

Abstract
Tumor-associated macrophages (TAMs) exert integrated effects in all aspects of tumor progression, including tumor cell proliferation, angiogenesis, invasion, and metastasis. Recently, considerable preclinical and clinical trials have demonstrated that TAM-targeted therapy is an effective antitumor therapeutic approach, especially as a complementary strategy in combination with conventional chemotherapy, radiotherapy, or emerging immunotherapy. Here, we review all of the current clinical trials targeting TAMs worldwide up to May 2021 and highlight instances of the synergetic therapeutic efficacy of TAM-targeted combined therapeutic strategies. In total, 606 clinical trials were conducted, including 143 tested products. There has been explosive growth in macrophage-targeted therapy around the world during the past decade. Most trials were at early phase, and two-thirds used macrophage-targeting therapy as part of a combination approach. The most common combination is that of traditional chemotherapy with TAM-targeted therapy, followed by immune checkpoint inhibitors and targeted drugs. TAM-targeted therapeutic approaches are a newly emerging but rapidly developing area of anticancer therapy, especially as a combinatorial therapeutic approach. Further investigation of promising combination strategies will pave the way to more effective anticancer therapies.
Keywords: tumor-associated macrophages, cancer, therapy, clinical trials, combination, CAR macrophages
Graphical abstract
Wang et al. reviewed all of the clinical trials targeting tumor-associated macrophages (TAMs) worldwide up to May 2021 and highlighted that TAM-targeted therapeutic approach is a newly emerging but rapidly developing area of anticancer therapy, especially as a combinatorial therapeutic approach, for which advanced exploration will pave the way to more effective anticancer therapies.
Introduction
The tumor microenvironment (TME) is a complex and continuously evolving entity in which tumor cells, stromal cells, and immune cells interact dynamically and reciprocally.1 As the critical infiltrated immune cell types in the TME, tumor-associated macrophages (TAMs) exert integrated effects in all aspects of tumor progression, including promoting tumor cell proliferation and tumor angiogenesis, participating in tumor invasion and metastasis, mediating immune suppression, shaping and remodeling the TME, and contributing to tumor immune escape.2,3 Hence, it is understandable that TAMs-targeted therapy has emerged as a major research area in the future of cancer therapy.
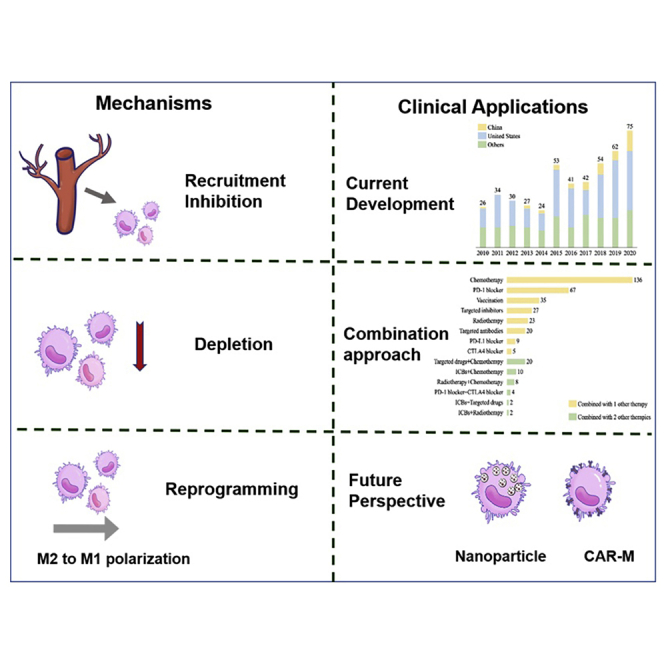
According to different stimulating factors and secreted products, macrophages can be polarized into two functional categories: a classically activated M1-like phenotype with pathogen-killing, proinflammatory abilities and an alternatively activated M2-like phenotype with tumor-promoting, anti-inflammatory functions.4 However, increasing evidences indicate that the M1/M2 dichotomy is an oversimplification, and that macrophages within each class are heterogeneous phenotypically, transcriptionally, and functionally.5 TAMs share markers of both M1 and M2 macrophages, but they have a unique transcriptional profile that is distinct from both M1 and M2 macrophages.4 Current potential therapeutic strategies to target TAMs can be roughly divided into two types: the inhibition of pro-tumor TAMs via eliminating existent TAMs or inhibiting further TAM recruitment, and the activation of antitumor TAMs via reprogramming pro-tumorigenic TAMs into antitumorigenic TAMs (Figure 1).5, 6, 7 Considerable preclinical and clinical trials have demonstrated that the TAM-targeted therapeutic approach is an effective antitumor strategy.5,7,8 However, more effective antitumor treatments are usually the result of a combination of multiple treatment strategies. In this vein, TAM-targeted therapy, as a complementary strategy in combination with conventional chemotherapy, radiotherapy, or emerging immunotherapy, has attained inspiring antitumor efficacy in recent studies.
Figure 1.

The anticancer mechanisms of TAM-targeted therapy
Here, we review the current clinical strategies for targeting TAMs, and highlight the synergetic therapeutic efficacy of TAM-targeted combined therapeutic strategies to lay the foundation for the clinical application of combined therapeutic strategies. We also discuss some promising potential strategies in the future of cancer immunotherapy.
Trends in the number of clinical trials of TAM-targeted therapies
We collected the details of clinical trials targeting TAMs for all kinds of tumors up to May 30, 2021 worldwide from the Pharmaprojects database, developed by a leading international research group known as INFORMAS (Table 1). Pharmaprojects indexes information from over 40,000 public sources. In total, 606 clinical trials with 143 tested products were analyzed. Most trials were Phase I, I/II, or II (570, 94.1%), and only 36 (5.9%) trials were in Phase II/III, III or IV development. Among all the trials, 260 (42.9%) have been completed, while 146 (24.1%) have been terminated and 37 (6.1%) closed. About two-thirds of the trials (406, 67.0%) focused on solid tumors, and the remaining one-third (226, 37.3%) focused on hematological tumors. A total of 353 (58.3%) trials were carried out to evaluate safety, and the remaining 253 (41.7%) were carried out to evaluate efficacy. The patient population had mostly stage III to IV tumors (315, 52.0%) and were receiving second- or later-line treatment (446, 73.6%). More than half of the trials (370, 61.1%) targeted macrophages as a combination therapy agent, while 236 (38.9%) studied macrophage-targeted therapy as a monotherapy.
Table 1.
Classification and characteristics of clinical trials on TAM-targeted therapy
| Characteristics and trial type | China, N (%) | USA, N (%) | Others, N (%) | Monotherapy, N (%) | Combination therapy, N (%) | Total N (%) |
|---|---|---|---|---|---|---|
| Phase I | 22 (45.8) | 147 (43.5) | 79 (35.9) | 105 (44.5) | 143 (38.6) | 248 (40.9) |
| Phase I/II | 11 (22.9) | 87 (25.7) | 56 (25.5) | 33 (14.0) | 121 (32.7) | 154 (25.4) |
| Phase II | 10 (20.8) | 94 (27.8) | 64 (29.1) | 78 (33.1) | 90 (24.3) | 168 (27.7) |
| Phase II/III | 1 (2.1) | 1 (0.3) | 4 (1.8) | 3 (1.3) | 3 (0.8) | 6 (1.0) |
| Phase III | 4 (8.3) | 6 (1.8) | 15 (6.8) | 7 (3.0) | 18 (4.9) | 25 (4.1) |
| Phase IV | 0 | 3 (0.9) | 2 (0.9) | 4 (1.7) | 1 (0.3) | 5 (0.8) |
| Trial status | ||||||
| Open | 24 (50.0) | 93 (27.5) | 42 (19.1) | 47 (19.9) | 112 (30.3) | 159 (26.2) |
| Closed | 2 (4.2) | 18 (5.3) | 17 (7.7) | 10 (4.3) | 27 (7.3) | 37 (6.1) |
| Completed | 17 (35.4) | 134 (39.6) | 109 (49.5) | 116 (49.2) | 144 (38.9) | 260 (42.9) |
| Terminated | 5 (10.4) | 93 (27.5) | 48 (21.8) | 63 (26.7) | 83 (22.4) | 146 (24.1) |
| Primary endpoint | ||||||
| Safety | 27 (56.3) | 208 (61.5) | 118 (53.6) | 132 (55.9) | 221 (59.7) | 353 (58.3) |
| Efficacy | 21 (43.8) | 130 (38.5) | 102 (46.4) | 104 (44.1) | 149 (40.3) | 253 (41.7) |
| Line of therapy | ||||||
| Neoadjuvant | 0 | 23 (6.8) | 7 (3.2) | 11 (4.7) | 19 (5.1) | 30 (5.0) |
| Adjuvant | 0 | 12 (3.6) | 7 (3.2) | 4 (1.7) | 15 (4.1) | 19 (3.1) |
| First line | 7 (14.6) | 67 (19.8) | 44 (20.0) | 20 (8.5) | 99 (26.8) | 119 (19.6) |
| Second line | 27 (56.3) | 193 (57.1) | 118 (53.6) | 131 (55.5) | 207 (55.9) | 338 (55.8) |
| Latter line | 13 (27.1) | 55 (16.3) | 40 (18.2) | 36 (15.3) | 72 (19.5) | 108 (17.8) |
| N/A | 7 (14.6) | 59 (17.5) | 54 (24.5) | 69 (29.3) | 51 (13.8) | 120 (19.8) |
| Stage of disease | ||||||
| Early stage | 0 | 38 (11.2) | 24 (10.9) | 21 (8.9) | 41 (11.1) | 62 (10.2) |
| III/IV | 21 (43.8) | 135 (39.9) | 88 (40.0) | 86 (36.4) | 158 (42.7) | 244 (40.3) |
| IV | 2 (4.2) | 34 (10.1) | 35 (15.9) | 23 (9.7) | 48 (13.0) | 71 (11.7) |
| N/A | 25 (52.1) | 135 (39.9) | 78 (35.5) | 96 (40.7) | 142 (38.4) | 238 (39.3) |
| Tumor category | ||||||
| Solid tumor | 30 (62.5) | 226 (66.9) | 150 (68.2) | 144 (61.0) | 262 (70.8) | 406 (67.0) |
| Hematological tumor | 22 (45.8) | 126 (37.3) | 78 (35.5) | 115 (48.7) | 111 (30.0) | 226 (37.3) |
| Unspecific tumor | 0 | 4 (1.2) | 1 (0.5) | 0 | 5 (1.4) | 5 (0.8) |
| Total | 48 (7.9) | 338 (55.8) | 220 (36.3) | 236 (38.9) | 370 (61.1) | 606 |
N/A, not applicable.
