

Abstract
SUMOylation is a posttranslational modification of lysine residues. Modification of proteins by small ubiquitin-like modifiers (SUMO)1, -2, and -3 can achieve varied, and often unique, physiological and pathological effects. We looked for SUMO2-specific effects on vascular endothelial function. SUMO2 expression was upregulated in the aortic endothelium of hypercholesterolemic low-density lipoprotein receptor-deficient mice and was responsible for impairment of endothelium-dependent vasorelaxation in these mice. Moreover, overexpression of SUMO2 in aortas ex vivo, in cultured endothelial cells, and transgenically in the endothelium of mice increased vascular oxidative stress and impaired endothelium-dependent vasorelaxation. Conversely, inhibition of SUMO2 impaired physiological endothelium-dependent vasorelaxation in normocholesterolemic mice. These findings indicate that while endogenous SUMO2 is important in maintenance of normal endothelium-dependent vascular function, its upregulation impairs vascular homeostasis and contributes to hypercholesterolemia-induced endothelial dysfunction.
NEW & NOTEWORTHY Sumoylation is known to impair vascular function; however, the role of specific SUMOs in the regulation of vascular function is not known. Using multiple complementary approaches, we show that hyper-SUMO2ylation impairs vascular endothelial function and increases vascular oxidative stress, whereas endogenous SUMO2 is essential for maintenance of normal physiological function of the vascular endothelium.
Keywords: oxidative stress, sumoylation, vascular endothelial function
INTRODUCTION
Vascular endothelial dysfunction promotes atherosclerosis, the underlying cause of cardiovascular pathologies such as myocardial infarction, stroke, unstable angina, and sudden cardiac death (24, 33). Recent studies have highlighted the importance of SUMOylation in the regulation of vascular endothelial function and atherosclerosis (15, 16). SUMOylation is a complex posttranslational modification that involves the conjugation of small ubiquitin-like modifiers (SUMOs). The process is mediated by the E1 activators known as SUMO-activating enzymes (SAE)-1 and SAE-2, and the E2-conjugating enzyme UBC9. Deconjugation of SUMOs is controlled by SUMO-specific protease (SENPs) (39). Similar to other posttranslational modifications, SUMOylation regulates target protein activity, stability, and localization (40).
Increased SUMOylation promotes vascular endothelial dysfunction and atherosclerosis. Knockout of SENP2 (SENP2−/−), which increases SUMO2/3 conjugation, is embryonically lethal, while SENP2+/− mice exhibit impaired vascular endothelial function and augmented atherosclerosis (15, 16). This suggests that, although SUMOylation is essential for development, increased SUMOylation is detrimental to the vasculature.
There are four isoforms of SUMO (SUMO1, SUMO2, SUMO3, and SUMO4), all of which are unique in their function (13, 32). Genetic deletion of SUMO2 is embryonically lethal, whereas deletions of SUMO1 or SUMO3 are not (38, 43). Furthermore, cardiac-specific overexpression of SUMO1 improves cardiac function (19, 36), whereas SUMO2 induces cardiomyopathy (4, 20). To define the role of SUMOs in the vasculature, we focused on SUMO2, the most abundantly expressed isoform (32, 38), and investigated its effect on vascular endothelial function. Using ex vivo and in vivo manipulation of SUMO2, we show that upregulation of SUMO2 promotes oxidative stress and impairs endothelium dependent vasorelaxation in hypercholesterolemic mice, whereas basal endogenous SUMO2 supports physiological endothelial function.
METHODS
Animals.
All experimental animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Iowa and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Low-density lipoprotein receptor-deficient (LDLr−/−) mice were purchased from Jackson Laboratory (stock no. 002207). To induce hyperlipidemia, both wild-type and LDLr−/− mice were fed a Western diet (TD.88137, Harlan Teklad) containing 21% (wt/wt) saturated fat extracted from milk, 48.5% (wt/wt) carbohydrate, 17.3% (wt/wt) protein, and 0.2% (wt/wt) cholesterol for 12 wk. Because sex hormones are known to regulate vascular function (5, 31), we used only male mice (18–20 wk old) for this study.
Generation of the endothelium-specific HA-SUMO2 (e-SUMO2) transgenic mouse model.
We generated a transgenic mouse selectively expressing a fusion protein of hemagglutinin (HA-amino acid sequence YPYDVPDYA) and SUMO2 in the endothelium. The transgene construct contained a 5′-HA-tagged human SUMO2 full-length open reading frame (288 bp) under the control of an ~2.5-kb mouse endothelium-specific Ve-Cad promoter. C57BL/6 × C57BL/6 fertilized embryos were injected (Cyagen, Santa Clara, CA) with a linearized plasmid containing the transgene. Pseudopregnant females were implanted with viable injected embryos, resulting in the birth of 53 pups. Pups were screened using two sets of primers to confirm incorporation of the entire promoter-insert cassette. Seven potential founders were identified (2 males and 5 females), and subsequent breeding of these founders to C57BL/6 partners confirmed germline transmission of the transgene in two lines, which were maintained separately (founders 1 and 2). Genotyping primer sequences were set 1 (512 bp product): forward 5′-TGA CAT CCA CTT TGC CTT TCT CTC-3′, reverse 5′-CAC TGC ATT CTA GTT GTG GTT TGT C-3′; and set 2 (535 bp product): forward 5′ AGG-CAG-CTC-ACA-AAG-GAA-CAA-TAA-C-3′, reverse 5′-TAA-ACT-GCA-CCA-CAG-AAC-CAT-CCT-G-3′.
siRNA transfection.
