

Summary
To identify cellular mechanisms responsible for pressure overload triggered heart failure, we isolated cardiomyocytes, endothelial cells, and fibroblasts as most abundant cell types from mouse hearts in the subacute and chronic stages after transverse aortic constriction (TAC) and performed RNA-sequencing. We detected highly cell-type specific transcriptional responses with characteristic time courses and active intercellular communication. Cardiomyocytes after TAC exerted an early and sustained upregulation of inflammatory and matrix genes and a concomitant suppression of metabolic and ion channel genes. Fibroblasts, in contrast, showed transient early upregulation of inflammatory and matrix genes and downregulation of angiogenesis genes, but sustained induction of cell cycle and ion channel genes during TAC. Endothelial cells transiently induced cell cycle and extracellular matrix genes early after TAC, but exerted a long-lasting upregulation of inflammatory genes. As we found that matrix production by multiple cell types triggers pathological cellular responses, it might serve as a future therapeutic target.
Subject areas: Biological sciences, Molecular biology, Systems biology, Transcriptomics
Graphical abstract
Highlights
-
•
TAC induces matrix and growth, but reduces contraction genes in cardiomyocytes
-
•
TAC induces genes related to matrix, inflammation, and cell cycle in endothelial cells
-
•
TAC induces matrix and inflammation, but reduces angiogenesis genes in fibroblasts
-
•
Matrix proteins trigger growth, proliferation, and migration in cardiac cells
Biological sciences; Molecular biology; Systems biology; Transcriptomics
Introduction
Despite modern therapies, chronic heart failure (CHF) remains associated with high mortality rates, similar as observed in many forms of cancer (Mamas et al., 2017). One common stimulus ultimately leading to heart failure is chronic pressure overload, for example, due to long-standing uncontrolled arterial hypertension or aortic stenosis (Heineke and Molkentin, 2006). At a certain point in disease progression, even removal of pressure overload cannot sufficiently ameliorate heart failure (Hein et al., 2003). In order to improve morbidity and mortality of CHF patients, new therapeutic targets need to be identified that enable therapies beyond the current treatment algorithms, addressing, for example, pathological molecular circuits in specific cardiac cell types that have so far not been recognized. In terms of numbers, cardiomyocytes, endothelial cells, and fibroblasts are the three main cell types in the heart (Pinto et al., 2016). When the heart is exposed to pressure overload, several processes simultaneously occur in different cell types: Cardiomyocytes undergo hypertrophic growth, cellular metabolism partially switches from fatty acid to glucose oxidation and a number of genes that are typically only expressed during embryonic development become re-expressed in ventricular cardiomyocytes (Heineke and Molkentin, 2006; Hill and Olson, 2008; Neubauer, 2007). In the chronic phase, cardiomyocyte dysfunction becomes stronger, energetic shortage prevails, and cellular hypertrophy reaches a plateau (Neubauer, 2007). Cardiac fibroblasts proliferate and produce extracellular matrix (Souders et al., 2009). Cardiac endothelial cells engage in enhanced angiogenic activity, but in the chronic phase, capillary rarefaction emerges (Hein et al., 2003; Heineke et al., 2007; Mohammed et al., 2015; Sano et al., 2007). The myocardial response to pressure overload is coordinated between the different compartments by intercellular communication, for example, via secreted proteins, but details remain largely unknown. To obtain an unbiased view of pathological mechanisms, gene expression has to be assessed in different cell types and at different time points during the development of heart failure due to different etiologies (e.g., myocardial infarction, pressure overload, diabetes mellitus). Initially, transcriptional profiling had been applied mainly to whole heart samples from mice and humans (Liu et al., 2015; Tan et al., 2002; Yang et al., 2014; Zhao et al., 2004). By this approach, predominantly the RNA expression of cardiomyocytes is determined, since these cells constitute by far the biggest portion of the heart’s volume (Zhou and Pu, 2016). Although these studies gave important insights, the gene-expression patterns in non-cardiomyocytes remained far less well explored. In addition, regulation of gene expression occurs at different levels, transcriptionally, but also post-transcriptionally, and therefore cannot solely be assessed at the mRNA stage. In this study, we compared the RNA transcriptome of cardiac myocytes, endothelial cells and fibroblasts in mice under baseline conditions and during short term and chronic pressure overload. Our study reveals the highly cell-specific nature and time course of gene-expression changes after TAC and computes potential responsible transcriptional regulators. Furthermore, increased and decreased ligand receptor interactions between the three cell types during TAC were analyzed. In the course of the study, we focused on the endothelial cell lineage and conducted single cell as well as Ribo-Tag-Sequencing, again under baseline condition and during pressure overload. We found a transient expression of collagen and matrix remodeling genes in endothelial cells, but also in fibroblasts and cardiomyocytes early after pressure overload. This is relevant for disease progression and intercellular communication because matrix proteins such as fibronectin trigger cardiomyocyte hypertrophy and promote endothelial proliferation and migration as well as fibroblast migration.
Results
The phenotype of subacute and chronic cardiac pressure overload in mice
Mice were analyzed 1 or 8 weeks after TAC as models of subacute or chronic pressure overload, respectively (Figure S1A). The heart weight to body weight ratio (HW/BW) as a sign of cardiac hypertrophy was significantly increased after one week of TAC versus sham operated mice and further increased significantly 8 weeks after TAC (Figure S1B). Echocardiography revealed a preserved left ventricular systolic function (measured as ejection fraction, EF) in the subacute state, but a strong decline occurred in the chronic phase. Similarly, an increase of left ventricular end-diastolic area, which indicates ventricular dilatation, was only noted 8 weeks after TAC. The left ventricular wall thickness was significantly increased one and 8 weeks after TAC. The heart rate during echocardiography did not significantly differ between all groups (Figures S1C–S1F). Histological examination after staining with Sirius red showed significantly more myocardial fibrosis after TAC, but no difference between both TAC time points was visible (Figures S1G and S1H). As determined by RNA sequencing and verified in part by qPCR, the expression of typical stress responsive genes (Nppa, Nppb, Myh7, Acta1) was markedly upregulated 1 and 8 weeks after TAC in cardiomyocytes (Figures S1I and S1J).
Purification of cardiomyocytes, fibroblasts and endothelial cells from mouse hearts
After enzymatic digestion of mouse hearts, endothelial cells were labeled with anti-Cd146 coupled microbeads and captured from the digestion solution by Miltenyi columns. In immunofluorescence staining, we verified that CD146 protein indeed co-localizes with Isolectin B4 labeled endothelial cells in mouse heart sections one and 8 weeks after sham or TAC surgery (Figure S2A). The endothelial cell depleted column flow-through was incubated with feeder cell removal microbeads to label and capture cardiac fibroblasts. Cardiomyocytes were isolated from separate hearts with a Langendorff apparatus and isolated from the non-myocytes by sedimentation. RNA from the three cardiac cell types was isolated from mice one week after sham or TAC surgery and 8 weeks after TAC surgery and was subjected to bulk RNA sequencing (Figure 1A). Interrogation of cell marker gene expression from this analysis revealed exclusive high expression of typical contractile genes (Myh6, Tnnt2, Actc1, Ttn and Myh7) in cardiomyocytes, but markedly less expression of typical fibroblast and endothelial genes after sham or TAC surgery at all time points in these cells. Fibroblasts are less well defined, but PDGFRα was suggested as cardiac fibroblast specific marker, and different collagen genes as well as Vimentin are often used to identify these cells (Pinto et al., 2016). Accordingly, the expression of Col3a1, Col1a1, Vim (Vimentin) and Pdgfrα was high in fibroblasts after sham and after 1 or 8 weeks of TAC surgery. Postn (Periostin), which is a marker of activated fibroblasts, was expressed at low levels in fibroblasts from sham operated mice, but was induced after TAC (Tallquist and Molkentin, 2017). Cardiac endothelial cells under all conditions exerted high expression levels of Cdh5 (VE-Cadherin), Pecam1 (CD31), Kdr (VEGF-receptor 2), Flt1 (VEGF-receptor 1) and Vwf (von Willebrandt factor), which are all highly specific endothelial cell markers. In addition, however, we noted that one week after TAC endothelial cells expressed Col3a1, Col1a1, Vim and Postn and therefore co-clustered with fibroblasts with regard to marker gene expression, although Pdgfrα expression remained low, suggesting that no complete transition to fibroblast gene-expression took place (Figure 1B). We verified the purity of the isolated cardiac endothelial cells by FACS analysis, which revealed around 93% of these cells were double-positive for CD31 (Pecam1) and CD102 (ICAM1) and around 95% were positive for CD31 alone (Figure 1C). Cardiac endothelial cells were largely negative for the pan-leukocyte marker CD45 (only 2.3% positive) and the fibroblast marker Mefsk4 (only 1.2% positive). Analysis of fibroblast purity revealed 96% Mefsk4 positivity, but also around 10–20% positivity for CD31. In addition, we found the fibroblasts to be 30.5% positive for CD45, which had previously been observed (Skelly et al., 2018). All this showed that fibroblasts (isolated by the specific marker Mefsk4) are a rather heterogeneous cell population. The FACS analyses results were almost identical after 8 weeks of TAC, but the endothelial cells showed a bit higher positivity for CD45 (11%) and Mefsk4 (around 7%) after one week TAC (Figures S2B and S2C). A principal component analysis from the bulk RNA sequencing data confirmed that global gene-expression patterns were strongly different between all cell types (Figure 1D). In cardiomyocytes, gene-expression was significantly different after one week TAC, but did not change much further 8 weeks after surgery. Endothelial and similarly fibroblast gene-expression, in contrast, became markedly different from sham after one week of TAC, but normalized back toward sham levels 7 weeks later. In the analysis of protein coding mRNAs one week after TAC versus sham, 1772 genes were significantly upregulated in cardiomyocytes, 4325 in fibroblasts and 1565 in endothelial cells. 620 genes were commonly upregulated in all cell types (Figure S3A). On the other hand, 7073 genes were significantly downregulated in cardiomyocytes, 3587 and 4199 genes were downregulated in fibroblasts and endothelial cells, respectively. 1098 genes were commonly downregulated in all cell types (Figure S3B). 8 weeks after TAC 2262 genes were significantly upregulated versus sham in cardiomyocytes, 3609 in fibroblasts and 816 in endothelial cells. 78 genes were commonly upregulated in all cell types (Figure S3C). On the other hand, 5189 genes were significantly downregulated in cardiomyocytes, 2757 and 754 genes were downregulated in fibroblasts and endothelial cells, respectively (Figure S3D). With regard to annotated non-coding (nc) RNAs, we identified mainly antisense RNAs and lincRNAs. microRNAs were rarely picked up in our analysis, because they were lost due to their small size in our RNA isolating procedure. In order to minimize false positive results in this often low expressed group of RNAs, we only considered RNAs with an expression level of at least 2 counts per million reads (cpm). We found that 41 annotated non-coding RNAs were significantly upregulated in cardiomyocytes, 107 in fibroblasts and 33 in endothelial cells one week after TAC versus sham (Figure S4). 180 ncRNAs were significantly downregulated in cardiomyocytes, 85 in endothelial cells and 50 in fibroblasts one week after TAC. Because most of the ncRNAs are functionally still not well characterized, we focused our subsequent analyses on protein-coding mRNAs, in order to identify so far unrecognized cellular processes that could affect pressure overload triggered myocardial disease. The complete gene-expression tables from cardiomyocytes, endothelial cells and fibroblasts after sham, and one and 8 weeks after TAC surgery are available in Table S2.Gene expression in cardiomyocytes one week after TAC, related to Figure 1, Table S3.Gene expression in cardiomyocytes 8 weeks after TAC, related to Figure 1, Table S4.Gene expression in fibroblasts one week after TAC, related to Figure 1, Table S5.Gene expression in fibroblasts 8 weeks after TAC, related to Figure 1, Table S6.Gene expression in endothelial cells one week after TAC, related to Figure 1, Table S7.Gene expression in endothelial cells 8 weeks after TAC, related to Figure 1.
Figure 1.

Endothelial, fibroblast and cardiomyocyte cell purification from mouse hearts
(A) Scheme on the experimental procedures to isolate the indicated cell types from mouse hearts.
(B) Heatmap of cell marker gene expression in RNA from isolated cardiomyocytes (CM), endothelial cells (EC) or fibroblasts (FB) after sham or the indicated time point after TAC surgery.
(C) Cardiac endothelial cells and fibroblasts were isolated from mouse hearts and stained for the endothelial markers CD31 and CD102, for the leukocyte marker CD45, and the fibroblast marker Mefsk4. Subsequently, flow cytometric analyses were performed and representative results are shown here. The numbers indicated in each quadrant indicates the percentage of cells localized in that particular quadrant.
(D) RNA from the different cell types after sham or 1 and 8 weeks after TAC was subjected to RNA sequencing. The differences in overall gene expression patterns were visualized by a principal component analysis.