These clinical trials were conducted mainly in the United States and 48 trials were conducted in China. The characteristics of the trials in China were similar to those worldwide, including 30 (62.5%) focused on solid tumors and 22 (45.8%) focused on hematological tumors. The trials in Phase I, I/II, II made up the majority (43, 89.6%); only 4 (8.3%) trials were in Phase III, and no trial had entered into Phase IV in China as of our endpoint. A total of 35.4% (17) of the trials have been completed, and 56.3% (27) aimed to evaluate safety as the primary endpoint. The patients in the China trials had mostly late stage cancers, and only 7 (14.6%) trials were in the first-line setting. We also reviewed clinical data on TAM-targeted therapy from January 1, 2013 to May 30, 2021 in the database from the Center for Drug Evaluation (CDE) of China (Table 2). Of these trials, 28 have been registered, 6 trials have been completed, and most trials are ongoing.
Table 2.
Clinical trials on TAM-targeted therapy from CDE database
| Trial ID | Mechanisms | Drugs | Tumors | Trial Phase | Trial status | Started year | Combination status |
|---|---|---|---|---|---|---|---|
| CTR20150824 | CSF-1/CSF-1R | Sulfatinib | thyroid carcinoma | II | completed | 2015 | monotherapy |
| CTR20160572 | Sulfatinib | biliary tract carcinoma | II | completed | 2016 | monotherapy | |
| CTR20150737 | Sulfatinib | pancreatic neuroendocrine | III | open | 2015 | monotherapy | |
| CTR20160448 | PLX3397 | melanoma | I/II | terminated | 2016 | monotherapy | |
| CTR20170936 | chiauranib | hepatocellular carcinoma | I | completed | 2017 | monotherapy | |
| CTR20170246 | chiauranib | non-Hodgkin’s lymphoma | I | completed | 2017 | monotherapy | |
| CTR20170765 | chiauranib | small cell lung cancer | I | open | 2017 | monotherapy | |
| CTR20170767 | chiauranib | ovarian cancer | I | completed | 2017 | monotherapy | |
| CTR20190609 | chiauranib | ovarian cancer | II | completed | 2019 | chemotherapy | |
| CTR20210658 | chiauranib | small-cell lung cancer | III | open | 2021 | monotherapy | |
| CTR20171427 | CM082 | gastric cancer | I | open | 2017 | chemotherapy | |
| CTR20160487 | CM082 | acute myeloid leukemia | I | open | 2016 | monotherapy | |
| CTR20210743 | ABSK021 | advanced solid tumor | I | open | 2021 | monotherapy | |
| CTR20201034 | surufatinib | advanced solid tumor | I | open | 2020 | PD-1 inhibitor | |
| CTR20181945 | surufatinib | biliary tract carcinoma | II/III | open | 2018 | monotherapy | |
| CTR20132583 | CXCL12/CXCR4 | plerixafor | non-Hodgkin’s lymphoma | III | completed | 2014 | chemotherapy |
| CTR20130291 | Burixafor | acute myeloid leukemia | I | completed | 2016 | chemotherapy | |
| CTR20200132 | PI3Kγ signal pathway | Duvelisib | follicular lymphoma | II | open | 2020 | monotherapy |
| CTR20182057 | CT365 | advanced solid tumor | I | open | 2018 | monotherapy | |
| CTR20200204 | TLR7 | TQ-A3334 | non-small cell lung cancer | I | open | 2020 | monotherapy |
| CTR20201728 | TLR8 | DN1508052-01 | advanced solid tumors | I | open | 2020 | monotherapy |
| CTR20192522 | CD47/SIRPα pathway | TJ011133 | acute myeloid leukemia | I/II | open | 2019 | chemotherapy |
| CTR20210555 | TJ011133 | acute myeloid leukemia | I/II | open | 2021 | chemotherapy | |
| CTR20192612 | IMM0306 | non-Hodgkin’s lymphoma | I | open | 2020 | monotherapy | |
| CTR20181964 | SHR-1603 | advanced solid tumor | I | terminated | 2018 | monotherapy | |
| CTR20200175 | IBI322 | advanced malignant tumors | I | open | 2020 | monotherapy | |
| CTR20191531 | IMM01 | lymphoma | I | open | 2019 | monotherapy | |
| CTR20202684 | AK117 | advanced solid tumors/lymphomas | I | open | 2020 | monotherapy | |
| CTR20200938 | IBI188 | acute myeloid leukemia | I | open | 2020 | chemotherapy | |
| CTR20210761 | IBI188 | advanced malignant tumors | I | open | 2021 | PD-1 inhibitor chemotherapy |
The number of trials involving TAM-targeted therapy is increasing worldwide, in the United States, and in China over the past 10 years. There has been explosive growth in the number of macrophage-targeted therapies worldwide since 2010, especially since 2017 (Figure 2).
Figure 2.

Trends in clinical trials on TAM-targeted therapy worldwide, the United States, China, and other countries in the past 10 years
Trends in clinical trials according to different mechanisms
To date, the clinical data with respect to TAM-targeted tumor therapy have been encouraging. Here, we summarize the therapeutic strategies according to their different mechanisms (Table 3). Broadly speaking, we have divided therapies into those that inhibit mononuclear macrophage recruitment, those that deplete TAMs, and those that reprogram TAMs (Figure 1).
Table 3.
Characteristics of clinical trials and drugs on TAM-targeted therapy stratified by targeting mechanisms
| Targeting pathways and mechanisms | Trials, N | Drugs, N | Active drugs | Trial status | Combination therapy |
|---|---|---|---|---|---|
| CSF-1/CSF-1R | 58 | 20 | ABSK-021 | I | monotherapy |
| AMB-051 | II | PD-1 | |||
| ARRY-382 | II | PD-1 | |||
| SNDX-6532 | II | monotherapy | |||
| chiauranib | III | chemotherapy | |||
| NMS-03592088 | II | monotherapy | |||
| pamufetinib | III | monotherapy | |||
| pexidartinib | launched | chemotherapy, radiotherapy, PD-1, targeted drugs | |||
| Q702 | I | PD-1 | |||
| surufatinib | launched | chemotherapy | |||
| TPX-0022 | I | monotherapy | |||
| vorolanib | III | monotherapy | |||
| CCL2/CCR2 | 14 | 5 | BMS-813160 | II | PD-1, chemotherapy, vaccination |
| CCX-872 | II | chemotherapy, radiotherapy | |||
| CCL5/CCR5 | 10 | 4 | maraviroc | I | PD-1, chemotherapy |
| leronlimab | II | chemotherapy | |||
| OB-002 | I | monotherapy | |||
| vicriviroc | II | PD-1 | |||
| CXCL12/CXCR4 | 115 | 15 | balixafortide | III | chemotherapy |
| burixafor | II | chemotherapy | |||
| GMI-1359 | I | monotherapy | |||
| mavorixafor | III | targeted inhibitor | |||
| motixafortide | III | chemotherapy, PD-1, PD-L1, targeted inhibitor | |||
| CD40/CD40L | 65 | 16 | ABBV-368 | I | PD-1, chemotherapy |
| ABBV-927 | II | PD-1, chemotherapy | |||
| CDX-1140 | I | radiotherapy, vaccination | |||
| GEN-1042 | II | monotherapy | |||
| mitazalimab | I | chemotherapy | |||
| NG-350A | I | monotherapy | |||
| selicrelumab | I | PD-1, CTLA-4, chemotherapy, vaccination | |||
| SL-172154 | I | monotherapy | |||
| sotigalimab | II | PD-1, chemotherapy, radiotherapy, vaccination | |||
| YH-003 | II | PD-1, chemotherapy | |||
| TLRs | 175 | 37 | |||
| TLR7 | AL-034 | II | targeted inhibitor | ||
| BDB-001 | I | PD-1, PD-L1 | |||
| BDC-1001 | II | PD-1 | |||
| BNT-411 | II | PD-L1, chemotherapy | |||
| DSP-0509 | II | PD-1 | |||
| RG-6115 | I | monotherapy | |||
| imiquimod | II | monotherapy | |||
| MBS-8 | I | monotherapy | |||
| resiquimod | II | PD-1, vaccination | |||
| TLR8 | DNA1 | I | monotherapy | ||
| SBT-6050 | I | PD-1 | |||
| DN-1508052 | I | monotherapy | |||
| motolimod | II | PD-1, chemotherapy, radiotherapy, targeted drugs | |||
| TLR9 | AST008 | II | monotherapy | ||
| CMP-001 | III | PD-1 | |||
| tilsotolimod | III | PD-1, CTLA4 | |||
| SD-101 | II | PD-1, radiotherapy | |||
| cavrotolimod | II | PD-1 | |||
| lefitolimod | III | CTLA4, chemotherapy | |||
| TLR5 | MRx-518 | II | radiotherapy | ||
| TLR3 | poly-ICLC | III | PD-1, vaccination, chemotherapy, radiotherapy | ||
| rintatolimod | launched | PD-1, vaccination, chemotherapy | |||
| PI3Kγ signal pathway | 90 | 12 | copanlisib | launched | PD-1, CTLA4, chemotherapy, targeted drug |
| CT-365 | I | monotherapy | |||
| duvelisib | launched | PD-1, chemotherapy, targeted drug | |||
| eganelisib | II | monotherapy | |||
| gedatolisib | I | chemotherapy | |||
| SF-112 | I | PD-1, targeted inhibitor | |||
| tenalisib | II | PD-1, targeted inhibitor | |||
| CD47/SIRPα pathway | 48 | 16 | AK-47 | II | monotherapy |
| ALX-148 | II | PD-1, chemotherapy | |||
| AO-176 | II | PD-1, chemotherapy | |||
| CC-90002 | I | targeted antibody | |||
| HX-009 | II | PD-1 | |||
| IBI-188 | II | chemotherapy | |||
| IBI-322 | I | monotherapy | |||
| IMC-002 | I | monotherapy | |||
| IMM-01 | I | monotherapy | |||
| IMM-0306 | I | monotherapy | |||
| lemzoparlimab | II | chemotherapy | |||
| magrolimab | III | PD-L1, chemotherapy, targeted drug | |||
| RRx-001 | III | chemotherapy, radiotherapy | |||
| TTI-622 | II | PD-1, chemotherapy | |||
| STING pathway | 39 | 17 | SNX-281 | I | PD-1 |
| BMS-986301 | I | PD-1, CTLA-4 | |||
| GSK-3745417 | I | PD-1 | |||
| E−7766 | I | monotherapy | |||
| SYNB-1891 | I | PD-L1 | |||
| TAK-676 | I | PD-1 | |||
| MK-2118 | I | PD-1 | |||
| ADU-S100 | II | PD-1, CTLA-4 | |||
| IMSA101 | II | PD-1, PD-L1 | |||
| CDK-002 | II | monotherapy | |||
| MK-1454 | II | PD-1 | |||
| NOX66 | II | PD-1, chemotherapy, radiotherapy | |||
| SB-11285 | II | PD-1 |
Inhibiting recruitment
Chemokine signaling is crucial for tumorigenesis, proliferation, angiogenesis, tumor progression, and metastasis through the recruitment of TAMs to the tumor site.9 Several chemokine signaling axes, for instance, C-C motif chemokine ligand 2 (CCL2)/C-C motif chemokine receptor 2 (CCR2), C-C motif chemokine ligand 5 (CCL5)/C-C motif chemokine receptor 5 (CCR5) and C-X-C motif chemokine ligand 12 (CXCL12)/C-X-C motif chemokine receptor 4 (CXCR4) axis were investigated in detail and appear to be promising therapeutic targets for targeting TAM.