Target-specific small interfering (si)RNAs and a nonspecific control siRNA (GC control) were purchased from Invitrogen. Sixty picomoles of siRNA was incubated with Lipofectamine 2000 (Invitrogen) at room temperature for 20 min before being added to cells prepared with OPTI-MEM medium (Invitrogen). Four hours later, the medium was replaced with fresh endothelial growth medium, and cells were cultured for an additional 18 h. For knockdown of SUMO2 in aortic rings, 500 nM siRNA for SUMO2 or scrambled control was transfected in serum-free EGM2 medium using Lipofectamine 2000 (siRNA for SUMO2: sense 5′-GCUGUUACAUGUAGGGCAATT-3′, antisense 5′-UUGCCCUACAUGUAACAGCTA-3′).
Cell culture.
Human umbilical vein endothelial cells (HUVECs) were purchased from Clonetics (San Diego, CA), cultured in endothelial growth medium 2 (Lonza, Walkersville, MD), and used until passage 6.
Adenoviral expression.
The optimum volume of the viral stock solution (sufficient for 1:100 multiplicity of infection) was added to the cells with the growth medium. After 4 h, fresh medium was added, and cells were allowed to grow for an additional 24 h before using for subsequent analysis.
Real-Time PCR.
Real-time PCR was performed for the target mRNA. GAPDH was used as an internal control. The reaction was assembled per the SuperScript II reverse transcriptase kit (Invitrogen) protocol. A reaction mixture containing 40 ng of cDNA was prepared with PCR buffer (200 mM Tris·HCl, 500 mM KCl), MgCl2, dNTP Mix, forward and reverse primer, Taq DNA polymerase and SyberGreen Mix (Applied Biosystems). The reaction was carried out for 40 cycles. Primer sequences were as follows: SUMO2 forward 5′-GGA TGG TTC TGT GGT GCA G-3′ and reverse 5′-CCT CCA TTT CCA ACT GTG C-3′; SUMO3 forward 5′-GCA GAT TCG ATT CCG GTT TG-3′ and reverse 5′-CTG GGC TGG AGT GTC TGT TTC-3′; manganese-superoxide dismutase (Mn-SOD) forward 5′-TGG CCA AGG GAG ATG TGT TAC-3′ and reverse 5′-CAG CAA CTC TCC TTT GGG TTC-3′; catalase forward 5′-ATT GCC GGC CAC CTG AAG GA-3′ and reverse 5′-CTT CAG AAG AGC CTG GAT-3′; GAPDH forward 5′-GCA ACA ATC TCC ACT TTG CCA C-3′ and reverse 5′-AAT GGT GAA GGT CGG TGT GAA C-3′. The data obtained in terms of average CT count were analyzed with ABI Prism 7000 SDS software.
Immunoblotting.
Cells or tissues were washed with PBS and lysed in an appropriate amount of protein lysis buffer, consisting of 1% Triton X-100, 50 mM Tris (pH 7.40), 5 mM EDTA, 150 mM NaCl, and 5% glycerol, containing protease inhibitor cocktail (Roche). Protein lysates were boiled in SDS-PAGE gel loading buffer, subjected to SDS-PAGE, transferred to a nitrocellulose filter, and probed with the specified primary antibody and the appropriate peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology). For detecting endothelial NO synthase (eNOS) dimers, cell lysates were mixed with loading dye without reducing agents and were maintained ice cold throughout the SDS-PAGE. Chemiluminescent signals were developed using Super Signal West Femto substrate (Pierce, Rockford, IL). Blots were imaged with a Gel Doc 2000 Chemi Doc system (Bio-Rad, Hercules, CA). Antibodies used were SUMO2/3 (Enzo and Cell Signaling Technologies; detects both SUMO2 and SUMO3); HA (BioLegend), eNOS, phospho-eNOS (Ser1177), Mn-SOD, and catalase (Cell Signaling Technologies), and GAPDH (Trevigen). Antibodies were validated using cell lysates, and the target protein was either overexpressed or knocked down. The correct protein band was always matched to the predicted size by its alignment to the molecular weight markers.
Whole cell reactive oxygen species assay by spectrophotometry.
HUVECs were plated in a clear-bottom black-walled 96 well plate and infected with adenoviruses encoding HA-SUMO2 or control virus encoding LacZ. For reactive oxygen species (ROS) detection and quantification, cells were incubated with H2- 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) (DCF 10 µM, Molecular Probes-Life Technologies) for 30 min. at 37°C. After incubation, cells were washed with prewarmed medium. DCF fluorescence was measured (excitation-emission 485/535 nm) using a spectrofluorometer (Spectra Max 300, Molecular Devices).
Histological processing and immunohistochemistry.
Tissues were stored in 10% buffered formalin. Paraffin blocks were prepared after routine processing. For immunohistochemical examinations, 5-μm-thick sections were prepared from paraffin blocks. Antigens were retrieved by heating the tissue sections (95°C, 20 min) in citrate buffer (10 mM, pH 6.0). For immunofluorescence, secondary antibodies tagged with fluorescent dye were used. Images were acquired on a confocal microscope (Zeiss 510). To prevent confounding autofluorescence of the elastic lamina (~488 nm), secondary antibodies with green fluorescence were not used. Images were pseudocolored after image acquisition. For better visualization of the endothelial location of the target proteins, the fluorescence signal of the target protein was converted to blue/green and merged with the vWF (von Willebrand factor) signal in red for colocalized antibodies. We used Alexa fluor 568 for visualization of the target protein and Alexa fluor 647 for vWF. Antibodies used were SUMO2/3 (Enzo and Cell Signaling Technologies); HA (BioLegend), 8-hydroxy-2′-deoxyguanosine (8-OHdG; Abnova), and vWF (Abcam).
