

Abstract
Cisplatin is one of the most effective chemotherapy drugs for ovarian cancer, but resistance is common. The initial response to platinum-based chemotherapy is as high as 80%, but in most advanced patients, final relapse and death are caused by acquired drug resistance. The development of resistance to therapy in ovarian cancer is a significant hindrance to therapeutic efficacy. The resistance of ovarian cancer cells to chemotherapeutic mechanisms is rather complex and includes multidrug resistance, DNA damage repair, cell metabolism, oxidative stress, cell cycle regulation, cancer stem cells, immunity, apoptotic pathways, autophagy and abnormal signaling pathways. The present review provided an update of recent developments in our understanding of the mechanisms of ovarian cancer platinum-based chemotherapy resistance, discussed current and emerging approaches for targeting these patients and presented challenges associated with these approaches, with a focus on development and overcoming resistance.
Keywords: molecular mechanisms, platinum-based chemotherapy resistance, ovarian cancer
1. Introduction
Ovarian cancer is one of the most common malignant tumors of the reproductive organs and has the highest mortality rate among all gynecological malignancies. At diagnosis, ~70% of patients present with advanced disease and most are resistant to platinum-based chemotherapy, resulting in a low five-year survival rate (1,2). Ovarian cancer can be subdivided into two main histological subtypes: Epithelial ovarian cancers (EOCs), which account for ~90% of ovarian cancers, and non-EOCs, which account for ~10% of ovarian cancers (3). Epithelia cancers include serous [high-grade serous carcinoma (HGSC) and low-grade serous carcinoma (LGSC)], endometrioid (high-grade endometrioid carcinoma and low-grade endometrioid carcinoma), clear-cell and mucinous carcinomas (2,4). LGSCs usually contain KRAS and BRAF mutations (5,6), whereas most HGSCs have TP53 mutations and exhibit severe aneuploidy genome aberrations. Clear cell carcinoma is characterized by mutations in the ARID1A, PIK3CA, PTEN and KRAS genes, while endometrioid carcinoma, similar to its uterine counterpart, has mutations in ARID1A, CTNNB1, and PTEN, as well as microsatellite instability (7,8).
Approximately 80% of patients with ovarian cancer are treated with cytoreductive surgery followed by adjuvant chemotherapy with carboplatin and paclitaxel or cisplatin and paclitaxel (2,9). However, ~70% of patients with this treatment regimen will relapse (10) and the recurring cancer is often resistant to standard platinum-based chemotherapy. In patients with advanced cancer, mortality is usually due to acquisition of drug resistance. Cisplatin is one of the most effective chemotherapy drugs for ovarian cancer, but resistance to cisplatin is common. Patient recurrence more than 6 months after front-line platinum-based therapy is considered platinum-sensitive, whereas platinum-resistant recurrence occurs after less than 6 months (2,11,12). During the six months after the completion of major platinum-based chemotherapy, disease progression is usually closely related to platinum resistance. Due to its significant impact on patient survival time and quality, improving the response to platinum is an important challenge. At present, the standard treatment for platinum-resistant or refractory ovarian cancer is pegylated liposomal adriamycin, weekly paclitaxel and topotecan, but the efficacy of this regimen is limited (13).
To provide a thorough understanding of the mechanism of drug resistance, in the present review, the important molecular mechanisms involved in the response of ovarian cancer patients to platinum-based chemotherapy, as well as methods to circumvent these mechanisms that have been studied extensively by clinical and laboratory researchers are explored (Fig. 1).
Figure 1.

Schematic overview of the mechanisms of platinum-based chemotherapy resistance in ovarian cancer.
2. Literature review methods
A systematic literature search was conducted via electronic search engine PubMed for eligible studies published until December 31, 2021. The keywords for searches were as follows: ‘Ovarian cancer’, ‘chemotherapy resistance’, ‘chemoresistance’. Furthermore, the referenes in retrieved articles were also manually reviewed for potentially relevant studies.
3. Molecular mechanisms of platinum-based chemotherapy resistance in ovarian cancer
Multidrug resistance (MDR)
Since the ovary is deep in the pelvic cavity and small in size, it is difficult to detect early ovarian cancer due to a lack of typical symptoms. Upon surgery, the tumor is confined to the ovary in less than 30% of patients with ovarian cancer (14); in most patients, the tumor has spread to the pelvic and abdominal organs. Although progress has been made in surgical techniques and chemotherapy drugs in recent years, the mortality rate of ovarian cancer has not decreased significantly. Chemotherapy MDR is the main cause of treatment failure (15). MDR is a drug-resistant phenotype in which cancer cells are simultaneously resistant to multiple drugs with different molecular targets and without obvious structural similarities (16). Overcoming MDR is a top priority in clinical and research oncology but remains elusive. In the present study, the latest literature on the main mechanisms of MDR were summarized and several new MDR reversal strategies were evaluated, including more effective and specific P-glycoprotein (P-gp) inhibitors.
Membrane-bound adenosine triphosphate (ATP)-dependent active drug efflux pumps can significantly decrease the intracellular concentration of the drug and thus the efficacy of treatment. P-gp uses the energy of ATP hydrolysis to transport various structural and functional drugs out of the cell. P-gp and multidrug-resistance-associated protein (MRP) are two main membrane proteins known to cause MDR in cancer. Inhibiting these proteins is a strategy to sensitize cancer cells to chemotherapy (17). Overexpression of P-gp can also lead to the development of MDR in human tumors (including ovarian cancer). Therefore, many years of extensive research have focused on overcoming P-gp-based MDR. To date, three generations of P-gp modulators have been developed. The second-generation P-gp modulator, valspodar, has exhibited the capacity to modulate ovarian cancer resistance in phase I, II and III clinical trials (18).
DNA damage repair (DDR)
As MDR is one of the most studied mechanisms of ovarian cancer drug resistance in the early stages of the research process, DDR is one of the most important mechanisms of ovarian cancer drug resistance. At present, the standard treatment for advanced ovarian cancer is surgical cytoreduction, followed by platinum and taxane-based chemotherapy. Current research focuses on new agents, particularly those that target the DDR pathway (19,20). A comprehensive understanding of the process of DDR in ovarian cancer and its working principle may promote future research on treatments and drug resistance.
Studies have revealed that more than 50% of HGSCs have homologous recombination repair (HR) pathway defects (21–23), which are mainly related to genetic and epigenetic changes in HR pathway genes. The tumor suppressors BRCA1 and BRCA2, which encode proteins involved in DDR via homologous recombination, have been associated with an increased risk of ovarian cancer (24). Mutations in BRCA1/2 are associated with high sensitivity to DNA-damaging agents, including poly-(ADP-ribose) polymerase (PARP) inhibitors and platinum (25–27). Patients with BRCA mutations have an improved overall response to platinum therapy, which is associated with a longer survival rate for ovarian cancer (28,29). Compared with patients who do not carry the mutation, patients with the mutation are less likely to have disease progression within 6 months after the end of the main treatment (28). Therefore, patients with BRCA1/2 mutations are more likely to have longer progression-free survival (PFS) than patients without mutations.
Concurrently, the study found that upon the first relapse of platinum-sensitive or platinum-resistant patients, the response rates for second-line platinum-based or nonplatinum-based chemotherapy were higher in patients carrying mutations than in those who did not (28). In BRCA-mutant cancers, BRCA reversion mutations that restore protein function are the key resistance mechanism of platinum-based chemotherapy (30).
Reversion mutations that may be caused by DNA-damaging chemotherapy or genome instability are base substitutions or insertions/deletions. Such mutations are usually close to the main protein truncation mutation and restore the open reading frame (ORF) of the gene to allow the production of functional protein, transforming tumor cells from HR defective to proficient. A total of 18% of platinum-refractory cancers and 13% of platinum-resistant cancers have BRCA mutations in circulating cell-free DNA (cfDNA) before treatment, compared with 2% of platinum-sensitive cancers (31). Before treatment, patients with no BRCA reversion mutations in cfDNA had significantly longer PFS after treatment with rucaparib compared with patients with reversion mutations (32).
In cancer, the restoration of HR function promotes drug resistance by repairing DNA damage induced by PARP inhibitors and/or platinum-based chemotherapy, destroying the basis of synthetic lethality and ultimately promoting cell survival (33). Reversion mutations in multiple HR pathway genes, including BRCA1, BRCA2, RAD51C, RAD51D (34) and PALB2, cause acquired resistance to platinum-based chemotherapies and PARP inhibitors (32).
Limited DNA end resection is the key to impaired HR in BRCA1-mutant cells. A loss-of-function CRISPR screen identified dynein light chain 1 protein (DYNLL1) as a factor responsible for platinum resistance in BRCA-defective patients with HGSC by facilitating DNA end resection (35). After platinum-based chemotherapy for BRCA1 mutant ovarian cancer, low expression of DYNLL1 was significantly associated with poor PFS.
