

Abstract
Dominant mutations in ubiquitously-expressed tRNA synthetase genes cause axonal peripheral neuropathy, accounting for at least six forms of Charcot-Marie-Tooth (CMT) disease. Genetic evidence in mouse and Drosophila models suggests a gain-of-function mechanism. Here, we used in vivo, cell-type-specific transcriptional and translational profiling to show that mutant tRNA synthetases activate the integrated stress response (ISR) through the sensor kinase GCN2. The chronic activation of the ISR contributed to the pathophysiology, and genetic deletion or pharmacological inhibition of Gcn2 alleviated the peripheral neuropathy. The activation of GCN2 by tRNA synthetase mutations suggests that their activity is still related to translation and that inhibiting GCN2 or the ISR may represent a therapeutic strategy in CMT.
One Sentence Summary:
Activation of GCN2 and the integrated stress response contributes to mutant tRNA synthetase-associated neurodegeneration.
In humans, dominant mutations in at least six aminoacyl tRNA-synthetases (aaRSs) cause the specific degeneration of peripheral motor and sensory axons (1, 2). A shared underlying mechanism is possible (3), and mouse studies support a neomorphic activity of mutant aaRSs (4–6). Impaired translation is attractive as a candidate disease mechanism. Overexpression of mutant glycyl- or tyrosyl-tRNA synthetase (GARS or YARS, respectively) in Drosophila causes decreased translation, independent of their normal tRNA charging activity (7). However, whether decreased translation is an effect of a neomorphic activity or secondary to cell stress, and whether cell stress contributes to disease have not been investigated.
To evaluate translation in vivo, we performed fluorescent non-canonical amino acid-tagging (FUNCAT, (8, 9)) in motor neurons of two mouse models of Charcot-Marie-Tooth (CMT) type 2D, caused by dominant mutations in Gars (Figures S1 and S2) (4, 10). Translation is impaired in motor neurons in both models. In pre-disease-onset mice, impairments in translation were present, and correlated with onset of neurodegeneration (Figure S2). Translation was not impaired in the liver or heart of mutant Gars mice using puromycin labeling (Figure S2).
To further examine changes in transcription and translation in motor neurons, we performed RiboTagging using Chat-Cre to activate the system in cholinergic neurons (11). We validated this system using spinal cord samples from wild-type mice by confirming an enrichment of known motor neuron markers and depletion of non-neuronal genes (Figure S3), as well as upregulation of known markers of axotomy such as Sprr1a, Atf3, and Npy (12–14) after unilateral sciatic nerve injury (Figure S3, and Supplementary Data S1).
We next compared ribosome-associated mRNA from Gars+/+ spinal cord to that from mutant GarsC201R/+ or GarsP278KY/+ spinal cord. GarsC201R/+ samples contain 1,976 upregulated and 126 downregulated transcripts and GarsP278KY/+ show 603 upregulated and 233 downregulated transcripts (Figure 1, A–C). Many of the same highly upregulated transcripts, including Fibroblast growth factor 21 (Fgf21), growth differentiation factor 15 (Gdf15), and corneodesmosin (Cdsn), were found in both genotypes. We also performed RNA sequencing from whole spinal cord in mutant Gars mice, including a third humanized CMT2D model, GarsΔETAQ/+ (6). Upregulated transcripts were similar between RiboTagging and whole spinal cord RNA sequencing experiments, and among all three mouse models (Figure 1 D, E, Jaccard similarity indices 0.12–0.16, p<0.002 across RNAseq experiments). The upregulated transcripts in mutant Gars datasets did not overlap with the upregulated transcripts after sciatic nerve injury, or with results from a mitofusin-2 mouse model of CMT2A (15). Only one differentially-expressed gene in the GarsC201R/+ whole spinal cord overlapped with an equivalent experiment in a mouse model of Ighmbp2-associated spinal muscular atrophy (16). We also performed RiboTagging and RNA sequencing from pre-disease-onset mutant Gars mice and found early upregulation of a subset of the same transcripts (Figure S3). Ingenuity Pathway Analysis of upregulated genes indicated forms of cell stress and activation of the integrated stress response (ISR) (Figure 1F). The ISR-associated transcription factor Atf4 and several known ATF4 targets were among the upregulated transcripts in our RiboTagging data (Figure 1C). We confirmed activation of the ISR using immunolabeling of phospho-eIF2α, which was elevated in all three Gars alleles, and was specific to a subset of spinal motor neurons (Figure 1G, H and Figure S4). These data indicate the ISR is activated in spinal motor neurons, this upregulation occurs prior to overt neuropathy symptoms, and this is seen in three Gars alleles.
