

Abstract
TxtE is a cytochrome P450 (CYP) homologue that mediates the nitric oxide (NO)-dependent direct nitration of l-tryptophan (Trp) to form 4-nitro-l-tryptophan (4-NO2-Trp). A recent report showed evidence that TxtE activity requires NO to react with a ferric-superoxo intermediate. Given this minimal mechanism, it is not clear how TxtE avoids Trp hydroxylation, a mechanism that also traverses the ferric-superoxo intermediate. To provide insight into canonical CYP intermediates that TxtE can access, electron coupling efficiencies to form 4-NO2-Trp under single-or limited-turnover conditions were measured and compared to steady-state efficiencies. As previously reported, Trp nitration by TxtE is supported by the engineered self-sufficient variant, TB14, as well as by reduced putidaredoxin. Ferrous (FeII) TxtE exhibits excellent electron coupling (70%), which is 50-fold higher than that observed under turnover conditions. In addition, two- or four-electron reduced TB14 exhibits electron coupling (∼6%) that is 2-fold higher than that of one-electron reduced TB14 (3%). The combined results suggest (1) autoxidation is the sole TxtE uncoupling pathway and (2) the TxtE ferric-superoxo intermediate cannot be reduced by these electron transfer partners. The latter conclusion is further supported by ultraviolet−visible absorption spectral time courses showing neither spectral nor kinetic evidence for reduction of the ferric-superoxo intermediate. We conclude that resistance of the ferric-superoxo intermediate to reduction is a key feature of TxtE that increases the lifetime of the intermediate and enables its reaction with NO and efficient nitration activity.
Graphical Abstract
Cytochrome P450s (CYPs) are thiolate-ligated heme enzymes that are widespread among all kingdoms of life; more than 300000 CYP sequences have been identified.1 While most CYPs exhibit monooxygenase activities, several natural and engineered CYPs catalyze a diverse range of nitrogen chemistries, including N-hydroxylations, nitrene transfers to promote amination or cyclization, reductive decomposition of nitramine functionalities in explosives, and nitrations.2–10 The canonical CYP monooxygenase mechanism has been intensely studied. However, less is understood about the mechanisms of CYP activities involving nitrogen, such as nitration. A better understanding of such mechanisms would be valuable for the development of engineered nitration biocatalysts.11–15
TxtE is a CYP homologue that catalyzes the direct nitration of l-tryptophan (Trp) to produce 4-nitro-l-tryptophan (4-NO2-Trp) using nitric oxide (NO) and O2 as co-substrates. This enzyme was discovered in the biosynthetic pathway for thaxtomins, virulence factors of the tuber disease scab, in Streptomyces scabies.16–20 More specifically, thaxtomin A inhibits cellulose production.21 The diketopiperazine core of thaxtomin A is formed from coupling 4-NO2-Trp with l-phenylalanine by two nonribosomal peptide synthetases (TxtA and TxtB).22 A tailoring enzyme, TxtC, completes the biosynthesis. The stereochemistry of the diketopiperazine core and the presence of the nitro group are critical for thaxtomin A’s virulence activity.20 A bacterial nitric oxide synthase (bNOS), TxtD, oxidizes l-arginine (Arg) to form l-citrulline (Cit) and the necessary NO for the direct nitration reaction by TxtE:23–25
| (1) |
A similar NO-dependent nitration pathway was found to be required for rufomycin biosynthesis by Streptomyces atratus.26 In this pathway, the enzyme RufO is a CYP homologue that nitrates l-tyrosine (Tyr) to the rufomycin precursor 3-nitro-l-tyrosine (3-NO2-Tyr). In addition to these native nitration systems, a self-sufficient TxtE fusion called TB14 has been engineered by the Ding lab.13,14 This variant links TxtE with the reductase domain of CYP102A1 at the C-terminus with a 14-amino acid linker, allowing for Trp nitration turnover without the need for separate electron transfer (ET) partners [e.g., spinach ferredoxin (Fd) and ferredoxin:NADP+ reductase (FNR)].
Early insight into the TxtE nitration mechanism was provided by 18O2 labeling studies that showed one oxygen atom of the nitro group of 4-NO2-Trp originates from O2.4 The second nitro oxygen atom likely originates from NO.23 Several mechanisms consistent with these early observations have been proposed, and each invoked FeIII-OONO as an intermediate.4,27,28 More recent work provided evidence for the feasibility of the proposed FeIII-OONO intermediate.29 The accumulated evidence of Louka et al. supports an ordered reaction mechanism in which the O2 binds to the ferrous (FeII) TxtE to form a ferric-superoxo intermediate (Figure 1). This intermediate was shown to rapidly react with NO to regenerate ferric (FeIII) TxtE. Their results support a pathway in which NO reacts with the ferric-superoxo intermediate to form the FeIII-OONO intermediate. Calculations of feasible Trp nitration pathways resulting from the FeIII-OONO intermediate that accounts for the observed regiospecificity were presented.
