

Abstract
Rickettsia species are endosymbionts hosted by arthropods and are known to cause mild to fatal diseases in humans. Here, we analyse the evolution and diversity of 34 Rickettsia species using a pangenomic meta-analysis (80 genomes/41 plasmids). Phylogenomic trees showed that Rickettsia spp. diverged into two Spotted Fever groups, a Typhus group, a Canadensis group and a Bellii group, and may have inherited their plasmids from an ancestral plasmid that persisted in some strains or may have been lost by others. The results suggested that the ancestors of Rickettsia spp. might have infected Acari and/or Insecta and probably diverged by persisting inside and/or switching hosts. Pangenomic analysis revealed that the Rickettsia genus evolved through a strong interplay between genome degradation/reduction and/or expansion leading to possible distinct adaptive trajectories. The genus mainly shared evolutionary relationships with α-proteobacteria, and also with γ/β/δ-proteobacteria, cytophagia, actinobacteria, cyanobacteria, chlamydiia and viruses, suggesting lateral exchanges of several critical genes. These evolutionary processes have probably been orchestrated by an abundance of mobile genetic elements, especially in the Spotted Fever and Bellii groups. In this study, we provided a global evolutionary genomic view of the intracellular Rickettsia that may help our understanding of their diversity, adaptation and fitness.
Subject terms: Evolution, Microbiology, Diseases, Pathogenesis
Introduction
Rickettsia species (spp.) (Order Rickettsiales, Family Rickettsiaceae) are obligate intracellular α-proteobacteria of both ecological and medical interest. These bacteria live in close association with a diverse range of hosts including arthropods, mostly, and vertebrates, plants, algae, annelids, amoebae, ciliate and medusae (as primary hosts and vectors)1–6. Rickettsia spp. are transmitted to humans or animals, mostly through arthropod bites, and are responsible for mild to fatal diseases such as epidemic typhus and Rocky Mountain spotted fever3,7.
Obligate intracellular bacterial endosymbionts and pathogens evolve in diverse host environments. These biological and ecological lifestyles make their genetic manipulations and understanding the mechanisms of their survival and pathogenicity8,9 difficult. To overcome this barrier, evolutionary biology using comparative genomics is a powerful tool to decipher genomic signatures that have shaped bacterial genomes and to understand their diversification, adaptation and fitness. Earlier investigations of Rickettsia spp. using comparative genomics have revealed several genetic and evolutionary features within these small (1.1–2.3 Mbp) genomic bacteria, such as (i) a high degree of inter-species genomic synteny; (ii) reductive evolution possibly in relation to their strict intracellular lifestyle; (iii) an enrichment in A + T content, polyA/T homopolymers; (iv) a lack of some metabolic pathways for which host cells provide the missing metabolites; (v) conjugative machinery and mechanisms of adhesion to host and motility; and (vi) a variable distribution of plasmids10–28.
Although initially thought to be devoid of plasmids, several Rickettsia spp. were demonstrated to harbour such mobile genetic elements (MGEs)27. While no obvious association between pathogenicity and acquisition of virulence factors was found in Rickettsia spp.20, other studies associated the increased virulence with genome reduction in this genus29. However, due to the absence of several genome sequences of the genus Rickettsia in public databases, all these comparative genomic investigations were carried out from subsets of species, sometimes drawing conclusions for a species by using a single strain.
In this study, we investigated the genomic evolution and diversity of the obligate intracellular Rickettsia spp. using phylogenomic and pangenomic meta-analyses from 34 arthropod-associated species which are represented by 80 genome sequences that we here reannotated. We investigated both the chromosomes and neglected plasmids, as both replicons are part of the historical life of this genus.
Results
In this study, we examined the diversity, evolution and adaptation of the genus Rickettsia from 34 species with validated names. The 80 studied strains had been isolated from diverse hosts between 1938 and 2012 worldwide (Table S1). In order to obtain a robust comparative genome analysis, we performed standard re-annotations of 61 complete genomes and 19 draft genomes sequences. Their general genomic features are summarised in Supplementary Table S1.
Phylogenomics and diversity
We first inferred a Maximum Likelihood phylogenomic tree from 461 concatenated core chromosomal proteins of the 80 studied strains (Fig. 1). Overall, our robust evolutionary tree showed that the Rickettsia genus has diverged into five distinct and well-supported major groups (BP = 100%): the Spotted Fever groups (SFGI and SFGII), the Typhus group (TG), the Canadensis group (CG) and the Bellii group (BG).
Figure 1.

Maximum likelihood phylogenomic tree of arthropod-associated Rickettsia spp. estimated from 461 core chromosomal genes containing 36,880 core genes of 34 species and 80 strains. For each strain, the columns show its chromosome size, gene content (pseudogenes in %), number of plasmids and their sizes (presented by the diameter of circles) and host (either familiar or taxonomic name), as well as the name of the disease that it causes in humans. A: Amblyomma, H: Homo sapiens, D: Dermacentor, R: Rhipicephalus, A. lagenoplastis: Argas lagenoplastis, H. leporispalustris/juxtacochi/sulcata: Haemaphysalis leporispalustris/juxtacochi/sulcata, H. asiaticum/marginatum/testudinarium/triguttatum: Hyalomma asiaticum/marginatum/testudinarium/triguttatum, I: Ixodes, A. sanguineus: Allodermanyssus sanguineus, C: Ctenocephalides, L: Liposcelis, C. capensis: Corvus capensis and B: Bandicota.
The SFGI rickettsiae group included several closely related species among which the core genome is highly conserved as reflected by short branches in the tree. This lineage is clearly split into two distinct sister sub-clades of rickettsiae (BP node supports = 100%), SFGIa and SFGIb, containing 20 and four species (e.g., R. conorii and R. endosymbiont of I. scapularis), respectively. The next well-resolved group is designated here as the SFGII rickettsiae group (also referred to as the transitional group, TRG), which includes five spotted fever rickettsial species (e.g., R. felis and R. australis). The core genome of this group appears to be more divergent compared with the SFGI group.
Another member of the SFG species, R. helvetica, was placed as an individual clade basal to the TG group but not the SFG group (Fig. 1). Examination of the multiple sequence alignments of Rickettsia core genes revealed that R. helvetica shares several non-synonymous mutations and large insertions/deletions with the SFGII, TG, CG and/or BG rickettsiae (Supplementary Fig. S1).
Remarkably, the TG group is clearly monophyletic and displays the longest and most diversified branch in the tree as compared with its close relatives (Fig. 1). This group accumulated several mutations in its core genome and then recently diverged into the R. prowazekii and R. typhi species. Last, the CG and BG groups were placed on two external branches of our phylogenomic tree. The former group includes R. canadensis and is clearly basal to the SFG and TG groups, whereas the latter contains R. bellii and is the earliest divergent outgroup among studied taxa.
Plasmids
Our genomic annotation detected 41 rickettsial plasmids, of which 11 were newly identified (Fig. 1, Table S1). These plasmids are present in 56% and 33% of examined species and strains, respectively. Thus, 44% and 67% of species and strains, respectively, had no plasmids. By mapping the current large-scale plasmid data set over the five rickettsial phylogroups (SFGI, SFGII, TG, CG and BG), we found that rickettsial plasmids were variably distributed between and within these lineages. They were present in only three phylogroups (SFGI, SFGII and BG), with between one and four plasmid(s) per strain. A large number of rickettsial plasmids (three or four) were only observed in three SFG species, R. raoultii, R. amblyommatis and R. buchneri.
Interestingly, we detected typical plasmidic genes in the chromosomes from the 11 plasmidless R. prowazekii strains and the two R. raoultii strains (one exhibiting three plasmids and the other being plasmidless). Indeed, each of the plasmidless R. prowazekii strains showed a cluster containing five pseudogenes (RprME0791 to RprME0807) that exhibited high homologies (ids = 67–92%) with rickettsial plasmid genes. Similarly, both R. raoultii strains harboured a cluster of six genes (Rra_909 to Rra_0916) that best matched (ids = 82–97%) six genes present exclusively in the R. peacockii plasmid (pRpe).
The phylogenetic trees computed from 5 selected genes common to several plasmids (Supplementary Fig. S2) and identified using the pan-plasmidome analysis revealed that each displayed two major ancestral nodes, one gathering all plasmids and the other all chromosomes of Rickettsia/Orientia. These results clearly show that these genes originated from a Rickettsia/Orientia ancestor harbouring one ancestral chromosome and one ancestral plasmid. Moreover, the supertree constructed from these five individual gene trees confirmed the distinction of both phylogenetic clades and their nodes, which were obtained in each of the 5 phylogenetic trees (Fig. 2).
Figure 2.
