

Abstract
C-terminal peptides (CAAPs) of the highly abundant serine protease alpha-1-antitrypsin (A1AT) have been identified at various lengths in several human materials and have been proposed to serve as putative biomarkers for a variety of diseases. CAAPs are enzymatically formed and these enzymatic activities are often associated with excessive immune responses (e.g. sepsis, allergies). However, most of those CAAPs have been either detected using in vitro incubation experiments or in human materials which are not easily accessible. To gain a comprehensive understanding about the occurrence and function of CAAPs in health and disease, a LC–MS/MS method for the simultaneous detection of nine CAAPs was developed and validated for human plasma (EDTA and lithium-heparin) and serum. Using this newly developed method, we were able to detect and quantify five CAAPs in healthy individuals thereby providing an initial proof for the presence of C36, C37, C40 and C44 in human blood. Concentrations of four CAAPs in a clinical test cohort of patients suffering from sepsis were significantly higher compared to healthy controls. These results reveal that in addition to C42 other fragments of A1AT seem to play a crucial role during systemic infections. The proposed workflow is simple, rapid and robust; thus this method could be used as diagnostic tool in routine clinical chemistry as well as for research applications for elucidating the diagnostic potential of CAAPs in numerous diseases. To this end, we also provide an overview about the current state of knowledge for CAAPs identified in vitro and in vivo.
Subject terms: Biomarkers, Diseases, Molecular medicine
Introduction
The endogenous balance between inhibitory active forms of the major human SERPIN alpha-1-antitrypsin (A1AT) and its target-proteinases (primarily human neutrophil elastase, HNE) is a crucial factor determining whether local inflammation processes will result in larger connective tissue damage1–3. Complex formation of A1AT with serine proteases does not only cause inactivation of both substrates but also cleavage of the SERPIN at the reactive site centered at Met382-Ser. However, several non-target enzymes are also able to cleave A1AT in the RCL (reactive center loop) region causing functional loss of the primary regulator towards human serine proteinases and thus increase the risk for the development of lung emphysema and other acute or chronic inflammatory conditions which are known pathophysiological consequences among A1AT deficient individuals4.
During the process of A1AT cleavage by certain enzymes (with or without complex formation), carboxyl-terminal peptides of A1AT (CAAPs) of various lengths can emerge. In addition to HNE which provokes the formation of C36 when complexed to A1AT5, many other enzymes have been identified exhibiting efficient A1AT degradation activity with simultaneous generation of CAAPs. Those enzymes include either endogenous human proteases (mainly matrix-metalloproteinases)5–25, proteases found in microbes causing infections26–32 or proteases that act as allergens for human allergic disorders33–35. However, CAAPs have been detected predominantly by using in vitro incubation experiments with purified A1AT. A few CAAPs have also been identified in vivo in human tissue and liquids such as urine36, bronchoalveolar lavage fluid37,38, gingival crevicular fluid39, spleen and bile40, lung tissue41, carotid artery tissue42, placenta43,44 and nipple aspiration fluids45. Some CAAPs were proposed to serve as putative biomarkers for glomerular kidney diseases36, pulmonary fibrosis38, gingivitis39 and carotid artery stenosis42. However, C42 (“CAAP48/47”) is the hitherto only CAAP detected in human blood46. In this previous study, C42 has been shown to function as inflammatory marker because C42 levels were significantly elevated in patients with severe sepsis compared with patients with systemic inflammatory response syndrome (SIRS) or HIV, respectively. Due to the higher concentrations of C42 found in the sepsis compared to SIRS group, a crucial role of microbes frequently triggering septic shock can be assumed. This is supported by the fact that extracellular microbial proteases, identified in vitro as CAAP-generating enzymes, serve as virulence factors for Staphylococcus aureus 27–29, Pseudomonas aeruginosa 27,30, Serratia marcescens27,31 and Candida albicans 47. In addition to the role of C42 as sepsis biomarker, CAAPs reveal immunomodulatory functions as well and thus might contribute themselves to the inflammatory process. Among the multiplicity of A1AT cleavage fragments, C36 and C42 are the best studied CAAPs regarding their inflammatory immune modulating functions. They act, for example, as chemoattractant for phagocytes and monocytes48, activators of human monocytes and neutrophils in vitro41,42,46,49, neutrophil extracellular trap inhibitory substances50 and alter hepatic functions and gene expression in vitro and in vivo51,52. From a pathophysiological perspective, CAAPs seem to be highly relevant as diagnostic tools for the identification of a variety of diseases. However, the diagnostic use of CAAP profiles is still associated with some challenges. First, CAAPs are likely not equally distributed among the human body. Second, many enzymes are capable to cleave A1AT at more than only one position in the RCL region. Third, knowledge gained from in vitro experiments cannot directly translated to natural and therefore more complex environments such as the human body.
To better understand the complexity of mechanisms leading to CAAP formation, we focused on the development of a diagnostic tool determining CAAP patterns. To this end, we implemented a fast, multiplexed LC–MS/MS method for the simultaneous quantification of nine CAAPs in human serum and plasma; readily available and widely-used biomaterial.
Results
A variety of cleavage products of the major plasma protease inhibitor alpha-1-antitrypsin (CAAPs) have been observed in vitro by incubation of alpha-1-antitrypsin with proteases as well as in vivo in several human tissues. With the exception of C42 (“CAAP48/47”)46, none of those CAAPs have been detected in human blood yet. Due to the vital role of CAAPs during inflammatory infection and possibly other diseases, a rapid method using readily available biomaterials such as blood-derived specimens is needed. Therefore, nine full-length C-terminal peptides were selected and synthesized compromising each CAAP amino acid lengths (from C45 to C22) in WT (wild type) and SNP (single nucleotide polymorphism) variant rs130353, respectively. In order to be able to quantitatively measure this peptide panel a multiplex MRM-based method was established and validated54,55 for human plasma (EDTA and lithium-heparin) and serum.
Method validation
Linearity, working range, carry-over and specificity
Nine calibrants were used in total ranging from 0.01 µM to 1.5 µM thereby covering more than two orders of magnitude in concentration. In eight independent analytical runs, mean coefficients of determination of each calibration curve from 0.01 to 1.5 µM were R2 ≥ 0.996 for each compound (CV < 0.4%, n = 8, respectively) except for C43WT and C43SNP which were R2 ≥ 0.992 (CV ≤ 0.31%, n = 8, respectively). These results demonstrate sufficient linearity throughout the entire calibration range. Working ranges for each of the CAAPs were set according to concentrations in healthy and critically ill patients and were optimized during validation procedure (Table 1).
Table 1.
Working ranges, QC sample concentrations and results of carry-over experiments. Carry-over is determined as the ratio of peak areas measured in blank albumin sample to peak areas of respective LLOQs (%).
| Analyte | Working range (no. of calibrants) |
Concentrations of QCs used for validation |
Carry-over |
|---|---|---|---|
| C36WT | 0.025–1.5 µM (8) |
QC2 (0.025 µM, LLOQ)/ QC3 (0.3 µM)/ QC4 (0.9 µM) |
4% |
| C36SNP | 2% | ||
| C42WT | 2% | ||
| C42SNP | 1% | ||
| C37WT | 0.01–1 µM (8) |
QC1 (0.01 µM, LLOQ) QC3 (0.3 µM) QC4 (0.9 µM) |
0% |
| C37SNP | 0% | ||
| C22WT | 0.01–0.5 µM (7) |
QC1 (0.01 µM, LLOQ) QC2 (0.025 µM) QC3 (0.3 µM) |
0% |
| C22SNP | 1% | ||
| C39WT | 0% | ||
| C39SNP | 0% | ||
| C40WT | 0% | ||
| C43WT | 0% | ||
| C43SNP | 2% | ||
| C44WT | 0% | ||
| C44SNP | 2% | ||
| C45WT | 0% | ||
| C45SNP | 0% | ||
| C40SNP | 0.0036–0.1785 µM (7) |
QC1 (0.0036 µM, LLOQ) QC2 (0.089 µM) QC3 (0.1073 µM) |
0% |
Carry-over was less than 5% of peak areas compared to respective LLOQ samples for all CAAPs and are thus in line with official recommendations (below 20% for analytes, Table 1)55. In none of the blank albumin samples measured directly after respective ULOQ samples internal standards were detected.
No interferences (test of specificity) above the LLOQ were detected (Additional file 2). We detected a few WT-variants above 20% of the LLOQ in the presence of respective SNP-variants. There was further no interference greater than 5% between CAAPs and internal standard detection (Additional file 2).
Accuracy and precision
Mean accuracies and precisions (within-run repeatability and between-run) were within recommended ranges (maximum ± 15% and ± 20% for LLOQ, respectively) (Table 2).
Table 2.