Strengthening DNA repair pathways is one of the ways that cancer cells resist the DNA-damaging effects of platinum (Fig. 2). Inhibiting these DNA repair pathways may restore the sensitivity of cancer cells to platinum. This is precisely the objective of several drugs under development. PARP inhibitors disrupt the mechanism by which damaged parts of DNA are removed and the drug trabectedin binds directly to and damages DNA (36). TOP1 initiates the DNA relaxation by cleaving one DNA strand. This in turn generates TOP1 cleavage complexes (TOP1ccs). The selective trapping of TOP1ccs by topotecan stabilizes TOP1ccs which covalently attach to the 3′-end of the breaks. These stalled TOP1ccs lead to lethal DNA double-strand breaks when they produce collisions with DNA replication (37). The drug topotecan blocks the action of the enzyme TOP1, thereby helping to cause DNA damage, improving the sensitivity of chemotherapy and is already licensed to treat recurrent ovarian cancer (38). As apical kinases, ATM (recruited to double-strand breaks) and ATR (recruited to single-stranded DNA) regulate the DNA damage response, and ATR inhibitors may restore platinum sensitivity for the treatment of patients with recurrent BRCA1/2 mutant ovarian cancer (39,40).
Figure 2.
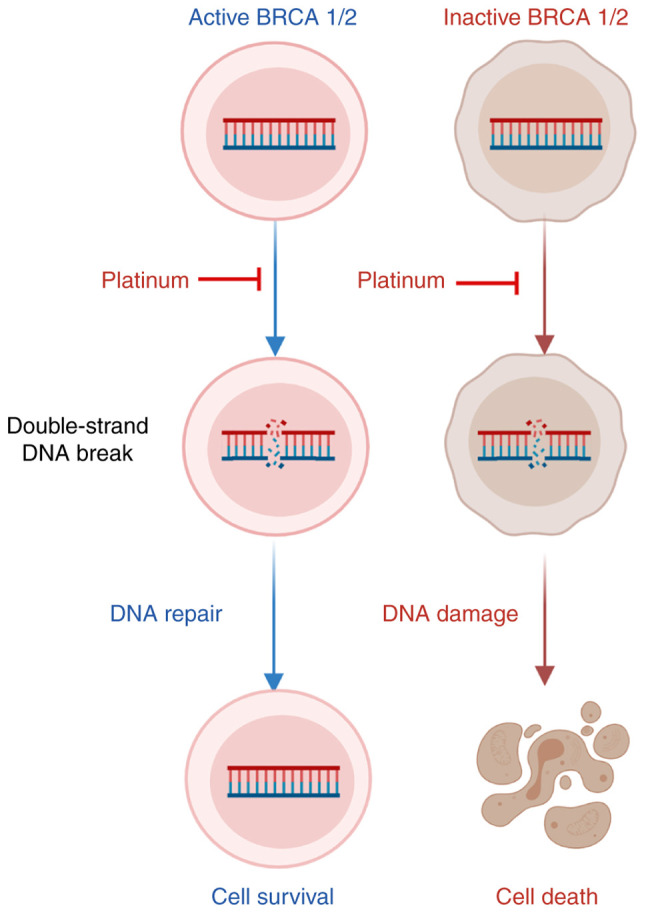
BRCA1/2 activity in ovarian cancer is associated with the response to platinum chemotherapy. BRCA1/2 functions in activating the repair of double-strand breaks and initiating homologous recombination, linking the maintenance of genomic integrity to tumor suppression. BRCA1/2 active cells can repair the platinum drug-caused double-strand DNA damage by homologous recombination, while BRCA1/2 inactive cells cannot, thus tumor cells with BRCA gene mutation are more sensitive to platinum drugs.
Cell cycle regulation
More than 50% of HGSCs are defective in the HR pathway (21). For HGSCs with intact HR, the expansion of cyclin E1 (CCNE1), which encodes the cell cycle regulator cyclin E1, is the best-characterized driving factor. Amplification or gain of CCNE1 is observed in 20% of HGSC tumors and is related to main treatment resistance and decreased overall survival (OS) (41). Patients with CCNE1 amplification are unlikely to benefit from PARP inhibitors and are unlikely to respond to platinum drugs due to the mutual exclusivity of CCNE1 amplification and BRCA1/2 mutations (42,43).
Cyclin-dependent kinase (CDKs) are proteins required for appropriate progression of the cell cycle and also play a central role in regulating DDR (44). CCNE1 is essential for CDK2 activation and its overexpression can lead to premature entry into the S phase, abrogating DNA repair during the G1 phase and leading to increased levels of replication stress. Checkpoint kinase 1 and 2 (CHK1 and CHK2) are responsible for regulating DNA replication and DNA damage response (45). Thus, CCNE1 overexpression may increase sensitivity to CHK1 inhibition (46). Promising targeted strategies using WEE1 kinase inhibitors, CHK1 inhibitors and CDK2 inhibitors are under review in clinical trials examining biomarkers (47). The combination of the CDK2 inhibitor dinaciclib and the AKT inhibitor MK2206 exhibited a selective synergistic effect in a CCNE1-expanded cell line in a xenograft model (42). Approximately 60% of patients with platinum-resistant or refractory diseases receive clinical benefit from the CHK1 and CHK2 inhibitor, prexasertib (LY2606368) (48). Recently, a study reported that the CCNE1-overexpressing HGSC model is markedly sensitive to combinations of cell cycle checkpoint kinase and immune checkpoint inhibitors (29).
CCNE1 and RB1 are cyclins related to cell cycle transition in the G1-S phase. Tumors with increased CCNE1 copy number are more resistant to platinum therapy, while RB1 loss is associated with high sensitivity to platinum therapy (49,50). Studies have shown that loss of RB1 protein expression is associated with longer OS and PFS (47,50,51).
ATM/ATR kinases play a central role in coordinating the DDR. Blocking the activity of key CDKs can signal DNA damage and cause cell cycle arrest. The combination of ATR inhibitor and PARP inhibitor (PARPi) has a synergistic effect in reducing the survival rate and colony formation in BRCA1/2 mutant PARPi/platinum-resistant cell models that are resistant to PARPi and platinum or exhibit de novo resistance (46).
CDK6 can bind to and phosphorylate FOXO3, thereby inducing the expression of ATR. CDK6 regulates ATR through FOXO3 to control platinum-induced cell death (52). In a model of advanced platinum-resistant tumors, silencing or pharmacological inhibition of CDK6 increased the sensitivity of EOC cells to platinum without affecting RB1 phosphorylation but increased platinum-induced DNA damage by increasing apoptosis (52). Notably, compared with other models, CDK6 is less involved in regulating G1-S transition and proliferation in EOC. When platinum and CDK6 inhibitor PD0332991 are combined, platinum induces significant cell cycle arrest in the S phase (52), while CDK6 inhibition induces more apoptosis (Fig. 3).
Figure 3.
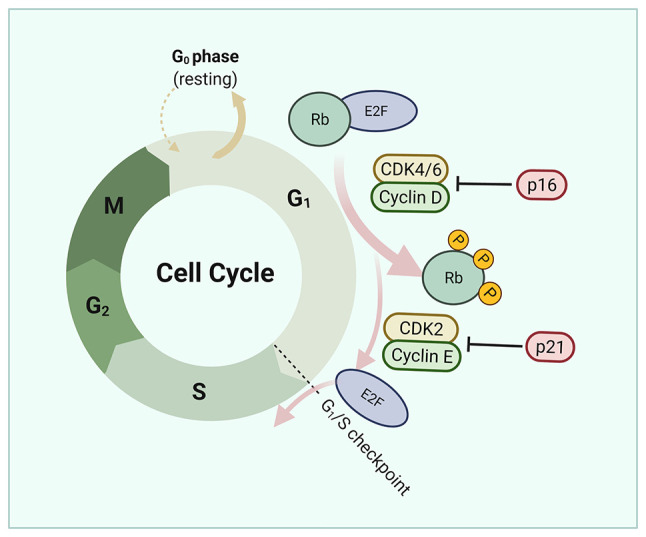
Regulation of the cell cycle by platinum treatment in ovarian cancer. CCNE1 and RB are cyclins associated to cell cycle transition in the G1-S phase. CCNE1 is essential for CDK2 activation and its overexpression can lead to premature entry into the S phase, abrogating DNA repair during the G1 phase and leading to increased levels of replication stress. Inhibition of CDK6 increases the sensitivity of epithelial ovarian cancers cells to platinum by increasing apoptosis.