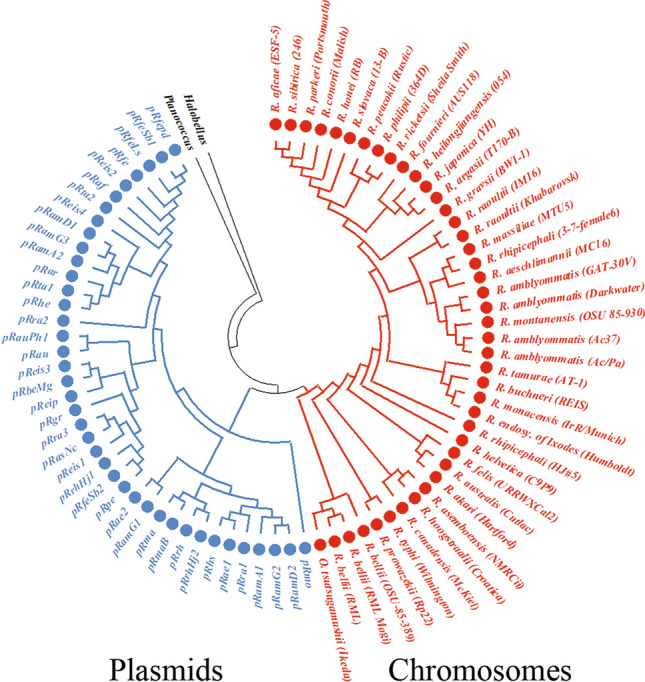
Supertree obtained from five genes of Rickettsia plasmids (n = 40) and chromosomes (n = 42) using the Robinson-Foulds algorithm (RF distance = 181). Orientia tsutsugamushi Ikeda (NC_010793.1), Planococcus antarcticus (ZP_10207867.1) and Halobellus rufus (WP_049986146.1) were used as best homologues to rickettsial genes. The list of genes used in the analysis includes dnaA-like replication initiator protein, helix-turn-helix DNA-binding domain, patatin-like phospholipase, cell surface antigen Sca12 and small heat shock protein (Supplementary Fig. S2).
Arthropod hosts
We compared the diversity of arthropods (as the primary hosts) associated with Rickettsia spp. across the five rickettsial phylogroups (Fig. 1). We found that most SFGI Rickettsia spp. were mainly associated with ticks (Acari), similarly to CG and BG rickettsiae. For example, the SFGI group was associated with seven genera and 27 species of ticks (Amblyomma, Argas, Dermacentor, Haemaphysalis, Hyalomma, Ixodes and Rhipicephalus). Moreover, some SFGI, CG and BG Rickettsia spp. were found to be associated with the same tick genus and/or species. However, SFGII rickettsiae were found in ticks, mites (Acari), fleas, and lice (Insecta). Furthermore, some rare strains of the four groups (SFGI, SFGII, CG and BG), as well as all those belonging to TG rickettsiae were identified in vertebrates such as humans or rats.
Pan-chromosome and pan-plasmidome
Using predicted genes from both chromosomes and plasmids, we separately inferred the Rickettsia pan-chromosome of 34 species (80 strains) and pan-plasmidome of 19 species (41 plasmids). The pan-chromosome of Rickettsia spp. included 4073 cRiGs (orthologous clusters of chromosomal rickettsial genes) clustered from 93,691 protein-coding genes (117,821 CDSs) (see an example of the pan-chromosome patterns across ten genomes in Supplementary Fig. S3), while the pan-plasmidome of Rickettsia spp. contained 457 pRiGs (orthologous clusters of plasmidic rickettsial genes) grouped from 1502 protein-coding genes (1893 CDSs).
Within the pan-chromosome, only 15% (599) of cRiGs represented the core chromosome (present in all strains), whereas the remaining 85% (3474) of cRiGs corresponded to the flexible chromosome (present in a subset of strains). In the pan-plasmidome, we did not find any pRiG depicting the core plasmidome (present in all plasmids) indicating that 100% (457) of pRiGs constituted the flexible plasmidome (present in a subset of plasmids). However, both the core and flexible chromosomes (or plasmidomes) exhibited proportions of the gene contents that were variably distributed across the five rickettsial phylogroups (SFGI, SFGII, TG, CG and BG) and their species. We found that between 31 and 53% of genes represented the core chromosome in SFGI group species (in which the smallest fractions were found in R. buchneri REIS and R. hoogstraalii RCCE3). These representations increased from 58 to 60% in CG species (e.g., in R. canadensis) and to 70% in TG species (e.g., in R. prowazekii) phylogroups. In BG species (R. bellii) core genes accounted for 43% of their gene contents.
We found that the saturation curve of the core chromosome quickly became relatively constant, indicating that the addition of more chromosomes would not influence its stability (Fig. 3A). However, the saturation curve of the pan-chromosome gradually increased with the increase in the number of chromosomes, but it seemed to reach a plateau at a later stage, suggesting that the Rickettsia pan-chromosome was comprehensively open, and that genomes from several additional strains would need to be included for it to be completely closed. When increasing the number of plasmids (from one to 28), the core plasmidome remained equal to zero, confirming the absence of genes common to all plasmids (Fig. 3B). However, the saturation curve of the pan-plasmidome was far from reaching a plateau, indicating that the Rickettsia pan-plasmidome is also open.
Figure 3.

Estimation of the core chromosome (y = 497 e−0.312x + 737; R2 = 0.95) and pan-chromosome (y = 1029 x0.23 + 87; R2 = 0.99) established through the analysis of 60 complete chromosomes of 28 Rickettsia spp. (A). Estimation of the core plasmidome (y = 144.2 e−1.48x + 0.36; R2 = 0.99) and pan-plasmidome (y = 100.7 x0.42—74.3; R2 = 0.99) simulated from 27 complete plasmids representing 15 Rickettsia spp. (B). Box plots show the median and quartiles.
Genome size, degradation and expansion
In our study, we examined the Rickettsia genome degradation (i.e., increase of pseudogenes or split genes) and expansion (i.e., increase in numbers of gene duplications, plasmids and prophages) together with their genome sizes across the five phylogroups in the chromosomes and/or plasmids.
Chromosomes
Rickettsia spp. displayed consistent differences in chromosome sizes between and within the five phylogroups: SFGI (from 1.2 to 1.7 Mb) to the SFGII (from 1.2 to 2.3 Mb), CG (1.15) and BG (1.5 Mb) (Fig. 1, Table S1). The TG phylogroup still exhibited the smallest chromosomes in the genus (1.1 Mb).
When we examined the chromosome degradations in these phylogroups, overall we found that the proportions of pseudogenes were comparable between four phylogroups (Fig. 1, Table S1): SFGI (from 29 to 42%, except in R. buchneri REIS), SFGII (from 25 to 35%, except in R. felis LSU), CG (from 27 to 29%) and BG (from 22 to 26%). However, we found consistent variations in the proportions of pseudogenes more within the SFGI and SFGII than in the CG, BG and TG rickettsiae. The latter phylogroup exhibited the smallest proportions of pseudogenes (from 13 to 18%). Two exceptional strains, R. buchneri REIS and R. felis LSU, exhibited the largest fractions of pseudogenes (50–52%) in the genus, suggesting that each may have chromosomes which are highly riddled by stop codons which may result from a neutral/adaptive event and/or an incomplete sequence assembly of highly repetitive chromosomes (see below).
Moreover, the analysis of the chromosome expansions across the five phylogroups revealed that the total numbers of chromosomal gene duplications were remarkably variable and higher in the SFGI (from 52 to 837) and SFGII (from 74 to 805) groups (Fig. 4). They decreased with low variations in the BG group (from 163 to 194) and much more in the CG (from 35 to 53) and TG (from 4 to 12) groups. Similarly, the frequency of chromosomal gene amplifications increased in the SFGI (from two to 186 copies) and SFGII (from 2 to 104 copies) groups and decreased in the BG group (from two to 27 copies) and much more in the CG (from two to nine copies) and TG (two copies) groups. Two strains of the SFGI and SFGII groups, R. buchneri REIS and R. hoogstraalii RCCE3 were a remarkable illustration of a massive genome expansion in the genus.
Figure 4.

Distributions of gene proliferations identified in the pan-chromosome across 34 Rickettsia spp. (80 strains or chromosomes). The x-axis represents the 80 Rickettsia chromosomes examined. The left y-axis shows the total number of gene copies present in each chromosome. For each chromosome, colours indicate the numbers of genes according to their copy counts (from 2 to 186). The right y-axis displays the total gene count per chromosome.
We then analysed the possible contribution of prophages in the chromosome sizes of some species representatives of each phylogroup that exhibited large variations in their chromosome sizes (Table S2). We detected no prophage in the smallest TG R. prowazekii Rp22 (1.1 Mbp) and CG R. canadensis (1.15 Mbp) chromosomes nor in the large SFGI R. asembonensis NMRCii chromosome (1.3 Mb). Moreover, all detected prophages were defective (either incomplete or questionable) and exhibited sizes ranging from 5.7 to 31.8 kb. When summing the sizes of these prophages for each species, we found that they represent less than 0.004% of the chromosome sizes in all phylogroups and species examined. As examples, the cumulated sizes of prophages represent only 0.0015%, 0.0004% and 0.0007% of the largest chromosomes, i.e., R. bellii RML369-C (1.5 Mb, BG), R. buchneri REIS (1.7 Mb, SFGI) and R. hoogstraalii RCCE3 (2.3 Mb, SFGII), respectively.