Accuracy and precision of repeated measurements.
| Analyte | Quality control | Within-run repeatability (technical replicates, n = 5) |
Between-run (independent runs, n = 5) |
||
|---|---|---|---|---|---|
| Accuracy (mean, %) |
Precision (mean CV, %) |
Accuracy (mean, %) |
Precision (mean CV, %) |
||
| C36WT |
QC2/ QC3/ QC4 |
110/ 111/ 107 | 5/ 2/ 1 | 113/ 108/ 106 | 2/ 3/ 2 |
| C36SNP | 113/ 108/ 104 | 1/ 3/ 1 | 113/ 107/ 104 | 1/ 2/ 2 | |
| C42WT | 109/ 107/ 104 | 2/ 2/ 3 | 110/ 106/ 105 | 3/ 2/ 3 | |
| C42SNP | 107/ 106/ 104 | 3/ 2/ 3 | 109/ 106/ 105 | 2/ 2/ 3 | |
| C37WT |
QC1/ QC3/ QC4 |
117/ 108/ 104 | 4/ 2/ 2 | 121/ 108/ 104 | 6/ 3/ 2 |
| C37SNP | 113/ 107/ 105 | 5/ 2/ 3 | 117/ 105/ 103 | 2/ 2/ 3 | |
| C22WT |
QC1/ QC2/ QC3 |
99/ 100/ 100 | 2/ 3/ 3 | 103/ 105/ 102 | 4/ 4/ 2 |
| C22SNP | 98/ 101/ 102 | 3/ 2 / 4 | 101/ 105/ 102 | 7/ 3/ 3 | |
| C39WT | 120/ 114/ 109 | 5/ 1/ 1 | 117/ 119/ 106 | 2/ 4/ 4 | |
| C39SNP | 111/ 111/ 103 | 3/ 4/ 2 | 117/ 110/ 103 | 7/ 3/ 1 | |
| C40WT | 115/ 112/ 106 | 3/ 4/ 2 | 117/ 112/ 106 | 2/ 3/ 2 | |
| C40SNP | 112/ 108/ 104 | 8/ 6/ 2 | 105/ 103/ 101 | 5/ 2/ 2 | |
| C43WT | 110/ 108/ 99 | 3/ 2/ 2 | 117/ 111/ 101 | 4/ 4/ 2 | |
| C43SNP | 109/ 107/ 100 | 5/ 2/ 2 | 117/ 112/ 103 | 5/ 3/ 3 | |
| C44WT | 117/ 113/ 103 | 1/ 2/ 2 | 118/ 112/ 103 | 10/ 7/ 4 | |
| C44SNP | 112/ 107/ 103 | 2/ 2/ 2 | 113/ 111/ 102 | 9/ 9/ 4 | |
| C45WT | 117/ 116/ 104 | 2/ 2/ 2 | 115/ 111/ 104 | 8/ 8/ 4 | |
| C45SNP | 112/ 111/ 102 | 5/ 1/ 2 | 111/ 111/ 102 | 6/ 5 /4 | |
Selectivity and investigation of matrix effects
Selectivity was determined in blank albumin samples and results revealed that no peaks (analyte or internal standards) were detected. Serial dilutions of CAAPs standard in pool (human) matrices verified that the responses of concentrations in albumin, serum and plasma (EDTA and lithium-heparin) are linear, respectively (Additional file 3). Due to the unavailability of analyte-free biological matrix, matrix effects were investigated by determining recovery rates and precisions of each compound in different human whole blood derived specimens which were compared to zero albumin samples with equal amount of CAAPs added (using area ratios instead of back-calculated concentrations). Most of the analytes spiked into EDTA plasma were within ± 20% of recovery compared to zero albumin sample (Table 3). Precisions were ≤ 10% between six individually analyzed human donors suggesting that matrix effects are low in EDTA plasma. Recovery rates of CAAPs in lithium-heparin as well as in serum were clearly lower than those in EDTA plasma revealing that effects of these matrices are considerably high. However, most of the CAAP concentrations in serum were consistently back-calculated among the six measured healthy individuals (precision ≤ 20%).
Table 3.
Recovery rates of spiked CAAP concentrations in different biological matrices. Accuracies and precisions are shown as mean of six voluntary donors. H: high CAAPs concentration; L: low CAAPs concentration.
| EDTA plasma | Lithium-heparin plasma | Serum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy (mean, n = 6) |
Precision (n = 6) |
Accuracy (mean, n = 6) |
Precision (n = 6) |
Accuracy (mean, n = 6) |
Precision (n = 6) |
|||||||
| H | L | H | L | H | L | H | L | H | L | H | L | |
| C22WT | 98% | 99% | 4% | 1% | 46% | 46% | 10% | 13% | 58% | 59% | 8% | 13% |
| C22SNP | 98% | 99% | 3% | 2% | 46% | 46% | 11% | 13% | 58% | 59% | 8% | 14% |
| C36WT | 99% | 91% | 2% | 5% | 25% | 20% | 21% | 68% | 49% | 50% | 10% | 18% |
| C36SNP | 105% | 109% | 4% | 4% | 28% | 29% | 24% | 38% | 52% | 53% | 12% | 14% |
| C37WT | 125% | 126% | 5% | 6% | 40% | 42% | 14% | 14% | 66% | 65% | 5% | 9% |
| C37SNP | 129% | 132% | 4% | 3% | 51% | 55% | 11% | 17% | 80% | 80% | 6% | 9% |
| C39WT | 84% | 78% | 6% | 8% | 31% | 33% | 16% | 21% | 47% | 44% | 11% | 17% |
| C39SNP | 83% | 80% | 9% | 10% | 19% | 21% | 25% | 30% | 30% | 29% | 21% | 27% |
| C40WT | 117% | 105% | 5% | 8% | 28% | 28% | 19% | 22% | 47% | 43% | 12% | 14% |
| C40SNP | 102% | 102% | 5% | 2% | 27% | 29% | 17% | 21% | 43% | 44% | 13% | 17% |
| C42WT | 100% | 97% | 2% | 7% | 36% | 34% | 13% | 31% | 52% | 52% | 9% | 17% |
| C42SNP | 99% | 103% | 4% | 7% | 28% | 28% | 21% | 31% | 39% | 39% | 17% | 21% |
| C43WT | 119% | 121% | 5% | 6% | 44% | 45% | 12% | 13% | 57% | 55% | 5% | 7% |
| C43SNP | 113% | 116% | 3% | 6% | 33% | 34% | 14% | 15% | 38% | 40% | 11% | 12% |
| C44WT | 81% | 79% | 8% | 5% | 18% | 18% | 19% | 28% | 25% | 23% | 11% | 20% |
| C44SNP | 82% | 80% | 8% | 6% | 15% | 17% | 25% | 39% | 21% | 22% | 15% | 23% |
| C45WT | 77% | 72% | 7% | 10% | 24% | 20% | 19% | 23% | 35% | 27% | 7% | 14% |
| C45SNP | 88% | 88% | 8% | 5% | 20% | 24% | 21% | 22% | 30% | 30% | 12% | 16% |
To clarify reasons for high variations of peak area ratios of CAAPs in lithium-heparin plasma, variations of internal standards were additionally analyzed (Table 4).
Table 4.
Mean peak areas and precisions of internal standards in different biological matrices. Different CAAP concentrations were spiked into each of the individual matrices: 0 = pure albumin; H: high CAAPs concentration; L: low CAAPs concentration.
| No. of values | Peak Area | ||||||
|---|---|---|---|---|---|---|---|
| C42IS | C37IS | C22IS | |||||
| mean | CV (%) | mean | CV (%) | mean | CV (%) | ||
| Albumin (H, L) | n = 4 | 9.2*106 | 2.7% | 7.0*106 | 3.0% | 7.9*106 | 1.2% |
| EDTA (0, H, L) | n = 34 | 7.7*106 | 6.4% | 5.7*106 | 9.6% | 7.6*106 | 5.4% |
| Li-Hep (0, H, L) | n = 35 | 6.7*106 | 18.5% | 4.7*106 | 19.4% | 7.3*106 | 14.1% |
| Serum (0, H, L) | n = 33 | 6.0*106 | 8.5% | 4.2*106 | 10.2% | 7.0*106 | 6.4% |
High variations in recovery of CAAPs in lithium-heparin plasma derived most likely from inconsistent responses of all three internal standards in lithium-heparin plasma.
Stability
Stability of analytes in processed samples was determined at six independent days for a time span of 20 h in the autosampler. Deviations of standing samples to freshly measured samples were below 19% at the LLOQ and below 9% for other QC levels for each analyte, respectively (Additional file 4). Stability of analytes in biological matrix was determined by consecutive cycles of freezing and thawing as well as extended intervals at room temperature from thawing to processing (approximately one hour). Deviations of either freeze/ thaw treatment or extended benchtop time at room temperature compared to freshly processed QCs did not exceed ± 10% (CV) and mean accuracies were within 98–117, respectively (Additional file 4).
Quantification of CAAPs in healthy humans and comparison between different whole blood-derived specimens
Using the current MRM-based method we were able to quantify concentrations at baseline levels from healthy donors (n = 6) for five out of nine CAAPs investigated (namely C36, C37, C40, C42 and C44; Fig. 1).
Figure 1.

Comparison of CAAP levels in EDTA plasma, lithium-heparin plasma and serum. Relative changes (compared to EDTA plasma) were calculated for each donor individually (n = 6) before mean accuracy (%) and CV (%) were calculated.
Absolute concentrations of C36, C37, C40, C42 and C44 in EDTA plasma were 0.105 ± 0.030 µM, 0.031 ± 0.011 µM, 0.017 ± 0.001 µM, 0.121 ± 0.007 µM and 0.023 ± 0.005 µM, respectively (mean of n = 6 ± standard derivation). Since accuracy and precision of recovery rates in EDTA plasma exhibit best results among three tested blood-derived specimens, CAAP concentrations in lithium-heparin and serum were compared relative to EDTA plasma. Concentrations of CAAPs found in different matrices from same healthy donors revealed that the relative change of concentrations is consistent for serum compared to EDTA plasma (range of CV for five detected CAAPs: 2.5–8.7%; Fig. 1). However, mean serum concentrations differ between 94 and 131% compared to EDTA, depending on the respective CAAP. C36, C37 and C42 measured in lithium-heparin plasma also showed acceptable variation in relative concentrations compared to EDTA plasma (11.1%, 5.3% and 4% CV, respectively) and comparable mean concentrations compared to EDTA (between 105 and 106%). However, C40 and C44 showed unacceptable high variations among individuals (43.4 and 23.1%) and mean concentrations were much lower in lithium-heparin plasma (82 and 55%) compared to EDTA.
