

Abstract
A 49-year-old Japanese man had shown developmental delay, learning difficulties, epilepsy, and slowly progressive gait disturbance in elementary school. At 46 years old, he experienced repeated drowsiness with or without generalized convulsions, and hyperammonemia was detected. Brain magnetic resonance imaging detected multiple cerebral white matter lesions. An electroencephalogram showed diffuse slow basic activities with 2- to 3-Hz δ waves. Genetic tests confirmed a diagnosis of hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Leukoencephalopathy was resolved following the administration of L-arginine and lactulose with a decrease in plasma ammonia levels and glutamine-glutamate peak on magnetic resonance spectroscopy. Leukoencephalopathy in HHH syndrome may be reversible with the resolution of hyperammonemia-induced glutamine toxicity.
Keywords: HHH syndrome, leukoencephalopathy, MRS, magnetic resonance spectroscopy, hyperammonemia, glutamine toxicity
Introduction
Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome (OMIM: 238970) is a rare autosomal recessive urea cycle disorder (UCD) caused by biallelic pathogenic variants in the SLC25A15 gene, which encodes ornithine translocase (1,2). The exact incidence of HHH syndrome is unclear but is estimated to be less than 1:2,000,000 (3). To our knowledge, only 15 Japanese patients with HHH syndrome have been reported in the literature (4-13). Patients with HHH syndrome show various neurological symptoms, such as intellectual disability, epilepsy, pyramidal dysfunction, and ataxia (1,2). They also develop encephalopathy associated with hyperammonemia, as seen in other types of UCDs (1). In HHH syndrome, brain magnetic resonance imaging (MRI) detects abnormalities, such as mild cerebral and cerebellar atrophy, white matter changes (14,15), cystic lesions and calcifications (14), and diffuse brain edema (16). However, to our knowledge, no studies using magnetic resonance spectroscopy (MRS) have investigated the effects of therapeutic intervention on abnormal radiological findings in patients with HHH syndrome.
We herein report the improvement of leukoencephalopathy by L-arginine and lactulose administration and reduction in the glutamine-glutamate (Glx) peak on MRS in a 49-year-old man with childhood-onset HHH syndrome.
Case Report
A 49-year-old Japanese man with healthy, consanguineous parents had displayed developmental delay, learning difficulties, and slowly progressive gait disturbance during elementary school. He had no history of perinatal complications or any neurological diseases in his family. He presented with convulsions and was diagnosed with epilepsy at 11 years old. Despite his dropping out of ambulatory treatment, the epileptic episodes disappeared.
At 18 years old, his intelligence quotient evaluated using the Tanaka-Binet test was 49. After graduating from a high school for disabled students, he started work but retired after a month because of poor relationships with his colleagues. He subsequently lived at home with his mother. At 46 years old, he entered a group home, where he experienced repeated drowsiness with or without generalized convulsions. He was admitted to another hospital, and antiepileptic drugs (levetiracetam and lacosamide) were administered.
At that time, brain MRI showed multiple high-intensity lesions in the cerebral white matter on fluid-attenuated inversion recovery (FLAIR) imaging sequence (Fig. 1A). Hyperammonemia was detected for the first time. However, no intervention for hyperammonemia was started at this time. Transient drowsiness, with or without convulsions, occurred about once a month. At 49 years old, he was admitted to our hospital.
Figure 1.

