

Abstract
The basal ganglia regulate a wide range of behaviors, including motor control and cognitive functions, and are profoundly affected in Parkinson’s disease (PD). However, the functional organization of different basal ganglia nuclei has not been fully elucidated at the circuit level. In this study, we investigated the functional roles of distinct parvalbumin-expressing neuronal populations in the external globus pallidus (GPe-PV) and their contributions to different PD-related behaviors. We demonstrate that substantia nigra pars reticulata (SNr)-projecting GPe-PV neurons and parafascicular thalamus (PF)-projecting GPe-PV neurons are associated with locomotion and reversal learning, respectively. In a mouse model of PD, we found that selective manipulation of the SNr-projecting GPe-PV neurons alleviated locomotor deficit, whereas manipulation of the PF-projecting GPe-PV neurons rescued the impaired reversal learning. Our findings establish the behavioral importance of two distinct GPe-PV neuronal populations and, thereby, provide a new framework for understanding the circuit basis of different behavioral deficits in the Parkinsonian state.
The basal ganglia are a group of subcortical nuclei that regulate motor and cognitive functions1,2. Recent identification of neuronal heterogeneity in the basal ganglia suggests that functionally distinct neural circuits defined by their efferent projections exist even within the same nuclei3–5. This distinction might account for a multitude of symptoms associated with basal ganglia disorders, such as PD6,7. However, incomplete understanding of the basal ganglia functional organization has hindered further investigation of individual circuits that might underlie different behavioral symptoms in disease states.
The GPe is a central basal ganglia nucleus that can influence many downstream brain regions through its strong inhibitory output8. Most GPe neurons express PV and innervate multiple basal ganglia and thalamic nuclei9–12. Through this anatomical connectivity, the GPe is well positioned to regulate basal ganglia function. Indeed, GPe neurons exhibit movement-entrained neural activity11, and aberrant GPe neuronal activity is highly associated with movement deficits in both patients with PD13 and Parkinsonian animal models14,15. Importantly, high-frequency deep-brain stimulation of the GPe in human patients and prolonged optogenetic activation of the GPe-PV neurons in dopamine-depleted mice have been shown to alleviate Parkinsonian motor symptoms16,17. However, contrary to the canonical basal ganglia model, these stimulation effects might be mediated through GPe neurons that innervate the SNr—an output nucleus of the basal ganglia that directly controls motor-related brain regions, including the motor thalamus and the mesencephalic locomotor region18,19. Moreover, a subset of GPe neurons were previously found to display robust activity during a task requiring a stimulus–action–outcome-based association20. This raises the intriguing possibility that control of non-motor behavior could also be achieved through the GPe-PV neurons innervating nuclei involved in cognitive function, such as the PF21,22. These results suggest that the GPe-PV neurons might influence a variety of behavioral functions through distinct neuronal populations, but incomplete understanding of the GPe circuit organization has precluded any investigation of a potential divergent control of motor and non-motor behaviors.
In this study, we investigated the projection-specific roles of the GPe-PV neurons in locomotion and reversal learning, as well as their contributions to locomotor and reversal learning impairments, in a Parkinsonian mouse model. We identified two distinct GPe-PV neuronal populations that are embedded in discrete neural pathways and established their contributions to different behavioral deficits in dopamine-depleted mice. Our findings provide a novel framework for understanding the circuit basis of varying behavioral symptoms of the Parkinsonian state, which could provide better strategies for the treatment of PD.
Results
Divergent efferent pathways of GPe-PV neurons.
We first examined the projection pattern and synaptic targets of the GPe-PV neurons by injecting an adeno-associated virus (AAV) expressing a red fluorescent protein, mRuby2, and synaptophysin fused with enhanced green fluorescent protein (eGFP) in a Cre-dependent manner into the GPe of PvCre mice. This approach restricts the eGFP expression to the pre-synaptic terminals, allowing us to discriminate between putative synaptic targets and the axon of passage4. Consistent with previous findings, we observed robust labeling of synaptic terminals in the subthalamic nucleus (STN), the internal globus pallidus (GPi; entopeduncular nucleus in rodents), the SNr, the PF and the dorsal striatum (Fig. 1a,b)12,23.
Fig. 1 |. Distinct subpopulations of GPe-PV neurons project to the SNr and PF.
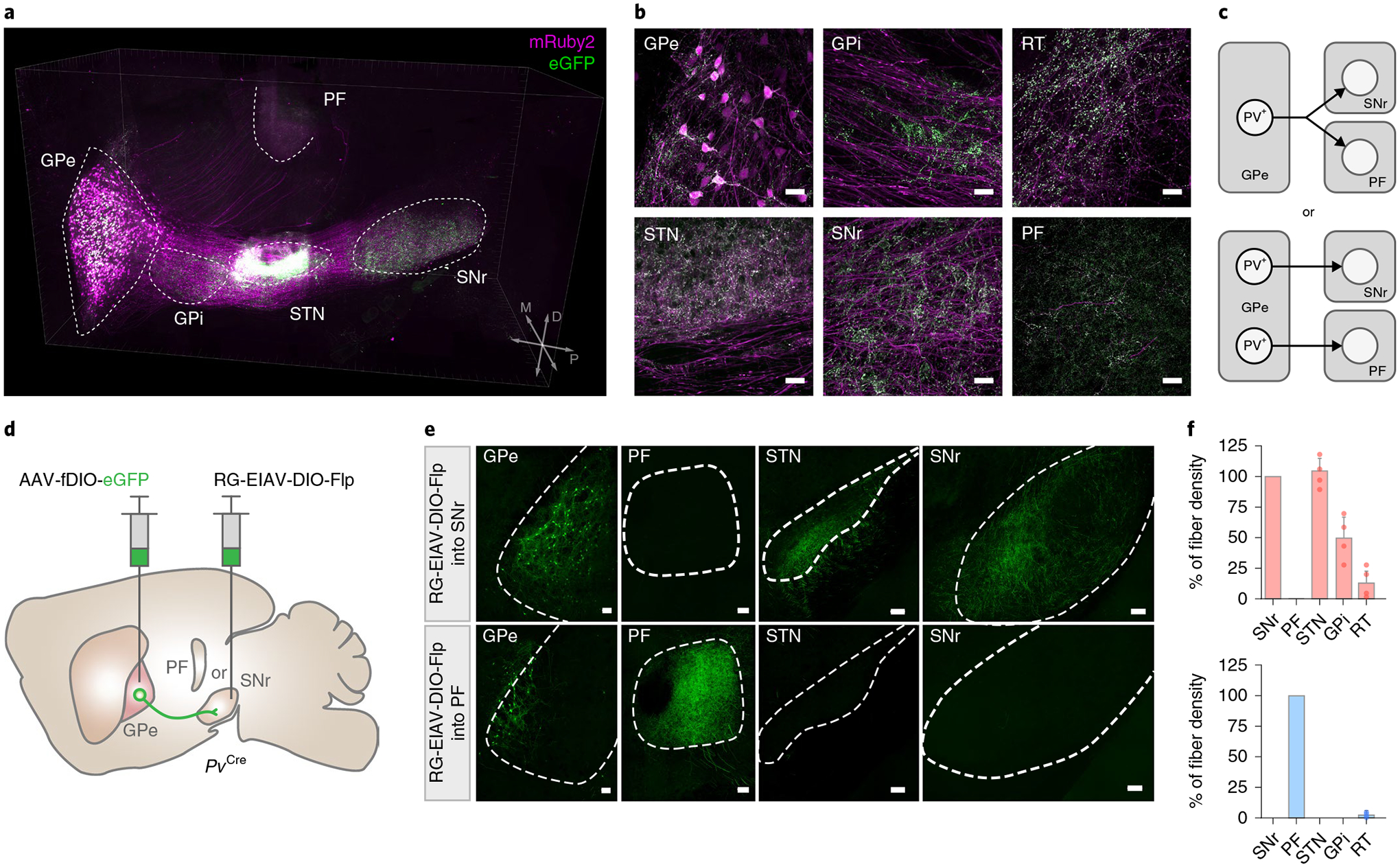
a, Three-dimensional rendering of a cleared mouse hemisphere showing brain-wide projection patterns of GPe-PV neurons labeled by mruby2 (soma, axonal fibers) and eGFP (pre-synaptic sites). b, representative confocal images of the injection site in GPe (top left) and target structures showing axonal fibers in magenta and synaptic puncta in green (repeated in n = 4 mice). Scale bar, 30 μm. c, Two possible projection patterns: GPe-PV neurons collateralize to multiple targets (left) or individual neurons project to distinct targets (right). d, Schematic of viral strategy using a pseudotyped equine infectious anemia lentivirus capable of neuron-specific retrograde infection (rG-EIAV). SNr or PF of PvCre mice is injected with rG-EIAV that expresses Flp recombinase in a Cre-dependent manner (rG-EIAV-DIO-Flp) and the GPe is injected with an AAV that expresses eGFP in a Flp-dependent manner (AAV-fDIO-eGFP). e, Confocal images of SNr- and PF-projecting GPe-PV neurons and their axons (repeated in n = 4 mice for each target area). Scale bar, 100 μm. f, The quantification of the axonal fibers of PVGPe-SNr neurons (top; n = 4 mice) and PVGPe-PF neurons (bottom; n = 4 mice) in projected areas measured by the fluorescent intensity. Data are presented as % mean ± s.e.m. of control sections.
Although GPe neurons project to many target regions, it remains unclear whether separate GPe neurons project to distinct targets or individual GPe neurons collateralize onto multiple targets (Fig. 1c). In particular, we focused on the GPe outputs to the SNr and the PF, two regions known to be involved in motor control17,18 and behavioral flexibility21,22, respectively. These distinct functions led us to hypothesize that discrete subclasses of the GPe-PV neurons project to either the SNr or the PF. To test this hypothesis, we used an intersectional genetic targeting approach to label GPe-PV neurons in a cell-type- and projection-specific manner. We adapted a novel viral vector capable of neuron-specific retrograde infection—a pseudotyped equine infectious anemia lentivirus (EIAV) carrying extracellular and transmembrane domains of the rabies glycoprotein conjugated to the cytoplasmic domain of vesicular stomatitis virus glycoprotein (RG-EIAV)4,24,25. RG-EIAV confers stable, retrograde expression of transgenes suitable for long-term studies without cytotoxicity. We injected RG-EIAV-inducing Flp recombinase in a Cre-dependent manner (RG-EIAV-DIO-Flp) into either the SNr or the PF along with an AAV-expressing Flp-dependent eGFP (AAV-fDIO-eGFP) into the GPe of PvCre mice (Fig. 1d). By restricting eGFP expression to a projection-defined subset of GPe-PV neurons, we found that SNr-projecting GPe-PV neurons (PVGPe-SNr) were present throughout the GPe and innervate multiple nuclei in the basal ganglia, including the STN and the GPi. By contrast, the PF-projecting GPe-PV neurons (PVGPe-PF) were clustered in the ventromedial region of the GPe and innervate only the PF (Fig. 1e,f). Thus, PVGPe-SNr and PVGPe-PF neurons are anatomically distinct neuronal populations that might represent separate neural pathways.
In addition, we examined the messenger RNA (mRNA) expression of the previously identified molecular markers of the GPe neurons in the PVGPe-SNr and PVGPe-PF neurons by using multiplex fluorescence in situ hybridization (mFISH) in conjunction with the intersectional viral genetic approach outlined above. We selected the following markers to determine if the PVGPe-SNr and PVGPe-PF neurons reconcile with any of the previously classified GPe neuronal populations26–29: Foxp2 (forkhead box protein P2), Nkx2-1 (NK2 homeobox 1), Lhx6 (LIM homeobox protein), Scn4b (sodium voltage-gated channel beta subunit 4) and Sst (somatostatin). We found that most PVGPe-SNr and PVGPe-PF neurons express Nkx2-1 and Scn4b mRNA, whereas Lhx6 is preferentially expressed in the PVGPe-SNr neurons (Extended Data Fig. 1a–c).
Inputs to PVGPe-SNr and PVGPe-PF neurons.
Because PVGPe-SNr and PVGPe-PF neurons are anatomically distinct, we reasoned that each subpopulation is a part of a separate pathway and receives inputs from different brain regions. To test this, we mapped the brain-wide inputs to each subpopulation by expressing the TVA receptor and an optimized rabies glycoprotein (oPBG)30 in either PVGPe-SNr or PVGPe-PF neurons, followed by an injection of the EnvA-pseudotyped, glycoprotein-deleted rabies virus (EnvA-RVΔG-eGFP) into the GPe (Fig. 2a). Whole-brain quantification of eGFP-labeled neurons revealed that the PVGPe-SNr neurons receive proportionally more inputs from the STN, whereas the PVGPe-PF neurons receive more inputs from the cortex and the midbrain (Fig. 2b–d). Although the main input of both populations originates in the striatum, the PVGPe-SNr and the PVGPe-PF neurons are preferentially innervated by the dorsolateral striatum (DLS) and the dorsomedial striatum (DMS), respectively (Fig. 2b,c). Furthermore, by determining the molecular identity of the striatal input neurons, we found that the PVGPe-SNr neurons receive proportionally more inputs from striatal projection neurons that express Drd2 mRNA, whereas the PVGPe-PF neurons receive more inputs from striatal projection neurons that express Drd1a mRNA (Fig. 2e,f). This contrasts with the classical model of the basal ganglia that posits GPe innervation by Drd2-expressing striatal projection neurons. Taken together, this suggests that PVGPe-SNr and PVGPe-PF neurons are part of distinct neural pathways that might underlie different behavioral functions.
Fig. 2 |. Whole-brain mapping of inputs to PVGPe-SNr and PVGPe-PF neurons.
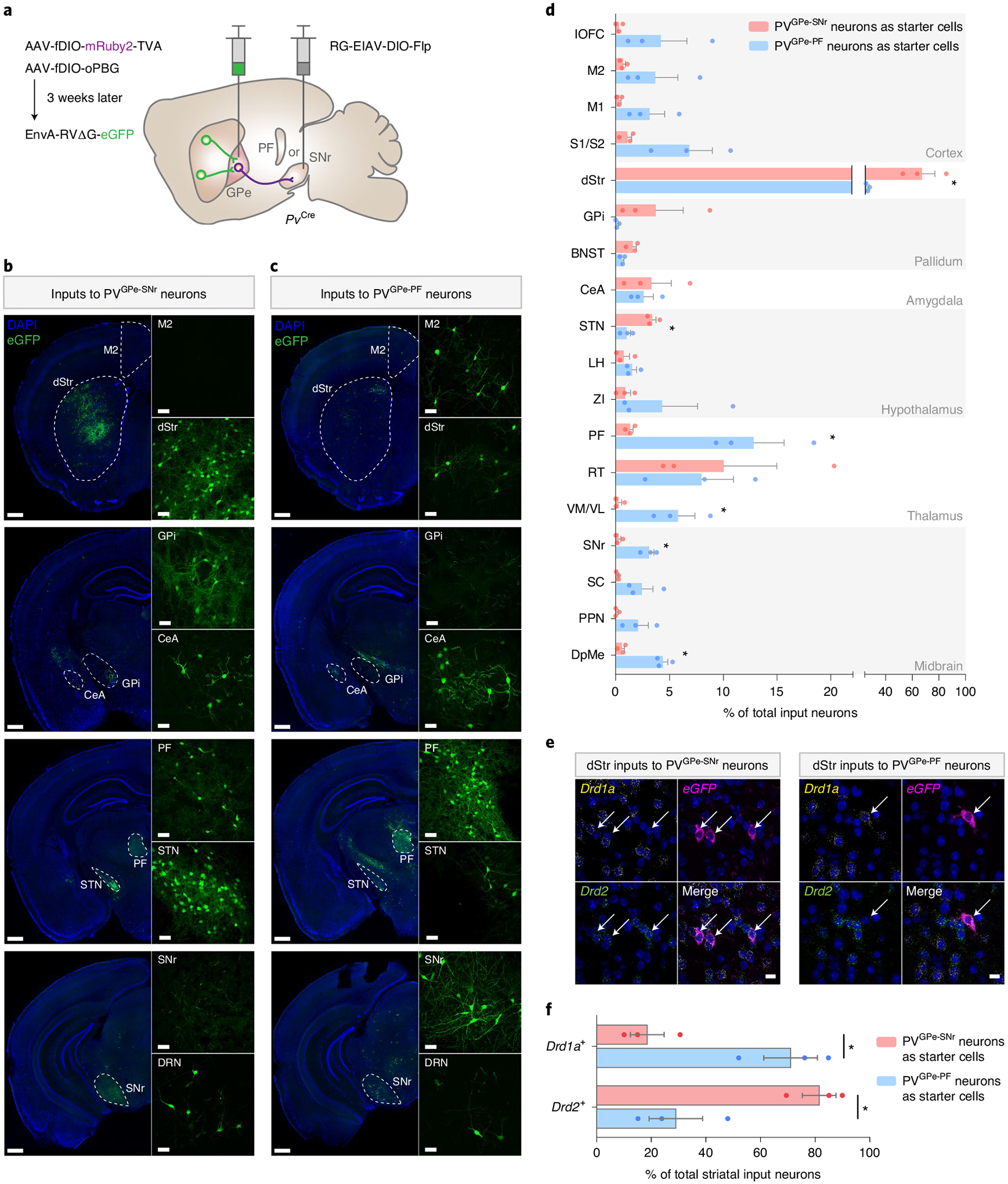
a, Viral strategy to map pseudotyped rabies-mediated monosynaptic inputs to PVGPe-SNr neurons. In PvCre mice, SNr or PF was injected with rG-EIAV-DIO-Flp, and GPe was injected with AAV-fDIO-TVA-mruby and AAV-fDIO-oPBG. EnvA-rVΔG-eGFP was subsequently injected into the GPe. b,c, representative images of select brain areas showing trans-synaptically labeled input neurons of PVGPe-SNr neurons (b) and PVGPe-PF neurons (c). repeated in n = 3 mice for each subpopulation. d, Whole-brain quantification of inputs to PVGPe-SNr and PVGPe-PF neurons. Data are presented as percentage of total cells in each brain area relative to the total number of brain-wide inputs. Unpaired t-tests performed for individual brain regions: t(4) = 4.212, *P = 0.0136 for dStr; t(4) = 4.883, *P = 0.0081 for STN; t(4) = 4.216, *P = 0.0135 for PF; t(4) = 3.588, *P = 0.0230 for VM/VL; t(4) = 5.992, *P = 0.0039 for SNr; t(4) = 7.299, *P = 0.0019 for DpMe (n = 3 mice for each subpopulation). *P < 0.05. e, representative images of striatal neurons sending input to PVGPe-SNr (left) and PVGPe-PF (right) neurons with cell-type-specific markers (repeated in n = 3 mice for each subpopulation). Arrows represent co-localization between striatal input neurons labeled by the rabies virus and mrNA labeled by cell-type-specific probes. f, Quantification of Drd1a+ or Drd2+ striatal inputs to PVGPe-SNr and PVGPe-PF neurons, showing that Drd1a+ striatal neurons project preferentially to PVGPe-PF neurons, whereas Drd2+ striatal neurons project preferentially to PVGPe-SNr neurons. Paired t-test, t(2) = 9.333; *P = 0.0113 (n = 3 mice for each subpopulation). *P < 0.05. Scale bars, 50 μm (b and c inset), 500 μm (b and c) and 10 μm (e). All data are presented as mean ± s.e.m. BNST, bed nucleus of stria terminalis; CeA, central amygdala; DpMe, deep mesencephalic nucleus; DrN, dorsal raphe nucleus; dStr, dorsal striatum; GPi, internal globus pallidus; lOFC, lateral orbitofrontal cortex; LH, lateral hypothalamus; M1, primary motor cortex; M2, secondary motor cortex; PPN, pedunculopontine nucleus; rT, reticular thalamus; S1/S2, somatosensory cortex; SC, superior colliculus; STN, subthalamic nucleus; VM/VL, ventromedial/ventrolateral thalamus; ZI, zona incerta.