Cardiomyocyte gene expression after TAC
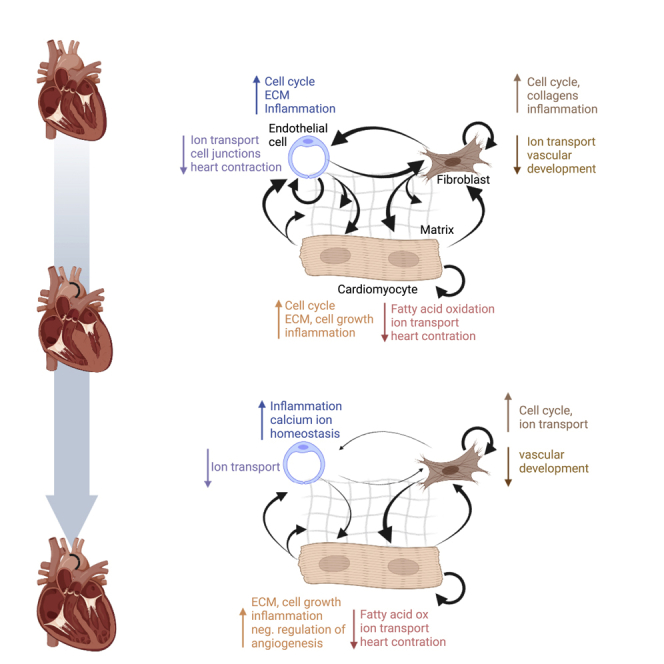
TAC induced a marked dysregulation of gene-expression in cardiomyocytes, which followed different patterns and was subdivided into eight different clusters (Figures 2A–2C). Genes in clusters 1, 2, 3 and 7 were mainly downregulated versus sham in response to TAC. Cluster 1 contained genes important for catabolic processes and fatty acid oxidation (Gene ontology, biological process), while Cluster 2 included genes involved in transmembrane ion transport, but also for adrenergic receptors. mRNAs of clusters 1 and 2 were suppressed already at one week, and stayed downregulated at the chronic 8 weeks time point after TAC. The dynamic regulation of clusters is shown in Figure 2A (with example genes) and C, while related gene-ontology classes are displayed in Figure 2B. Genes in cluster 3 (connected to basic cell function, e.g. mRNA processing, mitochondrial genes, DNA replication) were mildly downregulated only late in disease. Cluster 7 genes were only marginally regulated. We found upregulated genes in clusters 4, 5, 6 and 8. Genes in clusters 4 (related to inflammation) and 6 (related to extracellular matrix, cell growth and wound healing) were induced already after 1 week of TAC and stayed upregulated at the chronic stage. Cluster 5 mRNAs (related to negative regulation of cell migration and (negative) regulation of vessel morphogenesis) were mainly upregulated 8 weeks after TAC. Genes in cluster 8 were only transiently upregulated 1 week after TAC and encoded proteins involved in DNA replication and cell cycle control. We verified the mRNA upregulation of selected genes (Ccl2, Col1a1, Col3a1, Thbs1) in isolated cardiomyocytes after 1 and 8 weeks of TAC by qPCR, which compare well with the results from our RNAseq approach (compare Table S1), although no statistical significance was found for the induction of Col1a1 and Thbs1 after 8 weeks of TAC (Figures 2D–2G). To identify potential transcriptional regulators of the observed gene-expression patterns in the different clusters, we employed a motif search algorithm to identify transcription factor binding sites in the promoter region of the regulated genes within the clusters. The top identified motifs for each cluster are demonstrated in Figure 2H. Of note, the transcription factor MEF2, identified in cluster 2, was previously found to induce cardiomyopathy when overexpressed in mouse cardiomyocytes, in part by dysregulating ion channel genes as they are contained in cluster 2 (Xu et al., 2006). With regard to the promoters of inflammatory genes in cluster 4, we found overrepresentation of motifs binding Twist2, SpiB, PU and Ets1 transcription factors, which all induce inflammatory genes (Ghisletti et al., 2010; Mudry et al., 2015). SMAD2 and 3 binding sites were found in promoters of extracellular matrix genes in cluster 6, which fits to the known role of TGFβ/SMAD signaling for the induction of myocardial fibrosis (Khalil et al., 2017). Binding motifs for Jun and Fos (as part of the AP-1 complex, involved in cardiomyocyte hypertrophy) were found in promotors of cluster 5 and 6 genes. Genes in cluster 8 exerted an enrichment of E2F transcription factor binding sites, typically found in promotors of cell cycle genes (Muller et al., 2017). Many additional motifs were identified, which could be investigated in the future. We performed a ligand receptor analysis to investigate the potential interaction of cardiomyocyte derived, pressure overload regulated protein ligands with their receptors expressed in endothelial cells, fibroblasts and cardiomyocytes, according to our RNA-seq analysis. To identify these interactions, we considered ligands from cardiomyocytes that were significantly upregulated (“GAINED”) or downregulated (“LOSS”) in these cells by at least two-fold after one week of TAC. To prioritize among the ligand-receptor pairs in each category, a ligand-receptor score (that included expression levels of ligand and receptor and fold-change regulation of the ligand due to TAC) was calculated for each ligand-receptor pair. More than 400 cardiomyocyte derived interactions were gained after TAC (vs. sham),while 150 interactions were lost or reduced toward other cells (Figure S5A). The highest amount of ligand receptor interactions from cardiomyocytes was outgoing toward fibroblasts. At 8 weeks of TAC less cardiomyocyte derived interactions were initiated to endothelial cells and fibroblasts and even less interactions were lost, while signals gained from cardiomyocytes to other cardiomyocytes slightly increased (Figure S5B). As indicated in Figures S5C and S5D, after 1 week of TAC, cardiomyocytes induce collagens and matrix glycoproteins, but also growth factors (e.g. BMP10 and TGFβ2) to interact with other cells. In turn, cardiomyocytes withdrew, for instance, FGF13 and 16, Laminin and EphrinB3 based signals to other cells and Angiopoetin-1 was less released toward endothelial cells. After 8 weeks, the interaction of the natriuretic peptides (encoded by Nppa and Nppb) with their receptors Npr1 and Npr3 on cardiomyocytes became more relevant, because of the strong upregulation especially of Npr3 (10-fold) and Npr1 (2-fold). We also compared our analysis to the one performed by Zongna Ren et al., who performed a single cell analysis of cardiomyocytes and non-cardiomyocytes from hearts at different time points after TAC (Figures S5E–S5H)(Ren et al., 2020). When comparing our 8 cardiomyocyte subclusters (Figures 2A–2C) to the 9 clusters identified by Ren et al. in cardiomyocytes, we found that very large parts of the genes we identified were not detected by Ren et al. (“novel”, Figure S5E). These “novel” genes, belonged to GO terms shown in Figure S5G (e.g., fatty-acid beta oxidation, DNA repair, leukocyte migration, extracellular matrix organization). On the other hand, we also identified about 80% of the genes detected by Ren et al., which distributed to different clusters from their analysis (“known genes”). GO terms related to these genes are shown in Figure S5H. About 20% of these genes were not found in our analysis (Figure S5F), they were related to the GO-terms sarcomere organization and heart contraction (Figure S5H). Together, these results indicate that cardiomyocytes profoundly change their gene-expression during pressure overload, resulting in a long lasting upregulation of inflammatory, anti-angiogenic and extracellular matrix genes as well as a sustained downregulation of metabolic, ion channel and basic cell function genes. While cardiomyocytes engage in rich intercellular communications toward endothelial cells and fibroblasts and other cardiomyocytes early after TAC, this is markedly reduced toward interaction mainly with other cardiomyocytes in the chronic phase.
Figure 2.

Cardiomyocyte gene expression after TAC
The cardiomyocyte gene expression was analyzed with RNA sequencing
(A) A heatmap of differentially expressed genes in cardiomyocytes in sham vs. 1 week and vs. 8 weeks of TAC is shown. Eight different clusters were observed that each had a characteristic expression pattern. Example genes are indicated on the right.
(B) Gene ontology terms (GO, biological process) related to the 8 clusters are shown.
(C) Visualization of cluster gene regulation over time under the indicated conditions. Data are shown as mean ±SEM.
(D–G) Relative mRNA levels of the indicated genes measured by qPCR in isolated cardiomyocytes from mice treated as indicated. Data are shown as mean ±SEM. ∗p<0.05, ∗∗p<0.01 as determined by two-tailed unpaired t-test, except for (F) in the right panel, where a two-tailed Mann-Whitney test was used.
(H) Enrichment of top ten transcription factor binding motifs in the promoter region of regulated genes in the indicated clusters.
Fibroblast gene expression after TAC
The TAC induced gene-expression in cardiac fibroblasts occurred in different patterns and was divided into 8 clusters (Figures S6A–S6C). Genes in Cluster 1 (related to cell adhesion, cation transport and membrane potential) were first sharply downregulated one week and then strongly upregulated 8 weeks after TAC. Cluster 2 genes (cell division and cell cycle genes) were strongly induced 1 week after TAC and stayed upregulated at a lower level 8 weeks after TAC, while genes in cluster 6 (related to inflammation, angiogenesis and collagens) were upregulated 1 week after TAC, but normalized 7 weeks later. Cluster 3 genes (related to fluid homeostasis and nerve impulse transmission) and cluster 4 genes (blood circulation, cell fate specification) were only upregulated in the chronic phase of disease. In turn, genes in cluster 7 (related to vascular development) and 8 (related to blood circulation, vascular and connective tissue development) were downregulated one and one and eight weeks after TAC, respectively. Analysis of potential transcriptional regulators revealed that promoters of cluster 4 and cluster 6 genes were strongly enriched with ETS-transcription factor binding motifs (Figure S6D). From these, especially PU.1 was previously implicated in myofibroblast formation and fibrosis (Khalil et al., 2017; Wohlfahrt et al., 2019). Ligand/receptor analyses showed that fibroblast increased many ligands to interact with other cells, while decreasing a slightly smaller number of ligands after one week of TAC (Figures S7A andS7C). In the chronic phase, far less fibroblast derived ligand-receptor pairs were up- or downregulated (Figures S7B andS7D). The ligands that were still upregulated (versus sham) at that stage were mainly interacting with cardiomyocytes or other fibroblasts. Example ligand receptor pairs are shown in the Figure. When comparing our bulk RNAseq analysis in fibroblasts with the single cell based approach by Ren et al., we again found that very large parts of the genes we identified were not detected by Ren et al. (“novel”, Figure S7E)(Ren et al., 2020). These “novel” genes, belonged to GO terms shown in Figure S7G (e.g. sodium ion transport, cell cycle phase transition, leukocyte migration, sprouting angiogenesis, ossification). On the other hand, we also identified about 60% of the genes detected by Ren et al., which distributed to different clusters from their analysis (“known genes”). GO terms related to these genes are shown in Figure S7H. About 40% of these genes were not found in our analysis (Figure S7F), they were related to the GO-terms oxidative phosphorylation and generation of precursor metabolites and energy (Figure S7H). In summary, we observed a very versatile regulation of genes in cardiac fibroblasts, with only transient upregulation of extracellular matrix and inflammatory genes and transient downregulation of angiogenesis related genes in the subacute phase after TAC, but sustained upregulation of cell-cycle, cell adhesion and ion channel genes in the chronic phase.