Brain MRI and MRS. (A) FLAIR sequence image showing multiple high-intensity lesions in the cerebral white matter. (B) Twenty-eight months after the initial MRI evaluation, FLAIR detected cerebral white matter lesions with progression of cerebral atrophy. (C) FLAIR showing improvement in leukoencephalopathy with mild progression of cerebral atrophy two months after L-arginine and lactulose therapy. (D) MRS demonstrates a high Glx peak with normal peaks for NAA and GPC-PCh before treatment. (E) The decreased Glx peak on MRS is apparent eight months after initiating L-arginine and lactulose therapy. All 1H-MRS scans were obtained using a Prisma 3.0-T Siemens system (Siemens Medical Solutions, Erlangen, Germany). A single-voxel MRS scan was obtained with a point-resolved spectroscopy sequence with water suppression: TR/TE=2,000/30 ms, voxel size=8 mL (2×2×2 cm3). The voxel was placed in the right semioval center. A non-water-suppressed spectrum was also obtained for the eddy-current correction. All MRS metabolites were quantified by fitting the experimental data in the frequency domain using the LCModel method (22). Ratios of the metabolite concentrations were calculated with respect to the total Cr and PCr values. Cr: creatine, FLAIR: fluid-attenuated inversion recovery, Glx: glutamine plus glutamate, GPC: glycerophosphocholine, MRI: magnetic resonance imaging, MRS: magnetic resonance spectroscopy, NAA: N-acetyl aspartylglutamic acid, PCh: phosphocholine, PCr: phosphocreatine, TE: echo time, TR: repetition time
On a physical examination, no abnormal findings were noted. On a neurological examination, he was slightly disoriented (Glasgow Coma Scale, E4V4M6). The Mini-Mental State Examination (MMSE) score was moderately decreased (22/30). Apart from a small voice, the cranial nerve functions were normal. Asterixis, mild spasticity, and brisk tendon reflexes without weakness were observed in the upper limbs. A lower limb examination demonstrated spasticity and weakness [i.e., iliopsoas and hamstring muscles, 4/4; tibialis anterior muscle, 2/2; and other muscles, 5/5, on the Medical Research Council Scale (range, 0-5)] with exaggerated tendon reflexes, ankle clonus, and pathological reflexes. Cerebellar ataxia and dysautonomia were absent. While he was able to walk without aid, his gait was spastic and unstable.
Tests for a complete blood count, the hepatorenal function, glucose, and C-reactive protein showed no abnormalities. The plasma ammonia level was elevated (176 μg/dL; normal range, 12-66). A plasma amino-acid analysis showed high levels of glutamine, ornithine, and citrulline at 1031 (range, 420-700), 559 (range, 0-100), and 51.2 (range, 17-43) nmol/mL, respectively. Brain MRI showed cerebral white matter lesions with progression of cerebral atrophy (Fig. 1B). A high signal of globus pallidus on T1-weighted imaging was unapparent. MRS of the white matter lesion of the right semioval center detected high peaks of Glx (Fig. 1D). Spinal MRI showed no abnormalities. Single-photon emission computed tomography detected a diffuse decrease in perfusion, except to the cerebellum. An electroencephalogram (EEG) conducted while awake showed diffuse slow basic activities with 2- to 3-Hz δ waves without epileptic activities (Fig. 2A), suggestive of encephalopathy. No motor potentials were evoked in the upper or lower limbs on transcranial magnetic stimulation. Sanger sequencing revealed a homozygous nonsense variant in SLC25A15 (NM_014252.3:c.535C>T: p.Arg179*). This SLC25A15 gene variant has been reported as a pathogenic variant in the HHH syndrome (1), thereby confirming the diagnosis of HHH syndrome.
Figure 2.
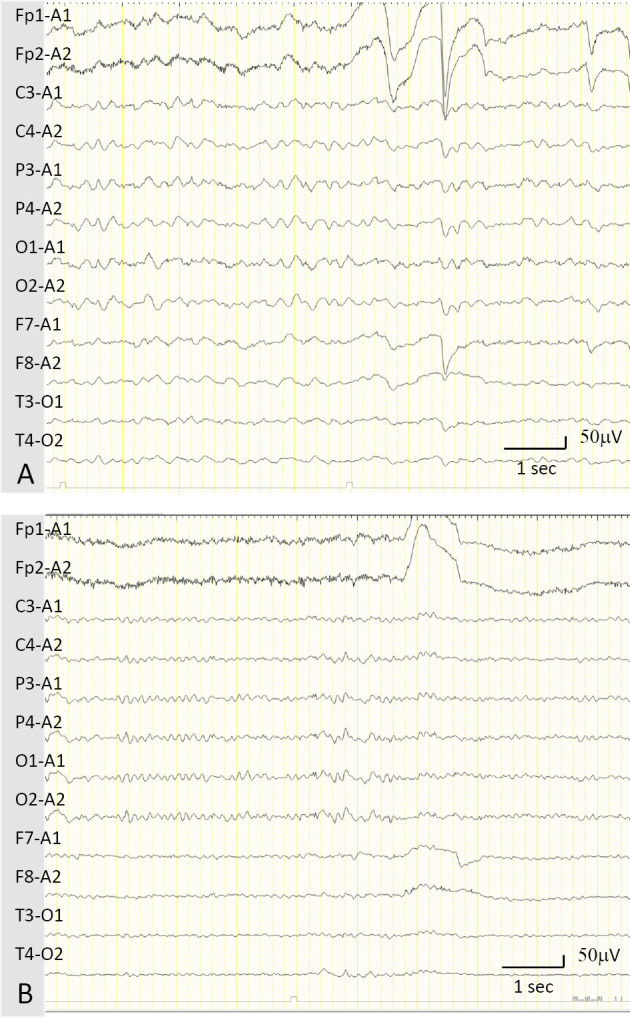
