

Abstract
Brain ischemia results from cardiac arrest, stroke or head trauma. These conditions can cause severe brain damage and are a leading cause of death and long-term disability. Neurons are far more susceptible to ischemic damage than neighboring astrocytes, but astrocytes have diverse and important functions in many aspects of ischemic brain damage. Here we review three main roles of astrocytes in ischemic brain damage. First, we consider astrocyte glycogen stores, which can defend the brain against hypoglycemic brain damage but may aggravate brain damage during ischemia due to enhanced lactic acidosis. Second, we review recent breakthroughs in understanding astrocytic mechanisms of transmitter release, particularly for those transmitters with known roles in ischemic brain damage: glutamate, d-serine, ATP and adenosine. Third, we discuss the role of gap-junctionally connected networks of astrocytes in mediating the spread of damaging molecules to healthy ‘bystanders’ during infarct expansion in stroke.
Ischemia severely compromises the brain’s energy supply, either focally by artery blockage (stroke) or globally (cardiac arrest). Within seconds of initiation of global brain ischemia, consciousness is lost and, if it persists for as little as 5 min, severe brain damage is induced1,2. Stroke is one of the leading causes of death and disability in humans. Despite the prevalence and consequences of brain ischemia, the only effective treatment is reinstating the blood supply. This problem has motivated much research into the cellular mechanisms of ischemic brain damage.
Historically, this research has focused on neurons, partly because neurons are more sensitive to ischemia, but also because glutamatergic transmission, thought to occur only in neurons, is central to ischemic brain damage1. However, we now know that astrocytes have diverse and significant roles in glutamatergic signaling. Astrocytic glutamate transporters are the primary controllers of ambient extracellular glutamate concentration ([Glu]o), and astrocytes express glutamate receptors and have a variety of mechanisms for releasing glutamate. Thus, astrocytes are substantially affected by ischemia and intimately involved in the neuronal response to ischemia.
Here we review some important roles of astrocytes in ischemic brain damage, particularly those with therapeutic potential, focusing on how astrocytes influence neuronal death and damage. We start with an overview of the cellular response to loss of energy substrates, with emphasis on astrocytic glycogen stores. We then review how astrocytes influence glutamatergic processes. Finally we discuss the role of astrocytic gap-junctional proteins, which may spread damaging signals from a focal ischemic core to surrounding regions (the penumbra) or conversely enable protective signals from penumbral regions to enter the core. We focus on these issues because most of our current understanding of them has been formed in the last decade.
Cellular response to loss of glucose and oxygen
The primary insult that ischemia brings to all cells is a loss of the energy substrates glucose and oxygen, which slows or stops the synthesis of ATP through glycolysis and oxidative phosphorylation. There are potentially large reserves of alternatives to glucose as substrates for both glycolysis and respiration, such as glycogen, lactate and fatty acids. Thus, whether or not an interruption of glucose supplied by the blood depletes cellular ATP depends on the availability of alternative glycolytic and oxidative substrates, and on the cell’s rate of ATP consumption3. In contrast to glucose, oxygen is an irreplaceable driver of mitochondrial respiration, the main source of cellular ATP. Consequently, lack of oxygen immediately and severely reduces cellular ATP production, which results in a rapid decline in cellular [ATP] because of ongoing consumption3. Interrupting oxidative phosphorylation also causes ATP synthase to run backward and consume ATP, thereby accelerating the loss of ATP4. Furthermore, by interrupting oxidative phosphorylation late in the respiratory chain, at complex IV, loss of oxygen causes electron leak to transiently generate an increase in reactive oxygen species (ROS)4. Finally, when respiration is inhibited but glycolysis persists, protons and lactate generated during glycolysis accumulate, causing rapid intracellular acidification3. Thus, ischemia invariably causes depletion of cellular ATP, intracellular acidification and generation of ROS. Intracellular acidification and transient generation of ROS are stressful for most cells, but it is the loss of ATP and the ensuing malfunction of ATP-dependent processes, in particular those that are specific to brain cells, that make the brain so sensitive to ischemic damage.
A pivotal event in severe or complete brain ischemia is the inhibition of the Na+-K+ ATPase upon loss of cellular ATP. (ATP falls to 0–25% of that in normally perfused tissue2,5.) Inhibition of the Na+-K+ ATPase during severe ischemia causes, after several minutes, a profound loss of ionic gradients and membrane depolarization in neurons and astrocytes (Fig. 1a)3,6,7. The degradation of ionic gradients is associated with a large, sustained rise in the extracellular concentration of glutamate ([Glu]o = 100–300 μM) and other neurotransmitters8. The release of glutamate initiates a positive feedback loop, with the activation of glutamate receptors further decreasing ionic gradients and consuming ATP, both of which promote further release of glutamate9. This cascade of events culminates in large elevations of intracellular calcium in neurons10 and astrocytes11, which triggers the death of neurons, and potentially of astrocytes, by a process known as excitotoxicity1. The mechanisms underlying glutamate-triggered, Ca2+-mediated excitotoxicity are complex, but primarily involve activation of proteases, generation of ROS, and mitochondrial Ca2+ overload acting synergistically to damage cellular proteins, membranes, DNA and mitochondrial function, resulting in necrosis, apoptosis or both2,12,13. If the duration of severe ischemia is short enough (~5–20 min), cell death tends to be delayed and selective for neurons, whereas longer durations lead to broader and more rapid cellular destruction, resulting in infarct in the core of focal ischemia or patient death for global ischemia2,14.
Figure 1.
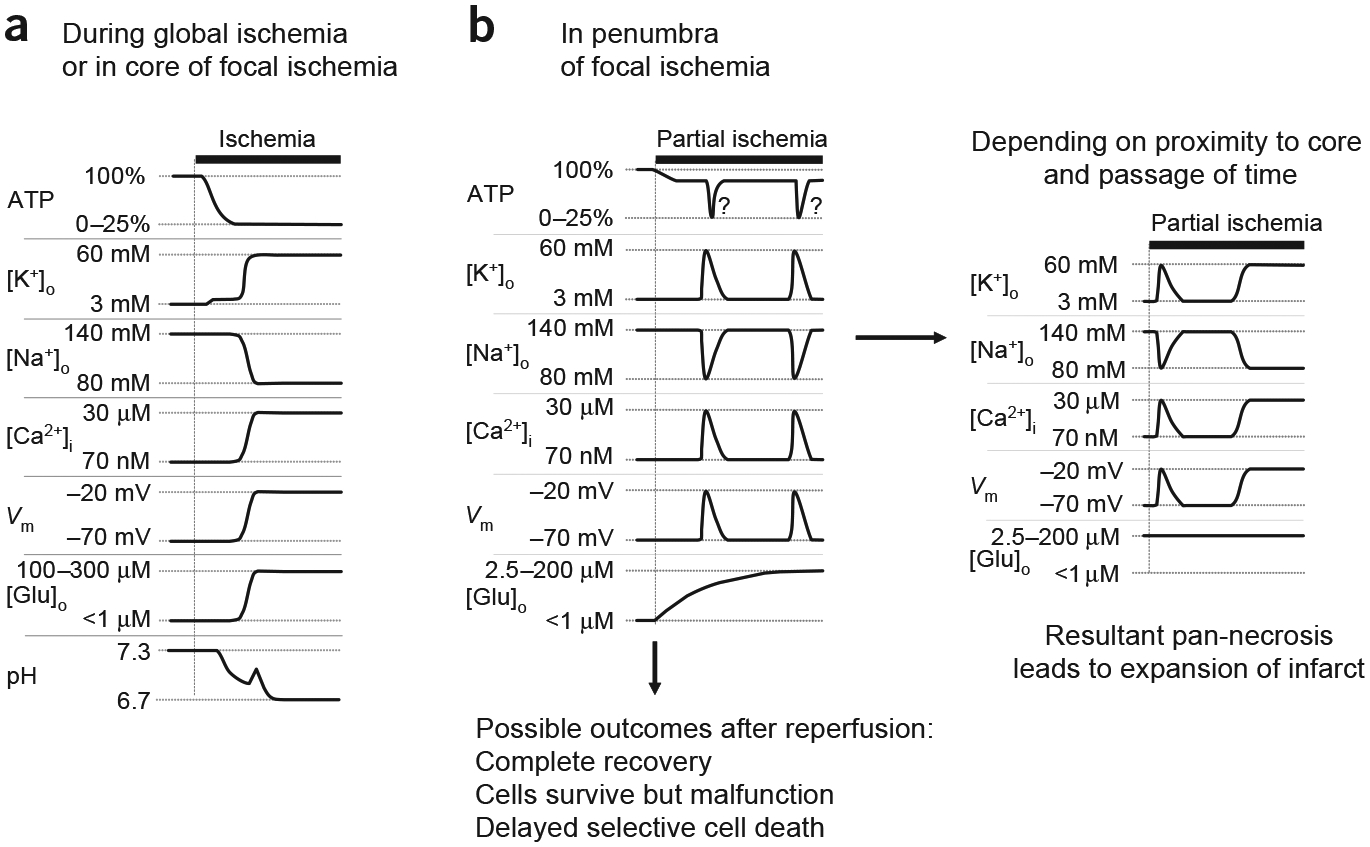
Events in brain ischemia. (a) Schematic illustration of events that occur during severe brain ischemia, as occurs in global ischemia and in the core of focal ischemia. Interruption of ATP production leads to inhibition of the Na+-K+ ATPase and to a consequent decrease of the transmembrane ion gradients. The disruption of ion homeostasis depolarizes cells and causes a large release of glutamate into the extracellular space. Ischemia also leads to extracellular acidification. (b) Events in the penumbra of focal ischemia. Initially, the drop in ATP is less severe, but triggers repeated transient depolarizations and associated ion shifts, while [Glu]o rises slowly but steadily (left panel). If reperfusion is initiated soon enough, complete recovery or selective damage may occur (bottom). With increasing duration of ischemia and proximity to the core, the transient ischemic depolarizations may evolve into terminal depolarization and ionic disruption, which leads to pan-necrosis and expansion of the infarct (right panel). Question marks next to transient ATP dips indicate our speculation that ischemic transient depolarizations may induce hypoxia, similarly to spreading depression in nonischemic brain tissue, and resultant transient enhanced ATP loss. Such an effect may act synergistically with ongoing partial ischemia to promote terminal disregulation and exacerbate damage.