Cell metabolism
Metabolic reprogramming is emerging as a proposed molecular mechanism of cisplatin resistance (53). Accumulating evidence has suggested that the metabolism of tumor tissues differs from that of matched normal tissues and metabolic reprogramming is likely to be an important cause of treatment resistance (54,55). Metabolic reprogramming involves a series of metabolic alterations involving all major pathways from glucose metabolism to glutamine and lipids as well as mitochondrial (mt) alterations (56). The pentose phosphate pathway (PPP) is an important component of glucose metabolism that uses glucose-6-phosphate as the primary substrate (57). The products of PPP biosynthesis are ribonucleotides and nicotinamide adenine dinucleotide phosphate; the latter is essential for reductive biosynthesis (57).
Overexpression and higher enzyme activity of glucose-6-phosphate dehydrogenase (G6PD) can increase cisplatin resistance. G6PD and transketolase have been identified as possible targets to overcome cisplatin resistance (53). The enzymes that regulate glycolysis flow are transcriptionally regulated by three major transcription factors: p53, hypoxia-inducible factor-1 (HIF-1) and Myc (58). HIF-1, a major hypoxia-induced transcription factor, promotes a dissociation of glycolysis and the tricarboxylic acid cycle (59). HIF-1 allows adaptation to hypoxia by increasing glucose transport, glycolysis and lactate production. In addition to stimulating glycolysis, HIF-1 inhibits the function of the mt respiratory chain in a variety of ways. Inhibition of HIF-1 may redirect aerobic glycolysis toward mt oxidative phosphorylation, which can sensitize cells to cisplatin through overproduction of reactive oxygen species (ROS), leading to apoptosis; the cisplatin response in ovarian cancer cells can be improved by targeting HIF-1-regulated cancer metabolism (60). Studies have demonstrated that metformin can modulate cell growth and metabolism by inhibiting mt activity, AMP/ATP balance disturbance and AMPK activation and can partially reverse platinum resistance in the PDX model (61–63). This provides a new direction for reducing resistance.
Oxidative stress
Cell metabolism induces the production of ROS; a variety of chemotherapeutic drugs, including cisplatin, also induce the production of large amounts of ROS in tumor cells. The effectiveness of chemotherapy depends on the induction of oxidative stress. Increased ROS can cause oxidative DNA damage, leading to genomic instability and promoting cellular apoptosis, senescence or autophagy. To withstand oxidative stress, cells activate the transcription factor Nrf2, the major regulator of antioxidant responsive element-mediated genes (64).
Activation of the Nrf2 pathway is involved in the development of ovarian cancer and platinum resistance (65). Thus, targeting redox regulation is a promising strategy to overcome drug resistance (66). In line with this, Nrf2 inhibition is expected to increase chemotherapy sensitivity. Combining Nrf2 inhibition with chemotherapy enhances cytotoxic effects but produces side effects such as chemotherapy-induced myelosuppression (67). Additionally, Nrf2 inhibition results in enhanced sensitivity toward ROS-induced DNA damage, whereas PARP inhibitors inhibit this DNA repair pathway (68). Furthermore, PARP inhibitors increase not only DNA damage but also ROS (69). Studies have revealed that combined treatment with Nrf2 inhibitors and PARP inhibitors improves therapeutic efficacy, particularly in BRCA1 mutant cancer cells and no severe side effects are expected (68,69).
Mitochondria are important sites of redox activity. Notably, compared with cisplatin-sensitive HGSC cells, cisplatin-resistant HGSC cells have a lower mt content and lower levels of mtROS, which induce cell death (70). The principle of anticancer treatment with chemotherapeutic drugs is usually to disrupt cell integrity by destroying nuclear DNA (nDNA) to induce cell death. In addition, mtDNA, similar to nDNA, is greatly affected by cisplatin. Therefore, mtDNA damage is evident in cisplatin-treated cells (71,72). Furthermore, the ATP synthase inhibitor oligomycin A can block mt function and prevent the induction of mtROS during cisplatin treatment, thereby reducing cisplatin-induced apoptosis (70).
In fact, several oxidative stress-related genes, such as ARHGEF6, TXNRD1, GLA, GSTZ1 (73), thioredoxin (12), E26 oncogene homolog 1 (74,75) and ALDH (76), have been linked to chemoresistance in ovarian cancer. Increasing ROS through pharmacological methods may render ovarian cancer cells sensitive to cisplatin and overcome drug resistance. An ALDH inhibitor named CM37, has been revealed to increase intracellular ROS levels in ovarian cancer cells, leading to DNA damage and inhibition of cell survival and proliferation (76).
Cancer stem cells (CSCs)
The current oncology hypothesis proposes that only a small percentage of cancer cells can spread into the tumor. These cells are called tumor promoter cells or CSCs and have pluripotent properties similar to those of normal stem cells (77,78). Previous studies have revealed that CSCs are a unique cell population that causes tumor recurrence and metastasis, leading to the formation of new tumors (79,80). CSCs also exist in ovarian tumors and are resistant to chemotherapy (81,82) Therefore, the development of new therapies for CSCs aims to improve the lives of patients with cancer, particularly those with metastatic disease, and to avoid the recurrence of chemotherapy-resistant tumors.
One understudied mechanism of chemoresistance is the persistence of quiescent cancer cells that are not eliminated by chemotherapy. According to previous studies, these residual tumors are enriched in CSCs (83), which are more resistant to chemotherapy (Fig. 4) (74,84,85). There are already several surface markers for ovarian CSC identification, including MyoD, CD44, CD117 (86), CD133 (87), ALDH (76) and nuclear factor of activated T cells, cytoplasmic 4 (NFATC4) (88,89).
Figure 4.

CSCs are more resistant to chemotherapy. The persistence of quiescent cancer cells that are hard to be eliminated by chemotherapy and these residual tumors are enriched in CSCs, which are more resistant to chemotherapy. CSCs are a unique cell population that causes tumor recurrence and metastasis, leading to the formation of new tumors. CSCs, cancer stem cells.
CD44 is a cell-surface glycoprotein of the hyaluronate receptor that plays a role in tumor stemness, recurrence and drug resistance in ovarian cancer. Compared with cells cultured under differentiation conditions, isolated CD44+/CD117+ ovarian CSCs could completely regenerate the original tumor phenotype in mice and were more resistant to cisplatin and paclitaxel (90). The presence of ALDH+CD133+ CSC-like cells in primary ovarian tumors is associated with shorter disease-free survival and OS. In addition, compared with parental cells, these cells exhibit enhanced chemoresistance to the human primary ovarian tumor phenotype (89,91). The aforementioned studies revealed that CSCs are closely related to the chemoresistance of ovarian cancer. Metformin treatment significantly reduces the stemness of cancer by reducing ALDH+CD133+ CSCs in patients with ovarian cancer (92). Phase III clinical trials have shown that metformin is a favorable adjunct in the treatment of EOC.
Studies have demonstrated that activating the PI3K/Akt/mTOR signaling pathway can enhance the expression of epithelial-mesenchymal transition (EMT) and CSC markers in chemoresistant EOC cells. Accordingly, the PI3K inhibitor BEZ235 renders EOC cells sensitive to cisplatin by inhibiting the expression of EMT and CSC markers (91). NFATC4 is enriched in ovarian CSC-like cells, which leads to chemotherapy resistance by downregulating MYC at an early stage, helping cells enter a quiescent state (88). The aforementioned studies revealed that NFATC4 is a significant therapeutic target for ovarian cancer that is worthy of in-depth study.
Immunity
There is increasing evidence that the immune response may affect the prognosis of patients with ovarian cancer. For patients with recurrent ovarian cancer, the immune system can be activated to identify and attack cancer cells to prevent recurrence. The tumor microenvironment is a potential factor for recurrence and chemotherapy resistance (93). Among them, the presence of tumor-infiltrating lymphocytes, particularly CD8+ tumor-infiltrating lymphocytes, often indicates an improved prognosis (94). In a study of patients receiving platinum-based chemotherapy, the 5-year OS rate of patients whose tumors contained significant T-cell infiltration was 38%, but the 5-year OS rate of patients whose tumors contained very few T cells was only 4.5% (95). CXCL9, CCL21 and CCL22 were more highly expressed in tumors with significant T-cell infiltrates than in tumors with few T cells (96,97).
Several studies have shown that high tumor-associated macrophage (TAM) density is closely related to poor prognosis and resistance to treatment in patients with ovarian cancer (98,99). The mechanism of drug resistance is as follows: i) Macrophages promote tumor polarization; ii) macrophages affect the pro-survival signaling pathways; and iii) macrophages upregulate MDR genes in cancer cells (10). In various ovarian cancer cell lines treated with cisplatin or carboplatin, macrophages are induced to differentiate into M2 macrophages through increased IL-10 production and enhanced activation of the STAT3 signaling factor (100,101). The understanding of the involvement of TAMs in tumor progression and chemoresistance provided by these studies has revealed new opportunities for the development of ovarian cancer therapies (10,99).