Histological image quantification.
Images were quantified using Zen Blue software (Zeiss). For endothelium-specific measurements, the endothelial layer was manually traced following the innermost elastic lamina and aided by staining for endothelial-specific vWF. Mean fluorescence intensity was calculated and expressed as fold differences among groups.
Ex vivo adenoviral infections.
Briefly, mice were anesthetized and euthanized by rapid cardiac excision. The descending thoracic aorta was carefully excised and placed in ice-cold Krebs buffer containing (in mM) 118.3 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 KH2PO4, 25 NaHCO3, 1.2 MgSO4, 11 glucose, and 0.0026 CaNa2EDTA. The aorta was cleaned of excess fat and cut transversely into 3- to 4-mm rings. Ad-LacZ or Ad-HA-SUMO2 adenoviral stocks at a final concentration of 2.5 × 107 pfu/mL in endothelial growth medium 2 were added to each aortic ring and incubated at 37°C for 24 h.
Vascular reactivity.
Vascular reactivity was performed as previously reported (22). Aortic rings were suspended between two wire stirrups (150 mm) in a four-chamber myograph system (DMT Instruments) in 6 mL of Krebs-Ringer (95% O2-5% CO2, pH 7.4, 37°C). The mechanical force signal was amplified, digitalized, and recorded (PowerLab 8/30). All concentration-effect curves were performed on arterial rings beginning at their optimum resting tone. Endothelium-dependent and -independent relaxations were determined by generating dose-response curves to acetylcholine (ACh; 10−9–10−5 M) and sodium nitroprusside (SNP; 10−9–10−5 M), respectively, on phenylephrine (PE; 10−6 M)-induced precontracted vessels. Vasorelaxation evoked by ACh and SNP was expressed as percent contraction with reference to PE (10−6 M) and determined by calculating the percentage of inhibition to the preconstricted tension.
NO bioavailability.
NO bioavailability was measured physiologically by determining the increase in contractile response to PE (10−9–10−5 M) in aortic rings in the presence of eNOS inhibitor (l-NAME). NO bioavailability was calculated as percent increase in contractile response to PE (10−9–10−5 M) in the presence of l-NAME.
Statistics.
All data met assumptions of the statistical test, and all statistical analysis was performed using GraphPad Prism 6. Data represent means ± SE of at least three independent assays. Significance of difference between two groups was determined using two-tailed independent-sample Student’s t-test. For more than two groups, one-way ANOVA, and for groups having more than one variable two-way ANOVA, was performed followed by post hoc analysis to determine the level of significance between two groups. The vascular reactivity data were analyzed by nonlinear regression analysis (curve fit). Results were considered significant if P values were ≤0.05.
RESULTS
Upregulation of SUMO2 mediates vascular endothelial dysfunction in hypercholesterolemic mice.
Prior studies investigating the role of SUMOylation in the regulation of vascular function demonstrated that promoting SUMOylation potentiates the deleterious effects of hypercholesterolemia on the vasculature (15). Therefore, we first evaluated the expression of SUMO2/3-conjugated proteins in aortas of LDLr−/− mice on a normal diet (ND) or high-fat diet (HFD). We noted a significant increase (P < 0.01) in the SUMO2/3-conjugated protein level in the aortic endothelium of LDLr−/− mice on HFD compared with ND (Fig. 1, A and B), whereas in the wild-type mice it was negligible (data not shown here).
Fig. 1.

Small ubiquitin-like modifier 2 (SUMO2) upregulation promotes vascular endothelial dysfunction in hypercholesterolemic mice. A: immunofluorescence for SUMO2/3 in aortas of low-density lipoprotein receptor-deficient (LDLr−/−) mice on a normal diet (ND) and high-fat diet (HFD). B: relative quantification of endothelial SUMO2/3 in mouse aortas (ND, n = 5 and HFD, n = 4). C: relative abundance of SUMO2 mRNA in aortas of LDLr−/− mice (n = 3). Endothelium-dependent relaxation of aortas of LDLr−/− mice on ND (D and E, n = 4) and HFD (G and H, n = 5), and endothelium-independent relaxation of aortas of LDLr−/− mice on ND (F, n = 4) and HFD (I, n = 4) transfected with small interfering RNA (siRNA) for SUMO2 or scrambled control (GC control). MFI, mean fluorescence intensity; vWF, von Willebrand factor; L, lumen; ACh, acetylcholine; PE, phenylephrine; SNP, sodium nitroprusside. Values are means ± SE. ***P < 0.001, **P < 0.01, unpaired t-test for B and C. E, F, H, and I: nonlinear regression analysis (curve fit).