Clinical applications
Since CAAPs can be generated from endogenous enzymatic activity associated with inflammation as well as by pathogens frequently causing septic shock, a clinical patient’s cohort suffering from sepsis was chosen to initially investigate CAAP profiles during infectious-induced systemic inflammation. EDTA plasma was used because it proved to be the most suitable blood-derived specimens during method validation. Concentrations of C36, C37, C40 and C42 were significantly higher in the sepsis cohort compared to healthy control samples (P < 0.001, respectively, Fig. 2). C44 in septic patient samples remains on healthy concentration level (P = 0.229). C22, C39, C43 and C45 were absent or below LLOQ.
Figure 2.
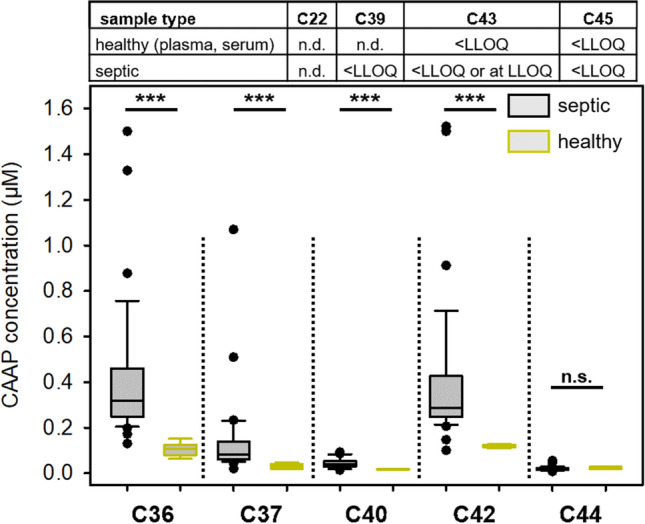
CAAPs abundance in healthy individuals (n = 6) and septic patients (n = 36, cohort A). Data are based on measurements of EDTA plasma samples, which were obtained from patients within the first three days after onset of sepsis. Statistical analysis was conducted using Mann–Whitney Rank Sum Test. *** P < 0.001, n.d. not detected, n.s. not significant.
Based on median values for each CAAP concentration, sepsis patients exhibit between 2.3 and 3.0 times higher concentrations of C36, C37, C40 and C42 than healthy individuals. Among the significantly increased CAAPs in sepsis samples compared to healthy individuals (Fig. 2), we have investigated the validity of C36, C37, C40 and C42 as potential biomarker for sepsis using ROC analysis (Table 5). C36 and C42 resulted in AUROC value of above 97% and high sensitives (97%) and specificities (100%), respectively. Classifications of C37 and C40 were less sensitive and specific for discriminating sepsis from healthy patients.
Table 5.
Accuracy of selected CAAPs for their potential to discriminate sepsis patients from healthy individuals. The data set used was the same as in Fig. 2.
| CAAP | AUC [95% CI] |
Cutoffa (µM) |
Sensitivity (n+/n) |
Specificity (n-/n) |
|---|---|---|---|---|
| C36 | 99% [98–101%] | 0.162 | 97% (35/36) | 100% (6/6) |
| C37 | 97% [92–102%] | 0.048 | 97% (34/36) | 71% (5/6) |
| C40 | 96% [91–102%] | 0.020 | 88% (35/36) | 50% (1/6) |
| C42 | 97% [92–103%] | 0.137 | 97% (35/36) | 100% (6/6) |
a Cutoffs are derived from Kolmogorov–Smirnov statistics. AUC: area under the receiver operating characteristics curve; CI: confidence interval; n+: measurements with positive test results; n-: measurements with negative test results; n: all measured values.
For the two most abundant CAAPs C36 and C42, we have also analyzed their plasma concentrations in septic patients during the first week of treatment at the ICU (Fig. 3, n = 27).
Figure 3.

Development of C36 und C42 concentrations in septic patients. To obtain trends over time, linear regression analyses were conducted for patients with at least three samples collected during their stay at the ICU (cohort B, n = 27). Patients exhibiting opposite trends for C36 and C42 are highlighted.
Over the course of the disease, changes in concentrations of C36 and C42 vary greatly within the patient cohort; with C36 concentrations tending to decrease in the majority of cases (Fig. 3). We have further analyzed the development of C36 and C42 concentrations towards striking features among the sepsis cohort and we found that three out of 27 patients exhibit opposite trends for C36 and C42 concentrations over time (patient ID 21, 31 and 49; Fig. 3). For these three cases, C42 increases while C36 decreases over time.
Discussion
Since the role of CAAPs as biomarker has been proposed for many diseases (36,38,39,42,46, see also Introduction), the proposed method is of important and broad clinical relevance. To this end, we also provide an overview about the current state of knowledge for CAAPs identified in vitro and in vivo (Table 6). In addition to CAAP generating conditions already described in the literature (Table 6), a vital role of CAAPs in other clinical contexts such as cancer progression18,45,56,57 and viral infections can be assumed due to increased MMP activities in the course of these diseases. For example, increased serum concentrations of neutrophil elastase (C36-generating enzyme) and MMP-12 (C42-generating enzyme) have been detected in severe COVID-19 patients58. Biomarkers that allow to quantify systemic inflammation can become an essential tool for diagnosis as well as for monitoring the therapy of systemic inflammatory disorders, but are rarely available yet59.
Table 6.
Summary of known sources for CAAPs. ain brackets cleavage site numbering according to 418 amino acid sequence length of A1AT including precursor molecule of 24 amino acids60 using one-letter-code for amino acids.
| CAAPa | in vivo (human) | in vitro human tissue/ cells | in vitro assays using purified enzymes | References | |
|---|---|---|---|---|---|
| Enzyme name | Enzyme hostb | ||||
| C45 (G373A) | Not detected in plasma or serum of healthy or septic individuals | Current study | |||
| Glycyl endopeptidase | Carica papaya | 29,61 | |||
| C44 (A374M) | Plasma & serum (no difference between healthy and septic individuals) | Current study | |||
| Placenta | 43,44 | ||||
| BALF (higher concentrations in premature infants with bronchopulmonary dysplasia) | 37 | ||||
| MMP-11 | human endogenous | 6 | |||
| Adamalysin II | Crotalus adamanteus | 29,62 | |||
| C43 (M375F) | Not detected in plasma or serum of healthy or septic individuals | Current study | |||
| Thermolysin | Bacillus thermoproteolyticus | 61 | |||
| C42 (F376L) | Plasma & serum (higher concentrations in sepsis cohort than healthy individuals) | Current study | |||
| EDTA plasma (higher concentrations in sepsis cohort than SIRS, HIV or neutropenia) | 46 | ||||
| NAF (only found in woman with breast cancer and among those only found in affected breast) | 45 | ||||
| MMP-1 | Human endogenous | 7,8 | |||
| MMP-7 | Human endogenous | 9–11 | |||
| MMP-8 | Human endogenous | 12–14 | |||
| MMP-9 | Human endogenous | 12,14 | |||
| MMP-12 | Human endogenous | 15,16 | |||
| MMP-13 | Human endogenous | 63 | |||
| MMP-25 | Human endogenous | 17 | |||
| MMP-26 | Human endogenous | 18,19 | |||
| Periodontain | Porphyromonas gingivalis | 26,27 | |||
| Aureolysin | Staphylococcus aureus | 27,28 | |||
| Thermolysin | Bacillus thermoproteolyticus | 27,28,61 | |||
| Papain | Carica papaya | 29,61 | |||
| C40 (E378A) | Plasma & serum (higher concentrations in sepsis cohort than healthy individuals) | Current study | |||
| Cathepsin L | Human endogenous | 20 | |||
| Periodontain | Porphyromonas gingivalis | 26,27 | |||
| Glutamyl endopeptidase | Staphylococcus aureus | 28 | |||
| Aureolysin | Staphylococcus aureus | 27 | |||
| Staphopain A | Staphylococcus aureus | 27–29 | |||
| Pseudolysin | Pseudonomas aeruginosa | 27 | |||
|
C39 (A379I) |
Not detected in plasma or serum of healthy or septic individuals | Current study | |||
| Der p1 allergen | Dermatophagoides pteronyssinus | 33 | |||
| C37 (P381M) | Plasma & serum (higher concentrations in sepsis cohort than healthy individuals) | Current study | |||
| MMP-1 | Human endogenous | 8 | |||
| MMP-3 | Human endogenous | 7,10,21 | |||
| MMP-7 | Human endogenous | 10,11 | |||
| MMP-8 | Human endogenous | 13,14 | |||
| MMP-9 | Human endogenous | 14 | |||
| MMP-12 (mice) | Human endogenous | 16 | |||
| MMP-13 | Human endogenous | 63 | |||
| MMP-25 | Human endogenous | 17 | |||
| MMP-26 | Human endogenous | 18,19 | |||
| Pseudolysin | Pseudomonas aeruginosa | 27,30 | |||
| C36 (M382S) | Plasma & serum (higher concentrations in sepsis cohort than healthy individuals) | Current study | |||
| Spleen & bile (not found in plasma) | 40 | ||||
| BALF. Upregulated in patients with acute exacerbations idiopathic pulmonary fibrosis | 38 | ||||
| Gingival crevicular fluid (putative biomarker for GCF). Higher concentration than in healthy group | 39 | ||||
| Supernatants of activated (LPS-stimulated) neutrophils (not detected in unstimulated controls) | 64 | ||||
| Lung tissue (however, not correlated to COPD) | 41 | ||||
| Carotid artery tissue (symptomatic carotid artery stenosis undergoing carotid endarterectomy) | 42 | ||||
| BALF of premature infants | 37 | ||||
| Neutrophil Elastase | Human endogenous | 5 | |||
| Cathepsin L | Human endogenous | 20 | |||
| MMP-3 | Human endogenous | 10 | |||
| Prostate-specific antigen | Human endogenous | 22,23 | |||
| Mesotrypsin (only Pittsburgh variant (M382 > R)) | Human endogenous | 24 | |||
| High temperature requirement A1 | Human endogenous | 25 | |||
| Serralysin | Serratia marcescens | 27,31 | |||
| EspPα | enterohemorrhagic Escherichia coli | 32 | |||
| Seaprose | Aspergillus melleus | 34 | |||
| Endopeptidase (ragweed) | Ambrosia artemisiifolia | 35 | |||
| Papain | Carica papaya | 27,29,61 | |||
| Trypsin | Streptomyces erythraeus | 65 | |||
| bovines trypsin | bovine | 65 | |||
| porcine pancreatic elastase | porcine | 66 | |||
| α-chymotrypsin | bovine | 61 | |||
| C22 (F396L) | Not detected in plasma or serum of healthy or septic individuals | Current study | |||
| Urine (higher concentration in glomerular kidney disease patients compared to healthy) | 36 | ||||
| BALF (higher concentrations in premature infants with bronchopulmonary dysplasia) | 37 | ||||
| MMP-7 | human endogenous | 11 | |||
bbold: human infectious microbes, underlined: not human infectious organism. A1AT: α1-Antitrypsin; CAAP: C-terminal region of α1-Antitrypsin; MMP: matrix metalloproteinase; BALF: bronchoalveolar lavage fluid. NAF: nipple aspiration fluids.