In addition, the immune checkpoint protein programmed death ligand (PD-L1) is often expressed by ovarian tumor cells, and PD-1 is a receptor often expressed by tumor-infiltrating lymphocytes. Studies have found that the interaction between PD-1 and PD-L1 is a key therapeutic target for reactivating the immune response against a variety of cancers. Therefore, blocking the PD-1/PD-L1 interaction with an antibody against the PD-L1 molecule is a new therapeutic opportunity for patients with advanced platinum-resistant ovarian cancer (102–104). The human immunoglobulin G1 monoclonal antibody avelumab has a wild-type Fc region that blocks PD-L1. Avelumab has shown antitumor activity in patients with relapsed or refractory ovarian cancer who have progressed after platinum-based chemotherapy (13).
Apoptotic pathways
The effectiveness of chemotherapy strongly depends on the ability of ovarian cancer cells to undergo drug-induced apoptosis (Fig. 5) (105–107). Platinum-based chemotherapeutics, such as carboplatin and cisplatin, are alkylating agents that bind to DNA to produce intra- and interstrand crosslinks, thereby inducing DNA damage that culminates in mitochondria-mediated apoptosis (108,109). The mt apoptotic pathway is controlled by proapoptotic (e.g., PUMA, Bim, Bid, Bad, Bik, Bax, Noxa and Bmf) and antiapoptotic (e.g., Bcl-2 Bcl-w, Mcl-1, Bfl-1 and Bcl-xL) proteins of the Bcl-2 family (110). Inhibition of apoptosis-related proteins in ovarian cancer cells increases cisplatin resistance and PI3K/mTOR inhibits the induction of pro-apoptotic and anti-apoptotic Bcl-2 family proteins, which are associated with treatment fragility in ovarian cancer cells (111).
Figure 5.
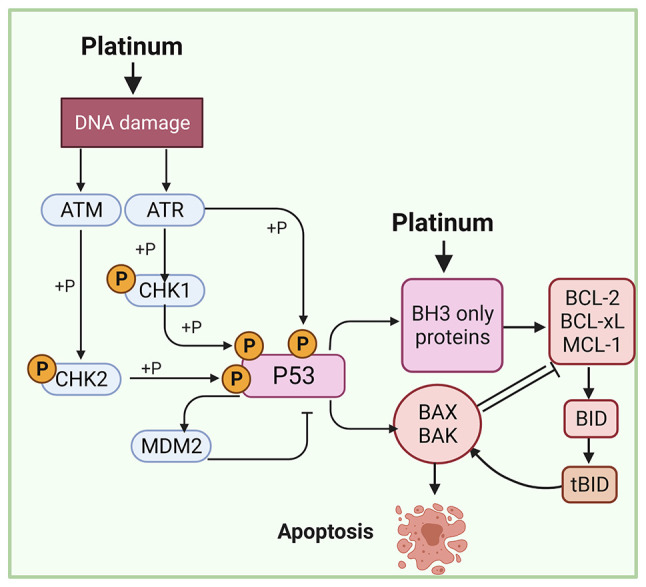
Effectiveness of chemotherapy strongly depends on the ability of ovarian cancer cells to undergo drug-induced apoptosis. Inactivation of p53 increase resistance to platinum-based chemotherapy by reducing the sensitivity of cells to activate apoptotic responses. The balance of proapoptotic and antiapoptotic proteins of the Bcl-2 family is a key factor to promote the sensitivity of platinum-based chemotherapy.
Approximately 40–80% of patients with EOC have TP53 mutations (112,113). In addition, a few cases of HGSCs lacking TP53 mutations have been reported with amplification of MDM2, which is a known alternative mechanism for inactivation of p53 (114). Mutations in the p53 gene can increase resistance to various DNA-damaging agents (including cisplatin) by reducing the sensitivity of cells to activate apoptotic responses (115). The introduction of wild-type p53 protein into A2780/CP cisplatin-resistant cells by adenovirus gene transfer renders these cells significantly sensitive to platinum-based cytotoxicity and further supports the participation of p53 in cisplatin resistance (112).
ARID1A mutation is a known genetic driver of ovarian cancer. Notably, ARID1A mutations are found in more than 50% of ovarian clear cell carcinomas and 30% of ovarian endometrioid carcinomas (116,117). ARID1A and TP53 mutations are typically mutually exclusive in ovarian cancer (118). ARID1A mutations lead to upregulation of HDAC6, which in turn inactivates the apoptosis-promoting function of p53, indicating that drug inhibition of HDAC6 is an effective treatment strategy for cancers with ARID1A mutations (119).
Autophagy
On the one hand, autophagy protects cells from genotoxic stress to prevent tumorigenesis and carcinogenic transformation. On the other hand, autophagy can be used as a survival strategy for cancer cells to overcome the stress caused by chemotherapy, radiotherapy or other treatments (120,121). Previous studies have demonstrated that autophagy in ovarian cancer cells can be induced by cisplatin through ubiquitin-binding protein p62 (SQSTM1) or HMGB1 (122,123). Autophagic flux in cisplatin-resistant ovarian cancer cells is caused by cisplatin. At present, the cytoprotective function of autophagy in cancer cells is considered a potential chemotherapy resistance mechanism.
Most studies have suggested that targeting autophagy-related molecules may increase the chemosensitivity of cancer cells (124,125). Knockdown of ATG7 and ATG14 can inhibit basic autophagy in ovarian cancer cells and promote cell death induced by cisplatin (126). As a negative regulator of autophagy, mTOR inhibitors are used in combination with cisplatin to make cancer cells sensitive to chemotherapy. However, certain studies have suggested that chemosensitivity can be promoted by autophagy (127,128). The enhanced sensitivity of autophagy to cisplatin depends on different mechanisms, and dormant autophagic cancer cells are still susceptible to cisplatin-based chemotherapy.
Abnormal signaling pathways
A total of 10% of serous ovarian cancer is LGSC, which is characterized by early onset (median age 46 years), slow growth and poor response to chemotherapy (4,129). BRAF and KRAS hotspot mutations are found in ~2/3 of patients with LGSC. Furthermore, almost all patients with LGSC harbor a mutation predicted to induce ERK activation (5). Notably, RAS and PI3K participate in intensive cross-talk to regulate each other and coregulate downstream functions (130). Therefore, blocking only one pathway will induce compensatory signaling in the other pathway, ultimately leading to treatment failure and relapse (131).
PI3K/Akt/mTOR signaling plays an important role in regulating the cell cycle, quiescence and proliferation. Various somatic mutations in PTEN, Akt1 and mTOR have been identified in ovarian cancer and can induce enhanced PI3K/Akt/mTOR signaling (132,133). Excessive activation of PI3K/Akt/mTOR signaling is associated to chemoresistance and cancer metastasis and inhibition of PI3K/Akt/mTOR signaling can restore the sensitivity of chemotherapy-resistant ovarian cancer cells to chemotherapy drugs (134). Furthermore, combination treatment using RAS and PI3K inhibitors in ovarian cancer cell lines carrying activated oncogenic KRASG12D and deletion of two copies of the PTEN gene is a promising strategy for tumors that are rapidly resistant to targeted therapy alone (135). In addition, Wnt receptor Frizzled 7 (FZD7) is expressed in tumors and platinum-resistant cells. Knockdown of FZD7 reduces spheroid formation, increases sensitivity to platinum, and delays tumor occurrence (83).
As a cell surface transmembrane glycoprotein, the folate receptor (FR) promotes the unidirectional transport of folates into the cell. FR has limited distribution, and aberrant overexpression of FR is a characteristic of numerous epithelial tumors, including non-small cell lung, endometrial and ovarian cancer. Specifically, ~80% of EOC tumors constitutively express FR (136). In addition, the increase in receptor expression may be a negative prognostic factor for the chemotherapy response of this malignant tumor (137). Preclinical studies have revealed that folate-conjugated vintafolide (EC145) (138) and mirvetuximab soravtansine (IMGN853) (139) are well tolerated and active against platinum-resistant ovarian cancer, and response rates and PFS are encouraging.
4. Conclusions
Despite advances in chemotherapy, the 5-year survival rate of patients with ovarian cancer remains less than 50%, mainly due to chemotherapy resistance (22). Both primary resistance (patients do not respond at all to treatment and the disease progresses) and acquired resistance (patients eventually develop acquired resistance after an initial response) to platinum are associated with a severely negative prognosis for EOC patients. For these patients, a thorough understanding of their resistance mechanisms and active drugs is an urgent unmet clinical need (140). The resistance of ovarian cancer cells to chemotherapeutic mechanisms is rather complex and includes MDR, DDR, cell metabolism, oxidative stress, cell cycle regulation, CSCs, immunity, apoptotic pathways, autophagy and abnormal signaling pathways. Therefore, a single mechanism cannot fully explain the resistance of ovarian cancer cells to treatment.