Distinct electrophysiological properties of PVGPe-SNr and PVGPe-PF neurons.
GPe neurons exhibit heterogeneous autonomous firing patterns and intrinsic membrane properties10,11,31. To determine whether PVGPe-SNr and PVGPe-PF neurons also differ in their electrophysiological properties, we performed ex vivo whole-cell recordings in acute brain slices. We selectively labeled PVGPe-SNr and PVGPe-PF neurons by injecting RG-EIAV-expressing Cre in a Flp-dependent manner (RG-EIAV-fDIO-Cre) into either the SNr or PF of PvFlp × Ai14 mice, resulting in projection-specific tdTomato expression in the GPe-PV neurons (Fig. 3a). Both populations were spontaneously active at rest, with the PVGPe-SNr neurons firing more regularly at a higher rate compared to the PVGPe-PF neurons (Fig. 3b–d). Further evaluation also revealed differences in driven activity and action potential waveforms of these two subpopulations (Fig. 3e–l). Thus, in addition to their different anatomical connectivity, PVGPe-SNr and PVGPe-PF neurons are distinguishable by their electrophysiological properties.
Fig. 3 |. PVGPe-SNr and PVGPe-PF neurons exhibit distinct electrophysiological properties.
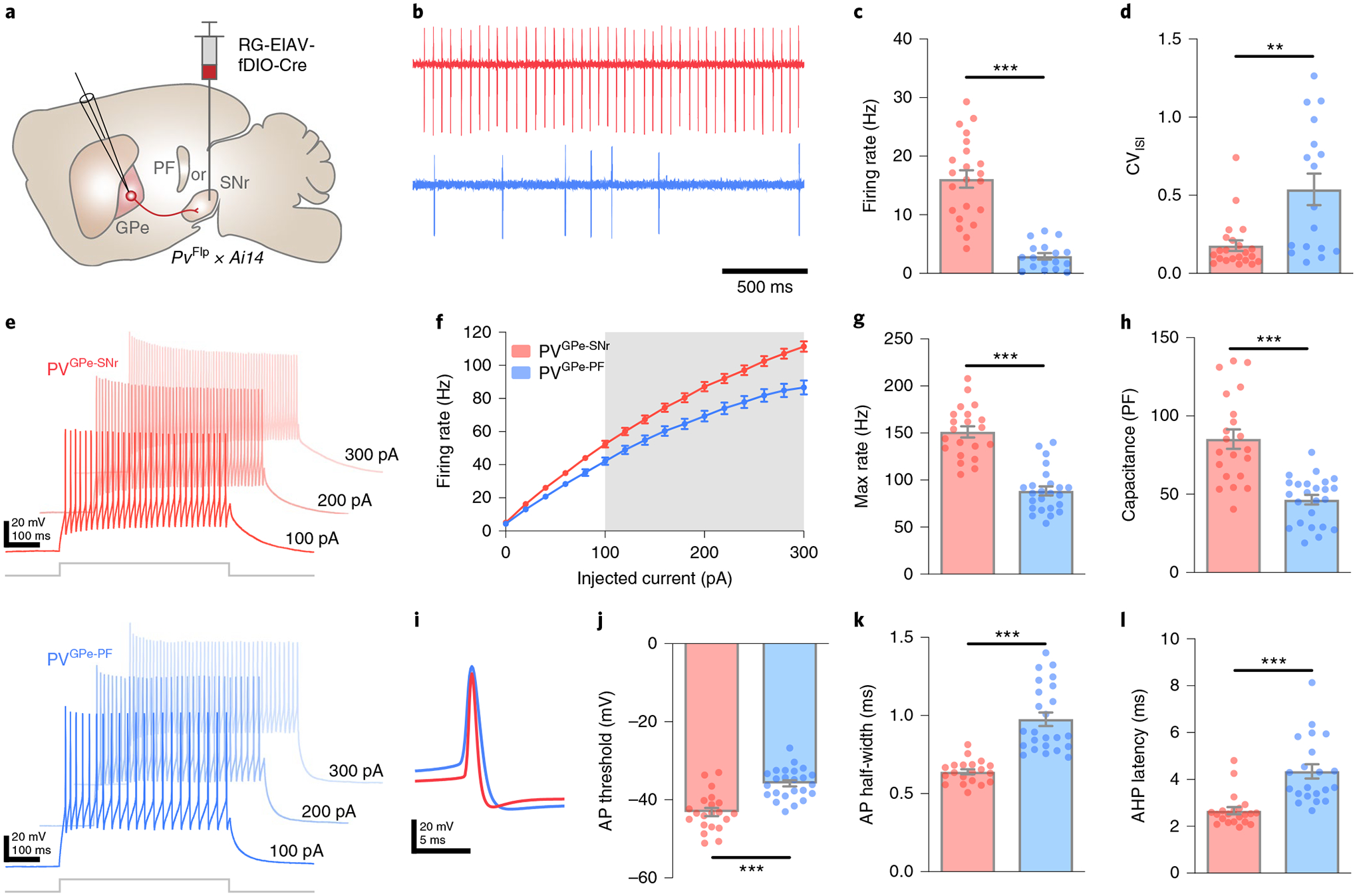
a, Strategy using PvFlp × Ai14 mice for labeling GPe-PV neurons in a projection-specific manner. rG-EIAV-fDIO-Cre is injected into either SNr or PF. b, representative cell-attached recordings from PVGPe-SNr and PVGPe-PF neurons. c, Autonomous firing rate of PVGPe-SNr and PVGPe-PF neurons. Mann–Whitney U-test, U = 7, ***P = 1.764 × 10−9 (n = 22 cells from eight mice for PVGPe-SNr and n = 17 cells from seven mice for PVGPe-PF neurons). d, Firing regularity of PVGPe-SNr and PVGPe-PF neurons. Mann–Whitney U-test, U = 7, **P = 0.0019 (n = 22 cells for PVGPe-SNr and n = 17 cells for PVGPe-PF neurons). CVISI, coefficent of variation of interspike interval. e, representative traces of neuronal firing in response to 100-pA, 200-pA and 300-pA current injections in PVGPe-SNr and PVGPe-PF neurons. f, Action potential (AP) firing frequency in response to a range of current injections for PVGPe-SNr and PVGPe-PF neurons. Gray shading shows a significant difference (P < 0.05). Multiple t-tests, corrected for multiple comparisons using the Holm–Sidak method (n = 21 cells from seven mice for PVGPe-SNr and n = 24 cells from 11 mice for PVGPe-PF neurons). g, Maximum firing rate of PVGPe-SNr and PVGPe-PF neurons. Mann–Whitney U-test, U = 23, ***P = 2.831×10−9 (n = 21 cells for PVGPe-SNr and n = 24 cells for PVGPe-PF neurons). h, Membrane capacitance of PVGPe-SNr and PVGPe-PF neurons. Mann–Whitney U-test, U = 23, ***P = 1.001 × 10−6 (n = 21 cells for PVGPe-SNr and n = 24 cells for PVGPe-PF neurons). i, representative traces of AP waveforms recorded from PVGPe-SNr and PVGPe-PF neurons. j, AP threshold of PVGPe-SNr and PVGPe-PF neurons. Mann–Whitney U-test, U = 57, ***P = 1.851 × 10−6 (n = 21 cells for PVGPe-SNr and n = 24 cells for PVGPe-PF neurons). k, AP half-width of PVGPe-SNr and PVGPe-PF neurons. Mann–Whitney U-test, U = 10, ***P = 1.579 × 10−10 (n = 21 cells for PVGPe-SNr and n = 24 cells for PVGPe-PF neurons). l, After-hyperpolarization (AHP) latency of PVGPe-SNr and PVGPe-PF neurons. Mann–Whitney U-test, U = 32, ***P = 1.597 × 10−7 (n = 21 cells for PVGPe-SNr and n = 24 cells for PVGPe-PF neurons). All data are presented as mean ± s.e.m.
PVGPe-SNr neurons mediate locomotion.
Given their distinct connectivity patterns and electrophysiological properties, PVGPe-SNr and PVGPe-PF neurons might convey separate information streams to their respective targets to mediate different behaviors. We next determined whether the activity of PVGPe-SNr and PVGPe-PF neurons displayed a time-locked response to locomotion initiation and termination. We expressed an axon-targeted, genetically encoded Ca2+ indicator (axon-GCaMP6s)32 and mRuby3 in the GPe-PV neurons and used fiber photometry to simultaneously measure the population calcium activity from their axon terminals in the SNr and the PF during locomotion (Fig. 4a and Extended Data Fig. 2a,b)33. Mice were head-fixed over a cylindrical treadmill, and GCaMP6s fluorescence from the SNr and the PF was measured during self-initiated locomotion (Fig. 4b). We observed an increase in fluorescence intensity in both target areas during the transition from rest to run. Although the activity of both subpopulations is correlated with locomotion bouts, the rise in the activity of the PVGPe-SNr neurons occurred slightly before the locomotion onset, whereas the PVGPe-PF neuronal activity rose after the movement onset (Fig. 4c,d and Extended Data Fig. 2c,d). This suggests that the activity of both populations might be differentially associated with locomotion.
Fig. 4 |. Activity of PVGPe-SNr neurons bidirectionally modulates locomotion.
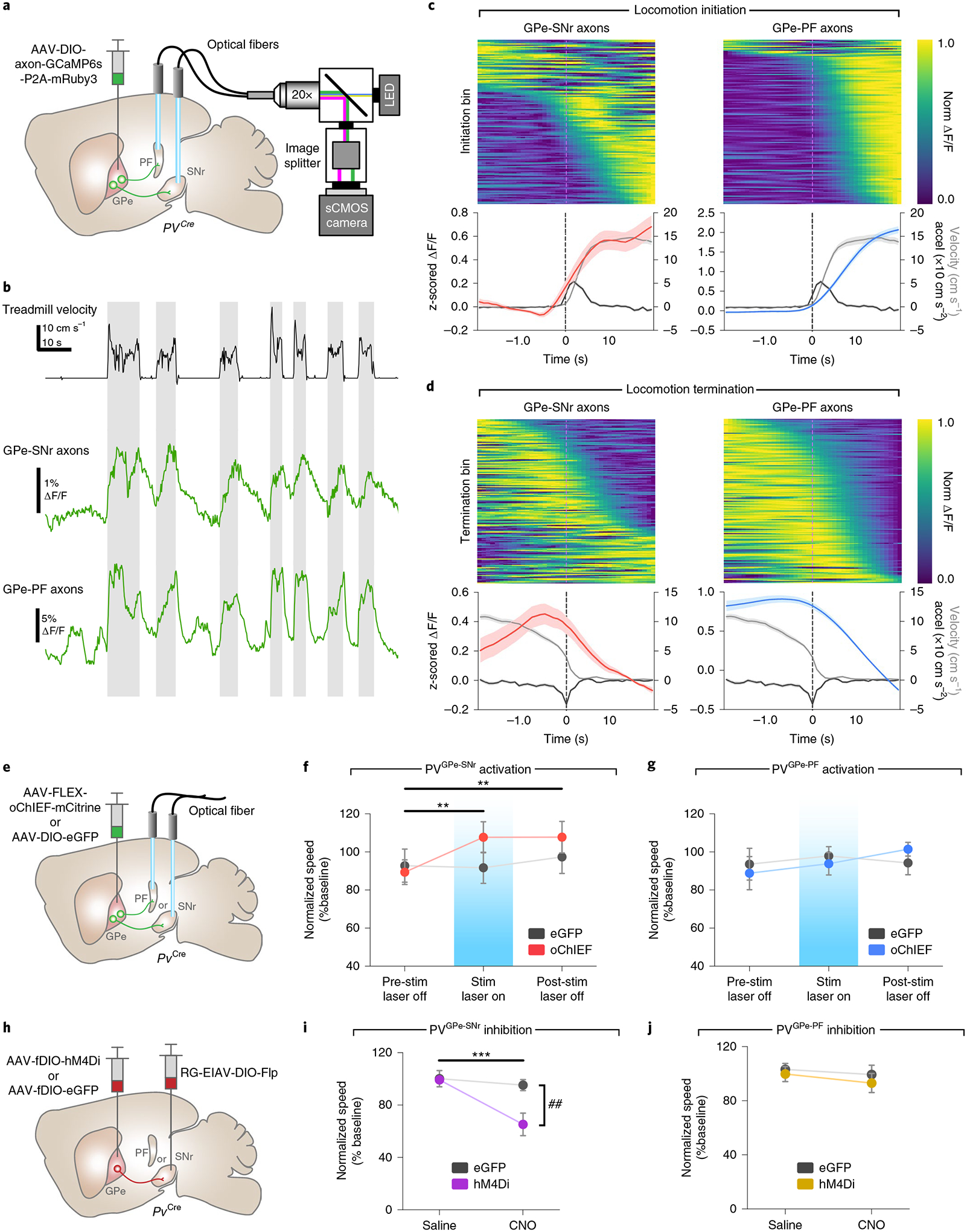
a, Schematic of fiber photometry setup. b, representative ΔF/F traces of GCaMP6s fluorescence recorded from axonal fibers of PVGPe-SNr and PVGPe-PF neurons from one mouse (n = 7 mice total) and corresponding treadmill velocity (top). Shaded areas indicated locomotion bins. c, Top: ΔF/F aligned to locomotion initiations from all mice (ΔF/F was normalized for each row and sorted by time to half-maximum). Bottom: mean acceleration (black), velocity (gray) and z-scored ΔF/F triggered at locomotion initiation (mean across all events). d, Same as c, but aligned to locomotion terminations. e, Schematic of viral injections and optic fiber implantations for optogenetic activation. f, Activation of GPe-PV terminal in the SNr increased locomotion during stimulation and post-stimulation periods. Two-way repeated-measures ANOVA, main effectstim×group: F2,36 = 3.863, P = 0.0302; main effectstim: F2,36 = 5.513, P = 0.0082; main effectgroup: F1,18 = 0.5161, P = 0.4817; Bonferroni’s post hoc test, **P = 0.0029 for pre-stim laser off versus stim laser on and **P = 0.0027 for pre-stim laser off versus post-stim laser off (n = 10 mice for both eGFP and oChIEF). g, Activation of GPe-PV terminal in the PF had no effect on locomotion. Two-way repeated-measures ANOVA, main effectstim×group: F2,26 = 0.6664, P = 0.5221; main effectstim: F2,26 = 0.6870, P = 0.5120; main effectgroup: F1,13 = 0.0065, P = 0.9368 (n = 7 mice for eGFP and n = 8 mice for oChIEF). h, Schematic of viral injections for cell-type- and projection-specific expression of inhibitory DrEADD (hM4Di) in PVGPe-SNr and PVGPe-PF neurons. i, Inhibition of PVGPe-SNr neurons suppresses locomotion. Two-way repeated-measures ANOVA, main effecttreatment×group: F1,18 = 9.644, P = 0.0061; main effecttreatment: F1,18 = 17.30, P = 0.0006; main effectgroup: F1,18 = 4.968, P = 0.0388; Bonferroni’s post hoc test, ***P = 1.381 × 10−4 for saline-hM4Di versus CNO-hM4Di and ##P = 0.002 for CNO-eGFP versus CNO-hM4Di (n = 10 mice for both eGFP and hM4Di). j, Inhibition of PVGPe-PF neurons had no effect on locomotion. Two-way repeated-measures ANOVA, main effecttreatment×group: F1,10 = 0.069, P = 0.7980; main effecttreatment: F1,10 = 0.9681, P = 0.3484; main effectgroup: F1,10 = 0.5119, P = 0.4911; n = 6 mice for both eGFP and hM4Di). Data in f–j are presented as mean speed (normalized to baseline) ± s.e.m. Shaded areas in c and d indicate s.e.m.
To unambiguously determine whether the activity of the PVGPe-SNr and the PVGPe-PF neurons can modulate locomotion, we first expressed oChIEF34 in the GPe-PV neurons and bilaterally photostimulated their axons in the SNr or the PF while measuring the locomotor activity in an open field (Fig. 4e). For each mouse, photostimulation lasted 10 min and consisted of 500-ms pulses that repeated every 1.5 s. The stimulation paradigm was determined by testing the efficacy of a range of stimulation frequencies that can reliably inhibit spiking activity of SNr neurons ex vivo and was used throughout this study (Extended Data Fig. 3c,d). Photostimulation of PVGPe-SNr axons significantly increased locomotor activity that persisted beyond the stimulation period (Fig. 4f). By contrast, photostimulation of the PVGPe-PF axons did not show any effect on locomotion (Fig. 4g).
Next, we tested whether the activity of the PVGPe-SNr and the PVGPe-PF neurons is necessary for locomotion. We examined the effect of silencing PVGPe-SNr and PVGPe-PF neurons on locomotion by expressing an inhibitory chemogenetic receptor (hM4Di) that can be activated by clozapine N-oxide (CNO) in each subpopulation (Fig. 4h). We used chemogenetic inhibition for behavioral testing to ensure prolonged inhibition of neural activity in projection-defined populations while avoiding the potential issue on the efficacy of optogenetic inhibition at the pre-synaptic terminals. We validated the efficacy of chemogenetic inhibition by recording from the PVGPe-SNr and the PVGPe-PF neurons expressing hM4Di in acute brain slices while CNO was being applied (Fig. 4h and Extended Data Fig. 3e–g). Inactivation of the PVGPe-SNr neurons induced a significant reduction in locomotor activity, whereas inactivation of the PVGPe-PF neurons did not show any effect on locomotion (Fig. 4i,j). Collectively, our results indicate that the activity of the PVGPe-SNr neurons, but not the PVGPe-PF neurons, can bidirectionally modulate locomotion and plays an essential role in motor control.
PVGPe-PF neurons are involved in reversal learning.