Surrounding the core of a focal ischemic locus is a moderately hypoperfused region termed the penumbra2. The milder loss of perfusion in the penumbra compromises energy production to a lesser extent, with ATP falling to about 50–70% of that in normally perfused tissue (Fig. 1b)2. The more moderate fall in ATP does not immediately cause the severe, terminal ionic disruption that occurs in the core, but it does trigger a series of repeated transient ischemic depolarizations (TIDs) and associated ion shifts (Fig. 1b)15,16. Depending on the proximity to the core and the duration of the ischemic episode, penumbral TIDs may eventually evolve into terminal depolarization similar to that in the core (Fig. 1b)15. Transition from TIDs to terminal depolarization is associated with expansion of the infarct, whereas regions of the penumbra that only show TIDs do not evolve into infarct, but show selective neuronal damage or recovery (Fig. 1b)15. Despite the transient nature of ischemic depolarizations and ionic shifts, [Glu]o rises steadily in the penumbra over the first hour of ischemia, and then declines to nearly control levels by 2–3 h of continued ischemia17. The exact relationship between penumbral TIDs, glutamate and brain damage are not yet fully understood, but TIDs are temporally and spatially associated with both a rise of [Glu]o in the penumbra and an expansion of the infarct15, and blocking glutamate receptors reduces the size of the ischemic infarct.
Eventually much of what is described above must happen to both neurons and astrocytes. However, astrocytes differ from neurons in several important aspects that make astrocytes less susceptible to ischemic damage. First, astrocytes are able to maintain ATP levels longer than neurons during ischemia, and so severe ionic disregulation proceeds more slowly3,18. Second, astrocytes have a lower density of ionotropic glutamate receptors than neurons, and may have better ionic buffering and antioxidant capacity. These attributes presumably underlie the well-known selective destruction of neurons over astrocytes. Importantly, these attributes also place astrocytes in the position of potentially being able to protect their ailing neighbors, much as they do in the healthy brain. But because astrocytes too are stressed by ischemia, they may be less magnanimous, and may contribute to their neighbors’ demise. Ascertaining these possibilities, pro and con, should help identify potential therapeutic interventions.
Potential roles of astrocyte glycogen stores
The rate of progression of the aforementioned catastrophic events depends on the availability of alternative energy substrates and the rate of consumption of ATP by ongoing cellular processes, and these two elements represent a first significant difference between neurons and astrocytes. First, because neurons have a higher density of ion channels, and a consequent greater energy demand imposed by maintaining ionic gradients, neurons deplete ATP supplies upon cessation of ATP production more rapidly than astrocytes3. Second, most glycogen in the adult brain is found in astrocytes, not neurons19. Despite this lopsided cellular compartmentalization, astrocyte glycogen stores can benefit neurons during both physiological and pathological circumstances. By converting glycogen to lactate and passing lactate to neighboring neurons, astrocytes supplement neuronal energy requirements during periods of intense activity and during pathological shortages of glucose (hypoglycemia)20–24. The role of glycogen during ischemia is less obvious and less well studied. Because the primary benefit of glycogen during intense activity or hypoglycemia is through oxidation of lactate23,24, the protective potential of glycogen should be diminished in the absence of O2 during complete ischemia. Nonetheless, it seems reasonable that with less severe ischemia, as occurs in the penumbra, some oxidation of glycogen-derived lactate could occur and preserve energy status in either astrocytes or neurons. Furthermore, because astrocytes can convert glycogen to glucose19, they could, in principle, delay ATP depletion even in complete ischemia through anaerobic glycolysis. Similarly, because both neurons and astrocytes express glucose transporters, and intracellular glucose is in near equilibrium with extracellular glucose22,25, astrocytes could supply neurons with glycogen-derived glucose, thereby allowing neurons to delay their own ATP depletion. In fact, during complete brain ischemia, the decline in [ATP] parallels the decline in [glycogen], both of which are delayed relative to the decline in [glucose], albeit only by a couple of minutes5.
In contrast to the potential benefit of astrocytic glycogen stores in delaying ATP loss, studies of hyperglycemia during stroke indicate that maintaining glycolysis in the absence of O2 can exacerbate ischemic damage because of enhanced lactic acidosis. Indeed, hyperglycemia in human stroke patients is correlated with adverse tissue and functional outcome26. Although hyperglycemia in humans could simply be indicative of other complications that contribute to worse outcome, animal studies in which hyperglycemia is experimentally induced in the absence of other complications suggest that hyperglycemia per se exacerbates ischemic brain damage. Although raising plasma glucose levels before inducing ischemia delays the decline in ATP27 and delays the onset of ischemic depolarization28, it does not reduce cell death, even if the duration of ischemia is short enough (2.5 min) that hyperglycemic animals do not show ischemic depolarization28. For longer episodes of ischemia (10–20 min), hyperglycemia actually results in greater cell death and damage, despite delaying the onset of ischemic depolarization and ionic disregulation29. In these studies, hyperglycemia was associated with increased lactic acid production27, and the susceptibility for increased damage to hyperglycemia showed a sharp threshold with respect to the ischemia-induced drop in pH, which, in turn, was highly correlated with the magnitude of hyperglycemia29. The tight correlation between the degree of hyperglycemia exacerbated damage and lower pH, coupled with studies showing that lowering pH through hypercapnia also exacerbates ischemic brain damage, suggests that hyperglycemia exacerbates ischemic damage by enhancing production of lactate by anaerobic glycolysis30. Although this assertion is widely accepted, alternative mechanisms involving glucose-induced release of glucocorticoids have been postulated31.
Given the results on hyperglycemia, we might predict that despite the potential for astrocytic glycogen stores to delay astrocytic or neuronal ionic disregulation, consequent damage by lactic acid would counteract any benefit gained. Conversely, the case for a beneficial role of astrocyte glycogen stores would be strengthened if it turns out that hyperglycemic damage is actually mediated by glucocorticoid release31. Even if hyperglycemic augmentation of ischemic damage is due to lactic acidosis, the influence of hyperglycemia on lactic acidosis may be different from that of metabolizing glycogen in astrocytes. In particular, the degree and rate of ischemia-induced lactic acidosis produced by hyperglycemia, which raises glucose in both astrocytes and neurons, is likely to be greater than the degree and rate of ischemia-induced lactic acidosis produced by astrocyte-restricted and more slowly metabolized glycogen. And, given the sharp pH threshold for enhanced damage29, if metabolizing astrocytic glycogen is able to delay loss of ATP while generating even slightly less lactic acidosis than hyperglycemia, a net benefit may be obtained. However, in vitro studies of cultured astrocytes indicate that lactic acidosis–induced damage of astrocytes may be mediated primarily by an intracellular Ca2+ overload that is generated by a sequence consisting of activated Na+-H+ exchange, cytosolic Na+ loading, and resultant reversal of Na+-Ca2+ exchange32. Thus, even if metabolizing astrocytic glycogen produces less total tissue lactic acidosis than hyperglycemia, it may still promote astrocyte Ca2+ overload, which could lead to greater damage, either by impairing astrocytic protection of neurons or by killing astrocytes, thereby leading to infarct.
Ultimately, determining whether astrocyte glycogen stores protect brain tissue in animal models of stroke will require direct experimental testing. There may be several methods for boosting astrocyte glycogen stores, such as increasing glycogen synthase expression or activity19, or using inhibitors that prevent glycogen breakdown under normoglycemic conditions but not during hypoglycemia, the latter of which is feasible in vivo and protects brain electrical activity during hypoglycemia20. Because of the potential complication of increased lactic acidosis, it would also be useful to determine whether inhibiting known ischemic acid damage pathways in conjunction with raising astrocyte glycogen stores provides even greater protection than either manipulation alone. Of particular interest would be blocking the mitochondrial permeability transition pore with cyclosporin A, which protects against hyperglycemic ischemia27, or blocking acid sensing ion channels that contribute to ischemic brain damage27,33. Finally, although it is now generally accepted that hyperglycemia exacerbates ischemic damage, a recent survey of clinical studies suggests that mild hyperglycemia is actually beneficial for lacunar strokes34, a subtype of stroke that accounts for 15–25% of all ischemic strokes. Thus, even if raising astrocyte glycogen stores is not beneficial in more traditional stroke models, it would be interesting to determine whether it is beneficial in animal models of lacunar stroke.