Figure 1.
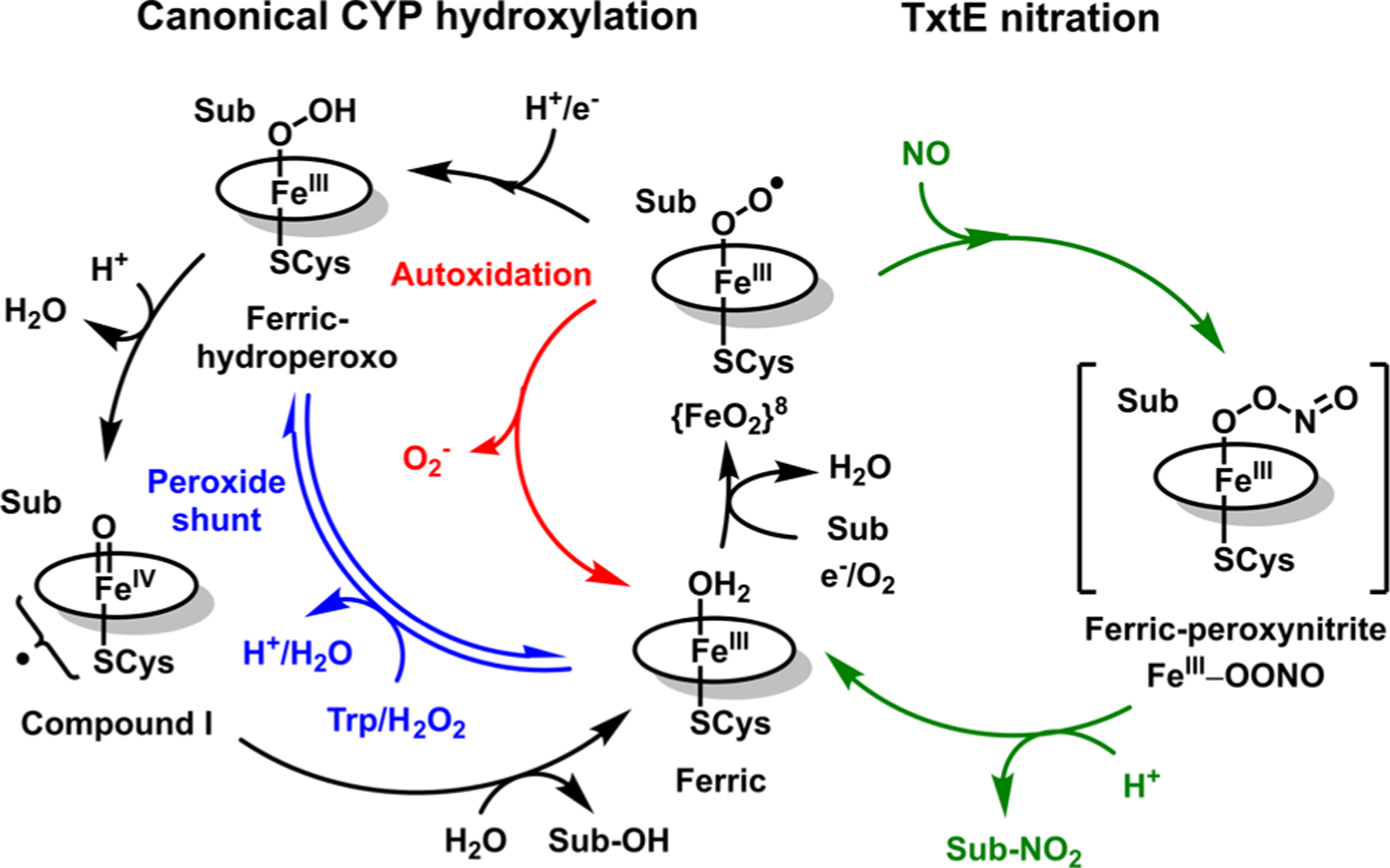
Reaction mechanism for canonical substrate (Sub) hydroxylation by CYPs (left cycle) and the minimal mechanism for nitration by the CYP homologue TxtE (right cycle).
The TxtE nitration mechanism and the hydroxylation mechanism by canonical CYPs both traverse a ferric-superoxo intermediate (Figure 1). In canonical CYPs, the ferric-superoxo intermediate is reduced en route to substrate hydroxylation. In principle, TxtE should exhibit poor efficiency for nitration turnover because the two pathways should compete for the same ferric-superoxo intermediate. Indeed, both TxtE and TB14 exhibit poor electron coupling during turnover (2.4% and 5.3%, respectively).13,14 In addition, low 3-NO2-Tyr yields by RufO strongly suggest a poor electron coupling efficiency for this nitration system.5 However, the hydroxylated product has not been observed,4 so the poor coupling efficiency is due to other reasons.