Plasmids
Plasmid sizes also varied between and within the SFGI (from 12.3 to 121.4 kb), SFGII (from 19.4 to 63.8 kb) and BG (28.7 kb) phylogroups (Fig. 1, Table S1). The pRaf, pRae1 and pRma plasmids were the smallest plasmids in the Rickettsia genus (about 12–15 kb in sizes). Moreover, the numbers of plasmids were also variable and greater in the SFGI (from zero to four plasmids) and SFGII (from zero to three) groups compared to the BG group (zero to one).
Examination of the plasmid degradation revealed that the proportion of pseudogenes was highly variable in the SFGI (from 0 to 64.7%) and SFGII (from 17.9 to 58.6%) phylogroups (Fig. 1, Table S1). The largest proportions (> 50) of pseudogenes were found in several SFG plasmids, either complete or draft (e.g., pRamA2, pReis4 and pRfeLs).
The distribution of the total numbers of gene duplications in plasmids varied within the SFGI (from two to 64) and SFGII (from four to 36) phylogroups (Fig. 5). However, the frequency of gene proliferations in plasmids was low (from two to seven copies).
Figure 5.

Distributions of gene proliferations identified in the pan-plasmidome across 19 Rickettsia spp. (26 strains, 41 plasmids). The x-axis represents the 26 Rickettsia strains, each harbouring from 1 to 4 plasmids. The left y-axis shows the total number of plasmidic gene copies present in each strain. For each plasmid, colours indicate the numbers of genes according to their copy counts (from 2 to 7). The right y-axis displays the total plasmidic gene count per strain.
Evolutionary networks
To infer the evolutionary relationships of the Rickettsia genus, we constructed an initial homology network of the pan-chromosome (4073 cRiGs, including 599 core and 3474 flexible genes) by identifying the first best hit of each cRiG in the pan-plasmidome and non-Rickettsia lineages (Fig. 6). This network excluded several plasmidic genes that did not have any sequence match with the pan-chromosome. Then, a network of the pan-plasmidome (457 flexible pRiGs) was constructed by detecting the first and second best hits of each pRiG in the pan-chromosome and non-Rickettsia lineages (Fig. 7). This latter analysis distinguished three gene sets: a first gene set in which the pan-plasmidome matched the pan-chromosome; a second gene set in which the pan-plasmidome matched other lineages; and a third gene set in which the pan-plasmidome matched both the pan-chromosome and other lineages.
Figure 6.

Evolutionary network of the Rickettsia pan-chromosome displaying best homologues from the Rickettsia pan-plasmidome and customised databases containing non-Rickettsia protein sequences of bacteria, archaea, eukarya and viruses. Nodes correspond to the pan-chromosome cRiGs. Node colours correspond to the core (in blue) or flexible (in red) cRiGs. Node sizes refer to sequence identities (from 30 to 100%) between a cRiG and its first best homologue, which are linked by blue edges. * unclassified bacteria.
Figure 7.

Evolutionary network of the Rickettsia pan-plasmidome exhibiting homologues from the Rickettsia pan-chromosome and customised databases containing non-Rickettsia protein sequences of bacteria, archaea, eukarya and viruses. Nodes represent the flexible pan-plasmidome pRiGs (in red). Node sizes refer to sequence identities (from 30 to 100%) between a pRiG and its first best homologue (blue edges). Some pRiGs also displayed second best homologues (red edges).
The pan-chromosome network
We found that 75% of the pan-chromosome displayed best sequence homologies (identities = 30–100%) with the pan-plasmidome and diverse bacterial, archaeal, eukaryotic and viral taxa (56 classes/105 orders/162 families) as well as other unclassified organisms (Fig. 6). Within this taxonomic pattern, the pan-chromosome shared best sequence similarities (34.6%, including 503 core and 906 flexible genes) with the α-proteobacteria (Fig. 6, see examples of genes in Table S3). Remarkably, a second large fraction of the pan-chromosome (25.3% including only three core and 1026 flexible genes) exhibited best sequence similarity matches with the pan-plasmidome. Interestingly, a third gene set of the pan-chromosome contained small proportions of genes (from 7.1 to 1.2%; including 60 core and 551 flexible genes) that exhibited a best hit with distant bacterial lineages largely belonging to γ-proteobacteria, cytophagia, and β/δ-proteobacteria (Fig. 6, Table S3). Smaller fractions of genes (from 0.9 to 0.02%) best matched 54 more distant taxonomic classes (either identified or unidentified) such as cyanobacteria, chlamydiia and viruses (Fig. 6, Table S3). The remaining pan-chromosome (25%) had no homologue with the pan-plasmidome and non-Rickettsia lineages available in the databases.
The pan-plasmidome network
Overall, 72% of the pan-plasmidome genes exhibited best similarities (ids = 30–100%) with the pan-chromosome, diverse lineages (15 classes/35 orders/47 families) and/or other unclassified organisms (Fig. 7), whereas the remaining 28% had no homologue with our datasets and databases. Among this taxonomic profile, 10% (as first gene set of the 72%) of the pan-plasmidome displayed best matches with the pan-chromosome only, and 18% (as second gene set of the 72%) exhibited best hits with α-proteobacteria (9%), followed by γ-proteobacteria (3.9%), cytophagia (0.9%), actinobacteria (1.3%), δ-proteobacteria (1.3%), β-proteobacteria (1.1%), and cyanobacteria (0.4%), and then with much smaller proportions with four lineages (0.2%) (Fig. 7, Table S3). The remaining 44% (as third gene set of the 72%) of the pan-plasmidome showed best sequence similarities with the pan-chromosome and several lineages including α-proteobacteria (19.1%), γ-proteobacteria (8.3%), cytophagia (5.4%), β-proteobacteria (2.4%), actinobacteria (2.2%), δ-proteobacteria (1.5%), cyanobacteria (0.7%) and other lineages (from 0.4 to 0.2%) (Fig. 7, Table S3).
Discussion
In this study, we examined the diversity, evolution and adaptation of arthropod-associated Rickettsia spp. using genomics, phylogenomics and pangenomics data analysis from 34 species (80 strains).
Overall, using phylogenomic analysis we found that the Rickettsia genus has diverged into five distinct major groups: two Spotted Fever groups (SFGI and SFGII), the Typhus group (TG), the Canadensis group (CG) and the Bellii group (BG). Our phylogenomic tree corroborates other rickettsial phylogenies estimated from subsets of species, strains and/or core genes/proteins2,21,22,26,30–36. However, although some of these phylogenies included a novel rickettsial clade (from Adalia)2,30,36 and four others (from Hydra spp., Torix spp., Rhizobius spp. and meloidae)2,4–6,30, they would not impact our robust five rickettsial phylogroups. Indeed, the Adalia clade was located between the CG and BG groups, while the other four clades were all external to the BG group. The rare specimens2,4–6,30,36 previously clustered in these new clades were not included in our study because no genomic sequence of these bacteria was available. Therefore, the BG phylogroup represents the outgroup of our data set, as it is the earliest divergent group among our taxa.
The 80 Rickettsia strains that we studied were isolated from diverse hosts worldwide, including Acari, Insecta and vertebrates. Examination of the diversity of arthropods of these bacterial species across the five rickettsial phylogroups revealed that the SFGI, CG and BG Rickettsia spp. were mainly associated with ticks (Acari). However, some species of these groups were also identified in mosquitoes, fleas and/or sheep keds (SFGI in Insecta)37–39, and in beetles, mosquitoes and/or flies (CG and BG in Insecta)2,40. The examined SFGII rickettsiae were detected in ticks, mites (Acari), fleas or lice (Insecta) and, in the case of R. felis, in ticks, mites (Acari) and mosquitoes (Insecta)3,39–43. Other Rickettsia spp. of our dataset were found to be associated with vertebrates2. For example, the TG species (R. prowazekii and R. typhi) were identified in humans and rats, respectively. However, these species are known to be associated with fleas and/or lice (Insecta)3, but were also detected in ticks44,45. On the other hand, our study found that 55% of Rickettsia strains were associated with ticks.
Previous data indicated that Rickettsia species are mainly associated with acarian and/or insect hosts, with a predominance in ticks. This entails two evolutionary scenarios that might have occurred for the Rickettsia-host association. First, an ancestor of Rickettsia spp. might have been associated with an acarian and/or an insect prior to its diversification. This suggests that Rickettsia spp. may have been maintained and have diverged inside diverse arthropod species through vertical transmissions (transovarial and trans-stadial). However, co-speciation between Rickettsia and arthropod species appears to be ruled out by the incongruence of phylogenetic trees31,46. Moreover, some Rickettsia spp. were found in association with various species of acarians and/or insects, suggesting that a strict co-speciation process is unlikely2. Episodic host changes between acarians, insects and/or vertebrates may have occurred by blood feeding or directly from infectious faeces of arthropods during the diversification of Rickettsia spp.2,33,42,47. Therefore, the large proportion of Rickettsia strains (i.e., 55%) detected in ticks does not necessarily indicate that they are more abundant in ticks than in other arthropods but may be related to the fact that ticks are more sampled than insects for medical but not environmental purposes.