Endothelial cell specific gene expression in response to TAC
One week after TAC, endothelial cells exerted mainly transient changes in gene-expression that were divided into 8 clusters with regard to regulation dynamics (Figures 3A–3C). Genes in cluster 1 (mitosis, cell cycle and extracellular matrix genes) and cluster 7 (extracellular matrix and matrix remodeling genes) were upregulated one week, but returned toward sham levels 8 weeks after TAC. We verified increased endothelial cell cycle activity by immunostaining for the cell proliferation marker Ki67 (together with endothelial labeling by Isolectin B4), which was increases in capillary endothelial cells one week, but not 8 weeks after TAC (Figure 3D). Cluster 3 genes (related to inflammation) were upregulated one week after TAC and remained induced seven weeks later. Genes in clusters 2 (cell junction, endothelial/vascular genes), 4 (mRNA processing/splicing genes) and 5 (heart contraction, potassium ion transport genes) were all downregulated subacutely after TAC, but returned to sham levels in the chronic phase. The expression of cardiomyocyte sarcomeric genes (“heart contraction”) in endothelial cells has been observed before, but its functional significance remains unclear (Li et al., 2019). Genes in cluster 6 (related to calcium ion homeostasis) were upregulated selectively 8 weeks after TAC, and genes in cluster 8 (ion transport genes) were downregulated one and 8 weeks after TAC. Binding sites of transcriptional regulators over-represented in the promoter regions of regulated genes in the different clusters are shown in Figure 3E. As an example, genes in cluster 1 exerted binding motifs for E2F transcription factors and CHR motifs (known regulate cell cycle genes) and SMAD (known to induce the expression of extracellular matrix genes) (Khalil et al., 2017; Muller et al., 2017). Accordingly, increased nuclear abundance of activated (phosphorylated) SMAD3 was detected in endothelial cells after 1 week of TAC in immunofluorescence staining (Figures 3F and 3G). Cluster 3 genes exerted promotor motifs for NFAT and RUNX and ETS transcription factors, which are typically found in promoter region of inflammatory genes (Lee et al., 2017; Wang et al., 2020b; Wohlfahrt et al., 2019). The ligand-receptor interaction analysis revealed that endothelial cells increased the expression of many paracrine and autocrine ligands after one week TAC compared to sham, whereas a much smaller number of ligands was downregulated (Figures 4A and 4C). 8 weeks after TAC, far less ligands were regulated (Figures 4B and4D). Endothelial cells increased the transcription of various matrix components and glycoproteins, which interacted with different integrins, but also other cell surface receptors on fibroblasts, cardiomyocytes and endothelial cells. We verified increased Fibronectin abundance after one week of TAC that overlapped strongly with endothelial cells at that time point by immunofluorescence staining. After 8 weeks of TAC, the Fibronectin increase was reduced compared to the one week time point and the overlap with endothelial cells was less evident (Figures 4E and 4F). Endothelial cells also enhanced the expression of growth factors (like Ctgf or Igf-1) and inflammatory stimuli (such as Il1b), interacting with their respective receptor on other cells. Example ligand receptor pairs are shown in the Figure. Because large parts of endothelial derived ligands were related to extracellular matrix, we assessed the functional impact of different matrix proteins on fibroblasts, endothelial cells and cardiomyocytes. As shown in Figures 4G and 4H, coating the cell culture dish with Fibronectin, but not Collagen or Gelatin (containing denatured collagen) induced cellular hypertrophy in neonatal rat cardiomyocytes compared with uncoated dishes. Culturing of C166 endothelial cells on Fibronectin or Collagen, but not on Gelatin, promoted their proliferation versus plating on uncoated dishes (Figure 4I). Similarly, Collagen, Fibronectin and even Gelatin triggered endothelial cell migration (in a scratch wound assay) (Figures 4J and 4K), while the migration of primary rat cardiac fibroblasts was only enhanced by Fibronectin. Therefore, matrix based proteins synthesized by cardiomyocytes, endothelial cells and fibroblasts after TAC strongly affect cell functions and will likely change the cardiac remodeling process. When comparing our bulk RNAseq analysis in endothelial cells with the single cell based approach by Ren et al., we again found that very large parts of the genes we identified were not detected by Ren et al. (“novel”, Figure S8A)(Ren et al., 2020). These “novel” genes, belonged to GO terms shown in Figure S8C (e.g., extracellular matrix organization, cell cycle phase transition, leukocyte migration, potassium ion transport). On the other hand, we also identified about 55% of the genes detected by Ren et al., which distributed to different clusters from their analysis (“known genes”, Figure S8B). GO terms related to these genes are shown in Figure S8D. About 45% of these genes were not found in our analysis (Figure S8B), they were related to the GO-terms cellular respiration and generation of precursor metabolites and energy (Figure S8D). In summary, cardiac endothelial cells transiently induced cell cycle and extracellular matrix genes and transiently downregulated cell junction genes one week after TAC, but induce inflammatory genes early that remain upregulated in the chronic phase.
Figure 3.

Endothelial cell gene expression after TAC
The endothelial gene expression was analyzed with RNA sequencing.
(A) A heatmap of differentially expressed genes in endothelial cells in sham vs. 1 week and vs. 8 weeks of TAC is shown. Eight different clusters were observed that each had a characteristic expression pattern. Example genes are indicated on the right.
(B) Gene ontology terms (GO, biological process) related to the 8 clusters are shown.
(C) Visualization of cluster gene regulation over time under the indicated conditions. Data are shown as mean ±SEM.
(D) Immunofluorescence staining for the indicated markers from mouse heart sections after treatment as indicated. The arrows point at Ki-67 positive endothelial cells. Scale bar: 50μm.
(E) Enrichment of top ten transcription factor binding motifs in the promoter region of regulated genes in the indicated clusters.
(F) Immunofluorescence staining for the indicated markers from mouse heart sections after treatment as indicated. The arrows point at phospho(p)-SMAD3 positive endothelial cells. Scale bar: 50μm.
(G) A quantitative analysis of pSMAD3 positive endothelial cells in hearts from mice treated as indicated. Data are shown as mean ±SEM. ∗p<0.05 as determined by one-way ANOVA with Sidak’s multiple comparisons test.
Figure 4.

Interaction of endothelial cell derived ligands in response to pressure overload
(A) Number of interactions between upregulated (gain) or downregulated (loss) endothelial cell (EC) derived ligands after 1 week (A) and 8 weeks (B) of TAC with receptors expressed in EC, fibroblasts (FB) and cardiomyocytes (CM). Example interactions gained or lost after 1 week (C) or 8 weeks (D) of TAC are shown.
(E and F) Immunofluorescence staining for the indicated markers from mouse heart sections after treatment as indicated. Scale bar: 50μm.
(G) Isolated rate neonatal cardiomyocytes plated on different matrices as shown and stained for markers as indicated. Scale bar: 50μm.
(H) Quantification from cardiomyocyte cell area from cells as shown in (G). Data are shown as mean ±SEM.
(I) C166 endothelial cell proliferation plated on different matrices as indicated measured as BrDU incorporation by ELISA. Data are shown as mean ±SEM.
(J and K) (J) Quantification and representative images (K) of endothelial cell migration after plating on different matrices as shown. Scale bar: 50μm. Data are shown as mean ±SEM.
(L) Migration of primary cardiac fibroblasts plated on different matrices as indicated. Data are shown as mean ±SEM. ∗p<0.05, ∗∗∗p<0.001, ∗∗∗∗p<0.0001 as determined by one-way ANOVA with Sidak’s multiple comparisons test.
Comparison of gene-expression between cell types
We noted in our analysis that despite a very different general gene-expression pattern between the three analyzed cell types (see Figure 1D), a subgroup of genes with similar function, mostly related to inflammation, cell cycle/mitosis, growth factors, cell adhesion and extracellular matrix were expressed and regulated in all cells, although with different time dynamics (see above). As shown in Table S1, cell cycle genes were expressed at low levels in cardiomyocytes, but were highly expressed in fibroblasts and endothelial cells. Growth factor genes were expressed at high levels in all cell types and were mainly upregulated early after TAC. Inflammatory genes were only expressed at low levels in cardiomyocytes, but very high in fibroblasts and endothelial cells, in which they even remained induced in the chronic phase. Unexpectedly, cell adhesion and collagen genes were robustly expressed in all cell types: Their mRNA levels were (as anticipated) highest in fibroblasts, where the expression was only mildly induced in response to one week of TAC, but changed back to sham level after 8 weeks. Cardiomyocyte had the lowest mRNA levels of these genes. Interestingly, the expression of cell adhesion genes in endothelial cells was about as high as in fibroblasts and the collagen genes were only slightly lower than in cardiac fibroblasts. Genes from both categories were upregulated after one week of TAC in endothelial cells, but their expression went down toward sham levels 8 weeks after TAC. It should be noted, however, that direct comparison of gene-expression between the cell types should be interpreted with caution, due to possible effects of differing isolation protocols.
Single cell sequencing of endothelial cells
Because we found the expression of matrix/adhesion, collagen and inflammatory genes in endothelial cells interesting, and because the impact of this cell type on cardiac remodeling was largely neglected in the field so far, we focused our subsequent analysis on cardiac endothelial cells. In theory, low-level contaminations for example by fibroblasts and inflammatory cells might have contributed to this finding in our bulk-sequencing analysis. Therefore, we employed single cell sequencing of cardiac endothelial cells (isolated as described in Figure 1A) after sham, and one and 8 weeks after TAC. Two biological replicates (1600 single cells) were analyzed per condition (in total 4800 single cells). As shown in Figure 5A, 12 different clusters were identified in cardiac endothelial cells. We observed clusters with high expression of angiogenesis genes (clusters 0 and 6), of collagen/or ECM organization genes (clusters 6 and 7), of mRNA translation related genes (clusters 8 and 9), of inflammatory and extravasation genes (clusters 4, 8, 9), of mitochondrial genes, and of erythrocyte genes (likely reflecting a contamination with erythrocytes) (Figure S9A). Genes representing characteristic GO terms (biological process) were enriched in the different clusters (Figure 5B). When comparing the clusters between sham and different time points after TAC, it became evident that the time course of gene-expression differed between clusters (Figure 5C). For instance, cluster 6 cells (containing ECM/collagens genes) were markedly more abundant after one week, but no longer after 8 weeks of TAC. In addition, cluster 8 cells (inflammatory and mRNA translation genes), became more abundant one week after TAC and remained elevated 8 weeks after TAC. Importantly, endothelial cell markers such as Vcam1 or Pecam1 were expressed in the same clusters, indicating that inflammatory and matrix genes were upregulated in endothelial cells (Figures 5A and S9A). To more directly analyze collagen and inflammatory gene expression in endothelial cells, we quantified their co-expression with endothelial marker genes within the same cell. As endothelial marker genes, we employed Kdr and Cdh5, as well as Fabp4 and Nrp, which were found to be highly enriched in cardiac endothelial cells, according to (Ren et al., 2020).Col1a2, Col3a1, Col4a2, Col5a2, Ccl2 or Il1b were strongly more expressed in endothelial cells one week, but not 8 weeks after TAC versus sham, indicating that they were transiently induced in endothelial cells during pressure overload (Figures 5D, S9B, and S9C).
Figure 5.

Single cell sequencing of cardiac endothelial cells
(A) UMAP plot from pooled single cell RNA sequencing data from isolated cardiac endothelial cells after sham, 1 and 8 weeks after TAC surgery (n = 2 samples/condition). 12 different subclusters were found. Main genes are shown on the right.
(B) Gene ontology terms (GO, biological process) related to the clusters as indicated in (A).
(C) Visualization of cluster gene regulation over time under the indicated conditions.
(D) Quantification of the expression of the indicated genes in sham and 1 and 8 weeks after TAC in endothelial cells. Cdh5, Kdr, Fabp4 and Nrp1 are cardiac endothelial marker genes. N = 2 samples/condition.
Ribo-Tag Sequencing in endothelial cells after TAC
To understand whether changes in the endothelial transcriptome were also visible at the posttranscriptional level, we performed endothelial cell specific Ribo-tag sequencing. For this purpose, we crossed the Ribo-Tag mouse with endothelial specific Cre driver mice (Cdh5-CreERT2). In the Ribo-Tag/Cdh5-CreERT2 mouse, activated Cre will trigger the recombination of a floxed Rpl22 allele leading to the synthesis HA-Rpl22-tagged ribosomes specifically in endothelial cells (Figure 6A). As reported in detail previously, hearts one week after sham or TAC surgery were homogenized, an RNAse digestion was performed, and endothelial monosomes were isolated by HA-directed immunoprecipitation (Doroudgar et al., 2019). RNA fragments that were protected from RNAse digestion by ribosomes (“Ribosome protected fragments”) were then forwarded to RNA-sequencing to determine which RNAs were being actively translated in cardiac endothelial cells. When considering genes that showed a significant, at least two-fold higher ribosomal density after one week of TAC versus sham, 473 genes were identified. From these, 287 (61%) had also been found increased in the RNA seq analysis from endothelial cells. Only 124 genes were significantly less abundant at the ribosome one week after TAC, and of these 39 (32%) were also downregulated after TAC at the mRNA level (RNA seq) (Figure 6B). A heatmap and the associated GO terms of genes with higher (Up) and lower (Down) association with ribosomes versus sham is shown in Figure 6C. Among the upregulated genes, similar functional categories (related to cell cycle, collagens and inflammatory response) were identified by the Ribo-tag compared to the RNA-seq approach one week after TAC. In addition to these terms, “nucleosome assembly” related genes were more translated after TAC. Here, mainly histone encoding mRNAs are listed, which indicates that histones were more produced, likely due to a higher need because of increased DNA synthesis. Histones were not detected at the mRNA level (see Figure 3), because they are lacking a poly-A tail, which precluded identification in our RNA sequencing approach (Marzluff et al., 2008). Among the downregulated genes in “metabolic process”, which was (with nine genes) the most significant group, mainly fatty acid metabolism associated regulators were included. We next identified translationally upregulated protein ligands in endothelial cells from our Ribo-Tag analysis and intersected the results with previously published Ribo-Tag data from cardiomyocytes early after TAC to determine upregulated endothelial cell ligands that interacted with receptors translated in cardiomyocytes (Doroudgar et al., 2019). We also identified increasingly translated ligands from cardiomyocytes early after TAC that interacted with translated receptors in endothelial cells. Like from the transcriptomic analysis (Figure 3) an interaction score was calculated that depended on the ribosomal abundance of ligand and receptor mRNAs and the increase of the ribosomal abundance of ligand mRNA versus sham (Figure 6D).Similar as seen in the transcriptome analysis, endothelial cells translated more collagens and cell adhesion proteins binding (integrin) receptors on cardiomyocytes and endothelial cells. Furthermore, endothelial cells translated more growth factors (Igf1 and Ptn) acting on cardiomyocytes as well as inflammatory signals (Anxa1, Bgn) acting on cardiomyocytes and endothelial cells. When analyzing translationally induced cardiomyocyte derived ligands after TAC, we noted, similar as in our transcriptome based analysis, that cardiomyocytes promote the production of angiogenesis regulating signals (Ctgf, Cyr61, Col18a1) and also matrix associated proteins (Col1a1, Fbn1, Fn1) toward endothelial cells. Example ligand receptor pairs are shown in the Figure. Comparing the ligand-receptor interactions identified after Ribo-Tag sequencing with the once found after RNA seq (see Figures 4C, 4D, S5C, and S5D), we found only a partial overlap, which was larger when ECs were considered as receiving, receptor expressing cell (Figure 6E).