Figure 1. Differential gene expression in Gars/CMT2D mice.

(A) 1976 up- and 126 down-regulated ribosome associated mRNAs were found in GarsC201R/+ motor neurons compared to Gars+/+. (B) 603 up- and 233 down-regulated ribosome-associated mRNAs were found in GarsP278KY/+ motor neurons compared to Gars+/+. (C) 174 upregulated genes were found in common between GarsC201R/+ and GarsP278KY/+, six genes relevant to subsequent studies are noted. (D) The intersections of upregulated gene lists from whole spinal cord RNAseq in three different dominant alleles of Gars. (E) The upregulated genes shared in RiboTagging data overlap the union upregulated gene list of whole spinal cord RNAseq. (F) Ingenuity Pathway Analysis of upregulated ribosome-associated mRNAs with log FC≥1.5 and FDR<.05 in GarsP278KY/+ motor neurons. (G) Activation of the ISR was confirmed with immunolabeling of phospho-eIF2α, which was found specifically in ventral horn motor neurons (arrowheads) in GarsP278KY/+ mutant spinal cord (right hand panel). (H) The ratio of phospho-eIF2α labelling intensity to that of the motor neuron marker ChAT is increased in all three alleles of Gars, and in GarsP278KY/+ compared to GarsC201R/+, points represent individual motor neurons, n=3 mice/genotype. Volcano plots show transcripts with log FC>|1.5|; P<.05. N = 4–6 mice (in A-C) or 6 mice (3 males, 3 females in D) per genotype at 8-weeks-of-age.
To address the cell type-specificity within the spinal cord, we examined five of the top upregulated ATF4-target genes using RNAscope in situ hybridization. Chat was used as a marker of motor neurons, which include Spp1-positive alpha motor neurons (~70%), and Spp1- negative gamma motor neurons (~30%) (Figure 2A) (17, 18). Robust and specific upregulation of all five transcripts was confirmed in Chat-positive cells in the ventral horn in mutant Gars motor neurons (Figure 2 B–D and figure S5). Approximately 70% of GarsP278KY/+ motor neurons expressed the ATF4 target genes (Figure 2E and figure S5). ISR activation was selective to alpha motor neurons because Spp1-positive cells were also Fgf21-postive, whereas Fgf21-negative cells were Spp1-negative, (Figure 2 F, G, H).
Figure 2: ATF4 target gene expression is restricted to alpha motor neurons within mutant Gars spinal cord.

(A) Motor neurons in the ventral horn of the spinal cord include alpha- (Spp1-positive) and gamma- (Spp1-negative) populations, but only Spp1-positive cells express Fgf21 in mutant mice. (B) RNAscope in situ hybridization (ISH) showed upregulation of the ATF4 target gene Fgf21 in a subset of ventral horn motor neurons in GarsΔETAQ/+ mice. (C) ISH in spinal cord of GarsP278KY/+ mice. Chat (red), Gdf15 (green), Fgf21 (yellow). Some Chat-positive mutant Gars motor neurons do not show expression of disease-related transcripts (arrows) (D) Quantification of background-subtracted ISH fluorescence intensity. ***p<.001, ****p<.0001 (two-tailed Student’s T-test, N=4 mice per genotype at 8 weeks of age.) (E) Percentage of GarsP278KY/+ motor neurons that expressed the 5 disease-associated transcripts. (F) RNAscope ISH in spinal cord with Chat and Spp1 revealed the same percentage of alpha- and gamma motor neurons in mutant mice. (G, H) Only Spp1-positive alpha motor neurons in mutant spinal cords express Fgf21. Arrows in (H) point to cells labeled with Chat that do not express Spp1 or Fgf21. N=3 mice per genotype at 8 weeks of age in F-H.
We next examined a related mouse model of dominant intermediate CMT type C, carrying a disease-associated knockin mutation in tyrosyl tRNA-synthetase, YarsE196K (19) (Figure S6). We identified activation of the ISR in alpha motor neurons in this model at 7-months-of-age by phospho-eIF2α labeling and by in situ hybridization for the same five ATF4 target genes. This was observed in YarsE196K/E196K, and to a lesser extent, YarsE196K/+ mice (Figure S7). Whole spinal cord RNAseq also indicated that ATF4 target genes were upregulated in YarsE196K/E196K mice (Figure S8, Jaccard similarity index 0.064, p<0.002, YarsE196K/E196K with the union of Gars gene lists), although the signal visible in YarsE196K/+ motor neurons by in situ hybridization was not detectable, which was confirmed by qRT-PCR (Figure S8).