The role of astrocytes in ischemic glutamate release
Despite the strong evidence for the role of excitotoxic damage in animal models of brain ischemia, human clinical trials of glutamate receptor antagonists have been remarkably unsuccessful at preventing brain damage in human ischemia. This failure has several likely explanations. First, because glutamate receptors have a pervasive role in all aspects of brain signaling, many clinical trials have either used antagonists at doses below those that are maximally effective in animal experiments to try to avoid side effects, or they have had to be terminated because of severe side effects35,36. Second, the interruption of the blood supply itself complicates the delivery of potentially therapeutic agents to the region in need37, unlike in animal models in which experimental agents can be delivered prophylactically. Circumventing these complications requires developing therapeutic compounds with few or no side effects, which would obviously reduce acute complications during an ischemic event, but would also enable prophylactic treatment for high-risk patients, including those with previous heart attack or stroke, those with vascular disease or those undergoing cardiac or brain surgery. There are two main avenues for interventions with minimal side effects aimed at preventing ischemia-induced glutamate-mediated excitotoxicity. The first is to develop glutamate receptor antagonists that are only active during pathological overactivation38, and the second is to determine the mechanisms mediating glutamate release during ischemia, in the hopes that such mechanisms may only be active during ischemia or may have more subtle physiological roles, and thus could be blocked or antagonized without significant side effects. The prospects for the latter avenue have been greatly enhanced by recent discoveries regarding astrocytic glutamate signaling. Accordingly, in the following section, we introduce the glutamate-mediated neuronal responses to ischemia to provide a framework for considering astrocytic contributions to those responses. Then we review recent advances in our understanding of astrocytic glutamate signaling not only during ischemia, but also under physiological conditions. In the latter case, even where there is no direct evidence for a role in ischemia, we speculate about potential roles.
Characteristics of glutamate release during ischemia.
As discussed above, excessive release of glutamate and activation of neuronal glutamate receptors is thought to be an important trigger for ischemia-induced neuronal damage. Early in vivo studies of electrical potential and extracellular ion concentrations identified two main phases of response to severe ischemia6,39. The first phase, lasting 1–2 min, is characterized by cessation of evoked electrical potentials, an isoelectric EEG and a gradual elevation of extracellular K+ to about 10 mM, with only minor changes in the concentration of other principal ions and in local electrical potential. The second phase is characterized by a rapid and large depolarization of the electrical potential, which is accompanied by a loss of ionic gradients (Fig. 1a). In vivo, the latter phase is accompanied by a pronounced elevation of extracellular glutamate and other neurotransmitters8. Because these early in vivo studies were unable to differentiate between neuronal and astrocytic roles in these gross electrical and ionic events, and they lacked adequate temporal resolution to resolve detailed properties of synaptic responses, many investigators have developed in vitro slice models of ischemia to record from individual neurons and astrocytes during simulated ischemia9,18,40. Voltage-clamp recordings from neurons in brain slices indicate that the neuronal response to simulated ischemia that is mediated by glutamate receptors occurs in two phases (Fig. 2a), which correspond to the two phases observed with in vivo studies. The earliest notable glutamate-mediated response is a large increase in the frequency of miniature excitatory postsynaptic currents (mEPSCs, Fig. 2a), presumably mediated by increased vesicle fusion. After a delay of several minutes (the exact timing depending on the experimental temperature, age of animal from which the slice was derived, and severity of the ischemia), during which mEPSC frequency increases progressively, the second phase of neuronal response is typified by a large, sustained glutamate receptor–mediated current (Fig. 2a), which, because of its magnitude, in unclamped cells drives membrane depolarization to near the reversal potential for nonspecific cation channels, which include glutamate receptor–gated channels (Fig. 2b). Paradoxically, during the period of enhanced mEPSC frequency, there is also a strong suppression of evoked EPSCs (Fig. 2a), which is mediated by adenosine acting at presynaptic adenosine receptors to suppress action potential–dependent vesicle release and is thought to protect neurons. It is likely that astrocytes are important in all three alterations of glutamatergic responses, as discussed in the following sections (Fig. 3).
Figure 2.
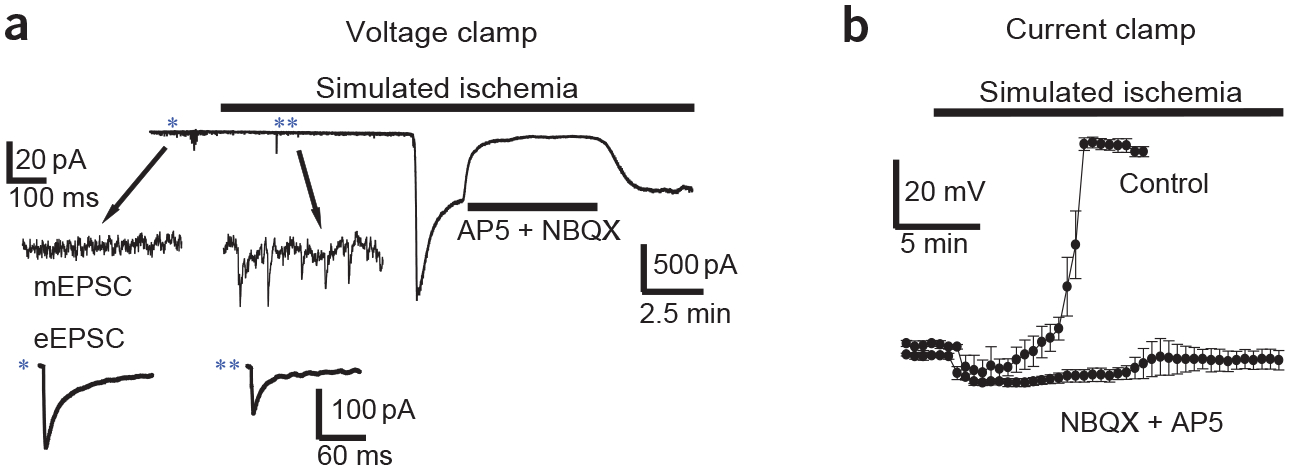
Simulated ischemia affects three aspects of glutamatergic signaling. (a) Voltage-clamp recording (holding voltage, Vh = −30 mV) of current in a CA1 pyramidal cell in a hippocampal slice during simulated ischemia. Blockade of large inward current by NMDA receptor antagonist AP5 (50 μM) and non-NMDA receptor antagonist NBQX (25 μM) indicates that it is generated by glutamate receptors. Inset shows typical examples of changes (*before ischemia compared with **after ischemia) in miniature EPSC (mEPSC) frequency (top) and suppression of evoked EPSC (eEPSC) amplitude (bottom) from different cells. Traces are adapted from data gathered in ref. 9. (b) Plot shows average (n = 3 or 4) membrane potential of Purkinje cells in cerebellar slices during simulated ischemia under control conditions or in the presence of glutamate receptor antagonists (as in a). Data from ref. 69.
Figure 3.