To investigate the involvement of the GPe-PV neurons in behavioral flexibility, we examined the activity of the PVGPe-SNr and PVGPe-PF neurons during a context-based foraging reversal discrimination task. In this task, food-restricted mice were placed in an arena with two bowls, each filled with digging medium of different context, and a food reward was placed in only one bowl. In each trial, mice were allowed to freely choose to dig in either bowl. During the ‘association phase’ of the task, mice quickly associated one context with a rewarding outcome after the food was initially found. Once the mice could reliably discriminate between the rewarding and non-rewarding contexts, they entered the ‘reversal phase’ in which the context–outcome association was reversed by switching the food placement into a bowl containing context not previously associated with a reward. The behavioral performance of this task was determined during the reversal phase in which the mice needed to switch their choice to obtain the food reward (Fig. 5a and Extended Data Fig. 4a,b; see details in Methods)35,36. Using fiber photometry, we found that the overall activity of the PVGPe-PF neurons was significantly higher during trials relative to the rest periods between trials (inter-trial intervals), whereas there was no difference in the activity of the PVGPe-SNr neurons between the two periods. This suggests that the activity of the PVGPe-PF neurons is selectively associated with the task (Extended Data Fig. 4d,e). Next, we examined the time course of the activity of both PVGPe-SNr and PVGPe-PF neurons event locked to when the trial started and the moment the mice started digging to signify a choice (Fig. 5b) in three different stages of the task: 1) association phase—a period in which the mice form an initial context–outcome association; 2) early reversal phase—a period immediately after the context–outcome association is reversed; and 3) late reversal phase—the final period of reversal learning in which the mice can reliably discriminate between the two contexts. Both GPe-PV subpopulations showed a brief response to trial onsets across all stages of the task, possibly due to an increase in locomotion at the start of each trial (Fig. 5c,f). Notably, we found that only the PVGPe-PF neurons showed a significant increase in activity when the mice started digging in the early stage of reversal learning relative to the association and late stage of reversal learning (Fig. 5d,g). The increased neural activity in the early reversal phase was not due to a change in locomotor activity, as we did not observe any difference in the average speed of the mice across all stages of the task (Extended Data Fig. 4c). Further analysis indicated that the activity of the PVGPe-PF neurons increased when mice made an incorrect choice (Fig. 5e,h). These results strongly suggest that the PVGPe-PF neurons are involved in context-based reversal learning.
Fig. 5 |. Activation of PVGPe-PF neurons impairs reversal learning.
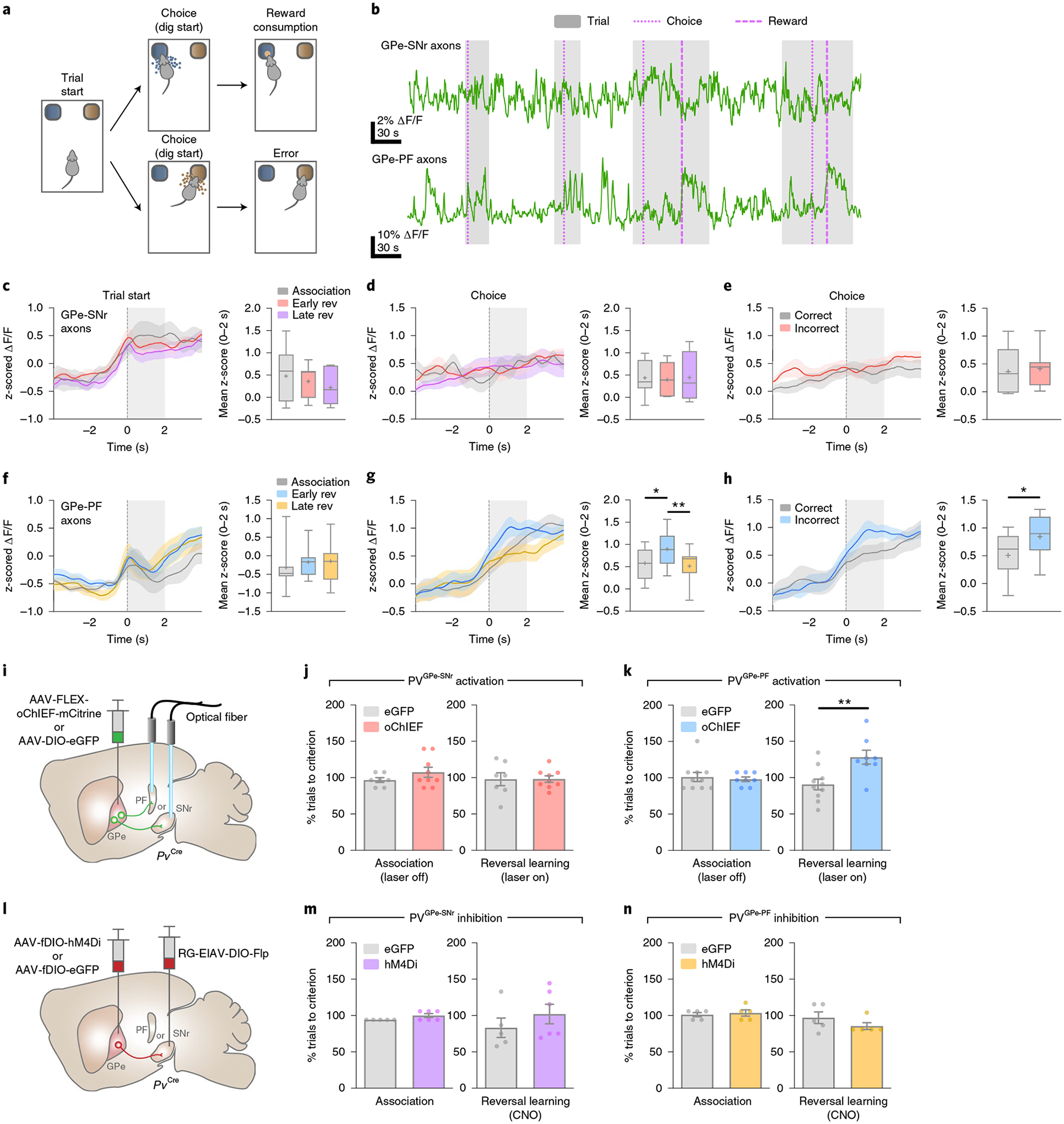
a, Behavioral task schematic showing the structure of each trial; after the trial starts, the mouse makes a choice by digging in one of the two bowls. Only the correct choice yields a food reward. b, representative fiber photometry ΔF/F traces showing the activity of PVGPe-SNr and PVGPe-PF axons from one mouse (n = 7 mice total) encompassing four trials in the reversal learning phase. Thin dotted line, thick dotted line and shaded area represent dig start, the beginning of food consumption and trial period, respectively. c,d, Left: traces of z-scored ΔF/F (averaged across seven mice) from PVGPe-SNr axons aligned to trial start (c) and timing of choice (d). right: mean z-score from 0–2 s after behavioral onset. Early rev, early stage of reversal learning; Late rev, late stage of reversal learning. One-way repeated-measures ANOVA, effectstage: F2,12 = 1.300, P = 0.3084 in c and effectstage: F2,12 = 0.0378, P = 0.9630 in d. e, Left: traces of z-scored ΔF/F (averaged across seven mice) from PVGPe-SNr axons aligned to the timing of correct or incorrect choices during the reversal learning phase. right: mean z-score from 0–2 s after behavioral onset. Paired t-test, t(6) = 0.5930, P = 0.5748. f,g, Left: traces of z-scored ΔF/F (averaged across seven mice) from PVGPe-PF axons aligned to trial start (f) and timing of choice (g). right: mean z-score from 0–2 s after behavioral onset. One-way repeated-measures ANOVA, effectstage: F2,12 = 1.280, P = 0.3135 in e and effectstage: F2,12 = 8.008, P = 0.0062 in f; Bonferroni’s post hoc test, *P = 0.0279 and **P = 0.0084. h, Left: traces of z-scored ΔF/F (averaged across seven mice) from PVGPe-PF axons aligned to the timing of correct or incorrect choices during the reversal learning phase. right: mean z-score from 0–2 s after behavioral onset. Paired t-test, t(6) = 3.128, *P = 0.0204. i, Schematic of viral injections and optic fiber implantations for optogenetic activation during reversal learning. j, Activation of PVGPe-SNr axons did not affect behavioral flexibility during reversal learning. Unpaired t-test, t(14) = 0.0273, P = 0.9786; n = 7 mice for eGFP and n = 9 mice for oChIEF (500-ms pulses repeated every 1.5 s). k, Activation of PVGPe-PF axons during reversal learning impaired behavioral flexibility. Unpaired t-test, t(16) = 3.142, **P = 0.0063; n = 10 mice for eGFP and n = 8 mice for oChIEF (500-ms pulses repeated every 1.5 s). l, Schematic of viral injections and experimental timeline for cell-type- and projection-specific expression of inhibitory DrEADD (hM4Di) in PVGPe-SNr and PVGPe-PF neurons. m, Inhibition of PVGPe-SNr neurons during reversal learning had no effect on behavioral flexibility. Unpaired t-test, t(9) = 0.9840, P = 0.3508; n = 5 mice for eGFP and n = 6 mice for hM4Di. n, Inhibition of PVGPe-PF neurons during reversal learning had no effect on behavioral flexibility. Unpaired t-test, t(8) = 1.245, P = 0.2483; n = 5 mice for eGFP and n = 5 mice for hM4Di. Shaded areas accompanying the z-scored ΔF/F traces in c–h indicate s.e.m. Box and whisker plots are used in the right panels of c–h where the central line of the box and ‘+’ represent the median and the mean, respectively. The box extends from the 25th to 75th percentiles, and the whiskers go down to the smallest value and up to the largest. Data in j, k, m and n are presented as mean ± s.e.m.
The changes in activity of the PVGPe-PF neurons when the mice signified a choice across different stages of the task and increased activity during incorrect choices raised the question of whether the activity dynamic of these neurons is involved in reversal learning or only in encoding errors. To answer this question, we manipulated the neural activity of both GPe-PV subpopulations during reversal learning by prolonged optogenetic activation or chemogenetic inhibition and assessed the behavioral consequences. Although photostimulation of the PVGPe-SNr axons did not affect reversal learning, activation of the PVGPe-PF axons significantly increased the number of trials to criterion (Fig. 5i–k). Further analysis revealed a higher number of regressive errors when the PVGPe-PF axons were activated, indicating that the mice had difficulty in establishing a new context–outcome association (Extended Data Fig. 5a–d). Chemogenetic inhibition of both PVGPe-SNr and PVGPe-PF neurons did not affect reversal learning (Fig. 5l–n). Interestingly, we found that the activity of the PVGPe-PF neurons was not associated with a spatial information-based operant reversal learning task (Extended Data Fig. 6). Our results indicated that, although not required during context-based reversal learning, sustained activation of the PVGPe-PF neurons can negatively affect the ability to maintain a new choice pattern after the context–outcome association is reversed.
Dopamine depletion alters synaptic output of GPe-PV neurons.
Patients with PD can exhibit a multitude of behavioral symptoms that likely reflect adaptations of different neural circuits to dopamine depletion. The differential effects of projection-specific manipulation of the GPe-Pv neurons on locomotion and reversal learning suggest that alteration in synaptic inputs, outputs or intrinsic properties of PVGPe-SNr and PVGPe-PF neurons might underlie different Parkinsonian behavioral deficits37,38. To induce dopamine depletion, we used the well-established 6-hydroxydopamine (6-OHDA)-lesioned Parkinsonian mouse model (Fig. 6a)37. We first recorded from the PVGPe-SNr and the PVGPe-PF neurons in acute brain slices and found that dopamine depletion had no effect on the autonomous firing rate or the ratio of excitatory and inhibitory synaptic inputs (E/I ratio) of both subpopulations (Fig. 6b–e). We subsequently examined whether synaptic outputs at the GPe-SNr and the GPe-PF synapses were altered. We replaced the extracellular Ca2+ with Sr2+ and photostimulated the GPe-PV axons while recording from SNr and PF neurons to measure quantal-like inhibitory post-synaptic currents (qIPSCs) from the simulated synapses (Fig. 6f,g)39. Interestingly, the frequency of qIPSCs significantly decreased in SNr neurons but markedly increased in PF neurons. On the contrary, dopamine depletion did not show any major effect on the amplitude of qIPSCs measured from both the SNr and PF neurons (Fig. 6h,i). Additionally, the paired-pulse ratios (PPRs) of IPSCs, measured while photostimulating the GPe-PV axons, were increased in the SNr neurons and decreased in the PF neurons (Fig. 6j,k). Taken together, our results indicate that dopamine depletion affects the synaptic outputs of the PVGPe-SNr and the PVGPe-PF neurons by differentially altering the probability of pre-synaptic neurotransmitter release.
Fig. 6 |. PVGPe-SNr and PVGPe-PF neurons exhibit distinct electrophysiological adaptations to dopamine depletion.
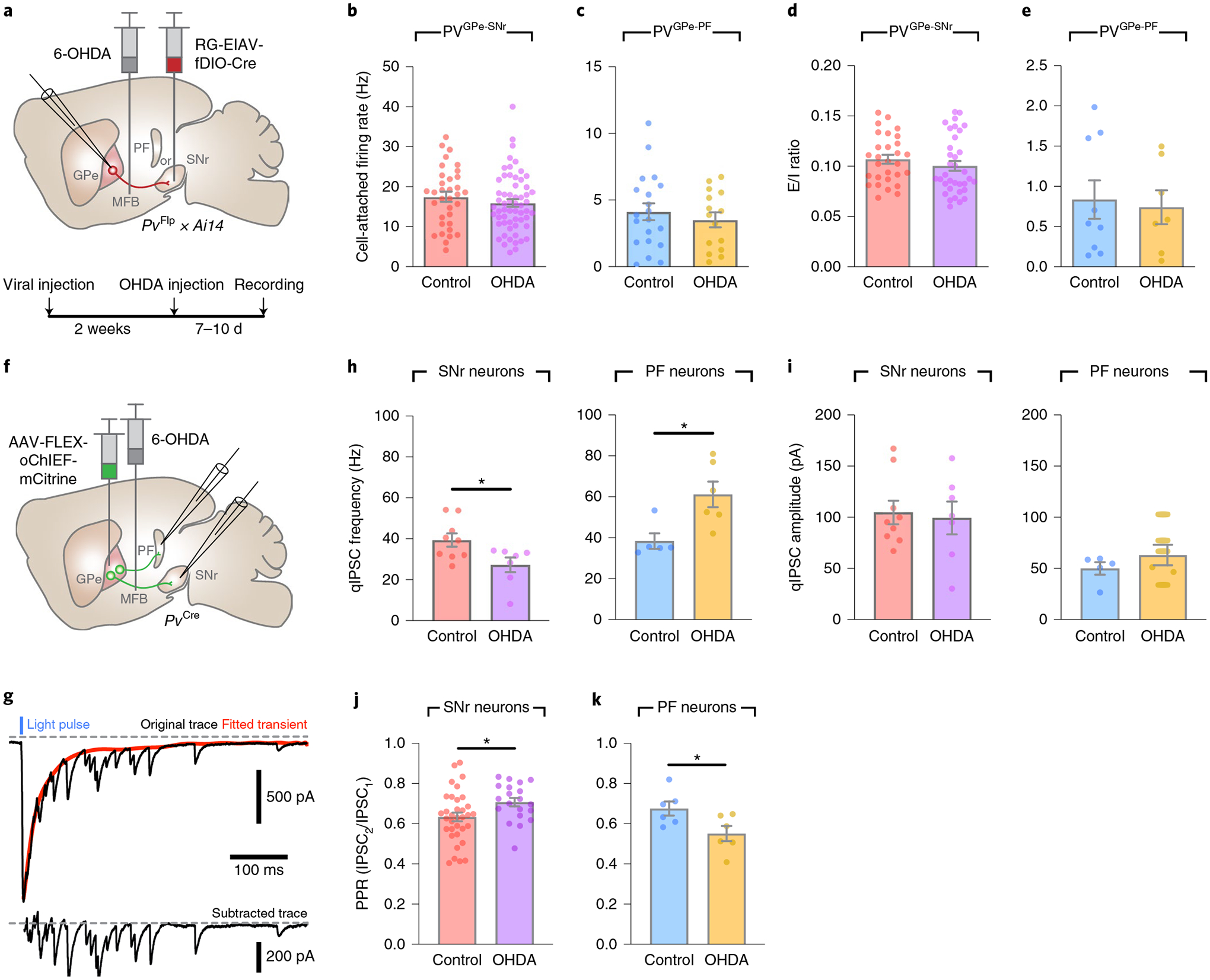
a, Schematic of viral and 6-OHDA injections and the experimental timeline for the recording of PVGPe-SNr and PVGPe-PF neurons in acute slices after dopamine depletion. b,c, Autonomous firing rate in PVGPe-SNr neurons (b; control n = 34 cells from nine mice, OHDA n = 59 cells from 12 mice) and PVGPe-PF neurons (c; control n = 20 cells from eight mice, OHDA n = 15 cells from seven mice) recorded in cell-attached configuration in the presence of synaptic transmission blockers, NBQX and PTX in extracellular solution. Mann–Whitney U test, U = 849, P = 0.2213 in b and U = 139, P = 0.7297 in c. d,e, E/I ratio onto PVGPe-SNr neurons (d; control n = 29, OHDA n = 35 cells) and PVGPe-PF neurons (e; control n = 7, OHDA n = 9 cells) from naive control and dopamine-depleted mice. Mann–Whitney U test, U = 419, P = 0.2371 in d and U = 29, P = 0.8371 in e. f, Schematic of viral and 6-OHDA injections for measuring qIPSCs in acute slices after dopamine depletion while photostimulating PVGPe-SNr and PVGPe-PF axons. g, Example trace (top) showing qIPSCs in SNr neurons elicited by photostimulation of GPe-PV terminals. red trace represents a fitted curve generated by Python script. qIPSC amplitudes were measured from subtracted trace (bottom) from 0–400 ms after stimulation. h, Frequency of optically evoked qIPSCs was decreased and increased in when recorded from SNr and PF neurons, respectively. Unpaired t-test, t(14) = 2.485, *P = 0.0262 for SNr and t(9) = 2.947, *P = 0.0163 for PF (SNr control n = 9 cells from three mice, SNr OHDA n = 7 cells from three mice; PF control n = 5 cells from two mice, PF OHDA n = 6 cells from two mice). i, Amplitudes of optically evoked qIPSCs in SNr and PF neurons were not altered by dopamine depletion. Unpaired t-test, t(14) = 0.2773, P = 0.7856 for SNr and t(9) = 1.052, *P = 0.3204 for PF (SNr control n = 9 cells, SNr OHDA n = 7 cells; PF control n = 5 cells, PF OHDA n = 6 cells). j,k, PPr (second IPSC peak/first IPSC peak amplitude) measured from SNr neurons (j; control n = 34 cells from eight mice, OHDA n = 20 cells from six mice) and PF neurons (k; control n = 6 cells from three mice, OHDA n = 6 cells from four mice) of naive control and dopamine-depleted mice. Unpaired t-test, t(52) = 2.244, *P = 0.0291 in j and t(10) = 2.418, *P = 0.0362 in k. All data are presented as mean ± s.e.m.
Roles of GPe-PV neurons in Parkinsonian behavioral deficits.
The selective modulation of motor and cognitive behaviors by PVGPe-SNr and PVGPe-PF neurons and their synaptic changes after dopamine depletion suggests that changes to these circuits underlie different Parkinsonian deficits. Our results strongly suggest that reduced synaptic output of the PVGPe-SNr neurons might lead to decreased locomotor activity. We reasoned that artificially enhancing the activity of the PVGPe-SNr neurons in dopamine-depleted mice might reverse locomotive deficits. In line with previously published studies, we observed a significant reduction in locomotion when more than 70% of nigrostriatal dopaminergic fibers were lesioned (Fig. 7a–d and Extended Data Fig. 7)40. Interestingly, photostimulation of the PVGPe-SNr neurons in dopamine-depleted mice induced a robust increase in locomotion that persisted beyond the stimulation period (Fig. 7b–d), suggesting that reduced GPe-SNr synaptic output might indeed contribute to immobility in PD.