The CCL2/CCR2 axis
Numerous studies have suggested that the CCL2/CCR2 axis can activate cancer cells through a variety of mechanisms, including recruiting monocytes expressing CCR2 from peripheral blood to the tumor site, promoting maturation of those monocytes into TAMs, and promoting cancer metastasis by enhancing TAM retention via a CCL2-induced chemokine cascade.10,11 The CCL2 levels in tumor tissue have been reported as potential biomarkers in a variety of cancer types.12 However, the agents targeting the CCL2/CCR2 axis are still clinically not available as anticancer approaches. Several clinical trials have been completed with a relative low objective response rate (<30%), whether single-agent therapy or the combination therapy of a CCR2 inhibitor with conventional chemotherapy.13, 14, 15 In total, 14 trials targeting the CCL2/CCR2 axis have been conducted. Five drugs have been studied, but only one drug is still active. BMS-813160 is now in Phase II trials for use in colorectal cancer and pancreatic cancer therapy.
There are many potential reasons for the disappointing clinical results of these agents. The most important explanation may be a lack of proper patient selection; patients whose tumors show high expression of CCR2 by immunohistochemistry or other methods have an improved chance of benefiting from these therapies. The selection of cancer types is also essential for trial success. The CCL2/CCR2 axis may play more important roles in cancers with high immunogenicity than in those with low immunogenicity. Finally, considering the multifunctional nature of CCL2 in various organs such as the lung and digestive tract, the blockage of the CCL2/CCR2 axis may cause unexpected side effects on cancer patients.12,13 Further studies are necessary to fully understand the complicated dynamics and functions of CCL2/CCR2 in the TME for different cancers.
The CCL5/CCR5 axis
More recently, CCR5 has attracted extensive attention as a new therapeutic target for metastatic cancer due to its strong associations with tumor progression and metastasis.16 Previous studies have demonstrated that elevated levels of CCR5 are indicative of poor prognosis in various cancers, including breast, pancreatic, cervical, prostate, and gastric cancers.17 CCR5 can induce tumor cell homing to metastatic sites, promote pro-tumorigenic and pro-metastatic inflammation, induce proliferative signaling, angiogenesis, and resistance to cell death, and also deregulate cellular energetics.18,19 CCR5 antagonists have been developed for HIV treatment, as this receptor is an essential co-receptor for entry of the virus into cells. Maraviroc, a small molecule inhibitor of CCR5, was approved by the US Food and Drug Administration (FDA) for HIV therapy in 2007, and leronlimab, a humanized monoclonal antibody, reached the preregistration status for HIV therapy in July 2021. Several clinical trials have attempted the repurposing of these CCR5-targeted agents for cancer therapy.18 Our database searches identified 10 trials targeting the CCL5/CCR5 axis in the cancer setting, with 4 active drugs, including maraviroc and leronlimab. More clinical trials are still needed to assess the efficacy and safety of this therapeutic approach in patients with metastatic tumors.
The CXCL12/CXCR4 axis
The role of CXCL12/CXCR4 in tumor cell proliferation, migration, and angiogenesis has been recognized for over 2 decades.19 Recent studies have confirmed the role of CXCR4 in promoting M2-like polarization of macrophages in various cancers, thus turning the TME toward an immunosuppressive environment.20 Strategies for the inhibition of CXCR4 have been developed and studied both preclinically and clinically in both hematologic and solid tumors, and thus far appear to be effective, safe, and well tolerated.21
TAM depletion and inhibition of activation
Colony-stimulating factor (CSF)-1/CSF-1 receptor (CSF-1R) signaling pathway
CSF-1/CSF-1R signaling plays important roles in TAM biology, including the recruitment of monocytes into tumors, and polarization of those cells toward a pro-tumor M2-like phenotype, which is usually linked with poor prognosis in cancer patients.22 CSF-1/CSF-1R axis blockade, including via kinase inhibitors and antibodies, can attenuate tumor cell proliferation, tumor-associated angiogenesis, reprogram TAMs toward M1-like phenotype, stimulate CD8+ cell activation, and eventually impede tumor progression.22,23 However, CSF-1R blockade alone achieves modest therapeutic benefit, leading to the delay of tumor growth at most. Among the 11 active drugs and 58 trials identified by our analysis, surprisingly, two drugs have launched. Pexidartinib (PLX-3397), a CSF-1R kinase inhibitor, received FDA approval for the treatment of adult patients with symptomatic tenosynovial giant cell tumor with severe morbidity or functional limitations that is not amenable to improvement with surgery as an orphan drug through the expedited review designation in 2019. Recently, surufatinib (HMPL-012), a CSF-1R kinase inhibitor, launched for the treatment of both pancreatic and non-pancreatic neuroendocrine tumors in China in June 2021. The launch of this therapy in the United States is expected in 2022. Although these kinase inhibitors have demonstrated exciting results, antibodies targeting CSF-1R, such as cabiralizumab and emactuzumab, are still under investigation in Phase I trials in combination with radiotherapy, immune checkpoint blockers (ICBs), or other TAM-targeted agents, such as CD40 agonists, in various solid tumors.24, 25, 26 Given the acceptable safety and modest antitumor activity of these agents, more clinical phases are expected in the future.
Other drugs for TAM depletion
Trabectedin is a marine-derived alkaloid that can induce the specific death of monocytes/macrophages by the tumor necrosis factor (TNF)-related apoptosis-inducing ligand-dependent pathway. It can selectively reduce the number of TAMs, but it does not affect neutrophils or lymphocytes in the TME. This depletion is accompanied by decreased angiogenesis and activated cytotoxic T lymphocytes, leading to promising clinical outcomes.27 Trabectedin has been approved by the FDA for the treatment of unresectable or metastatic liposarcoma and leiomyosarcoma.
Bisphosphonates can deplete TAMs selectively by suppressing the migration, invasion, and tumor infiltration of TAMs, and can reduce tumor cell growth in preclinical breast cancer models. Bisphosphonates are generally packaged with liposomes in the current clinical trials due to their macrophage-killing efficacy, usually relying on the intrinsic phagocytic activities.28
Reprogramming TAMs
CD47/signal regulatory protein alpha (SIRP-α) axis
CD47, a transmembrane protein highly expressed in various tumor cells, can inhibit the phagocytosis of TAMs via binding to SIRP-α and transducing a “don't eat me” (antiphagocytic) signal to TAMs, leading to immune escape.29 Blocking the CD47/SIRP-α interaction enhances the phagocytotic abilities of antigen-presenting cells, and has been investigated as a promising anticancer approach in preclinical and clinical studies.30 Blockade of CD47 is also under study as an immune checkpoint therapy. In earlier studies, CD47 antibodies were designed to inhibit tumor growth by restoring the phagocytic function of TAMs.31 However, there is more evidence that CD47 blockade triggers the adaptive antitumor immune response, and most of the antitumor effect is mediated by cross-priming antigens to CD8+ T cells by CD47-activated dendritic cells.32,33 Furthermore, it should be noted that the ability of CD47 blockade to promote an immune response is dependent upon the cytosolic sensing of tumor DNA by dendritic cells (DCs). Although this does not negate the approach, the degree to which efficacy is macrophage dependent is unclear. The first-in-human Phase I trial of an anti-CD47 antibody, Hu5F9-G4, in patients with advanced cancers was completed in 2019 and demonstrated that the therapy is well tolerated.34 There are currently 14 active drugs targeting CD47 in 48 trials. However, most of them are ongoing in Phase I or Phase II, and it is too early to evaluate the safety or clinical efficacy profiles.