Alternatively, a rickettsial ancestor might have originally been associated with other eukaryotic hosts, as suggested by recent studies that identified two novel rickettsial clades within the genus Rickettsia: the torix clade associated with amoeba, leeches, amphipods and arthropods, and the hydra clade associated with protists and unidentified environmental hosts2,4–6. Both clades are ancestral outgroups to our five phylogroups, suggesting other possible host changes over time2.
Thus, all these data indicate that Rickettsia-host associations, diversity and ecology are complex and may reflect complex adaptations and trajectories. While arthropods are the most diverse animal phylum living in diverse ecological ecosystems48,49, using a limited diversity of both partners may seriously influence and undermine knowledge of the evolutionary history of Rickettsia and their hosts. Further studies on the evolution, diversity and ecology of Rickettsia and relatives, together with their partners, would improve our understanding not only of Rickettsia-host relationships but also of Rickettsia phylogeny and classification.
Regarding plasmids, which are mobile genetic elements harboured by prokaryotic cells, they replicate independently of the chromosome and act as a major driving force in prokaryotic diversity, evolution and fitness50,51. In this study, we identified 41 plasmids in members of the Rickettsia genus. The presence of these plasmids suggests two possible evolutionary scenarios regarding their origin. First, the ancestor of rickettsiae harboured one plasmid system (or more), that was(were) inherited and vertically transmitted to its progeny. Indeed, the 41 studied plasmids are present in three rickettsial phylogroups (SFGI, SFGII, and BG). In addition to these SFG species, R. asiatica and R. akari also harbour plasmids52–54, respectively. Moreover, although, it has been reported that there were no instances of plasmid-associated genes within TG rickettsial genomes22, our pangenomic study showed that the plasmidless TG R. prowazekii species (11 strains) harboured a gene cluster integrated in its chromosomes that best matched rickettsial plasmidic genes. Similarly, the SFG species R. raoultii (two strains, one with three plasmids and the other being plasmidless) exhibited a gene cluster that best matched a R. peacockii plasmid in its chromosomes. These results indicated that the TG R. prowazekii and the SFG R. raoultii species likely had one and a fourth plasmid, respectively, that were disrupted and integrated in their chromosomes during rickettsial evolution28. Overall, we bring evidence that rickettsial plasmids were present in four of the five rickettsial phylogroups as either individual replicons or plasmid genes integrated in the chromosomes. In addition, our supertree gathered plasmids into a single phylogenetic clade which is distinct from that of the chromosomes, suggesting that rickettsial plasmids originated from a common plasmid ancestor. Altogether, these results on 41 plasmids confirmed our previous work performed on 20 plasmids28. A potential ancestral plasmid was also suggested for the genus Chlamydia, another strictly intracellular pathogen of humans and animals55.
The alternative hypothesis that rickettsial plasmids may have been acquired laterally from other organisms17,19–21,54 does not appear to be relevant. Indeed, we identified several orthologous gene clusters that were common to most plasmids, and our supertree did not display any evidence of plasmid horizontal transfer, thus supporting only the vertical inheritance hypothesis28.
Rickettsial plasmids were identified in 56% and 33% of studied species and strains, respectively, at a rate of one to four plasmids. In contrast, 44% and 67% of studied species and strains had no plasmids, respectively. This result indicated that, during rickettsial diversification from ancestral lineages and a likely vertical transmission, plasmids were probably retained, multiplied and/or lost between ancestral and present lineages. The retention, multiplication and loss of plasmids may be due to evolutionary adaptations of rickettsiae to either single or several eukaryotic hosts during their obligate intracellular lifestyle. The rickettsial adaptation by the persistence and multiplication of plasmids (> = one replicon) in some strains suggests that most plasmids may harbour functionally indispensable genes (either laterally acquired and/or duplicated, see “Discussion” below) conferring upon them beneficial traits that could contribute to their survival and/or fitness in arthropod and/or vertebrate hosts. Moreover, the retention, multiplication and potential key roles of plasmids in several Rickettsia spp. make this genus remarkable in comparison with several closely related genera in the order Rickettsiales (families Rickettsiaceae [genera Orientia, Occidentia, Phycorickettsia], Anaplamataceae [Anaplasma, Ehrlichia, Neorickettsia] and Candidatus Midichloriaceae [Candidatus Midichloria, Candidatus Aquarickettsia]), which are devoid of plasmids21,56–59. Moreover, only a single putative plasmid was identified in the intracellular genus Wolbachia, another member of the family Anaplamataceae60. This plasmid is smaller (9.2 Kb) than rickettsial plasmids (> 12.3 kb) and does not match their most common gene, the DnaA-like gene. This suggests a possible distinct origin from that of rickettsial plasmids.
On the other hand, the rickettsial adaptation by the loss of plasmids in some strains may result from the fact that plasmids became non-essential and represented a biological cost for these bacteria. This adaptive loss of plasmids may be total and due to the reductive evolutionary process, which may confer to rickettsiae another strategy of survival and fitness inside their hosts. The adaptive loss of plasmids may also be partial by maintaining, after a reductive disruption, some gene clusters via lateral gene incorporation into the chromosomes, as shown for example, in the plasmidless R. prowazekii, and in the R. raoultii species28, as well as in a Coxiella burnetti strain61. The loss of plasmids may also be accidental or result from numerous passages of the bacteria in selective cell culture, as observed for R. felis62. Some species, such as R. parkeri, R. typhi and R. canadensis, which are represented by a small number of strains (< = four), all plasmidless, require exhaustive population genomic investigation to confirm their status, as has been carried out for R. japonica63 and Orientia tustsugamushi, the closest relative genus to Rickettsia56. Therefore, more genomic data are needed to improve our knowledge of the diversity and distribution of plasmids in Rickettsia spp.
Genome reduction (or shrinkage) and expansion are other evolutionary processes that shape bacterial evolution, adaptation and diversity, leading to either their simplification or complexification11,19,64. Genome reduction is known to have occurred during the transition of bacteria from a free-living to obligate intracellular lifestyle11,64, and can result from both degradation or pseudogenisation and the loss of non-essential genes (either neutral ratchet or adaptive) under relaxed selective pressure and genetic drift65–70. However, genome expansion may take place by gene duplication and amplification (GDA forming paralogues) and horizontal gene transfer (HGT forming xenologues)71,72. Several studies showed that the genus Rickettsia undergoes reductive evolution10–14,19,20,28,29,31,34, but genome degradation and expansions remained less investigated in both the chromosomes and plasmids.
This study revealed consistent variations in the genome degradation and expansion between and within the five rickettsial phylogroups. Indeed, the SFG rickettsiae, followed by the BG rickettsiae, displayed the largest genomes in the genus and a high and differential degree of degradation and/or expansion by duplication in both the chromosomes and plasmids and/or an increase in plasmid numbers. It has been suggested that most gene expansions arrive via HGT and not via duplication71. The genome expansion identified in the SFG and BG species originated more from gene duplications and proliferations of both orthologous and/or xenologous genes. In contrast, the TG and CG rickettsiae have the smallest genomes in the genus and exhibited the lowest degree of genome degradation and expansions. These rickettsiae have the most reduced genomes in the genus10–14,19,20, and the loss of plasmids in these phylogroups may have contributed to this evolutionary process. Overall, these results show that the genus Rickettsia clearly fits with a general biphasic model of genomic evolution68 including genomic reduction by pseudogenisation and loss of genes and/or plasmids13,14,19,20,28,29,31,34, as well as genomic expansion by gene amplification73,74 and/or plasmid multiplication28. The SFG and BG species promote expansion rather than contraction, whereas the TG and CG species undergo a reduction rather than amplification. This suggests that members of the genus Rickettsia may have followed two distinct adaptive trajectories during its evolutionary history. Similar genomic evolutions were also described in several intracellular bacteria that underwent a genomic expansion (e.g., Orientia tsutsugamushi56,58, Wolbachia strain wFol75, Burkholderia mallei67 and Cardinium strain76) and/or a genomic contraction (e.g., Candidatus Fokinia solitaria77, Buchnera aphidicola, Mycobacteium leprae67,78, and Chlamydia spp.79).