The poor electron coupling of both TxtE and RufO during nitration turnover suggests that the observed uncoupling is a side effect related to the mechanisms of their unique nitration activities. Therefore, identifying the TxtE uncoupling pathways will provide insight into the TxtE nitration mechanism. Uncoupling pathways have been well established in CYPs and their variants to result from nonproductive dissociation of superoxide (O2 −) or hydrogen peroxide (H2O2 ) from on-pathway CYP intermediates. For reference within this paper, the canonical CYP catalytic cycle and some typical autoxidation pathways are summarized in Figure 1. For further discussion of the catalytic cycle and these pathways, we direct the reader to several thorough reviews.2,30–32 Important to this report are two typical CYP uncoupling pathways: (1) the autoxidation pathway, in which superoxide (O2 −) is dissociated from the ferric-superoxo intermediate to regenerate the high-spin FeIII CYP (Figure 1, red pathway), and (2) the peroxide shunt, from which diffusible hydrogen peroxide (H2O2) and the FeIII CYP are generated from the FeIII-OOH intermediate (Figure 1, blue pathway).
In the following, we characterize TxtE uncoupling pathways to probe the canonical CYP intermediates accessible to the enzyme. Electron coupling efficiencies of TxtE and TB14 under limited-turnover conditions show these conditions are highly efficient compared to steady-state conditions. The combined results suggest that the only TxtE uncoupling pathway is autoxidation of the TxtE ferric-superoxo intermediate. This observation also suggests that the TxtE ferric-superoxo intermediate is resistant to reduction and cannot access more reduced intermediates, such as a ferric-(hydro)peroxo species. Kinetic and spectral data from stopped-flow ultraviolet−visible (UV−vis) absorption time courses monitoring the lifetime of the TxtE ferric-superoxo intermediate in the presence of excess reducing equivalents are consistent with this conclusion. Our results suggest this resistance of ferric-superoxo to reduction is a key feature of TxtE that enables efficient nitration by avoiding competing hydroxylation activity. These results highlight the high evolutionary versatility of CYPs.
METHODS
Materials and General Protocols.
The plasmid for putidaredoxin expression (pPdX) was obtained from AddGene (plasmid 85084). PdX was purified and expressed as previously described.33 DEA- and PROLI-NONOates were purchased from Cayman Chemicals. DEA and PROLI-NONOate stocks were dissolved in 10 mM NaOH and quantified by measuring the absorbance at 250 (ε250 = 6500 M−1 cm−1) and 252 nm (ε = 8400 M−1 cm−1), respectively. General reagents and medium components were purchased from Fisher Scientific or VWR. Isopropyl β-d-1-thiogalactopyranoside (IPTG) and 5-amino-levulinic acid (5-ALA) were purchased from GoldBio. Water used for all solutions was of 18.2 MΩ cm resistivity from a Barnstead Nanopure system (Thermo Fisher Scientific). Solvents for LC-MS experiments were of at least HPLC grade and contained 0.3% (v/v) formic acid.
TxtE Expression and Purification.
TxtE and TB14 were expressed in Escherichia coli Lemo21 (DE3) or BL21 (DE3) using a modified protocol previously reported.13 Briefly, pTxtE was grown at 37 °C in 4 × 1 L of Terrific Broth (TB) cultures containing 50 μg/mL kanamycin and 30 μg/mL chloramphenicol (Lemo21 only) to an OD of 0.6−1.0. At this time, expression was induced with 1 mM IPTG and supplemented with 100 mg each of (NH4)5[Fe(C6H4O7)2] [ferric ammonium citrate (FAC)] and 5-aminolevulinic acid (5-ALA). The induced cultures were incubated at 21 °C for 40 h before the cells were harvested. As previously described, cells were lysed and protein was purified from the lysate.
Preparation of LC-MS Samples.
Samples containing 100 μM TxtE and 500 μM Trp were titrated with sodium dithionite in an anaerobic glovebox until the 412 nm UV−vis absorbance of FeII TxtE no longer increased. An aliquot of the PROLI-NONOate stock in 10 mM NaOH was pipetted onto the cap of a microcentrifuge tube containing the FeII TxtE sample. The samples were removed from the box, exposed to air, capped, and mixed by inversion to simultaneously introduce O2 into the FeII TxtE and to initiate NO generation by the PROLI-NONOate. Samples were incubated at 21 °C for 30 min. Samples were centrifuged in 30 kDa molecular weight cutoff Microcon centrifugal filters (Millipore) and filtered using 0.45 μM syringe filters to remove TxtE and particulates prior to HPLC analysis. Samples containing FeIII TxtE and PdXred were prepared in a similar fashion. Samples of PdXred were prepared by titration with a dithionite solution. The reduction was monitored by the disappearance of peaks in the 400−450 region and the appearance of a new peak at 550 nm.34 Samples of TB14 were prepared in a similar fashion except with 10−40 μM NADPH (0.5, 1.0, and 2.0 equiv of NADPH per protein monomer) as the electron source.
