

Abstract
Developing an efficient, concise synthesis of the fungal natural product illudalic acid has been a long-standing challenge, made more pressing by the recent discovery that illudalic acid and analogs are selective phosphatase inhibitors. Syntheses of illudalic acid have become progressively more efficient over the decades yet remain strategically grounded in a 17-step synthesis reported in 1977. Here we validate a two-step process — convergent [4+2] benzannulation and one-pot coordinated functional group manipulations — for preparing the key trifunctional pharmacophore of illudalic acid. The modular building blocks are readily available in 2–3 steps, for a longest linear sequence (LLS) of 5 steps to illudalic acid from 3,3-dimethylcyclopentanone. A small collection of analogous indanes and tetralins featuring the same pharmacophore were prepared by a similar route. These compounds potently and selectively inhibit the human leukocyte common antigen-related (LAR) subfamily of protein tyrosine phosphatases (PTPs). Evidence supporting a postulated covalent ligation mechanism is provided herein.
Graphical Abstract
A convergent 5-step synthesis (LLS) of illudalic acid allows for concise preparation of analogues for pharmacological evaluation.
Introduction
Originally identified as an “inactive acidic compound” from the toxic eastern jack o’lantern mushroom (Omphalotus illudens, formerly Clitocybe illudens) in 19521 and characterized in 1969,2 iludalic acid (Fig. 1A) emerged from high-throughput screening in 20053 as the “first potent, selective small molecule inhibitor”4 of the leukocyte common antigen-related (LAR)5 protein tyrosine phosphatase (PTP).
Fig. 1.
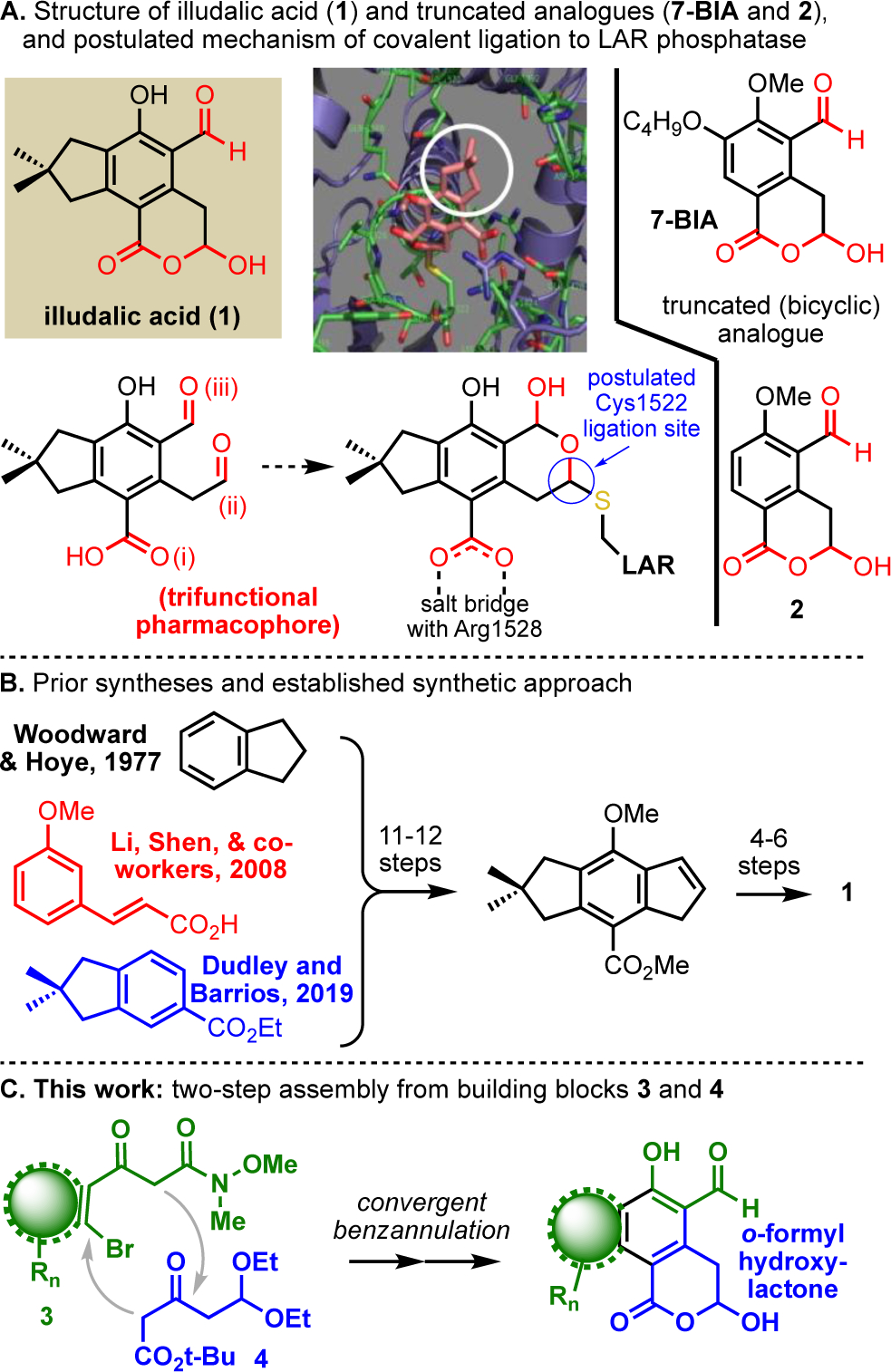
(A) Structure and postulated function of illudalic acid (1) and prior analogues, with the neopentylene ring-fusion of 1 circled in the molecular docking simulation reproduced from ref 4. (B) Prior syntheses. (C) This work.