Due to the role of C42 as biomarker in systemic infection46 and thus its potential application as sepsis biomarker in routine diagnostics, method development had focused on a rapid workflow. In contrast to the original LC–MS/MS method only targeting C42WT and C42SNP46, the current method possesses a rapid workflow and optimized chromatographic conditions for eighteen CAAP analytes. The entire process from sample preparation to quantitative results can be completed in less than one hour (30 min. sample preparation, 12 min. LC run). Working ranges were optimized based on results for carryover and specificity as well as concentrations detected in healthy and septic individuals. Concentration-dependent responses of all investigated CAAPs were linear across more than two orders of magnitude. Therefore, time-consuming steps for sample preparation (e.g. extraction or dilution), which are usually applied, are not necessary. In addition to the more efficient workflow, quantification of CAAPs in healthy individuals can now also be achieved (Fig. 1) due to increased sensitivity of the new method compared to the previous method46. Finally, tests determining accuracy and precision (within-run repeatability and between-runs) of back-calculated CAAP concentrations using QC samples were successfully passed (Table 2).
Even though results of specificity between WT and SNP of some CAAPs are higher than 20% of the respective LLOQ (Additional file 2), we assume that the chance of incorrectly determined concentrations due to interferences from other CAAPs is negligible due to several reasons. First, concentrations below the LLOQ are generally not taken for final data analysis. Second, WT- and SNP-variant would both considered in one sample in case of heterozygote genotype of rs1303 polymorphism which is true for only 37% of European American individuals67). Third, among the analytes which exceed limits of specificity measures according to guidelines (maximum 20% of the LLOQ)54,55, C36 is only one for which we have actually measured concentrations near the ULOQ. Fourth, back-calculated concentrations of WT and SNP which differ greater than 30% from each other are not considered as heterozygote genotype.
Combining results from recovery rates and absolute concentrations in healthy donors (Table 3, Fig. 1), EDTA plasma and serum are appropriate matrices to quantitatively measure CAAPs, whereas lithium-heparin plasma revealed results with high variations among individuals. Using lithium-heparin plasma for CAAP measurements is hence not suggested. Irrespective of the matrix, comparison of respective CAAP concentrations among different clinical entities (e.g. healthy versus diseased) should be performed within the same matrix, if possible. Nevertheless, for data analysis it is recommended to define reference ranges for each CAAP concentration in each matrix.
The presence of C36, C37, C40, C42 and C44 in healthy individuals (Fig. 2) is in agreement with knowledge from the literature as those CAAPs have been reported to be generated endogenously in humans (Table 6). However, apart from our own previous study46, this is to the best of our knowledge the first report about quantitatively measured C-terminal cleavage products of alpha-1-antitrypsin in blood-derived samples. Johansson and colleagues analyzed CAAPs in plasma, bile and spleen using liquid–liquid extraction and amino acid sequencing techniques40. However, they have detected C36 in bile and spleen only but not in plasma. Four peptides (C22, C39, C43, C45) were not detected or below the LLOQ (Fig. 2). Their absence in plasma is partly in agreement with knowledge from the literature (Table 6). C39, C43, and C45 have indeed never been detected in vivo; and in vitro identifications of C4361 and C4529,61 have been correlated only to enzymes that are not associated with human diseases yet. C39, however, has only been found in vitro as potential cleavage product associated with allergic reactions33 whereas C22 has been identified in vivo in urine36 and BALF37. In addition to the higher concentrations of C4246 in septic patients compared to healthy individuals (Fig. 2) our results demonstrate that C36, C37 and C40 seem to play a crucial role in infectious-induced systemic inflammation as well and thus might also have the potential to serve as sepsis biomarkers in blood-derived samples. And indeed, the validity of C36 and C42 as sepsis biomarker was confirmed by their high sensitivities and specificities, respectively (Table 5). C36 experiences the greatest change among CAAPs measured in septic patients compared to concentrations measured in healthy individuals (n = 36, Fig. 2). This observation might be related to MMP activity since it has been shown that higher plasma levels of MMP-3 are associated with septic shock and mortality68. C22, C39, C43 and C45 seem to play only a minor, if any, role during systemic inflammation because they were not detected or below the LLOQ in septic patients (Fig. 2). C36 and C42 concentrations of samples from septic patients (n = 27) change over time (Fig. 3) suggesting that enzymatic activities and/or elimination kinetics are related to the course of infectious-induced systemic inflammation. It is in agreement with knowledge from the literature that formations of C36 and C42 are mediated through distinct enzymatic activities, respectively (Table 6). Of note, for three patients (ID 21, 31 and 49) we detected clearly opposite trends for C36 (decrease) and C42 (increase) concentrations over time (Fig. 3). Decrease of C36 is followed by high initial C36 concentrations; moreover all three cases were affected by pneumonia (site of sepsis infection) with very high SOFA scores at admission (patient 21: 11; patient 31: 15; patient 49: 15). It is well known that increased activity of neutrophil elastase is involved in the pathogenesis of various lung diseases such as pneumonia (see reviews for example69,70). C36 can be formed by neutrophil elastase5 and has already been found in vivo in lung-correlated tissues37,38,41. Further investigation and studies are needed to uncover potential correlations of CAAPs with, for example, site of infection, organ failure or specific enzymatic activities.
Conclusion
Since the generation of CAAPs is multifaceted and they have been found in a variety of biomaterials and in response to a wide range of pathological changes, our method provides a useful clinical tool for many diseases. We are confident that the presented method can be easily adopted towards a variety of biological materials in order to specifically study CAAP levels at sites of generation such as placenta, BALF, bile or urine as well.
Material and methods
Selection of target compounds
The scientific literature search was conducted manually for known cleavage sites of A1AT. Among the variety of C-terminal peptides of A1AT (CAAPs) a panel of nine full-length CAAPs was selected (Fig. 4) according to their experimental evidence. Fragments that were only predicted as well as intermediate fragments in the C-terminal region of A1AT (such as VIRIP) were not collected.
Figure 4.

Cleavage sites of selected CAAPs within the C-terminal amino acid sequence of A1AT. CAAPs are named after their respective amino acid lengths after cleavage. Yellow: 25 residue reactive centre loop of A1AT (UniProtKB). Red: single nucleotide polymorphism rs130353 in the CAAP sequence (E > D376).
Due to a minor allele frequency for the single nucleotide polymorphisms (SNP) of A1AT (rs1303, 28%) within the investigated C-terminal amino acid sequence, the final CAAP panel consists of eighteen analytes (wild type [WT] and SNP for each CAAP length). Authentic CAAPs and internal standards were obtained from sb-PEPTIDE (SmartBioscience SAS, Saint Egrève, FRANCE) as lyophilized peptides in aliquots. C22, C37 and C42 containing isotopic labelled lysine (13C15N) were used as internal standards (IS) for quantification (C22IS, C37IS, C42IS).