Figure 6.

Ribo-Tag Sequencing in endothelial cells after TAC
(A) Scheme of the endothelial Ribo-Tag sequencing approach.
(B) Venn diagrams to show the overlap between significantly upregulated genes and downregulated genes between endothelial RNA-sequencing (from previous Figures) and RiboTag sequencing. The numbers in the different sections indicate the number of genes (co-) regulated in the indicated sections of the diagram.
(C) Heatmap of differentially translated genes in sham vs. 1 week TAC is shown (n = 4 sham, n = 5 TAC). Related gene ontology terms (GO, biological process) among the up- and downregulated genes with example genes is shown on the right.
(D) Ligand-receptor analysis of gained new interactions between endothelial cells and cardiomyocytes is shown. The first two columns address endothelial cell derived ligands, the last two columns describe cardiomyocyte derived ligands. The ligand receptor pairs are shown on the right.
(E) Venn diagrams to show the overlap of gained interaction between the indicated cell types computed from our RNA-seq versus the Ribo-Tag Seq results. The numbers in the different sections indicate the number of genes (co-) regulated in the indicated sections of the diagram.
Collagen protein production by cardiac endothelial cells
Since we had indirect evidence from our Ribo-Seq that the increased collagen mRNA levels might also lead to more collagen protein production, we wanted to analyze this directly. Immunofluorescence studies revealed a partial co-localization of collagen-1 and collagen-3 with endothelial cells labeled by isolectin B4 (IB4) in mouse cardiac cryosections. This co-localization was especially enhanced after TAC (compared to sham), when collagen staining became stronger (Figures 7A and 7B). We finally investigated whether human endothelial cells can express and produce collagen. The purity of human umbilical vein endothelial cells (HUVECs) and human coronary microvascular endothelial cells (HCMECs) was confirmed by confluent immunostaining for VE-Cadherin (CDH5) (Figure 7C). A qPCR analysis revealed significantly higher Col1a1 mRNA expression in HCMECs compared to HUVECs. An ELISA from the cellular supernatant showed a strongly increased collagen 1 protein production in HCMECs compared to HUVECs (Figure 7D). To decipher whether collagen 1 production could be triggered by TGFβ, we cultured HUVECs and HCMECs in growth factor depleted media with or without the addition of TGFβ. As shown in Figure 7E, TGFβ increased collagen 1 production in HCMECs, but not in HUVECs. Therefore, cardiac microvascular endothelial cells might have a specific ability to produce collagen, at least when compared to umbilical vein endothelial cells.
Figure 7.

Collagen protein is produced by cardiac endothelial cells
(A) Immunofluorescence staining of mouse heart sections for Isolectin B4 (IB4) to label endothelial cells and for Collagen 1 and 3 (Scale bar =50μm).
(B and C) Immunostaining for the endothelial marker CDH5 and nuclear staining for DAPI in cultured confluent human umbilical vein endothelial cells (HUVECs) and human cardiac microvascular endothelial cells (HCMECs). Scale bar= 50μm.
(D) Col1 mRNA and Collagen 1 (Col1) protein from the indicated cells and their supernatant, respectively. Data are shown as mean ±SEM.
(E) Collagen 1 protein from the supernatant of the indicated cells with and without TGF-β stimulation as indicated. (D) and (E), data are shown as mean ±SEM. The numbers indicated in the graph indicate the number of samples analyzed in that particular condition. ∗p<0.05, ∗∗p<0.01, ∗∗∗∗p<0.0001 as determined by two-tailed unpaired t-test (panel D) or by one-way ANOVA with Sidak’s multiple comparisons test.
Discussion
Although a cardiomyocyte centric view has long prevailed with regard to the mechanisms of heart failure development, the advent of sophisticated cell isolation technologies, next-generation sequencing and especially single cell sequencing has expanded the view and shifted attention at least in part to the non-myocyte compartment. Indeed, multiple studies have addressed transcriptomic changes in myocytes and non-myocytes using bulk or single-cell sequencing during development, under homeostatic conditions and after ischemic injury, but the cardiac response to pressure overload has-at least in non-myocytes- so far not been addressed in a similar manner (Cleuren et al., 2019; DeLaughter et al., 2016; Farbehi et al., 2019; Gladka et al., 2018; Li et al., 2019; Litvinukova et al., 2020; Lother et al., 2018; McLellan et al., 2020; Paik et al., 2020; Ren et al., 2020; Skelly et al., 2018; Vigil-Garcia et al., 2021; Wang et al., 2020a, 2020c). We observed strongly differing global gene-expression patterns between cardiomyocytes, endothelial cells and fibroblasts, with much smaller changes induced by TAC versus sham surgery. The timely pattern of the response to pressure overload and the nature of the gene changes was highly cell type specific. Cardiomyocytes transiently induce genes related to the cell cycle in the subacute phase after TAC, but these genes were downregulated in the chronic phase despite persisting pressure overload. Activation of cell cycle genes, which might be associated with cardiomyocyte proliferation, appears to be adaptive during pressure overload (Angelis et al., 2008; Malek Mohammadi et al., 2019; Toischer et al., 2017). Enhanced cardiomyocyte renewal could counteract myocyte drop-out by necrosis or apoptosis due to long term pressure overload (Hein et al., 2003). The transient nature of cell cycle gene induction could therefore be responsible for insufficient cardiac regeneration in chronic pressure overload and could possibly be overcome by therapeutic induction of cell cycle regulators. Cardiomyocytes also induced a profound and sustained upregulation of extracellular matrix and inflammatory genes, although the mRNA expression levels of inflammatory mediators were much lower in cardiomyocytes compared to fibroblast or endothelial cells. Collagen production in cardiomyocytes was previously detected, but its relative contribution to heart injury related fibrosis remains unclear (Kanisicak et al., 2016; Schram et al., 2010). We also found that angiogenic regulators, among them several anti-angiogenic genes, were predominantly induced in the chronic phase of pressure overload, when capillary rarefaction is known to emerge, which likely contributes to cardiac dysfunction (Hein et al., 2003; Heineke et al., 2007; Mohammed et al., 2015; Sano et al., 2007). Pressure overload entailed an early and prolonged downregulation of multiple different functional gene classes. Downregulation of fatty acid metabolism genes was in line with increasing importance of glucose metabolism in hypertrophy, while downregulation of axonogenesis genes could account for the previously observed cardiac hypoinnervation after TAC (Muhlfeld et al., 2013). Among the downregulated genes in cardiomyocytes were many ion channels and cardiac contractility related genes, which might predispose to arrhythmia and cardiac dysfunction. Compared to cardiomyocytes with their long-term and profound dysregulation of genes, cardiac endothelial cells and fibroblasts exerted more transiently regulated gene-clusters, predominantly one week after TAC. Accordingly, matrix and inflammatory genes were transiently upregulated and angiogenesis genes transiently downregulated after TAC in fibroblasts. Early upregulation of cell cycle and extracellular matrix genes in our study are in line with previous reports showing fibroblast proliferation and matrix synthesis within one week after the induction of pressure overload (Kanisicak et al., 2016; Moore-Morris et al., 2014). Impact of fibroblasts on cardiac angiogenesis occurs in cardiac aging and in tumors, but had only recently been found in the myocardium in pressure overload (Dittrich et al., 2021; Kalluri and Zeisberg, 2006; Vidal et al., 2019). Remarkably, fibroblasts strongly upregulated multiple inflammatory mediators one week after TAC, some of which exerted the highest levels compared to the other cells. In line with our results, previous work had suggested that mechanical loading and TAC induces expression of the chemokine Ccl2 in cardiac fibroblasts (and in a lower amount in endothelial cells) (Lindner et al., 2014; Unudurthi et al., 2020). Enhanced myocardial angiogenesis occurs within 1–2 weeks after the induction of pressure-overload, which was reflected by endothelial upregulation of cell cycle and vascular genes in our analysis. The functional impact of endothelial cells to the cardiac response to pressure overload, however, has remained less clear: on one hand, the inhibition of angiogenesis entailed reduced heart function, while on the other hand, endothelial to mesenchymal transition (EndoMT) from endothelial cells to fibroblasts was suggested to underlie cardiac fibrosis (Zeisberg et al., 2007). Although complete transdifferentiation of cardiac endothelial cells into fibroblasts during pressure overload or myocardial infarction was disputed by other groups using lineage tracing techniques, Stefanie Dimmeler et al. recently suggested that an incomplete EndoMT might occur after myocardial infarction, whereby endothelial cells maintain their identity, but upregulate mesenchymal genes (Col3a1, Fn1 or others) (Kanisicak et al., 2016; Manavski et al., 2018; Moore-Morris et al., 2014). In our study, we report partial EndoMT early after pressure overload, which was characterized mainly by the upregulation of collagen and matrix remodeling genes. EndoMT appeared to be transient in nature, as it was no longer observed 8 weeks after TAC surgery. Evidence of partial EndoMT in our study was supported by bulk and single cell RNA sequencing: the latter revealed co-expression of endothelial and collagen genes, which occurred in 10–20% of endothelial cells after one week of TAC. A similar percentage of partial EndoMT has been previously observed after myocardial infarction (Glaser et al., 2020; Tombor et al., 2021). In addition to analysis at the mRNA level, we detected translation of collagens and matrix remodeling genes in endothelial cells one week after TAC versus sham, and verified co-localization of collagen 1 and 3 proteins with endothelial cells specifically after one week of TAC. Furthermore, we detected collagen 1 production by cultured cardiac endothelial cells (but not by HUVECs), which was even more stimulated by the EndoMT inducer TGFβ. Beside the mesenchymal genes, endothelial cells upregulated a range of inflammatory regulators after TAC. Their expression was much higher than in cardiomyocytes, but a little less compared to fibroblasts. In the single-cell sequencing analysis, the fraction of endothelial cells expressing inflammatory proteins was different from the ones expressing mesenchymal genes, although some overlap existed. Expression of cytokines by endothelial cells will likely promote the recruitment of monocytes into the myocardium during pressure overload. According to our motif analysis, the transcription factor Runx1 might drive inflammatory gene expression, while SMAD and PU.1 could trigger collagen expression in endothelial cells (Michaud et al., 2008). Like fibroblasts, endothelial cells expressed and/or upregulated different growth factors (such as prominently Igf-1) on a higher level than cardiomyocytes and thereby supported their own growth, but likely also that of cardiomyocytes in an autocrine and paracrine manner. Indeed, the amount of endothelial cells has previously been positively correlated with the degree of myocardial hypertrophy (Tirziu et al., 2007). Our computed interaction analysis of TAC induced cellular ligands and their expressed receptors suggested that all three cell-types initiated rich communication to other cells, which strongly diminished 8 weeks after TAC. In this chronic stage, cardiac myocytes were the most “talkative” cells and also received far more signals than the other cells. It is interesting to note that beside growth factors and cytokines, intercellular communication after TAC appears to strongly involve extracellular matrix proteins, like collagens and fibronectin. It was suggested that extracellular matrix does not only constitute the basic structure of the heart, but also serves as intercellular signaling mediator by binding to receptors on target cells (mainly integrins) predominantly during development and strain or injury, in order to trigger growth and cell proliferation or migration via outside-in signaling (Ieda et al., 2009; Lu et al., 2016; Ogawa et al., 2000; Zeltz and Gullberg, 2016). Indeed, we found, for instance, that fibronectin promotes cardiomyocyte hypertrophy, endothelial cell proliferation and migration and fibroblast migration, which might critically affect disease progression. Our study reveals insights into the contribution of the three main resident cardiac cell types to early and late pressure overload, which because of the use of bulk-sequencing of purified cell populations with a higher sequencing depth goes beyond the results of previous studies that relied mainly on single cell sequencing (Ren et al., 2020). It also constitutes a deep resource and a reference for cardiac cellular coding and non-coding gene-expression under baseline conditions and during pressure overload.