EEG findings. (A) An EEG obtained while awake showed diffuse slow basic activity with 2- to 3-Hz δ waves before treatment. (B) The EEG findings improved and demonstrated an α wave of approximately 8 Hz in frequency 1 month after initiating L-arginine and lactulose therapy. EEG: electroencephalogram
A protein-restricted meal of 1 g/kg/day did not decrease the plasma ammonia levels [i.e. 205 μg/dL (before breakfast), 282 μg/dL (1 hour after breakfast), and 287 μg/dL (1 hour after dinner)]. Upon increasing the dose of oral L-arginine (from 3 to 6 g/day) and lactulose, the plasma ammonia levels decreased to the normal range [57.2±9.4 μg/dL (mean±standard deviation)] with disappearance of disorientation, seizure, and asterixis. The apparent slow basic activities in the EEG were resolved with treatment, although the frequency of the α wave was approximately 8 Hz (Fig. 2B). After 2 months of treatment with L-arginine and lactulose, brain MRI showed resolving leukoencephalopathy associated with mild progression of cerebral atrophy (Fig. 1C). Furthermore, MRS detected a decreased Glx peak associated with an improvement in leukoencephalopathy, eight months after starting the treatment (Fig. 1E). The MMSE score increased slightly (24/30).
Discussion
We herein report an adult patient with childhood-onset HHH syndrome who demonstrated reversible leukoencephalopathy following the administration of L-arginine and lactulose, despite a lack of appropriate therapy for over 35 years. Furthermore, this is the first report of a decrease in the Glx peak on MRS with the resolution of hyperammonemic leukoencephalopathy in HHH syndrome.
The pathophysiology of abnormal brain MRI findings in HHH syndrome has not been elucidated. White matter lesions, similar to those observed in HHH syndrome, have been described in patients with other UCDs, such as ornithine transcarbamylase deficiency (OTCD) (17,18). Takahashi et al. reported an increased Glx peak on MRS in four of six patients with OTCD (18). Furthermore, they showed decreased and increased Glx peaks on MRS accompanied by improved and exacerbated clinical severity, respectively (18). However, they did not describe changes in the white matter lesions on MRI in detail (18). An increased Glx peak supposedly reflects hyperammonemia-induced glutamine accumulation in the brain, which induces astrocyte enlargement (19) and cerebral edema (20). In our patient, improvement in both the cerebral white matter lesions observed on MRI and the encephalopathic symptoms following intervention for hyperammonemia may have been due to the resolution of hyperammonemia-induced glutamine toxicity. The mild progression in cerebral atrophy, despite an improvement in leukoencephalopathy, may be partly due to the resolution of both astrocyte swelling and brain edema. The deterioration of cerebral atrophy prior to intervention for hyperammonemia may have been due to intense hyperammonemia-induced glutamine toxicity.
The prognosis of HHH syndrome varies remarkably from mild neurological involvement to severely disabling disease, with or without treatment (1,21). Treatment outcomes have not been elucidated, especially in patients who receive delayed intervention in adulthood. In our experience, even adult HHH syndrome patients with long-standing disease may respond to treatment and show a decrease in leukoencephalopathy.
Conclusion
We encountered an adult patient with childhood-onset HHH syndrome who showed improvement in leukoencephalopathy, associated with a decrease in the Glx peak on MRS, following treatment with L-arginine and lactulose. Leukoencephalopathy in HHH syndrome may be reversible with the resolution of hyperammonemia-induced glutamine toxicity.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
The authors thank the staff at the Center for Medical Genetics and Laboratory Medicine of Shinshu University Hospital for performing the genetic analyses.