Sample preparation
Lyophilized authentic peptide standards were dissolved and diluted in a 70 g/L Albumin (from human serum, lyophilized, ≥ 99%, Sigma-Aldrich) in PBS (Sigma-Aldrich, St. Louis, MO, USA) solution. Internal standards C37IS and C42IS were dissolved in Milli-Q water (Millipore, Brussels, Belgium) containing 0.1% (v/v) formic acid (≥ 95%, Sigma-Aldrich) whereas C22IS was only dissolved in ultrapure water. Mixtures of CAAPs used as calibration standards (calibrants) and quality control samples (QCs) were prepared as stock solution to a final concentration of 14 µM, respectively, (expect C40SNP which was 5 µM) with subsequent serial dilutions. Calibrants and QCs were obtained from separate serial dilutions. Internal standard solution was prepared by adding equal volumes of each IS yielding a final concentration of 0.8 µM, respectively. All samples used for method development and validation were prepared and stored in 0.5 mL Protein LoBind Tubes (Eppendorf AG, Hamburg, Germany) in aliquots at − 80 °C until use.
For LC–MS/MS analyses, 10 µL of internal standard solution was added to 35 µL of calibrant, QC or study sample and gently mixed by pipetting. This mixture was then briefly centrifuged (10 s; 16,000 g; + 10 °C; model 5415R, Eppendorf AG) before 90 µL of cooled methanol (≥ 99.9%, Carl Roth, Karlsruhe, Germany) was added and briefly vortexed subsequently. After centrifugation (10 min; 16,000 g; 10 °C) 100 µL of supernatant was transferred into 2 mL brown glass autosampler vials using 200 µL glass inserts (Wicom, Heppenheim, Germany).
LC–MS/MS analyses
For chromatographic separation, a Shimadzu HPLC system (Duisburg, Germany) was used which is equipped with a binary pump (LC20AB), a thermostatic autosampler (SIL20AC) and a thermostatic column compartment (CTO20AC). 5 µL sample was injected from the autosampler (maintained at 10 °C) and loaded on the UHPLC column (bioZen 3.6 µM Intact C4, 2.1 ˣ 100 mm, Phenomenex, Aschaffenburg, Germany). The column oven was maintained at 45 °C; solvent A was 0.1% formic acid in ultrapure water (v/v) and solvent B was 0.1% formic acid in LC–MS Grade acetonitrile (v/v, ≥ 99.95%, Carl Roth). A step gradient was applied as follows: 5–25% B from 0–1 min., 25–46% B from 1–6 min., 46–85%B from 6–6.5 min., 85% B from 6.5–8.5 min., 85–5% B from 8.5–9.0 min. and 5% B from 9–12 min. The flow rate was 350 µL/min from start to 6.5 min. and 10.6 to 12 min., respectively, and 600 µL/min from 7.0 to 10.5 min.
For mass spectrometry detection, a Triple Quad 5500 + (AB SCIEX, Framingham, MA) in multiple reaction monitoring (MRM) scan type was used with Q1 at low and Q3 at unit resolution. The instrument was equipped with a Turbo V™ ion source and operated in positive electrospray ionization mode with source parameters as follows: curtain gas = 35, collision gas = 8, ionSpray voltage = 5500, temperature = 700 °C, nebulization gas (GS1) = 70, drying gas (GS2) = 70.
Two transitions were monitored for each compound (quantifier, qualifier) and one for the internal standards using an intensity threshold of 50 cps. Corresponding compound-dependent parameters were optimized for each transition (Table 7) whereas entrance potential was 10 V for all transitions. Dwell time was 35 ms for all quantifier and 10 ms for all qualifier. A representative chromatogram is shown in Additional file 1.
Table 7.
Compound-dependent parameters for the detection and quantification of CAAPs.
| Compound (massa) |
RT-window [min] |
Precursor | Product [m/z] |
DP [V] |
CE [V] |
CXP [V] |
IS | ||
|---|---|---|---|---|---|---|---|---|---|
| m/z | charge | ||||||||
|
C22WT (2502.33) |
Quantifier | 3.3–3.5 | 626.8 | [M + 4H]4+ | 754.1 | 110 | 25 | 7 | C22IS |
| Qualifier | 626.7 | 217.3 | 125 | 30 | 20 | ||||
|
C22SNP (2488.32) |
Quantifier | 3.3–3.5 | 623.3 | 749.4 | 110 | 25 | 7 | ||
| Qualifier | 623.3 | 217.3 | 125 | 30 | 20 | ||||
|
C36WT (4132.23) |
Quantifier | 4.2–4.7 | 689.84 | [M + 6H]6+ | 787.8 | 105 | 20 | 6 | C37IS |
| Qualifier | 689.84 | 173.4 | 125 | 50 | 15 | ||||
|
C36SNP (4118.21) |
Quantifier | 4.1–4.6 | 687.49 | 785 | 105 | 20 | 6 | ||
| Qualifier | 687.49 | 173.4 | 125 | 20 | 15 | ||||
|
C37WT (4263.27) |
Quantifier | 4.3–4.8 | 711.74 | [M + 6H]6+ | 787.8 | 105 | 30 | 6 | |
| Qualifier | 854.09 | [M + 5H]5+ | 984.8 | 125 | 40 | 20 | |||
|
C37SNP (4249.25) |
Quantifier | 4.3–4.8 | 709.5 | [M + 6H]6+ | 785.1 | 105 | 30 | 6 | |
| Qualifier | 851.26 | [M + 5H]5+ | 981.1 | 125 | 40 | 20 | |||
|
C39WT (4473.40) |
Quantifier | 4.4–5.0 | 747.06 | [M + 6H]6+ | 728.2 | 105 | 30 | 6 | |
| Qualifier | 896.05 | [M + 5H]5+ | 984.5 | 125 | 40 | 15 | |||
|
C39SNP (4459.39) |
Quantifier | 4.3–4.9 | 744.63 | [M + 6H]6+ | 785.1 | 105 | 35 | 6 | |
| Qualifier | 744.63 | 129.2 | 125 | 40 | 15 | ||||
|
C40WT (4544.44) |
Quantifier | 4.4–5.0 | 758.25 | [M + 6H]6+ | 728.2 | 105 | 20 | 6 | C42IS |
| Qualifier | 910.38 | [M + 5H]5+ | 873.6 | 125 | 30 | 20 | |||
|
C40SNP (4530.42) |
Quantifier | 4.3–4.9 | 756.45 | [M + 6H]6+ | 725.9 | 105 | 20 | 6 | |
| Qualifier | 907.51 | [M + 5H]5+ | 870.8 | 125 | 30 | 10 | |||
|
C42WT (4786.57) |
Quantifier | 4.6–5.2 | 799.2 | [M + 6H]6+ | 873.6 | 105 | 25 | 6 | |
| Qualifier | 958.6 | [M + 5H]5+ | 1091.9 | 125 | 40 | 20 | |||
|
C42SNP (4772.55) |
Quantifier | 4.5–5.1 | 796.75 | [M + 6H]6+ | 870.7 | 105 | 25 | 6 | |
| Qualifier | 956 | [M + 5H]5+ | 1088.4 | 125 | 40 | 15 | |||
|
C43WT (4933.64) |
Quantifier | 4.9–5.5 | 823.65 | [M + 6H]6+ | 873.4 | 105 | 25 | 6 | |
| Qualifier | 988.28 | [M + 5H]5+ | 1091.9 | 125 | 40 | 15 | |||
|
C43SNP (4919.62) |
Quantifier | 4.8–5.4 | 821.3 | [M + 6H]6+ | 870.8 | 105 | 25 | 6 | |
| Qualifier | 985.4 | [M + 5H]5+ | 1088.2 | 120 | 40 | 15 | |||
|
C44WT (5064.68) |
Quantifier | 5.0–5.6 | 845.35 | [M + 6H]6+ | 873.9 | 105 | 35 | 6 | |
| Qualifier | 1014.5 | [M + 5H]5+ | 1091.9 | 125 | 40 | 15 | |||
|
C44SNP (5050.66) |
Quantifier | 5.0–5.5 | 843.08 | [M + 6H]6+ | 870.7 | 100 | 35 | 6 | |
| Qualifier | 1011.59 | [M + 5H]5+ | 1088.1 | 125 | 40 | 15 | |||
|
C45WT (5135.71) |
Quantifier | 5.1–5.7 | 857.59 | [M + 6H]6+ | 873.3 | 100 | 35 | 6 | |
| Qualifier | 1028.58 | [M + 5H]5+ | 1091.9 | 125 | 40 | 15 | |||
|
C45SNP (5121.70) |
Quantifier | 5.1–5.6 | 855.19 | [M + 6H]6+ | 871.0 | 100 | 35 | 6 | |
| Qualifier | 1025.85 | [M + 5H]5+ | 1088.1 | 120 | 40 | 15 | |||
|
C22IS (2516.37) |
Quantifier | 3.3–3.5 | 630.06 | [M + 4H]4+ | 756.4 | 110 | 25 | 7 | - |
| Qualifier | 630.06 | [M + 4H]4+ | 224.4 | 125 | 30 | 20 | |||
|
C37IS (4277.30) |
Quantifier | 4.3–4.8 | 714.01 | [M + 6H]6+ | 790.6 | 105 | 30 | 6 | |
| Qualifier | 856.76 | [M + 5H]5+ | 988.0 | 125 | 40 | 15 | |||
|
C42IS (4807.62) |
Quantifier | 4.7–5.2 | 802.72 | [M + 6H]6+ | 876.3 | 105 | 25 | 6 | |
| Qualifier | [M + 5H]5+ | 1095.1 | 125 | 40 | 15 | ||||
a Theoretical monoisotopic mass (g/mol). DP = declustering potential, RT = retention time, CE = collision energy, CXP = collision cell exit potential, SNP = single nucleotide polymorphism, IS = internal standard.