Reversible protein tyrosine phosphorylation, mediated cooperatively by kinases (PTKs)6 and phosphatases (PTPs),7 plays a critical role in cellular signalling. PTPs were long overlooked as loosely regulated (perhaps “undruggable”8) signal suppressors, but they are now recognized as important targets for drug discovery.9,10 Dysregulation of the 38 classical, cysteine-dependent, tyrosine-specific PTPs is broadly linked to various cancers and metabolic and nervous system disorders.11 The LAR subfamily12 of receptor-type transmembrane PTPs comprises LAR, PTPRσ, and PTPRδ, each of which is an important chemotherapeutic target. LAR overexpression causes insulin resistance;13 PTPRσ suppression improves cardiac reinnervation after a heart attack;14 and decreasing PTPRδ activity correlates with lower propensity for substance abuse and addiction.8,15 LAR-PTP inhibitors are of interest because they would help elucidate the roles of these enzymes in disease and provide therapeutic leads. However, the highly conserved, positively charged PTP active site has been difficult to target selectively with cell-permeable small molecules.16 Given its potency and selectivity, the illudalic acid scaffold offers important insights into LAR PTP inhibition.
Synthesis of illudalic acid (1) has been a challenge and a barrier to pharmacological development. The first synthesis was reported by Woodward and Hoye in 1.1% overall yield over 17 steps (Fig. 1B).17 Modified versions of this same approach were reported by Shen in 20084 (16 steps. 2.4% overall) and us in 201918 (20 steps, 7.7% overall) to support some preliminary molecular pharmacology. Truncated analogues of illudalic acid have been identified that were moderately more accessible (by the prior synthetic approach) but are less potent. For example, analog 2 was largely inactive in our assays.18 Shen reported that some efficacy can be restored by incorporating acyclic substituents in place of the neopentylene ring fusion.19 In 2018, Uhl and coworkers reported that one of these truncated analogues, 7-BIA, is active in in vivo mouse models of cocaine abuse, and is a lead compound for PTPRδ inhibition.15 7-BIA is prepared in 13 steps; the first step introduces the 7-butoxy substituent, and the other 12 steps are dedicated to crafting the key pharmacophore. Innovation in the synthesis of illudalic acid is needed to realize its chemotherapeutic potential for LAR-PTP inhibition.
Here we report a two-step annulation of readily available building blocks to produce the natural product and analogues (Fig. 1C). The annulation was inspired by a 2010 naphthol synthesis reported by Fu, Jiang, and coworkers,20 and the building blocks are crafted to deliver the target structures with minimal additional processing. We have made other illudalane sesquiterpenes,21 driven in part by an interest in the challenges and opportunities of illudalic acid. Illudalic acid and analogues are now available in as few as 5 linear steps from simple cycloalkanones.
Results and Discussion
Synthesis of illudalic acid.
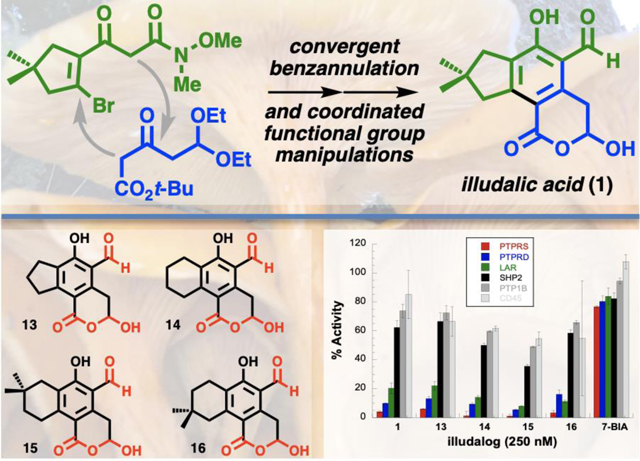
The synthesis of our two key building blocks is outlined in Fig. 2. Pinnick oxidation of known bromo enal 522 (prepared here by Vilsmeier–Haack-type formylation23 of 3,3-dimethylcyclopentanone) set up CDI-mediated Claisen-type condensation to provide β-keto amide 3a. A similar CDI-mediated decarboxylative coupling24 of acids 7 and 8 produced β-keto ester 4 in 72% yield.