Peripheral sensory neurons were examined for ISR activation by probing for upregulation of the ATF4 target genes in lumbar dorsal root ganglia (DRGs, Figure S9). Robust upregulation of Fgf21 was observed in a subset of mutant Gars DRG neurons, especially medium-large neurons, co-labeled with Nefh (20). We included parvalbumin (Pvalb) as an additional marker (21), and subsets of both mechanosensitive (Nefh+, Pvalb−) and proprioceptive (Nefh+, Pvalb+) sensory neurons expressed Fgf21 (Figure S9). Thus, the ISR was activated specifically in alpha motor neurons and in a subset of sensory neurons.
The ISR is activated by four discrete kinases, which all phosphorylate eIF2α to suppress cap-dependent translation and activate ATF4 target gene expression (22). ISR activation through GCN2 kinase is observed with impairments in translation and ribosome stalling (23, 24). We tested for the involvement of GCN2 by breeding GarsP278KY/+ mice into a Gcn2 knockout background (Gcn2KO/KO). The GarsP278KY/+ mice have a disease onset at ~2 weeks-of-age and actively lose motor and sensory axons until about 2 months-of-age, followed by very slow progression (Fig 3A) (4). We assayed body weight, grip strength and endurance using a wire hang test, and performed neurophysiology and histopathological examination in a cohort of mice aged to 8-weeks and in a second aged to 16-weeks. The homozygous removal of Gcn2 significantly alleviated neuropathy in GarsP278KY/+ mice. The neuropathy started, but the progression was prevented, not merely delayed, as demonstrated by the improvement in phenotypes at 8 weeks-of-age (Figure S10), which was even greater by 16 weeks (Figure 3 B–F and Figure S11). At 16-weeks-of-age, body weights and motor performance of mutant Gars mice lacking GCN2 (Gcn2KO/KO;GarsP278KY/+) were restored to nearly Gars+/+ levels. Gcn2KO/KO;GarsP278KY/+ mice also had improved sciatic motor nerve conduction velocity, no significant motor axon loss, larger diameter motor axons, and less denervation at the neuromuscular junction (NMJ) compared to mutant Gars mice with GCN2 (Gcn2+/+ or +/KO;GarsP278KY/+). There was no expression of ATF4 target genes in motor neurons of Gcn2KO/KO ;GarsP278KY/+ mice by in situ hybridization or RNA sequencing (Figure 3G and figure S11). Upon removal of Gcn2 only 85 genes were differentially expressed between Gars+/+ and GarsP278KY/+ mice (Data S1 and Figure S11), which no longer included the ATF4 target genes and included many pseudogenes. IPA suggested that these 85 genes show a translation signature. Thus, genetic deletion of Gcn2 alleviates the ISR and neuropathy.
Figure 3. Deletion of Gcn2 alleviates the GarsP278KY/+ phenotype.