Bioanalytical method validation
Working range of calibrants was defined based on CAAPs detected in samples from healthy donors (set to approximate lower limit of quantification) and in samples from septic patients (set to middle to high calibration range) which were re-analyzed from a previously work46. Nine calibrants were used in total ranging from 0.01 µM to 1.5 µM thereby covering more than two orders of magnitude in concentration. Linearity was assessed determining mean linear regression coefficients of each calibration curve from 0.01 to 1.5 µM in eight independent analytical runs. However, respective LLOQs (lower limit of quantification) and ULOQs (upper limit of quantification) were defined individually based on natural occurrence (see above) and results from validation parameter directly affected individual working ranges (carry-over, specificity). Four QC samples were prepared and measured in total (QC1, QC2, QC3, and QC4) to cover the entire range of calibration. However, three QCs for each of the respective analytes were used for method validation. Carry-over was determined measuring pure albumin solutions prepared without internal standards (but equal amount and concentration of water/formic acid solution as regular internal standard solution, “blank albumin sample”) directly after the respective highest calibrants. Peak areas were compared to peak areas from respective LLOQ samples (prepared with regular internal standard solution). Specificity was assessed using single compounds at respective highest calibrant level which were prepared and calculated as described for carry-over. The specificity of analytes towards internal standards was determined using pure albumin solution with internal standard (“zero albumin sample”). Accuracy and precision was determined by back-calculated concentrations from repeated measurements of the same QC samples during one analytical run (within-run repeatability, n = 5) and from freshly prepared samples during five independent analytical runs (between-run, n = 5). Due to the unavailability of analyte-free biological matrix, selectivity of analytes were assessed in pure albumin solution containing internal standards. Matrix effects were determined calculating recovery rates and precision of each compound in human blood samples. Direct comparison between different anticoagulants (EDTA, lithium-heparin) and serum was performed using whole blood from the same human donor. Whole blood was collected in EDTA K3, Lithium-Heparin Plasma-Gel and Serum Gel Z S-Monovettes (Sarstedt, Nümbrecht, Germany) from six healthy donors (3f./ 3 m; age from 27 to 45 years), respectively, and prepared using standard centrifugal conditions (10 min., 2762 g, room temperature; model 5804R, Eppendorf AG) before storage at -80 °C. To minimize pre-analytical interferences, HIL indices (hemolysis, icterus, lipemia) were measured in lithium-heparin plasma as well as serum (Architect ci8200, Abbott Laboratories, Abbott Park, IL, USA) and results from both measures were valued as within normal ranges (data not shown). To determine recovery rates in different blood matrices spiked plasma was obtained by adding 5 µL of the spiking solution (peptide-mix standard in albumin: high [7 µM] and low [1 µM]) to 30 µL of different biological matrices from six healthy donors (EDTA plasma, lithium-heparin plasma and serum), respectively. Control samples (“0”) were pure albumin solutions added to biological matrices. Measurements were performed in duplicates. To determine recovery rates, accuracy was calculated by peak area ratios of analyte to internal standard using following equation: (area ratio of spiked sample) – (area ratio of endogenous concentration))/ (area ratio of spiked albumin sample). To evaluate the consistence of matrix effect among individual donors, precision of peak area ratios of analyte to internal standard was investigated for six individual donors in each of the biological matrix. To verify the linear response of CAAPs in different matrices, pool matrices consisting of equal volumes from each of the six donors was prepared and spiked with standard peptide mix (14 µM stock) before serial dilutions in the same pool matrix were conducted. Investigations of analyte stabilities were carried out using QC samples. Autosampler stability was determined by injecting each QC at the beginning of the run and 20 h later during six independent runs. The CV (%) of the back-calculated concentration (n = 2) was used to ensure autosampler stability. To determine stability of analytes after two cycles of freezing and thawing (“freeze/ thaw”) as well as after chilling for 60 min. at room temperature (“benchtop”) three QCs at two concentrations levels were used (QC2 and QC3), respectively. Samples were analyzed against freshly prepared QCs.
Study cohort
To initially determine CAAP profiles in sepsis patients, a well-characterized set of study samples provided by the Department of Anesthesiology and Intensive Care Therapy (Jena University Hospital) was chosen71. All Patients were treated at the Intensive Care Unit (ICU) and blood samples were taken within 24 h after septic symptoms emerged and within the following seven days. For data analysis and interpretation of CAAPs profiles, two subgroups were selected (cohort A and cohort B). Above all, we were interested in CAAP profiles to investigate their potential as early biomarkers. Therefore, we grouped all patients of whom blood samples were available within three days after sepsis onset (cohort A). Next, we were interested in CAAPs development. To this end, we grouped all patients of whom blood samples were available at least three different days after sepsis onset and during their stay at the ICU (cohort B). Patient characteristics of the subgroups are listed in Table 8.
Table 8.
Patient characteristics. Severe sepsis/ septic shock was diagnosed according to the ACCP/SCCM criteria72 and corresponds to the Sepsis-3 definition73.
| Cohort A, n = 36 | Cohort B, n = 27 | |
|---|---|---|
| Age, median (range) | 66 (31–84) | 66 (31–84) |
| Female, n (%) | 14 (39) | 8 (30) |
| 28-day-survival, n (%) | 27 (75) | 22 (81) |
| SOFA at day of study admissiona, median (25–75 percentile range) | 10 (7–13) | 10 (9–13) |
| Site of infection, n (%) | ||
| Abdominal | 16 (44.4) | 9 (33.3) |
| Pneumonia | 10 (27.8) | 11 (40.7) |
| Soft tissue | – | 2 (7.4) |
| Primary bacteremia | 5 (13.9) | 1 (3.7) |
| Endocarditis | 3 (8.3) | 3 (11.1) |
| Urogenital | 2 (5.6) | 1 (3.7) |
| Microbial-positive infection, n (%) | 24 (67) | 16 (59) |
| Positive blood culture | 9 (37.5) | 5 (31) |
| Multiple microbial-positive | 5 (20.8) | 5 (31) |
| S. aureus | 4 (16.7) | 3 (18.8) |
| E. coli | 5 (20.8) | 4 (25) |
| P. aeruginosa | 3 (12.5) | – |
| Enterococci | – | – |
| others | 5 (20.8) | 3 (18.8) |
Cohort A includes all patients of whom blood samples were available within three days after sepsis onset. Cohort B includes patients of whom blood samples were available at least three different days after sepsis onset and during their stay at the ICU.
a When SOFA was not available at the day of study admission, SOFA was taken from the next day (n = 4).
Data processing and statistical analysis
Analyst Software (version 1.6.2 and 1.7.1) was used for mass spectrometer data acquisition and processing. The raw data were imported into the quantification wizard tool, peak integration was reviewed individually and results were generated in relation to the respective internal standard. A 1/x*x weighted quadratic regression was used to calculate the concentrations of each of the eighteen compounds.
To calculate final concentrations for each of the nine CAAP lengths, concentrations of C42WT and C42SNP were used as reference compounds for determining individual phenotypes of rs1303 polymorphisms (homozygote WT, homozygote SNP or heterozygote). A tolerance of maximum 30% coefficient of variance was accepted between concentrations of C42WT and C42SNP for the heterozygote type.
Graphs and statistical analysis were performed using SigmaPlot 14.0. Measurements are presented as boxplots (median, 10/90th percentile) with error bars. For the classification of CAAPs as biomarkers, receiver operating characteristics analyses and cross tables were performed using SPSS statistics version 27 (IBM, New Armonk, NY).
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Jena University Hospital (protocol numbers: 2018–1145-Material, 2018-1145_1-Material, 4619–11/15) and conducted in accordance with the Declaration of Helsinki. All participants (or their legal guardians) gave their written informed consent for analyses and data collection on a consent documentation form.
Supplementary Information
Acknowledgements
The authors thank Kerstin Stein for her support in sample preparation. We are grateful to all the study participants for their valuable contribution.
Author contributions
A.B. design of the study; method development; acquisition, analyses and interpretation of the data; writing the manuscript. C.S.P. analyses, discussion, interpretation and critical reviewing of data; sepsis cohort: acquisition of study samples, patient inclusion, and survey of clinical data. C.S.C. initial involvement in the selection of peptides and albumin matrix. M.B. discussion, interpretation and critical reviewing of data; sepsis cohort: conception of the study and survey of clinical data. M.K. conception and design of the study; discussion, interpretation and critical reviewing of data. All authors contributed to the submitted version of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
PD Dr. Dr. Kiehntopf and Dr. Bigalke are inventors of a patent covering the presented method as a tool for characterizing systemic inflammation (applicant: University Hospital Jena; inventors: Arite Bigalke and Michael Kiehntopf; application number: 22154836.5; status of application: submitted). The University Hospital Jena is also applicant of other published patents which are related to methods determining the origin of an infection (EP17719610.2; EP16167699.4). A patent covering the initial identification of C42 is published as well (published as CN104204808, EP2592421, EP2780719, US20170242035; Applicant: University Hospital Jena; Inventors: Michael Kiehntopf, Diana Schmerler, Thomas Deufel, Frank Brunkhorst). C. Schnabel, C. Sponholz and M. Bauer declare no potential competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Arite Bigalke, Email: arite.bigalke@gmail.com.
Michael Kiehntopf, Email: michael.kiehntopf@med.uni-jena.de.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-07752-w.