Numerous new strategies are being studied to try to overcome this chemical resistance, including combining platinum-based chemotherapy with new molecularly targeted drugs, such as bevacizumab or olaparib. Combining the vascular endothelial growth factor A-neutralizing antibody bevacizumab with chemotherapy has been revealed to reduce or slow the growth of advanced EOC, but this combination does not appear to extend survival (11). Olaparib is a PARP inhibitor that is only used for cancer patients with BRCA gene mutations since the drug only works on cells where the BRCA pathway is blocked (36). However, only a small percentage of patients with ovarian cancer have mutations in the BRCA gene (141). More detailed mechanistic insights and the development of biomarkers, particularly non-invasive biomarkers, are required to accurately select patients for therapy and facilitate the evaluation of therapeutic efficacy in real time.
Acknowledgements
Not applicable.
Funding Statement
The present study was supported by the West China Second University Hospital Clinical Research Fund (grant no. KL105) and the Sichuan Provincial Key Research and Development Projects (grant. no. 2020YFS0081).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Authors' contributions
JM designed the review and edited the manuscript. LY and HJX wrote the manuscript. HJX, YYL, XXL, and XW collected and analyzed data. Data authentication is not applicable. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers. 2016;2:16061. doi: 10.1038/nrdp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orr B, Edwards RP. Diagnosis and treatment of ovarian cancer. Hematol Oncol Clin North Am. 2018;32:943–964. doi: 10.1016/j.hoc.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Gershenson DM, Bodurka DC, Lu KH, Nathan LC, Milojevic L, Wong KK, Malpica A, Sun CC. Impact of age and primary disease site on outcome in women with low-grade serous carcinoma of the ovary or peritoneum: Results of a large single-institution registry of a rare tumor. J Clin Oncol. 2015;33:2675–2682. doi: 10.1200/JCO.2015.61.0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grisham RN, Sylvester BE, Won H, McDermott G, DeLair D, Ramirez R, Yao Z, Shen R, Dao F, Bogomolniy F, et al. Extreme outlier analysis identifies occult mitogen-activated protein kinase pathway mutations in patients with low-grade serous ovarian cancer. J Clin Oncol. 2015;33:4099–4105. doi: 10.1200/JCO.2015.62.4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chui MH, Chang JC, Zhang Y, Zehir A, Schram AM, Konner J, Drilon AE, Da Cruz Paula A, Weigelt B, Grisham RN. Spectrum of BRAF mutations and gene rearrangements in ovarian serous carcinoma. JCO Precis Oncol. 2021;5 doi: 10.1200/PO.21.00055. PO.21.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidson B, Tropé CG. Ovarian cancer: Diagnostic, biological and prognostic aspects. Women's Health (Lond) 2014;10:519–533. doi: 10.2217/WHE.14.37. [DOI] [PubMed] [Google Scholar]
- 8.Reavis HD, Drapkin R. The tubal epigenome-An emerging target for ovarian cancer. Pharmacol Ther. 2020;210:107524. doi: 10.1016/j.pharmthera.2020.107524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman RL, Duska LR, Ramirez PT, Heymach JV, Kamat AA, Modesitt SC, Schmeler KM, Iyer RB, Garcia ME, Miller DL, et al. Phase 1–2 study of docetaxel plus aflibercept in patients with recurrent ovarian, primary peritoneal, or fallopian tube cancer. Lancet Oncol. 2011;12:1109–1117. doi: 10.1016/S1470-2045(11)70244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nowak M, Klink M. The Role of Tumor-associated macrophages in the progression and chemoresistance of ovarian cancer. Cells. 2020;9:1299. doi: 10.3390/cells9051299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, Sovak MA, Yi J, Nycum LR. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30:2039–2045. doi: 10.1200/JCO.2012.42.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz IN, Coley HM, Kramer HB, Madhuri TK, Safuwan NA, Angelino AR, Yang M. Proteomics analysis of ovarian cancer cell lines and tissues reveals drug resistance-associated proteins. Cancer Genomics Proteomics. 2017;14:35–51. doi: 10.21873/cgp.20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Disis ML, Taylor MH, Kelly K, Beck JT, Gordon M, Moore KM, Patel MR, Chaves J, Park H, Mita AC, et al. Efficacy and safety of avelumab for patients with recurrent or refractory ovarian cancer: Phase 1b results from the JAVELIN solid tumor trial. JAMA Oncol. 2019;5:393–401. doi: 10.1001/jamaoncol.2018.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moufarrij S, Dandapani M, Arthofer E, Gomez S, Srivastava A, Lopez-Acevedo M, Villagra A, Chiappinelli KB. Epigenetic therapy for ovarian cancer: Promise and progress. Clin Epigenetics. 2019;11:7. doi: 10.1186/s13148-018-0602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren F, Shen J, Shi H, Hornicek FJ, Kan Q, Duan Z. Novel mechanisms and approaches to overcome multidrug resistance in the treatment of ovarian cancer. Biochim Biophys Acta. 2016;1866:266–275. doi: 10.1016/j.bbcan.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T, He H. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small. 2009;5:2673–2677. doi: 10.1002/smll.200900621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zalewski M, Kulbacka J, Saczko J, Drag-Zalesinska M, Choromanska A. Valspodar-modulated chemotherapy in human ovarian cancer cells SK-OV-3 and MDAH-2774. Bosn J Basic Med Sci. 2019;19:234–241. doi: 10.17305/bjbms.2019.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baekelandt M, Lehne G, Tropé CG, Szántó I, Pfeiffer P, Gustavssson B, Kristensen GB. Phase I/II trial of the multidrug-resistance modulator valspodar combined with cisplatin and doxorubicin in refractory ovarian cancer. J Clin Oncol. 2001;19:2983–2993. doi: 10.1200/JCO.2001.19.12.2983. [DOI] [PubMed] [Google Scholar]
- 19.Gee ME, Faraahi Z, McCormick A, Edmondson RJ. DNA damage repair in ovarian cancer: Unlocking the heterogeneity. J Ovarian Res. 2018;11:50. doi: 10.1186/s13048-018-0424-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sengupta D, Mukhopadhyay A, Sengupta K. Emerging roles of lamins and DNA damage repair mechanisms in ovarian cancer. Biochem Soc Trans. 2020;48:2317–2333. doi: 10.1042/BST20200713. [DOI] [PubMed] [Google Scholar]
- 21.Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer. 2016;60:49–58. doi: 10.1016/j.ejca.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Christie EL, Bowtell DDL. Acquired chemotherapy resistance in ovarian cancer. Ann Oncol. 2017;28((Suppl 8)):viii13–viii15. doi: 10.1093/annonc/mdx446. [DOI] [PubMed] [Google Scholar]
- 23.Karakashev S, Fukumoto T, Zhao B, Lin J, Wu S, Fatkhutdinov N, Park PH, Semenova G, Jean S, Cadungog MG, et al. EZH2 inhibition sensitizes CARM1-high, homologous recombination proficient ovarian cancers to PARP Inhibition. Cancer Cell. 2020;37:157–167.e6. doi: 10.1016/j.ccell.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27:1449–1455. doi: 10.1093/annonc/mdw142. [DOI] [PubMed] [Google Scholar]
- 25.Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, Bowman-Colin C, Li Y, Greene-Colozzi A, Iglehart JD, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2:366–375. doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GR, IV, et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201. doi: 10.1038/nm1016-1192c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 2017;7:20–37. doi: 10.1158/2159-8290.CD-16-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alsop K, Fereday S, Meldrum C, DeFazio A, Emmanuel C, George J, Dobrovic A, Birrer MJ, Webb PM, Stewart C, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30:2654–2663. doi: 10.1200/JCO.2011.39.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iyer S, Zhang S, Yucel S, Horn H, Smith SG, Reinhardt F, Hoefsmit E, Assatova B, Casado J, Meinsohn MC, et al. Genetically defined syngeneic mouse models of ovarian cancer as tools for the discovery of combination immunotherapy. Cancer Discov. 2021;11:384–407. doi: 10.1158/2159-8290.CD-20-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Domchek SM. Reversion mutations with clinical use of PARP inhibitors: Many genes, many versions. Cancer Discov. 2017;7:937–939. doi: 10.1158/2159-8290.CD-17-0734. [DOI] [PubMed] [Google Scholar]