SFG and BG rickettsiae as well as several closer or distant bacteria seemingly defied the reductive force which is characteristic of several symbiotic and/or pathogenic bacteria living in bottleneck intracellular niches. Moreover, SFG rickettsiae showed the highest core genome conservation in the genus as observed in the phylogenomic tree. All these results suggest that SFG and BG rickettsiae may be less dependent upon host cell resources for their survival and be less pathogenic than TG and CG rickettsiae. It may also explain why virulence in SFG rickettsiae may be multifactorial80. However, although the increase in virulence was associated with strong reductive evolution in TG rickettsiae29, the high level of core gene mutations observed in this phylogroup suggests that adaptive substitutions may also contribute to the pathogenicity and deadly diseases caused by these bacteria25, which requires further investigation.
Although they are intracellular bacteria exhibiting small genomes (from about 1.1 to 2.3 Mb), Rickettsia spp. exhibited a comprehensively open pan-chromosome and a completely open pan-plasmidome. Moreover, both Rickettsia “pans” shared genes with each other (25.3% and 39%, respectively) and/or α-proteobacteria (34.6% and 15%, respectively) as well as with distantly related and unrelated lineages largely belonging to γ/β/δ-proteobacteria, cytophagia, actinobacteria cyanobacteria, chlamydia, viruses and/or unknown taxa. These results suggest that rickettsial genomes harbour an important genomic diversity, and like several free-living prokaryotes81, Rickettsia spp. exhibited several genes which may have been laterally acquired (and duplicated or not) between their chromosomes, plasmids, some close α-proteobacteria relatives and/or distant lineages. Moreover, Rickettsia spp. contained an important proportion of genes (25–28%) that have no significant non-Rickettsia homologue in databases, suggesting the occurrence of more potential xenologues, whose origins remain to be discovered. More computational and experimental investigations are needed to determine the functions of critical genes for rickettsial survival and/or pathogenicity.
In addition, it is likely that arthropod-associated Rickettsia spp. have exchanged genetic material with microbiome members of these arthropods or other reservoirs in order to acquire genetic advantages and/or to amplify existing or new functions profitable for their survival, competition and/or fitness. Indeed, several lineages that shared homologous genes with Rickettsia spp. were previously identified among microbial communities in these ecological niches82–87. Among these taxa, Wolbachia (Rickettsiales), Candidatus Odyssella (Holosporales), Maritalea (Rhizobiales), Francisella spp., Legionella massiliensis, Pseudomonas, Candidatus Hamiltonella and/or Berkiella (γ-proteobacteria), Chromobacterium spp. (β-proteobacteria), Lawsonia intracellularis (δ-proteobacteria), Cardinium spp. (Cytophagia), Spirulina major (Cyanobacteria), Candidatus Protochlamydia amoebophila (Chlamydiia) and viruses (Caudovirales) can be cited as examples. Moreover, the mobile and integrative genetic elements found in rickettsial genomes, including 41 plasmids, 1036 transposase and integrase genes (23% of the pan-chromosome and 23% of the pan-plasmidome) and 178 conjugative tra genes (3.8% of the pan-chromosome and 5% of the pan-plasmidome)19,36,58,73,74, may have greatly orchestrated the strong cross-exchanges (or crosstalk), particularly for the SFG and BG phylogroups, and consequently the adaptation and fitness of Rickettsia spp. Thus, understanding the structure and dynamics of microbial communities in arthropods through metagenomic studies together with the mobile genetic elements will provide new insights into the dynamics of gene exchanges and interactions between Rickettsia and these micro-organisms.
Conclusion
During their diversification and adaptation to eukaryotic hosts (mainly arthropods), rickettsial genomes may have been shaped by diverse evolutionary processes. This study is the first large comparative evolutionary genomic investigation of members of the genus Rickettsia carried out on fully re-annotated chromosomes (80) and plasmids (41) of 34 identified species. The study shows that rickettsial phylogroups, as well as species or strains, may follow common and/or specific adaptive trajectories for their survival inside arthropods and/or probably their pathogenicity for vertebrate hosts. Rickettsia evolved through genomic degradation and reduction, and/or genomic expansion punctuated by probably short, explosive and innovative episodes of complexification. This trade-off evolution in the genus Rickettsia corroborates the general model of contraction/expansion of organisms proposed by Wolf and Koonin68 and may be due to their long-term intracellular lifestyle and partnerships as well as to the probable switching of arthropod hosts and interaction with their microbiomes.
Using recently updated public databases, our current results provide a robust insight into the evolutionary relationships between the genus Rickettsia and diverse distant lineages, thus improving the results of similar previous studies performed on some rickettsial species such as R. prowazekii13, R. bellii18, R. felis17,22,31,33,88 and R. massiliae89, or groups of Rickettsia species19,90. Deeper investigations on population genomics, novel Rickettsia species and metagenomics of arthropods are needed to improve our knowledge of their diversity, ecology and evolution.
Our work has shed light on the complex genomic history of the obligate intracellular Rickettsia spp. It represents a comprehensive overview into post-genomic and experimental studies such as RNAseq and small regulatory RNAs91–94, proteomics27,95–98, genetic transformation9 and gene functions16,99–101 aiming at better understanding the molecular mechanisms of critical genes driven by evolutionary processes, which have influenced the adaptation, survival and virulence of Rickettsia spp.
Material and methods
Genome sequences and reannotation
A total of 34 Rickettsia species depicted by 80 strains were investigated in this study. Studied strains were isolated from diverse arthropod hosts, animals or humans (clinical patients) around the world (Table S1). Their publicly available genomic sequences were downloaded from the ftp server (ftp://ftp.ncbi.nih.gov/Genome/) of the National Center for Biotechnology Information (NCBI). Of all studied genomes, 61 were complete and 19 were draft sequences. To avoid potential biases across the originally data deposited in NCBI that were obtained by different gene prediction methods and annotation pipelines, we fully re-analysed the raw genomic sequences. First, we distinguished between unidentified contigs of plasmids and chromosomes. This was performed by aligning each sequenced draft genome against the plasmid contigs of the complete genomes using the MAUVE software102,103. Second, all separated chromosomes and plasmid contigs (27 previously and 14 newly identified plasmids) were subjected to a standard CDS (CoDing Sequence > = 40aa) prediction with the AMIgene software104. Manual curation was then performed to reject false predicted CDSs. Automated functional re-annotation was performed against the RickBase19 and/or non-redundant protein databases using pipRick (an in-house annotation pipeline written in Perl language) according to the MicroScope Genome Annotation platform criteria105 and using the BLASTp algorithm106. Thus, two standardised databases from Rickettsia genomes and plasmids were constructed for further investigation. The prediction of the presence or absence of prophages in Rickettsia chromosomes was investigated using PHASTER (Phage Search Tool Enhanced release)107.
Pangenome inference
To construct the Rickettsia pangenome, we separately inferred the pan-chromosome and pan-plasmidome in the 34 Rickettsia species (80 strains). For each “pan”, we first performed an all-against-all protein search using both OrthoFinder108 (with default parameters) and Proteinortho109 (Cutoffs: E-value = 10–6 and coverage ≥ 60%) software programmes. We then extracted consensus pan-chromosome and pan-plasmidome, each containing clusters of orthologues of chromosomal and plasmidic rickettsial genes (that we refer to here as cRiGs and pRiGs, respectively) but not ORFs or CDSs as follows:
-
(i)
for both “pans”, we checked and uncollapsed each orthogroup (from OrthoFinder) with the help of cluster(s) of orthologous/paralogous genes (from Proteinortho) by performing visual/manual/scrutiny inspection and comparison of their accession numbers and/or protein sequence similarities and identifying clear cases of segregating paralogues which may be co-orthologues and not in-paralogues within a single orthogroup30. This strategy greatly helps the automatic and robust inference of pangenomic statistics of clusters of orthologous genes.
-
(ii)
retrieve pseudogenes defined as either split genes or gene fragments (i.e., genes interrupted by frameshifts or internal stop codons with at least one CDS having a match coverage < 80%) using a reference protein having the longest aa sequences within each cRiG or pRiG, BLAST best matches and annotation18,19,27,28. Indeed, in highly degraded genomes, such as Rickettsia genomes, the recognition of orthology/paralogy for pseudogenes remains a difficult task for pangenomic prediction programmes19,27. They can mistakenly place orthologous/paralogous genes into distinct or single orthology groups, consequently leading to an overestimation of the statistics of orthologous/paralogous clusters and the gene contents of taxa in a pan-genome study.
-
(iii)
discard some clusters displaying short (< 60 aa) and/or chimeric (< 100 aa) genes having no match or no significant hits with the GenBank database19.
Last, we estimated the pan-chromosome and pan-plasmidome sizes using the distance method implemented in the PanGP software110.