Limitations of the study
A limitation of our study was that we did not include less abundant, but likely still functionally very important-cell types such as different leukocyte populations, nerve cells, smooth muscle cells and others. Further work will be needed to investigate these cells and add them to the existing analysis. As we focused on endothelial cells in the course of the study, one of our main findings is the transient release of collagens by endothelial cells during pressure overload. Further studies are needed to determine the relative contribution of endothelial cells, but also other cells to tissue fibrosis in the heart. This could be achieved, for instance, by cell specific ablation of central transcriptional regulators (such as SMADs) during experimental pressure overload and quantification of the resulting myocardial fibrosis. When a significant impact on tissue fibrosis by endothelial cells or another cell type prevails and if this is confirmed in human heart failure, this cell type might be targeted by anti-fibrotic therapies in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified Rat Anti-Mouse CD31 | BD Pharmingen | #553370 |
| Purified Rat anti Mouse CD102 | BD Pharmingen | # 553326 |
| Anti-Collagen I antibody | Abcam | ab34710 |
| Anti-Collagen 3 antibody | Abcam | Ab7778 |
| Purified Rat Anti-Mouse CD144 | BD Biosciences | #555289 |
| Isolectin B4 | Vector Laboratories | FL-1201 |
| CD146 (LSEC) MicroBeads | Miltenyi Biotec | 130-092-007 |
| Purified Rat Anti-Mouse CD16/CD32 (Mouse BD Fc Block™) | BD Biosciences | # 553141 |
| CD45 Antibody, anti-mouse | Miltenyi Biotec | Clone 30F11 |
| CD31 Antibody, anti-mouse | Miltenyi Biotec | Clone 390 |
| Feeder Cells Antibody, anti-mouse | Miltenyi Biotec | Clone mEF-SK4 |
| FITC anti-mouse CD102 Antibody | BioLegend | Clone 3C4 |
| Anti-HA Magnetic Beads | Thermo Fisher | 88837 |
| Rabbit monoclonal anti-CD146 | Abcam | Cat#ab75769, |
| Rabbit monoclonal anti-phospho-smad3 (S423+S425) | Abcam | Cat# ab52903, |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab15580, |
| Mouse monoclonal anti-Cardiac Troponin T | Abcam | Cat# ab8295, |
| Rabbit polyclonal anti-Fibronectin | Sigma-Aldrich | Cat# F3648, |
| GSL I - isolectin B4 antibody | Vector Laboratories | Cat# FL-1201, |
| VECTASHIELD HardSet Mounting Medium with DAPI antibody | Vector Laboratories | Cat# H-1500, |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase I | Worthington | #4196 |
| ‘Seq B’ microparticles | Chemgenes Corp | PMID 26000488 |
| Recombinant human TGF-β1 | Peprotech | 100-21 |
| Fibronectin human plasma | Sigma-Aldrich | F2006; CAS:86088-83-7 |
| Collagen I, Rat Tail, 100 mg | Corning | 354236 |
| Gelatine extra pure | Carl Roth | 4582; CAS: 9000-70-8 |
| Cycloheximide (CHX) | Sigma-Aldrich/Merck | C1988 |
| 5X Mammalian Polysome Buffer | Epicentre/Illumina | ASBHMR1212 |
| Dithiothreitol (DTT) | Sigma-Aldrich | 43819-5G |
| Triton-X100 | Sigma-Aldrich | X100-500ML |
| cOmplete, EDTA free Protease inhibitor cocktail tablets | Sigma-Aldrich | 11873580001 |
| SUPERase⋅In™ RNase Inhibitor | Invitrogen™ | AM2696 |
| Invitrogen Ambion RNase I | Thermo Fisher Scientific | AM2295 |
| Novex™ TBE-Urea Gels 15% | Thermo Fisher Scientific | EC68855BOX |
| TRIzol™ Reagent | Thermo Fisher Scientific | 15596018 |
| Ambion™ DNase I | Thermo Fisher Scientific | AM2222 |
| TBE (10X), RNase-free | AM9865 | Thermo Fisher Scientific |
| Novex™ TBE-Urea Sample Buffer | Thermo Fisher Scientific | LC6876 |
| SYBR™ Gold | Thermo Fisher Scientific | S11494 |
| Liberase DH Blendzyme | Roche Applied Sciences | 5401089001 |
| Trypsin-Stock 10x | Gibco | 15090046 |
| BDM | Sigma-Aldrich | B0753-100 g |
| Collagenasetype I | Worthington | LS004197 |
| DNAse I | Worthington | LS002139 |
| Collagenasetype II | Worthington | LS004176 |
| Pancreatin | Sigma-Aldrich | P3292 |
| Percoll | GE-Healthcare | 17-0891-01 |
| RPMI Media 1640 | Gibco | 21870076 |
| Horse Serum | Sigma-Aldrich | H1270 |
| FBS superior | Sigma-Aldrich | S0615 |
| Bovine calf serum | Hyclone | SH30073.03 |
| Critical commercial assays | ||
| miRNeasy Mini Kit | QIAGEN | 217004 |
| NucleoSpin RNA II | Macherey-Nagel | 740.955.250 |
| Dynabeads® mRNA DIRECT™ MicroPurification Kit | ThermoFisher Scientific | #61012 |
| Maxima H Minus First Strand cDNA Synthesis Kit | ThermoFisher Scientific | K1652 |
| Maxima SYBR Green qPCR Master Mix | ThermoFisher Scientific | K0253 |
| Cell Proliferation ELISA, BrdU (colorimetric) | Roche | 11647229001 |
| Human Pro-Collagen I alpha 1 DuoSet | R&D Systems | DY6220-05 |
| DuoSet ELISA Ancillary Reagent Kit 2 | R&D Systems | DY008 |
| RNA Clean & Concentrator 5 | Zymo Research | R1013 |
| TruSeq Ribo Profile (Mammalian) Library Prep Kit | Illumina | ASLPA1212 |
| RNA Clean & Concentrator-25 | Zymo Research | R1017 |
| Bioanalyzer High Sensitivity DNA | Agilent | 5067-4626 |
| NextSeq 500/550 High Output Kit v2.5 | Illumina | 2024906 |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 |
| ScriptSeq™ v2 RNA-Seq | ||
| Library Preparation Kit | Epicentre | SSV21106 |
| TruSeq SBS Kit v3-HS | Illumina | FC-401-3001 |
| Deposited data | ||
| GSE180794 | CM,FB and EC | Bulk-RNA-seq |
| GSE180794 | Only EC | Ribo-seq |
| GSE180794 | Only EC | scDROP-seq |
| Experimental models: Cell lines | ||
| HUVEC single donor | Promo Cell | C-12200 |
| Human Cardiac Microvascular Endothelial cells (HCMEC) | Promo Cell | C-12285 |
| Mouse endothelial cell line: C166 | ATCC | CRL-2581 |
| Experimental models: Organisms/strains | ||
| C57Bl6N mice, male, aged 8-10 weeks old | Charles River | Strain 027 |
| Mouse: Ribo-tag | Jackson Laboratory | JAX ID 011029 |
| Mouse: Cdh5-CreERT2:Ribo-tag | This paper | N/A |
| Oligonucleotides | ||
| Primer for human Gapdh, forward: TCGACAGTCAGCCGCATCTTCTTT | This paper | N/A |
| Primer for human Gapdh, reverse: ACCAAATCCGTTGACTCCGACCTT | This paper | N/A |
| Primer for human Col1, forward: ACCTGGTCAAACTGGTCCTG | This paper | N/A |
| Primer for human Col1, reverse: CCTGTGGTCCAACAACTCCT | This paper | N/A |
| Primer for mouse Nppa, forward: ATTGACAGGATTGGAGCCCAGAGT | This paper | N/A |
| Primer for mouse Nppa, reverse: TGACACACCACAAGGGCTTAGGAT | This paper | N/A |
| Primer for mouse Myh7, forward: AGGCAAGGCAAAGAAAGGCTCATC | This paper | N/A |
| Primer for mouse Myh7, reverse: GCGTGGAGCGCAAGTTTGTCATAA | This paper | N/A |
| Primer for mouse Gapdh, forward: CGTCCCGTAGACAAAATGGT | This paper | N/A |
| Primer for mouse Gapdh, reverse: GAATTTGCCGTGAGTGGAGT | This paper | N/A |
| Primer for mouse Col1a1, forward: CCGCTGGTCAAGATGGTC | This paper | N/A |
| Primer for mouse Col1a1, reverse: CCTCGCTCTCCAGCCTTT | This paper | N/A |
| Primer for mouse Col3a1, forward: ATAAGCCCTGATGGTTCTCG | This paper | N/A |
| Primer for mouse Col3a1, reverse: ATGCATGTTTCCCCAGTTTC | This paper | N/A |
| Primer for mouse Ccl2, forward: AGCACCAGCCAACTCTCACT | This paper | N/A |
| Primer for mouse Ccl2, reverse: TCATTGGGATCATCTTGCTG | This paper | N/A |
| Primer for mouse Thbs1, forward: GACTCGGGACCCATCTATGA | This paper | N/A |
| Primer for mouse Thbs1, reverse: GCAAGAAGAGAGGCAAGGAA | This paper | N/A |
| Software and algorithms | ||
| STAR | https://code.google.com/p/rna-star/ | STAR |
| Rsubread | https://bioconductor.org/packages/release/bioc/html/Rsubread.html | FeatureCounts |
| EdgeR | https://bioconductor.org/packages/release/bioc/html/edgeR.html | EdgeR |
| clusterProfiler | https://guangchuangyu.github.io/software/clusterProfiler/ | enrichGO |
| Complexheatmap | https://jokergoo.github.io/ComplexHeatmap-reference/book/ | Complexheatmap |
| findMotifs.pl from homer | http://homer.ucsd.edu/homer/microarray/index.html | findMotifs.pl |
| R.4.1.1 for Windows | https://cran.r-project.org/bin/windows/base/ | Base |
| dropSeqPipe | https://github.com/Hoohm/dropSeqPipe | dropSeqPipe |
| Seurat (version 4.0.2) | https://satijalab.org/seurat/articles/get_started.html | Seurat |
| BioRender | Gallery (biorender.com) | For Graphical Abstract |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Joerg Heineke (Joerg.Heineke@medma.uni-heidelberg.de)
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Male C57Bl6N mice (Charles River Laboratories) at the age of 8-10 weeks were used for cardiac sham and TAC surgery. The animals had free access to water and a standard diet and were maintained on a 12-h light and dark cycle at a room temperature of 22 ± 2°C with a humidity of 35-60%. All animal procedures described in this study were approved by the local state authorities (LAVES-Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, 33.12-42502-04-17/2706). All procedures including the use and care of animals were carried out according to the Guide for the Care and Use of Laboratory Animals published by the National Research Council (NIH Publication No. 85-23, revised 1996) and the German animal protection code.
Transverse aortic constriction
TAC or sham surgery was performed by subjecting the aorta to a defined 26-gauge constriction in 8-9 weekold mice as described previously (Appari et al., 2017; Grund et al., 2019) (Heineke, 2013; Riehle and Bauersachs, 2019). Mice were subcutaneously (s.c.) injected with 0.02 mg/kg atropine and 0.1 mg/kg buprenorphine. Anesthesia was induced with 3-4% isoflurane in an induction chamber. After oral intubation, mice were connected to a small animal ventilator (MiniVent Type 845, Harvard Apparatus), and anesthesia was maintained with 1.5-2% isoflurane. A left sternal thoracotomy was performed, the thymus partially removed, and the transverse aorta was tied around a 26G needle (Heineke, 2013). In sham-operated control mice, the same procedure was conducted, but no ligature was placed around the aorta. Sham samples for all RNA-seq approaches was harvested one week after surgery. During surgery, the animals were placed on a heating pad connected to a temperature controller (Föhr Medical Instruments) to maintain body temperature at 37 °C.
Transthoracic echocardiography
Mice were anesthetized with 4% isoflurane in an induction chamber and placed on a heating pad to keep body temperature at 37°C. Anesthesia was maintained with 1-2% isoflurane via mask. Non-invasive echocardiography was performed in parasternal long- and short axis with a 30MHz transducer (Vevo 770, Visualsonics), as previously described (Appari et al., 2017; Grund et al., 2019). LV end-diastolic diameter (LVEDD), enddiastolic average wall thickness (Wth), heart rate and enddiastolic volume (EDV) as well as endsystolic volume (ESV) were recorded or calculated. Ejection fraction was calculated as [(EDV-ESV)/EDV] x 100.