CD40/CD40L
CD40, a transmembrane cell surface glycoprotein, is a co-stimulatory receptor belonging to the TNF receptor superfamily that is expressed mainly on antigen-presenting cells. CD40-CD40L interaction is responsible for the activation and proliferation of B cells, immunoglobulin (Ig) isotope switch, and humoral immune memory.35 The activation of CD40 can re-educate TAMs to promote anticancer effects independent of T cells.36 Several studies have indicated that anti-CD40 antibodies can redirect the monocyte and macrophage population to infiltrate tumors, upregulate matrix metalloproteinases, and degrade fibrosis, and then convert TAM into activated macrophages with an anticancer phenotype.37 However, dacetuzumab, a humanized anti-CD40 monoclonal antibody, failed to demonstrate an improvement in complete response (CR) rates or objective response rates (ORRs) in combination with rituximab ± chemotherapy for patients with diffuse large B cell lymphoma (DLBCL) in a Phase II trial (NCT00529503), leading to a recommendation for termination. With respect to the safety profile, dacetuzumab increased the incidence and severity of hematopoietic cytopenias and febrile neutropenia, which can be explained by the interaction of the antibody with non-cancerous cells, including blood cells.38 The absence of significant clinical activity in early agents has resulted in a number of studies focusing on modifications, including new epitopes and new Fc region designs to improve clinical efficacy. In addition, anti-CD40 antibody development has been historically challenging due to previous clinical results. There is a further therapeutic opportunity to enhance the delivery of chemotherapy and achieve even greater tumor regression. A recently developed agent called NG-350A is an oncolytic adenoviral vector designed to deliver gene therapy that encodes for an anti-CD40 monoclonal antibody. It can selectively deliver antibodies to tumor cells locally and allows the antibodies to be well tolerated and more effective. A first-in-human Phase I trial will evaluate the safety and tolerability of NG-350A by intravenous infusion as monotherapy or by combination with a checkpoint inhibitor in 125 patients with metastatic or advanced epithelial tumors.
Phosphoinositide 3-kinase (PI3K) γ signal pathway
PI3K is a lipid kinase involved in intracellular signal transduction. Activation of the PI3K pathway has been linked to tumor cell proliferation, survival, and apoptosis through regulating various downstream molecules.39 Accumulating evidence both in vitro and in vivo has indicated that PI3K signaling is a critical factor for driving M2 polarization and M2-like TAMs-induced tumor cell invasion.40 Among the PI3K families, PI3Kγ has been found to be involved in various steps of macrophage activation and function.41 Inhibition of PI3Kγ can block the immunosuppressive effects of TAMs, enhancing the activation of cluster of CD8+ T cells and responses to immune checkpoint inhibitors.41,42 Loss of PI3Kγ expression also blocks TAM-mediated tumor metastasis in a number of tumor models.43 A total of 90 trials have focused on the PI3K pathway associated with TAMs, studying 7 active drugs. All of them are currently in Phase I or II trials. From the onset, these drugs were developed mainly to target the PI3K/mammalian target of rapamycin (mTOR) dual pathways, which leads to excess on-target adverse events without adding significant benefits due to the full blockade of PI3K/mTOR signaling. In the ROVER study (NCT01442090), the first Phase II trial to compare the dual inhibition by apitolisib against single mTOR inhibition by everolimus in metastatic renal cell carcinoma, apitolisib demonstrated limited efficacy, while half of the patients who have been treated with apitolisib required treatment modifications and nearly one-third discontinued treatment permanently.44 Therefore, the development of single pan-class PI3K inhibitors and even highly selective isoform-specific PI3K inhibitors were eagerly expected. The trials of some agents have been discontinued—namely pictilisib for insufficient efficacy, buparlisib for excessive toxicity, and taselisib for lack of a clinically meaningful benefit; fortunately, two PI3K inhibitors have received approval for clinical use.45,46 Copanlisib, a pan-PI3K inhibitor, has been approved to treat adult patients with follicular B cell non-Hodgkin’s lymphoma who have relapsed disease following at least two prior systemic therapies. In 2021, it was also designated as an orphan drug for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma. In addition, duvelisib (IPI-145), a PI3Kγ and -δ inhibitor, received FDA approval in 2018 for use in adult patients with relapsed or refractory follicular lymphoma after at least 2 prior systemic therapies.
Toll-like receptors (TLRs)
TLRs are key regulators of the innate immune system, usually expressed in distinct cellular locations of macrophages. TLRs can drive the polarization process toward M1-like phenotypes by interacting with their corresponding agonists in tumor cells.47 Importantly, the efficiency of polarization driven by the TLR agonists is comparable to some common strong induction signals, such as standard lipopolysaccharide (LPS) and IFN-γ triggers.48 Therefore, strategies to more precisely reprogram TAMs have relied on the use of TLR agonists, such as TLR3 agonists, TLR4 agonists, TLR7/8 agonists, or TLR9 agonists. Due to the diversity, the repertoire of TLR agonists is rapidly expanding. Although the success of preclinical models has demonstrated the potential for TAM re-education, unfortunately, most of the early clinical trials have been closed, citing a lack of efficacy concerns as the primary reason, and safety issues also contributing to the decision. The systemic administration of the agonists in patients presents significant toxic effects considering its indiscriminate biodistribution, which leads to unrestrained immune activation and systemic inflammation, undermining its potential effect.49 Another major issue is related to its rapid systemic degradation. In this sense, the potential clinical strategies targeting TLRs focus on the association with an efficient delivery system that can maintain the drug stability and mitigate the toxicity. Recent studies have highlighted the development of TLR agonist-loaded nanocomplexes, which led to effective drug delivery to TAMs with limited off-target effects. Several studies have used poly-ICLC (polyinosinic-polycytidylic acid mixed with the stabilizers carboxymethylcellulose and polylysine), a synthetic double-stranded RNA targeting TLR3 to treat various advanced solid tumors. A Phase II trial in which poly-ICLC was administered by intramuscular injections in patients with advanced solid tumors, including melanoma, head and neck squamous cell carcinoma, skin sarcoma, and breast cancer, has been completed, with positive outcomes.50 The average progression-free survival (PFS) was 52 weeks and the therapeutic effect in treated patients lasted 24 months without serious adverse events. Poly-ICLC has additionally been investigated in the immunotherapy of coronavirus disease 2019 (COVID-19) infection. Poly-ICLC therefore represents a promising therapy for both cancer and other conditions. A Phase II/III study of first-line intratumoral CMP-001, a virus-like particle containing a TLR9 agonist, in combination with programmed death ligand-1 (PD-1) blocker compared to nivolumab monotherapy, in subjects with unresectable or metastatic melanoma was initiated in 2021, with orphan drug designation granted by the FDA previously. Several Phase III trials targeting TLR9 are also under investigation.
Stimulator of interferon gene (STING) pathway agonist
Cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)-STING signaling is critical for sensing cytosolic DNA and triggering innate immune responses.51 Damaged nuclear or mitochondrial DNA from tumor cells activates cGAS and further becomes a strong initiator of STING pathway activation. The activated cGAS-STING pathway in macrophages induces the massive production of downstream type I interferons, leading to an innate and adaptive antitumor response. Recently, the development of STING agonists has become a hotspot in cancer therapies. At present, most of them are still in the preclinical or early clinical stages. According to previous studies, STING agonists promote the M1-like polarization of tumor-infiltrating macrophages and the re-education of M2 cells toward an M1 phenotype.52,53 5,6-Dimethylxanthenone-4-acetic acid (DMXAA), or vadimezan, a direct ligand for murine STING, renders significant therapeutic effects in preclinical studies and shows excellent clinical prospects in a randomized Phase II clinical trial.54 However, it failed to show clinical efficacy in a larger Phase III trial in patients with advanced non-small cell lung cancer.55 The reasons for the disappointing outcomes in the Phase III trial have been carefully investigated. First, due to stringent species selectivity, DMXAA could not activate human STING, which may have caused the significant dropoff in efficacy between the preclinical and clinical stages. Second, the selection of cancer types for the Phase III trial may have played a role in the failed trial. DMXAA targets sensitive, highly vascularized cancers, but not those with normal vasculature. In addition, following the vascular disruption, DMXAA causes hypoxia, produces vascular endothelial growth factor (VEGF), and induces angiogenesis, which imply that the combination therapy of DMXAA and anti-angiogenic agents may be considerably effective. In addition, due to its immunostimulatory activity, DMXAA may have the potential for combination therapy with immunotherapy agents.56 Based on the studies outlined above, an artificial ligand of human STING, ADU-S100, has been clinically tested in combination with the anti-PD1 agent spartalizumab in patients with advanced/metastatic solid tumors or lymphoma, as well with pembrolizumab in PD-L1+ recurrent or metastatic squamous cell carcinoma of head and neck cancer patients. Preliminary data from this ongoing study shows that ADU-S100 is well tolerated and a partial response was observed.57 To date, other clinical trials of STING agonists, including MK-1454, SB11285, GSK3745417, BMS-986301, and BISTING, have been approved by agencies. Their safety and efficacy need to be evaluated. It is expected that as the design of these agents improves, we will obtain better clinical benefits.
To summarize, of the 606 trials included in this analysis, the trials targeting TLRs accounted for 28.9% (175), followed by those targeting the CXCL12/CXCR4 axis (115, 19.0%), the PI3K signal pathway (90, 14.8%), CD40 (65, 10.7%), the CSF-1/CSF-1R axis (58, 9.6%), CD47 (48, 7.9%), and the STING pathway (27, 4.5%). The distribution of corresponding targeted products is a little different compared to the trials (Figure 3; Table 3). The most common products studied were those targeting TLRs, followed by the CSF-1/CSF-1R axis, CD40, CD47, the CXCL12/CXCR4 axis, and the PI3K pathway. Up to the present, only 5 agents targeting TAMs for anticancer therapy have achieved agency approval.
Figure 3.