Network analysis
To delineate networks of evolutionary relationships between the Rickettsia pan-chromosome or Rickettsia pan-plasmidome and microbial and non-microbial organisms, we first searched homologies between the pan-chromosome (4073 reference protein sequences of cRiGs) and the non-redundant protein database (excluding Rickettsia species) using BLASTp (E-value = 10–6, coverage > = 60 and identity > = 30%). The same approach was then performed for the pan-plasmidome using the same criteria. All best matches were visually examined. After that, the pan-chromosome was searched for homologies against a customised database (i.e., a database constructed from the best NR and plasmid homologues packaged into single protein FASTA files) using the same BLASTp criteria to identify the first and/or the second best homologues that could be either plasmids and/or other microbial or non-microbial genes. Finally, results of the last homology searches were summarised into evolutionary networks between the pan-chromosome, pan-plasmidome and microbial as well as non-microbial protein sequences using the CYTOSCAPE software111. A similar strategy was used for the construction of the evolutionary network of the pan-plasmidome (457 reference protein sequences of each pRiG) as compared to the pan-chromosome, as well as microbial and non-microbial protein sequences (excluding Rickettsia species) from the non-redundant protein databases using the same thresholds.
Phylogenomic and supertree analysis
Multiple sequence alignment of the core chromosome (152,764 aligned sites of 461 cRiGs) was carried out using the MAFFT software112. The phylogenomic tree was obtained using the Maximum Likelihood method and the –m PROTGAMMAJTT parameter using raxmlHPC-PTHREADS, and then plotted with the iTOL113 and MEGA114 software programmes. Node robustness was estimated using Bootstrap (BP) analysis of 300 replicates.
To perform plasmid phylogenetic analysis and investigate their evolutionary origins, we first selected 5 genes common to several plasmids and identified using the pan-plasmidome analysis. Overall, we searched for complete genes that are present in plasmids. However, we removed genes that are degraded, as Rickettsia species evolved by reductive evolution through progressive gene degradation. Degraded genes or short gene fragments cannot be used in the construction of phylogenies as they can blur the signals. The list of the 5 plasmidic genes used in our analysis includes dnaA-like replication initiator protein, patatin-like phospholipase, helix-turn-helix DNA-binding domain, cell surface antigen Sca12 and heat shock protein. Then, we searched for the best homologous genes of these five genes using the Blast tool against our Rickettsia pan-chromosome gene and GenBank databases, as previously described28. Sequences of each of the 5 genes were subjected to multiple sequence alignment, and phylogenetic analyses using Neighbour-joining (NJ) or Maximum Likelihood (ML) methods. NJ and ML trees were performed using the JTT amino acid substitution matrix and the WAG model plus the Nearest-Neighbour-Interchange (NNI) under gamma (Γ) distribution, respectively. We then constructed a supertree from Neighbour-joining (NJ) or Maximum Likelihood (ML) newik trees of five selected genes using the Robinson-Foulds algorithm115. Supertree methods enable synthesizing collections of phylogenetic trees with incomplete taxon overlap into comprehensive trees, or supertrees, that combine the information contained in a set of input trees and include all taxa found in the input trees115,116.
Supplementary Information
Acknowledgements
The study was funded by the Fondation Méditerranée Infection.
Author contributions
K.E. and P.F. conceived the project. K.E. performed data analysis, wrote and edited the manuscript. K.E., E.G., P.F. and D.R. revised the manuscript. All authors approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Khalid El Karkouri, Email: khalid.elkarkouri@univ-amu.fr.
Pierre-Edouard Fournier, Email: pierre-edouard.fournier@univ-amu.fr.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-07725-z.
References
- 1.Perlman SJ, Hunter MS, Zchori-Fein E. The emerging diversity of Rickettsia. Proc. Biol. Sci. 2006;273:2097–2210. doi: 10.1098/rspb.2006.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinert LA, Werren JH, Aebi A, Stone GN, Jiggins FM. Evolution and diversity of Rickettsia bacteria. BMC Biol. 2009;7:6. doi: 10.1186/1741-7007-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parola P, et al. Update on tick-borne rickettsioses around the world: A geographic approach. Clin. Microbiol. Rev. 2013;26:657–702. doi: 10.1128/CMR.00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galindo LJ, et al. Combined cultivation and single-cell approaches to the phylogenomics of nucleariid amoebae, close relatives of fungi. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019 doi: 10.1098/rstb.2019.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pilgrim J, et al. Torix Rickettsia are widespread in arthropods and reflect a neglected symbiosis. Authorea. 2020 doi: 10.22541/au.159534851.19125003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park E, Poulin R. Widespread Torix Rickettsia in New Zealand amphipods and the use of blocking primers to rescue host COI sequences. Sci. Rep. 2020;10:16842. doi: 10.1038/s41598-020-73986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raoult D, Roux V. Rickettsioses as paradigms of new or emerging infectious diseases. Clin. Microbiol. Rev. 1997;10:694–719. doi: 10.1128/cmr.10.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renesto P, Ogata H, Audic S, Claverie JM, Raoult D. Some lessons from Rickettsia genomics. FEMS Microbiol. Rev. 2005;29:99–117. doi: 10.1016/j.femsre.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Burkhardt NY, et al. Development of shuttle vectors for transformation of diverse Rickettsia species. PLoS ONE. 2011;6:e29511. doi: 10.1371/journal.pone.0029511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersson SG, Eriksson AS, Naslund AK, Andersen MS, Kurland CG. The Rickettsia prowazekii genome: A random sequence analysis. Microb. Comp. Genom. 1996;1:293–315. [PubMed] [Google Scholar]
- 11.Andersson SGE, Kurland CG. Reductive evolution of resident genomes. Trends Microbiol. 1998;6:263–268. doi: 10.1016/s0966-842x(98)01312-2. [DOI] [PubMed] [Google Scholar]
- 12.Andersson SGE, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- 13.Andersson JO, Andersson SGE. Insights into the evolutionary process of genome degradation. Curr. Opin. Genet. Dev. 1999;9:664–671. doi: 10.1016/s0959-437x(99)00024-6. [DOI] [PubMed] [Google Scholar]
- 14.Ogata H, et al. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science. 2001;293:2093–2098. doi: 10.1126/science.1061471. [DOI] [PubMed] [Google Scholar]
- 15.McLeod MP, et al. Complete genome sequence of Rickettsia typhi and comparison with sequences of other rickettsiae. J. Bacteriol. 2004;186:5842–5855. doi: 10.1128/JB.186.17.5842-5855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gouin E, et al. The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature. 2004;427:457–461. doi: 10.1038/nature02318. [DOI] [PubMed] [Google Scholar]
- 17.Ogata H, et al. The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. PLoS Biol. 2005;3:248. doi: 10.1371/journal.pbio.0030248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogata H, et al. Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PLoS Genet. 2006;2:76. doi: 10.1371/journal.pgen.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blanc G, et al. Reductive genome evolution from the mother of Rickettsia. PLoS Genet. 2007;3:14. doi: 10.1371/journal.pgen.0030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darby AC, Cho NH, Fuxelius HH, Westberg J, Andersson SG. Intracellular pathogens go extreme: Genome evolution in the Rickettsiales. Trends Genet. 2007;23:511–520. doi: 10.1016/j.tig.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Gillespie JJ, et al. Plasmids and rickettsial evolution: Insight from Rickettsia felis. PLoS ONE. 2007;2:266. doi: 10.1371/journal.pone.0000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillespie JJ, et al. Rickettsia phylogenomics: Unwinding the intricacies of obligate intracellular life. PLoS ONE. 2008;3:e2018. doi: 10.1371/journal.pone.0002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellison DW, et al. Genomic comparison of virulent Rickettsia rickettsii Sheila Smith and avirulent Rickettsia rickettsii Iowa. Infect. Immun. 2008;76:542–550. doi: 10.1128/IAI.00952-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Felsheim RF, Kurtti TJ, Munderloh UG. Genome sequence of the endosymbiont Rickettsia peacockii and comparison with virulent Rickettsia rickettsii: identification of virulence factors. PLoS ONE. 2009;4:e8361. doi: 10.1371/journal.pone.0008361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bechah Y, et al. Genomic, proteomic, and transcriptomic analysis of virulent and avirulent Rickettsia prowazekii reveals its adaptive mutation capabilities. Genome Res. 2010;20:655–663. doi: 10.1101/gr.103564.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillespie JJ, et al. Secretome of obligate intracellular Rickettsia. FEMS Microbiol. Rev. 2015;39:80. doi: 10.1111/1574-6976.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El Karkouri K, et al. Multi-omics analysis sheds light on the evolution and the intracellular lifestyle strategies of spotted fever group Rickettsia spp. Front. Microbiol. 2017;8:1363. doi: 10.3389/fmicb.2017.01363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El Karkouri K, Pontarotti P, Raoult D, Fournier PE. Origin and evolution of rickettsial plasmids. PLoS ONE. 2016;11:e0147492. doi: 10.1371/journal.pone.0147492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fournier PE, et al. Analysis of the Rickettsia africae genome reveals that virulence acquisition in Rickettsia species may be explained by genome reduction. BMC Genom. 2009;10:166. doi: 10.1186/1471-2164-10-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murray GG, Weinert LA, Rhule EL, Welch JJ. The phylogeny of rickettsia using different evolutionary signatures: How tree-like is bacterial evolution? Syst. Biol. 2016;65:265–279. doi: 10.1093/sysbio/syv084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merhej V, Raoult D. Rickettsial evolution in the light of comparative genomics. Biol. Rev. Camb. Philos. Soc. 2011;86:379–405. doi: 10.1111/j.1469-185X.2010.00151.x. [DOI] [PubMed] [Google Scholar]