Methods details
Immunofluorescence and histological analysis
For organ harvesting, mice were sacrificed and whole hearts were immediately removed, washed in cold PBS and 0.5% KCl in PBS and dissected at the mid-ventricular level. Basal parts of the ventricle were embedded in paraffin or OCT. Immunofluorescence staining was performed in 7μm OCT cryosections. Slides were fixed in 4% PFA for 20 minutes, permeabilized in 0.3% Triton X for 20 minutes and blocked with 3% BSA for at least 1 hour at room temperature. Eachstep was followed by repeated washing in PBS. Slides were then serially incubated with primary and secondary antibodies. The following primary antibodies were used for immunostaining: anti-Collagen 1 (ab34710, Abcam), anti-Collagen 3 (ab7778, Abcam), anti-CD146 (ab75769, Abcam), anti-phospho SMAD3 (S423+S425, ab52903, Abcam), anti-Ki67 (ab15580, Abcam), anti-Fibronectin (F3648, Sigma-Aldrich), anti-CDH5 (2150-1470, BD-Pharmingen). Nuclei were visualized with 4',6-diamidino-2-phenylindole (DAPI, Vector Laboratories), and Fluorescein-labelled GSL I-isolectin B4 (IB4, Vector Laboratories) was used to label cardiac endothelial cells. For light microscopic histological analysis, cryosections of 12μm thickness were prepared. Sections were stained with Sirius Red following standard procedures to demonstrate the fibrotic area. The scar area was determined as the percentage of fibrotic tissue area in relation to total LV area in midventricular sections (Adobe Photoshop CS6), as described (Appari et al., 2017; Grund et al., 2019).
Cardiac cell isolation for RNA sequencing
Adult ventricular cardiomyocytes were prepared from adult mice as previously described using a Langendorff system (Appari et al., 2017; Grund et al., 2019). In brief, the heart was removed from the chest, cannulated via the ascending aorta, and mounted on a modified Langendorff perfusion apparatus. The heart was first retrogradely perfused with perfusion buffer (NaCl 113mM, KCl 4.7mM, KH2PO4 600μM, Na2HPO4 x 2H2O 600μM, MgSO4 x 7H2O 1.2mM, NaHCO3 12mM, KHCO3 10mM, HEPES 10mM, Taurine 30mM, Glucose 5.5mM, BDM 10mM) at 37°C. Subsequently, the perfusion was switched to digestion buffer (containing 0.042 mg/mL Liberase DH Blendzyme, Roche Applied Science, Trypsin, Gibco, 12.5μM CaCl2 in perfusion buffer) for 10 minutes. The heart was then disassembled from the Langendorff system and transferred to the petri dish containing the digestion solution with cells. An equal amount of stop solution (10% bovine calf serum, 12.5μM CaCl2 in perfusion buffer) was added. Then the heart was cut into small pieces and carefully homogenized through a cut off P1000 pipet-tip before the solution was filtered through a 100μm cell strainer and transferred to a 15mL conical tube, where cardiomyocytes were allowed to sediment for 8 minutes. After removal of the supernatant (containing dead cells and non-myocytes), the cardiomyocytes were washed twice with cold PBS, before being subjected to the RNA isolation procedure.
Cardiac endothelial cells and fibroblasts were isolated from hearts of adult mice. The hearts were cut into small pieces and digested with a combination of collagenase I (Worthington 500U/ml) and DNAse I (150U/ml) in RPMI media for one hour at 37°C. The solution was then filtered through a 70μm cell strainer and incubated with anti-CD146 coated microbeads (Miltenyi) for 30minat 4°C in MACS buffer (0.5% bovine serum albumin, 2mM EDTA in PBS). The microbeads were subsequently captured by Miltenyi columns. After extensive washing with MACS buffer and subsequent elution, this step was repeated once with a fresh column and then the endothelial cell-coupled beads were eluted and directly forwarded to RNA extraction. The column flow through was then incubated with feeder-removal-beads (Miltenyi) to bind to fibroblasts. The bead-coupled fibroblasts were captured by double incubation with Miltenyi columns. After elution, the fibroblasts were forwarded to RNA extraction.
Flow cytometry
After MACS separation, the cells were washed with PBS containing 2% FCS and 2 mM EDTA and incubated for 5minat 4°C with Fc block solution (Purified rat anti-mouse CD16/CD32, Clone 2.4G2) from BD Biosciences. The cells were then incubated for 20minat 4 °C with the following directly conjugated anti-mouse antibodies: CD45-PerCP (clone 30F11), CD31-PE (clone 390) and anti-feeder cells-APC (clone mEF-SK4) (all antibodies from Milteny Biotec) and CD102/ICAM-2-FITC (clone 3C4) from BioLegend. After washing, the cells were counted by FACS LSR II von BD. Data were analyzed using FlowJo software (version 10).
Drop-seq single cell sequencing
Dead cells and multi-cell cluster were excluded from the analysis by cell sorting after staining with 4′,6-Diamidin-2-phenylindol (DAPI). Individual cells were then packaged in picoliter droplets together with the barcoded ‘Seq B’ microparticles (Chemgenes Corp; Wilmington, MA; referenced in the Macosko et al. (PMID 26000488)) following drop-seq laboratory protocol v. 3.1 from McCarrol Lab (http://mccarrolllab.org/dropseq/; accessed last time March 3rd, 2021). For packaging, the high-throughput single-cell RNA sequencing system from Dolomite Bio (Royston, UK) was used. In brief, the beads were resuspended in lysis buffer at a concentration of 600 beads/μl and then loaded into a sample loop. The cells were resuspended at a concentration of 300 cells/μl in PBS, including 0.01% bovine serum albumin (BSA), and then loaded into an agitated tube. Monodisperse droplets were generated in droplet generation oil (Bio-Rad) by pumping the suspensions through glass single cell RNA seq chips (Dolomite Bio) at a speed of 200 μL/min for the oil and 30 μL/min for beads and cells. The aqueous droplets containing single cells and/or beads in oil were collected in 50 mL falcon tubes. We immediately proceeded with droplet breakage and cDNA synthesis according to the drop-seq laboratory protocol v. 3.1 from McCarrol Lab. Briefly, droplet contents (mRNA bound on the beads' oligos) were released and washed from oil using 6x SSC buffer and perfluorooctanol, and cDNA was generated by reverse transcription using Maxima H-RTase (Thermofisher Scientific). Subsequently, beads were subjected to exonuclease I treatment prior cDNA amplification by SMART PCR (4 + 14-18 cycles), purification, and tagmentation for sequencing exactly according to the drop-seq laboratory protocol v. 3.1. After final analysis on Bioanalyzer to validate tagmented library size and concentration, the libraries were sequenced on the Next Seq 550 (Illumina) at the Research Core Unit Genomics, Hannover Medical School with 75 cycles High Output, paired end, read1 26bp with custom, read2 58bp und Single index 8bp.
Ribo-Tag Sequencing
The Ribo-Taq mouse model and ribosome-protected fragments immunoprecipitation were described previously (Doroudgar et al., 2019). The Ribo-tag mice were purchased from Jackson Laboratory (JAX ID 011029). For endothelial cell-specific tagging of ribosomes, Ribo-tag mice were crossed with Cdh5-CreERT2 mice expressing the Cre recombinase under the control of the Cadherin 5 (Cdh5) promoter (Cdh5-CreERT2:Ribo-tag). At 9 weeks of age, male mice underwent transverse aortic constriction or sham operation. Ribosome footprints were generated after immunoprecipitation of endothelial cell-specific monosomes with anti-HA magnetic beads after treating the lysate with RNase I. Libraries were generated according to the mammalian Ribo-seq kit (Illumina). Barcodes were used to perform multiplex sequencing and create sequencing pools containing at least eight different samples. Sample pools were sequenced on the NextSeq550 platform using 75-bp sequencing chemistry. For heart homogenates, mice were sacrificed, and their hearts were quickly excised, washed in PBS containing 100 μg/mL cycloheximide (CHX), and snap frozen in liquid nitrogen. Left ventricular tissue was homogenized using a tissue homogenizer in 5 volumes of ice-cold polysome buffer (20 mM Tris pH 7.4, 10 mM MgCl, 200 mM KCl, 2 mM DTT, 1% Triton X-100, 1U DNAse/μl) containing 100 μg/mL CHX. The tissue was homogenized further by passing the lysate through a 23-gauge syringe needle ten times. For complete lysis, the samples were kept on ice for 10 min and subsequently centrifuged at 20,000×g to precipitate cell debris and the supernatant was immediately used in the further steps. From the lysate, 100 μL was used as input, from which RNA was extracted using Trizol. The remaining lysate was used for RPF generation after RNAse digestion for 45minat room temperature. RNAse digestion was stopped by adding SUPERase-In (Ambion). The lysate was used for anti-HA IP of monosomes. Anti-HA magnetic beads (Thermo Fisher, 88836; 25μL per heart) were washed with 1000 μL polysome lysis buffer three times. The lysate was then added to anti-HA magnetic beads and incubated with rotation at 4°C for 4h. Beads were then washed five times with 500 μL of high salt buffer (20 mM Tris pH 7.4, 10 mM MgCl, 300 mM KCl, 2 mM DTT, 1% Triton X-100). The washed beads were subjected to RNA extraction using TRIZOL for library construction. Ribo-Seq libraries were generated according the to the mammalian Ribo-seq kit (Illumina) after size selection using Urea gels.
RNA isolation
RNA was isolated using RNeasy mini kit (Qiagen) and the NucleoSpin RNA II kit (Macherey Nagel) according to the manufacturer's protocol.
Quantitative real-time PCR
cDNA was generated using the Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) and quantitative PCR was performed following standard procedures. Gene-expression was normalized to Gapdh mRNA expression. Primer sequences are displayed in the key resources table.
Bulk RNA sequencing
RNA from different cardiac cell types was analyzed by bulk sequencing at the Helmholtz Center for Infection Research in Braunschweig. Quality and integrity of total RNA was controlled on the Agilent Technologies 2100 Bioanalyzer (Agilent Technologies; Waldbronn, Germany). The RNA sequencing library was generated from 100ng total RNA using Dynabeads® mRNA DIRECT™ MicroPurification Kit (Thermo Fisher) for mRNA purification followed by ScriptSeq v2 RNA-Seq Library Preparation Kit (Epicentre) according to the manufacturer’s protocols. The libraries were sequenced on Illumina HiSeq2500 using TruSeq SBS Kit v3-HS (50 cycles, single ended run) with an average of 3 x107 reads per RNA sample.
Cell culture of rat and mouse heart cells
Neonatal rat ventricular cardiomyocytes and fibroblasts were isolated from 50-80 1-3 day old Sprague-Dawley rats. Hearts were minced into small pieces, before they were digested 6-7 times in digestion solution (0.116M NaCl, 5.4mM KCl, 5.6mM dextrose, 10.9mM NaH2PO4, 0.41 mM MgSO4, 20mM HEPES, pH7.3 with 0.44mg/ml collagenase type 2, Worthington, and 0.1mg/ml pancreatin, Sigma Aldrich). Cardiomyocytes and fibroblasts were then separated by Percoll density gradient centrifugation (Heineke, 2013). Neonatal rat cardiomyocytes were plated overnight in plating media (Media 199, containing 5% fetal bovine serum and 10% horse serum), before being switched to serum free media (Grund et al., 2019). Rat neonatal fibroblast were cultured in DMEM Media with 10% FBS (Scharf et al., 2019).
DNA synthesis was measured to analyze endothelial cell proliferation by performing a BrdU incorporation assay with a commercially available kit (Roche).
A scratch assay was performed to study fibroblast and endothelial cell migration. The cell monolayer of growing fibroblasts was scraped in a straight line to create a “scratch injury” with a p200-pipet tip. Migration of cells was analyzed 18 hours later.
Human endothelial cell culture
Human umbilical vein endothelial cells (HUVECs) and human cardiac microvascular endothelial cells (HCMECs) were purchased from PromoCell. They were cultured according to manufacturer's suggestions. In brief, HUVECs were cultured in Endothelial Cell Growth Medium (PromoCell) containing a growth factor cocktail with ECGS/H 0.4% (sterile-filtered, aqueous extract from mixed-sex bovine hypothalamic tissue), FCS (fetal calf serum) 2%, EGF (epidermal growth factor) 0.1 ng/mL, hydrocortisone 1 μg/mL, bFGF (basic fibroblast growth factor ) 1 ng/mL. HCMECs were cultured in Endothelial Cell Growth Medium MV for microvascular endothelial cells (PromoCell) containing a growth factor cocktail with ECGS/H 0.4%, FCS 5%, EGF 10 ng/mL, hydrocortisone 1 μg/mL. Endothelial cells were used for experiments at passage 2. TGFβ (Peprotech) was used at a concentration of 10 ng/mL.
Collagen 1 ELISA
Collagen 1 levels were determined from the endothelial supernatant using the Human Pro-Collagen I alpha 1 DuoSet ELISA kit according to the manufacturer's protocol (R&D Systems, DY6220-05).