Fig. 7 |. PVGPe-SNr and PVGPe-PF neurons mediate different behavioral deficits in dopamine-depleted mice.
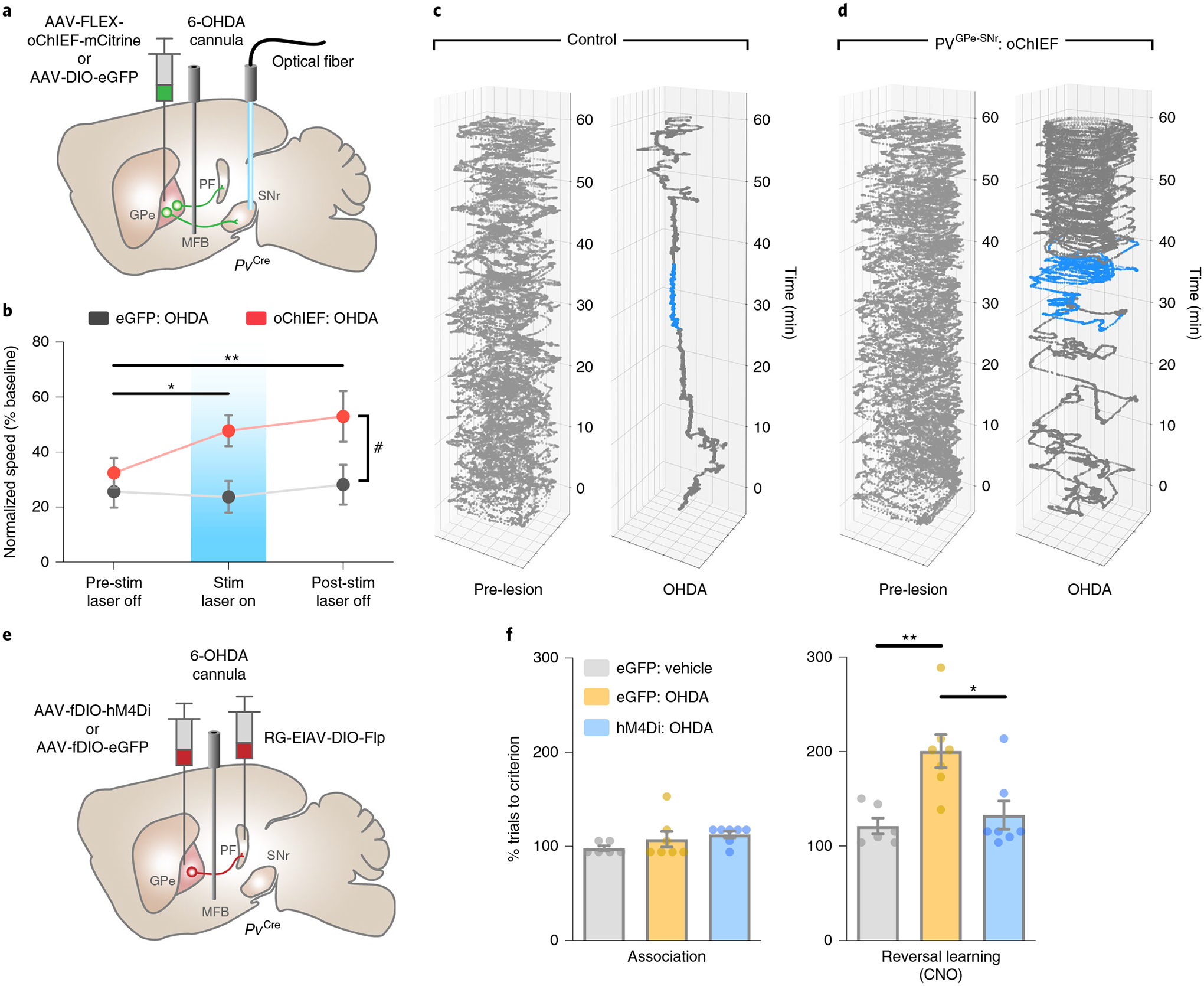
a, Schematic of viral injections, optical fiber and drug cannula implantations for optogenetic activation during locomotion in dopamine-depleted mice. b, Activation of PVGPe-SNr axons restored locomotor activity during stimulation and post-stimulation periods in dopamine-depleted mice. Two-way repeated-measures ANOVA, main effectstim×group: F2,38 = 2.691, P = 0.0807; main effectstim: F2,38 = 3.460, P = 0.0417; main effectgroup: F1,19 = 5.436, P = 0.0309; Bonferroni’s post hoc test, *P = 0.0476 for oChIEF pre-stim laser off versus oChIEF stim laser on, **P = 0.0050 for oChIEF pre-stim laser off versus oChIEF post-stim laser off, #P = 0.0409 for eGFP stim laser on versus oChIEF stim laser on and #P = 0.0327 for eGFP post-stim laser off versus oChIEF post-stim laser off (eGFP n = 10 mice, oChIEF n = 11 mice). c, Position of a control mouse in an open field over 60 min before (left) and after (right) dopamine depletion. The mouse received bilateral photostimulation in the SNr at 30–40 min after session start (blue line). d, Same as in b, but for a mouse with oChIEF expression in GPe-PV neurons. e, Schematic of viral injections and drug cannula implantations for cell-type- and projection-specific expression of inhibitory DrEADD (hM4Di) in PVGPe-PF neurons. f, Performance of mice that received chemogenetic inhibition in PVGPe-PF neurons after dopamine depletion. Mice in all groups received CNO injections (5 mg kg−1) in the reversal learning phase (n = 7 mice for eGFP-vehicle, n = 7 mice for eGFP-OHDA and n = 7 mice for hM4Di-OHDA). Left: dopamine depletion did not affect performance in the association phase. right: inhibition of PVGPe-PF neurons during reversal learning improved behavioral flexibility in dopamine-depleted mice. One-way ANOVA, F2,17 = 8.642, P = 0.0026; Bonferroni’s post hoc test, *P = 0.0114 and **P = 0.0046. All data are presented as mean ± s.e.m.
Besides displaying motor deficits, patients with PD and Parkinsonian animal models display cognitive flexibility impairments in early stages of the disease38,41,42. Because the PVGPe-PF neurons showed increased synaptic output after dopamine depletion, we hypothesized that inactivating these neurons might rescue behavioral inflexibility. As a first step toward testing this hypothesis, we induced a mild dopamine depletion by a bilateral infusion of low-dose 6-OHDA into the medial forebrain bundle (MFB) to mimic the early stage of dopamine loss. This treatment lesioned approximately 50% of the dopaminergic fibers in the striatum while preserving overall locomotor function of the mice (Extended Data Fig. 7b,d). This allowed us to assess the performance in the reversal learning task. We found that the formation of the context–outcome association was not affected by dopamine depletion, whereas reversal learning was profoundly impaired. The mice that received the treatment struggled to maintain a new choice pattern after initially shifting away from the previous correct choice, as reflected by an increase in regressive errors (Extended Data Fig. 8a). In a separate group of mice, we suppressed the activity of the PVGPe-PF neurons during the reversal learning phase with chemogenetic inactivation. We selectively expressed hM4Di in the PVGPe-PF neurons and administered CNO during the reversal learning phase (Fig. 7e). Remarkably, the CNO treatment significantly improved reversal learning performance after mild dopamine depletion (Fig. 7f and Extended Data Fig. 8a). By contrast, optogenetic activation of the PVGPe-SNr neurons after mild dopamine depletion did not show any effect on reversal learning (Extended Data Fig. 8b,c). Collectively, these experiments revealed that each of the two subpopulations underwent specific adaptation in response to dopamine loss, which contributed to a separate behavioral deficit observed at a different stage of dopamine depletion.
Discussion
Current understanding of the circuit-specific roles of GPe-PV neurons is hindered by the lack of knowledge of their anatomical and functional organization. Using complementary electrophysiology, viral-mediated tracing and behavioral approaches, we demonstrate that distinct PV neuronal projections from the GPe are selectively involved in discrete basal ganglia-related behaviors. We further show that the physiological adaptation of each circuit to dopamine loss might contribute to different behavioral deficits and that manipulation of these circuits restores motor and cognitive function in a Parkinsonian mouse model.
Diversity of GPe neurons and their distinct roles in behavioral deficits.
Previous studies have focused mainly on how the basal ganglia output controls motor functions through the thalamocortical loop and motor-control nuclei in the midbrain, whereas fewer studies have focused on the functional role of the direct output from the GPe to the thalamic nuclei. Using viral-mediated tracing, we were able to differentiate GPe-PV populations based on their projections to either SNr or PF. These two subpopulations display topographical preference within the GPe, with the PVGPe-PF neurons located more medial than the PVGPe-SNr neurons. In contrast to classical models of the basal ganglia, we found that the PVGPe-PF and PVGPe-SNr projection neurons are preferentially innervated by D1R-SPNs and D2R-SPNs, respectively. These discrete input and output organizations suggest that GPe subpopulations signal within distinct information streams. Specifically, we confirmed, using a reversal learning paradigm, that the GPe-PF pathway is selectively involved in behavioral flexibility. Our results suggest that, although the activity of basal ganglia circuitry is associated with a wide range of behaviors, each microcircuit within the basal ganglia might control unique behaviors. However, few studies have elucidated the contribution of distinct microcircuitry to different behaviors associated with basal ganglia. Consistent with this notion, a recent study demonstrated that the striatum can be subdivided into more than 20 different regions based on the anatomical organization of input and outputs43, which further emphasizes the need for investigation of the role of individual microcircuits in the basal ganglia. It has been suggested that DMS and DLS preferentially regulate cognitive function and motor control, respectively, but their downstream pathways are not clearly delineated. Here we provide the evidence that PVGPe-PF neurons might be one of the major downstream pathways of DMS contributing to the cognitive functions. Our findings provide a novel anatomical and functional framework of GPe to understand the role of different basal ganglia circuitries mediating distinct behavioral functions.
The engagement of distinct basal ganglia circuitry in different PD states.
Recent studies showed that prolonged optogenetic stimulation of the GPe-PV neurons can restore spontaneous movement in dopamine-depleted mice17. Although these studies demonstrated the role of GPe-PV neurons in motor function, we were able to further isolate the GPe-SNr pathway as a key mediator in locomotor deficits and show that activation of this microcircuit was sufficient to restore movement. Notably, the behavioral relevance of the GPe-PF projection in PD has not been previously addressed. Strong innervation of the PF by the GPe-PV neurons suggests that these neurons could play a role in regulating the thalamostriatal pathway12, which is involved in behavioral flexibility and motor control.
The progression of PD is commonly evaluated by the severity of motor symptoms; however, non-motor symptoms, such as cognitive impairments, are also common and often appear in an early phase of the disease42,44. It is plausible that the distinct basal ganglia subcircuits are involved in the distinct symptoms of PD-related behavioral changes during the progression of this disease. Nevertheless, the potential involvement of different neural circuits in the development of both motor and non-motor deficits of PD is poorly understood, mainly because of the lack of knowledge in the anatomical and functional organization of circuits underlying different behavioral impairments. We demonstrated that, in 6-OHDA-injected animals, dopamine depletion decreased the synaptic efficacy of the PVGPe-SNr microcircuit but increased the synaptic activity of the PVGPe-PF microcircuit. By artificially compensating for these changes in synaptic transmission using optogenetic and chemogenetic manipulations, we were able to separately rescue locomotor deficits and behavioral inflexibility in animal models of the varying degrees of dopamine and neuronal loss found in PD. Our results indicate that differences in how these GPe-PV subpopulations adapt to decreasing levels of dopamine might underlie the progressive nature of PD. Indeed, several studies reported anti-Parkinsonian and anti-dyskinetic effects of DBS in the PF of human patients, further supporting the idea that GPe-PF connections might have important roles in the development of PD45,46. Importantly, we also found that the GPe-PF pathway is critical for maintaining a new action–outcome association during reversal learning. By manipulating the activity of the GPe-PF microcircuit, future treatments might also be able to ameliorate the behavioral inflexibility of PD.
Our findings establish the differential contributions from two distinct GPe-PV microcircuits in specific Parkinsonian-like behaviors linked to early and late stages of the disease. Our results suggest that further elucidation of the detailed connectivity of GPe subpopulations to their downstream targets, including PF, STN and SNr, is needed to fully define the function of each microcircuit and design better therapeutic strategies for the various behavioral impairments of PD.
Methods
Animals.
All procedures to maintain and use mice were approved by the Institutional Animal Care and Use Committee at the University of California, San Diego. Mice were maintained on a 12-hour light/dark cycle with regular mouse chow and water available ad libitum except when placed under food restriction. All behavioral experiments were performed during the dark cycle. PvCre, Ai14 (Rosa26-CAG-LSL-tdTomato) and PvFlp transgenic mice were obtained from the Jackson Laboratory (JAX strain nos. 008069, 007914 and 22730, respectively) and were maintained on a C57BL/6J background. PvFlp × Ai14 transgenic mice were generated by crossing PvFlp mice with Ai14 mice. For all experiments, male and female heterozygous mice, aged 8–20 weeks, were randomly picked and used. Not all behavioral experiments were performed blinded, but most behavioral experiments were confirmed by the repeated experiments performed and analyzed blinded without knowledge of the treatment history of the animals.
Viral vectors.
AAV plasmids were constructed using standard molecular cloning methods. Synaptophysin-eGFP, oPBG, hM4D(Gi)-mCherry, axon-GCaMP6s-P2A-mRuby3 and TVA DNA fragments were obtained from pAAV-phSyn1(S)-FLEX-tdTomato-T2A-SypEGFP-WPRE (a gift from Hongkui Zeng; Addgene plasmid no. 51509), pAAV-hSyn-DIO-hM4D(Gi)-mCherry (a gift from Bryan Roth; Addgene plasmid no. 44362), pAAV-EF1α-DIO-oPBG (a gift from Edward M. Callaway), pAAV-hSynapsin1-axon-GCaMP6s-P2A-mRuby3 (a gift from Lin Tian; Addgene plasmid no. 112005) and pAAV-EF1α-FLEX-GTB (a gift from Edward M. Callaway; Addgene plasmid no. 26197), respectively. pAAV-FLEX-oChIEF-mCitrine was a gift from Roger Tsien (Addgen plasmid no. 50973). A Flp-dependent, double-floxed, inverted open reading frame (fDIO) was constructed with two heterospecific pairs of FRT and FRT5 sequences based on pAAV-EF1α-fDIO-hChR2(H134R)-eYFP (a gift from Karl Deisseroth; Addgene plasmid no. 55639). We used EF1α promoter to drive the expression of target constructs for all AAV vectors, except for AAV-DIO-mRuby2-T2A-Synaptophysin-eGFP, AAV-fDIO-hM4D(Gi)-mCherry, AAV-DIO-axon-GCaMP6s-P2A-mRuby3 and AAV-FLEX-oChIEF-mCitrine, which are driven by the human synapsin1 promoter.
All AAV vectors used in this study were packaged as serotype DJ and generated as previously described45. In brief, AAV vectors were produced by transfection of AAV293 cells (Agilent) with three plasmids: an AAV vector plasmid carrying target constructs (DIO-mRuby2-T2A-Synaptophysin-eGFP, DIO-eGFP, fDIO-eGFP, fDIO-oPBG, fDIO-mRuby2-P2A-TVA, fDIO-hM4D(Gi)-mCherry or FLEX-oChIEF-mCitrine), AAV helper plasmid (pHELPER, Agilent) and AAV rep-cap helper plasmid (pRC-DJ, a gift from M. Kay). At 72 h after transfection, the cells were collected and lysed by a repeated freeze–thaw procedure. Viral particles were then purified by an iodixanol step-gradient ultracentrifugation and subsequently concentrated using a 100-kDa molecular cutoff ultrafiltration device (Millipore). The genomic titer was determined by quantitative polymerase chain reaction. The AAV vectors were diluted in PBS to a working concentration of approximately 1013 viral particles per ml.
EIAV genomic vector plasmids were constructed from pEIAV-SIN6.1-CBGFPW (a gift from John Olsen; Addgene plasmid no. 44173) by replacing eGFP coding sequence with DNA fragments containing either DIO-FlpO or fDIO-Cre. RG-EIAV vectors were generated by a modified version of a published protocol24. Briefly, HEK293T cells were transfected with three plasmids: an EIAV genomic vector (pEIAV-CAG-DIO-Flp or pEIAV-CAG-fDIO-Cre), a helper packaging plasmid (pEV53B; a gift from John Olsen) and a pseudotyping plasmid-encoding fusion protein, FuG-B2, an envelope protein carrying extracellular and transmembrane domain of rabies virus glycoprotein and conjugated to the cytoplasmic domain of vesicular stomatitis virus (a gift from Kazuto Kobayashi)46. At 72 h after transfection, viral particles were harvested from the media by centrifugation using a SureSpin 630 swinging bucket rotor (Thermo Fisher Scientific) at 5,700 r.p.m. and 16,200 r.p.m. for 16 h and 2 h, respectively. EIAV viral particles were reconstituted from the pellets with PBS and immediately stored at −80 °C.
Rabies virus was generated as previously described47 from a full-length complementary DNA (cDNA) plasmid containing all components of the virus except the coding sequence of the rabies glycoprotein, which was replaced with eGFP (a gift from Karl-Klaus Conzelmann). In brief, B7GG cells were transfected with a total of five plasmids: four plasmids expressing the viral components pcDNA-SADB16N, pcDNA-SADB16P, pcDNA-SADB16L and pcDNA-SADB16G and the rabies virus genomic vector. The virus-containing media was collected 3–4 d after transfection and used for further amplification. Viral particles were harvested from the media by centrifugation using a SureSpin 630 rotor at 20,000 r.p.m. for 2 h. Rabies viral particles were reconstituted from the pellets with PBS and immediately stored at −80 °C. To generate EnvA-pseudotyped, glycoprotein-deleted rabies virus expressing eGFP (EnvA-RVΔG-eGFP), we used a modified version of a published protocol48. BHK-EnvA cells were grown in four 15-cm dishes to 50–60% confluency. The cells were transduced with the rabies virus expressing eGFP generated in the previous step. After 3 h, the media was removed, and the cells were rinsed with PBS, trypsinized for 5 min at 37 °C and isolated by pipetting repeatedly with a P1000 pipette. Each dish was observed carefully under the microscope to ensure that the cells did not aggregate. The cells were then pelleted by centrifugation and plated in four new dishes. Trypsinization was repeated two more times at 9 h and 21 h after transduction to ensure complete removal of the original rabies glycoprotein. The virus-containing media was collected 4 d after transduction and concentrated as described above. Plasmids expressing the rabies viral components and B7GG, BHK-EnvA and HEK-TVA cells were gifts from Edward M. Callaway. All cells used were identified as mycoplasma negative (tested by MycoAlert LT07-118, Lonza).
Stereotaxic viral injections and optic fiber/cannula implantation.