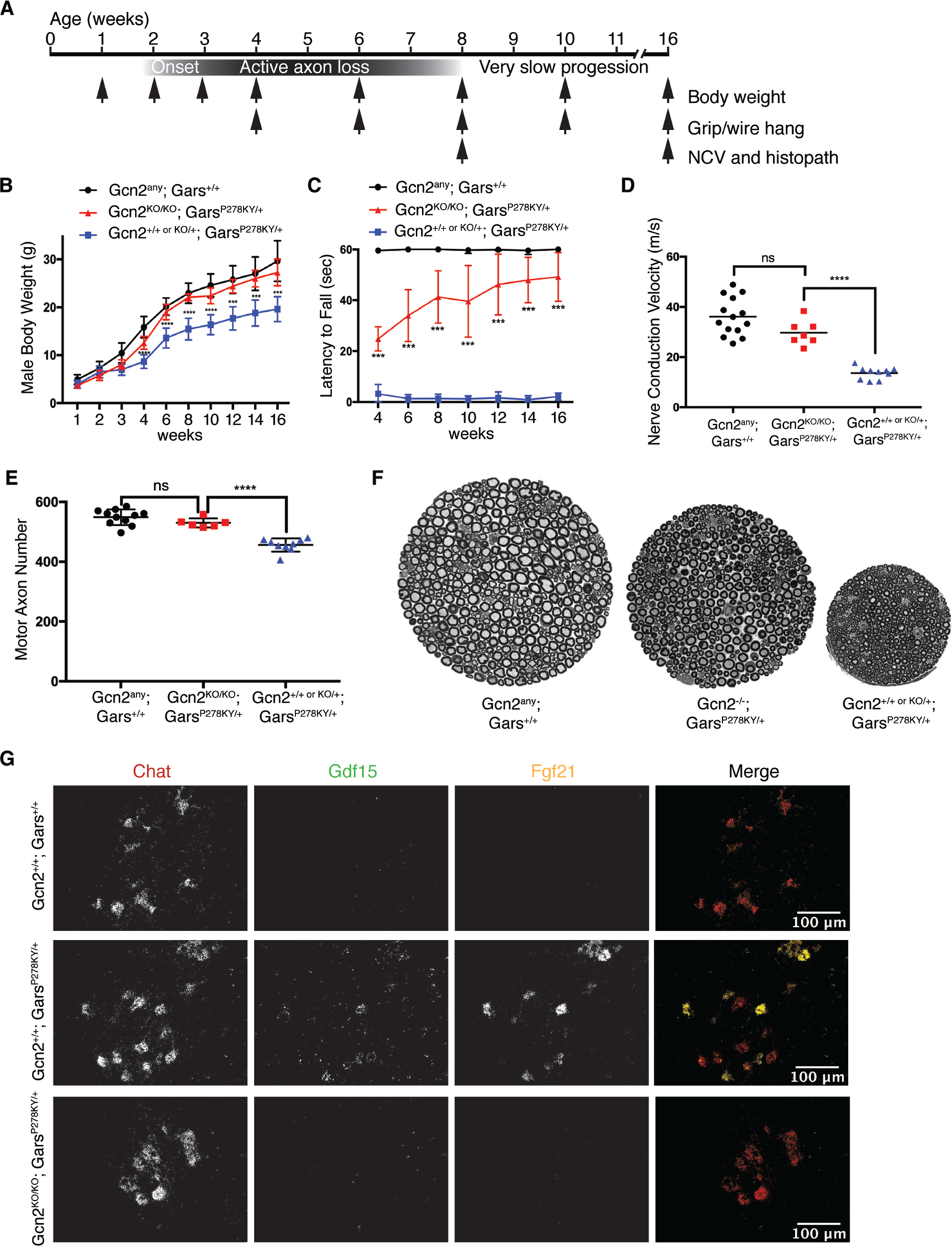
(A) GarsP278KY/+ mice have a phenotype by 2-weeks-of-age, and actively lose peripheral axons until ~8-weeks-of-age, after which the phenotype progresses very slowly. (B) Body weight of male Gcn2KO/KO;GarsP278KY/+ mice is significantly increased over Gcn2+/+ or +/KO;GarsP278KY/+ (one-way ANOVA, Gcn2any denotes +/+, +/KO, or KO/KO genotype). (C) Gcn2KO/KO;GarsP278KY/+ mice have increased latency to fall from an inverted wire grid (one-way ANOVA). The test is stopped after 60 seconds and the mean of 3 trials is reported for each day. Body weight and wire hang analyses performed with 18–22 mice per grouped genotype. (D) Motor nerve conduction velocity of the sciatic nerve is increased in Gcn2KO/KO;GarsP278KY/+ mice compared to Gcn2+/+ or +/−;GarsP278KY/+, and no longer different from Gars+/+ regardless of Gcn2 genotype (one-way ANOVA). (E) The average number of motor axons in the femoral nerve of 16-week-old Gcn2KO/KO;GarsP278KY/+ mice is greater compared to Gcn2+/+ or +/−;GarsP278KY/+. Gcn2KO/KO;GarsP278KY/+ axon counts are no longer different from Gars+/+ mice regardless of Gcn2 genotype (one-way ANOVA). (F) The motor branch of the femoral nerve in Gcn2KO/KO;GarsP278KY/+ mice is intermediate in size compared to Gars+/+ and Gcn2+/+ or +/−;GarsP278KY/+. (G) Gcn2+/+;Gars+/+motor neurons show no expression of ATF4 target genes Gdf15 (green) or Fgf21 (yellow), while Gcn2+/+;GarsP278KY/+ motor neurons show robust upregulation. ATF4 target gene expression is eliminated in Gcn2KO/KO;GarsP278KY/+ motor neurons. Data points in D and E represent individual mice of both sexes.
