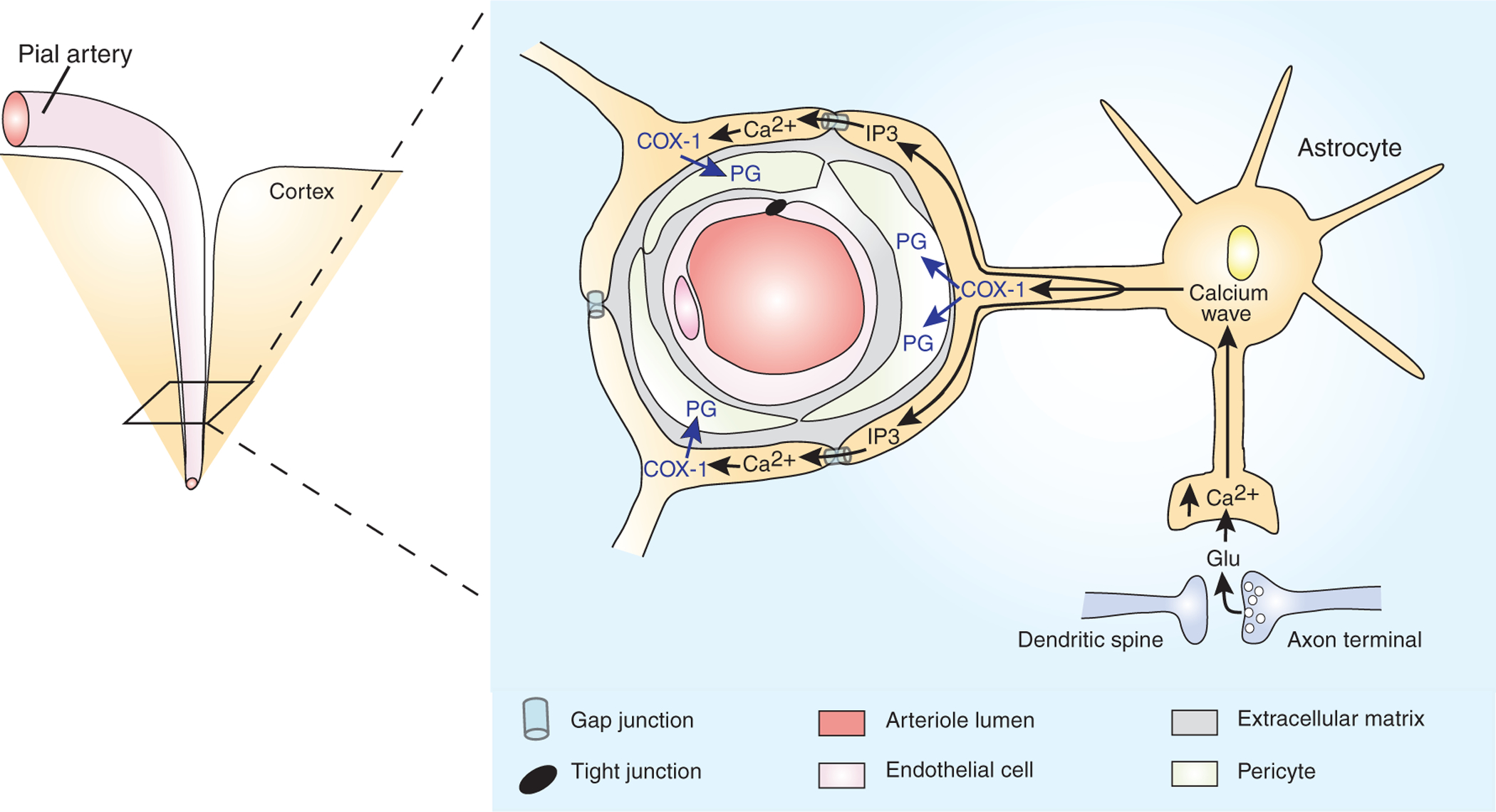
Astrocytes couple increased local blood flow to neuronal activity. By sending specialized processes both to arterioles (astrocyte endfeet) and to glutamatergic synapses, cortical astrocytes form an anatomical link between neurons and the blood supply. During neuronal activity, glutamate spills out of glutamatergic synapses, and, by activating astrocytic metabotropic glutamate receptors, triggers an astrocytic calcium wave. As the calcium wave invades an astrocytic endfoot, Ca2+ stimulates phospholipase A2 to produce arachidonic acid (AA). The AA is metabolized by the cyclooxygenase, COX-1, into a vasodilating prostaglandin (PG). Coordinated vasodilation is facilitated by Ca2+-induced production of IP3, which passes through gap junctions to trigger synchronous calcium waves and PG production in neighboring endfeet. For simplicity, PG is depicted as inducing vasodilation by acting in pericytes that surround the arteriole4. However, the site of action of PG is unknown, and there are other arteriole-associated cell types.