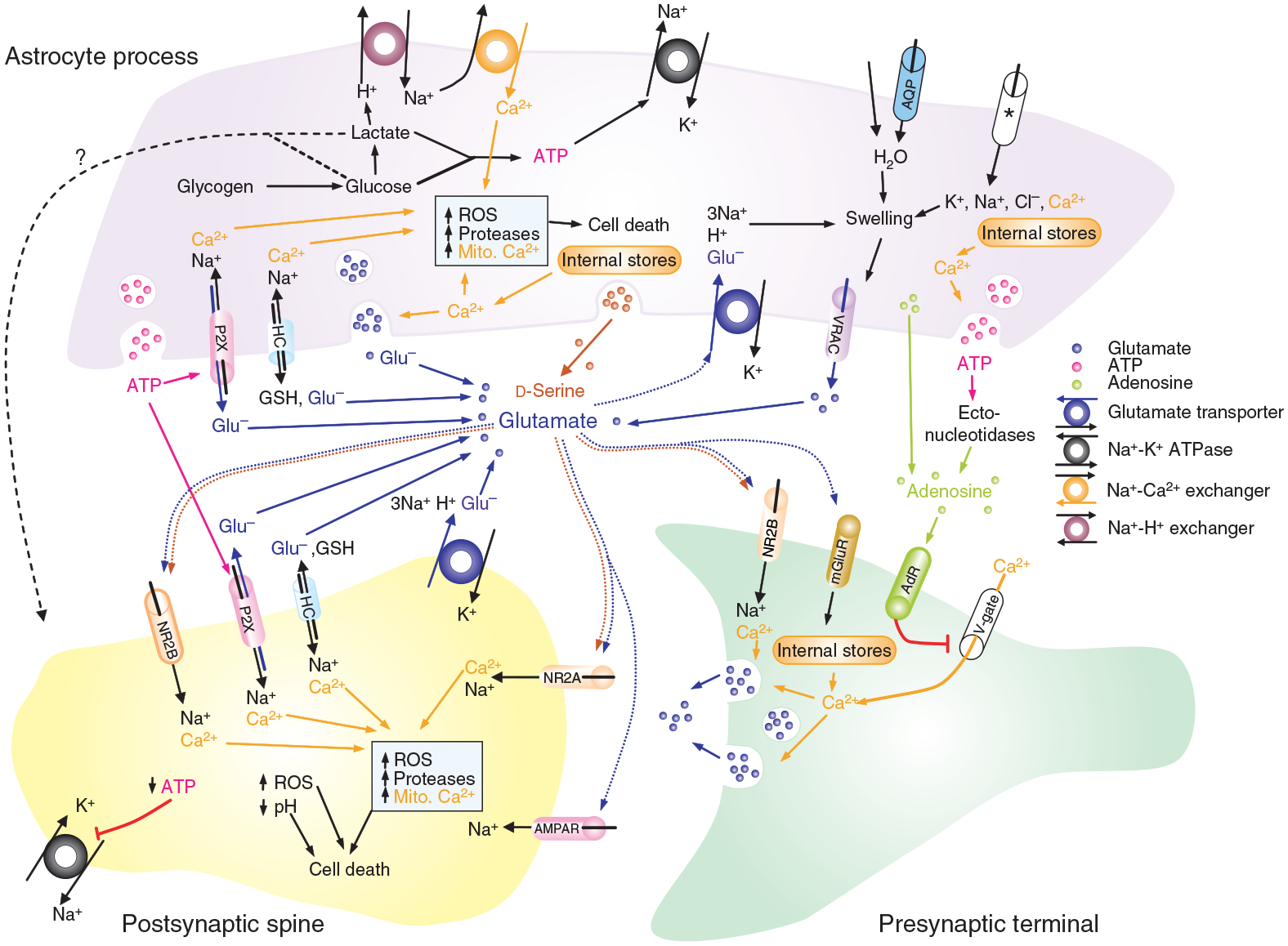
Summary diagram of the processes in neurons and astrocytes which have been shown to or could in principle contribute to the rise in [Glu]o and [Ca2+]i. AdR, adenosine receptor; AMPAR, AMPA receptor; AQP, aquaporin channel; ASIC, acid-sensing ion channels; GSH, glutathione; HC, connexin hemichannel; mGluR, metabotropic glutamate receptor; mito. Ca2+, mitochondrial calcium; NR2A/B, NMDA receptor with subunit 2A or B, respectively; P2XR, P2X receptor; V-gate, voltage-gated calcium channel; *ion flux through various unidentified ion channels. Details of evidence and our speculations in the text.
Purine release may modulate ischemic glutamate release.
Over the past decade, it has become clear that, contrary to previous dogma, astrocytes, like neurons, have and can release vesicles that contain glutamate or ATP41,42. ATP release from astrocytes is fundamental to the intercellular propagation of astrocyte Ca2+ waves, with ATP being released in response to elevations of intracellular Ca2+ and stimulating Ca2+ waves in neighboring cells by activating P2Y purinergic receptors43. The mechanisms by which astrocytes release ATP are still contentious and are likely to be diverse41, but there is good evidence that elevated cytoplasmic Ca2+ can trigger vesicular release of ATP from astrocytes44,45. Therefore, because ischemia raises cytoplasmic Ca2+ in astrocytes3,4,11, it is likely that ATP is released by astrocytes during ischemia46,47. ATP can cause glutamate release from astrocytes through activation and opening of P2X (particularly P2X7) receptors48,49. Thus, it is possible that ATP released by astrocytes during ischemia could contribute to the sustained rise in [Glu]o (Fig. 2a) through activation of glutamate-permeable P2X receptor channels, located on astrocytes themselves, or perhaps on neurons, which also express P2X receptors. Regardless of whether P2X receptors make a significant contribution to ischemic glutamate release, because of their high permeability to Ca2+, their activation during ischemia is likely to be damaging. Indeed, blocking P2X receptors reduces infarct volume and improves functional recovery in an in vivo model of stroke50.
By contrast, ATP released by astrocytes during ischemia is rapidly hydrolyzed to adenosine by ectonucleotidases47. Thus, ATP released by astrocytes contributes to the rise in extracellular adenosine that occurs during ischemia8,47, which suppresses evoked EPSCs and protects neurons51. Astrocytes can also directly release adenosine during hypoxia, through an as-yet-unknown but nonvesicular mechanism52. Therapeutically, it would seem useful to develop mechanisms that enhance conversion of ATP to adenosine, favor adenosine release over ATP release, and block P2X receptors.
Vesicular release of glutamate from astrocytes.
Astrocytes can also release vesicles that contain glutamate, which has a variety of actions on neurons, many of them possibly relevant to ischemia41,42. The most relevant are the generation of large, slow EPSCs, mediated by extrasynaptic NMDA receptors of the NR2B subtype41,53, and the increase in mEPSC frequency, mediated by activation of either presynaptic NMDA receptors of the NR2B subtype54 or metabotropic glutamate receptors (mGluRs)55. The targeting of extrasynaptic NR2B NMDA receptors is intriguing because ischemic activation of NR2B NMDA receptors (including but not limited to extrasynaptic receptors) is damaging to neurons, but activation of NR2A NMDA receptors is protective56. The ability of astrocytic vesicular glutamate release to drive an increase in mEPSC frequency in neurons through activation of mGluRs is also interesting because the ischemia-induced increase in mEPSC frequency (Fig. 2a) is independent of extracellular Ca2+ (ref. 40), and astrocytic vesicle release can be easily triggered by Ca2+ release from intracellular stores42,57,58, which presumably underlies mGluR-induced increased mEPSC frequency also.
Although recent evidence casts doubt on the ability of physiological astrocytic Ca2+ waves to elicit glutamatergic currents in neighboring neurons59, it seems likely that the severe rise in astrocytic Ca2+ that occurs during ischemia would induce vesicle release. Given the potential of astrocytic vesicular glutamate release to contribute to the ischemia-induced increased mEPSC frequency (Fig. 2a) and sustained glutamate currents (Fig. 2a), and given that astrocyte glutamate release activates NR2B NMDA receptors, it would be useful to determine whether blocking vesicle release from astrocytes protects neurons from ischemic damage. These issues could be investigated using a transgenic mouse in which SNARE-dependent vesicular release of gliotransmitters is selectively impaired in astrocytes60.
Roles of astrocytic and neuronal glutamate transporters.
Under physiological conditions, the extracellular concentration of glutamate is controlled by a family of five high-affinity, Na+-dependent plasma-membrane glutamate transporters (Fig. 4a), which power the uptake of glutamate against its electrochemical gradient by coupling transport of glutamate with the cotransport of three sodium ions and a proton and the countertransport of a potassium ion down their respective electrochemical gradients61. Although there are other types of glutamate transporters, the high-affinity Na+-dependent transporters are dominant in terminating the action of synaptically released glutamate and maintaining the ambient [Glu]o below that at which significant activation of glutamate receptors occurs. Accordingly, blocking glutamate uptake by these transporters leads to rapid accumulation of [Glu]o62 and consequent activation of glutamate receptors63,64.
Figure 4.