In a more translational experiment, we treated GarsΔETAQ/+ mutant and littermate control mice with a GCN2-inhibitor (GCN2iB) (25). Treatment began at disease-onset at 2-weeks-of-age and continued for three weeks (Figure 4A). GCN2iB improved body weight, motor performance, and sciatic nerve conduction velocity, although improvement in compound muscle action potential (CMAP) amplitude did not reach significance (Figure 4B–E). A decrement in CMAP amplitude with repetitive nerve stimulation, indicative of synaptic transmission problems at the NMJ (26), was also improved (Figure 4F). The effects of GCN2iB were seen in both sexes, but were of greater magnitude in males (Figure S12). GCN2iB effectively reduced ISR activation, assayed by phospho-eIF2α labeling and by qRT-PCR for ATF4 target genes (Figure 4G and Figure S13), with a larger effect in males than females.
Figure 4. Pharmacological inhibition of GCN2 is efficacious.

(A) Study design: Cohorts of GarsΔETAQ/+ or littermate control (Gars+/+) mice were treated with GCN2iB or vehicle starting at 2-weeks-of-age for three weeks, 8–9 mice per sex per genotype per treatment group were enrolled. (B) GCN2iB improved body weight in GarsΔETAQ/+ male mice. (C) GCN2iB improved strength and endurance in the wire hang test. (D) GCN2iB improved sciatic motor nerve conduction velocity in GarsΔETAQ/+ mice, although values were still reduced compared to controls. (E) Compound muscle action potential amplitude was not significantly improved over vehicle-treated mutant mice. (F) The decrement in CMAP amplitude following bouts of repetitive nerve stimulation was alleviated in GarsΔETAQ/+ mice with GCN2iB treatment. Data from male and female mice is pooled in C-F, although improvement was generally greater in males. (G) Expression of the ATF4 target gene B4galnt2 was reduced with GCN2iB treatment; effects were greater in males. Statistical analysis: B, C, F, G were tested with 2-way ANOVA and correction for multiple comparisons, D, E were tested with 1-way ANOVA and correction for multiple comparisons. Asterisks in B, C, F indicate significance between GCN2iB-treated GarsΔETAQ/+ and vehicle-treated Gars+/+ mice, and the pairwise comparisons noted in D, E and G.
We tested for the presence of the secreted ATF4 targets FGF21 and GDF15 in the blood of neuropathy patients with mutations in GARS, alanyl-ARS (AARS), YARS, and tryptophanyl-ARS (WARS). Mutant Gars mice have elevated levels of FGF21 in blood (Figure S14). Neuropathy patients had highly variable levels of FGF21; however, GDF15 was significantly increased in both aaRS patients and in CMT1A patients. The cellular source of the circulating GDF15 is unknown, but it may serve as a biomarker of neuropathy.
Our findings indicate that neuropathy-causing mutations in aaRSs activate the ISR through GCN2 in alpha motor and a subset of sensory neurons, and that inhibiting GCN2 may be an effective therapeutic strategy. The activation of GCN2 may be explained by translation defects downstream of the sequestration of tRNAs by the mutant synthetases (27). However, the highly specific activation of the ISR remains puzzling, and is not explained by codon bias, as we did not find evidence of this in motor neuron RiboTagging data (Figure S15). Furthermore, we cannot distinguish whether the ISR exacerbates neuropathy through a defect in translation that is compounded by eIF2α phosphorylation, which further decreases translation, or whether expression of ATF4 target genes in peripheral neurons is deleterious. However, the residual phenotype is very mild when the ISR is blocked.
Data and materials availability:
Animal models, reagents and protocols are available upon request, the GarsC201R mice were generated Harwell, UK. Data are presented here and in supplementary materials. Sequencing data is available through the GEO database (28). Code for codon usage analysis is available at https://github.com/cgob/codon_usage/ (29).
Supplementary Material
Acknowledgments:
We would like to thank the patients who participated in this study, as well as the Scientific Services at the Jackson Laboratory and Drs. Alex Rossor and Gregory Cox for input and assistance.