General LC-MS Protocols.
LC-MS analysis was performed using an Agilent 1260 LC stack connected to an Agilent 6230 TOF mass spectrometer with electrospray ionization (ESI). Analyses used a gradient of water (solvent A) and acetonitrile (solvent B) for the following times; percentages are with respect to solvent B: 1% from 0 to 1 min, 1% to 20% from 1 to 8 min, 20% to 99% from 8 to 10 min, 99% from 10 to 12 min, and 1% from 12 to 13 min. The flow rate was set at 1.0 mL/min. The mass spectrometer was run in negative ion mode with a probe voltage of 4500 V and a fragmentation voltage of 175 V. To monitor Trp, Trp-OH, and NO2-Trp, extracted ion chromatograms were obtained with ions at m/z 203 (203.0180), m/z 219 (219.0793), and m/z 248 (247.9937), respectively.
The quantitation of 4-NO2-Trp was performed with a Shimadzu (Kyoto, Japan) Prominence UHPLC system with an Agilent Poroshell 120 EC-C18 column (2.7 μm, 4.6 mm × 50 mm) equipped with a photodiode array (PDA) detector. The HPLC method included column equilibration first with 10% solvent B (acetonitrile with 0.1% formic acid) for 2 min and then with a linear gradient from 10% to 50% solvent B over 8 min, followed by another linear gradient from 50% to 99% solvent B over 5 min. The column was further cleaned with 99% solvent B for 3 min and then reequilibrated with 10% solvent B for 1 min. Solvent A consisted of water with 0.1% FA. The flow rate was set at 0.5 mL/min, and 4-NO2-Trp was detected at 380 nm.
Stopped-Flow Spectrophotometry.
Stopped-flow measurements were performed on an SX20 stopped-flow spectrophotometer (Applied Photophysics) equipped with a xenon arc lamp, monochromator, and a photodiode array detector. For anaerobic experiments, enzyme samples and reagents were prepared within a glovebox containing a nitrogen atmosphere. The stopped-flow lines were made anaerobic by flushing the drive syringes and mixing circuit with sodium dithionite and rinsed with deoxygenated working buffer [100 mM Tris (pH 8.0)] immediately before experimental samples were loaded. All single-mix experiments were mixed in a 1:1 (v/v) ratio. For sequential-mixing experiments, A and B lines were mixed at a 1:1 (v/v) ratio in the premix line. The premix line was then mixed at a 1:1 (v/v) ratio with the C line. Conditions after mixing are listed in the figure captions. Analytical fits of sums of exponents to single-wavelength data were performed using nonlinear regression in Origin 2018b (OriginLab). Global fitting of spectral time courses was performed using KinTek explorer (KinTek Corp.).
TxtE, TB14, and PdXred in 100 mM Tris (pH 8.0) were reduced by titration with dithionite as described for preparation of the LC-MS samples. Protein samples loaded into the stopped-flow instrument contained either FeII TxtE alone, FeII TxtE with PdXred, or TB144e‑red with Trp as noted. These samples were mixed against either degassed or air-saturated (∼260 μM O2) 100 mM Tris buffer (pH 8.0).
Sequential-mixing stopped flow was used to monitor the reaction of TxtE ferric-superoxo versus NO. A sample of FeII TxtE with Trp was prepared as described above. In the first mixing step, this solution was mixed with air-saturated (∼260 μM O2) working buffer and aged in the premix circuit for 1 s. After aging, this mixture was mixed with either 1.33 mM DEA NONOate or 1.2 mM buffered NO.
RESULTS
Verification That Trp Is Not Hydroxylated.
We first verified that the poor electron coupling observed by TxtE cannot be attributed to Trp hydroxylation. To this end, a series of TB14 LC-MS samples were prepared (Figure 2A). First, TB14 samples containing all of the components necessary for nitration turnover except for NO were prepared. These conditions are hereafter termed O2-turnover conditions. Under negative ion mode, LC-MS of these samples showed the presence of a compound with an ion at m/z 219 (Figure 2A and Figure S1), which is consistent with the [M − H]− ion for Trp-OH. The poor electron coupling exhibited by TB14 strongly suggests that uncoupled electrons are diverted from product formation to reduction of O2 to form diffusible H2O2 either directly or by disproportionation of O2−. Therefore, the observed hydroxylation could be H2O2-dependent. To test this hypothesis, the TB14 O2-turnover experiments were repeated in the presence of 5 μM catalase, a scavenger of H2O2. In these samples, no signal at m/z 219 was observed, indicating that Trp hydroxylation requires formation of diffusible H2O2, and therefore, hydroxylation is an off-pathway activity. This conclusion is also supported by the appearance of Trp-OH in TB14 samples containing H2O2 and Trp. However, this Trp-OH is also formed when free hemin or boiled TxtE was incubated with Trp and H2O2. Therefore, it is inconclusive if TxtE mediates the observed H2O2-dependent Trp hydroxylation. Regardless, these LC-MS data indicate that TxtE cannot hydroxylate Trp via O2 reduction, and consequently, electron uncoupling during TxtE nitration turnover cannot be attributed to Trp hydroxylation.