- 32.Vitorino L, Chelo IM, Bacellar F, Zé-Zé L. Rickettsiae phylogeny: A multigenic approach. Microbiol. Read. 2007;153:160–168. doi: 10.1099/mic.0.2006/001149-0. [DOI] [PubMed] [Google Scholar]
- 33.Gillespie JJ, et al. Genomic diversification in strains of Rickettsia felis Isolated from different arthropods. Genome Biol. Evol. 2014;7:35–56. doi: 10.1093/gbe/evu262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merhej V, Angelakis E, Socolovschi C, Raoult D. Genotyping, evolution and epidemiological findings of Rickettsia species. Infect. Genet. Evol. 2014;25:122–137. doi: 10.1016/j.meegid.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 35.Stothard DR, Clark JB, Fuerst PA. Ancestral divergence of Rickettsia bellii from the spotted fever and typhus groups of Rickettsia and antiquity of the genus Rickettsia. Int. J. Syst. Bacteriol. 1994;44:798–804. doi: 10.1099/00207713-44-4-798. [DOI] [PubMed] [Google Scholar]
- 36.Weinert LA, Welch JJ, Jiggins FM. Conjugation genes are common throughout the genus Rickettsia and are transmitted horizontally. Proc. Biol. Sci. 2009;276:3619–3627. doi: 10.1098/rspb.2009.0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo W, Tian J, Lin X. Extensive genetic diversity of Rickettsiales bacteria in multiple mosquito species. Sci. Rep. 2016;6:38770. doi: 10.1038/srep38770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu D, et al. First report of Rickettsia raoultii and R. slovaca in Melophagus ovinus, the sheep ked. Parasit. Vect. 2016;9:600. doi: 10.1186/s13071-016-1885-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radzijevskaja J, et al. Prevalence and diversity of Rickettsia species in ectoparasites collected from small rodents in Lithuania. Parasit. Vect. 2018;11:375. doi: 10.1186/s13071-018-2947-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, et al. Molecular detection of Rickettsia felis and Rickettsia bellii in mosquitoes. Vect. Borne Zoon. Dis. 2019 doi: 10.1089/vbz.2019.2456. [DOI] [PubMed] [Google Scholar]
- 41.Oliveira KA, et al. Molecular identification of Rickettsia felis in ticks and fleas from an endemic area for Brazilian spotted fever. Mem. Inst. Oswaldo Cruz. 2008;103:191–194. doi: 10.1590/s0074-02762008000200011. [DOI] [PubMed] [Google Scholar]
- 42.Choi YJ, et al. Molecular detection of various rickettsiae in mites (acari: trombiculidae) in southern Jeolla Province, Korea. Microbiol. Immunol. 2007;51:307–312. doi: 10.1111/j.1348-0421.2007.tb03912.x. [DOI] [PubMed] [Google Scholar]
- 43.Brown LD, Macaluso KR. Rickettsia felis, an emerging flea-borne rickettsiosis. Curr. Trop. Med. Rep. 2016;3:27–39. doi: 10.1007/s40475-016-0070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medina-Sanchez A, et al. Detection of a typhus group Rickettsia in Amblyomma ticks in the state of Nuevo Leon, Mexico. Ann. N. Y. Acad. Sci. 2005;1063:327–332. doi: 10.1196/annals.1355.052. [DOI] [PubMed] [Google Scholar]
- 45.Sprong H, et al. Ixodes ricinus ticks are reservoir hosts for Rickettsia helvetica and potentially carry flea-borne Rickettsia species. Parasit. Vect. 2009;2:41. doi: 10.1186/1756-3305-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Modeo L, et al. Candidatus Trichorickettsia mobilis, a Rickettsiales bacterium, can be transiently transferred from the unicellular eukaryote Paramecium to the planarian Dugesia japonica. PeerJ. 2020;8:e8977. doi: 10.7717/peerj.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng-Nguyen D, et al. Domestic dogs are mammalian reservoirs for the emerging zoonosis flea-borne spotted fever, caused by Rickettsia felis. Sci. Rep. 2020;10:4151. doi: 10.1038/s41598-020-61122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgecombe GD, Legg DA. Origins and early evolution of arthropods. Palaeontology. 2014;57:457–468. [Google Scholar]
- 49.Giribet G, Edgecombe GD. The phylogeny and evolutionary history of arthropods. Curr. Biol. 2019;29:R592–R602. doi: 10.1016/j.cub.2019.04.057. [DOI] [PubMed] [Google Scholar]
- 50.Tamminen M, Virta M, Fani R, Fondi M. Large-scale analysis of plasmid relationships through gene-sharing networks. Mol. Biol. Evol. 2012;29:1225–1240. doi: 10.1093/molbev/msr292. [DOI] [PubMed] [Google Scholar]
- 51.Hülter NF, Wein T, Effe J, Garoña A, Dagan T. Intracellular competitions reveal determinants of plasmid evolutionary success. Front. Microbiol. 2020;11:2062. doi: 10.3389/fmicb.2020.02062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thu MJ, et al. Complete genome sequence of Rickettsia asiatica strain Maytaro1284, a member of spotted fever group Rickettsiae isolated from an Ixodes ovatus tick in Japan. Microbiol. Resour. Announc. 2019;8:00886–919. doi: 10.1128/MRA.00886-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baldridge GD, Burkhardt NY, Felsheim RF, Kurtti TJ, Munderloh UG. Plasmids of the pRM/pRF family occur in diverse Rickettsia species. Appl. Environ. Microbiol. 2008;74:645–652. doi: 10.1128/AEM.02262-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baldridge GD, et al. Wide dis-persal and possible multiple origins of low-copy-number plasmids in rickettsia species associated withblood-feeding arthropods. Appl. Environ. Microbiol. 2010;76:1718–1731. doi: 10.1128/AEM.02988-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szabo KV, O’Neill CE, Clarke IN. Diversity in chlamydial plasmids. PLoS ONE. 2020;15:e0233298. doi: 10.1371/journal.pone.0233298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fleshman A, et al. Comparative pan-genomic analyses of Orientia tsutsugamushi reveal an exceptional model of bacterial evolution driving genomic diversity. Microb. Genom. 2018;4:000199. doi: 10.1099/mgen.0.000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mediannikov O, et al. High quality draft genome sequence and description of Occidentia massiliensis gen. nov., sp. Nov., a new member of the family Rickettsiaceae. Stand. Genomic. Sci. 2014;9:9. doi: 10.1186/1944-3277-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cho NH, et al. The Orientia tsutsugamushi genome reveals massive proliferation of conjugative type IV secretion system and host-cell interaction genes. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7981–7986. doi: 10.1073/pnas.0611553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klinges JG, et al. Phylogenetic, genomic, and biogeographic characterization of a novel and ubiquitous marine invertebrate-associated Rickettsiales parasite, Candidatus Aquarickettsia rohweri, gen. nov., sp. Nov. ISME J. 2019;13:2938–2953. doi: 10.1038/s41396-019-0482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reveillaud J, et al. The Wolbachia mobilome in Culex pipiens includes a putative plasmid. Nat. Commun. 2019;10:1051. doi: 10.1038/s41467-019-08973-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Willems H, Ritter M, Jäger C, Thiele D. Plasmid-homologous sequences in the chromosome of plasmidless Coxiella burnetii Scurry Q217. J. Bacteriol. 1997;179:3293–3297. doi: 10.1128/jb.179.10.3293-3297.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fournier PE, et al. Variations of plasmid content in Rickettsia felis. PLoS ONE. 2008;3:2289. doi: 10.1371/journal.pone.0002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akter A, et al. Extremely low genomic diversity of Rickettsia japonica distributed in Japan. Genome Biol. Evol. 2017;9:124–133. doi: 10.1093/gbe/evw304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryant J, Chewapreecha C, Bentley SD. Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Fut. Microbiol. 2012 doi: 10.2217/fmb.12.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wixon J. Featured organism: reductive evolution in bacteria: Buchnera sp., Rickettsia prowazekii and Mycobacterium leprae. Comparat. Funct. Genom. 2001;2:44–48. doi: 10.1002/cfg.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khachane AN, Timmis KN, Santos VA. Dynamics of reductive genome evolution in mitochondria and obligate intracellular microbes. Mol. Biol. Evol. 2007;24:449–456. doi: 10.1093/molbev/msl174. [DOI] [PubMed] [Google Scholar]