References
- 1.Camacho J, Rioseco-Camacho N. Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. In: GeneReviews™ [Internet]. Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, Eds. University of Washington, Seattle, 2012: 1993-2021. [Google Scholar]
- 2.Camacho JA, Obie C, Biery B, et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet 22: 151-158, 1999. [DOI] [PubMed] [Google Scholar]
- 3.Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab 110: 179-180, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oyanagi K, Tsuchiyama A, Itakura Y, et al. The mechanism of hyperammonaemia and hyperornithinaemia in the syndrome of hyperornithinaemia, hyperammonaemia with homocitrullinuria. J Inherit Metab Dis 6: 133-134, 1983. [DOI] [PubMed] [Google Scholar]
- 5.Koike R, Fujimori K, Yuasa T, Miyatake T, Inoue I, Saheki T. Hyperornithinemia, hyperammonemia, and homocitrullinuria: case report and biochemical study. Neurology 37: 1813-1815, 1987. [DOI] [PubMed] [Google Scholar]
- 6.Inoue I, Saheki T, Kayanuma K, et al. Biochemical analysis of decreased ornithine transport activity in the liver mitochondria from patients with hyperornithinemia, hyperammonemia and homocitrullinuria. Biochim Biophys Acta 964: 90-95, 1988. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu H, Maekawa K, Eto Y. Abnormal urinary excretion of polyamines in HHH syndrome (hyperornithinemia associated with hyperammonemia and homocitrullinuria). Brain Dev 12: 533-535, 1990. [DOI] [PubMed] [Google Scholar]
- 8.Shigeto H, Yamada T, Kobayashi T, Goto I. A case of hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome with spastic paraparesis and severe distal muscle atrophy of lower limbs. Rinsho Shinkeigaku 32: 729-732, 1992. (in Japanese, Abstract in English). [PubMed] [Google Scholar]
- 9.Tsujino S, Kanazawa N, Ohashi T, et al. Three novel mutations (G27E, insAAC, R179X) in the ORNT1 gene of Japanese patients with hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome. Ann Neurol 47: 625-631, 2000. [PubMed] [Google Scholar]
- 10.Miyamoto T, Kanazawa N, Kato S, et al. Diagnosis of Japanese patients with HHH syndrome by molecular genetic analysis: a common mutation, R179X. J Hum Genet 46: 260-262, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Miyamoto T, Kanazawa N, Hayakawa C, Tsujino S. A novel mutation, P126R, in a Japanese patient with HHH syndrome. Pediatr Neurol 26: 65-67, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Torisu H, Kira R, Kanazawa N, et al. A novel R275X mutation of the SLC25A15 gene in a Japanese patient with the HHH syndrome. Brain Dev 28: 332-335, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Ono H, Tamada T, Shigematsu Y. Lactate/pyruvate in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Pediatr Int 60: 762-764, 2018. [DOI] [PubMed] [Google Scholar]
- 14.Kim SZ, Song WJ, Nyhan WL, Ficicioglu C, Mandell R, Shih VE. Long-term follow-up of four patients affected by HHH syndrome. Clin Chim Acta 413: 1151-1155, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Filosto M, Alberici A, Tessa A, Padovani A, Santorelli FM. Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome in adulthood: a rare recognizable condition. Neurol Sci 34: 1699-1701, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Silfverberg T, Sahlander F, Enlund M, Oscarson M, Hårdstedt M. Late onset hyperornithinemia-hyperammonemia-homocitrullinuria syndrome - how web searching by the family solved unexplained unconsciousness: a case report. J Med Case Rep 12: 274, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gropman A. Brain imaging in urea cycle disorders. Mol Genet Metab 100: S20-S30, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takanashi J, Kurihara A, Tomita M, et al. Distinctly abnormal brain metabolism in late-onset ornithine transcarbamylase deficiency. Neurology 59: 210-214, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Willard-Mack CL, Koehler RC, Hirata T, et al. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience 71: 589-599, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi H, Koehler RC, Brusilow SW, Traystman RJ. Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol 261: H825-H829, 1991. [DOI] [PubMed] [Google Scholar]
- 21.Martinelli D, Diodato D, Ponzi E, et al. The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Orphanet J Rare Dis 10: 29, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 30: 672-679, 1993. [DOI] [PubMed] [Google Scholar]