LDLr−/− mice have inherent vascular endothelial dysfunction, which is exacerbated by hypercholesterolemia (2, 30). Therefore, we asked whether upregulation of SUMO2 in the vasculature plays a role in endothelial dysfunction of LDLr−/− mice. We knocked down SUMO2 in aortic rings of LDLr−/− mice ex vivo by transfection of siRNA targeting SUMO2 (Fig. 1C) and evaluated endothelium-dependent and -independent vasorelaxation. Knockdown of SUMO2 significantly increased (P < 0.001) endothelium-dependent relaxation of aortic rings from LDLr−/− mice on ND (Fig. 1, D and E) as well as those on HFD (Fig. 1, G and H) without affecting endothelium-independent relaxation (Fig. 1, F and I). Thus, SUMO2 mediates hypercholesterolemia-induced endothelial dysfunction in mice.
Overexpression of endothelial SUMO2 impairs endothelium-dependent vasorelaxation.
To examine the endothelium-specific effects of SUMO2 in vivo, we generated transgenic mice with endothelium-specific overexpression of SUMO2 (e-SUMO2; Fig. 2A). Transgene-positive founders were identified by amplifying the HA-tagged SUMO2 transgene from genomic DNA (Fig. 2B). In e-SUMO2 mice, we visualized SUMO2ylated proteins in aortic endothelium by immunofluorescence using a HA antibody and noted that the expression of HA-SUMO2 was specific to the endothelial layer (Fig. 2C). We also measured mRNA levels of SUMO2 and SUMO3 in aortas of e-SUMO2 mice and noted a significant increase (P < 0.001) in the level of total SUMO2 mRNA without any change in the level of SUMO3 expression (Fig. 2D). Next, we asked whether this increase in endothelial SUMO2 increases the pool of SUMO2/3-conjugated proteins. We performed immunofluorescence for SUMO2/3 in aortic sections from wild-type and e-SUMO2 transgenic mice and noted a significant increase (P < 0.05) in endothelial SUMO2/3-conjugated proteins in e-SUMO2 mice compared with wild-type controls (Fig. 2, E and F). Collectively, these findings show that SUMO2 overexpression in the endothelium boosts SUMO2ylation.
Fig. 2.

Overexpression of small ubiquitin-like modifier 2 (SUMO2) impairs endothelium-dependent relaxation. A: schematic of transgene showing the PCR primer location for identifying founders. B: PCR genotyping of founders. C: representative immunofluorescence showing expression of HA-SUMO2 in aortic endothelium of e-SUMO2 mice. D: real-time PCR showing total mRNA of SUMO2 and SUMO3 in wild-type and e-SUMO2 mouse aortas (n = 3). E: representative immunofluorescence showing SUMO2/3-conjugated proteins. F: quantification of SUMO2/3 immunofluorescence in aortic endothelium of wild-type and e-SUMO2 mice (n = 5). G–I: endothelium-dependent relaxation (wild-type, n = 7; e-SUMO2, n = 8; G), endothelium-independent relaxation (wild-type, n = 8; e-SUMO2, n = 8; H), and nitric oxide (NO) bioavailability (wild-type n = 8; e-SUMO2 n = 8; I) of aortas of e-SUMO2 mice from founder 1. J–L: endothelium-dependent relaxation (wild-type, n = 12; e-SUMO2, n = 9; J), endothelium-independent relaxation (wild-type, n = 12; e-SUMO2, n = 10; K), and NO bioavailability (wild-type, n = 12; e-SUMO2, n = 10; L) of aortas of e-SUMO2 mice from founder 2. M: immunoblot showing expression of HA-SUMO2 in aortic rings. N: endothelium-dependent relaxation (Ad-LacZ, n = 4; Ad-HA-SUMO2, n = 5). O: endothelium-independent relaxation (Ad-LacZ, n = 5; Ad-HA-SUMO2, n = 6) of wild-type aortas infected with Ad-LacZ or Ad-HA-SUMO2. L, lumen; MFI, mean fluorescence intensity; T, transgenic; MW, DNA ladder; MT, pure transgene equivalent to 5 copies of transgene; WT, wild type; NTC, no template control; vWF, von Willebrand factor; IB, immunoblot; ACh, acetylcholine; PE, phenylephrine; SNP, sodium nitroprusside. Values are means ± SE. *P < 0.05, ***P < 0.001. Two-way ANOVA for D, Student’s t-test for F, and nonlinear regression analysis (curve fit) for G–L, N, and O.
Vascular reactivity studies in aortic rings from one line of e-SUMO2 mice (founder 1 lineage) showed significant impairment (P < 0.001) of endothelium-dependent relaxation (Fig. 2G). However, endothelium-independent relaxation and NO bioavailability were not affected (Fig. 2, H and I). We also examined vascular endothelial function in progeny from another line of e-SUMO2 mice (founder 2 lineage). We noted an exaggeration of endothelial dysfunction in these mice along with impaired endothelium-independent relaxation and reduced NO bioavailability (Fig. 2, J–L). Thus, we show that overexpression of SUMO2 in endothelium impairs endothelium-dependent vasorelaxation.
To complement these studies, we assessed the effect of SUMO2 overexpression ex vivo on vascular reactivity. We generated an adenovirus expressing a fusion protein of HA and SUMO2 (Ad-HA-SUMO2; Fig. 2M). Aortic rings of wild-type mice were infected ex vivo with either Ad-HA-SUMO2 or Ad-LacZ (control virus), and vascular reactivity was evaluated. Aortic rings expressing SUMO2 had impaired endothelium-dependent relaxation (Fig. 2N); however, endothelium-independent relaxation did not change (Fig. 2O). Collectively, these data show that SUMO2 overexpression in vivo or ex vivo impairs endothelium-dependent relaxation of blood vessels.