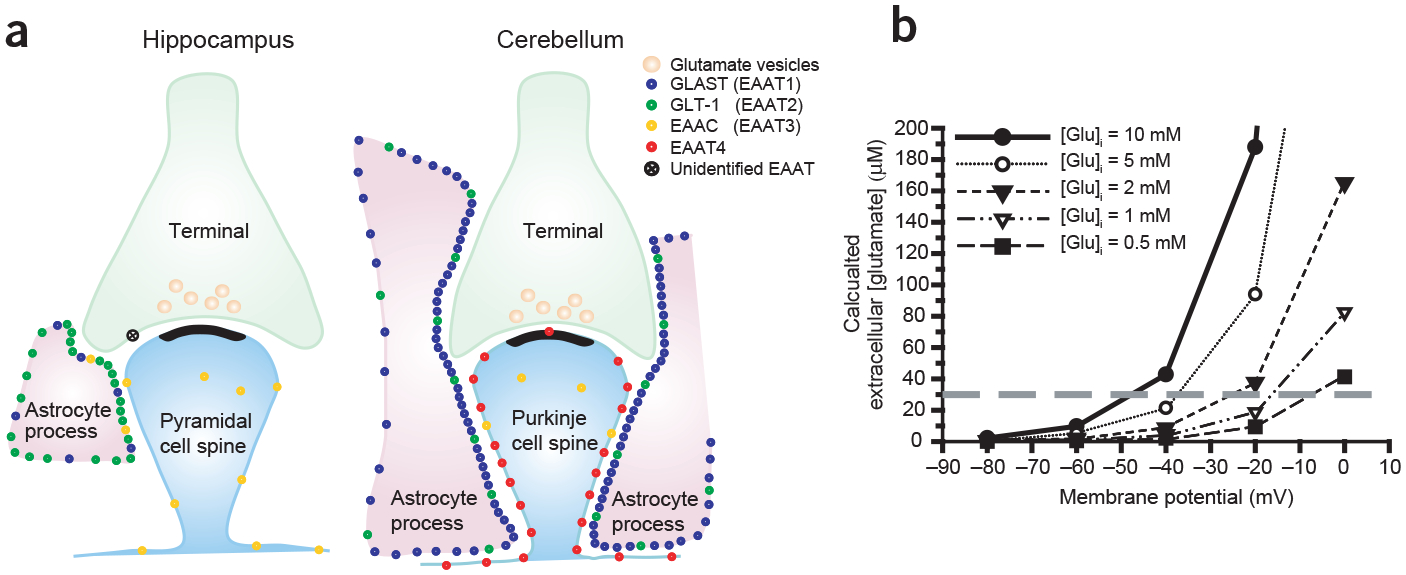
Expression and operation of Na+-dependent plasma-membrane glutamate transporters. (a) Schematic representation of distribution and relative densities of different glutamate transporter subtypes (based on quantitative immunocytochemistry, adapted from ref. 61). EAAT1–4 are members of the family of five high-affinity Na+-dependent plasma-membrane glutamate transporters. (EAAT1 is the human homolog of GLAST, and EAAT2 is the human homolog of GLT-1.) (b) Plot showing predicted (from equation (1)) extracellular glutamate concentration at equilibrium for ionic gradients during ischemia, given different membrane potentials and different intracellular concentrations of glutamate. The concentrations of co- and countertransported ions used were (in mM) [Na+]o = 87, [Na+]i = 39, [K+]o = 60, [K+]i = 131, [H+]o = 2 and [H+]i = 4. Gray dashed line shows concentration below which cultured neurons can tolerate 5 min exposure without detectable damage67.
During brain ischemia, the ionic gradients and membrane potential that power glutamate uptake are disrupted, with [Na+]o dropping to ~80 mM, [K+]o rising to ~60 mM and membrane potential dropping to ~–20 mV (Fig. 1a). In principle, the shift in ion gradients and loss of membrane potential can cause the Na+-dependent glutamate transporters to run backward and release glutamate65. However, whether this occurs also depends on the intracellular glutamate concentration. We and others have demonstrated experimentally that glutamate release by transporter reversal is a main source of [Glu]o accumulation during ischemia in vivo62 and simulated ischemia in vitro9. However, these studies, which used glutamate transporter antagonists that block all members of the transporter family, leave open the question of which cell type, neurons or astrocytes, is doing the releasing by reverse transport.
Astrocytes have a much higher density of glutamate transporters (Fig. 4a), and so have a higher capacity, but under physiological conditions, neurons have a much higher concentration of intracellular glutamate and depolarize more rapidly than astrocytes3,18, and thus their transporters are more likely to reverse. The minimum [Glu]o that the glutamate transporter can maintain depends on the membrane potential and on the gradients of the transported ions and is given by the equation65
| (1) |
where V is the membrane potential, F is Faraday’s constant, R is the gas constant and T is the absolute temperature. From this relationship, we can predict [Glu]o for different intracellular glutamate concentrations and membrane potentials (Fig. 4b). It is difficult to know precisely what [Glu] is in different cellular compartments, but estimates from quantitative electron immunocytochemistry suggest that the glutamate concentration is about 4–6 fold higher in glutamatergic axon terminals than in astrocytic processes and GABAergic neuronal processes, with intermediate levels found in glutamatergic neuronal somas66. A reasonable estimate from the quantitative data suggests that astrocyte processes have between 0.5 and 3 mM glutamate, whereas the concentration in neuronal axon terminals exceeds 10 mM. From these estimates, and the values we derived from equation (1) (Fig. 4b), we can predict that unless astrocytes depolarize beyond −20 mV, it is not likely that they will release glutamate to excitotoxic levels (>30 μM, from ref. 67), whereas neurons almost certainly will. Indeed, quantitative electron immunocytochemical studies of glutamate concentration indicate that ischemia causes a loss of glutamate from neuronal somas and dendrites and a corresponding increase in astrocytic processes, suggesting that during ischemia neurons release glutamate and astrocytes take it up66. In accordance with this prediction, by monitoring glutamate release with membrane currents in voltage-clamped neurons (as in Fig. 2a), we have found that genetic deletion of the glial glutamate transporters GLT-1 (predominant in the cortex and hippocampus, Fig. 4a) or GLAST (predominant in the cerebellum, Fig. 4a) does not reduce the glutamate release that is sensed by neurons during the first 10–15 min of simulated severe ischemia in hippocampal or cerebellar slices68,69.
An in vivo study in the hippocampus found that genetic deletion of GLT-1 increases bulk glutamate buildup during the first 5 min of severe global ischemia, but that it decreases bulk glutamate buildup during longer periods of ischemia70. Thus, during the early stages of severe ischemia, astrocytes continue taking up glutamate, presumably because of their low initial glutamate content66 and relative resistance to membrane depolarization3,11. However, as evidenced by the decreased buildup of [Glu]o in GLT-1 knockout mice during longer periods of severe ischemia70, eventually astrocytes succumb and release glutamate by reversed transport by GLT-1. Yet this thermodynamic inevitability develops at a time when irrevocable damage is already done, because genetic deletion of GLT-1 results in greater delayed cell death for short periods of global ischemia, but has no effect on cell death for longer periods of ischemia. Also, although astrocytes may maintain the driving force for glutamate uptake, the function of the transporter can be impaired by oxidative stress during ischemia71, and so it may not function as well as it could based on thermodynamics alone.
If during the early phases of severe ischemia astrocytes are still able to take up glutamate, it would imply that it is neurons that release glutamate by reverse transport. This proposal is supported by a study of hippocampal slices showing that genetic deletion of the primary neuronal Na+-dependent glutamate transporter, EAAC1, significantly delays the onset of the anoxic depolarization72. Taken together, the data support the idea that during the early stages of severe ischemia, glutamate is released by neuronal glutamate transporters running backward (and by vesicle exocytosis), and that astrocytic glutamate transporters continue taking up glutamate released by neurons, thus protecting neurons from more extensive damage.
We suggest that the reason astrocyte glutamate transporters continue taking up glutamate when neuronal transporters are releasing it is that because, compared to neurons, astrocytes have lower concentrations of cytoplasmic glutamate66, and they are more resistant to membrane depolarization18 (Fig. 4b). However, because conversion of glutamate to glutamine by glutamine synthetase in astrocytes is an ATP-dependent process, glutamate that is taken up by astrocytes under ischemic conditions may not be converted as efficiently73, and thus glutamate will accumulate in the astrocytic cytoplasm66. Then if ischemia persists, when astrocytes eventually do depolarize, the accumulated glutamate is released, albeit at a time when neuronal damage is beyond repair.
The dynamics of astrocytic glutamate transport could be quite different during focal ischemia, where astrocytes in the penumbral region may, as discussed earlier, be better able to maintain ATP levels. If astrocytes in the penumbra can slow or prevent their loss of ATP by consuming glycogen, they may be able to maintain conversion of glutamate to glutamine, thus preventing glutamate from accumulating, and they may be able to maintain a better membrane potential. Both of these factors would promote the ability of astrocytes to continue to take up glutamate (Fig. 4b) for prolonged periods. In support of this idea, in an in vivo study of the penumbral region of focal ischemia, blocking GLT-1 resulted in elevated [Glu]o that stayed elevated, relative to that in ischemic controls without GLT-1 blockade, for up to an hour of ischemia, and lasted for nearly two hours during reperfusion17. In another focal ischemia study, genetic suppression of GLT-1 increased the size of infarct, even when the focal ischemic episode lasted for a full hour (ref. 74). Thus, in the penumbral region of a focal ischemic event, astrocytes are able to take up glutamate, and they protect neurons for prolonged periods.
Given the ability of astrocytes to protect neurons in global or focal ischemia models by maintaining uptake activity of Na+-dependent plasma-membrane glutamate transporters, methods for increasing expression or activity of the glial glutamate transporters, GLT-1 and GLAST, should be therapeutically useful. Indeed, the protection against ischemic damage that is provided by ischemic preconditioning is mediated, in part, by upregulation of GLT-1 expression75, and β-lactam antibiotics, which can increase GLT-1 expression or activity, are protective in in vitro models of ischemia71,76. The latter finding is exciting in light of the ease with which some β-lactam antibiotics cross the blood-brain barrier, an important secondary attribute for therapeutic approaches to any brain-based pathology. Similarly, given the potential role of astrocytic glutamine synthetase in keeping cytoplasmic glutamate concentrations low, and consequently improving the ability of astrocytes to take up glutamate during mild ischemia, increasing expression or activity of astrocytic glutamine synthetase77 might be therapeutic for stroke. In fact, chemical preconditioning–induced tolerance to focal ischemia is associated with upregulation of glutamine synthetase78.