Mice were anesthetized with a mixture of ketamine (100 mg kg−1) and dexmedetomidine (1 mg kg−1) and placed on a stereotaxic frame (David Kopf Instruments). Body temperature was maintained with a heating pad during surgery and recovery from anesthesia. All stereotactic coordinates were derived from the Paxinos and Franklin Mouse Brain Atlas. Viruses were infused into the brain by using pulled glass micropipettes coupled with a syringe pump (PHD ULTRA, Harvard Apparatus) at a rate of 100 nl min−1. For the tracing experiment to visualize output regions of GPe PV neurons, 150 nl of AAV-FLEX-mRuby2-T2A-Synaptophysin-eGFP was injected into the GPe of PvCre mice (anteroposterior −0.35 mm, mediolateral 1.95 mm from bregma and depth −3.5 mm from brain surface). For cell-type- and projection-specific tracing, 500 nl of RG-EIAV-DIO-Flp was unilaterally injected into either SNr (anteroposterior −3.25 mm, mediolateral 1.6 mm from bregma and depth −4.4 mm from brain surface) or PF (anteroposterior −2.15 mm, mediolateral 0.625 mm from bregma and depth −3.1 mm from brain surface) of PvCre mice along with 350 nl of AAV-fDIO-eGFP into ipsilateral GPe. After allowing 3 weeks of expression, mice were euthanized for circuit mapping analysis.
For mapping of input–output relationship, 500 nl of RG-EIAV-DIO-Flp was unilaterally injected into either the SNr or PF of PvCre mice along with 350 nl of a 1:1 mixture of AAV-fDIO-oPBG and AAV-fDIO-mRuby2-P2A-TVA into ipsilateral GPe. After allowing 3 weeks of expression, animals were again anesthetized as previously described, and 350 nl of RVΔG-eGFP(EnvA) was injected into the ipsilateral GPe. Mice were euthanized after 7 d for quantification of input neurons and in situ hybridization.
For electrophysiological recordings of GPe neurons, 500 nl of RG-EIAV-fDIO-Cre was unilaterally injected into either the SNr or PF of PvFlp × Ai14 mice. For recordings of post-synaptic neurons in the SNr and PF, 350 nl of AAV-FLEX-oChIEF-mCitrine was unilaterally injected into the GPe of PvCre mice. After allowing 3 weeks of expression, acute slices were prepared for recordings.
For optogenetic behavioral experiments, PvCre mice were bilaterally injected with 350 nl of AAV-FLEX-oChIEF-mCitrine (or AAV-DIO-eGFP for controls) into the GPe. Subsequently, a bilateral 26-gauge guide cannula (cut length 4.5 mm, center-to-center distance 2 mm, Plastic One) was implanted over the MFB (anteroposterior −0.5 mm, mediolateral 1.0 mm from bregma) for 6-hydroxydopamine infusion and was temporally secured in place with cyanoacrylate glue. Optic fibers (200 μm, 0.22 NA, Doric Lenses) were cut to length (4.5 mm for SNr and 3.5 mm for PF) and implanted bilaterally above the SNr (depth 4.0 mm from brain surface) or PF (mediolateral angle 15°, anteroposterior −2.15 mm, mediolateral −1.65 from bregma, depth −3.0 mm from brain surface) during the same surgery session. The optic fibers and the guide cannula were secured in place with a layer of adhesive cement (Radiopaque L-powder for C&B METABOND, Parkell). Once dried, absorbable sutures and a second layer of dental cement (Ortho-Jet, Lang Dental) were used to seal the head incision. For chemogenetic behavioral experiments, 500 nl of RG-EIAV-DIO-Flp was bilaterally injected into either the SNr or PF of PvCre mice along with 350 nl AAV-fDIO-hM4D(Gi)-mCherry (or AAV-fDIO-eGFP for controls) into the GPe. During the same surgery session, a bilateral guide cannula was implanted over the MFB as described above. Behavioral experiments were performed 4 weeks after surgery to allow time for recovery and optimal viral expression.
For fiber photometry, PvCre mice were unilaterally injected with 350 nl of AAV-DIO-axon-GCaMP6s-P2A-mRuby3 into the GPe. Optic fibers (400 μm, 0.48 NA, Doric Lenses) were cut to length (4.5 mm for SNr and 3.5 mm for PF) and unilaterally implanted above the ipsilateral SNr (depth 4.0 mm from brain surface) and PF in conjunction with a head bar during the same surgery session, as described above. Behavioral experiments and recordings were performed 3 weeks after surgery to allow time for recovery and optimal viral expression.
Upon completion of behavioral experiments, viral injections and optic fiber placements were confirmed with histology to ensure proper targeting (Extended Data Figs. 2a,b and 3a,b).
Dopamine depletion.
6-OHDA solution was freshly prepared by adding 6-OHDA HCl (Sigma-Aldrich) to an appropriate volume of sterile saline (0.9% NaCl) with 0.02% sodium ascorbate (Sigma-Aldrich) to achieve the desired concentration. To prevent lesioning of non-dopamine monoaminergic neurons, desipramine (25 mg kg−1) was injected intraperitoneally 30 min before 6-OHDA injection48.
6-OHDA injection for electrophysiological recordings was performed with the same method mentioned above 2 weeks after viral injection. Mice were anesthetized with isoflurane (2% in O2), and 750 nl of 6-OHDA solution (2.5 μg μl−1) was unilaterally injected into the MFB (anteroposterior −0.5 mm, mediolateral 1.0 mm from bregma, depth −4.8 mm from brain surface) at a rate of 150 nl min−1. For behavioral experiments with dopamine-depleted animals, mice were bilaterally injected with viral vectors and implated with guide cannula over medial forebrain bundle (MFB; 4.8 mm from brain surface). Two to 3 weeks after the surgery, a bilaeral injector (33 gauge, 5 mm; Plastic One) attached to a syringe pump (PHD ULTRA, Harvard Apparatus) was lowered into the guide cannula for OHDA infusion while mice were anesthetized with isoflurane (2% in O2). Then, 750 nl of 6-OHDA solution (high dose, 2.5 μg μl−1 for locomotion; low dose, 1.25 μg μl−1 for reversal learning task) was bilaterally infused into the MFB at a rate of 150 nl min−1. Sham-operated control mice underwent similar procedures and were injected with vehicle (saline with 0.02% sodium ascorbate) instead of 6-OHDA. After infusion, the body mass of each animal was carefully monitored, and a nutritionally fortified water gel (DietGel Recovery, ClearH2O) was provided in conjunction with a shallow water dish and moist mouse chow to aid recovery. Recordings and locomotion measurements were performed 7–10 d after 6-OHDA infusion. Reversal learning task was performed 3 d after 6-OHDA infusion.
SHIELD-MAP tissue processing.
PvCre mice injected with AAV-FLEX-mRub y2-T2A-Synaptophysin-eGFP were processed for SHIELD-MAP as previously described49. In brief, mice were deeply anesthetized with isoflurane and transcardially perfused with 10 ml of ice-cold PBS followed by 20 ml of freshly made ice-cold SHIELD perfusion solution. The brains were carefully extracted and post-fixed in the same solution at 4 °C for 48 h. The brains were cut parasagittally into 3-mm blocks containing the GPe, SNr and PF and transferred to a new conical tube with 20 ml of ice-cold SHIELD Off solution and incubated at 4 °C for 24 h. Brain blocks were then incubated in SHIELD On solution at 37 °C for 24 h and washed in PBS containing 0.02% sodium azide (Sigma-Aldrich) overnight at room temperature. Subsequently, the samples were cleared with SDS clearing buffer for 10 d at 45 °C while shaking and then washed overnight at room temperature with PBS containing 1% (vol/vol) Triton-X 100 (Sigma-Aldrich) and then with PBS. The samples were then incubated in monomer solution at 4 °C for 3 d and were gel-embedded under nitrogen gas purge at 33 °C for 4 h. The samples were rehydrated in PBS for several hours and transferred to deionized water for tissue expansion. A light-sheet microscope (SmartSPIM, LifeCanvas Technologies) with a ×10, 0.6 NA objective (Olympus) was used to obtain images from expanded samples. Formulas for all solutions and a detailed protocol can be found at http://chunglabresources.com/.
Histology.
After the designated time for viral expression in tracing experiments or immediately after behavioral experiments, mice were deeply anesthetized with isoflurane and transcardially perfused with ice-cold 4% paraformaldehyde in PBS. Brains were carefully extracted and post-fixed in the same fixative at 4 °C for at least 12 h. Brains were sliced at 60-μm thickness with a vibratome (VT1000, Leica). Brain sections were mounted on glass slides (SuperFrost Plus, Thermo Fisher Scientific) or collected and stored in cryoprotectant (30% ethylene glycol, 30% glycerol in PBS) at −20 °C for immunohistochemistry. Mounted slides were coverslipped with DAPI Fluoromount-G (Southern Biotech) and imaged with a ×10 objective on an Olympus VS120 virtual slide microscope.
Tyrosine hydroxylase immunoreactivity.
Quantification of striatal tyrosine hydroxylase (TH) immunoreactivity was used to assess the degree of dopamine depletion. Immunohistochemistry was carried out in free-floating fixed brain sections containing the dorsal striatum. The sections were washed with PBS for three times (10 min each) and incubated at room temperature for 1 h in blocking solution containing 10% normal horse serum (Abcam), 0.2% BSA (Sigma-Aldrich) and 0.5% Triton-X 100 (Sigma-Aldrich) in PBS. The sections were then incubated at 4 °C for 20 h in carrier solution containing 1% normal horse serum, 0.2% BSA and 0.5% Triton-X 100 in PBS with anti-TH primary antibody (1:2,000, Millipore AB152). The sections were washed in PBS for three times and incubated in carrier solution containing Alexa Fluor 647-conjugated donkey anti-rabbit secondary antibody (1:1,000, Life Technologies A-31573) at room temperature for 2 h. Finally, the sections were washed two times in PBS, mounted, coverslipped and imaged as described above. Each slide always included sections from naive control animals that were processed and imaged in parallel to use as a reference. We used the pixel intensity measuring tool in Fiji (ImageJ) to analyze fluorescence intensity from a 500 × 500-μm area taken from both hemispheres of each section. The fluorescence intensity for each section was normalized to the intensity of the naive control tissue from the same slide. For locomotion measurements, all data included in the analysis were from mice with <35% TH immunoreactivity. For the reversal learning task, all data included in analysis were from mice with <70% TH immunoreactivity (Extended Data Fig. 7).
mFISH.
Mice that underwent surgeries for mapping of the input–output relationship were deeply anesthetized with isoflurane and transcardially perfused with ice-cold PBS. The brains were extracted, submerged in embedding medium (Tissue-Tek O.C.T., Sakura) and frozen with 2-methylbutane (Sigma-Aldrich) chilled with dry ice in 70% ethanol. The frozen brain blocks were stored at −20 °C for at least 1 d and then sliced with a cryostat microtome (Thermo Fisher Scientific) to obtain 20-μm coronal sections. For each brain, eight sections regularly sampled across the entire dorsal striatum were mounted on SuperFrost Plus slides (Thermo Fisher Scientific) and processed exactly as described in the RNAscope assay online protocol (Advanced Cell Diagnostics). We used a probe against eGFP mRNA to label striatal neurons infected with EnvA-RVΔG-eGFP that spread trans-synaptically from either PVGPe-SNr or PVGPe-PF starter cells. Probes against Drd1a and Drd2 mRNAs were used to determine whether the labeled striatal neurons were direct pathway spiny projection neurons (D1R-SPNs) or indirect pathway SPNs (D2R-SPNs). Probes against Pvalb, Sst, Lhx6, Scn4b, Nkx2-1 and Foxp2 mRNA were used for labeling GPe neurons. All probes were purchased from Advanced Cell Diagnostics. Because the labeling of eGFP mRNA was the strongest, we assigned the Atto-647 secondary probe to the eGFP probe to prevent bleed-through of the fluorescent signal into neighboring channels during imaging. Images of the entire dorsal striatum and GPe were acquired through a ×30 silicone-immersion objective on a confocal microscope (FluoView FV1200, Olympus). All eGFP-labeled neurons were manually quantified.
Quantification of input–output relationship.
All sections except the olfactory bulb and the cerebellum were collected and processed as described in the ‘Histology’ section. All eGFP-labeled neurons from all brain regions except the GPe (injection site of the rabies virus) were quantified and presented as a percentage of total eGFP-labeled neurons from each brain. Brain regions were assigned based on the Paxinos and Franklin Mouse Brain Atlas. Regions with <1% of total eGFP-labeled neurons were not included in the summary plot in Fig. 2.
Ex vivo electrophysiology.
Mice were anesthetized with isoflurane and transcardially perfused with ice-cold choline-based slicing solution containing (in mM): 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 7 MgCl2, 25 glucose, 0.5 CaCl2, 110 choline chloride, 11.6 sodium ascorbate and 3.1 sodium pyruvate. Brains were carefully extracted and transferred to a chamber filled with the same solution on a vibratome (VT1200, Leica). Brains were sliced at 250 μm (coronally for recording of SNr and PF neurons, parasagittally for PVGPe-SNr neurons and horizontally for PVGPe-PF neurons) and incubated at 35 °C for 15–20 min in recovery solution containing (in mM): 118 NaCl, 2.6 NaHCO3, 11 glucose, 15 HEPES, 2.5 KCl, 1.25 NaH2PO4, 2 sodium pyruvate, 0.4 sodium ascorbate, 2 CaCl2 and 1 MgCl2. Slices were maintained at room temperature for at least 1 h until transferred to a recording chamber an Olympus BX51WI upright microscope. The chamber was continuously superfused with artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 11 glucose, 1.3 MgCl2 and 2.5 CaCl2, maintained at 30 ± 2 °C by a feedback temperature controller. Slicing solution, recovery solution and ACSF were constantly bubbled with 95% O2 and 5% CO2. All compounds were purchased from Tocris or Sigma-Aldrich.
For all recordings, patch pipettes (3–5 MΩ) were pulled from borosilicate glass (G150TF-4, Warner Instruments) with a DMZ Universal Electrode Puller (Zeitz Instruments) and filled with appropriate intracellular solutions. Liquid junction potential was not corrected for any experiments. Neurons were visualized with differential interference contrast optics or epifluorescence (Olympus). Recordings were made with a MultiClamp 700B amplifier and pClamp10 software (Molecular Devices). Data were low-pass filtered at 1 kHz and digitized at 10 kHz with a digitizer (Digidata 1440, Molecular Devices). Series resistance was monitored, and cells that displayed >20% change over the duration of recording were excluded.
For current-clamp recording, pipettes were filled with an intracellular solution containing (in mM): 125 K+-gluconate, 4 NaCl, 10 HEPES, 0.5 EGTA, 20 KCl, 4 Mg2+-ATP, 0.3 Na+-GTP and 10 Na2-phosphocreatine (290–300 mOsm, pH 7.2). Autonomous firing activity was acquired in a cell-attached configuration for 1 min before break-in. To measure the firing capacity of identified PVGPe-SNr and PVGPe-PF neurons, baseline firing was maintained at 5 Hz, and currents of increasing intensity (20-pA increments, duration = 500 ms) were injected until neurons went into depolarization block. All current-clamp recordings were performed in the presence of 5 μM NBQX and 50 μM picrotoxin (PTX) to block synaptic transmission. Detection of spikes and measurements of action potential characteristics were performed using a custom Python script with the following criteria. Action potential threshold was measured as a change in voltage from rest at which the slope = 20 mV ms−1. Peak amplitude was measured as a change in voltage from action potential threshold to the peak of action potential, and after-hyperpolarization was measured as a change in voltage and time from threshold to minimum after the peak. Half-width was calculated as full-width at half-maximum amplitude. All measurements were quantified while neurons were firing at 5 Hz.
Passive membrane properties were calculated from voltage-clamp recordings with a custom Python script based on the previously described method10. Membrane resistance (Rm) was calculated by Rm = ΔVtest/ΔI where ΔVtest is a 10-mV step (50 ms), and ΔI is the difference between steady-state current and baseline during the last 10 ms of the voltage step. Membrane capacitance (Cm) was calculated by Cm = Qt * ΔVtest where Qt is the integral of the transient current elicited by ΔVtest, a 10-mV voltage step (50 ms).
To measure the E/I ratio, pipettes were filled with a Cs-based intracellular solution containing (in mM): 115 Cs+-methanesulphonate, 10 HEPES, 1 EGTA, 1.5 MgCl2, 4 Mg2+-ATP, 0.3 Na+-GTP, 10 Na2-phosphocreatine, 2 QX 314-Cl and 10 BAPTA-tetracesium (295 mOsm, pH 7.35). Electrically evoked excitatory post-synaptic currents and inhibitory post-synaptic currents (EPSCs and IPSCs, respectively) were recorded from identified PVGPe-SNr and PVGPe-PF neurons at −60 mV (for EPSCs) and 0 mV (for IPSCs). The E/I ratio was calculated by dividing the amplitude of EPSCs by the amplitude of IPSCs. Spontaneous EPSCs and IPSCs were also recorded at −60 mV and 0 mV, respectively.
For the recording of Sr2+-induced asynchronous optogenetically evoked post-synaptic currents, pipettes were filled with high-chloride intracellular solution containing (in mM): 122 CsCl, 8 NaCl, 10 glucose, 1 CaCl2, 10 HEPES, 10 EGTA, 2 Mg2+-ATP, 0.3 Na+-GTP and 2 QX 314-Cl (280–290 mOsm, pH 7.2). Slices were incubated with Ca2+-free ACSF containing 4 mM SrCl2 for 30 min before recording. oChIEF-expressing axon terminals were stimulated with a 5-ms blue light pulse emitted from a collimated light-emitting diode (473 nm, Thorlabs) driven by a T-Cube LED Driver (Thorlabs) under the control of Digidata 1440A Data Acquisition System and pClamp10 software (Molecular Devices). Light was delivered through the reflected light fluorescence illuminator port and the ×40 objective at maximum intensity (13.45 mW). Recordings of optogenetically evoked IPSCs were obtained from SNr or PF neurons located close to mCitrine-labeled axons in the presence of 5 μM NBQX. IPSC events were analyzed using a custom Python script based on a previously described method50. The best fit curve of the largest evoked transient was subtracted from the original trace, and the amplitude and frequency of IPSC events were measured between 0 and 400 ms after photostimulation.
To measure the PPR, pipettes were filled with a Cs-based intracellular solution. Optogenetically evoked IPSCs were recorded from SNr or PF neurons located close to mCitrine-labeled axons while the GPe-PV axons were stimulated with two 5-ms light pulses separated by a 100-ms interval. PPR was calculated as the ratio of 2nd IPSC peak amplitude/1st IPSC peak amplitude. Target recordings in the SNr (Extended Data Fig. 3c,d) were performed similarly with trains of 5-, 10-, 20- and 50-Hz light pulses or a constant 500-ms light pulse.
Behavioral assay: locomotion.
Locomotion measurements in freely moving mice were performed in an open-field arena (opaque white acrylic; W: 30 cm, L: 30 cm, H: 30 cm). To acclimate mice to the arena, we allowed them to freely explore the arena for 1 h per day for three consecutive days. Baseline locomotor activity was obtained on the next day from 10–60 min after start during a 1-h session. On subsequent days, the mice were tested based on their behavioral manipulation. The activity was recorded as a movie from overhead with a 15-Hz frame rate. The center point of each mouse in each frame was tracked offline using Viewer II software (Biobserve) and was exported as raw data. Locomotor activity was then analyzed using a custom Python script to calculate speed in centimeters per second.
Behavioral assay: context-based reversal learning task.