- 31.Pietragalla A, Arcieri M, Marchetti C, Scambia G, Fagotti A. Ovarian cancer predisposition beyond BRCA1 and BRCA2 genes. Int J Gynecol Cancer. 2020;30:1803–1810. doi: 10.1136/ijgc-2020-001556. [DOI] [PubMed] [Google Scholar]
- 32.Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, Helman E, Radke MR, Say C, Vo LT, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9:210–219. doi: 10.1158/2159-8290.CD-18-0715. [DOI] [PubMed] [Google Scholar]
- 33.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, Kuiper MJ, Ho GY, Barker H, Jasin M, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7:984–998. doi: 10.1158/2159-8290.CD-17-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He YJ, Meghani K, Caron MC, Yang C, Ronato DA, Bian J, Sharma A, Moore J, Niraj J, Detappe A, et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature. 2018;563:522–526. doi: 10.1038/s41586-018-0670-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penson RT, Valencia RV, Cibula D, Colombo N, Leath CA, III, Bidziński M, Kim JW, Nam JH, Madry R, Hernández C, et al. Olaparib versus nonplatinum chemotherapy in patients with platinum-sensitive relapsed ovarian cancer and a germline BRCA1/2 mutation (SOLO3): A randomized phase III trial. J Clin Oncol. 2020;38:1164–1174. doi: 10.1200/JCO.19.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marzi L, Szabova L, Gordon M, Weaver Ohler Z, Sharan SK, Beshiri ML, Etemadi M, Murai J, Kelly K, Pommier Y. The indenoisoquinoline TOP1 inhibitors selectively target homologous recombination-deficient and schlafen 11-positive cancer cells and synergize with olaparib. Clin Cancer Res. 2019;25:6206–6216. doi: 10.1158/1078-0432.CCR-19-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan S, Xuan J, Brajanovski N, Tancock MRC, Madhamshettiwar PB, Simpson KJ, Ellis S, Kang J, Cullinane C, Sheppard KE, et al. The RNA polymerase I transcription inhibitor CX-5461 cooperates with topoisomerase 1 inhibition by enhancing the DNA damage response in homologous recombination-proficient high-grade serous ovarian cancer. Br J Cancer. 2021;124:616–627. doi: 10.1038/s41416-020-01158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yap TA, O'Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, Tunariu N, Perez-Lopez R, Rodrigues DN, Riisnaes R, et al. Phase I trial of first-in-class ATR Inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol. 2020;38:3195–3204. doi: 10.1200/JCO.19.02404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konstantinopoulos PA, Cheng SC, Wahner Hendrickson AE, Penson RT, Schumer ST, Doyle LA, Lee EK, Kohn EC, Duska LR, Crispens MA, et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:957–968. doi: 10.1016/S1470-2045(20)30180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakayama N, Nakayama K, Shamima Y, Ishikawa M, Katagiri A, Iida K, Miyazaki K. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer. 2010;116:2621–2634. doi: 10.1002/cncr.24987. [DOI] [PubMed] [Google Scholar]
- 42.Au-Yeung G, Lang F, Azar WJ, Mitchell C, Jarman KE, Lackovic K, Aziz D, Cullinane C, Pearson RB, Mileshkin L, et al. Selective targeting of cyclin E1-Amplified high-grade serous ovarian cancer by cyclin-dependent kinase 2 and AKT Inhibition. Clin Cancer Res. 2017;23:1862–1874. doi: 10.1158/1078-0432.CCR-16-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Etemadmoghadam D, Weir BA, Au-Yeung G, Alsop K, Mitchell G, George J, Australian Ovarian Cancer Study Group. Davis S, D'Andrea AD, Simpson K, et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc Natl Acad Sci USA. 2013;110:19489–19494. doi: 10.1073/pnas.1314302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campbell GJ, Hands EL, Van de Pette M. The Role of CDKs and CDKIs in murine development. Int J Mol Sci. 2020;21:5343. doi: 10.3390/ijms21155343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Angius G, Tomao S, Stati V, Vici P, Bianco V, Tomao F. Prexasertib, a checkpoint kinase inhibitor: From preclinical data to clinical development. Cancer Chemother Pharmacol. 2020;85:9–20. doi: 10.1007/s00280-019-03950-y. [DOI] [PubMed] [Google Scholar]
- 46.Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, Medvedev S, Kinose Y, Devins K, Verma P, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun. 2020;11:3726. doi: 10.1038/s41467-020-17127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gorski JW, Ueland FR, Kolesar JM. CCNE1 amplification as a predictive biomarker of chemotherapy resistance in epithelial ovarian cancer. Diagnostics (Basel) 2020;10:279. doi: 10.3390/diagnostics10050279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gralewska P, Gajek A, Marczak A, Rogalska A. Participation of the ATR/CHK1 pathway in replicative stress targeted therapy of high-grade ovarian cancer. J Hematol Oncol. 2020;13:39. doi: 10.1186/s13045-020-00874-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garsed DW, Alsop K, Fereday S, Emmanuel C, Kennedy CJ, Etemadmoghadam D, Gao B, Gebski V, Garès V, Christie EL, et al. Homologous recombination DNA repair pathway disruption and retinoblastoma protein loss are associated with exceptional survival in high-grade serous ovarian cancer. Clin Cancer Res. 2018;24:569–580. doi: 10.1158/1078-0432.CCR-17-1621. [DOI] [PubMed] [Google Scholar]
- 50.da Costa AABA, do Canto LM, Larsen SJ, Ribeiro ARG, Stecca CE, Petersen AH, Aagaard MM, de Brot L, Baumbach J, Baiocchi G, et al. Genomic profiling in ovarian cancer retreated with platinum based chemotherapy presented homologous recombination deficiency and copy number imbalances of CCNE1 and RB1 genes. BMC Cancer. 2019;19:422. doi: 10.1186/s12885-019-5622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi M, Whorton AE, Sekulovski N, Paquet M, MacLean JA, Song Y, Van Dyke T, Hayashi K. Inactivation of TRP53, PTEN, RB1, and/or CDH1 in the ovarian surface epithelium induces ovarian cancer transformation and metastasis. Biol Reprod. 2020;102:1055–1064. doi: 10.1093/biolre/ioaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dall'Acqua A, Sonego M, Pellizzari I, Pellarin I, Canzonieri V, D'Andrea S, Benevol S, Sorio R, Giorda G, Califano D, et al. CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol Med. 2017;9:1415–1433. doi: 10.15252/emmm.201607012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giacomini I, Ragazzi E, Pasut G, Montopoli M. The pentose phosphate pathway and its involvement in cisplatin resistance. Int J Mol Sci. 2020;21:937. doi: 10.3390/ijms21030937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morandi A, Indraccolo S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim Biophys Acta Rev Cancer. 2017;868:1–6. doi: 10.1016/j.bbcan.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 55.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Butler EB, Zhao Y, Muñoz-Pinedo C, Lu J, Tan M. Stalling the engine of resistance: Targeting cancer metabolism to overcome therapeutic resistance. Cancer Res. 2013;73:2709–2717. doi: 10.1158/0008-5472.CAN-12-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 58.Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis-the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65:3981–3999. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist Updat. 2018;38:1–11. doi: 10.1016/j.drup.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Ai Z, Lu Y, Qiu S, Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016;373:36–44. doi: 10.1016/j.canlet.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ricci F, Brunelli L, Affatato R, Chilà R, Verza M, Indraccolo S, Falcetta F, Fratelli M, Fruscio R, Pastorelli R, Damia G. Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts. Ther Adv Med Oncol. 2019;11:1758835919839543. doi: 10.1177/1758835919839543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Urpilainen E, Puistola U, Boussios S, Karihtala P. Metformin and ovarian cancer: The evidence. Ann Transl Med. 2020;8:1711. doi: 10.21037/atm-20-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim TH, Suh DH, Kim MK, Song YS. Metformin against cancer stem cells through the modulation of energy metabolism: Special considerations on ovarian cancer. Biomed Res Int. 2014;2014:132702. doi: 10.1155/2014/132702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 65.Wu X, Han LY, Zhang XX, Wang L. The study of Nrf2 signaling pathway in ovarian cancer. Crit Rev Eukaryot Gene Expr. 2018;28:329–336. doi: 10.1615/CritRevEukaryotGeneExpr.2018020286. [DOI] [PubMed] [Google Scholar]