Fig. 2.
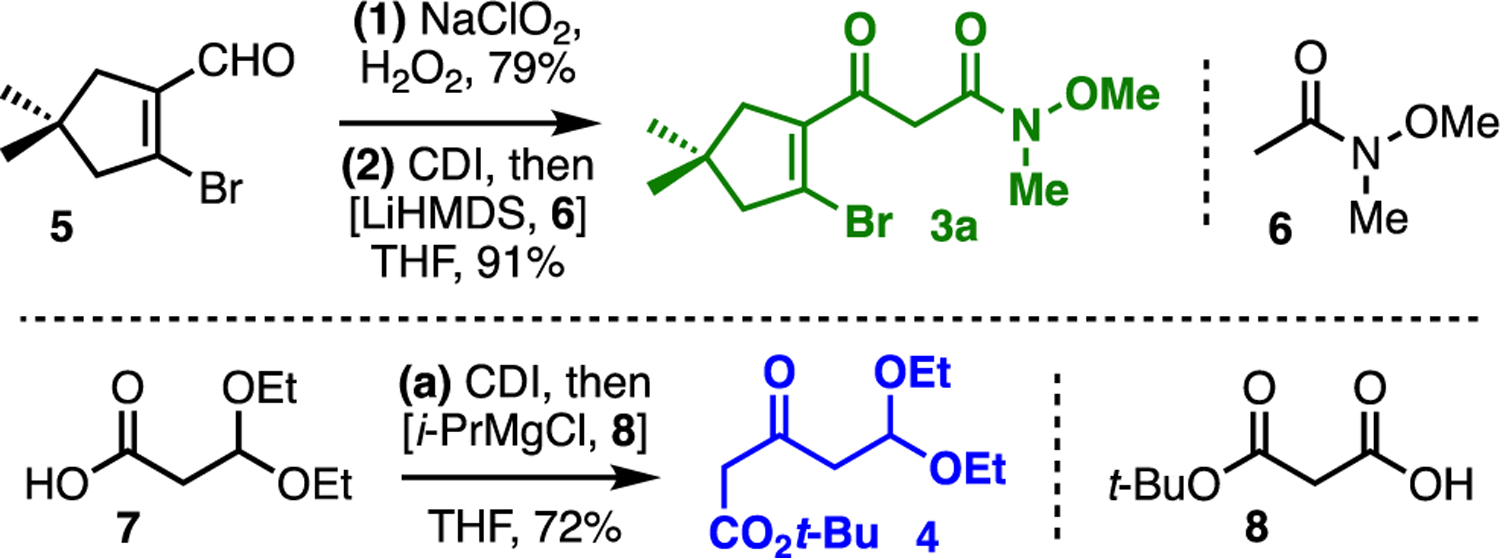
Synthesis of building blocks 3 and 4; see ESI for details.
The Cu-catalyzed, base-mediated [4 + 2] benzannulation of β-keto amide 3a with β-keto ester 4 provided indane 9a in 79% yield (Fig. 3). Excess β-keto ester 4 is recoverable by chromatography and can be recycled. The postulated mechanistic pathway outlined in Fig. 3 is consistent with the Fu/Jiang methodology20 and our observations: Cu-catalyzed coupling of enolate Cs•4 with vinyl bromide 3a triggers aldol cyclization to produce cyclohexenol intermediate 10, which persists in solution. Dehydration of 10 can produce either of two isomeric cyclohexadienones (not shown) but is slow, presumably because A-strain disfavors formation of enol tautomers of 10. Excess Cs2CO3 is advantageous for promoting dehydration, whereas less Cs2CO3 and/or other bases were less effective. Homocoupling of β-keto amide 3a occurs in the absence of β-keto ester 4 but is effectively suppressed by the desired benzannulation. Deprotonation of ester 4 is presumably more facile under the mildly basic conditions compared to amide 3a, favoring the desired coupling. Replacing the Weinreb amide of 3a with ester or nitrile functionality here was deleterious to the reaction outcome, although we have not thoroughly explored these alternatives.