Trends in clinical trials on TAM-targeted therapy based on targeting mechanisms
Trends in the clinical trials of monotherapy
Some preclinical models and early-stage clinical trials have revealed that TAM-targeted monotherapy is advantageous; however, other studies have suggested only limited efficacy when macrophages are used as a single agent. In our analysis, we found that 38.9% (236) trials focused on monotherapy. Almost half have been completed, 26.7% have terminated, and fewer than 20% are open. Most of the trials (216, 91.5%) are in Phase I, I/II, or II, and focus on safety as the primary endpoint. A total of 70.8% of the trials are in second line or later; 46.2% focus on patients with stage III or IV tumors. Encouragingly, 7 trials have reached Phase III. The trials in Phase III mainly target CSF-1R, the PI3K pathway, or TLRs. Two trials of these Phase III trials target the PI3K pathway in lymphoma using 2 approved drugs mentioned above, copanlisib and duvelisib. Two trials targeting TLRs have been terminated. SD-101, an intratumoral TLR9 agonist, was evaluated to be efficacious for metastatic melanoma in combination with anti-PD-1 therapy (pembrolizumab) in a Phase Ib/II trial in advanced melanoma.58 The study reported that an ORR of 70% (21 of 31) and a 6-month PFS rate of 76% in patients naive to anti-PD-1 treatment for patients who received SD-101. Finally, there are 3 trials targeting CSF1-R inhibitor, including TAS-115 for osteosarcoma, chiauranib for progressed or relapsed small-cell lung cancer, and surufatinib for advanced pancreatic neuroendocrine tumor, which has achieved agency approval.
While targeting TAMs is an attractive strategy, most immunotherapies are not powerful enough when administrated as solo therapies. Therefore, there is a need to develop combination therapies that synergize and prolong antitumor immunity through the activation of immune cells via multiple different mechanisms.
Trends in the clinical trials of combination therapy
Recent studies have suggested that TAMs play an important role in modulating responses to conventional therapies and immunotherapies. We reviewed all of the combination trials using these agents. In total, 370 trials (representing 61.1% of all TAM-related trials) have been carried out to evaluate the macrophage-targeted therapeutic approach combined with other therapies. Similar to monotherapy, almost all of the trials (353, 95.7%) are in Phase I, I/II, or II, with 38.9% completed, 30.3% open, and 22.4% terminated. A total of 26.8% are in first line, which is higher than those in monotherapy.
The combined therapies under study include chemotherapy, targeting therapy, radiation therapy, and ICBs. According to the different combined therapy approaches, we categorized the trials into several classes (Figure 4). Most of the trials combine 2 therapeutic approaches (87.6%) and 12.4% (46) combine 3 approaches. The most common combination type is the combination of traditional chemotherapy with TAM-targeted therapy (174, 28.7%), followed by the combination of ICBs (99, 16.3%), including PD-1, PD-L1, cytotoxic T lymphocyte antigen-4 (CTLA-4), or even both PD-1 and CTLA-4 added to TAM-targeted therapy, the combination of targeted drugs (44, 7.3%), the combination of vaccinations (35, 5.8%), and the combination of radiotherapies (33, 5.4%).
Figure 4.

Trends in clinical trials on TAM-targeted combination therapy
TAM-targeted therapy combined with chemotherapy
The most common combination approach is chemotherapy. Chemotherapeutics mostly reprogram TAMs toward an M1-like phenotype; however, they can also promote an M2-like phenotype, depending on the tumor type and therapy schedule. Moreover, chemotherapeutics induce the recall of TAMs to a tumor site as a consequence of the damaging effects after chemotherapy.59 The combination of TAM-targeted therapy with chemotherapy can eliminate the impairment.
Of the chemotherapy-TAM combination trials we analyzed, 126 (95.5%) are in Phase I, I/II, or II and 6 are in Phase III. Of the Phase III trials, 2 targeted TLRs and had negative outcomes, 1 targeted CXCR4 and showed a positive outcome, and 1 targeting CD47 is still active. Tilsotolimod (IMO-2125), a synthetic TLR9 agonist, has attracted much attention in the treatment of melanoma. A Phase I/II study confirmed the safety and efficacy of intratumoral IMO-2125 in combination with ipilimumab or pembrolizumab in patients with anti-PD-1 refractory metastatic melanoma, with a median overall survival (OS) of 21.0 months, ORR 22.4%, and stable disease (SD) 49%.60 The drug received both fast track designation and orphan drug designation by the FDA in 2017. More potential approaches should be explored for combination therapy in the future.
TAM-targeted therapy combined with radiotherapy
Although approximately half of all cancer patients undergo radiotherapy for local tumor control, various neoplasms do not respond to the therapy.61 Radiotherapy can paradoxically promote tumor growth and invasion through the recruitment and repolarization of TAMs at the tumor site, limiting the efficacy of radiotherapy.62 This can be highlighted by the use of liposomal clodronate to deplete macrophages before ionizing radiation. The effect on TAMs depends on the dosage used, whether low doses (below 1 Gy) or high doses (above 10 Gy). These can favor the M2-like phenotype, prompting angiogenesis and tumor growth.63 Therefore, it is of interest to investigate potential combination-therapy approaches of TAM-targeted agents and radiotherapy to abolish radiotherapy-induced immunosuppressive effects. All of the trials of combined radiotherapy included in our analysis were Phases I, I/II, or II. A total of 12 trials have been completed, 6 are active, and 8 have been terminated. The strategies of combination therapy included CXC4 inhibitor, CSF-1R inhibitor, anti-CD47 antibody, anti-CD40 antibody, and TLR agonists.
Several signaling pathways, including CSF-1/CSF-1R and CXCL12/CXCR4, are involved in the radiation-induced recruitment of TAMs, leading to the depletion of CD4+ cell infiltration and delay of tumor regrowth after radiotherapy through the depletion of TAMs.64,65 The inhibition of CXCL12/CXCR4 interaction with AMD3100 efficiently abrogated tumor vasculogenesis and tumor regrowth in preclinical studies and clinical trials.66 An ex vivo study showed that low-dose radiation of monocytes increased TLR expression.67 A Phase II trial assessed the feasibility of using the intratumoral injection PF-3512676, a TLR9 agonist, in combination with local radiation as a therapy for low-grade B -cell lymphomas. In this study, 7 of 30 patients were evaluated as partial response (PR) and 19 of 30 as SD through 1 cycle treatment at the 12 months after initiation, and 2 patients were evaluated as CR, 4 as PR, and 14 as SD. Encouragingly, no patient was evaluated as progressive disease,. The median PFSs were 41 and 35 weeks, respectively, among treatment-naive and relapsed/refractory patients. Importantly, in response to in situ vaccination, all of the patients made tumor-specific immune responses within 2–4 weeks post-vaccination.68
TAM-targeted therapy combined with targeted drugs
Of all 44 trials of combined therapy with targeted drugs, including antibodies and inhibitors, more than half (25, 56.8%) combined with anti-CD20 antibody. Of these, 1 Phase III trial has been completed with an anti-CD47 antibody, Hu5F9-G4, combining with rituximab involving patients with relapsed or refractory non-Hodgkin’s lymphoma on the basis of the promising outcome in its Phase Ib trial. A total of 50% of the patients treated with the combination therapy had objective response (OR), with 36% having a CR among enrolled 22 patients. The rates of OR and CR were 40% and 33% among patients with DLBCL and 71% and 43% among those with follicular lymphoma. At a median follow-up of 6.2 months among patients with DLBCL and 8.1 months among those with follicular lymphoma, 91% of the responses were ongoing.69 A positive result is speculated for the Phase III trial.
Platinum-based chemotherapy, fluorouracil, and cetuximab combination treatment is the standard of care for first-line recurrent and/or metastatic squamous cell carcinoma of the head and neck, with unsatisfactory PFS and OS. The effect of adding motolimod, a TLR8 agonist, to the standard combination therapy was evaluated in a Phase II trial with 195 patients. Although adding motolimod did not improve PFS or OS in the intent-to-treat population, significant benefit was observed in human papillomavirus (HPV)+ patients in the prespecified subgroup analysis with longer PFS (7.8 versus 5.9 months) and OS (15.2 versus 12.6 months). Meanwhile, in an exploratory analysis, patients with injection site reactions had longer PFS (7.1 versus 5.9 months) and OS (18.7 versus 12.6 months).70 This suggests that TAM-targeted therapy may benefit subset- and biomarker-selected patients.
TAM-targeted therapy combined with immune checkpoint inhibitors
In recent years, ICB therapeutic strategies have provided new treatment options for cancer patients and demonstrated significant benefit in some patients across a wide range of cancers. However, the vast majority of patients still fail to benefit from ICB therapy, indicating that the monotherapy is not potent enough. Recent evidence supports a role played by TAMs in limiting the response to ICB therapy by robustly increasing the expression of inhibitory checkpoint molecules, including PD-1, PD-L1, and CTLA-4. However, TAMs can also release soluble factors that orchestrate tissue remodeling events to restrict the tumor access of CD8+ T cells.71 The addition of TAM-targeted therapy to ICB has proven to be a promising approach to overcome the limited response rates for ICB monotherapy.
We reviewed 99 trials of ICB + TAM-targeted combination therapy. The most common combined approach is the combination of TAM-targeted agents with PD-1 (67, 67.7%). There were 9 (9.1%) trials with PD-L1, 5 (5.1%) with CTLA-4, and 4 (4.0%) combined with both PD-1 and CTLA-4. Two trials targeting the ICB + TAM-targeted combination therapy added to chemotherapy, 2 added to radiotherapy, and 2 added to targeting drugs. Almost all of the trials are early phase; the most advanced is the Phase II/III trial investigating the combination of CMP-001 with nivolumab described above. Several Phase II trials have been completed, and one of those yielded a positive outcome. That study, a Phase IIb pilot study, assessed the effect of BL-8040, a CXCR4 inhibitor, combined with pembrolizumab in patients with metastatic pancreatic cancer. The disease control rate (DCR) was 34.5% in the evaluable population, including 9 patients (31%) with SD and 1 (3.4%) with PR. The median OS was 3.3 months in the intent-to-treat population. Notably, in patients receiving combination therapy as second-line therapy, the median OS was 7.5 months. Meanwhile, in the expansion cohort of this study, 22 patients received the combination therapy added to chemotherapy, with an ORR, DCR, and median duration of response of 32%, 77%, and 7.8 months, respectively. This suggests that combined CXCR4 and PD-1 blockade may expand the benefit of chemotherapy and warrants confirmation in subsequent randomized trials.72 Several other Phase II studies are openly targeting mainly CD47 and TLRs.