Figure 2.
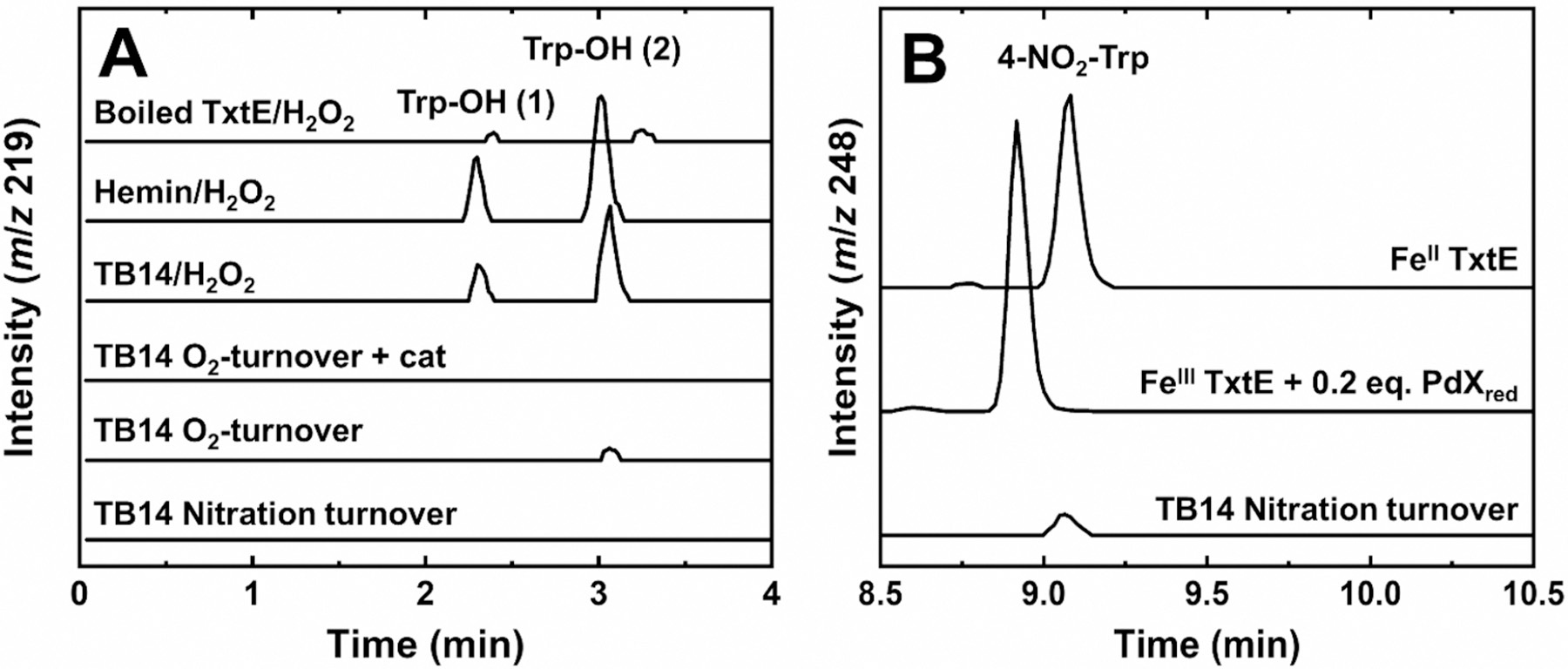
Representative LC-MS extracted ion chromatograms (EICs) of TxtE and TB14 reactions monitoring (A) Trp-OH (m/z 219) or (B) 4-NO2-Trp (m/z 248) formation. All reaction mixtures contained 500 μM Trp in 100 or 200 mM Tris buffer (pH 8.0) with additional components: 3 mM H2O2 and denatured TxtE (Boiled TxtE/H2O2); 5 μM hemin and 3 mM H2O2 (Hemin/H2O2); 5 μM TB14 and 5 mM H2O2 (TB14/H2O2); 5 μM TB14, 500 μM NADPH, and 5 μM catalase (TB14 O2-turnover + cat); 5 μM TB14 and 500 μM NADPH (TB14 O2-turnover); 0.5 μM TB14, 2 mM NADPH, and 1.33 mM DEA NONOate (TB14 Nitration turnover); 100 μM FeII TxtE and 0.9 mM PROLI-NONOate (FeIITxtE); and 100 μM FeIII TxtE, 200 μM PdXred, and 0.75 mM PROLI-NONOate (FeIIITxtE + 0.2 eq. PdXred). All samples were incubated at room temperature for 120 min (TB14 samples) or 30 min (all others) prior to analysis.