Mice were tested on a modified version of naturalistic foraging reversal discrimination task originally developed for rats35,51 and adapted for mice52,53. Mice were food deprived to reduce body weight to 80–85% of the ad libitum feeding weight and habituated with a small animal food bowl (Lixit Nibble Food Bowl) for 7 d in their home cage before testing. During the same 7-d period, each mouse was handled for 1 min per day to minimize stress associated with handling during testing. One day before testing, mice were acclimated to a testing chamber (opaque white acrylic; W: 30 cm, L: 60 cm, H: 30 cm). Each mouse was placed in a waiting area separated from the rest of the testing chamber by a removable start gate. At the start of each trial, the gate was lifted, and each mouse was allowed to explore two bowls placed at the opposite end of the chamber until a food reward was found and consumed. Both bowls did not contain digging media, and only one bowl contained a food reward (a piece of Honey Nut Cheerios, ~50 mg).
The task consisted of two consecutive phases that were performed over a 2-d period. First, in the association phase, mice were trained to associate a certain context with a rewarding outcome and a different context without reward until they could discriminate reliably. In the subsequent reversal phase, the context–outcome association was reversed, and, thus, the mice needed to switch their actions to obtain the reward. The contexts were either olfactory (odor of the digging medium) or somatosensory (texture of the digging medium that hides the bait). When the olfactory contexts were used, one bowl contained coconut-scented soft bedding (National Geographic), and the other contained lavender-scented soft bedding (National Geographic). When the somatosensory contexts were used, one bowl contained aquarium sand (National Geographic), and the other contained aquarium gravel (National Geographic). Only one type of context was used with each mouse throughout the entire period of the task. All contexts were presented in small animal food bowls that were identical in color and size. Digging media were mixed with the Honey Nut Cheerios powder (0.1% by volume).
On the first testing day (association phase), during each trial, the mouse was kept in the waiting area, and two bowls were present at the opposite end of the chamber separated from the mouse by the start gate. At the start of each trial, the gate was lifted, and the mouse was allowed to explore two bowls until digging in one bowl to signify a choice. The food reward was paired with a certain context throughout the task and was hidden in the digging medium. The baited bowl was pseudorandomly presented on either side of the testing cage. The mouse was required to choose the correct context that would result in getting a food reward. The association phase was complete when a criterion of eight correct trials in a block of ten trials was met (80% accuracy). On the second day (reversal phase), the mouse was presented with the same set of contexts as in the association phase, but the context–reward pairing was reversed. A similar criterion was required to complete the reversal phase. For analysis of event-locked activity in Fig. 5c–h, the reversal phase was broken down into early reversal (all trials before reaching the last ten-trial block, representing a period before the criterion was met) and late reversal (all trials in the last ten-trial block, representing a period at which the criterion was met).
The number of correct and incorrect choices during each phase was scored manually, and the performance of each mouse was reflected by the number of trials needed per phase to meet the criterion. Similarly to previous findings, we observed an increase in the number of trials to criterion when odor was the relevant context54–56. Therefore, the performance data for each mouse was normalized to an average number of trials to criterion from all naive control mice (control mice that did not receive photostimulation or CNO) that were presented with a similar type of context (odor or somatosensory) during the task. In addition, error analysis was performed in the reversal phase to measure the ability to maintain new context–reward association after switching. Incorrect trials were classified as perseverative errors when the incorrect bowl was chosen more than three times in a block of four consecutive trials. Once a mouse started making fewer than three errors in a block, all subsequent incorrect trials were considered regressive errors38.
Behavioral assay: the spatial information-based operant reversal learning task.
For operant conditioning experiments, mice were food deprived to ~85% of free-feeding body weight throughout the experiment. Experiments were conducted using a standard mouse operant chamber (Med-Associates, MED-307W-D1) equipped with two retractable levers and a food reward port. The operant conditioning experiments consisted of three phases: 1) habituation phase, 2) association phase and 3) reversal phase.
During the habituation phase, both levers were exposed, and pressing either lever was rewarded with a single food pellet delivered through the reward port. Mice were habituated for three sessions (one session per day) for 30 min per session. Most mice exhibited a preference for one of the levers, and this lever was then considered the ‘rewarded’ lever for the subsequent association phase. During the association phase, each session comprised a maximum of 30 trials per session. Each trial began with both levers simultaneously exposed and consisted of a maximum of 10 s. When either lever was pressed, both levers were simultaneously retracted, and the trial immediately stopped. Between each trial was a random inter-trial interval (5–20 s). Each session was roughly 15 min, and the mice underwent two sessions per day. The entire association phase consisted of six sessions (or 3 d of training). Each animal proceeded to the subsequent trial if over 75% of attempted trials were rewarded. During the reversal phase, the parameters were identical to the association phase, with the exception that the ‘rewarded’ lever was switched to the other lever (Extended Data Fig. 6).
The fiber photometry measurement and optogenetic/chemogenetic stimulation were performed as described below. Briefly, for fiber photometric measurement of GCaMP6 signaling during the operant reversal learning task was performed as described below (see “Fiber Photometry” section). The following behavioral time points were automatically marked during the task: trial start, lever pressing and reward. z-scored ΔF/F from each recording session was aligned to these behavioral time points. These traces were then averaged across all mice to generate summary traces. Performance on a reversal learning task also reflects the subject’s ability to make decisions. Therefore, one possible interpretation of our results is that GPe neurons participate in reversal learning by affecting decision-making processes. If true, we would expect to see changes in GPe neural activity during decision-making. Therefore, we sought to compare the fiber photometry activity during different stages of our reversal learning paradigm: initial association, early reversal and late reversal. In line with previous literature, we define the decision-making period as the interval between trial onset and choice. However, on individual trials, mice pressed the lever at different times after trial onset, and, therefore, we interpolated the signal between onset and press to ~200 arbitrary units (frames). This interpolated signal was then trial averaged and compared between different reversal learning stages.
For optogenetic stimulation during the reversal learning task, light pulses were delivered for the entire duration of each trial during the reversal learning phase. For chemogenetic stimulation during the reversal learning task, mice were injected with the working solution 1 h before the first trial of the reversal learning phase.
Fiber photometry.
GCaMP6s and mRuby3 fluorescence was collected simultaneously at 20 Hz through a patchcord of bundled 400-μm diameter, 0.48-NA fibers (Doric Lenses) coupled to a setup similar to that previously described33. We used Lumencor SPECTRA X light engine as an excitation light source for cyan (470-nm) and green (550-nm) light. At the start of each recording session, light intensity was adjusted to 120 μW (measured at the tips of the patchcord). Photometry data were analyzed with a custom Python script. First, data from the red channel were scaled to the green channel using a linear fit and were used as baseline (F0) to correct for motion artifacts. ΔF was then calculated by subtracting this baseline. ΔF/F0 was then smoothed by filtering with a 2nd-order Savitzky–Golay filter. Finally, ΔF/F0 from each recording session was z-scored relative to the entire session and aligned to behavioral events.
To determine the relationship of fluorescence signals to locomotion, recordings were performed while the mice were head-fixed on a cylindrical treadmill (15-cm diameter). The treadmill velocity was sampled at 20 Hz by a rotary encoder (TRD-SH Series, AutomationDirect) coupled to the axle of the treadmill. An Arduino was used to send triggers to the sCMOS camera (Orca-Flash4, Hamamatsu) to synchronize the treadmill velocity and fluorescence data stream. Locomotion initiation is the time point at which the treadmill accelaeration was greater than 10 cm s−2. This acceleration was not preceded within a 1-s window by an average velocity lower than 1 cm s−1, indicating a clear transition from rest to locomotion. Termination was identified as the time point at which the treadmill acceleration was lower than −10 cm s−2 and the average velocity in the 1-s window succeeding that time point was lower than 1 cm s−1, indicating a rapid transition from locomotion to rest. z-scored ΔF/F0 from each recording session was aligned to locomotion initiations and terminations. These traces were then averaged across all sessions and mice to generate summary traces.
In the reversal learning task, recordings were performed while the mice were performing the reversal learning task as described above. The following behavioral time points were manually marked offline from the videos recorded during the task: trial start, choice (the moment the mouse started digging) and trial end. z-scored ΔF/F0 from each recording session was aligned to these behavioral time points. These traces were then averaged across all mice to generate summary traces. An average of z-scored ΔF/F0 was calculated from 0–2 s after behavioral onsets to compare between different stages of the task.
Optogenetic stimulation.
A 473-nm blue laser diode (OEM Laser Systems) was connected to the ferrule of the optic fiber previously implanted on mice though a plastic sleeve. Laser power was measured before each experiment and adjusted to ~8 mW. For all experiments with optogenetic activation, photostimulation consisted of 500-ms pulses repeated every 1.5 s. Light pulses were generated with a TTL pulse generator (OPTG-4, Doric Lenses). For locomotion assay, light pulses were delivered for 10 min starting at 30 min after session start during a 1-h session (Fig. 7c,d). For the reversal learning task, light pulses were delivered for the entire duration of each trial during the reversal learning phase.
Chemogenetic inhibition.
CNO (Enzo) was dissolved in water to obtain a stock concentration of 5 mg ml−1 and stored in small aliquots at −20 °C. The working solution was freshly prepared before each use by diluting the stock with 0.9% saline to obtain a concentration of 0.5 mg ml−1. Mice were injected with 0.01 ml g−1 body weight of the working solution to achieve a dose of 5 mg kg−1. For locomotion assay, on the first test day, mice were injected with saline 1 h before testing. On the second day, mice were injected with the working solution 1 h before testing. For the reversal learning task, mice were injected with the working solution 1 h before the first trial of the reversal learning phase.
Statistics and reproducibility.
Statistical analyses were performed using Prism 8 (GraphPad Software). All statistical data can be found in the figure legends with corresponding sample sizes. No statistical methods were used to pre-determine sample sizes. Applied statistics were conducted on data from three or more biologically independent experimental replicates. Our sample sizes were chosen based on common practice in mouse anatomical and behavioral experiments, which are similar to those reported in previous publications4,17. All data were tested for normality with the Shapiro–Wilk test. Then, the appropriate parametric or non-parametric tests, including Student’s t-test, Mann–Whitney U test, Wilcoxon signed-rank test and one- or two-way analysis of variance (ANOVA), were applied. Correction for multiple comparisons for ANOVA was performed using the Bonferroni method. All statistical tests were two tailed. Statistical significance levels were set at *P < 0.05, **P < 0.01 and ***P < 0.001. All data are presented as mean ± s.e.m. unless otherwise noted in the figure legend.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Molecular identity of PVGPe-SNr and PVGPe-PF neurons.
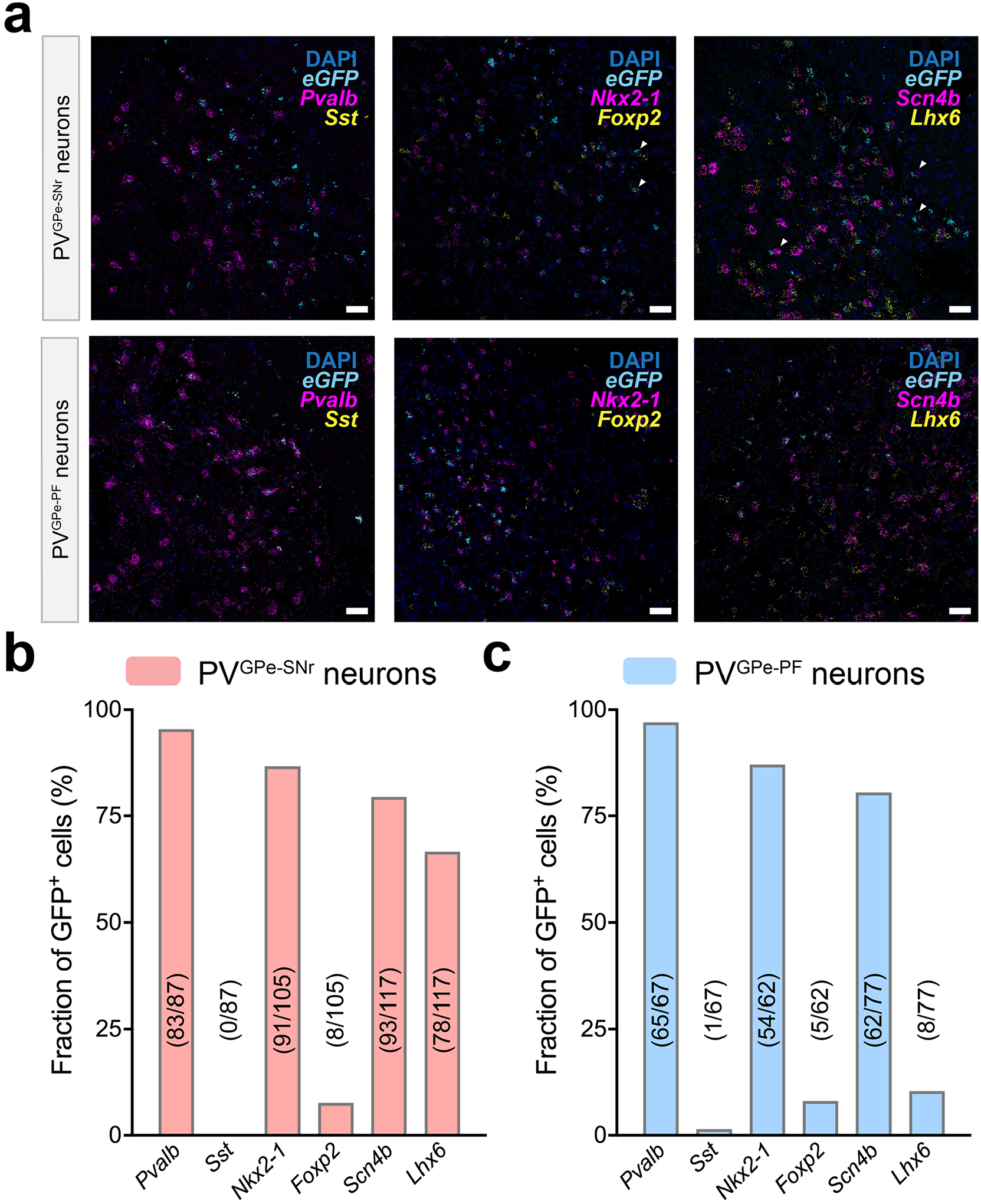
a. mFISH experiments for PVGPe-SNr neurons (top row) and PVGPe-PF neurons (bottom row) with several molecular markers for GPe neurons including parvalbumin (Pvalb), somatostatin (Sst), sodium voltage-gated channel beta subunit 4 (Scn4b), LIM homeobox protein (Lhx6), NK2 homeobox 1 (Nkx2-1) and Forkhead box protein P2 (Foxp2). PVGPe-SNr and PVGPe-PF neurons are labelled by the probe against eGFP. Scale bar in a, 20 mm. b, c, Quantification of GFP-labelled PVGPe-SNr neurons (b, n = 3 mice) and GFP-labelled PVGPe-PF neurons (c, n = 4 mice) expressing specific molecular markers. Fractions show the number of neurons expressing each molecular marker out of total GFP positive neurons in sections.
Extended Data Fig. 2 |. Controls for fiber photometry recordings.
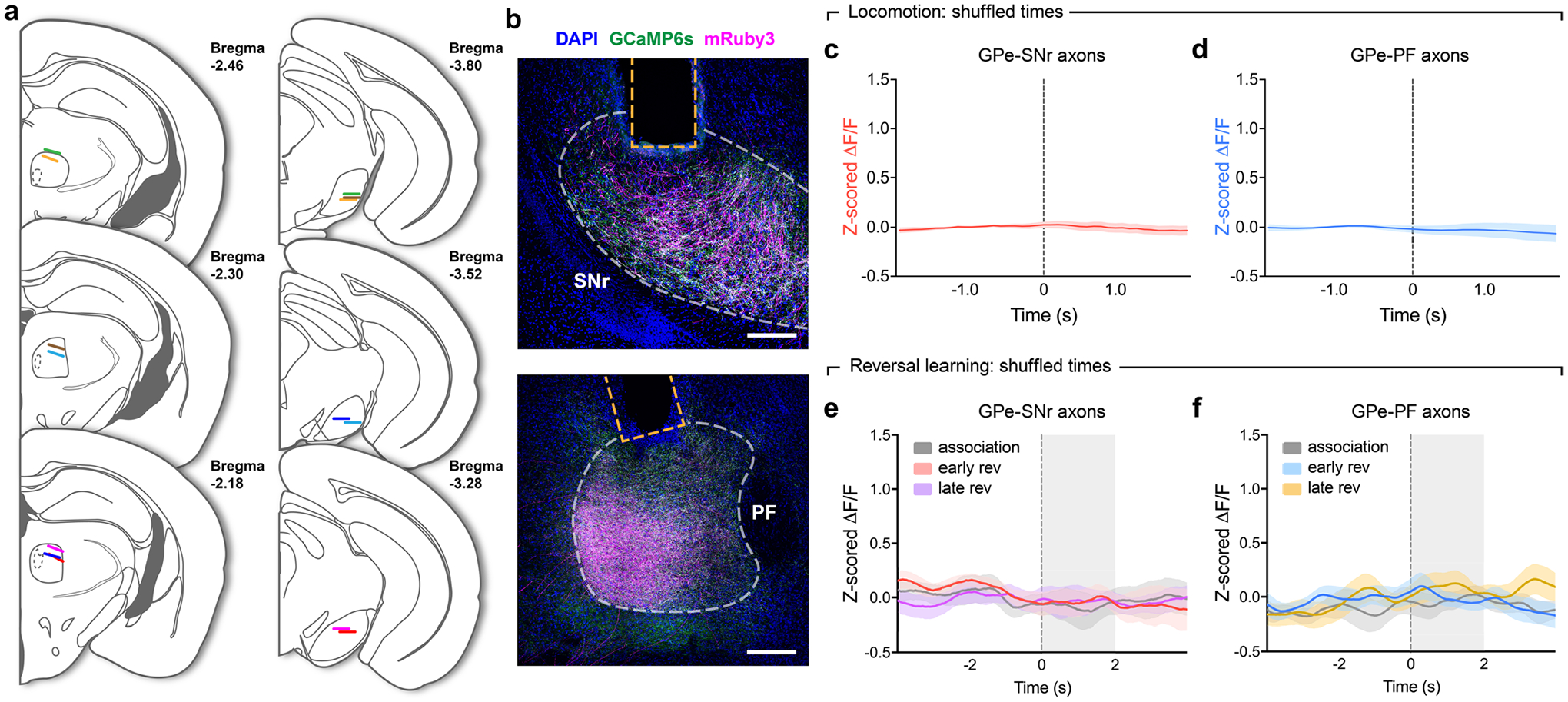
a, Optic fiber placements in PF and SNr for fiber photometry recordings. Different colors denote fibers from the same mouse (n = 7 mice). b, representative images from n = 7 mice used in photometry recordings showing mruby3 and axon-GCaMP6s expression in axons of GPe-PV neurons at implantation sites for optic fibers. Scale bars, 200 μm. c, Z-scored ΔF/F (averaged across all events) representing the activity of PVGPe-SNr axons at randomly chosen time points during treadmill locomotion. Number of events was determined based on the number of locomotion onsets in each recording session (n = 135 events). d, same as in c, but showing the activity of PVGPe-PF axons. e, Z-scored ΔF/F (averaged across 7 mice) representing the activity of PVGPe-SNr axons at randomly chosen time points during different stages of reversal-learning task. Number of events was determined based on the number of trials in each stage of the task. f, same as in e, but showing the activity of PVGPe-PF axons. Shaded areas accompanying the z-scored ΔF/F traces in c-f indicate SEM.