TAM-targeted therapy combined with vaccination
In recent years, anticancer vaccines have become a new way to stimulate immune activation by administrating specific tumor-associated antigens conjugated with co-stimulatory molecules or loaded on the immune cells of patients, inducing the regression in premalignant lesions in tumor patients.73 Recently, data from animal models have suggested that TAMs critically contribute to vaccine-induced tumor regressions.74 We identified 35 trials focused on the combination of vaccination with TAM-targeted therapies, whether as an adjuvant or combined into the vaccine to enhance the therapeutic benefit of the vaccine and reduce the adverse effects. Most of these trials are in Phase I or I/II; only 5 have reached Phase II, all of which are evaluating a TLR3 agonist, poly-ICLC.
A Phase II clinical trial was conducted to evaluate the poly-ICLC matured DCs as an adjuvant for NY-ESO-1 and Melan0A/MART-1 peptides (dendritic cell vaccine) with or without montanide, both with systemic administration of poly-ICLC in patients with melanoma in complete clinical remission but at high risk for disease recurrence. This vaccine trial reached the primary endpoint of safety and tolerability. NY-ESO-1 protein in combination with poly-ICLC induced integrated antibodies and CD4+ T cell response in most patients with no treatment-related grade 3/4 adverse events.75 Further trials should consider testing NY-ESO-1 antigens with poly-ICLC in combination with ICB therapy in the high-risk resected melanoma patient, as this is now the standard of care in this setting.76 Wang and Steinmetz identified the combination immunotherapy using cowpea mosaic virus in situ vaccination and CD47-blocking antibodies in the 4T1 breast tumor model.77 Cowpea mosaic virus in situ vaccination can activate the innate immune system to cause the recruitment and activation of macrophages. The blockade of CD47 can inhibit antiphagocytic signals to induce macrophage phagocytosis of cancer cells. Therefore, the combination therapy boosts the ability of cancer cell phagocytosis for macrophages, in turn priming the adaptive immune system leading to a potential antitumor immune response. However, to our knowledge, no clinical trial has been initiated to further confirm the results of these preclinical studies. Several Phase I/II trials have evaluated the effect of combination therapy, including CD40 and other TLR agonists.
In this regard, the combination of TAM-targeted agents with vaccination in situ may be a superior approach. More effort should be made to maximize the benefits through the personalized vaccine-based combination therapies.
Conclusions and future perspectives
TAM-targeted therapeutic approaches are a newly emerging but rapidly developing area of anticancer therapy. In spite of great clinical interest, the optimal therapeutic approach has been yet to be identified. First, TAMs exhibit extensive phenotypic diversity such that the categorization of these cells along the M1/M2 spectrum is controversial. Due to the integral role of TAMs in the innate immune system, targeting one specific function is significantly difficult. Some pan-macrophage-targeted therapy may cause systemic toxicities. Second, different tumor types have different degrees of macrophage infiltration and polarization. The available clinical parameters that could indicate the specific populations who will derive maximal benefit from TAM-targeted therapy is still lacking. With respect to the complexity of TAMs, a greater understanding of interactions between macrophages and tumor cells is needed. More clinical data regarding the correlation between TAMs and patient outcomes are also needed to guide patient selection.
Due to the limited efficacy of monotherapies, a combinatorial therapeutic approach has the potential to compensate for the shortcomings of each therapy, not only for TAM-targeted agents but also for conventional therapies and other immunotherapeutics. Current preclinical and clinical trials have suggested that TAM-targeted therapy could significantly improve the efficacy of these previous approaches. However, future studies should focus on precise combination therapy, including the selection of combined therapeutic agents or modes to maximize immunostimulatory activities and minimize potential toxicities.
Treatment delivery methods for inducing tumor-localized therapy response may be an effective means to eliminate the additional adverse effects caused by the combination. Nanoparticles (NPs) are promising carriers for the delivery of a variety of anticancer agents, which are expected to enhance anticancer treatment.78 Recently, TAMs have attracted much attention as a promising nanotherapeutic agent due to their powerful ability to detect and absorb NPs through interfering with the intended delivery of drugs in the NPs. However, the cellular backpack, a polymeric particle, may be an alternative approach for NPs attaching to the surface of TAMs but not undergoing phagocytosis due to its size, disk-like shape, and flexibility. The backpacks on TAMs can accumulate in the target organ, maintaining a relative long-term cell effect without harming the carrier cells.79 In addition, NPs have been used to reprogram TAMs through direct interaction with macrophages in recent studies.80 Research into the use of NPs as a new TAM-associated strategy need to be explored further in preclinical and clinical trials.
Given the success of chimeric antigen receptor (CAR)-T cell therapy, some studies have tried to develop CAR macrophages (CAR-M) for tumor immunotherapy. CAR-M therapy aims to modify macrophages with specific CARs to improve the abilities of phagocytosis and antigen presentation (Figure 5). The transduction of chimeric receptors will acquire an M1 phenotype and convert bystander M2 cells into M1.81,82 Compared with CAR-T, CAR-M has its unique advantages and leads to less non-tumor toxicity due to its limited time in circulation. Thus far, 2 clinical trials based on CAR-M approaches have been approved by the FDA. One is for the treatment of relapsed or refractory human epidermal growth factor receptor 2 (HER2) overexpressed tumor patients with anti-HER2 CAR-M. The other is for the treatment of relapsed or refractory ovarian cancer and peritoneal mesothelioma with mRNA-targeted CAR-M. CAR-M therapy has shown obvious advantages in solid tumors compared to activated macrophage reinfusion therapy.81 However, the manufacturing of CAR-M has become a bottleneck in the application of this technology due to the limits of macrophage expansion and the transduction efficiency by viral vectors. Pluripotent stem cell-derived macrophages could be one solution. Zhang et al. developed induced pluripotent stem cell-derived CAR-expressing macrophage cells (CAR-iMac) with some advantages, including antigen-dependent macrophage functions, polarization toward an M1 state, enhanced phagocytosis of tumor cells, and continuous expansion and anticancer cell activity in vivo.81 However, it is still far from clinical application. In the future, more targeted genes regulating macrophage activation and polarization in CAR-M could be modified to improve the TME for cancer immunotherapy. In addition, combination therapy with CAR-M and other TAM-targeted therapeutic agents may provide a new therapeutic option to improve the treatment of solid tumors. Although the safety and efficacy of CAR-M have been confirmed by preclinical studies, further clinical trials are urgently needed to investigate the efficacy and safety of CAR-M.
Figure 5.

The anticancer mechanism of CAR-M therapy
In spite of some outstanding difficulties, targeting TAMs remains a promising therapeutic approach combined with other therapies to improve outcomes for cancer patients. Continued exploration of promising combination strategies will pave the way for more effective anticancer therapies.
Acknowledgments
This work was supported by the Chinese Academy of Medical Sciences (grant no. 2019XK320068), the Chinese Academy of Medical Sciences (grant no. 2020-I2M-2-007), the Beijing Municipal Science and Technology Commission (International Pharmaceutical Clinical Research and Development Platform 2015), and the Beijing Municipal Health Commission, Beijing Demonstration Research Ward (grant no. BCRW20200303).
Author contributions
N.L. and Y.Z. conceived the review; S.W. and Y.Y. drafted the manuscript; P.M., H.H., Q.T., and H.M. analyzed and interpreted the manuscript; Y.F., N.J., Y.L., Q.Z., and W.T. collected and analyzed the data. All of the authors approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Yan Zha, Email: zhayan72@126.com.
Ning Li, Email: lining@cicams.ac.cn.