Nitration Observed by TxtE under Limited-Turnover Conditions.
Anaerobic samples containing 100 μM FeII TxtE and excess Trp were simultaneously exposed to air and PROLI-NONOate, an the NO generator. Analysis by LC-MS showed a peak in the extracted ion chromatogram at m/z 248. This result is consistent with the formation of 4-NO2-Trp and matches the peak observed in the LC-MS spectrum of TB14 steady-state nitration samples (Figure 2B). The observation of 4-NO2-Trp without the need for additional reducing equivalents indicates that only one electron is needed for TxtE nitration. This observation is consistent with recently reported data,29 and all proposed mechanisms, in which NO reacts with the ferric-superoxo intermediate.
Putidaredoxin is a well-characterized redox partner for CYP101A1 that could function as a redox partner for TxtE.35,36 The reduction potential of PdX is −240 mV versus an estimated reduction potential of 50 mV for CYP ferric-superoxo species.37,38 While PdX has not yet been reported to support TxtE activity, it has been shown to support RufO activity to form 3-NO2-Tyr.5 Furthermore, the TxtE crystal structure was shown to have a structurally homologous arginine residue needed to support ET between PdXred and CYP101A1.39 To test if PdX supported TxtE turnover, samples of 100 μM FeIII TxtE with 2 equiv of reduced PdX (PdXred) and excess Trp were simultaneously exposed to air and PROLI-NONOate. By LC-MS, the samples showed a peak at m/z 248 consistent with the formation of 4-NO2-Trp (Figure 2B). Because the reaction was initiated with FeIII TxtE, the results verify that PdXred can reduce TxtE to initiate the nitration reaction. In addition, the results indicate that PdXred supports nitration turnover by TxtE.
High Electron Coupling Efficiencies Observed under Limited-Turnover Conditions.
Electron uncoupling may occur by a noncanonical uncoupling pathway that is unique to the nitrogen-containing intermediates formed along the TxtE nitration pathway. For example, the putative FeIII-OONO intermediate proposed for TxtE is also a proposed intermediate for NO dioxygenase reactivity by hemoglobin and flavohemoglobin.40–42 The NO dioxygenase product is nitrate (NO3 −). Thus, a TxtE uncoupling pathway that diverts electrons to formation of NO3− instead of 4-NO2-Trp can be envisioned. To test for a unique TxtE pathway, we quantified the 4-NO2-Trp produced in single-turnover TxtE samples.
The single-turnover FeII TxtE samples discussed above (Figure 2) contained 70 ± 8 μM 4-NO2-Trp (Figures S2 and S3). Because nitration requires only one electron, electron efficiencies were calculated on a per reducing equivalent basis. By this calculation, TxtE under single-turnover conditions exhibited an electron coupling efficiency of 70 ± 8% (Table 1). For comparison, a single turnover of TxtE is 50- and 25-fold more efficient than steady-state turnover of TxtE (1.1%) and TB14 (2.3%), respectively.14 The high efficiency under single-turnover conditions strongly suggests against an uncoupling pathway after the NO reaction of the TxtE ferric-superoxo with the ferric-superoxo intermediate.
Table 1.
Electron Coupling Efficiencies for Limited- and Steady-State Nitration Turnover of TxtE and TB14
| condition | [4-NO2-Trp] (μM)a | RE (μM)b | coupling efficiency (%)c |
|---|---|---|---|
| FeIITxtEd | 70 (8) | 100 | 70 (8) |
| TB141e‑rede | 0.58 (0.02) | 20 | 2.9 (0.1) |
| TB142e‑rede | 2.4 (0.8) | 40 | 6.1 (1.9) |
| TB144e‑rede | 4.7 (0.8) | 80 | 6.0 (1.0) |
| TxtE steady state (Fd/FNR) | 1.1 (0.2)f | ||
| TB14 steady state | 2.3 (0.3)f |
Average of three trials; standard deviation reported in parentheses.
Reducing equivalents (RE).
Calculated as [4-NO2-Trp]/[reducing equivalents].
Reaction conditions: 100 μM FeII TxtE, 500 μM Trp, and 0.86 mM PROLI-NONOate in buffer (pH 8.0).
Reaction conditions: 20 μM TB14, 0.5 mM Trp, and 1 mM PROLI-NONOate in buffer (pH 8.0) with either 10 μM (TB141e‑red), 20 μM (TB142e‑red), or 40 μM (TB144e‑red) NADPH.
Converted to [4-NO2-Trp]/[reducing equivalents] from ref 14.