References
- 1.Janciauskiene S. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim. Biophys. Acta. 2001;1535(3):221–235. doi: 10.1016/S0925-4439(01)00025-4. [DOI] [PubMed] [Google Scholar]
- 2.Mkaouar H, et al. Serine protease inhibitors and human wellbeing interplay: new insights for old friends. PeerJ. 2019;7:e7224. doi: 10.7717/peerj.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janciauskiene SM, et al. The discovery of alpha1-antitrypsin and its role in health and disease. Respir. Med. 2011;105(8):1129–1139. doi: 10.1016/j.rmed.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Oriano M, et al. Protease-Antiprotease Imbalance in Bronchiectasis. Int. J. Mol. Sci. 2021;22(11):15. doi: 10.3390/ijms22115996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banda MJ, et al. The inhibitory complex of human alpha 1-proteinase inhibitor and human leukocyte elastase is a neutrophil chemoattractant. J. Exp. Med. 1988;167(5):1608–1615. doi: 10.1084/jem.167.5.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pei D, Majmudar G, Weiss SJ. Hydrolytic inactivation of a breast carcinoma cell-derived serpin by human stromelysin-3. J. Biol. Chem. 1994;269(41):25849–25855. doi: 10.1016/S0021-9258(18)47324-3. [DOI] [PubMed] [Google Scholar]
- 7.Mast AE, et al. Kinetics and physiologic relevance of the inactivation of alpha 1-proteinase inhibitor, alpha 1-antichymotrypsin, and antithrombin III by matrix metalloproteinases-1 (tissue collagenase), -2 (72-kDa gelatinase/type IV collagenase), and -3 (stromelysin) J. Biol. Chem. 1991;266(24):15810–15816. doi: 10.1016/S0021-9258(18)98480-2. [DOI] [PubMed] [Google Scholar]
- 8.Desrochers PE, Jeffrey JJ, Weiss SJ. Interstitial collagenase (matrix metalloproteinase-1) expresses serpinase activity. J. Clin. Investig. 1991;87(6):2258–2265. doi: 10.1172/JCI115262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sires UI, et al. Matrilysin is much more efficient than other matrix metalloproteinases in the proteolytic inactivation of alpha 1-antitrypsin. Biochem. Biophys. Res. Commun. 1994;204(2):613–620. doi: 10.1006/bbrc.1994.2503. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z, et al. Proteolysis of human native and oxidised alpha 1-proteinase inhibitor by matrilysin and stromelysin. Biochim. Biophys. Acta. 1994;1199(2):224–228. doi: 10.1016/0304-4165(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 11.Kramer HB, et al. Elevation of intact and proteolytic fragments of acute phase proteins constitutes the earliest systemic antiviral response in HIV-1 infection. PLoS Pathog. 2010;6(5):e1000893. doi: 10.1371/journal.ppat.1000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vissers MC, et al. Cleavage and inactivation of alpha 1-antitrypsin by metalloproteinases released from neutrophils. J. Clin. Investig. 1988;82(2):706–711. doi: 10.1172/JCI113651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knauper V, Reinke H, Tschesche H. Inactivation of human plasma alpha 1-proteinase inhibitor by human PMN leucocyte collagenase. FEBS Lett. 1990;263(2):355–357. doi: 10.1016/0014-5793(90)81412-H. [DOI] [PubMed] [Google Scholar]
- 14.Desrochers PE, et al. Proteolytic inactivation of alpha 1-proteinase inhibitor and alpha 1-antichymotrypsin by oxidatively activated human neutrophil metalloproteinases. J. Biol. Chem. 1992;267(7):5005–5012. doi: 10.1016/S0021-9258(18)42931-6. [DOI] [PubMed] [Google Scholar]
- 15.Gronski TJ, Jr, et al. Hydrolysis of a broad spectrum of extracellular matrix proteins by human macrophage elastase. J. Biol. Chem. 1997;272(18):12189–12194. doi: 10.1074/jbc.272.18.12189. [DOI] [PubMed] [Google Scholar]
- 16.Banda MJ, et al. Interaction of mouse macrophage elastase with native and oxidized human alpha 1-proteinase inhibitor. J. Clin. Investig. 1987;79(5):1314–1317. doi: 10.1172/JCI112955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nie J, Pei D. Rapid inactivation of alpha-1-proteinase inhibitor by neutrophil specific leukolysin/membrane-type matrix metalloproteinase 6. Exp. Cell Res. 2004;296(2):145–150. doi: 10.1016/j.yexcr.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Park HI, et al. Peptide substrate specificities and protein cleavage sites of human endometase/matrilysin-2/matrix metalloproteinase-26. J. Biol. Chem. 2002;277(38):35168–35175. doi: 10.1074/jbc.M205071200. [DOI] [PubMed] [Google Scholar]
- 19.Li W, et al. Matrix metalloproteinase-26 is associated with estrogen-dependent malignancies and targets alpha1-antitrypsin serpin. Cancer Res. 2004;64(23):8657–8665. doi: 10.1158/0008-5472.CAN-04-3019. [DOI] [PubMed] [Google Scholar]
- 20.Johnson DA, Barrett AJ, Mason RW. Cathepsin L inactivates alpha 1-proteinase inhibitor by cleavage in the reactive site region. J. Biol. Chem. 1986;261(31):14748–14751. doi: 10.1016/S0021-9258(18)66935-2. [DOI] [PubMed] [Google Scholar]
- 21.Winyard PG, et al. Proteolytic inactivation of human alpha 1 antitrypsin by human stromelysin. FEBS Lett. 1991;279(1):91–94. doi: 10.1016/0014-5793(91)80258-5. [DOI] [PubMed] [Google Scholar]
- 22.Zhang WM, et al. Prostate-specific antigen forms a complex with and cleaves alpha 1-protease inhibitor in vitro. Prostate. 1997;33(2):87–96. doi: 10.1002/(SICI)1097-0045(19971001)33:2<87::AID-PROS2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 23.Zhang WM, et al. Characterization and determination of the complex between prostate-specific antigen and alpha(1)-protease inhibitor in benign and malignant prostatic diseases. Scand. J. Clin. Lab. Investig. 2000;60:51–58. doi: 10.1080/713783586. [DOI] [PubMed] [Google Scholar]
- 24.Szepessy E, Sahin-Toth M. Human mesotrypsin exhibits restricted S1' subsite specificity with a strong preference for small polar side chains. FEBS J. 2006;273(13):2942–2954. doi: 10.1111/j.1742-4658.2006.05305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frochaux V, et al. Alpha-1-antitrypsin: a novel human high temperature requirement protease A1 (HTRA1) substrate in human placental tissue. PLoS ONE. 2014;9(10):e109483. doi: 10.1371/journal.pone.0109483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson D, et al. Purification and characterization of a novel cysteine proteinase (periodontain) from Porphyromonas gingivalis. Evidence for a role in the inactivation of human alpha1-proteinase inhibitor. J. Biol. Chem. 1999;274(18):12245–12251. doi: 10.1074/jbc.274.18.12245. [DOI] [PubMed] [Google Scholar]
- 27.Rapala-Kozik M, et al. Comparative cleavage sites within the reactive-site loop of native and oxidized alpha1-proteinase inhibitor by selected bacterial proteinases. Biol. Chem. 1999;380(10):1211–1216. doi: 10.1515/BC.1999.153. [DOI] [PubMed] [Google Scholar]
- 28.Potempa J, Watorek W, Travis J. The inactivation of human plasma alpha 1-proteinase inhibitor by proteinases from Staphylococcus aureus. J. Biol. Chem. 1986;261(30):14330–14334. doi: 10.1016/S0021-9258(18)67022-X. [DOI] [PubMed] [Google Scholar]
- 29.Mast AE, Enghild JJ, Salvesen G. Conformation of the reactive site loop of alpha 1-proteinase inhibitor probed by limited proteolysis. Biochemistry. 1992;31(10):2720–2728. doi: 10.1021/bi00125a012. [DOI] [PubMed] [Google Scholar]
- 30.Morihara K, et al. Purification of human plasma alpha 1-proteinase inhibitor and its inactivation by Pseudomonas aeruginosa elastase. J. Biochem. 1984;95(3):795–804. doi: 10.1093/oxfordjournals.jbchem.a134671. [DOI] [PubMed] [Google Scholar]
- 31.Virca GD, et al. Inactivation of human plasma alpha 1-proteinase inhibitor by a metalloproteinase from Serratia marcescens. Biochim. Biophys. Acta. 1982;704(2):267–271. doi: 10.1016/0167-4838(82)90155-8. [DOI] [PubMed] [Google Scholar]
- 32.Weiss A, Joerss H, Brockmeyer J. Structural and functional characterization of cleavage and inactivation of human serine protease inhibitors by the bacterial SPATE protease EspPalpha from enterohemorrhagic E. coli. PLoS ONE. 2014;9(10):e111363. doi: 10.1371/journal.pone.0111363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalsheker NA, et al. The house dust mite allergen Der p1 catalytically inactivates alpha 1-antitrypsin by specific reactive centre loop cleavage: a mechanism that promotes airway inflammation and asthma. Biochem. Biophys. Res. Commun. 1996;221(1):59–61. doi: 10.1006/bbrc.1996.0544. [DOI] [PubMed] [Google Scholar]
- 34.Korzus E, Luisetti M, Travis J. Interactions of alpha-1-antichymotrypsin, alpha-1-proteinase inhibitor, and alpha-2-macroglobulin with the fungal enzyme, seaprose. Biol. Chem. Hoppe Seyler. 1994;375(5):335–341. doi: 10.1515/bchm3.1994.375.5.335. [DOI] [PubMed] [Google Scholar]