Extended Data Fig. 3 |. Validation of optogenetic activation and chemogenetic inhibition.
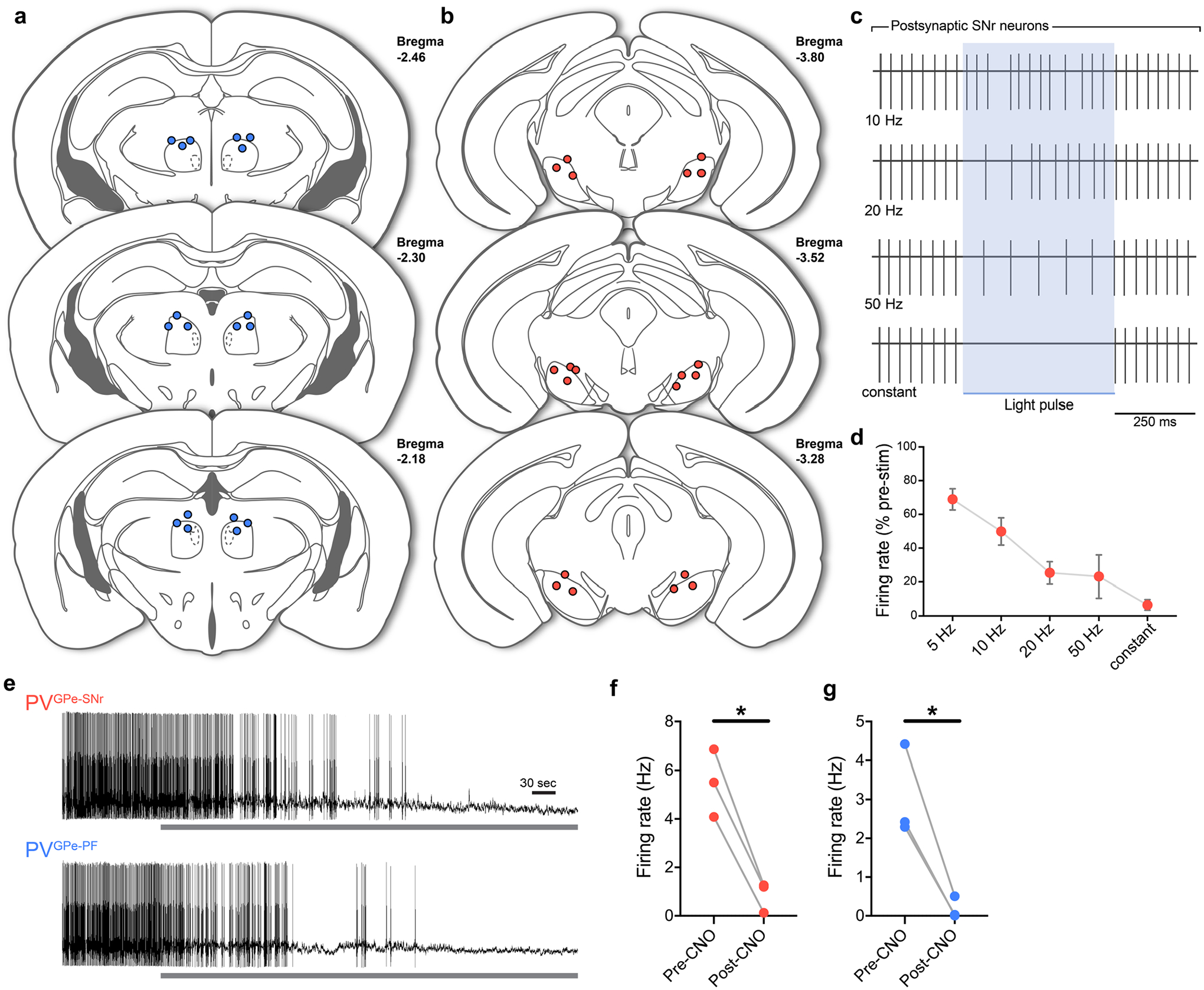
a-b, Fiber tip locations in PF (a) and SNr (b) for data in Figs. 4f–g, 5j–k, 7b and Extended Data Fig. 5, 8b–c. c, representative traces from cell-attached recording showing inhibition of firing activity in SNr neurons during photostimulation of PVGPe-SNr axons at different frequencies. d, Firing rates of SNr neurons during 5–50 Hz and constant photostimulation of PVGPe-SNr axons (n = 21 cells). Data presented as mean ± SEM. e. representative ex vivo cell-attached recording from PVGPe-SNr neurons (top) and PVGPe-PF neurons (bottom) expressing hM4Di. Gray bars show the application of CNO during recording. f-g. Summary data showing firing rates before and 2 min after CNO bath application of both PVGPe-SNr neurons (f; n = 3 cells; Paired t-test, t(2) = 9.259; *p = 0.0115) and PVGPe-PF neurons (g; n = 3 cells; Paired t-test, t(2) = 5.459; *p = 0.0320).
Extended Data Fig. 4 |. Structure of the reversal-learning task and comparison of overall neural activity during trials and inter-trial intervals.
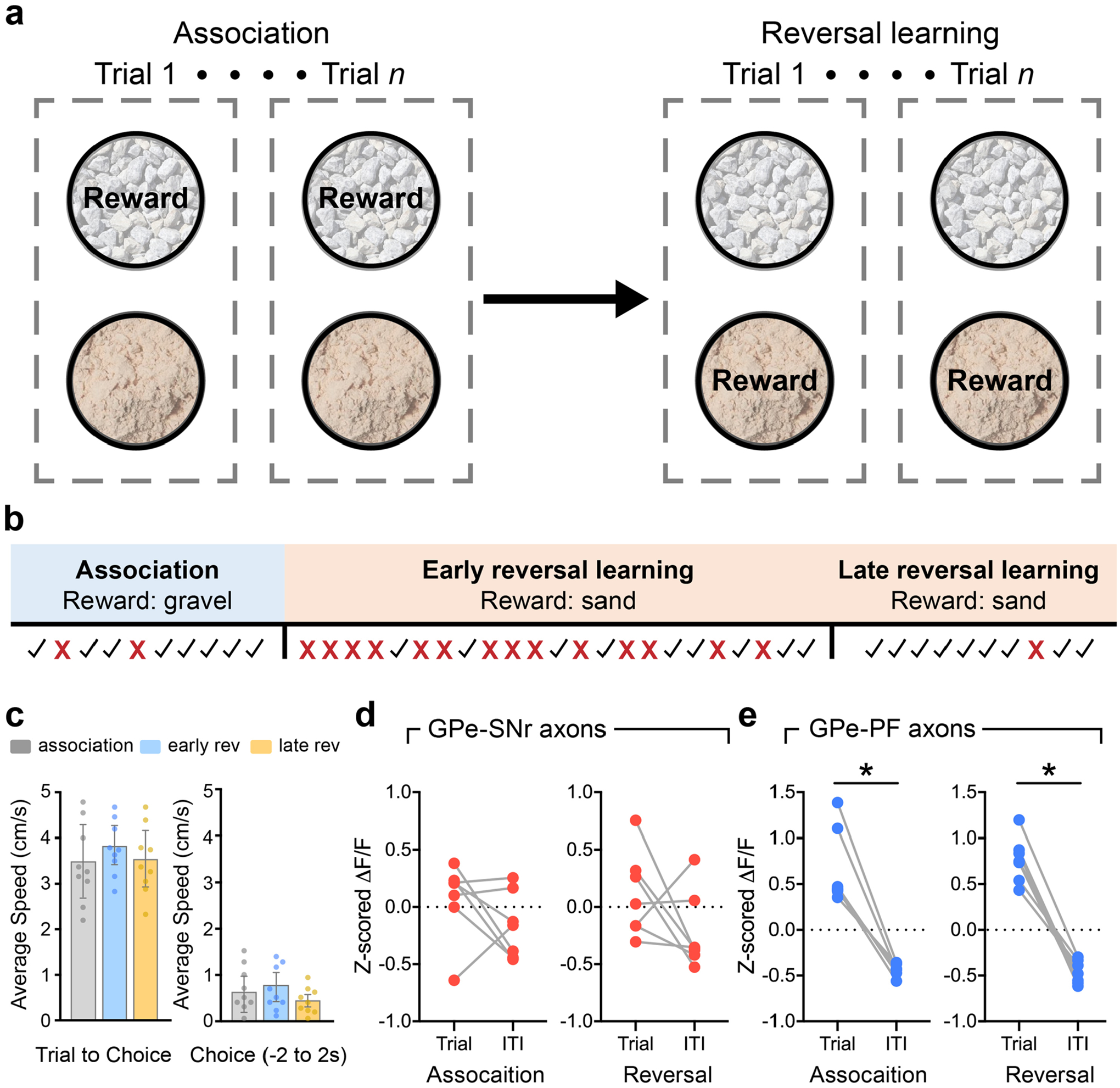
a, Example task structure using gravel and sand as digging media to provided two different contexts. The food reward is paired with sand in the association phase and is later switched to gravel in the reversal-learning phase. b, Timeline of the task showing how the association, early, and late reversal-learning stages are defined for a representative mouse. Check marks and crosses represent correct and incorrect trials, respectively. c, Average speed of animals across different stages of the reversal learning task. Note that no significant difference in locomotion was observed at different stages of the task. (Left: Duration of Trial to Choice, n = 9 mice, One-way ANOVA, F(2, 24) = 0.4743, p = 0.6280; right: Duration of Choice, n = 9, One-way ANOVA, F(2, 24) = 0.7019, p = 0.5055). All data presented as mean ± SEM. d, Comparison of mean z-scored ΔF/F for PVGPe-SNr axons between trial periods and inter-trial intervals (ITI) during the association phase (left; Wilcoxon sign-rank test, W = −12; p = 0.3750, n = 7 mice) and reversal-learning phase (right; Wilcoxon sign rank test, W = −18; p = 0.1562, n = 7 mice). e, same as in d, but for PVGPe-PF axons during the association phase (left; Wilcoxon sign-rank test, W = −28; *p = 0.0156, n = 7 mice) and reversal-learning phase (right; Wilcoxon sign rank test, W = −28; *p = 0.0156, n = 7 mice).
Extended Data Fig. 5 |. Activation of PVGPe-PF neurons increased number of regressive errors made during reversal learning.
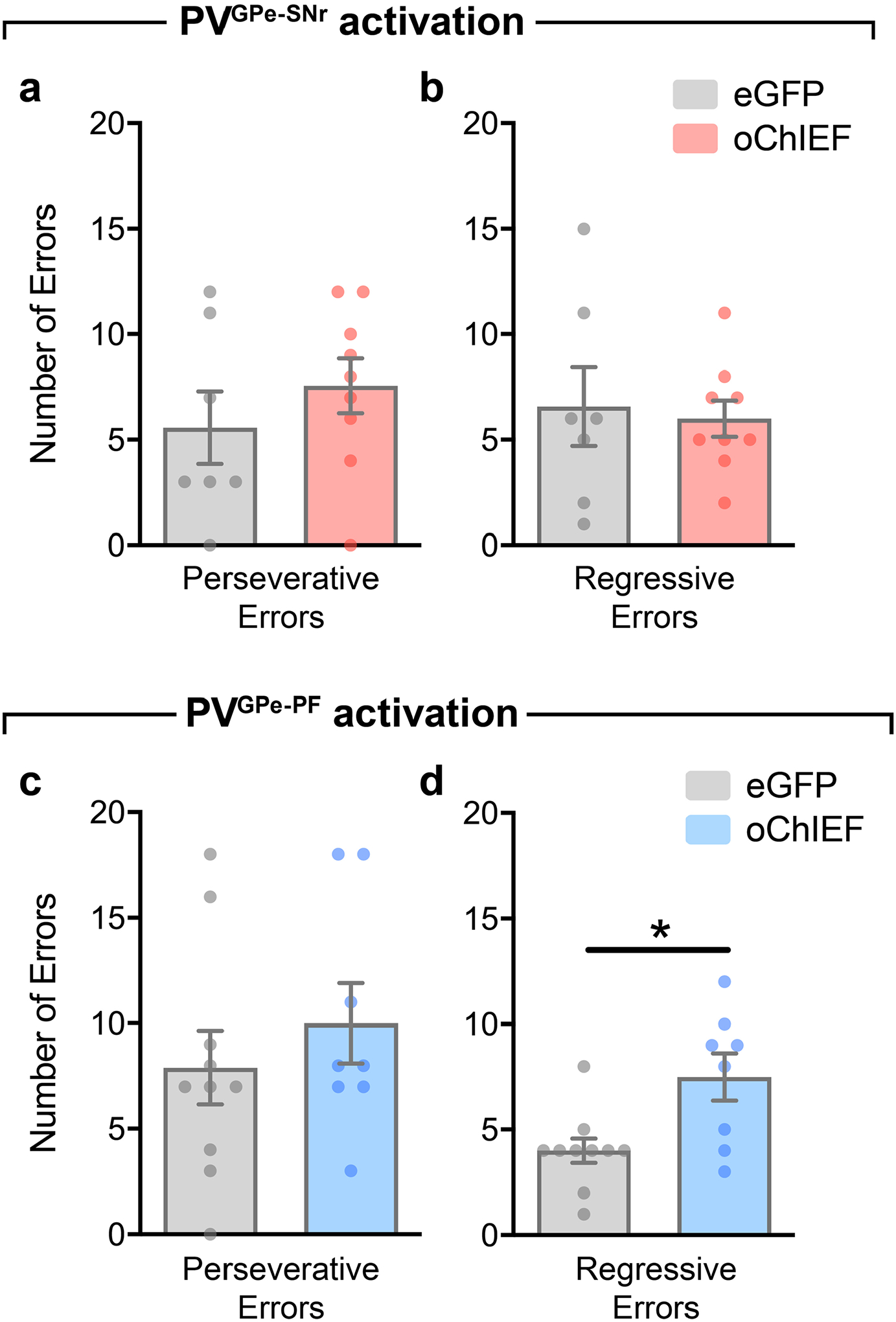
a-b, Number of errors during the reversal-learning phase made by mice that received photostimulation in PVGPe-SNr neurons (n = 7 mice for eGFP, n = 9 mice for oChIEF). a, Perseverative errors; Unpaired t-test, t(14) = 0.9432, p = 0.3616. b, regressive errors; Unpaired t-test, t(14) = 0.3002, p = 0.7684. c-d, Number of errors during the reversal-learning phase made by mice that received photostimulation in PVGPe-PF neurons (n = 10 mice for eGFP, n = 8 mice for oChIEF). c, Perseverative errors; Unpaired t-test, t(16) = 0.8109, p = 0.4293. d, regressive errors; Unpaired t-test, t(16) = 2.951, **p = 0.0094. All data presented as mean ± SEM.
Extended Data Fig. 6 |. Role of PVGPe-SNr and PVGPe-PF in reversal learning test for discriminated operant response in lever-pressing system.
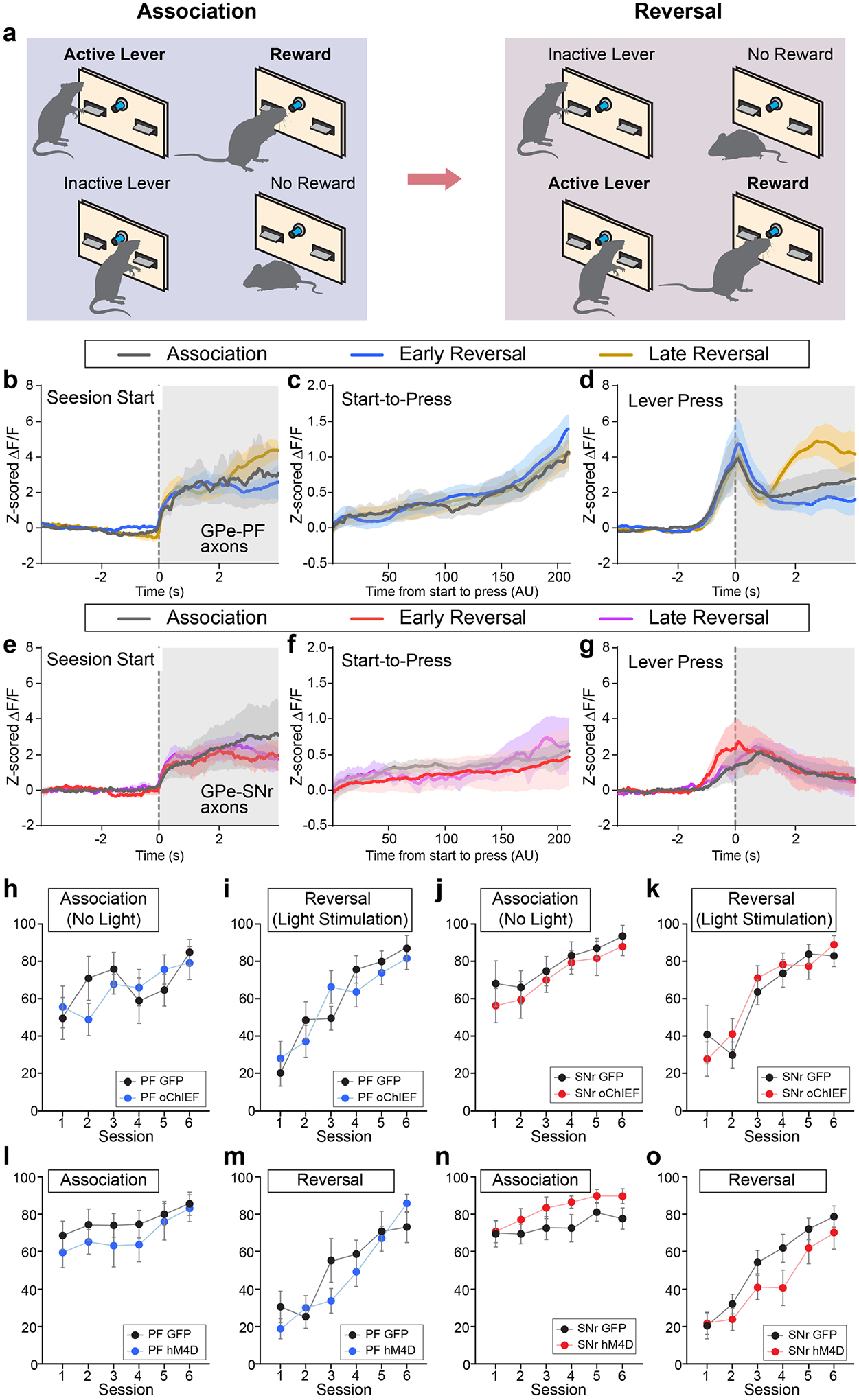
a, A schematic diagram for behavioral task. After the rule switch, active lever becomes inactive, and vice versa. b-d, Traces of z-scored ΔF/F (averaged across 5 mice) from PVGPe-PF axons during session start (b), from start to lever pressing (c) and during lever pressing (d) at the different behavioral stage. Note that the fiber photometry signals for interval between session start and lever pressing was interpolated because of the difference in interval. e-g, Traces of z-scored ΔF/F (averaged across 4 mice) from PVGPe-SNr axons during session start (e), from start to lever pressing (f) and during lever pressing (g) at the different behavioral stage. Note that the fiber photometry signals for interval between session start and lever pressing was interpolated because of the difference in interval. h-k, Activation of PVGPe-SNr and PVGPe-PF axons did not affect association (h, j) and reversal (i, k) in operant discrimination tasks. l-o, Inhibition of PVGPe-SNr and PVGPe-PF axons did not affect association (l, n) and reversal (m, o) in operant discrimination tasks. Shaded areas accompanying the z-scored ΔF/F traces in b-g indicate SEM. All other data are presented as mean ± SEM.