These results suggest that poor coupling under steady-state conditions is related to canonical CYP uncoupling pathways such as autoxidation or the peroxide shunt (Figure 1). The latter can be accessed only in the presence of excess electrons. To compare uncoupling in the absence and presence of excess electrons, 20 μM samples of one-electron (TB141e‑red), two-electron (TB142e‑red), or four-electron (TB144e‑red) reduced TB14 were treated with air and PROLI-NONOate. Quantitative analysis of these samples by HPLC showed that the TB14le-red, TB142e-red, and TB144e-red samples generated 0.58 ± 0.02, 2.4 ± 0.8, and 4.7 ± 0.8 μM 4-NO2-Trp, respectively (Table 1 and Figure S4). The calculated electron coupling efficiency for the TB141e‑red samples was 2.9 ± 0.1%. The TB142e‑red and TB144e‑red samples exhibited 6.1 ± 1.9% and 6.0 ± 1.0% electron coupling efficiencies, respectively, which are both 2-fold higher than that of the TB141e‑red sample. If TB142e-red or TB144e-red enabled reduction of the ferric-superoxo intermediate to the ferric-peroxo or –hydroperoxo species, then subsequent dissociation of H2O2 would be expected because TxtE cannot hydroxylate Trp. Therefore, TB142e‑red or TB144e‑red should exhibit decreased electron coupling efficiencies compared to that of TB141e‑red. Instead, the observed increase in coupling efficiency in the presence of additional reducing equivalents suggested that the ferric-superoxo intermediate is not reduced.
Kinetics of Autoxidation versus NO Reaction with the Ferric-Superoxo Intermediate.
If the ferric-superoxo intermediate is resistant to reduction, then the only accessible TxtE uncoupling pathway would be autoxidation. To test if ferric-superoxo autoxidation can kinetically compete with its reaction with NO, the intermediate decay kinetics were compared in the absence and presence of NO. First, the observed rate constant (kobs) for ferric-superoxo autoxidation was measured (Figure 3A). A reference spectrum of FeII TxtE was obtained by stopped-flow mixing an anaerobic solution of 10 μM FeII TxtE and 500 μM Trp with deoxygenated buffer. The resulting spectrum displayed absorbance features with maxima at 412 and 553 nm, consistent with previously reported spectra of FeII TxtE.29 An additional 318 nm peak in the spectrum was attributed to a small excess of dithionite in the protein samples. Mixing the FeII TxtE solution with air-saturated buffer resulted in the complete disappearance of the FeII TxtE absorption features within 2 ms. In its place, absorption features with maxima at 365, 425, and 561 nm were observed. These features are consistent with previously reported spectra for the TxtE ferric-superoxo intermediate.29 This intermediate spectrum decayed over several minutes to a species with spectral features centered at 402, 412, and 542 nm consistent with previously reported spectra for high-spin FeIII TxtE.4,11,14,29 Formation of the high-spin FeIII TxtE was expected due to the large excess of Trp used in these experiments.
Figure 3.
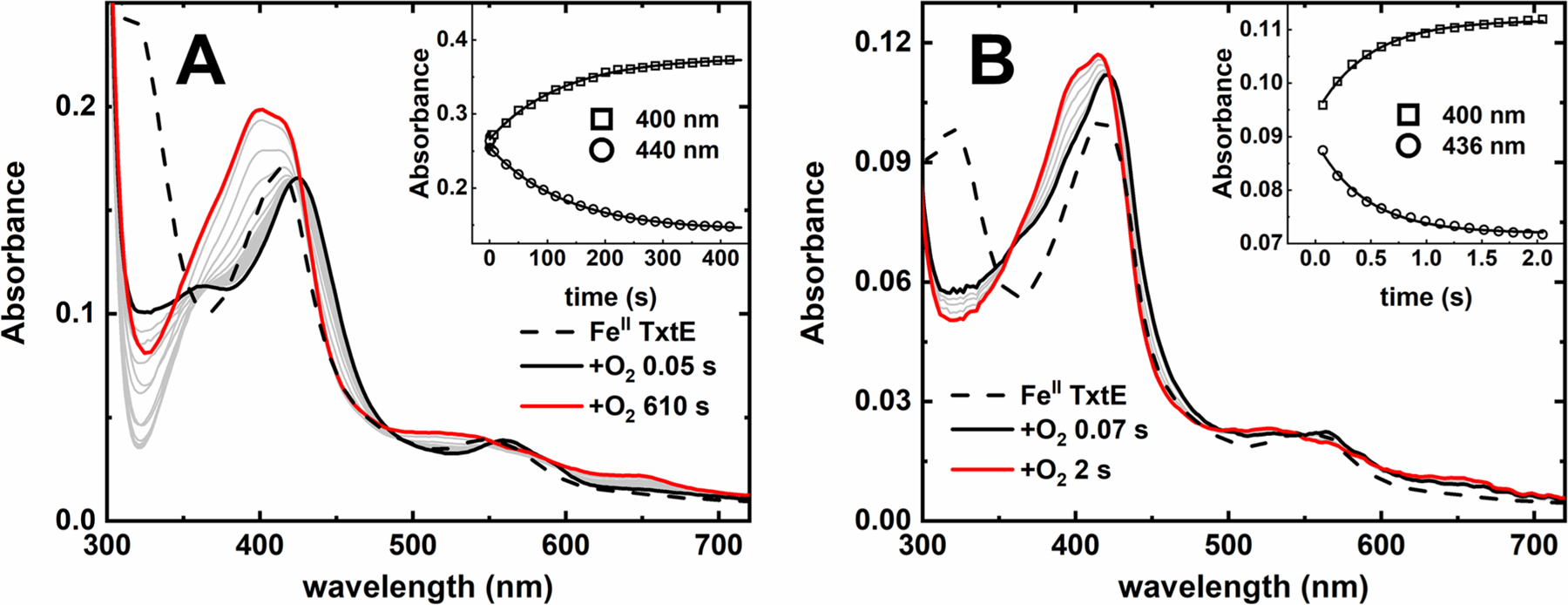