- 66.Gentric G, Kieffer Y, Mieulet V, Goundiam O, Bonneau C, Nemati F, Hurbain I, Raposo G, Popova T, Stern MH, et al. PML-regulated mitochondrial metabolism enhances chemosensitivity in human ovarian cancers. Cell Metab. 2019;29:156–173.e10. doi: 10.1016/j.cmet.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lister A, Nedjadi T, Kitteringham NR, Campbell F, Costello E, Lloyd B, Copple IM, Williams S, Owen A, Neoptolemos JP, et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol Cancer. 2011;10:37. doi: 10.1186/1476-4598-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Wijst MG, Huisman C, Mposhi A, Roelfes G, Rots MG. Targeting Nrf2 in healthy and malignant ovarian epithelial cells: Protection versus promotion. Mol Oncol. 2015;9:1259–1273. doi: 10.1016/j.molonc.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hou D, Liu Z, Xu X, Liu Q, Zhang X, Kong B, Wei JJ, Gong Y, Shao C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018;17:99–111. doi: 10.1016/j.redox.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kleih M, Böpple K, Dong M, Gaissler A, Heine S, Olayioye MA, Aulitzky WE, Essmann F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019;10:851. doi: 10.1038/s41419-019-2081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Podratz JL, Knight AM, Ta LE, Staff NP, Gass JM, Genelin K, Schlattau A, Lathroum L, Windebank AJ. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol Dis. 2011;41:661–668. doi: 10.1016/j.nbd.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: Possible role in apoptosis. Clin Cancer Res. 2006;12:5817–5825. doi: 10.1158/1078-0432.CCR-06-1037. [DOI] [PubMed] [Google Scholar]
- 73.Zhang J, Yang L, Xiang X, Li Z, Qu K, Li K. A panel of three oxidative stress-related genes predicts overall survival in ovarian cancer patients received platinum-based chemotherapy. Aging (Albany NY) 2018;10:1366–1379. doi: 10.18632/aging.101473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verschoor ML, Singh G. Ets-1 regulates intracellular glutathione levels: Key target for resistant ovarian cancer. Mol Cancer. 2013;12:138. doi: 10.1186/1476-4598-12-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilson LA, Yamamoto H, Singh G. Role of the transcription factor Ets-1 in cisplatin resistance. Mol Cancer Ther. 2004;3:823–832. [PubMed] [Google Scholar]
- 76.Nwani NG, Condello S, Wang Y, Swetzig WM, Barber E, Hurley T, Matei D. A Novel ALDH1A1 inhibitor targets cells with stem cell characteristics in ovarian cancer. Cancers (Basel) 2019;11:502. doi: 10.3390/cancers11040502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 78.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–738. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 79.Carnero A, Garcia-Mayea Y, Mir C, Lorente J, Rubio IT, LLeonart ME. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat Rev. 2016;49:25–36. doi: 10.1016/j.ctrv.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 80.Maugeri-Saccà M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17:4942–4947. doi: 10.1158/1078-0432.CCR-10-2538. [DOI] [PubMed] [Google Scholar]
- 81.Hu L, McArthur C, Jaffe RB. Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br J Cancer. 2010;102:1276–1283. doi: 10.1038/sj.bjc.6605626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, Yan PS, Huang TH, Nephew KP. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–4320. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Zhao G, Condello S, Huang H, Cardenas H, Tanner EJ, Wei J, Ji Y, Li J, Tan Y, et al. Frizzled-7 identifies platinum-tolerant ovarian cancer cells susceptible to ferroptosis. Cancer Res. 2021;81:384–399. doi: 10.1158/0008-5472.CAN-20-1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen J, Cao X, An Q, Zhang Y, Li K, Yao W, Shi F, Pan Y, Jia Q, Zhou W, et al. Inhibition of cancer stem cell like cells by a synthetic retinoid. Nat Commun. 2018;9:1406. doi: 10.1038/s41467-018-03877-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Muñoz-Galván S, Felipe-Abrio B, Verdugo-Sivianes EM, Perez M, Jiménez-García MP, Suarez-Martinez E, Estevez-Garcia P, Carnero A. Downregulation of MYPT1 increases tumor resistance in ovarian cancer by targeting the Hippo pathway and increasing the stemness. Mol Cancer. 2020;19:7. doi: 10.1186/s12943-020-1130-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Keyvani V, Farshchian M, Esmaeili SA, Yari H, Moghbeli M, Nezhad SK, Abbaszadegan MR. Ovarian cancer stem cells and targeted therapy. J Ovarian Res. 2019;12:120. doi: 10.1186/s13048-019-0588-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T, Huang Z, Bentley RC, Mori S, Fujii S, et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene. 2009;28:209–218. doi: 10.1038/onc.2008.374. [DOI] [PubMed] [Google Scholar]
- 88.Cole AJ, Iyengar M, Panesso-Gómez S, O'Hayer P, Chan D, Delgoffe GM, Aird KM, Yoon E, Bai S, Buckanovich RJ. NFATC4 promotes quiescence and chemotherapy resistance in ovarian cancer. JCI Insight. 2020;5:e131486. doi: 10.1172/jci.insight.131486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D, Ginestier C, Johnston C, Kueck A, Reynolds RK, et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011;71:3991–4001. doi: 10.1158/0008-5472.CAN-10-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li SS, Ma J, Wong AST. Chemoresistance in ovarian cancer: Exploiting cancer stem cell metabolism. J Gynecol Oncol. 2018;29:e32. doi: 10.3802/jgo.2018.29.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deng J, Bai X, Feng X, Ni J, Beretov J, Graham P, Li Y. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer. 2019;19:618. doi: 10.1186/s12885-019-5824-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brown JR, Chan DK, Shank JJ, Griffith KA, Fan H, Szulawski R, Yang K, Reynolds RK, Johnston C, McLean K, et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight. 2020;5:e133247. doi: 10.1172/jci.insight.133247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bogani G, Lopez S, Mantiero M, Ducceschi M, Bosio S, Ruisi S, Sarpietro G, Guerrisi R, Brusadelli C, Dell'Acqua A, et al. Immunotherapy for platinum-resistant ovarian cancer. Gynecol Oncol. 2020;158:484–488. doi: 10.1016/j.ygyno.2020.05.681. [DOI] [PubMed] [Google Scholar]
- 94.Nelson BH. The impact of T-cell immunity on ovarian cancer outcomes. Immunol Rev. 2008;222:101–116. doi: 10.1111/j.1600-065X.2008.00614.x. [DOI] [PubMed] [Google Scholar]
- 95.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 96.Shen GH, Ghazizadeh M, Kawanami O, Shimizu H, Jin E, Araki T, Sugisaki Y. Prognostic significance of vascular endothelial growth factor expression in human ovarian carcinoma. Br J Cancer. 2000;83:196–203. doi: 10.1054/bjoc.2000.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen CA, Cheng WF, Lee CN, Chen TM, Kung CC, Hsieh FJ, Hsieh CY. Serum vascular endothelial growth factor in epithelial ovarian neoplasms: Correlation with patient survival. Gynecol Oncol. 1999;74:235–240. doi: 10.1006/gyno.1999.5418. [DOI] [PubMed] [Google Scholar]
- 98.Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 99.An Y, Yang Q. Tumor-associated macrophage-targeted therapeutics in ovarian cancer. Int J Cancer. 2021;149:21–30. doi: 10.1002/ijc.33408. [DOI] [PubMed] [Google Scholar]
- 100.Liang R, Chen X, Chen L, Wan F, Chen K, Sun Y, Zhu X. STAT3 signaling in ovarian cancer: A potential therapeutic target. J Cancer. 2020;11:837–848. doi: 10.7150/jca.35011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lamichhane P, Karyampudi L, Shreeder B, Krempski J, Bahr D, Daum J, Kalli KR, Goode EL, Block MS, Cannon MJ, Knutson KL. IL10 Release upon PD-1 blockade sustains immunosuppression in ovarian cancer. Cancer Res. 2017;77:6667–6678. doi: 10.1158/0008-5472.CAN-17-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wan C, Keany MP, Dong H, Al-Alem LF, Pandya UM, Lazo S, Boehnke K, Lynch KN, Xu R, Zarrella DT, et al. Enhanced efficacy of simultaneous PD-1 and PD-L1 immune checkpoint blockade in high-grade serous ovarian cancer. Cancer Res. 2021;81:158–173. doi: 10.1158/0008-5472.CAN-20-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kalim M, Iqbal Khan MS, Zhan J. Programmed cell death ligand-1: A dynamic immune checkpoint in cancer therapy. Chem Biol Drug Des. 2020;95:552–566. doi: 10.1111/cbdd.13677. [DOI] [PubMed] [Google Scholar]