The ability of astrocytes to take up glutamate while neurons are releasing it pertains only to glutamate transport by Na+-dependent glutamate transporters, where the direction of glutamate flux is primarily determined by the electrochemical gradient of the three sodium ions that are cotransported with each glutamate molecule. The driving force for glutamate flux through unfacilitated passive pathways is outward for both neurons and astrocytes, because even the relatively low glutamate concentrations in astrocytes of 0.5 to 3 mM represent a huge concentration gradient, given the much lower extracellular concentration of glutamate (<1 μM). And, because glutamate is negatively charged, the more negative membrane potential of astrocytes18 actually promotes glutamate efflux through passive flux pathways. Thus, astrocytes may take up glutamate through Na+-dependent glutamate transporters while simultaneously releasing glutamate through the vesicular or passive pathways discussed in other sections. Such a futile cycle may accelerate astrocyte energy depletion and astrocytic Na+ accumulation, which may, in turn, exacerbate astrocytic swelling and Ca2+ overload. Accordingly, the therapeutic approaches described above, involving enhancing glutamate uptake by Na+-dependent glutamate transporters, may be further improved by preventing glutamate release through the passive efflux pathways or the vesicular mechanisms discussed in other sections.
Release of glutamate via volume-regulated anion channels.
Brain ischemia causes astrocytes to swell, probably through the interaction of a variety of potential mechanisms79. Swelling can be damaging on its own, but in addition, astrocytes are endowed with volume-regulated anion channels (VRACs) that open in response to swelling and allow the efflux of glutamate and other amino acids, as part of the process of regulatory volume decrease80. In in vivo focal ischemia models, drugs that block VRACs reduce glutamate efflux in both the ischemic core81 and the penumbra17, and they provide significant neuroprotection79. However, there is some ambiguity regarding the mechanism of neuroprotection and block of glutamate release provided by VRAC antagonists because most of them are not very specific, and some have other potential neuroprotectant properties, such as scavenging ROS79. Indeed, tamoxifen, which significantly reduces glutamate release during in vivo ischemia17, potently blocks swelling-induced VRAC currents in cultured astrocytes, but does not very effectively prevent swelling-induced amino acid release from the same cultured astrocytes82. Furthermore, L-644,711, which inhibits glutamate release from VRACs in cultured astrocytes, does not reduce glutamate release in an in vitro slice model of severe ischemia9. Nevertheless, the apparent ability of a variety of VRAC antagonists to reduce ischemic glutamate release during ischemia in vivo indicates that swelling-induced opening of astrocytic VRACs may be a significant source of ischemia-induced excitotoxic glutamate release, and the clear neuroprotective effect of tamoxifen is promising, regardless of the underlying mechanism(s). Further study of the role of VRACs in ischemic brain damage would be facilitated by the development of more specific VRAC antagonists and by the molecular identification of the proteins underlying VRAC currents.
d-Serine release in ischemia
Although activation of any subtype of glutamate receptor may contribute to neuronal damage, activation of the NMDA subtype of glutamate-gated channel is considered to be a primary cause of excitotoxicity because of its high permeability to Ca2+. Unlike other glutamate receptors, opening the NMDA receptor-gated channel requires activating two distinct obligatory co-agonist binding sites, one that binds glutamate and one that binds glycine or d-serine83,84. As glycine was the first co-agonist discovered, it was assumed until recently to be the endogenous NMDA receptor co-ligand. However, recent work indicates that d-serine is the endogenous co-ligand in many cases83. Although in some instances the glycine/serine site is saturated by ambient concentrations of agonist, in most brain regions the site is not saturated, and so it is available for modulation of NMDA receptor conductance magnitudes and, hence, of Ca2+ influx. Consequently, d-serine release during brain ischemia could potentiate excitotoxic actions of glutamate acting at NMDA receptors. Degrading extracellular d-serine protects neurons from damage produced by applying exogenous NMDA or by simulated ischemia83. However, this result only demonstrates that endogenous d-serine is required for NMDA receptor–mediated excitotoxicity; it does not prove that extra d-serine is released during ischemia. Although d-serine can be released from neurons or astrocytes, release from astrocytes seems to be through Ca+2-dependent vesicle exocytosis that can be triggered by activation of astrocytic AMPA receptors84. Thus, it seems likely that release of d-serine from astrocytes during ischemia contributes to neuronal damage.
Diverse roles of gap junctions and unopposed hemichannels
Gap junctions are specialized connections between two cells; they are made up of two symmetrical transmembrane pores that enable relatively nonselective passage of molecules smaller than ~1 kDa, including ions and hence current flow85. Each cell provides its own half of the gap junction connection, in the form of a hexamer of subunits called connexins, a multigene family of about 20 members (a similar gene family called pannexins also forms gap junctions)86. In the brain, both neurons and astrocytes express connexins and form gap junctions, primarily with their own cell type85. Thus, gap junctions enable the formation of electrically and metabolically connected networks of neurons or astrocytes. Although both cell types express connexins, gap junctional networks of astrocytes are more common and generally more extensive.
In addition to the role of gap junction hexamers in enabling coupled networks, both neurons87 and astrocytes88 also express fully formed connexin or pannexin hexamers that are not connected to a neighboring cell. These unopposed half gap junctions, or hemichannels, are functional in being able to open and allow small molecules (<1 kDa) to pass between the cytoplasm of the cell and the extracellular space87,88. Although the permeability of gap junctions and hemichannels is relatively nonselective, both are gated by a variety of factors, many of which, such as membrane potential, divalent cations, phosphorylation, pH and ROS, may be affected by ischemic conditions85. Some ischemic signals are expected to promote channel opening (membrane depolarization), but others are expected to promote channel closure (dephosphorylation, lowered pH, elevated [Ca2+]), so it is not easy to predict a priori what will happen to gap junctions and hemichannels during ischemia. Empirically, hemichannels on both astrocytes and neurons open during ischemia87,89, whereas astrocyte gap junctional coupling is reduced but not abolished89,90.
Given their nonselective permeability, it seems most likely that opening unopposed hemichannels during ischemia is harmful to both neurons and astrocytes. Unregulated influx of Na+ and Ca2+ is clearly harmful, and available evidence indicates that hemichannels may be permeable to glutamate49,88, the release of which is also clearly harmful. Glutathione, a primary endogenous antioxidant, can also be released through astrocytic hemichannels91. Again, loss of a primary antioxidant during ischemia should be harmful when excessive ROS are being generated.
Although it seems fairly obvious that opening hemichannels during ischemia will be harmful, the situation is more complex when considering gap junctions—and we must consider both because gap junctions and hemichannels tend to be made up of the same subunits in a given cell85, so it may be difficult to preferentially antagonize just hemichannels. It is not obvious what the role of gap junctions will be during global ischemia because all cells in a given region are likely to be in a similar state. However, a general principal emerges for focal ischemia: gap junctions allow toxic molecules from astrocytes in the ischemic core to diffuse to progressively healthier cells in the penumbra, and conversely allow health-promoting molecules from astrocytes in the penumbra to diffuse into dying cells in the ischemic core (Fig. 5a)85. Such a situation can be good or bad. Allowing toxic molecules, such as Ca2+, to diffuse away from the source might be good for the source cells, but it can clearly cause damage in the cells that would otherwise be healthy92, including possibly spreading signals that drive the transient, spreading depression–like ischemic depolarizations in the penumbra that are thought to contribute to the expansion of the infarct93,94. This ‘bystander’ effect has been studied most extensively in cells that do not normally express gap junctions, which in these studies are transfected with controlled numbers and subtypes of gap junction proteins and injected with defined toxic molecules92,95–97. Whether or not there is significant interaction between dying cells and healthy cells depends on the magnitude of the conductance (Fig. 5b)97 and the type of connexins expressed in each cell96. Further, whether dying cells kill healthy bystanders, or healthy bystanders protect dying cells, depends on the concentration of the toxic molecule and on the proportions of the respective cells, with a fairly high ratio of healthy cells to dying cells (95 to 5 for the toxin gangcyclovir triphosphate) required for protection (Fig. 5c,d)95. Much less is known about the preponderance of pro versus con relationships for astrocytes in an ischemic core and penumbra, partly owing to the present inability to block gap junctions without blocking hemichannels, and partly because there are many independent toxic and health-promoting molecules to consider85.
Figure 5.

Schematic illustrations of possible interactions between healthy and dying cells through gap junctions. (a) Toxic substances from dying cells in a focal ischemic core can diffuse through gap junctions to healthy cells in the penumbra, thereby spreading damage. Simultaneously, protective substances from healthy cells in the penumbra can diffuse into and protect damaged cells in the core. (b) The number of gap junctions determines the rate and extent of equilibration of substances between gap-junctionally coupled cells. (c,d) The ratio of toxic cells to healthy ‘bystanders’ determines whether toxic substances are safely diluted (c) or spread damage (d).