Downregulation of basal SUMO2 impairs endothelium-dependent vasorelaxation.
Given the indispensable role of SUMO2 in embryonic development (38), we asked whether endogenous basal SUMO2 expression has any physiological role in regulating vascular function. Treatment of healthy mouse aortic rings with SUMO2 siRNA ex vivo impaired endothelium-dependent relaxation without affecting endothelium-independent relaxation (Fig. 3, A and B). These data reinforce a role for basal SUMO2 in maintenance of physiological vascular endothelial function.
Fig. 3.
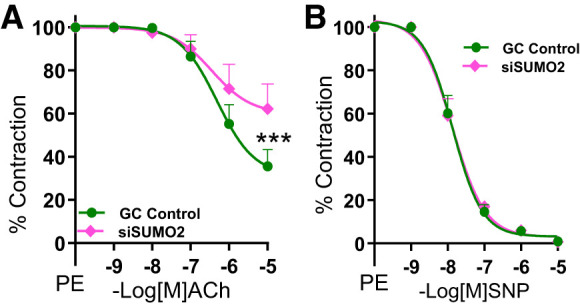
Basal small ubiquitin-like modifier 2 (SUMO2) is important for physiological endothelium-dependent relaxation. A and B: endothelium-dependent (GC, n = 7; siSUMO2, n = 7; A) and endothelium-independent (GC, n = 7; siSUMO2, n = 7; B) relaxation of aortic rings of wild-type mice transfected with siRNA of SUMO2 or scrambled control (GC control). Nonlinear regression analysis (curve fit). si, small interfering; ACh, acetylcholine; PE, phenylephrine; SNP, sodium nitroprusside. Values are means ± SE. ***P < 0.001.
SUMO2 overexpression increases endothelial ROS and uncouples eNOS.
Sumoylation increases ROS production (6, 23), and oxidative stress impairs vascular endothelial function and promotes atherosclerosis (12, 18, 37). Therefore, we asked whether SUMO2 increases cellular ROS. HUVECs were infected with adenoviruses expressing SUMO2 (Ad-HA-SUMO2) or the control virus (Ad-LacZ). HUVECs expressing SUMO2 showed a significant increase (P < 0.001) in ROS compared with cells infected with Ad-LacZ (Fig. 4, A and B). This increase in ROS was likely not due to a decrease in cellular antioxidant systems, since catalase and Mn-SOD were not downregulated with SUMO2 overexpression (Fig. 4, C–F). We also measured the oxidative stress in aortic rings of wild-type and e-SUMO2 mice by immunostaining for 8-OHdG, a marker of oxidative modification of DNA. We noted a significant increase (P < 0.001) in 8-OHdG in the endothelial layer of e-SUMO2 mouse aortas compared with wild-type mice (Fig. 4, G and H). In aortic lysates, Mn-SOD protein expression was not different between the groups, although there was a trend toward upregulation of catalase (P < 0.05; Fig. 4, I and J).
Fig. 4.

SUMO2 increases oxidative stress and uncouples endothelial NO synthase (eNOS). A: reactive oxygen species (ROS) measured as dichlorofluorescein fluorescence in human umbilical vein endothelial cells (HUVECs) infected with Ad-LacZ (n = 15) or Ad-HA-SUMO2 (n = 15). B: immunoblot (IB) of SUMO2 in HUVECs infected with Ad-LacZ or Ad-HA-SUMO2. MRNA level of Mn-superoxide dismutase (Mn-SOD; Ad-LacZ n = 3, Ad-SUMO2 n = 3) (C), and catalase (Ad-LacZ, n = 3 and Ad-HA-SUMO2, n = 3) (D) in HUVECs infected with Ad-HA-SUMO2 or Ad-Lac Z. E: immunoblot of Mn-SOD and catalase in HUVECs infected with Ad-HA-SUMO2 (n = 4) or Ad-Lac Z (n = 4) and their densitometric quantification (F). G: representative immunofluorescence image of 8-hydroxydeoxyguanosine (8-OHdG) in aortas of wild-type and e-SUMO2 mice and the quantification of endothelial 8-OHdG (n = 6) (H). I: immunoblot of Mn-SOD and catalase in aortas of wild-type (n = 3) and e-SUMO2 (n = 3) mice and their relative densitometric quantification (J). K: immunoblot of total, monomeric, dimeric, and phospho-S1177 eNOS in HUVECs infected with Ad-HA-SUMO2 (n = 4) or Ad-Lac Z (n = 4) and the relative quantification of monomeric and dimeric eNOS (L). MFI, mean fluorescence intensity; vWF, von Willebrand factor. Values are means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001. Student’s t-test for A, C, D, and H; two-way ANOVA for F, J, and L.
We next determined whether SUMO2 regulates eNOS. Overexpression of SUMO2 in HUVECs increased the expression of eNOS as well as the Ser1177 phosphorylation of eNOS (Fig. 4K). This was in contrast to the noted decrease in the NO bioavailability in e-SUMO2 mice (founder 2 lineage). Because eNOS can produce superoxide or NO depending on whether it is uncoupled or coupled, we asked how SUMO2 affects both forms of eNOS. SUMO2 overexpression increased the monomeric (uncoupled) form of eNOS (P < 0.01) Fig. 4L) but did not change the dimer of eNOS.