Extended Data Fig. 7 |. Quantification of TH immunoreactivity.
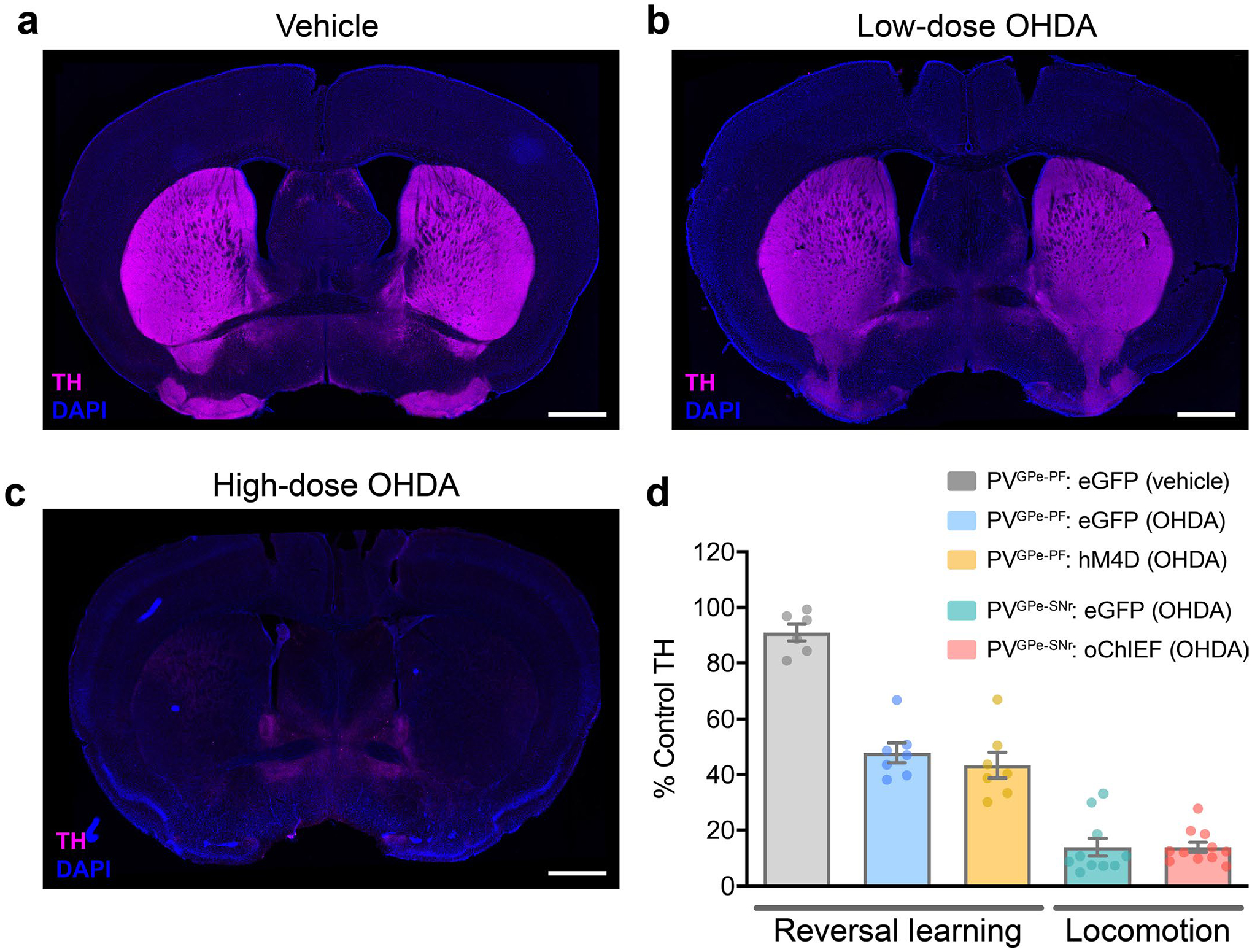
a, representative image of TH immunoreactivity in the striatum of a mouse injected with vehicle (0.02% sodium ascorbate in 0.9% saline). b, representative image of TH immunoreactivity in the striatum 3 days after bilateral injection of low-dose 6-OHDA (1.25 μg/μl). c, representative image of TH immunoreactivity in the striatum 10 days after bilateral injection of high-dose 6-OHDA (2.5 μg/μl). d, Quantification of TH immunoreactivity at different stages of dopamine depletion in rescue experiments for reversal learning and locomotion. Data presented as % mean ± SEM of naïve control striatal sections (n = 6 mice for PVGPe-PF: eGFP (vehicle), n = 7 mice for PVGPe-PF: eGFP (OHDA), n = 7 mice for PVGPe-PF: hM4D (OHDA), n = 10 mice for PVGPe-SNr: eGFP (OHDA), and n = 11 mice for PVGPe-SNr: oChIEF (OHDA)). Scale bar, 1 mm (a-c).
Extended Data Fig. 8 |. Manipulation of PVGPe-PF but not PVGPe-SNr neurons rescues behavioral flexibility deficit in dopamine-depleted mice.
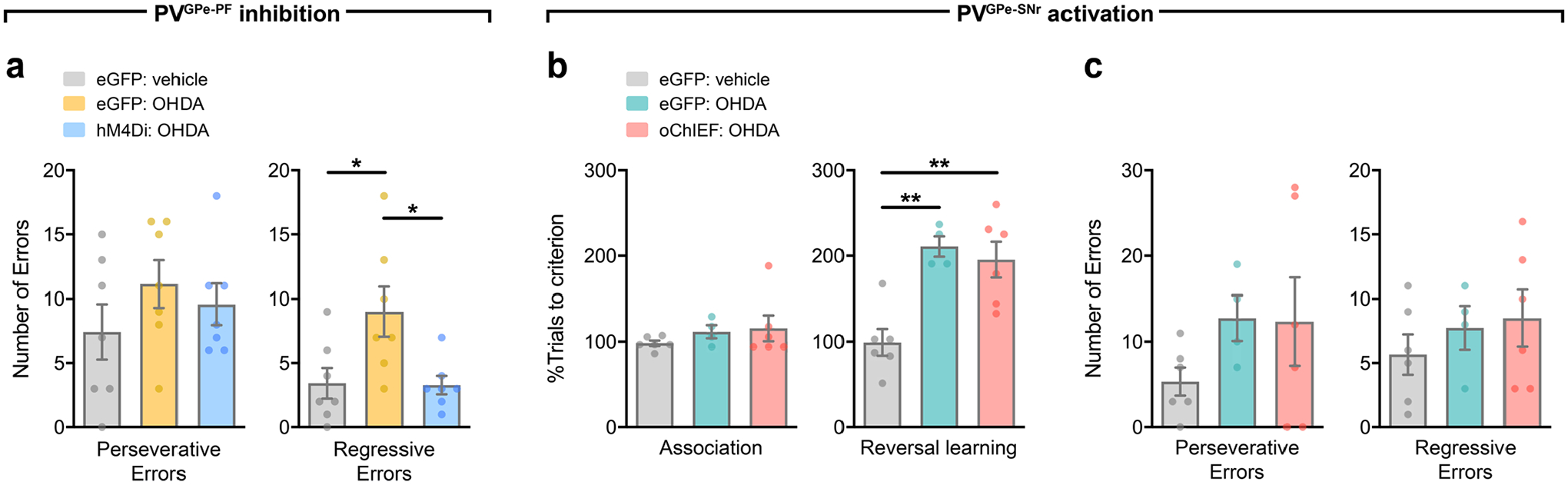
a, Number of errors during the reversal-learning phase made by mice that received chemogenetic inhibition in PVGPe-PF neurons after dopamine depletion (n = 7 mice for eGFP-vehicle, n = 7 mice for eGFP-OHDA, and n = 7 mice for hM4Di-OHDA). Left, perseverative errors; One-way ANOVA, F(2,18) = 0.9771, p = 0.3955. right, regressive errors; One-way ANOVA, F(2,18) = 5.595, p = 0.0129; Bonferroni’s post hoc test, *p = 0.0312 (eGFP-vehicle vs. eGFP-OHDA) and 0.0267 (eGFP-OHDA vs. hM4Di-OHDA). b, Performance of mice that received photostimulation in PVGPe-SNr neurons after dopamine depletion (n = 6 mice for eGFP-vehicle, n = 4 mice for eGFP-OHDA, and n = 6 mice for oChIEF-OHDA). Left, dopamine depletion did not affect performance in the association phase. One-way ANOVA, F(2,13) = 0.8222, p = 0.4611. right, activation of PVGPe-SNr neurons during reversal learning did not improved behavioral flexibility in dopamine-depleted mice. One-way ANOVA, F(2,13) = 11.69, p = 0.0012; Bonferroni’s post hoc test, **p = 0.0032 (eGFP-vehicle vs. eGFP-OHDA) and 0.0042 (eGFP-vehicle vs. oChIEF-OHDA). c, Number of errors during the reversal-learning phase made by mice in c. Left, perseverative errors; One-way ANOVA, F(2,13) = 1.308, p = 0.3038. right, regressive errors; One-way ANOVA, F(2,13) = 0.6464, p = 0.54. All data presented as mean ± SEM.
Acknowledgements
We thank D. Knowland and C. Santiago for their comments on the manuscript. S. Lilascharoen helped with quantification in Fig. 1 and with illustrations. We thank C. Gremel for essential comments on the reversal learning behavioral experiment. We thank the members of the Lim laboratory for support and discussions. V.L. was supported by the Anandamahidol Foundation Fellowship. This work was supported by grants from the National Institutes of Health (U01NS094342, R01DA049787, R01NS097772, R01MH108594 and U01MH114829).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41593-021-00810-y.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
The code that supports the findings of this study is available from the corresponding author upon reasonable request.
Competing interests
K.C. is a co-inventor on a patent application owned by MIT covering the SHIELD technology and is a co-founder of LifeCanvas Technologies.
Extended data is available for this paper at https://doi.org/10.1038/s41593-021-00810-y.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41593-021-00810-y.
References
- 1.Graybiel AM Habits, rituals, and the evaluative brain. Annu. Rev. Neurosci 31, 359–387 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Nelson AB & Kreitzer AC Reassessing models of basal ganglia function and dysfunction. Annu. Rev. Neurosci 37, 117–135 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J et al. Inhibitory basal ganglia inputs induce excitatory motor signals in the thalamus. Neuron 95, 1181–1196(2017). [DOI] [PubMed] [Google Scholar]
- 4.Knowland D et al. Distinct ventral pallidal neural populations mediate separate symptoms of depression. Cell 170, 284–297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephenson-Jones M et al. A basal ganglia circuit for evaluating action outcomes. Nature 539, 289–293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albin RL, Young AB & Penney JB The functional anatomy of basal ganglia disorders. Trends Neurosci 12, 366–375 (1989). [DOI] [PubMed] [Google Scholar]
- 7.Chaudhuri KR, Healy DG, Schapira AH & National Institute for Clinical, E. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 5, 235–245 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Kita H Globus pallidus external segment. Prog. Brain Res 160, 111–133 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Kita H & Kitai ST The morphology of globus pallidus projection neurons in the rat: an intracellular staining study. Brain Res 636, 308–319 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Hernández VM et al. Parvalbumin+ neurons and Npas1+ neurons are distinct neuron classes in the mouse external globus pallidus. J. Neurosci 35, 11830–11847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dodson PD et al. Distinct developmental origins manifest in the specialized encoding of movement by adult neurons of the external globus pallidus. Neuron 86, 501–513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mastro KJ, Bouchard RS, Holt HA & Gittis AH Transgenic mouse lines subdivide external segment of the globus pallidus (GPe) neurons and reveal distinct GPe output pathways. J. Neurosci 34, 2087–2099 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutchison WD et al. Differential neuronal activity in segments of globus pallidus in Parkinson’s disease patients. Neuroreport 5, 1533–1537 (1994). [DOI] [PubMed] [Google Scholar]
- 14.Pan HS & Walters JR Unilateral lesion of the nigrostriatal pathway decreases the firing rate and alters the firing pattern of globus pallidus neurons in the rat. Synapse 2, 650–656 (1988). [DOI] [PubMed] [Google Scholar]
- 15.Filion M & Tremblay L Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced Parkinsonism. Brain Res 547, 142–151 (1991). [PubMed] [Google Scholar]
- 16.Vitek JL, Hashimoto T, Peoples J, DeLong MR & Bakay RA Acute stimulation in the external segment of the globus pallidus improves Parkinsonian motor signs. Mov. Disord 19, 907–915 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Mastro KJ et al. Cell-specific pallidal intervention induces long-lasting motor recovery in dopamine-depleted mice. Nat. Neurosci 20, 815–823 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hikosaka O, Nakamura K & Nakahara H Basal ganglia orient eyes to reward. J. Neurophysiol 95, 567–584 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Caggiano V et al. Midbrain circuits that set locomotor speed and gait selection. Nature 553, 455–460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arkadir D, Morris G, Vaadia E & Bergman H Independent coding of movement direction and reward prediction by single pallidal neurons. J. Neurosci 24, 10047–10056 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown HD, Baker PM & Ragozzino ME The parafascicular thalamic nucleus concomitantly influences behavioral flexibility and dorsomedial striatal acetylcholine output in rats. J. Neurosci 30, 14390–14398 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradfield LA, Bertran-Gonzalez J, Chieng B & Balleine BW The thalamostriatal pathway and cholinergic control of goal-directed action: interlacing new with existing learning in the striatum. Neuron 79, 153–166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saunders A, Huang KW & Sabatini BL Globus pallidus externus neurons expressing parvalbumin interconnect the subthalamic nucleus and striatal interneurons. PLoS ONE 11, e0149798 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cetin A & Callaway EM Optical control of retrogradely infected neurons using drug-regulated ‘TLoop’ lentiviral vectors. J. Neurophysiol 111, 2150–2159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato S & Kobayashi K Improved transduction efficiency of a lentiviral vector for neuron-specific retrograde gene transfer by optimizing the junction of fusion envelope glycoprotein. J. Neurosci. Methods 227, 151–158 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Saunders A et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pamukcu A et al. Parvalbumin+ and Npas1+ pallidal neurons have distinct circuit topology and function. J. Neurosci 40, 7855–7876 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gittis AH et al. New roles for the external globus pallidus in basal ganglia circuits and behavior. J. Neurosci 34, 15178–15183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abrahao KP & Lovinger DM Classification of GABAergic neuron subtypes from the globus pallidus using wild-type and transgenic mice. J. Physiol 596, 4219–4235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim EJ, Jacobs MW, Ito-Cole T & Callaway EM Improved monosynaptic neural circuit tracing using engineered rabies virus glycoproteins. Cell Rep 15, 692–699 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saunders A et al. A direct GABAergic output from the basal ganglia to frontal cortex. Nature 521, 85–89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broussard GJ et al. In vivo measurement of afferent activity with axon-specific calcium imaging. Nat. Neurosci 21, 1272–1280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim CK et al. Simultaneous fast measurement of circuit dynamics at multiple sites across the mammalian brain. Nat. Methods 13, 325–328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin JY, Lin MZ, Steinbach P & Tsien RY Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys. J 96, 1803–1814 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Birrell JM & Brown VJ Medial frontal cortex mediates perceptual attentional set shifting in the rat. J. Neurosci 20, 4320–4324 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bissonette GB et al. Double dissociation of the effects of medial and orbital prefrontal cortical lesions on attentional and affective shifts in mice. J. Neurosci 28, 11124–11130 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blesa J, Phani S, Jackson-Lewis V & Przedborski S Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol 2012, 845618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grospe GM, Baker PM & Ragozzino ME Cognitive flexibility deficits following 6-OHDA lesions of the rat dorsomedial striatum. Neuroscience 374, 80–90 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Oliet SH, Malenka RC & Nicoll RA Bidirectional control of quantal size by synaptic activity in the hippocampus. Science 271, 1294–1297 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Willard AM et al. State transitions in the substantia nigra reticulata predict the onset of motor deficits in models of progressive dopamine depletion in mice. eLife 8, e42746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cools R, Barker RA, Sahakian BJ & Robbins TW Enhanced or impaired cognitive function in Parkinson’s disease as a function of dopaminergic medication and task demands. Cereb. Cortex 11, 1136–1143 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Peterson DA et al. Probabilistic reversal learning is impaired in Parkinson’s disease. Neuroscience 163, 1092–1101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hintiryan H et al. The mouse cortico-striatal projectome. Nat. Neurosci 19, 1100–1114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solari N, Bonito-Oliva A, Fisone G & Brambilla R Understanding cognitive deficits in Parkinson’s disease: lessons from preclinical animal models. Learn. Mem 20, 592–600 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Krauss JK, Pohle T, Weigel R & Burgunder JM Deep brain stimulation of the centre median-parafascicular complex in patients with movement disorders. J. Neurol. Neurosurg. Psychiatry 72, 546–548 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fasano A, Daniele A & Albanese A Treatment of motor and non-motor features of Parkinson’s disease with deep brain stimulation. Lancet Neurol 11, 429–442 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Lim BK, Huang KW, Grueter BA, Rothwell PE & Malenka RC Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 487, 183–189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kato S et al. A lentiviral strategy for highly efficient retrograde gene transfer by pseudotyping with fusion envelope glycoprotein. Hum. Gene Ther 22, 197–206 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Osakada F & Callaway EM Design and generation of recombinant rabies virus vectors. Nat. Protoc 8, 1583–1601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breese GR & Traylor TD Depletion of brain noradrenaline and dopamine by 6-hydroxydopamine. Br. J. Pharmacol 42, 88–99 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park YG et al. Protection of tissue physicochemical properties using polyfunctional crosslinkers. Nat. Biotechnol 10.1038/nbt.4281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanimura A, Du Y, Kondapalli J, Wokosin DL & Surmeier DJ Cholinergic interneurons amplify thalamostriatal excitation of striatal indirect pathway neurons in Parkinson’s disease models. Neuron 101, 444–458 (2019). [DOI] [PubMed] [Google Scholar]
- 53.McAlonan K & Brown VJ Orbital prefrontal cortex mediates reversal learning and not attentional set shifting in the rat. Behav. Brain Res 146, 97–103 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Colacicco G, Welzl H, Lipp HP & Würbel H Attentional set-shifting in mice: modification of a rat paradigm, and evidence for strain-dependent variation. Behav. Brain Res 132, 95–102 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Bissonette GB, Schoenbaum G, Roesch MR & Powell EM Interneurons are necessary for coordinated activity during reversal learning in orbitofrontal cortex. Biol. Psychiatry 77, 454–464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhawan SS, Tait DS & Brown VJ More rapid reversal learning following overtraining in the rat is evidence that behavioural and cognitive flexibility are dissociable. Behav. Brain Res 363, 45–52 (2019). [DOI] [PubMed] [Google Scholar]