Unfortunately, the complication does not stop here: in addition to the established role of gap junctions and hemichannels as intercellular and cellular channels, evidence is accumulating that gap junctional proteins interact with multiprotein signaling complexes and can influence numerous unexpected cellular processes, such as cytoskeletal dynamics and transcription98. Notably, with respect to ischemia, astrocytic gap junction coupling transcriptionally upregulates the expression of the primary astrocytic Na+-dependent glutamate transporter, GLT-1 (ref. 99), perhaps explaining why connexins can provide protection against metabolic inhibition independent of channel function100.
Perhaps not surprisingly, studies of the impact of blocking gap junctional function on ischemic brain damage have been mixed, with many showing protection but others showing exacerbation85. It is, however, clear that astrocyte gap junctions and hemichannels have diverse and important roles in ischemic brain damage. Improvements in specific antagonists and the development of techniques for selectively manipulating gap junctions and hemichannels will be important steps toward understanding and capitalizing on those roles.
Conclusions
The very nature of brain ischemia, a lack of blood, restricts the ability to deliver pharmacological agents to the site of need, thereby constraining the application of pharmacological interventions to two main arenas. The first is prophylaxis, whereby relevant pharmacological agents could be given in advance to high-risk patients, such as stroke and heart attack survivors (who have high risk of recurrence), patients with vascular diseases and heart surgery patients. The second is partial ischemia, represented primarily by the penumbra of focal ischemic events. Both situations require pharmaceuticals with few side effects.
Luckily, many of the potential therapeutic targets that are implicated in the astrocytic contribution to ischemic brain damage promise low side effects. First, as a prophylactic, increasing astrocytic glycogen stores does not have any obvious drawbacks for nervous system function, but in the event of an ischemic episode could boost the helpful aspects of astrocyte function and retard the harmful functions. Another promising prophylactic approach would be to increase astrocyte Na+-dependent glutamate transporter expression or activity. Similarly, boosting astrocytic antioxidant capacity could prevent oxidative suppression of glutamate uptake and would be likely to benefit numerous other functions not covered in this review. Along similar lines, increasing astrocytic glutamine synthetase capacity should, in partial ischemic conditions, improve astrocytes’ ability to degrade intracellular glutamate, thereby promoting protective glutamate uptake.
For many of the harmful processes that we have covered, it is more difficult to predict what side effects might be produced by inhibiting them, because we are still in the infancy of our understanding of their physiological functions. However, it is likely that blocking any of the various astrocytic release pathways for glutamate, ATP and d-serine would be protective during ischemia. Perhaps fortuitously, many of the release processes interact in a feedforward fashion, with, for example, ATP release inducing glutamate release inducing swelling, and so on. Thus, it may be possible to reduce the activity of several harmful pathways during ischemia simply by blocking one pathway. Future efforts at understanding the physiological function of the various astrocytic release pathways, their underlying mechanisms and their interactions during ischemia will provide valuable insight into potential therapies.
ACKNOWLEDGMENTS
We would like to thank D. Attwell for discussions. This work was supported by US National Institutes of Health grant 5R01NS051561.
References
- 1.Choi DW & Rothman SM The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci 13, 171–182 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Lipton P Ischemic cell death in brain neurons. Physiol. Rev 79, 1431–1568 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Silver IA, Deas J & Erecinska M Ion homeostasis in brain cells: differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience 78, 589–601 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Abramov AY, Scorziello A & Duchen MR Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci 27, 1129–1138 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowry OH, Passonneau JV, Hasselberger FX & Schulz DW Effect of ischemia on known substrates and cofactors of the glycolytic pathway in brain. J. Biol. Chem 239, 18–30 (1964). [PubMed] [Google Scholar]
- 6.Hansen AJ & Nedergaard M Brain ion homeostasis in cerebral ischemia. Neurochem. Pathol 9, 195–209 (1988). [DOI] [PubMed] [Google Scholar]
- 7.Kraig RP, Ferreira-Filho CR & Nicholson C Alkaline and acid transients in cerebellar microenvironment. J. Neurophysiol 49, 831–850 (1983). [DOI] [PubMed] [Google Scholar]
- 8.Phillis JW, Smith-Barbour M & O’Regan MH Changes in extracellular amino acid neurotransmitters and purines during and following ischemias of different durations in the rat cerebral cortex. Neurochem. Int 29, 115–120 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Rossi DJ, Oshima T & Attwell D Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 316–321 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Schubert P, Keller F, Nakamura Y & Rudolphi K The use of ion-sensitive electrodes and fluorescence imaging in hippocampal slices for studying pathological changes of intracellular Ca2+ regulation. J. Neural Transm. Suppl 44, 73–85 (1994). [DOI] [PubMed] [Google Scholar]
- 11.Duffy S & MacVicar BA In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J. Neurosci 16, 71–81 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arundine M & Tymianski M Molecular mechanisms of glutamate-dependent neuro-degeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci 61, 657–668 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholls DG, Johnson-Cadwell L, Vesce S, Jekabsons M, & Yadava N Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J. Neurosci. Res 23 April 2007. (doi: 10.1002/jnr.21290). [DOI] [PubMed] [Google Scholar]
- 14.Pulsinelli WA Selective neuronal vulnerability: morphological and molecular characteristics. Prog. Brain Res 63, 29–37 (1985). [DOI] [PubMed] [Google Scholar]
- 15.Higuchi T, Takeda Y, Hashimoto M, Nagano O & Hirakawa M Dynamic changes in cortical NADH fluorescence and direct current potential in rat focal ischemia: relationship between propagation of recurrent depolarization and growth of the ischemic core. J. Cereb. Blood Flow Metab 22, 71–79 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Nedergaard M & Hansen AJ Characterization of cortical depolarizations evoked in focal cerebral ischemia. J. Cereb. Blood Flow Metab 13, 568–574 (1993). [DOI] [PubMed] [Google Scholar]
- 17.Feustel PJ, Jin Y & Kimelberg HK Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 35, 1164–1168 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Xie M, Wang W, Kimelberg HK & Zhou M Oxygen and glucose deprivation-induced changes in astrocyte membrane potential and their underlying mechanisms in acute rat hippocampal slices. J. Cereb. Blood Flow Metab 22 August 2007. (doi: 10.1038/5j.jcbfm.9600545). [DOI] [PubMed] [Google Scholar]
- 19.Brown AM Brain glycogen re-awakened. J. Neurochem 89, 537–552 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Suh SW et al. Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide). J. Pharmacol. Exp. Ther 321, 45–50 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Brown AM et al. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J. Neurosci. Res 79, 74–80 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Ransom BR & Fern R Does astrocytic glycogen benefit axon function and survival in CNS white matter during glucose deprivation? Glia 21, 134–141 (1997). [PubMed] [Google Scholar]
- 23.Magistretti PJ Neuron-glia metabolic coupling and plasticity. J. Exp. Biol 209, 2304–2311 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Tekkok SB, Brown AM, Westenbroek R, Pellerin L & Ransom BR Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res 81, 644–652 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Silver IA & Erecinska M Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J. Neurosci 14, 5068–5076 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindsberg PJ & Roine RO Hyperglycemia in acute stroke. Stroke 35, 363–364 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Folbergrova J, Li PA, Uchino H, Smith ML & Siesjo BK Changes in the bio-energetic state of rat hippocampus during 2.5 min of ischemia, and prevention of cell damage by cyclosporin A in hyperglycemic subjects. Exp. Brain Res 114, 44–50 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Li PA, Kristian T, Shamloo M & Siesjo K Effects of preischemic hyperglycemia on brain damage incurred by rats subjected to 2.5 or 5 minutes of forebrain ischemia. Stroke 27, 1592–1601 (1996). [DOI] [PubMed] [Google Scholar]
- 29.Li PA, Shamloo M, Katsura K, Smith ML & Siesjo BK Critical values for plasma glucose in aggravating ischaemic brain damage: correlation to extracellular pH. Neurobiol. Dis 2, 97–108 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Li PA & Siesjo BK Role of hyperglycaemia-related acidosis in ischaemic brain damage. Acta Physiol. Scand 161, 567–580 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Schurr A Bench-to-bedside review: a possible resolution of the glucose paradox of cerebral ischemia. Crit. Care 6, 330–334 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chesler M Failure and function of intracellular pH regulation in acute hypoxic-ischemic injury of astrocytes. Glia 50, 398–406 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Xiong ZG et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118, 687–698 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Uyttenboogaart M et al. Moderate hyperglycaemia is associated with favourable outcome in acute lacunar stroke. Brain 130, 1626–1630 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Dawson DA, Wadsworth G & Palmer AM A comparative assessment of the efficacy and side-effect liability of neuroprotective compounds in experimental stroke. Brain Res 892, 344–350 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Devuyst G & Bogousslavsky J Update on recent progress in drug treatment for acute ischemic stroke. J. Neurol 248, 735–742 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Lo EH, Singhal AB, Torchilin VP & Abbott NJ Drug delivery to damaged brain. Brain Res. Brain Res. Rev 38, 140–148 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Chen HS & Lipton SA The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem 97, 1611–1626 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Astrup J, Symon L, Branston NM & Lassen NA Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke 8, 51–57 (1977). [DOI] [PubMed] [Google Scholar]