Fig. 3.

[4 + 2] Indane benzannulation and postulated mechanistic pathway; see ESI for details.
Selective reduction of the Weinreb amide can be accomplished with LiAlH4 in THF (Fig. 4). Chemoselectivity for reduction of the amide over the ester was our largest concern at the outset, but in practice the greater difficulty is in stopping reduction at the aldehyde stage. The phenol of 9a is positioned to direct hydride reduction of the amide, but phenoxide intermediate 11 is prone to decomposition in situ, which leads to overreduced byproducts. Dropwise addition of LiAlH4 to a solution of 9a in THF at 0 °C immediately produces bubbles, presumably of hydrogen gas, which subside before a full equivalent of LiAlH4 is added. Reduction of the amide is complete within 1 h, producing aldehyde 12 as the major product (46%) along with alcohol and amine by-products from overreduction of the amide. Reduction of the t-butyl ester was not observed. Hydrolysis of the diethyl acetal and t-butyl ester can be accomplished simultaneously with 6M aqueous HCl in acetone, producing illudalic acid (1) in 81% yield. Alternatively, acidic hydrolysis can be incorporated into the workup of the Weinreb amide reduction step. Thus, reduction of indane 9a with LiAlH4 followed by workup with strong acid produces illudalic acid in 47% yield from 9a, or 27% over four steps from enal 5.
Fig. 4.
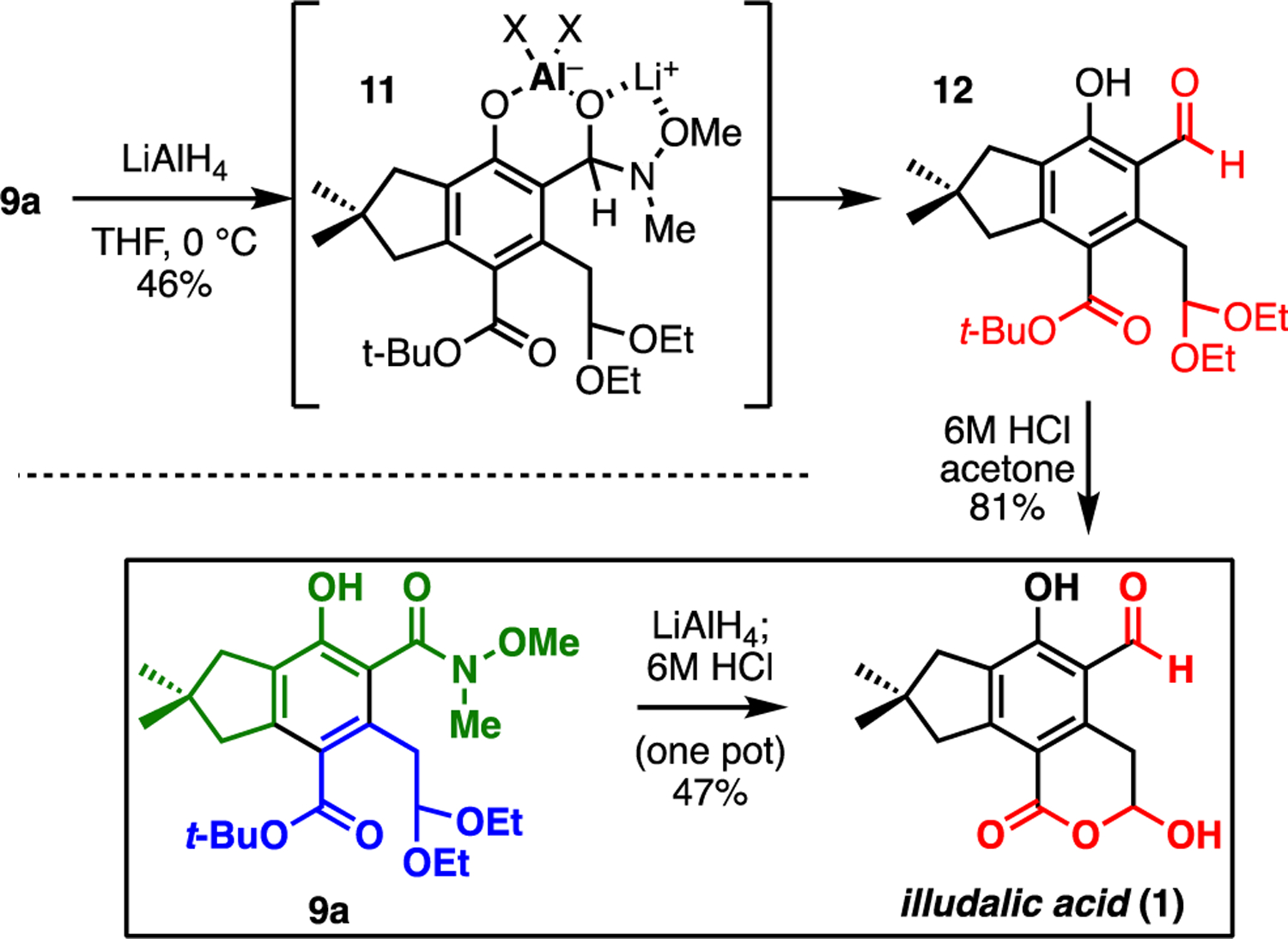
Reduction and hydrolysis to illudalic acid; see ESI for details.