In this issue, Takano et al.4 present an elegant new approach to studying functional hyperemia in vivo, where all elements of the neurovascular unit are intact and functioning under normal physiological conditions. They used multi-photon confocal microscopy on anesthetized mice with transgenically labeled astrocytes and perfused the arteries with FITC-conjugated dextran. This approach enabled the authors to selectively stimulate individual astrocytes while monitoring astrocytic Ca2+ signals and vascular dynamics in vivo. The authors report that activation of astrocytic Ca2+ waves, either directly by focal uncaging of caged Ca2+ in individual astrocytes or in response to stimulation of neural activity, rapidly induced local vasodilation and increased blood flow. Notably, the authors observed vasodilation in response to astrocytic Ca2+ signals regardless of the type of anesthetic used. This consistency across type of anesthetic is important because different anesthetics have disparate influences on vascular tone and responses to stimulation13. Thus, under normal physiological conditions, elevations in astrocytic Ca2+ trigger vasodilation (Fig. 1).
How do Ca2+ signals in astrocytic endfeet induce vasodilation? There are numerous vasoactive molecules that may be released by elevations of Ca2+ in astrocytes1, such as metabolites of arachidonic acid (AA) generated by Ca2+-activated phospholipase A2 (PLA2), NO generated by Ca2+-activated NO-synthase, ATP and/or glutamate released by Ca2+-induced vesicle exocytosis14 and K+ released through Ca2+-activated K+ channels. Takano et al.4 used an array of pharmacological agents to determine which, if any, of these potential mediators underlies astrocyte-induced vasodilation. Surprisingly, they found no role for NO or adenosine, both potent vasodilators, released from astrocytes during Ca2+ signaling14. They also found no role for the epoxygenase pathway of AA metabolism, but they did find that the cyclooxygenase (COX) pathway of AA metabolism was critical. In particular, preventing either arachidonic acid production (by blocking phospholipase A2) or conversion of arachidonic acid to prostanoids (by blocking COX-1, but not COX-2) inhibited astrocyte-induced vasodilation by about 70%.
One drawback of the otherwise powerful in vivo approach is that it is very difficult to accurately control what concentration of exogenously applied drug is achieved within various compartments of the intact brain. Thus, a negative result may simply reflect inadequate penetration of the tested agent to the intended target and, conversely, a positive result may be achieved artifactually if the high concentration of drug used to ensure proper penetration results in non-specific activity. However, in support of their findings, a previous in vitro study8, in which well-controlled drug delivery was possible, also found that astrocyte-induced vasodilation was blocked by COX inhibitors. And, in further support of a prominent role of COX-1, Takano et al.4 observed dense immunohistochemical staining for COX-1 in the astrocytic endfeet that ensheath arterioles, but little or no COX-2 expression.
Taken together, the results indicate that elevated Ca2+ concentration in astrocytic endfeet activates PLA2 and that the resultant arachidonic acid is converted by COX-1 into a vasodilating prostanoid (Fig. 1). A next important step will be to determine how and where prostanoids act to induce vasodilation. Do prostanoids act on arteriolar pericytes, endothelial cells or even simply in astrocytes to elicit some other transcellular signaling molecule? What mechanism underlies the vasodilation that persists (~30%) when COX signaling is blocked? In both regards, it would also be useful to determine the role of other potential astrocyte-derived, vasoactive molecules, such as glutamate and K+.