- 67.Song H, et al. The early stage of bacterial genome-reductive evolution in the host. PLoS Pathog. 2010;6:e1000922. doi: 10.1371/journal.ppat.1000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolf YI, Koonin EV. Genome reduction as the dominant mode of evolution. BioEssays. 2013;35:829–837. doi: 10.1002/bies.201300037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. RELAX: detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 2015;32:820–832. doi: 10.1093/molbev/msu400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weinert LA, Welch JJ. Why might bacterial pathogens have small genomes? Trends Ecol. Evol. 2017;32:936–947. doi: 10.1016/j.tree.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 71.Treangen TJ, Rocha EP. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011;7:e1001284. doi: 10.1371/journal.pgen.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elliott KT, Cuff LE, Neidle EL. Copy number change: Evolving views on gene amplification. Fut. Microbiol. 2013;8:887–899. doi: 10.2217/fmb.13.53. [DOI] [PubMed] [Google Scholar]
- 73.Gillespie JJ, et al. A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle. J. Bacteriol. 2012;194:376–394. doi: 10.1128/JB.06244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hagen R, Verhoeve VI, Gillespie JJ, Driscol TP. Conjugative Transposons and Their Cargo Genes Vary across Natural Populations of Rickettsia buchneri Infecting the Tick Ixodes scapularis. Genome Biol. Evol. 2018;10:3218–3229. doi: 10.1093/gbe/evy247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kampfraath AA, et al. Genome expansion of an obligate parthenogenesis-associated Wolbachia poses an exception to the symbiont reduction model. BMC Genom. 2019;20:106. doi: 10.1186/s12864-019-5492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brown AMV, et al. Comparative genomics of Wolbachia-Cardinium dual endosymbiosis in a plant-parasitic nematode. Front. Microbiol. 2018;9:2482. doi: 10.3389/fmicb.2018.02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Floriano AM, et al. The genome sequence of ‘Candidatus Fokinia solitaria’: Insights on reductive evolution in Rickettsiales. Genome Biol. Evol. 2018;10:1120–1126. doi: 10.1093/gbe/evy072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chong RA, Park H, Moran NA. Genome evolution of the obligate endosymbiont Buchnera aphidicola. Mol. Biol. Evol. 2019;36:1481–1489. doi: 10.1093/molbev/msz082. [DOI] [PubMed] [Google Scholar]
- 79.Sigalova OM, et al. Chlamydia pan-genomic analysis reveals balance between host adaptation and selective pressure to genome reduction. BMC Genom. 2019;20:710. doi: 10.1186/s12864-019-6059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Clark TR, et al. Comparative genome sequencing of Rickettsia rickettsii strains that differ in virulence. Infect. Immun. 2015;83:1568–1576. doi: 10.1128/IAI.03140-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Galperin MY, Kristensen DM, Makarova KS, Wolf YI, Koonin EV. Microbial genome analysis: The COG approach. Brief Bioinform. 2019;20:1063–1070. doi: 10.1093/bib/bbx117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Greay TL, et al. Recent insights into the tick microbiome gained through next-generation sequencing. Parasit. Vect. 2018;11:12. doi: 10.1186/s13071-017-2550-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ravi A, et al. Metagenomic profiling of ticks: Identification of novel rickettsial genomes and detection of tick-borne canine parvovirus. PLoS Negl. Trop. Dis. 2019;13:e0006805. doi: 10.1371/journal.pntd.0006805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhuang L, et al. Identification of tick-borne pathogen diversity by metagenomic analysis in Haemaphysalis longicornis from Xinyang China. Infect. Dis. Poverty. 2018;7:45. doi: 10.1186/s40249-018-0417-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tokarz R, et al. Microbiome analysis of Ixodes scapularis ticks from New York and connecticut. Ticks Tick Borne Dis. 2019;10:894–900. doi: 10.1016/j.ttbdis.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 86.Thoendel M. Targeted metagenomics offers insights into potential tick-borne pathogens. J. Clin. Microbiol. 2020;58:e01893–e1920. doi: 10.1128/JCM.01893-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Duan DY, Liu GH, Cheng TY. Microbiome analysis of the saliva and midgut from partially or fully engorged female adult Dermacentor silvarum ticks in China. Exp. Appl. Acarol. 2020;80:543–558. doi: 10.1007/s10493-020-00478-2. [DOI] [PubMed] [Google Scholar]
- 88.Merhej V, Notredame C, Royer-Carenzi M, Pontarotti P, Raoult D. The rhizome of life: the sympatric Rickettsia felis paradigm demonstrates the random transfer of DNA sequences. Mol. Biol. Evol. 2011;28:3213–3223. doi: 10.1093/molbev/msr239. [DOI] [PubMed] [Google Scholar]
- 89.Blanc G, et al. Lateral gene transfer between obligate intracellular bacteria: Evidence from the Rickettsia massiliae genome. Genome Res. 2007;17:1657–1664. doi: 10.1101/gr.6742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duron O. Lateral transfers of insertion sequences between WolbachiaCardinium and Rickettsia bacterial endosymbionts. Heredity. 2013;111:330–337. doi: 10.1038/hdy.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schroeder CLC, et al. Transcriptional profiling of Rickettsia prowazekii coding and non-coding transcripts during in vitro host-pathogen and vector-pathogen interactions. Ticks Tick Borne Dis. 2017;8:827–836. doi: 10.1016/j.ttbdis.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Narra HP, et al. Small Regulatory RNAs of Rickettsia conorii. Sci. Rep. 2016;6:36728. doi: 10.1038/srep36728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Narra HP, et al. Comparative transcriptomic analysis of Rickettsia conorii during in vitro infection of human and tick host cells. BMC Genom. 2020;21:665. doi: 10.1186/s12864-020-07077-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mika-Gospodorz B, et al. Dual RNA-seq of Orientia tsutsugamushi informs on host-pathogen interactions for this neglected intracellular human pathogen. Nat. Commun. 2020;11:3363. doi: 10.1038/s41467-020-17094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Renesto P, et al. Rickettsia conorii and R. prowazekii proteome analysis by 2DE-MS: a step toward functional analysis of rickettsial genomes. Ann. N. Y. Acad. Sci. 2005;1063:90–93. doi: 10.1196/annals.1355.014. [DOI] [PubMed] [Google Scholar]
- 96.Ogawa M, Renesto P, Azza S, Moinier D, Fourquet P. Proteome analysis of Rickettsia felis highlights the expression profile of intracellular bacteria. Proteomics. 2007;7:1232–1248. doi: 10.1002/pmic.200600721. [DOI] [PubMed] [Google Scholar]
- 97.Martins LA, et al. The intracellular bacterium Rickettsia rickettsii exerts an inhibitory effect on the apoptosis of tick cells. Parasit. Vectors. 2020;13:603. doi: 10.1186/s13071-020-04477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Csicsay F, et al. Proteomic analysis of Rickettsia akari proposes a 44 kDa-OMP as a potential biomarker for Rickettsialpox diagnosis. BMC Microbiol. 2020;20:200. doi: 10.1186/s12866-020-01877-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rennoll-Bankert KE, et al. Which way in? The RalF Arf-GEF orchestrates Rickettsia host cell invasion. PLoS Pathog. 2015;11:e1005115. doi: 10.1371/journal.ppat.1005115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Engström P, et al. Evasion of autophagy mediated by Rickettsia surface protein OmpB is critical for virulence. Nat. Microbiol. 2019;4:2538–2551. doi: 10.1038/s41564-019-0583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Voss OH, et al. Risk1, a phosphatidylinositol 3-kinase effector promotes Rickettsia typhi intracellular survival. MBio. 2020;11:e00820–e920. doi: 10.1128/mBio.00820-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rissman AI, et al. Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics. 2009;25:2071–2073. doi: 10.1093/bioinformatics/btp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bocs S, Cruveiller S, Vallenet D, Nuel G, Medigue C. AMIGene: Annotation of microbial genes. Nucleic Acids Res. 2003;31:3723–3726. doi: 10.1093/nar/gkg590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vallenet D, et al. MicroScope: a platform for microbial genome annotation and comparative genomics. Database. 2009;21:2. doi: 10.1093/database/bap021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Altschul SF, et al. Gapped BLAST and PSIBLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Arndt D, Marcu A, Liang Y, Wishart DS. PHAST, PHASTER and PHASTEST: Tools for finding prophage in bacterial genomes. Brief Bioinform. 2019;19:1560–1567. doi: 10.1093/bib/bbx121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Emms DM, Kelly S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lechner M, et al. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhao Y, et al. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics. 2014;30:1297–1299. doi: 10.1093/bioinformatics/btu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shannon P, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Katoh K, Rozewicki J, Yamada KD. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Letunic I, Bork P. Interactive tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019;47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bansal MS, Burleigh JG, Eulenstein O, Fernández-Baca D. Robinson-foulds supertrees. Algorithms Mol. Biol. 2010;5:18. doi: 10.1186/1748-7188-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Akanni WA, Wilkinson M, Creevey CJ, Foster PG, Pisani D. Implementing and testing Bayesian and maximum-likelihood supertree methods in phylogenetics. R. Soc. open sci. 2015;2:140436–140436. doi: 10.1098/rsos.140436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.