Funding:
RWB was supported by the National Institutes of Health (R37 NS054154, R01 NS113583, U54 OD020351) and the Paul E. Kelly Foundation. ELS was supported by NIH F31 NS100328. MES and MR were supported by NIH U54 NS065712-08. MR is also supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. EMS received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (#743216). ES was supported by the Muscular Dystrophy Association (MDA479773), the EU Joint Programme - Neurodegenerative Disease Research (JPND; ZonMW 733051075 (TransNeuro) and ZonMW 733051073 (LocalNMD)), an ERC consolidator grant (ERC-2017-COG 770244) and the Radala Foundation for ALS Research. RH is a Wellcome Trust Investigator (109915/Z/15/Z), supported by the Medical Research Council (UK) (MR/N025431/1 and MR/V009346/1), the European Research Council (309548), the Wellcome Trust Pathfinder Scheme (201064/Z/16/Z), the Newton Fund (MR/N027302/1), the Evelyn Trust (19/14), the Lily Foundation, an MRC strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) MR/S005021/1, and by the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Footnotes
Competing interests: RWB is on the scientific board of the Hereditary Neuropathy Foundation, RWB, MR and MES are on the scientific board of the Charcot-Marie-Tooth Association. ELS and RWB are co-inventors on the pending patent “GCN2 inhibitors to treat peripheral neuropathy.”
References and notes:
- 1.Storkebaum E, Bioessays 38, 818–829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonellis A, Green ED, Annu Rev Genomics Hum Genet 9, 87–107 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Wei N, Zhang Q, Yang XL, The Journal of biological chemistry 294, 5321–5339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW, Neuron 51, 715–726 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Motley WW et al. , PLoS Genet 7, e1002399 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morelli KH et al. , J Clin Invest 129, 5568–5583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niehues S et al. , Nat Commun 6, 7520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dieterich DC et al. , Nature protocols 2, 532–540 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Alvarez-Castelao B et al. , Nature biotechnology 35, 1196–1201 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Achilli F et al. , Disease models & mechanisms 2, 359–373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanz E et al. , Proceedings of the National Academy of Sciences of the United States of America 106, 13939–13944 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Starkey ML et al. , The Journal of comparative neurology 513, 51–68 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linda H, Skold MK, Ochsmann T, Frontiers in neurology 2, 30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Meister B, Elde R, Verge VM, Hokfelt T, Eur J Neurosci 5, 1510–1519 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Zhou Y et al. , J Clin Invest 130, 1756–1771 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox GA, Mahaffey CL, Frankel WN, Neuron 21, 1327–1337 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Stifani N, Front Cell Neurosci 8, 293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Misawa H et al. , J Neurosci Res 90, 732–742 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Jordanova A et al. , Nature genetics 38, 197–202 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Ferri GL et al. , Brain Res 515, 331–335 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Le Pichon CE, Chesler AT, Front Neuroanat 8, 21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pakos-Zebrucka K et al. , EMBO Rep 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishimura R, Nagy G, Dotu I, Chuang JH, Ackerman SL, eLife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inglis AJ et al. , Proceedings of the National Academy of Sciences of the United States of America 116, 4946–4954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura A et al. , Proceedings of the National Academy of Sciences of the United States of America 115, E7776–E7785 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spaulding EL et al. , The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 3254–3267 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuko A et al. , Science, (submitted).
- 28.GEO accession number GSE145006.
- 29.Codon usage analysis. available at https://github.com/cgob/codon_usage/.
- 30.Ishimura R et al. , Science 345, 455–459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Copeland NG, Jenkins NA, Court DL, Nat Rev Genet 2, 769–779 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Lewandoski M, Meyers EN, Martin GR, Cold Spring Harb Symp Quant Biol 62, 159–168 (1997). [PubMed] [Google Scholar]
- 33.tom Dieck S et al. , Nat Methods 12, 411–414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schindelin J et al. , Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Salzberg SL, Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B, Dewey CN, BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson MD, McCarthy DJ, Smyth GK, Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson MD, Oshlack A, Genome Biol 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Paramita P, Choi KP, Karuturi RK, Biol Direct 6, 27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munger SC et al. , Genetics 198, 59–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker EJ, Jay JJ, Bubier JA, Langston MA, Chesler EJ, Nucleic acids research 40, D1067–1076 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scherer SS et al. , The Journal of neuroscience : the official journal of the Society for Neuroscience 25, 1550–1559 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomez CM et al. , The Journal of neuroscience : the official journal of the Society for Neuroscience 17, 4170–4179 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rafael JA, Nitta Y, Peters J, Davies KE, Mammalian genome : official journal of the International Mammalian Genome Society 11, 725–728 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.