Taken together, these data show that overexpression of SUMO2 promotes oxidative stress in endothelial cells in vitro and vascular endothelium in vivo without reducing endogenous antioxidant defenses. It also suggests that uncoupling of eNOS may be one mechanism by which hyper-SUMO2ylation induces endothelial oxidative stress.
DISCUSSION
In this work, we identify a hitherto unappreciated role of SUMO2 in the regulation of vascular endothelial function. Using complementary techniques, we show that upregulation of SUMO2 impairs endothelium-dependent vasorelaxation and increases oxidative stress, while basal SUMO2 expression is essential for normal physiological endothelium-dependent relaxation. It is noteworthy that the absence of both, SUMO2 and hyper-SUMOylation (with knockout of de-SUMOylating enzymes), is embryonically lethal (17, 38). Thus, the essential role of SUMO2 in normal endothelial physiology reported herein corroborates the vital role of SUMO2 in embryonic development.
Previous studies investigating the cardiovascular effects of SUMO2 have noted a pathological function of the protein. In failing human hearts, there is an increase of SUMO2/3-conjugated proteins, and cardiac-specific transgenic expression of SUMO2 induces cardiomyopathy leading to increased mortality (20). Moreover, pathological oxidative stimuli such as hydrogen peroxide and advanced glycation end products promote SUMOylation (41). Furthermore, SUMOylation is increased in disease states such as diabetes and disturbed vascular flow (15). Our finding that upregulation of SUMO2 is responsible for vascular endothelial dysfunction in LDLr−/− mice indicates that hypercholesterolemia is another disease state in which SUMO2/3 plays a pathological role. In addition, the finding that SUMOylation in the vasculature is upregulated in hypercholesterolemic mice indicates that the process of SUMOylation and de-SUMOylation in the vasculature is highly dynamic. It would be interesting to determine whether normalization of lipid levels in hypercholesterolemic mice also reduces vascular SUMO2ylation.
Interestingly, one of the transgenic e-SUMO2 mice (founder 2 lineage) had impaired relaxation of aortic rings in response to both endothelium-dependent (ACh) and independent (SNP) agonists, but another (founder 1 lineage) did not show any impairment with the endothelium-independent agonist. In addition, impaired endothelium-dependent vasorelaxation of the aortas of founder 1 lineage mice was not associated with a reduction in the bioavailable NO. It is possible that the difference in vascular phenotype between these two lines of transgenic mice is due to varying levels of transgene expression.
Impairment of endothelium-independent vasorelaxation noted in the founder 2 lineage suggests that endothelial SUMO2 overexpression also affects the function of vascular smooth muscle cells, possibly via an indirect mechanism. Since SUMO2 increases ROS production, it is possible that SUMO2-induced ROS impairs vascular smooth muscle function, as previously reported (8, 34). It is important to note that there was a trend for catalase upregulation with SUMO2 overexpression. Catalase, although being an endogenous antioxidant, also impairs endothelium-dependent vasorelaxation, by reducing the level of hydrogen peroxide, an endothelium-derived relaxing factor (25, 28). Therefore, the increase in catalase by SUMO2 may be compensatory or contributes to the vascular phenotype of e-SUMO2 transgenic mice.
SUMO2 overexpression stimulated eNOS expression and phosphorylation at Ser1177 (S1177; Fig. 4K). In addition to producing NO, eNOS when uncoupled can produce superoxide (42). Phosphorylation of eNOS at Ser1177 is essential for both superoxide and NO production, depending on the state of eNOS coupling (7). SUMO2-induced upregulation of monomeric (uncoupled) eNOS but not dimeric (coupled) eNOS suggests that hyper-SUMO2ylation promotes uncoupling of eNOS. Superoxide produced by uncoupled eNOS quenches the NO to generate peroxynitrite (3) and decreases bioavailable NO. This may explain the reduction in bioavailable NO observed in the aortas of one line of e-SUMO2 mice. Although we cannot provide proof, the magnitude of transgene expression in the two founder lines may also determine the degree of eNOS uncoupling, perhaps explaining normal bioavailable NO in the first line.
Increased vascular oxidative stress and reduced NO bioavailability promote age-related endothelial dysfunction in the peripheral and cerebral vasculature (35). This dysfunction is further exacerbated by the absence of NF-E2-related factor 2 (Nrf2), a transcriptional factor that regulates oxidative homeostasis (11). The endothelial dysfunction can promote age-related diseases such as vascular cognitive impairment, Alzheimer’s disease, heart diseases, and sarcopenia (1, 9, 10). SUMOylation regulates age-associated changes in cellular processes such as autophagy, redox homeostasis, and growth factor signaling (29). Furthermore, SUMOylation and the SUMO-modifying enzymes (Ubc9 and SENP1) are altered with aging (27). It would thus be interesting to examine the role of hyper-SUMO2ylation in changes in vascular function of organs impacted by aging (14, 21, 26).
In conclusion, we show that hyper-SUMO2ylation plays an important part in vascular endothelial dysfunction associated with hypercholesterolemia, while at the same time, physiological expression of SUMO2 is essential for vascular homeostasis.
LIMITATIONS OF THE STUDY
This study does not delineate the mechanism(s) responsible for hyper-SUMO2ylation-induced changes in vascular function. Because SUMO2 overexpression did not reduce the expression of antioxidant enzymes (Mn-SOD and catalase), it is unlikely that the effect of hyper-SUMO2ylation on endothelial oxidative stress is secondary to changes in antioxidant defenses. Given the diversity of SUMOylated proteins, it is likely that multiple parallel mechanisms are responsible for the effects of SUMO2 on endothelial ROS. In addition, this study does not define the mechanism responsible for SUMO2-induced eNOS uncoupling, a topic that deserves further study.