References
- 1.Anderson N.M., Simon M.C. The tumor microenvironment. Curr. Biol. 2020;30:R921–R925. doi: 10.1016/j.cub.2020.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li C., Xu X., Wei S., Jiang P., Xue L., Wang J. Tumor-associated macrophages: potential therapeutic strategies and future prospects in cancer. J. Immunother. Cancer. 2021;9:e001341. doi: 10.1136/jitc-2020-001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez H., Hagerling C., Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;1:1267–1284. doi: 10.1101/gad.314617.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray P.J., Allen J.E., Biswas S.K., Fisher E.A., Gilroy D.W., Goerdt S., Gordon S., Hamilton J.A., Ivashkiv L.B., Lawrence T., et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeNardo D.G., Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019;19:369–382. doi: 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerriero J.L. Macrophages: the road less traveled, changing anticancer therapy. Trends Mol. Med. 2018;24:472–489. doi: 10.1016/j.molmed.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X., Liu R., Su X., Pan Y., Han X., Shao C., Shi Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer. 2019;5:177. doi: 10.1186/s12943-019-1102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urbantat R.M., Vajkoczy P., Brandenburg S. Advances in chemokine signaling pathways as therapeutic targets in glioblastoma. Cancers (Basel) 2021;13:2983. doi: 10.3390/cancers13122983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izumi K., Fang L.Y., Mizokami A., Namiki M., Li L., Lin W.J., Chang C.S. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 2013;5:1383–1401. doi: 10.1002/emmm.201202367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitamura T., Qian B.Z., Soong D., Cassetta L., Noy R., Sugano G., Hato Y., Li J.F., Pollard J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015;212:1043–1059. doi: 10.1084/jem.20141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwamoto H., Izumi K., Mizokami A. Is the C-C motif ligand 2-C-C chemokine receptor 2 axis a promising target for cancer therapy and diagnosis? Int. J. Mol. Sci. 2020;21:9328. doi: 10.3390/ijms21239328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pienta K.J., Machiels J.P., Schrijvers D., Alekseev B., Shkolnik M., Crabb S.J., Li S., Seetharam S., Puchalski T.A., Takimoto C., et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest. New Drugs. 2013;31:760–768. doi: 10.1007/s10637-012-9869-8. [DOI] [PubMed] [Google Scholar]
- 14.Nywening T.M., Wang-Gillam A., Sanford D.E., Belt B.A., Panni R.Z., Cusworth B.M., Toriola A.T., Nieman R.K., Worley L.A., Yano M., et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–662. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noel M., O'Reilly E.M., Wolpin B.M., Ryan D.P., Bullock A.J., Britten C.D., Linehan D.C., Belt B.A., Gamelin E.C., Ganguly B., et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest. New Drugs. 2020;38:800–811. doi: 10.1007/s10637-019-00830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiao X., Velasco-Velázquez M.A., Wang M., Li Z., Rui H., Peck A.R., Korkola J.E., Chen X., Xu S., DuHadaway J.B., et al. CCR5 governs DNA damage repair and breast cancer stem cell expansion. Cancer Res. 2018;78:1657–1671. doi: 10.1158/0008-5472.CAN-17-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiao X., Nawab O., Patel T., Kossenkov A.V., Halama N., Jaeger D., Pestell R.G. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. 2019;79:4801–4807. doi: 10.1158/0008-5472.CAN-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Upadhyaya C., Jiao X., Ashton A., Patel K., Kossenkov A.V., Pestell R.G. The G protein coupled receptor CCR5 in cancer. Adv. Cancer Res. 2020;145:29–47. doi: 10.1016/bs.acr.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huynh C., Dingemanse J., Meyer Z., Schwabedissen H.E., Sidharta P.N. Relevance of the CXCR4/CXCR7-CXCL12 axis and its effect in pathophysiological conditions. Pharmacol. Res. 2020;161:105092. doi: 10.1016/j.phrs.2020.105092. [DOI] [PubMed] [Google Scholar]
- 20.Song Z.Y., Wang F., Cui S.X., Gao Z.H., Qu X.J. CXCR7/CXCR4 heterodimer-induced histone demethylation: a new mechanism of colorectal tumorigenesis. Oncogene. 2019;38:1560–1575. doi: 10.1038/s41388-018-0519-2. [DOI] [PubMed] [Google Scholar]
- 21.Shi Y., Riese D.J., 2nd, Shen J. The role of the CXCL12/CXCR4/CXCR7 chemokine Axis in cancer. Front. Pharmacol. 2020;11:574667. doi: 10.3389/fphar.2020.574667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blondy T., d'Almeida S.M., Briolay T., Tabiasco J., Meiller C., Chéné A.L., Cellerin L., Deshayes S., Delneste Y., Fonteneau J.F., et al. Involvement of the M-CSF/IL-34/CSF-1R pathway in malignant pleural mesothelioma. J. Immunother. Cancer. 2020;8:e000182. doi: 10.1136/jitc-2019-000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magkouta S.F., Vaitsi P.C., Pappas A.G., Iliopoulou M., Kosti C.N., Psarra K., Kalomenidis I.T. CSF1/CSF1R axis blockade limits mesothelioma and enhances efficiency of anti-PDL1 immunotherapy. Cancers (Basel) 2021;13:2546. doi: 10.3390/cancers13112546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Machiels J.P., Gomez-Roca C., Michot J.M., Zamarin D., Mitchell T., Catala G., Eberst L., Jacob W., Jegg A.M., Cannarile M.A., et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J. Immunother. Cancer. 2020;8:e001153. doi: 10.1136/jitc-2020-001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiss S.A., Djureinovic D., Jessel S., Krykbaeva I., Zhang L., Jilaveanu L., Ralabate A., Johnson B., Levit N.S., Anderson G., et al. A phase I study of APX005M and cabiralizumab with or without nivolumab in patients with melanoma, kidney cancer, or non-small cell lung cancer resistant to anti-PD-1/PD-L1. Clin. Cancer Res. 2021;27:4757–4767. doi: 10.1158/1078-0432.CCR-21-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kindler H.L., Hseu R., Janisch L.A., Dai J., Hoffman M.D., Weichselbaum R.R., Pitroda S.P., Chmura S.J., Luke J.J. Phase I study of stereotactic body radiotherapy plus nivolumab and urelumab or cabiralizumab in advanced solid tumors. Clin. Cancer Res. 2021;27:5510–5518. doi: 10.1158/1078-0432.CCR-21-0810. [DOI] [PubMed] [Google Scholar]
- 27.Borgoni S., Iannello A., Cutrupi S., Allavena P., D’Incalci M., Novelli F., Cappello P. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating T cells and restores their anti-tumor phenotype. Oncoimmunology. 2018;7:e1393596. doi: 10.1080/2162402X.2017.1393596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holen I., Coleman R.E. Anti-tumour activity of bisphosphonates in preclinical models of breast cancer. Breast Cancer Res. 2010;12:214. doi: 10.1186/bcr2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiskopf K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur. J. Cancer. 2017;76:100–109. doi: 10.1016/j.ejca.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Liu X., Pu Y., Cron K., Deng L., Kline J., Frazier W.A., Xu H., Peng H., Fu Y.X., Xu M.M. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 2015;21:1209–1215. doi: 10.1038/nm.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sockolosky J.T., Dougan M., Ingram J.R., Ho C.C., Kauke M.J., Almo S.C., Ploegh H.L., Garcia K.C. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. U S A. 2016;113:E2646–E2654. doi: 10.1073/pnas.1604268113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu M.M., Pu Y., Han D., Shi Y., Cao X., Liang H., Chen X., Li X.D., Deng L., Chen Z.J., et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity. 2017;47:363–373.e5. doi: 10.1016/j.immuni.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pourakbari R., Hajizadeh F., Parhizkar F., Aghebati-Maleki A., Mansouri S., Aghebati-Maleki L. Co-stimulatory agonists: an insight into the immunotherapy of cancer. EXCLI J. 2021;20:1055–1085. doi: 10.17179/excli2021-3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sikic B.I., Lakhani N., Patnaik A., Shah S.A., Chandana S.R., Rasco D., Colevas A.D., O'Rourke T., Narayanan S., Papadopoulos K., et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019;37:946–953. doi: 10.1200/JCO.18.02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rakhmilevich A.L., Buhtoiarov I.N., Malkovsky M., Sondel P.M. CD40 ligation in vivo can induce T cell independent antitumor effects even against immunogenic tumors. Cancer Immunol. Immunother. 2008;57:1151–1160. doi: 10.1007/s00262-007-0447-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Long K.B., Gladney W.L., Tooker G.M., Graham K., Fraietta J.A., Beatty G.L. IFNγ and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 2016;6:400–413. doi: 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vonderheide R.H. CD40 agonist antibodies in cancer immunotherapy. Annu. Rev. Med. 2020;71:47–58. doi: 10.1146/annurev-med-062518-045435. [DOI] [PubMed] [Google Scholar]
- 38.Fayad L., Ansell S.M., Advani R., Coiffier B., Stuart R., Bartlett N.L., Forero-Torres A., Kuliczkowski K., Belada D., Ng E., et al. Dacetuzumab plus rituximab, ifosfamide, carboplatin and etoposide as salvage therapy for patients with diffuse large B-cell lymphoma relapsing after rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone: a randomized, double-blind, placebo-controlled phase 2b trial. Leuk. Lymphoma. 2015;9:2569–2578. doi: 10.3109/10428194.2015.1007504. [DOI] [PubMed] [Google Scholar]
- 39.Lv J., Liu C., Chen F.K., Feng Z.P., Jia L., Liu P.J., Yang Z.X., Hou F., Deng Z.Y. M2-like tumour-associated macrophage-secreted IGF promotes thyroid cancer stemness and metastasis by activating the PI3K/AKT/mTOR pathway. Mol. Med. Rep. 2021;24:604. doi: 10.3892/mmr.2021.12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H.Y., Zhang Y.Y., Xing T., Tang S.Q., Wen Q., Lyu Z.S., Lv M., Wang Y., Xu L.P., Zhang X.H., et al. M2 macrophages, but not M1 macrophages, support megakaryopoiesis by upregulating PI3K-AKT pathway activity. Signal Transduct. Target. Ther. 2021;6:234. doi: 10.1038/s41392-021-00627-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S., Woo G., Nguyen A.V., Figueiredo C.C., Foubert P., et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Henau O., Rausch M., Winkler D., Campesato L.F., Liu C., Cymerman D.H., Budhu S., Ghosh A., Pink M., Tchaicha J., et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443–447. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X., Luo G., Zhang K., Cao J., Huang C., Jiang T., Liu B., Su L., Qiu Z. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic cancer metastasis. Cancer Res. 2018;78:4586–4598. doi: 10.1158/0008-5472.CAN-17-3841. [DOI] [PubMed] [Google Scholar]