Molecular pharmacology.
Improved access to illudalic acid facilitates further exploration of its biological activity as a covalent inhibitor of LAR phosphatase. We find that illudalic acid is a pH-dependent and time-dependent LAR inhibitor. The IC50 value for illudalic acid-mediated inhibition of LAR at pH 6.5 is 2.1 ± 0.2 μM as compared to 52 ± 10 nM at pH 7.5. However, IC50 values are not the best measure of potency for a time-dependent, covalent inhibitor, so we determined KI and kinact values as described before.18,25 We measured the illudalic acid time-dependent inhibition of LAR at pH 7.5 and 37 °C (Fig. 5), and thereby determined that KI = 8 ± 3 μM for initial binding, and once bound, kinact = 2.3 ± 0.4 min−1 for covalent ligation. The kinact/KI = 3×105 corresponds to a protein deactivation half-life of t∞1/2 = 0.2 min under pseudo-first order conditions at 37 °C and pH 7.5. As compared to the values we originally obtained at 22 °C and pH 6.5 (KI = 130 ± 50 μM, kinact = 1.3 ± 0.4 min−1, and t∞1/2 = 0.5 min), the revised values are consistent with more potent inhibition at the higher pH (and temperature). We measured pKa with this pH-dependence in mind; illudalic acid is a weak diprotic acid with pKa values of 5.42 and 6.60, respectively (see ESI).
Fig. 5.

Illudalic acid (IA1) is a time-dependent inhibitor of LAR activity (inset). The kobs values were obtained and used to determine KI and kinact values (8 ± 3 μM and 2.3 ± 0.4 min−1, respectively) as described in refs 18 and 25.
Synthesis of tricyclic analogues.
Alternative β-keto amides 3 provide access to novel tricyclic analogues of illudalic acid (Fig. 6), with β-keto ester 4 serving as a common building block.26 Bromo enals 5 are available by Vilsmeier–Haack-type formylation of cyclic ketones.23 Oxidation and coupling with Weinreb acetamide (6, cf. Fig. 2) provides β-keto amides 3. Once amides 3 are prepared separately, parallel benzannulations with β-keto ester 4 furnish indanes and tetralins 9. Reduction and acidic hydrolysis complete the assembly of analogues 13-16. A broader range of β-keto amides 3 are envisioned for future work aimed at probing structure-activity relationships for selective phosphatase inhibition.
Fig. 6.
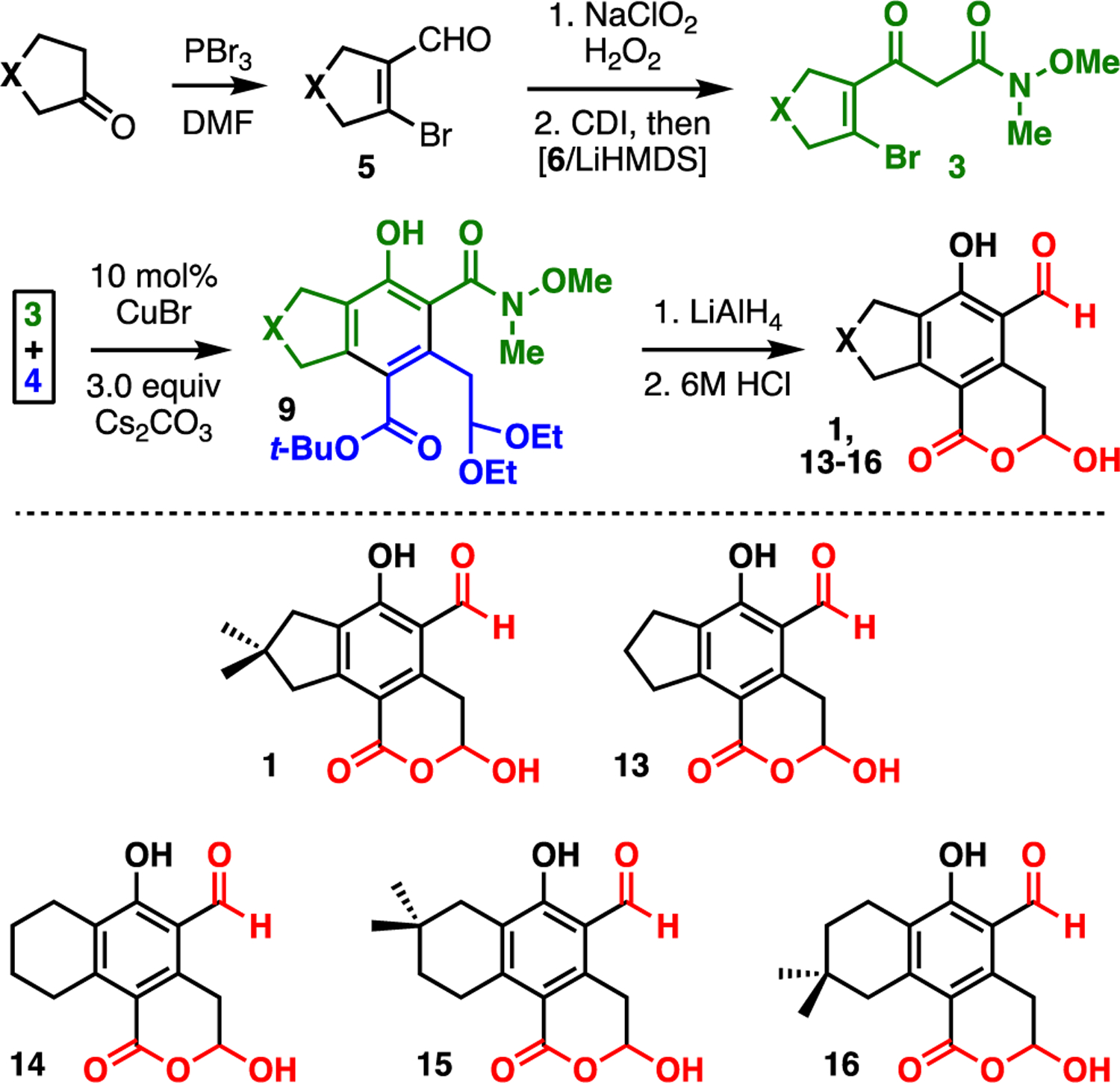
Synthesis of illudalic acid and analogues; see ESI for details.