- 35.Bagarozzi DA, Jr, et al. Purification and characterization of a novel endopeptidase in ragweed (Ambrosia artemisiifolia) pollen. J. Biol. Chem. 1996;271(42):26227–26232. doi: 10.1074/jbc.271.42.26227. [DOI] [PubMed] [Google Scholar]
- 36.Navarro-Munoz M, et al. Uromodulin and alpha(1)-antitrypsin urinary peptide analysis to differentiate glomerular kidney diseases. Kidney Blood Press. Res. 2012;35(5):314–325. doi: 10.1159/000335383. [DOI] [PubMed] [Google Scholar]
- 37.Tirone C, et al. Oxidative and proteolytic inactivation of alpha-1 antitrypsin in bronchopulmonary dysplasia pathogenesis: a top-down proteomic bronchoalveolar lavage fluid analysis. Front. Pediatr. 2021;9:7. doi: 10.3389/fped.2021.597415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carleo A, et al. Proteomic characterization of idiopathic pulmonary fibrosis patients: stable versus acute exacerbation. Monaldi Arch. Chest Dis. 2020 doi: 10.4081/monaldi.2020.1231. [DOI] [PubMed] [Google Scholar]
- 39.Preiano M, et al. An analytical method for assessing optimal storage conditions of gingival crevicular fluid and disclosing a peptide biomarker signature of gingivitis by MALDI-TOF MS. Proteomics Clin. Appl. 2018;12(5):e1800005. doi: 10.1002/prca.201800005. [DOI] [PubMed] [Google Scholar]
- 40.Johansson J, et al. Identification of hydrophobic fragments of alpha 1-antitrypsin and C1 protease inhibitor in human bile, plasma and spleen. FEBS Lett. 1992;299(2):146–148. doi: 10.1016/0014-5793(92)80234-8. [DOI] [PubMed] [Google Scholar]
- 41.Subramaniyam D, et al. C-36 peptide, a degradation product of alpha1-antitrypsin, modulates human monocyte activation through LPS signaling pathways. Int. J. Biochem. Cell. Biol. 2006;38(4):563–575. doi: 10.1016/j.biocel.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 42.Dichtl W, et al. The carboxyl-terminal fragment of alpha1-antitrypsin is present in atherosclerotic plaques and regulates inflammatory transcription factors in primary human monocytes. Mol. Cell. Biol. Res. Commun. 2000;4(1):50–61. doi: 10.1006/mcbr.2000.0256. [DOI] [PubMed] [Google Scholar]
- 43.Niemann MA, Baggott JE, Miller EJ. Inhibition of human serine proteases by SPAAT, the C-terminal 44-residue peptide from alpha1-antitrypsin. Biochim. Biophys. Acta. 1997;1340(1):123–130. doi: 10.1016/S0167-4838(97)00034-4. [DOI] [PubMed] [Google Scholar]
- 44.Niemann MA, Narkates AJ, Miller EJ. Isolation and serine protease inhibitory activity of the 44-residue, C-terminal fragment of alpha 1-antitrypsin from human placenta. Matrix. 1992;12(3):233–241. doi: 10.1016/S0934-8832(11)80066-1. [DOI] [PubMed] [Google Scholar]
- 45.Zhou J, et al. A unique proteolytic fragment of alpha1-antitrypsin is elevated in ductal fluid of breast cancer patient. Breast Cancer Res. Treat. 2010;123(1):73–86. doi: 10.1007/s10549-009-0625-5. [DOI] [PubMed] [Google Scholar]
- 46.Blaurock N, et al. C-terminal alpha-1 antitrypsin peptide: a new sepsis biomarker with immunomodulatory function. Mediators Inflamm. 2016;2016:6129437. doi: 10.1155/2016/6129437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gogol M, et al. Inactivation of alpha1-proteinase inhibitor by Candida albicans aspartic proteases favors the epithelial and endothelial cell colonization in the presence of neutrophil extracellular traps. Acta Biochim. Pol. 2016;63(1):167–175. doi: 10.18388/abp.2015_1163. [DOI] [PubMed] [Google Scholar]
- 48.Banda MJ, et al. Alpha 1-proteinase inhibitor is a neutrophil chemoattractant after proteolytic inactivation by macrophage elastase. J. Biol. Chem. 1988;263(9):4481–4484. doi: 10.1016/S0021-9258(18)68951-3. [DOI] [PubMed] [Google Scholar]
- 49.Janciauskiene S, Moraga F, Lindgren S. C-terminal fragment of alpha 1-antitrypsin activates human monocytes to a pro-inflammatory state through interactions with the CD36 scavenger receptor and LDL receptor. Atherosclerosis. 2001;158(1):41–51. doi: 10.1016/S0021-9150(00)00767-X. [DOI] [PubMed] [Google Scholar]
- 50.Campbell RA, et al. Placental HTRA1 cleaves alpha-1-antitrypsin to generate a NET-Inhibitory Peptide. Blood. 2021;138:977–988. doi: 10.1182/blood.2020009021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blaurock-Möller N, et al. CAAP48, a new sepsis biomarker, induces hepatic dysfunction in an in vitro liver-on-chip model. Front. Immunol. 2019;10(273):273. doi: 10.3389/fimmu.2019.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerbod-Giannone MC, et al. Suppression of cholesterol 7alpha-hydroxylase transcription and bile acid synthesis by an alpha1-antitrypsin peptide via interaction with alpha1-fetoprotein transcription factor. J. Biol. Chem. 2002;277(45):42973–42980. doi: 10.1074/jbc.M205089200. [DOI] [PubMed] [Google Scholar]
- 53.Graham A, et al. Characterisation of the alpha-1-antitrypsin M3 gene, a normal variant. Hum. Genet. 1990;85(3):381–382. doi: 10.1007/BF00206766. [DOI] [PubMed] [Google Scholar]
- 54.Agency, E. M., Guideline on bioanalytical method validation. 2011, Committee for Medicinal Products for Human Use (CHMP).
- 55.Agency, E. M., draft: ICH guideline M10 on bioanalytical method validation (Step 2b). 2019.
- 56.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2002;2(3):161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 57.Kataoka H, et al. Enhanced tumor growth and invasiveness in vivo by a carboxyl-terminal fragment of alpha1-proteinase inhibitor generated by matrix metalloproteinases: a possible modulatory role in natural killer cytotoxicity. Am. J. Pathol. 1999;154(2):457–468. doi: 10.1016/S0002-9440(10)65292-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zerimech F, et al. Protease-antiprotease imbalance in patients with severe COVID-19. Clin. Chem. Lab. Med. 2021;59(8):E330–E334. doi: 10.1515/cclm-2021-0137. [DOI] [PubMed] [Google Scholar]
- 59.Bauer M, et al. A transcriptomic biomarker to quantify systemic inflammation in sepsis - a prospective multicenter phase II diagnostic study. EBioMedicine. 2016;6:114–125. doi: 10.1016/j.ebiom.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Long GL, et al. Complete sequence of the cDNA for human alpha 1-antitrypsin and the gene for the S variant. Biochemistry. 1984;23(21):4828–4837. doi: 10.1021/bi00316a003. [DOI] [PubMed] [Google Scholar]
- 61.Chang WS, et al. Probing serpin reactive-loop conformations by proteolytic cleavage. Biochem. J. 1996;314:647–653. doi: 10.1042/bj3140647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kress LF, et al. Characterization of the inactive fragment resulting from limited proteolysis of human alpha-1-proteinase inhibitor by Crotalus-adamanteus proteinase-Ii. J. Biol. Chem. 1979;254(12):5317–5320. doi: 10.1016/S0021-9258(18)50597-4. [DOI] [PubMed] [Google Scholar]
- 63.Wilkinson DJ, et al. Matrix metalloproteinase-13 is fully activated by neutrophil elastase, and inactivates its serpin inhibitor, alpha-1 antitrypsin: Implications for osteoarthritis. Febs j. 2021;289:121–139. doi: 10.1111/febs.16127. [DOI] [PubMed] [Google Scholar]
- 64.Matamala N, et al. Identification of novel short C-terminal transcripts of human SERPINA1 gene. PLoS ONE. 2017;12(1):e0170533. doi: 10.1371/journal.pone.0170533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vukoti KM, Kadiyala CS, Miyagi M. Streptomyces erythraeus trypsin inactivates alpha1-antitrypsin. FEBS Lett. 2011;585(24):3898–3902. doi: 10.1016/j.febslet.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ossanna PJ, et al. Oxidative regulation of neutrophil elastase-alpha-1-proteinase inhibitor interactions. J. Clin. Investig. 1986;77(6):1939–1951. doi: 10.1172/JCI112523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/) [accessed 2021 July].
- 68.Jones TK, et al. Elevated Plasma Levels of Matrix Metalloproteinase-3 and Tissue-Inhibitor of Matrix Metalloproteinases-1 Associate with Organ Dysfunction and Mortality in Sepsis. Shock. 2021;57:41–47. doi: 10.1097/SHK.0000000000001833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Polverino E, et al. The role of neutrophil elastase inhibitors in lung diseases. Chest. 2017;152(2):249–262. doi: 10.1016/j.chest.2017.03.056. [DOI] [PubMed] [Google Scholar]
- 70.Domon H, Terao Y. The role of neutrophils and neutrophil elastase in pneumococcal pneumonia. Front. Cell Infection Microbiol. 2021 doi: 10.3389/fcimb.2021.615959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kramer M, et al. Alternative splicing of SMPD1 in human sepsis. PLoS ONE. 2015;10(4):e0124503. doi: 10.1371/journal.pone.0124503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bone RC, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 73.Singer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3) Jama-J. Am. Med. Assoc. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.