- 40.Katchman AN & Hershkowitz N Early anoxia-induced vesicular glutamate release results from mobilization of calcium from intracellular stores. J. Neurophysiol 70, 1–7 (1993). [DOI] [PubMed] [Google Scholar]
- 41.Haydon PG & Carmignoto G Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev 86, 1009–1031 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Montana V, Malarkey EB, Verderio C, Matteoli M & Parpura V Vesicular transmitter release from astrocytes. Glia 54, 700–715 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Gallagher CJ & Salter MW Differential properties of astrocyte calcium waves mediated by P2Y1 and P2Y2 receptors. J. Neurosci 23, 6728–6739 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Queiroz G, Meyer DK, Meyer A, Starke K & von Kügelgen I A study of the mechanism of the release of ATP from rat cortical astroglial cells evoked by activation of glutamate receptors. Neuroscience 91, 1171–1181 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Bal-Price A, Moneer Z & Brown GC Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia 40, 312–323 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Melani A et al. ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem. Int 47, 442–448 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Parkinson FE & Xiong W Stimulus- and cell-type-specific release of purines in cultured rat forebrain astrocytes and neurons. J. Neurochem 88, 1305–1312 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Duan S et al. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci 23, 1320–1328 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parpura V, Scemes E & Spray DC Mechanisms of glutamate release from astrocytes: gap junction “hemichannels”, purinergic receptors and exocytotic release. Neurochem. Int 45, 259–264 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Lammer A et al. Neuroprotective effects of the P2 receptor antagonist PPADS on focal cerebral ischaemia-induced injury in rats. Eur. J. Neurosci 23, 2824–2828 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Rudolphi KA, Schubert P, Parkinson FE & Fredholm BB Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol. Sci 13, 439–445 (1992). [DOI] [PubMed] [Google Scholar]
- 52.Martin ED et al. Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia 55, 36–45 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Fellin T et al. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43, 729–743 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Jourdain P et al. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci 10, 331–339 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Fiacco TA & McCarthy KD Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci 24, 722–732 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci 27, 2846–2857 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hua X et al. Ca2+-dependent glutamate release involves two classes of endoplasmic reticulum Ca2+ stores in astrocytes. J. Neurosci. Res 76, 86–97 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Montana V, Ni Y, Sunjara V, Hua X & Parpura V Vesicular glutamate transporter-dependent glutamate release from astrocytes. J. Neurosci 24, 2633–2642 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fiacco TA et al. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Pascual O et al. Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Danbolt NC Glutamate uptake. Prog. Neurobiol 65, 1–105 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Phillis JW, Ren J & O’Regan MH Transporter reversal as a mechanism of glutamate release from the ischemic rat cerebral cortex: studies with dl-threo-β-benzyloxyaspar-tate. Brain Res 868, 105–112 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Jabaudon D et al. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc. Natl. Acad. Sci. USA 96, 8733–8738 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cavelier P & Attwell D Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J. Physiol. (Lond.) 564, 397–410 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takahashi M et al. The role of glutamate transporters in glutamate homeostasis in the brain. J. Exp. Biol 200, 401–409 (1997). [DOI] [PubMed] [Google Scholar]
- 66.Ottersen OP, Laake JH, Reichelt W, Haug FM & Torp R Ischemic disruption of glutamate homeostasis in brain: quantitative immunocytochemical analyses. J. Chem. Neuroanat 12, 1–14 (1996). [DOI] [PubMed] [Google Scholar]
- 67.Choi DW, Maulucci-Gedde M & Kriegstein AR Glutamate neurotoxicity in cortical cell culture. J. Neurosci 7, 357–368 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamann M, Rossi DJ, Marie H & Attwell D Knocking out the glial glutamate transporter GLT-1 reduces glutamate uptake but does not affect hippocampal glutamate dynamics in early simulated ischaemia. Eur. J. Neurosci 15, 308–314 (2002). [DOI] [PubMed] [Google Scholar]
- 69.Hamann M, Rossi DJ, Mohr C, Andrade AL & Attwell D The electrical response of cerebellar Purkinje neurons to simulated ischaemia. Brain 128, 2408–2420 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mitani A & Tanaka K Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J. Neurosci 23, 7176–7182 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ouyang YB, Voloboueva LA, Xu LJ & Giffard RG Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci 27, 4253–4260 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gebhardt C, Korner R & Heinemann U Delayed anoxic depolarizations in hippocampal neurons of mice lacking the excitatory amino acid carrier 1. J. Cereb. Blood Flow Metab 22, 569–575 (2002). [DOI] [PubMed] [Google Scholar]
- 73.Gorovits R, Avidan N, Avisar N, Shaked I & Vardimon L Glutamine synthetase protects against neuronal degeneration in injured retinal tissue. Proc. Natl. Acad. Sci. USA 94, 7024–7029 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rao VL et al. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J. Neurosci 21, 1876–1883 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang M et al. The upregulation of glial glutamate transporter-1 participates in the induction of brain ischemic tolerance in rats. J. Cereb. Blood Flow Metab 27, 1352–1368 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Rothstein JD et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433, 73–77 (2005). [DOI] [PubMed] [Google Scholar]
- 77.Shaked I, Ben Dror I & Vardimon L Glutamine synthetase enhances the clearance of extracellular glutamate by the neural retina. J. Neurochem 83, 574–580 (2002). [DOI] [PubMed] [Google Scholar]
- 78.Hoshi A, Nakahara T, Kayama H & Yamamoto T Ischemic tolerance in chemical preconditioning: possible role of astrocytic glutamine synthetase buffering glutamate-mediated neurotoxicity. J. Neurosci. Res 84, 130–141 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Kimelberg HK Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 50, 389–397 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Kimelberg HK, Goderie SK, Higman S, Pang S & Waniewski RA Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J. Neurosci 10, 1583–1591 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Seki Y, Feustel PJ, Keller RW Jr., Tranmer BI & Kimelberg HK Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke 30, 433–440 (1999). [DOI] [PubMed] [Google Scholar]
- 82.Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK & Mongin AA Pharmacological comparison of swelling-activated excitatory amino acid release and Cl− currents in cultured rat astrocytes. J. Physiol. (Lond.) 572, 677–689 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martineau M, Baux G & Mothet JP d-Serine signalling in the brain: friend and foe. Trends Neurosci 29, 481–491 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Wolosker H d-Serine regulation of NMDA receptor activity. Sci. STKE [online] 2006, e41 (2006). [DOI] [PubMed] [Google Scholar]
- 85.Contreras JE et al. Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res. Brain Res. Rev 47, 290–303 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Connors BW & Long MA Electrical synapses in the mammalian brain. Annu. Rev. Neurosci 27, 393–418 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Thompson RJ, Zhou N & MacVicar BA Ischemia opens neuronal gap junction hemichannels. Science 312, 924–927 (2006). [DOI] [PubMed] [Google Scholar]
- 88.Ye ZC, Wyeth MS, Baltan-Tekkok S & Ransom BR Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci 23, 3588–3596 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Contreras JE et al. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 99, 495–500 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cotrina ML et al. Astrocytic gap junctions remain open during ischemic conditions. J. Neurosci 18, 2520–2537 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rana S & Dringen R Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett 415, 45–48 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Lin JH-C et al. Gap-junction-mediated propagation and amplification of cell injury. Nat. Neurosci 1, 494–500 (1998). [DOI] [PubMed] [Google Scholar]
- 93.Martins-Ferreira H, Nedergaard M & Nicholson C Perspectives on spreading depression. Brain Res. Brain Res. Rev 32, 215–234 (2000). [DOI] [PubMed] [Google Scholar]
- 94.Rawanduzy A, Hansen A, Hansen TW & Nedergaard M Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J. Neurosurg 87, 916–920 (1997). [DOI] [PubMed] [Google Scholar]
- 95.Wygoda MR et al. Protection of herpes simplex virus thymidine kinase-transduced cells from ganciclovir-mediated cytotoxicity by bystander cells: the Good Samaritan effect. Cancer Res 57, 1699–1703 (1997). [PubMed] [Google Scholar]
- 96.Andrade-Rozental AF et al. Gap junctions: the “kiss of death” and the “kiss of life”. Brain Res. Brain Res. Rev 32, 308–315 (2000). [DOI] [PubMed] [Google Scholar]
- 97.Cusato K et al. Gap junctions mediate bystander cell death in developing retina. J. Neurosci 23, 6413–6422 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Herve JC, Bourmeyster N, Sarrouilhe D & Duffy HS Gap junctional complexes: From partners to functions. Prog. Biophys. Mol. Biol 94, 29–65 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Figiel M, Allritz C, Lehmann C & Engele J Gap junctional control of glial glutamate transporter expression. Mol. Cell. Neurosci 35, 130–137 (2007). [DOI] [PubMed] [Google Scholar]
- 100.Lin JH et al. Connexin mediates gap junction-independent resistance to cellular injury. J. Neurosci 23, 430–441 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]