- 104.Constantinidou A, Alifieris C, Trafalis DT. Targeting programmed cell death −1 (PD-1) and Ligand (PD-L1): A new era in cancer active immunotherapy. Pharmacol Ther. 2019;194:84–106. doi: 10.1016/j.pharmthera.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 105.Fraser M, Leung B, Jahani-Asl A, Yan X, Thompson WE, Tsang BK. Chemoresistance in human ovarian cancer: The role of apoptotic regulators. Reprod Biol Endocrinol. 2003;1:66. doi: 10.1186/1477-7827-1-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Janzen DM, Tiourin E, Salehi JA, Paik DY, Lu J, Pellegrini M, Memarzadeh S. An apoptosis-enhancing drug overcomes platinum resistance in a tumour-initiating subpopulation of ovarian cancer. Nat Commun. 2015;6:7956. doi: 10.1038/ncomms8956. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 107.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, Deng J, Anderson KC, Richardson P, Tai YT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Baekelandt M, Kristensen GB, Nesland JM, Tropé CG, Holm R. Clinical significance of apoptosis-related factors p53, Mdm2, and Bcl-2 in advanced ovarian cancer. J Clin Oncol. 1999;17:2061. doi: 10.1200/JCO.1999.17.7.2061. [DOI] [PubMed] [Google Scholar]
- 109.Baekelandt M, Holm R, Nesland JM, Tropé CG, Kristensen GB. Expression of apoptosis-related proteins is an independent determinant of patient prognosis in advanced ovarian cancer. J Clin Oncol. 2000;18:3775–3781. doi: 10.1200/JCO.2000.18.22.3775. [DOI] [PubMed] [Google Scholar]
- 110.Binju M, Amaya-Padilla MA, Wan G, Gunosewoyo H, Suryo Rahmanto Y, Yu Y. Therapeutic inducers of apoptosis in ovarian cancer. Cancers (Basel) 2019;11:1786. doi: 10.3390/cancers11111786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zervantonakis IK, Iavarone C, Chen HY, Selfors LM, Palakurthi S, Liu JF, Drapkin R, Matulonis U, Leverson JD, Sampath D, et al. Systems analysis of apoptotic priming in ovarian cancer identifies vulnerabilities and predictors of drug response. Nat Commun. 2017;8:365. doi: 10.1038/s41467-017-00263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reles A, Wen WH, Schmider A, Gee C, Runnebaum IB, Kilian U, Jones LA, El-Naggar A, Minguillon C, Schönborn I, et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res. 2001;7:2984–2997. [PubMed] [Google Scholar]
- 113.Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee MJ, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018;19:207–215. doi: 10.1016/S1470-2045(18)30009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chui MH, Momeni Boroujeni A, Mandelker D, Ladanyi M, Soslow RA. Characterization of TP53-wildtype tubo-ovarian high-grade serous carcinomas: Rare exceptions to the binary classification of ovarian serous carcinoma. Mod Pathol. 2021;34:490–501. doi: 10.1038/s41379-020-00648-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lavarino C, Pilotti S, Oggionni M, Gatti L, Perego P, Bresciani G, Pierotti MA, Scambia G, Ferrandina G, Fagotti A, et al. p53 gene status and response to platinum/paclitaxel-based chemotherapy in advanced ovarian carcinoma. J Clin Oncol. 2000;18:3936–3945. doi: 10.1200/JCO.2000.18.23.3936. [DOI] [PubMed] [Google Scholar]
- 116.Jones S, Wang TL, Shih IeM, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Jr, Vogelstein B, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Guan B, Wang TL, Shih IeM. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71:6718–6727. doi: 10.1158/0008-5472.CAN-11-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, Zhai Y, Kossenkov AV, Vara-Ailor A, Rauscher FJ, III, et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol. 2017;19:962–973. doi: 10.1038/ncb3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Saha S, Panigrahi DP, Patil S, Bhutia SK. Autophagy in health and disease: A comprehensive review. Biomed Pharmacother. 2018;104:485–495. doi: 10.1016/j.biopha.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 121.Hu YL, Jahangiri A, Delay M, Aghi MK. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012;72:4294–4299. doi: 10.1158/0008-5472.CAN-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yu H, Su J, Xu Y, Kang J, Li H, Zhang L, Yi H, Xiang X, Liu F, Sun L. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur J Cancer. 2011;47:1585–1594. doi: 10.1016/j.ejca.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 123.Zhang Y, Cheng Y, Ren X, Zhang L, Yap KL, Wu H, Patel R, Liu D, Qin ZH, Shih IM, Yang JM. NAC1 modulates sensitivity of ovarian cancer cells to cisplatin by altering the HMGB1-mediated autophagic response. Oncogene. 2012;31:1055–1064. doi: 10.1038/onc.2011.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang Z, Zhou L, Chen Z, Nice EC, Huang C. Stress management by autophagy: Implications for chemoresistance. Int J Cancer. 2016;139:23–32. doi: 10.1002/ijc.29990. [DOI] [PubMed] [Google Scholar]
- 125.Follo C, Cheng Y, Richards WG, Bueno R, Broaddus VC. Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol Carcinog. 2018;57:319–332. doi: 10.1002/mc.22757. [DOI] [PubMed] [Google Scholar]
- 126.He J, Yu JJ, Xu Q, Wang L, Zheng JZ, Liu LZ, Jiang BH. Downregulation of ATG14 by EGR1-MIR152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophagy. Autophagy. 2015;11:373–384. doi: 10.1080/15548627.2015.1009781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shteingauz A, Boyango I, Naroditsky I, Hammond E, Gruber M, Doweck I, Ilan N, Vlodavsky I. Heparanase enhances tumor growth and chemoresistance by promoting autophagy. Cancer Res. 2015;75:3946–3957. doi: 10.1158/0008-5472.CAN-15-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ashrafizadeh M, Zarrabi A, Orouei S, Kiavash Hushmandi, Hakimi A, Amirhossein Zabolian, Daneshi S, Samarghandian S, Baradaran B, Najafi M. MicroRNA-mediated autophagy regulation in cancer therapy: The role in chemoresistance/chemosensitivity. Eur J Pharmacol. 2021;892:173660. doi: 10.1016/j.ejphar.2020.173660. [DOI] [PubMed] [Google Scholar]
- 129.Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, Chen X, Vellano CP, Jeong KJ, Ng PK, et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med. 2017;9:eaal5148. doi: 10.1126/scitranslmed.aal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Aksamitiene E, Kiyatkin A, Kholodenko BN. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem Soc Trans. 2012;40:139–146. doi: 10.1042/BST20110609. [DOI] [PubMed] [Google Scholar]
- 132.Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 134.Choi HJ, Heo JH, Park JY, Jeong JY, Cho HJ, Park KS, Kim SH, Moon YW, Kim JS, An HJ. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol Oncol. 2019;153:135–148. doi: 10.1016/j.ygyno.2019.01.012. [DOI] [PubMed] [Google Scholar]
- 135.Kim MJ, Lee SJ, Ryu JH, Kim SH, Kwon IC, Roberts TM. Combination of KRAS gene silencing and PI3K inhibition for ovarian cancer treatment. J Control Release. 2020;318:98–108. doi: 10.1016/j.jconrel.2019.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gupta S, Nag S, Aggarwal S, Rauthan A, Warrier N. Maintenance therapy for recurrent epithelial ovarian cancer: Current therapies and future perspectives-a review. J Ovarian Res. 2019;12:103. doi: 10.1186/s13048-019-0579-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lorusso PM, Edelman MJ, Bever SL, Forman KM, Pilat M, Quinn MF, Li J, Heath EI, Malburg LM, Klein PJ, et al. Phase I study of folate conjugate EC145 (Vintafolide) in patients with refractory solid tumors. J Clin Oncol. 2012;30:4011–4016. doi: 10.1200/JCO.2011.41.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Naumann RW, Coleman RL, Burger RA, Sausville EA, Kutarska E, Ghamande SA, Gabrail NY, Depasquale SE, Nowara E, Gilbert L, et al. PRECEDENT: A randomized phase II trial comparing vintafolide (EC145) and pegylated liposomal doxorubicin (PLD) in combination versus PLD alone in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2013;31:4400–4406. doi: 10.1200/JCO.2013.49.7685. [DOI] [PubMed] [Google Scholar]
- 139.Moore KN, Martin LP, O'Malley DM, Matulonis UA, Konner JA, Perez RP, Bauer TM, Ruiz-Soto R, Birrer MJ. Safety and activity of mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: A Phase I expansion study. J Clin Oncol. 2017;35:1112–1118. doi: 10.1200/JCO.2016.69.9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Holmes D. Ovarian cancer: Beyond resistance. Nature. 2015;527:S217. doi: 10.1038/527S217a. [DOI] [PubMed] [Google Scholar]
- 141.Huber D, Seitz S, Kast K, Emons G, Ortmann O. Use of oral contraceptives in BRCA mutation carriers and risk for ovarian and breast cancer: A systematic review. Arch Gynecol Obstet. 2020;301:875–884. doi: 10.1007/s00404-020-05458-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.