With vasodilator capacity clearly established for astrocytes, a crucial question remains—are astrocytes the mediators of functional hyperemia? Takano et al.4 found that electrical stimulation of cortical neural activity induced robust Ca2+ elevations in astrocytic endfeet, vasodilation and consequently increased blood flow (as assessed with Doppler flowmetry). Inhibiting COX-1 activity did not affect synaptic field potential magnitude or astrocyte Ca2+ signals, but it reduced vasodilation and the consequent increase in blood flow by 50%. Similarly, blocking the mGluR5 subtype of metabotropic glutamate receptor, which is expressed on astrocytic processes near synapses and can mediate neurally triggered astrocytic Ca2+ signals8, had no effect on synaptic potentials but reduced the vascular response by 50%. Blocking mGluRs also reduces functional hyperemia induced by peripheral sensory stimulation8.
As a whole, the results suggest that astrocytes indeed mediate about half the vasodilation that is triggered by neural activity (Fig. 1). Astrocytes ‘sense’ neural activity when synaptically released glutamate activates mGluRs on the astrocytic processes that surround synapses. Activation of mGluRs triggers a Ca2+ wave that spreads to the arteriole ensheathing endfeet, where it activates PLA2. The consequent production of AA and conversion to prostanoids by COX-1, located in the endfeet, triggers vasodilation by a mechanism that has not yet been identified. The central role of astrocytes in mediating functional hyperemia fits well with their proposed role in coupling neuronal activity to increased energy consumption9,10.
This study also raises a number of interesting questions. What mechanisms underlie the functional hyperemia that is not mediated by the astrocyte COX-1 pathway? How do other vasculature control mechanisms, such as auto-regulation and brainstem control, interact with astrocytic control? Notably, noradrenergic fibers from the brainstem influence cerebral blood flow, and most of these noradrenergic fibers form contacts with astrocytic endfeet15. Furthermore, if signals other than local neuronal activity can trigger astrocytes to induce vasodilation, how do such signals influence the BOLD signal used in fMRI studies? Finally, an obvious remaining issue is to determine whether the astrocyte-induced vasoconstriction observed in vitro11 can occur under any conditions, possibly pathological, in vivo.
The methodology developed by Takano et al.4 is ideally suited to addressing these questions and other questions regarding the function of the intact and functioning neurovascular unit. Application of their methodology should greatly facilitate future studies of the dynamic control of brain microcirculation and relevant pathophysiological conditions.
References
- 1.Iadecola C Nat. Rev. Neurosci 5, 347–360 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Vavilava MS, Lee LA & Lam AM Anesthesiol. Clin. North America 20, 247–264 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Attwell D & Iadecola C Trends Neurosci 25, 621–625 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Takano T et al. Nat. Neurosci 9, 260–267 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Simard M, Arcuino G Takano T, Liu OS & Nedergaard M J. Neurosci 23, 9254–9262 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ventura R & Harris KM J. Neurosci 19, 6897–6907 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haydon PG Nat. Rev. Neurosci 2, 185–193 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Zonta M et al. Nat. Neurosci 6, 43–50 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Pellerin PJ & Magistretti JJ Cereb. Blood Flow Metab 23, 1282–1286 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Kasischke KA, Vishwasrao HD, Fischer PJ, Zipfel WR & Webb WW Science 305, 99–106 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Mulligan SJ & MacVicar BA Nature 431, 195–199 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Peppiatt C & Attwell D Nature 431, 137–138 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Gordon EL, Meno JR, Ngai AC, Lam AM & Winn HR J. Neurosurg 83, 875–877 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Pascual O et al. Science 310, 113–116 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Cohen Z, Molinatti G & Hamel E J. Cereb. Blood Flow Metab 17, 894–904 (1997). [DOI] [PubMed] [Google Scholar]