Like illudalic acid (1) itself, these novel tricyclic analogues are potent and selective for the LAR-PTP subfamily. Results from preliminary inhibitory assays of 1, 13-16, and 7-BIA15,19 against the LAR-PTP subfamily (LAR, PTPRσ, and PTPRδ)12 and selected receptor-type and nonreceptor-type PTPs (PTP1B, SHP2, and CD45) are shown in Fig. 7. The natural product and these novel tricyclic analogues appear to offer significant advantages compared to 7-BIA in terms of potency and also now in terms of ease of access through synthesis. Compound 15 appears to be the most potent based on this preliminary screen, so we accumulated ~1 g of 15 by the route outlined in Fig. 6 in anticipation of future work. While 15 is selective for the LAR subfamily, its >50% inhibition of SHP2 at 250 nM puts 15 in league with some of the leading SHP2 inhibitors currently under development,27 which may warrant further investigation. Compound 16 shows a different selectivity profile within the LAR subfamily, which may hint at molecular design approaches for reversing selectivity between LAR and PTPRδ.
Fig. 7.

Screening assays: Compounds 1, 13, 14, 15, 16, and 7-BIA (referenced on the X-axis as “illudalogs”) as inhibitors of the activity of various PTPs at 250 nM.
Conclusions
We report a concise, convergent synthesis of illudalic acid, insights into its molecular pharmacology as a PTP inhibitor, and synthesis of analogous indanes and tetralins bearing the same trifunctional pharmacophore. The synthesis features benzannulation of the fully substituted arene core and phenol-directed hydride reduction, with acidic hydrolysis then revealing the target tricyclic structures. The compounds are potent, selective, and covalent inhibitors of the LAR subfamily of tyrosine phosphatase enzymes, and as such are promising lead compounds for therapeutic development.
Supplementary Material
Acknowledgements
Supported by NIH Grant P20GM103434 to GBD via the West Virginia IDeA Network for Biomedical Research Excellence (INBRE) and by NIH Grant R01GM135295 to AMB. Additional support from the West Virginia University and a generous contribution to the University of Utah from the ALSAM foundation is gratefully acknowledged.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: Experimental procedures, compound characterization data, copies of 1H and 13C NMR spectra, pKa and IC50 data (PDF).