Quantification and statistical analysis
All data are presented as mean ± standard error of the mean (SEM), unless indicated otherwise in the Figure legend. The investigators were blinded for mouse treatment during surgeries, echocardiography, organ weight determination and all histological quantifications. An unpaired, two-tailed Student's t-test was used to determine statistical significance when two groups were compared. If data were not normally distributed, the two-tailed Mann-Whitney test was used as non-parametric test. For comparison of more than two groups, statistical significance was determined by using one-way ANOVA (with Sidak’s multiple comparisons test, Prism 7 software). P-values <0.05 were considered as statistically significant.
Bulk RNA seq bioinformatics
Before alignment to the reference (mm10) each sequence was trimmed on base call quality and sequencing adapter contamination using the Trim Galore! wrapper tool. Reads shorter 20nt were removed from FASTQ files. Trimmed reads were aligned to the reference using the short read aligner STAR (https://code.google.com/p/rna-star/). Feature counts were determined using R package “Rsubread”. Only genes showing counts greater than 5 at least two times across all samples were considered for further analysis (data cleansing). Differentially expressed Genes were selected with EdgeR (0.75<FC>1.5 and pvalue <0.05) using the Wald test. Gene ontology biological processes and KEGG pathway analysis from different groups of genes was performed by compareCluster using the enrichGO databases with a p-value <0.05 Bonferroni as a method to correct the p-value. A Heatmap of differentially regulated genes was generated using the Complexheatmap program, values are the z-score of the log2CPM per gene. Motifs analyses were performed using the sequence of the significantly upregulated genes from -500 bp to 100bp around their transcriptional start site (TSS) by the help of findMotifs.pl from homer (settings-start -500-end 100-len 8,10-p 8). Motifs with p<0.05 were considered significant. Receptor-Ligands interaction analysis was performed by crossing the different gene sets from our RNA-seq with the ligand-receptor list from (Skelly et al., 2018). In order to find the most relevant ligand-receptor interaction, a LIG_REC_Score was calculated according to the following formula: LIG_REC_Score= as.numeric(scale(LIG_REC, center=F, scale=T)), where LIG_REC= (UP_TAC_LIG+EXP_TAC_REC)∗Log2fc_TAC_LIG, where UP_TAC_LIG is the sum of count per millions (CPM) of the ligands from the donor cells that were up-regulated after TAC (Log2FC>1 and P<0.05), EXP_TAC_REC is the CPM of the matched receptor from the receiver cell. Receptors were required to be expressed in TAC more than sham (Log2FC>0.1). The resulted LIG_REC value was normalized by the z-score with the help of the function scale from base (options, center=F, scale=T). To compare our results with the published data by Ren et al. 2020, we crossed the list of expressed genes in our clusters from each cell type with the expressed genes in the clusters of fibroblasts, endothelial cells and cardiomyocytes as detected by (Ren et al., 2020). Furthermore, the function ggalluvial from R was used to create the alluvial plots. EnrichGO from clusterprofiler was employed to identify the enriched biological function of each group.
Drop-seq single-cell bioinformatics
Analyses were performed via the snakemake pipeline dropSeqPipe version 0.4 (https://github.com/Hoohm/dropSeqPipe) and drop-seq-tools version 1.13. 800 cells per sample were defined as the number of input cells. Cell barcode was set to positions 1-12, while UMI was set to 13-20. UMI edit distance was set to 4. TruSeq adapters were trimmed using default parameters. Alignments were performed against the mouse mm10 reference genome, with quantification performed against the mm10.gtf annotation. UMAP and clusters analysis and plots were performed by the help of Seurat (version 4.0.2) (https://satijalab.org/seurat/articles/get_started.html). Line plots from the EC clusters were perfomed using a custom scripts in R.
Ribo tag bioinformatics
Translation prediction using Ribo-seq data was performed with Rp-Bp v2.0, based on Ensembl release 96 (Malone et al., 2017). Adapters removal and quality filtering was done with flexbar v3.0 using standard filtering parameters implemented in Rp-Bp (Dodt et al., 2012). Reads aligning to a custom bowtie2 v2.3.0 ribosomal index were discarded. Remaining reads were then aligned in genomic coordinates to the mouse genome with STAR v2.5.3a. Differential gene expression was performed using DESeq2 based on the translation predictions. Briefly, in-frame counts from open reading frames (ORFs) were aggregated at the gene level, and the resulting matrix was used as input for differential expression. We used periodic fragment lengths that showed a distinctive triplet periodicity, as determined with the automatic Bayesian selection of read lengths and ribosome P-site offsets (BPPS) method. We required the ORFs to have evidence from uniquely mapping reads, at least 10 in-frame P-sites, and to be detected as translated in at least 2 out of 9 biological replicates.
Datasets of our analyses are deposited in the GEO database under accession number GSE180794.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft through the Cluster of Excellence Rebirth (EXC 62/1 and EXC 62/3 to J.B. and J.H.), the Heisenberg Program and the SFB1366/1 (HE 3658/6-1&2 and SFB1366/1-A6 to J.H. and SFB1366/1-A3 to G.D.). M.V. acknowledges the DFG (German Research Foundation, DFG VO 1659 2/1, DFG VO 1659 2/2, DFG VO 1659 4/1, DFG VO 1659 6/1) and the Boehringer Ingelheim Foundation (Plus 3 Program).
Author contributions
NF, JC, AA, FAT, SG, MS, YW, AG, MKK, BB, ER, LJ and YL conducted experiments and analyzed data. JC, CFD, LW, RG, EB, CD and JH conducted bioinformatics analyses. RF, JB, GD and MV provided critical advice and infrastructure for the study. JC, RO, GD and MV provided important ideas and methodological concepts for the study and critically edited the manuscript. JH wrote the manuscript and supervised the study. All authors read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: March 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103965.
Supplemental information
Gene expression in isolated cardiomyocytes from mouse hearts 1 week after sham (CM_Sham) or TAC surgery (CM_TAC_1Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between CM_Sham and CM_TAC_1Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated cardiomyocytes from mouse hearts 1 week after sham (CM_Sham) and 8 weeks after TAC surgery (CM_TAC_8Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between CM_Sham and CM_TAC_8Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated fibroblasts from mouse hearts 1 week after sham (FB_Sham) or TAC surgery (FB_TAC_1Wo) as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between FB_Sham and FB_TAC_1Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated fibroblasts from mouse hearts 1 week after sham (FB_Sham) and 8 weeks after TAC surgery (FB_TAC_8Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between FB_Sham and FB_TAC_8Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated endothelial cells from mouse hearts 1 week after sham (EC_Sham) or TAC surgery (EC_TAC_1Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between EC_Sham and EC_TAC_1Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated endothelial cells from mouse hearts 1 week after sham (EC_Sham) and 8 weeks after TAC surgery (EC_TAC_8Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between EC_Sham and EC_TAC_8Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Data and code availability
-
•
Bulk RNA-seq, Ribo-Seq and single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not contain an original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Angelis E., Garcia A., Chan S.S., Schenke-Layland K., Ren S., Goodfellow S.J., Jordan M.C., Roos K.P., White R.J., MacLellan W.R. A cyclin D2-Rb pathway regulates cardiac myocyte size and RNA polymerase III after biomechanical stress in adult myocardium. Circ. Res. 2008;102:1222–1229. doi: 10.1161/CIRCRESAHA.107.163550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appari M., Breitbart A., Brandes F., Szaroszyk M., Froese N., Korf-Klingebiel M., Mohammadi M.M., Grund A., Scharf G.M., Wang H., et al. C1q-TNF-Related protein-9 promotes cardiac hypertrophy and failure. Circ. Res. 2017;120:66–77. doi: 10.1161/CIRCRESAHA.116.309398. [DOI] [PubMed] [Google Scholar]
- Cleuren A.C.A., van der Ent M.A., Jiang H., Hunker K.L., Yee A., Siemieniak D.R., Molema G., Aird W.C., Ganesh S.K., Ginsburg D. The in vivo endothelial cell translatome is highly heterogeneous across vascular beds. Proc. Natl. Acad. Sci. U S A. 2019;116:23618–23624. doi: 10.1073/pnas.1912409116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaughter D.M., Bick A.G., Wakimoto H., McKean D., Gorham J.M., Kathiriya I.S., Hinson J.T., Homsy J., Gray J., Pu W., et al. Single-cell resolution of temporal gene expression during heart development. Dev.Cell. 2016;39:480–490. doi: 10.1016/j.devcel.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich G.M., Froese N., Wang X., Kroeger H., Wang H., Szaroszyk M., Malek-Mohammadi M., Cordero J., Keles M., Korf-Klingebiel M., et al. Fibroblast GATA-4 and GATA-6 promote myocardial adaptation to pressure overload by enhancing cardiac angiogenesis. Basic Res. Cardiol. 2021;116:26. doi: 10.1007/s00395-021-00862-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt M., Roehr J.T., Ahmed R., Dieterich C. FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology (Basel) 2012;1:895–905. doi: 10.3390/biology1030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudgar S., Hofmann C., Boileau E., Malone B., Riechert E., Gorska A.A., Jakobi T., Sandmann C., Jurgensen L., Kmietczyk V., et al. Monitoring cell-type-specific gene expression using ribosome profiling in vivo during cardiac hemodynamic stress. Circ. Res. 2019;125:431–448. doi: 10.1161/CIRCRESAHA.119.314817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farbehi N., Patrick R., Dorison A., Xaymardan M., Janbandhu V., Wystub-Lis K., Ho J.W., Nordon R.E., Harvey R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;8:e43882. doi: 10.7554/eLife.43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.L., et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Gladka M.M., Molenaar B., de Ruiter H., van der Elst S., Tsui H., Versteeg D., Lacraz G.P.A., Huibers M.M.H., van Oudenaarden A., van Rooij E. Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation. 2018;138:166–180. doi: 10.1161/CIRCULATIONAHA.117.030742. [DOI] [PubMed] [Google Scholar]
- Glaser S.F., Heumuller A.W., Tombor L., Hofmann P., Muhly-Reinholz M., Fischer A., Gunther S., Kokot K.E., Hassel D., Kumar S., et al. The histone demethylase JMJD2B regulates endothelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. U S A. 2020;117:4180–4187. doi: 10.1073/pnas.1913481117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grund A., Szaroszyk M., Korf-Klingebiel M., Malek Mohammadi M., Trogisch F.A., Schrameck U., Gigina A., Tiedje C., Gaestel M., Kraft T., et al. TIP30 counteracts cardiac hypertrophy and failure by inhibiting translational elongation. EMBO Mol. Med. 2019;11:e10018. doi: 10.15252/emmm.201810018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein S., Arnon E., Kostin S., Schonburg M., Elsasser A., Polyakova V., Bauer E.P., Klovekorn W.P., Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- Heineke J. Screening for novel calcium-binding proteins that regulate cardiac hypertrophy: CIB1 as an example. Methods Mol. Biol. 2013;963:279–301. doi: 10.1007/978-1-62703-230-8_17. [DOI] [PubMed] [Google Scholar]
- Heineke J., Auger-Messier M., Xu J., Oka T., Sargent M.A., York A., Klevitsky R., Vaikunth S., Duncan S.A., Aronow B.J., et al. Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J. Clin.Invest. 2007;117:3198–3210. doi: 10.1172/JCI32573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J., Molkentin J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol.Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Hill J.A., Olson E.N. Cardiac plasticity. N. Engl. J. Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- Ieda M., Tsuchihashi T., Ivey K.N., Ross R.S., Hong T.T., Shaw R.M., Srivastava D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev.Cell. 2009;16:233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R., Zeisberg M. Fibroblasts in cancer. Nat. Rev. Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- Kanisicak O., Khalil H., Ivey M.J., Karch J., Maliken B.D., Correll R.N., Brody M.J., SC J.L., Aronow B.J., Tallquist M.D., Molkentin J.D. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016;7:12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil H., Kanisicak O., Prasad V., Correll R.N., Fu X., Schips T., Vagnozzi R.J., Liu R., Huynh T., Lee S.J., et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin.Invest. 2017;127:3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Manandhar S., Lee Y.M. Roles of RUNX in hypoxia-induced responses and angiogenesis. Adv. Exp. Med. Biol. 2017;962:449–469. doi: 10.1007/978-981-10-3233-2_27. [DOI] [PubMed] [Google Scholar]