GRANTS
K. Irani was supported by Veterans Affairs Merit Award I01 BX002940 and a University of Iowa Endowed Professorship in Cardiovascular Medicine. The e-SUMO2 mice were generated with a microfinance grant to S. Kumar from the Pappajohn Biomedical Institute.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.I. and S.K. conceived and designed research; Y.-R.K., J.S.J., Q.L., R.R.G., A.V., J.L., M.K., and S.K. performed experiments; Y.-R.K. and S.K. analyzed data; S.K. prepared figures; S.K. drafted manuscript; S.K. edited and revised manuscript; Y.-R.K., J.S.J., Q.L., R.R.G., A.V., J.L., M.K., K.I., and S.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Sanjana Dayal for the Mn-SOD and catalase antibodies.
REFERENCES
- 1.Balasubramanian P, Hall D, Subramanian M. Sympathetic nervous system as a target for aging and obesity-related cardiovascular diseases. Geroscience 41: 13–24, 2019. doi: 10.1007/s11357-018-0048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bar A, Targosz-Korecka M, Suraj J, Proniewski B, Jasztal A, Marczyk B, Sternak M, Przybyło M, Kurpińska A, Walczak M, Kostogrys RB, Szymonski M, Chlopicki S. Degradation of glycocalyx and multiple manifestations of endothelial dysfunction coincide in the early phase of endothelial dysfunction before atherosclerotic plaque development in apolipoprotein E/low-density lipoprotein receptor-deficient mice. J Am Heart Assoc 8: e011171, 2019. doi: 10.1161/JAHA.118.011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol Cell Physiol 271: C1424–C1437, 1996. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 4.Bernt A, Rangrez AY, Eden M, Jungmann A, Katz S, Rohr C, Müller OJ, Katus HA, Sossalla ST, Williams T, Ritter O, Frank D, Frey N. Sumoylation-independent activation of Calcineurin-NFAT-signaling via SUMO2 mediates cardiomyocyte hypertrophy. Sci Rep 6: 35758, 2016. doi: 10.1038/srep35758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boese AC, Kim SC, Yin KJ, Lee JP, Hamblin MH. Sex differences in vascular physiology and pathophysiology: estrogen and androgen signaling in health and disease. Am J Physiol Heart Circ Physiol 313: H524–H545, 2017. doi: 10.1152/ajpheart.00217.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- 7.Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem 283: 27038–27047, 2008. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi C, Sellak H, Brown FM, Lincoln TM. cGMP-dependent protein kinase and the regulation of vascular smooth muscle cell gene expression: possible involvement of Elk-1 sumoylation. Am J Physiol Heart Circ Physiol 299: H1660–H1670, 2010. doi: 10.1152/ajpheart.00677.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csipo T, Lipecz A, Fulop GA, Hand RA, Ngo BN, Dzialendzik M, Tarantini S, Balasubramanian P, Kiss T, Yabluchanska V, Silva-Palacios F, Courtney DL, Dasari TW, Sorond F, Sonntag WE, Csiszar A, Ungvari Z, Yabluchanskiy A. Age-related decline in peripheral vascular health predicts cognitive impairment. Geroscience 41: 125–136, 2019. doi: 10.1007/s11357-019-00063-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Csiszar A, Tarantini S, Fülöp GA, Kiss T, Valcarcel-Ares MN, Galvan V, Ungvari Z, Yabluchanskiy A. Hypertension impairs neurovascular coupling and promotes microvascular injury: role in exacerbation of Alzheimer’s disease. Geroscience 39: 359–372, 2017. doi: 10.1007/s11357-017-9991-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fulop GA, Kiss T, Tarantini S, Balasubramanian P, Yabluchanskiy A, Farkas E, Bari F, Ungvari Z, Csiszar A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience 40: 513–521, 2018. doi: 10.1007/s11357-018-0047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 118: 620–636, 2016. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong L, Ji WK, Hu XH, Hu WF, Tang XC, Huang ZX, Li L, Liu M, Xiang SH, Wu E, Woodward Z, Liu YZ, Nguyen QD, Li DW. Sumoylation differentially regulates Sp1 to control cell differentiation. Proc Natl Acad Sci USA 111: 5574–5579, 2014. doi: 10.1073/pnas.1315034111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460: 392–395, 2009. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heo KS, Chang E, Le NT, Cushman H, Yeh ET, Fujiwara K, Abe J. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res 112: 911–923, 2013. doi: 10.1161/CIRCRESAHA.111.300179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heo KS, Le NT, Cushman HJ, Giancursio CJ, Chang E, Woo CH, Sullivan MA, Taunton J, Yeh ET, Fujiwara K, Abe J. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest 125: 1299–1310, 2015. doi: 10.1172/JCI76453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang X, Qi Y, Zuo Y, Wang Q, Zou Y, Schwartz RJ, Cheng J, Yeh ET. SUMO-specific protease 2 is essential for suppression of polycomb group protein-mediated gene silencing during embryonic development. Mol Cell 38: 191–201, 2010. doi: 10.1016/j.molcel.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol 24: 998–1005, 2004. doi: 10.1161/01.ATV.0000125114.88079.96. [DOI] [PubMed] [Google Scholar]