Notes and references
- 1.Anchel M, Hervey A, and Robbins WJ, Production of Illudin M and of a Fourth Crystalline Compound by Clitocybe Illudens. Proc Natl Acad Sci U S A. 1952, 38, 927–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nair MSR, Takeshita H, McMorris TC, and Anchel M, Metabolites of Clitocybe illudens. IV. Illudalic acid, a sesquiterpenoid, and illudinine, a sesquiterpenoid alkaloid. J. Org. Chem 1969, 34, 240–243. [DOI] [PubMed] [Google Scholar]
- 3.Yang X, Li J, Zhou Y, Shen Q, Chen J, and Li J, Discovery of novel inhibitor of human leukocyte common antigen-related phosphatase. Biochim. Biophys. Acta 2005, 1726, 34–41. [DOI] [PubMed] [Google Scholar]
- 4.Ling Q, Huang Y, Zhou Y, Cai Z, Xiong B, Zhang Y, Ma L, Wang X, Li X, Li J, and Shen J, Illudalic acid as a potential LAR inhibitor: Synthesis, SAR, and preliminary studies on the mechanism of action. Bioorg. Med. Chem 2008, 16, 7399–7409. [DOI] [PubMed] [Google Scholar]
- 5.(a) Mooney RA and LeVea CM, The leukocyte common antigen-related protein LAR: candidate PTP for inhibitory targeting. Curr. Top. Med. Chem 2003, 3, 809–819. [DOI] [PubMed] [Google Scholar]; (b) Kulas DT, Goldstein BJ, and Mooney RA, The transmembrane protein-tyrosine phosphatase LAR modulates signaling by multiple receptor tyrosine kinases. J. Biol. Chem 1996, 271, 748–754. [DOI] [PubMed] [Google Scholar]
- 6.(a) Cohen P, Protein kinases—the major drug targets of the twenty-first century? Nat. Rev. Drug Discov 2002, 1, 309–315. [DOI] [PubMed] [Google Scholar]; (b) Gross S, Rahal R, Stransky N, Lengauer C, and Hoeflich KP, Targeting cancer with kinase inhibitors. J. Clin. Invest 2015, 125, 1780–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He R, Yu Z-H, Zhang R, and Zhang Z-Y, Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin 2014, 35, 1227–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang ZY, Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases. Acc. Chem. Res 2017, 50, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Stanford SM and Bottini N, Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol. Sci 2017, 38, 524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lazo JS, McQueeney KE, and Sharlow ER, New Approaches to Difficult Drug Targets: The Phosphatase Story. SLAS Discov. 2017, 22, 1071–1083. [DOI] [PubMed] [Google Scholar]; (c) Tautz L and Mustelin T, Strategies for developing protein tyrosine phosphatase inhibitors. Methods 2007, 42, 250–260. [DOI] [PubMed] [Google Scholar]
- 10.Tonks NK, Protein tyrosine phosphatases—from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Tonks NK, Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006, 7, 833–846. [DOI] [PubMed] [Google Scholar]; (b) Cheng A, Uetani N, Lampron C, and Tremblay ML, Protein tyrosine phosphatases as therapeutic targets. In Inhibitors of Protein Kinases and Protein Phosphatases. Springer, Berlin, Heidelberg, 2005, pp. 191–214. [Google Scholar]
- 12.Chagnon MJ, Uetani N, and Tremblay ML, Functional significance of the LAR receptor protein tyrosine phosphatase family in development and diseases. Biochem. Cell. Biol 2004, 82, 664–675. [DOI] [PubMed] [Google Scholar]
- 13.Zabolotny JM, Kim YB, Peroni OD, Kim JK, Pani MA, Boss O, Klaman LD, Kamatkar S, Shulman GI, Kahn BB, and Neel BG, Overexpression of the LAR (leukocyte antigen-related) protein-tyrosine phosphatase in muscle causes insulin resistance. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 5187–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner RT, Wang L, Lang BT, Cregg JM, Dunbar CL, Woodward WR, Silver J, Ripplinger CM, and Habecker BA, Targeting protein tyrosine phosphatase σ after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nature Commun. 2015, 6, 6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uhl GR, Martinez MJ, Paik P, Sulima A, Bi GH, Iyer MR, Gardner E, Rice KC, and Xi ZX, Cocaine reward is reduced by decreased expression of receptor-type protein tyrosine phosphatase D (PTPRD) and by a novel PTPRD antagonist. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 11597–11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Tautz L, Pellecchia M, and Mustelin T, Targeting the PTPome in human disease. Expert Opi.n Ther. Targets 2006, 10, 157–177. [DOI] [PubMed] [Google Scholar]; (b) Barr AJ, Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future Med. Chem 2010, 2, 1563–1576. [DOI] [PubMed] [Google Scholar]