- Li Z., Solomonidis E.G., Meloni M., Taylor R.S., Duffin R., Dobie R., Magalhaes M.S., Henderson B.E.P., Louwe P.A., D'Amico G., et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019;40:2507–2520. doi: 10.1093/eurheartj/ehz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner D., Zietsch C., Tank J., Sossalla S., Fluschnik N., Hinrichs S., Maier L., Poller W., Blankenberg S., Schultheiss H.P., et al. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res. Cardiol. 2014;109:428. doi: 10.1007/s00395-014-0428-7. [DOI] [PubMed] [Google Scholar]
- Litvinukova M., Talavera-Lopez C., Maatz H., Reichart D., Worth C.L., Lindberg E.L., Kanda M., Polanski K., Heinig M., Lee M., et al. Cells of the adult human heart. Nature. 2020;588:466–472. doi: 10.1038/s41586-020-2797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Morley M., Brandimarto J., Hannenhalli S., Hu Y., Ashley E.A., Tang W.H., Moravec C.S., Margulies K.B., Cappola T.P., et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015;105:83–89. doi: 10.1016/j.ygeno.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lother A., Bergemann S., Deng L., Moser M., Bode C., Hein L. Cardiac endothelial cell transcriptome. Arterioscler. Thromb.Vasc. Biol. 2018;38:566–574. doi: 10.1161/ATVBAHA.117.310549. [DOI] [PubMed] [Google Scholar]
- Lu Y.Y., Lin Y.K., Kao Y.H., Chung C.C., Yeh Y.H., Chen S.A., Chen Y.J. Collagen regulates transforming growth factor-beta receptors of HL-1 cardiomyocytes through activation of stretch and integrin signaling. Mol. Med. Rep. 2016;14:3429–3436. doi: 10.3892/mmr.2016.5635. [DOI] [PubMed] [Google Scholar]
- Malek Mohammadi M., Abouissa A., Azizah I., Xie Y., Cordero J., Shirvani A., Gigina A., Engelhardt M., Trogisch F.A., Geffers R., et al. Induction of cardiomyocyte proliferation and angiogenesis protects neonatal mice from pressure overload-associated maladaptation. JCI Insight. 2019;5:e128336. doi: 10.1172/jci.insight.128336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone B., Atanassov I., Aeschimann F., Li X., Grosshans H., Dieterich C. Bayesian prediction of RNA translation from ribosome profiling. Nucleic Acids Res. 2017;45:2960–2972. doi: 10.1093/nar/gkw1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamas M.A., Sperrin M., Watson M.C., Coutts A., Wilde K., Burton C., Kadam U.T., Kwok C.S., Clark A.B., Murchie P., et al. Do patients have worse outcomes in heart failure than in cancer? A primary care-based cohort study with 10-year follow-up in Scotland. Eur. J. Heart Fail. 2017;19:1095–1104. doi: 10.1002/ejhf.822. [DOI] [PubMed] [Google Scholar]
- Manavski Y., Lucas T., Glaser S.F., Dorsheimer L., Gunther S., Braun T., Rieger M.A., Zeiher A.M., Boon R.A., Dimmeler S. Clonal expansion of endothelial cells contributes to ischemia-induced neovascularization. Circ. Res. 2018;122:670–677. doi: 10.1161/CIRCRESAHA.117.312310. [DOI] [PubMed] [Google Scholar]
- Marzluff W.F., Wagner E.J., Duronio R.J. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet. 2008;9:843–854. doi: 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan M.A., Skelly D.A., Dona M.S.I., Squiers G.T., Farrugia G.E., Gaynor T.L., Cohen C.D., Pandey R., Diep H., Vinh A., et al. High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation. 2020;142:1448–1463. doi: 10.1161/CIRCULATIONAHA.119.045115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud J., Simpson K.M., Escher R., Buchet-Poyau K., Beissbarth T., Carmichael C., Ritchie M.E., Schutz F., Cannon P., Liu M., et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genomics. 2008;9:363. doi: 10.1186/1471-2164-9-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed S.F., Hussain S., Mirzoyev S.A., Edwards W.D., Maleszewski J.J., Redfield M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore-Morris T., Guimaraes-Camboa N., Banerjee I., Zambon A.C., Kisseleva T., Velayoudon A., Stallcup W.B., Gu Y., Dalton N.D., Cedenilla M., et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin.Invest. 2014;124:2921–2934. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudry J.M., Massart J., Szekeres F.L., Krook A. TWIST1 and TWIST2 regulate glycogen storage and inflammatory genes in skeletal muscle. J. Endocrinol. 2015;226:X1. doi: 10.1530/JOE-14-0474e. [DOI] [PubMed] [Google Scholar]
- Muhlfeld C., Schipke J., Schmidt A., Post H., Pieske B., Sedej S. Hypoinnervation is an early event in experimental myocardial remodelling induced by pressure overload. J. Anat. 2013;222:634–644. doi: 10.1111/joa.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller G.A., Stangner K., Schmitt T., Wintsche A., Engeland K. Timing of transcription during the cell cycle: protein complexes binding to E2F, E2F/CLE, CDE/CHR, or CHR promoter elements define early and late cell cycle gene expression. Oncotarget. 2017;8:97736–97748. doi: 10.18632/oncotarget.10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- Ogawa E., Saito Y., Harada M., Kamitani S., Kuwahara K., Miyamoto Y., Ishikawa M., Hamanaka I., Kajiyama N., Takahashi N., et al. Outside-in signalling of fibronectin stimulates cardiomyocyte hypertrophy in cultured neonatal rat ventricular myocytes. J. Mol.Cell Cardiol. 2000;32:765–776. doi: 10.1006/jmcc.2000.1119. [DOI] [PubMed] [Google Scholar]
- Paik D.T., Tian L., Williams I.M., Rhee S., Zhang H., Liu C., Mishra R., Wu S.M., Red-Horse K., Wu J.C. Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. 2020;142:1848–1862. doi: 10.1161/CIRCULATIONAHA.119.041433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto A.R., Ilinykh A., Ivey M.J., Kuwabara J.T., D'Antoni M.L., Debuque R., Chandran A., Wang L., Arora K., Rosenthal N.A., Tallquist M.D. Revisiting cardiac cellular composition. Circ. Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z., Yu P., Li D., Li Z., Liao Y., Wang Y., Zhou B., Wang L. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation. 2020;141:1704–1719. doi: 10.1161/CIRCULATIONAHA.119.043053. [DOI] [PubMed] [Google Scholar]
- Riehle C., Bauersachs J. Small animal models of heart failure. Cardiovasc. Res. 2019;115:1838–1849. doi: 10.1093/cvr/cvz161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M., Minamino T., Toko H., Miyauchi H., Orimo M., Qin Y., Akazawa H., Tateno K., Kayama Y., Harada M., et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- Scharf G.M., Kilian K., Cordero J., Wang Y., Grund A., Hofmann M., Froese N., Wang X., Kispert A., Kist R., et al. Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight. 2019;5:e126721. doi: 10.1172/jci.insight.126721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schram K., De Girolamo S., Madani S., Munoz D., Thong F., Sweeney G. Leptin regulates MMP-2, TIMP-1 and collagen synthesis via p38 MAPK in HL-1 murine cardiomyocytes. Cell Mol. Biol. Lett. 2010;15:551–563. doi: 10.2478/s11658-010-0027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly D.A., Squiers G.T., McLellan M.A., Bolisetty M.T., Robson P., Rosenthal N.A., Pinto A.R. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018;22:600–610. doi: 10.1016/j.celrep.2017.12.072. [DOI] [PubMed] [Google Scholar]
- Souders C.A., Bowers S.L., Baudino T.A. Cardiac fibroblast: the renaissance cell. Circ. Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallquist M.D., Molkentin J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017;14:484–491. doi: 10.1038/nrcardio.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan F.L., Moravec C.S., Li J., Apperson-Hansen C., McCarthy P.M., Young J.B., Bond M. The gene expression fingerprint of human heart failure. Proc. Natl. Acad. Sci. U S A. 2002;99:11387–11392. doi: 10.1073/pnas.162370099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirziu D., Chorianopoulos E., Moodie K.L., Palac R.T., Zhuang Z.W., Tjwa M., Roncal C., Eriksson U., Fu Q., Elfenbein A., et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J. Clin.Invest. 2007;117:3188–3197. doi: 10.1172/JCI32024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toischer K., Zhu W., Hunlich M., Mohamed B.A., Khadjeh S., Reuter S.P., Schafer K., Ramanujam D., Engelhardt S., Field L.J., Hasenfuss G. Cardiomyocyte proliferation prevents failure in pressure overload but not volume overload. J. Clin.Invest. 2017;127:4285–4296. doi: 10.1172/JCI81870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombor L.S., John D., Glaser S.F., Luxan G., Forte E., Furtado M., Rosenthal N., Baumgarten N., Schulz M.H., Wittig J., et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat. Commun. 2021;12:681. doi: 10.1038/s41467-021-20905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unudurthi S.D., Nassal D.M., Patel N.J., Thomas E., Yu J., Pierson C.G., Bansal S.S., Mohler P.J., Hund T.J. Fibroblast growth factor-inducible 14 mediates macrophage infiltration in heart to promote pressure overload-induced cardiac dysfunction. Life Sci. 2020;247:117440. doi: 10.1016/j.lfs.2020.117440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal R., Wagner J.U.G., Braeuning C., Fischer C., Patrick R., Tombor L., Muhly-Reinholz M., John D., Kliem M., Conrad T., et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight. 2019;4:e131092. doi: 10.1172/jci.insight.131092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil-Garcia M., Demkes C.J., Eding J.E.C., Versteeg D., de Ruiter H., Perini I., Kooijman L., Gladka M.M., Asselbergs F.W., Vink A., et al. Gene expression profiling of hypertrophic cardiomyocytes identifies new players in pathological remodelling. Cardiovasc. Res. 2021;117:1532–1545. doi: 10.1093/cvr/cvaa233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Yu P., Zhou B., Song J., Li Z., Zhang M., Guo G., Wang Y., Chen X., Han L., Hu S. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat.Cell Biol. 2020;22:108–119. doi: 10.1038/s41556-019-0446-7. [DOI] [PubMed] [Google Scholar]
- Wang Y., Hu J., Liu J., Geng Z., Tao Y., Zheng F., Wang Y., Fu S., Wang W., Xie C., et al. The role of Ca(2+)/NFAT in dysfunction and inflammation of human coronary endothelial cells induced by sera from patients with Kawasaki disease. Sci. Rep. 2020;10:4706. doi: 10.1038/s41598-020-61667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Cui M., Shah A.M., Tan W., Liu N., Bassel-Duby R., Olson E.N. Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution. Cell Rep. 2020;33:108472. doi: 10.1016/j.celrep.2020.108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfahrt T., Rauber S., Uebe S., Luber M., Soare A., Ekici A., Weber S., Matei A.E., Chen C.W., Maier C., et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature. 2019;566:344–349. doi: 10.1038/s41586-019-0896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Gong N.L., Bodi I., Aronow B.J., Backx P.H., Molkentin J.D. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J. Biol. Chem. 2006;281:9152–9162. doi: 10.1074/jbc.M510217200. [DOI] [PubMed] [Google Scholar]
- Yang K.C., Yamada K.A., Patel A.Y., Topkara V.K., George I., Cheema F.H., Ewald G.A., Mann D.L., Nerbonne J.M. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 2014;129:1009–1021. doi: 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg E.M., Tarnavski O., Zeisberg M., Dorfman A.L., McMullen J.R., Gustafsson E., Chandraker A., Yuan X., Pu W.T., Roberts A.B., et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Zeltz C., Gullberg D. The integrin-collagen connection - a glue for tissue repair? J.Cell Sci. 2016;129:1284. doi: 10.1242/jcs.188672. [DOI] [PubMed] [Google Scholar]
- Zhao M., Chow A., Powers J., Fajardo G., Bernstein D. Microarray analysis of gene expression after transverse aortic constriction in mice. Physiol. Genomics. 2004;19:93–105. doi: 10.1152/physiolgenomics.00040.2004. [DOI] [PubMed] [Google Scholar]
- Zhou P., Pu W.T. Recounting cardiac cellular composition. Circ. Res. 2016;118:368–370. doi: 10.1161/CIRCRESAHA.116.308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene expression in isolated cardiomyocytes from mouse hearts 1 week after sham (CM_Sham) or TAC surgery (CM_TAC_1Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between CM_Sham and CM_TAC_1Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated cardiomyocytes from mouse hearts 1 week after sham (CM_Sham) and 8 weeks after TAC surgery (CM_TAC_8Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between CM_Sham and CM_TAC_8Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated fibroblasts from mouse hearts 1 week after sham (FB_Sham) or TAC surgery (FB_TAC_1Wo) as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between FB_Sham and FB_TAC_1Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated fibroblasts from mouse hearts 1 week after sham (FB_Sham) and 8 weeks after TAC surgery (FB_TAC_8Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between FB_Sham and FB_TAC_8Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated endothelial cells from mouse hearts 1 week after sham (EC_Sham) or TAC surgery (EC_TAC_1Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between EC_Sham and EC_TAC_1Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Gene expression in isolated endothelial cells from mouse hearts 1 week after sham (EC_Sham) and 8 weeks after TAC surgery (EC_TAC_8Wo), as determined by bulk RNA sequencing. Gene expression (shown in columns N-P and S-U in CPM, log2 counts per million reads) was compared between EC_Sham and EC_TAC_8Wo, n = 3 were analyzed per condition. AV denotes average, SD denotes standard deviation.
Data Availability Statement
-
•
Bulk RNA-seq, Ribo-Seq and single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not contain an original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.