- 19.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 477: 601–605, 2011. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim EY, Zhang Y, Ye B, Segura AM, Beketaev I, Xi Y, Yu W, Chang J, Li F, Wang J. Involvement of activated SUMO-2 conjugation in cardiomyopathy. Biochim Biophys Acta 1852: 1388–1399, 2015. doi: 10.1016/j.bbadis.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Kiss T, Balasubramanian P, Valcarcel-Ares MN, Tarantini S, Yabluchanskiy A, Csipo T, Lipecz A, Reglodi D, Zhang XA, Bari F, Farkas E, Csiszar A, Ungvari Z. Nicotinamide mononucleotide (NMN) treatment attenuates oxidative stress and rescues angiogenic capacity in aged cerebromicrovascular endothelial cells: a potential mechanism for the prevention of vascular cognitive impairment. Geroscience, 2019. doi: 10.1007/s11357-019-00074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S, Kim YR, Vikram A, Naqvi A, Li Q, Kassan M, Kumar V, Bachschmid MM, Jacobs JS, Kumar A, Irani K. Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proc Natl Acad Sci USA 114: 1714–1719, 2017. doi: 10.1073/pnas.1614112114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le NT, Corsetti JP, Dehoff-Sparks JL, Sparks CE, Fujiwara K, Abe J. Reactive oxygen species, SUMOylation, and endothelial inflammation. Int J Inflamm 2012: 1, 2012. doi: 10.1155/2012/678190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lusis AJ. Atherosclerosis. Nature 407: 233–241, 2000. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest 106: 1521–1530, 2000. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, Roth GS, Ingram DK, Weindruch R, de Cabo R, Anderson RM. Caloric restriction improves health and survival of rhesus monkeys. Nat Commun 8: 14063, 2017. doi: 10.1038/ncomms14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nisticò R, Ferraina C, Marconi V, Blandini F, Negri L, Egebjerg J, Feligioni M. Age-related changes of protein SUMOylation balance in the AβPP Tg2576 mouse model of Alzheimer’s disease. Front Pharmacol 5: 63, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol 292: H93–H100, 2007. doi: 10.1152/ajpheart.00819.2006. [DOI] [PubMed] [Google Scholar]
- 29.Princz A, Tavernarakis N. The role of SUMOylation in ageing and senescent decline. Mech Ageing Dev 162: 85–90, 2017. doi: 10.1016/j.mad.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Rabelo LA, Cortes SF, Alvarez-Leite JI, Lemos VS. Endothelium dysfunction in LDL receptor knockout mice: a role for H2O2. Br J Pharmacol 138: 1215–1220, 2003. doi: 10.1038/sj.bjp.0705164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riedel K, Deussen AJ, Tolkmitt J, Weber S, Schlinkert P, Zatschler B, Friebel C, Müller B, El-Armouche A, Morawietz H, Matschke K, Kopaliani I. Estrogen determines sex differences in adrenergic vessel tone by regulation of endothelial β-adrenoceptor expression. Am J Physiol Heart Circ Physiol 317: H243–H254, 2019. doi: 10.1152/ajpheart.00456.2018. [DOI] [PubMed] [Google Scholar]
- 32.Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem 275: 6252–6258, 2000. doi: 10.1074/jbc.275.9.6252. [DOI] [PubMed] [Google Scholar]
- 33.Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol 209: 13–22, 2015. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ungvari Z, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith K, Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-κB activation in aged rat arteries. Am J Physiol Heart Circ Physiol 293: H37–H47, 2007. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 35.Ungvari Z, Tarantini S, Kiss T, Wren JD, Giles CB, Griffin CT, Murfee WL, Pacher P, Csiszar A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol 15: 555–565, 2018. doi: 10.1038/s41569-018-0030-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vejpongsa P, Yeh ET. Wrestling with heart failure: SUMO-1 to the rescue. Circ Res 114: 1561–1563, 2014. doi: 10.1161/CIRCRESAHA.114.304125. [DOI] [PubMed] [Google Scholar]
- 37.Wadley AJ, Veldhuijzen van Zanten JJ, Aldred S. The interactions of oxidative stress and inflammation with vascular dysfunction in ageing: the vascular health triad. Age (Dordr) 35: 705–718, 2013. doi: 10.1007/s11357-012-9402-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Wansleeben C, Zhao S, Miao P, Paschen W, Yang W. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep 15: 878–885, 2014. doi: 10.15252/embr.201438534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Dasso M. SUMOylation and deSUMOylation at a glance. J Cell Sci 122: 4249–4252, 2009. doi: 10.1242/jcs.050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woo CH, Abe J. SUMO--a post-translational modification with therapeutic potential? Curr Opin Pharmacol 10: 146–155, 2010. doi: 10.1016/j.coph.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woo CH, Shishido T, McClain C, Lim JH, Li JD, Yang J, Yan C, Abe J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res 102: 538–545, 2008. doi: 10.1161/CIRCRESAHA.107.156877. [DOI] [PubMed] [Google Scholar]
- 42.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 43.Zhang FP, Mikkonen L, Toppari J, Palvimo JJ, Thesleff I, Jänne OA. Sumo-1 function is dispensable in normal mouse development. Mol Cell Biol 28: 5381–5390, 2008. doi: 10.1128/MCB.00651-08. [DOI] [PMC free article] [PubMed] [Google Scholar]