- 17.Woodward RB and Hoye TR, Total synthesis of illudinine, illudalic acid, and illudacetalic acid. J. Am. Chem. Soc 1977, 99, 8007–8014. [Google Scholar]
- 18.McCullough BS, Batsomboon P, Hutchinson KB, Dudley GB, and Barrios AM, Synthesis and PTP Inhibitory Activity of Illudalic Acid and Its Methyl Ether, with Insights into Selectivity for LAR PTP over Other Tyrosine Phosphatases Under Physiologically Relevant Conditions. J. Nat. Prod 2019, 12, 3386–3393. [DOI] [PubMed] [Google Scholar]
- 19.Ling Q, Zhou Y-Y, Cai Z-L, Zhang Y-H, Xiong B, Ma L-P, Wang X, Li X, Li J, and Shen J-K, Synthesis and LAR inhibition of 7-alkoxy analogues of illudalic acid. Acta Pharmacol. Sin 2010, 45, 1385–97. [PubMed] [Google Scholar]
- 20.Xu H, Li S, Liu H, Fu H, and Jiang Y, Simple and efficient copper-catalyzed cascade synthesis of naphthols containing multifunctional groups under mild conditions. Chem. Commun 2010, 46, 7617–7619. [DOI] [PubMed] [Google Scholar]
- 21.(a) Hoang TT, Birepinte M, Kramer NJ, and Dudley GB, Six-step synthesis of alcyopterosin A, a bioactive illudalane sesquiterpene with a gem-dimethylcyclopentane ring. Org. Lett 2016, 18, 3470–3473. [DOI] [PubMed] [Google Scholar]; (b) Morrison AE, Hoang TT, Birepinte M, and Dudley GB, Synthesis of illudinine from dimedone. Org. Lett 2017, 19, 858–861. [DOI] [PubMed] [Google Scholar]; (c) Gaston R Jr. Geldenhuys WJ, and Dudley GB, Synthesis of illudinine from dimedone and identification of activity as a monoamine oxidase inhibitor (MAOI). J. Org. Chem 2020, 85, 13429–13437. [DOI] [PubMed] [Google Scholar]; (d) Tavakoli A and Dudley GB, Synthesis of 4,4-dimethyl-1,6-heptadiyne and alcyopterosin O. Org. Lett 2020, 22, 8947–8951. [DOI] [PubMed] [Google Scholar]
- 22.Aungst RA Jr, Chan C, and Funk RL, Total synthesis of the sesquiterpene (±)-illudin C via an intramolecular nitrile oxide cycloaddition. Org. Lett 2001, 3, 2611–2613. [DOI] [PubMed] [Google Scholar]
- 23.Binder RJ, Hatfield MJ, Chi L, and Potter PM, Facile synthesis of 1, 2-dione-containing abietane analogues for the generation of human carboxylesterase inhibitors. Eur. J. Med. Chem 2018, 149, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanari T, Shimada N, Kurosaki Y, Thrimurtulu N, Nambu H, Anada M, and Hashimoto S, Asymmetric Total Sythesis of (−)-Englerin A through Catalytic Diastereo- and Enantioselective Carbonyl Ylide Cycloaddition. Chem.-Eur. J 2015, 21, 11671–11676. [DOI] [PubMed] [Google Scholar]
- 25.Liu S, Zhou B, Yang H, He Y, Jiang ZX, Kumar S, Wu L, and Zhang ZY, Aryl vinyl sulfonates and sulfones as active site-directed and mechanism-based probes for protein tyrosine phosphatases. J. Am. Chem. Soc 2008, 130, 8251–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dudley GB, Batsomboon P, Gaston R Jr., and Fulo HF, Selective Phosphatase Inhibitors Based On Illudalic Acid. International Patent Application Number PCT/US2021/043713, July 29, 2021. [Google Scholar]
- 27.(a) Chen YNP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CHT, Chen Z, Cooke VG, and Dobson JR, Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [DOI] [PubMed] [Google Scholar]; (b) LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, Chen CHT, Chen YN, Chen Z, Deng Z, and Dore M, Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem 2020, 63, 13578–13594. [DOI] [PubMed] [Google Scholar]; (c) Yuan X, Bu H, Zhou J, Yang CY, and Zhang H, Recent advances of SHP2 inhibitors in cancer therapy: Current development and clinical application. J. Med. Chem 2020, 63, 11368–11396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.