- 44.Powles T., Lackner M.R., Oudard S., Escudier B., Ralph C., Brown J.E., Hawkins R.E., Castellano D., Rini B.I., Staehler M.D., et al. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2016;34:1660–1668. doi: 10.1200/JCO.2015.64.8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanacker H., Cassier P.A., Bachelot T. The complex balance of PI3K inhibition. Ann. Oncol. 2021;32:127–128. doi: 10.1016/j.annonc.2020.10.597. [DOI] [PubMed] [Google Scholar]
- 46.Dent S., Cortés J., Im Y.H., Diéras V., Harbeck N., Krop I.E., Wilson T.R., Cui N., Schimmoller F., Hsu J.Y., et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: the SANDPIPER trial. Ann. Oncol. 2021;32:197–207. doi: 10.1016/j.annonc.2020.10.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cen X., Liu S., Cheng K. The role of toll-like receptor in inflammation and tumor immunity. Front. Pharmacol. 2018;9:878. doi: 10.3389/fphar.2018.00878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodell C.B., Arlauckas S.P., Cuccarese M.F., Garris C.S., Li R., Ahmed M.S., Kohler R.H., Pittet M.J., Weissleder R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018;2:578–588. doi: 10.1038/s41551-018-0236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dajon M., Iribarren K., Cremer I. Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology. 2017;222:89–100. doi: 10.1016/j.imbio.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 50.Dacoba T.G., Anfray C., Mainini F., Allavena P., Alonso M.J., Torres Andón F., Crecente-Campo J. Arginine-based poly(I:C)-Loaded nanocomplexes for the polarization of macrophages toward M1-antitumoral effectors. Front. Immunol. 2020;11:1412. doi: 10.3389/fimmu.2020.01412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng J., Mo J., Zhu T., Zhuo W., Yi Y., Hu S., Yin J., Zhang W., Zhou H., Liu Z. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer. 2020;19:133. doi: 10.1186/s12943-020-01250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohkuri T., Kosaka A., Nagato T., Kobayashi H. Effects of STING stimulation on macrophages: STING agonists polarize into "classically" or "alternatively" activated macrophages? Hum. Vaccin. Immunother. 2018;14:285–287. doi: 10.1080/21645515.2017.1395995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Downey C.M., Aghaei M., Schwendener R.A., Jirik F.R. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2'3'-cGAMP, induces M2 macrophage repolarization. PLoS One. 2014;9:e99988. doi: 10.1371/journal.pone.0099988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKeage M.J., Von Pawel J., Reck M., Jameson M.B., Rosenthal M.A., Sullivan R., Gibbs D., Mainwaring P.N., Serke M., Lafitte J.J., et al. Randomised phase II study of ASA404 combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Br. J. Cancer. 2008;99:2006–2012. doi: 10.1038/sj.bjc.6604808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lara P.N., Jr., Douillard J.Y., Nakagawa K., von Pawel J., McKeage M.J., Albert I., Losonczy G., Reck M., Heo D.S., Fan X., et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J. Clin. Oncol. 2011;29:2965–2971. doi: 10.1200/JCO.2011.35.0660. [DOI] [PubMed] [Google Scholar]
- 56.Daei Farshchi Adli A., Jahanban-Esfahlan R., Seidi K., Samandari-Rad S., Zarghami N. An overview on Vadimezan (DMXAA): the vascular disrupting agent. Chem. Biol. Drug Des. 2018;91:996–1006. doi: 10.1111/cbdd.13166. [DOI] [PubMed] [Google Scholar]
- 57.Meric-Bernstam F., Sandhu S., Hamid O., Spreafico A., Kasper S., Dummer R., Shimizu T., Steeghs N., Lewis N., Talluto C., et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol. 2019;37:2507. [Google Scholar]
- 58.Ribas A., Medina T., Kummar S., Amin A., Kalbasi A., Drabick J.J., Barve M., Daniels G.A., Wong D.J., Schmidt E.V., et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov. 2018;8:1250–1257. doi: 10.1158/2159-8290.CD-18-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Y., Jin H., Song Y., Huang T., Cao J., Tang Q., Zou Z. Targeting tumor-associated macrophages: a potential treatment for solid tumors. J. Cell. Physiol. 2021;236:3445–3465. doi: 10.1002/jcp.30139. [DOI] [PubMed] [Google Scholar]
- 60.Haymaker C., Johnson D.H., Murthy R., Bentebibel S.E., Uemura M.I., Hudgens C.W., Safa H., James M., Andtbacka R.H.I., Johnson D.B., et al. Tilsotolimod with ipilimumab drives tumor responses in anti-PD-1 refractory melanoma. Cancer Discov. 2021;11:1996–2013. doi: 10.1158/2159-8290.CD-20-1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mavragani I.V., Nikitaki Z., Kalospyros S.A., Georgakilas A.G. Ionizing radiation and complex DNA damage: from prediction to detection challenges and biological significance. Cancers (Basel) 2019;11:1789. doi: 10.3390/cancers11111789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Younes A.I., Barsoumian H.B., Sezen D., Verma V., Patel R., Wasley M., Hu Y., Dunn J.D., He K., Chen D., et al. Addition of TLR9 agonist immunotherapy to radiation improves systemic antitumor activity. Transl. Oncol. 2021;14:100983. doi: 10.1016/j.tranon.2020.100983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malfitano A.M., Pisanti S., Napolitano F., Di Somma S., Martinelli R., Portella G. Tumor-associated macrophage status in cancer treatment. Cancers (Basel) 2020;12:1987. doi: 10.3390/cancers12071987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stafford J.H., Hirai T., Deng L., Chernikova S.B., Urata K., West B.L., Brown J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016;18:797–806. doi: 10.1093/neuonc/nov272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kioi M., Vogel H., Schultz G., Hoffman R.M., Harsh G.R., Brown J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest. 2010;120:694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Q., Allouch A., Martins I., Modjtahedi N., Deutsch E., Perfettini J.L. Macrophage biology plays a central role during ionizing radiation-elicited tumor response. Biomed. J. 2017;40:200–211. doi: 10.1016/j.bj.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El-Saghire H., Michaux A., Thierens H., Baatout S. Low doses of ionizing radiation induce immune-stimulatory responses in isolated human primary monocytes. Int. J. Mol. Med. 2013;32:1407–1414. doi: 10.3892/ijmm.2013.1514. [DOI] [PubMed] [Google Scholar]
- 68.Kim Y.H., Gratzinger D., Harrison C., Brody J.D., Czerwinski D.K., Ai W.Z., Morales A., Abdulla F., Xing L., Navi D., et al. In situ vaccination against mycosis fungoides by intratumoral injection of a TLR9 agonist combined with radiation: a phase 1/2 study. Blood. 2012;119:355–363. doi: 10.1182/blood-2011-05-355222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Advani R., Flinn I., Popplewell L., Forero A., Bartlett N.L., Ghosh N., Kline J., Roschewski M., LaCasce A., Collins G.P., et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin's lymphoma. N. Engl. J. Med. 2018;379:1711–1721. doi: 10.1056/NEJMoa1807315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferris R.L., Saba N.F., Gitlitz B.J., Haddad R., Sukari A., Neupane P., Morris J.C., Misiukiewicz K., Bauman J.E., Fenton M., et al. Effect of adding motolimod to standard combination chemotherapy and cetuximab treatment of patients with squamous cell carcinoma of the head and neck: the Active8 randomized clinical trial. JAMA Oncol. 2018;4:1583–1588. doi: 10.1001/jamaoncol.2018.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang E., Pelosof L., Lemery S., Gong Y., Goldberg K.B., Farrell A.T., Keegan P., Veeraraghavan J., Wei G., Blumenthal G.M., et al. Systematic review of PD-1/PD-L1 inhibitors in oncology: from personalized medicine to public health. Oncologist. 2021;26:e1786–e1799. doi: 10.1002/onco.13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bockorny B., Semenisty V., Macarulla T., Borazanci E., Wolpin B.M., Stemmer S.M., Golan T., Geva R., Borad M.J., Pedersen K.S., et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat. Med. 2020;26:878–885. doi: 10.1038/s41591-020-0880-x. [DOI] [PubMed] [Google Scholar]
- 73.Surolia I., Gulley J., Madan R.A. Recent advances in the use of therapeutic cancer vaccines in genitourinary malignancies. Expert Opin. Biol. Ther. 2014;14:1769–1781. doi: 10.1517/14712598.2014.955010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maiorano B.A., Schinzari G., Ciardiello D., Rodriquenz M.G., Cisternino A., Tortora G., Maiello E. Cancer vaccines for genitourinary tumors: recent progresses and future possibilities. Vaccines (Basel). 2021;9:623. doi: 10.3390/vaccines9060623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pavlick A., Blazquez A.B., Meseck M., Lattanzi M., Ott P.A., Marron T.U., Holman R.M., Mandeli J., Salazar A.M., McClain C.B., et al. Combined vaccination with NY-ESO-1 protein, poly-ICLC, and montanide improves humoral and cellular immune responses in patients with high-risk melanoma. Cancer Immunol. Res. 2020;8:70–80. doi: 10.1158/2326-6066.CIR-19-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gibney G.T., Kudchadkar R.R., DeConti R.C., Thebeau M.S., Czupryn M.P., Tetteh L., Eysmans C., Richards A., Schell M.J., Fisher K.J., et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015;21:712–720. doi: 10.1158/1078-0432.CCR-14-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang C., Steinmetz N.F. CD47 blockade and cowpea mosaic virus Nanoparticle in situ vaccination triggers phagocytosis and tumor killing. Adv. Healthc. Mater. 2019;8:e1801288. doi: 10.1002/adhm.201801288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gun S.Y., Lee S.W.L., Sieow J.L., Wong S.C. Targeting immune cells for cancer therapy. Redox Biol. 2019;25:101174. doi: 10.1016/j.redox.2019.101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anselmo A.C., Gilbert J.B., Kumar S., Gupta V., Cohen R.E., Rubner M.F., Mitragotri S. Monocyte-mediated delivery of polymeric backpacks to inflamed tissues: a generalized strategy to deliver drugs to treat inflammation. J. Control. Release. 2015;199:29–36. doi: 10.1016/j.jconrel.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 80.Zhu G., Mei L., Vishwasrao H.D., Jacobson O., Wang Z., Liu Y., Yung B.C., Fu X., Jin A., Niu G., et al. Intertwining DNA-RNA nanocapsules loaded with tumor neoantigens as synergistic nanovaccines for cancer immunotherapy. Nat. Commun. 2017;8:1482. doi: 10.1038/s41467-017-01386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang L., Tian L., Dai X., Yu H., Wang J., Lei A., Zhu M., Xu J., Zhao W., Zhu Y., et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020;13:153. doi: 10.1186/s13045-020-00983-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen Y., Yu Z., Tan X., Jiang H., Xu Z., Fang Y., Han D., Hong W., Wei W., Tu J. CAR-macrophage: a new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 2021;139:111605. doi: 10.1016/j.biopha.2021.111605. [DOI] [PubMed] [Google Scholar]