Stopped-flow spectral time courses of anaerobic FeII TxtE mixed with O2 in the (A) presence or (B) absence of Trp at pH 8.0. Dashed traces obtained by mixing an FeII TxtE solution against deoxygenated buffer. Solid black and red traces are the first and last collected spectra in the time courses, respectively. Gray traces were collected at intermediate times. The inset shows representative single-wavelength traces collected with low-intensity light time courses and fit with single-exponent functions. Final conditions: (A) 5 μM FeII TxtE, 250 μM Trp, and 130 μM O2 and (B) 5 μM FeII TxtE and 130 μM O2. All solutions were in 100 mM Tris (pH 8.0), and all reactions performed at 20 °C in a 1 cm path length cuvette.
Conversion of the TxtE ferric-superoxo intermediate to high-spin FeIII TxtE is consistent with autoxidation; however, this process should occur in one step and, therefore, the time course should exhibit isosbestic points. However, the time course in Figure 3A lacks isosbestic points. Meanwhile, time courses collected with low-intensity light exhibit clear isosbestic points (Figure S5). While these time course spectra exhibit more noise, the major spectral features are clearly consistent with conversion of the TxtE ferric-superoxo intermediate to the high-spin FeIII TxtE product and TxtE autoxidation with no additional processes. The additional kinetic phase observed in time courses collected with high-intensity light is likely a photochemical process unrelated to TxtE autoxidation. Single-wavelength traces monitoring decay of the TxtE ferric-superoxo intermediate (A442) and formation of high-spin FeIIITxtE (A400) collected with low-intensity light both were well fit to single-exponential functions (Figure 3A, inset) with a kobs of approximately 0.008 (0.001) s−1. This kobs was assigned as the TxtE autoxidation rate constant [kautox (Table 2)].
Table 2.
Rate Constants for TxtE and TB14
| condition | kautox (s−1)a | source |
|---|---|---|
| TxtE with Trp | 0.008 (0.001) | this work |
| TxtE with Trp (with PdXred) | 0.0072 (0.0015) | this work |
| TxtE without Trp | 3.7 (0.3) | this work |
| TB14 with Trp | 0.0064 (0.0006) | this work |
| kon (M−1 s−1) | source | |
| kon(O2) | 4.43 (0.03) × 106 | 29 |
| kon(NO) | 1.82 (0.03) × 106 | 29 |
Average of four to five trials; standard deviation reported in parentheses.
Omitting Trp increases the rate of TxtE autoxidation. Anaerobic mixing of FeII TxtE with air-saturated buffer (pH 8.0) (Figure 3B) resulted in a dead-time spectrum with absorption features centered at 365, 421, and 561 nm. This spectrum resembles the TxtE ferric-superoxo intermediate spectrum recorded in the presence of Trp (Figure 3A). The decay of this intermediate to a species with absorbance features centered at 400, 415, and 534 nm was complete within 2 s, which is consistent with high-spin FeIII TxtE. Several isosbestic points are observed in the time course, which is consistent with a one-kinetic step transition and assignment of the time course as autoxidation of TxtE. The A400 and A436 traces were both well fit to a single exponent with a kobs of 3.7 (0.3) s−1 (Figure 3B, inset, and Table 2). The stopped-flow UV−vis absorption analysis thus revealed that kautox of the TxtE ferric-superoxo intermediate is >400 times faster in the absence of Trp. This more rapid autoxidation in the absence of substrate is also observed for several other CYP homologues.43,44 It is unclear why in the absence of Trp, the protein decays to high-spin FeIII TxtE; however, the more rapid autoxidation observed in this experiment suggests that there is no Trp contamination in the experiment.
Sequential-mixing stopped flow was used to estimate the kobs of the TxtE ferric-superoxo intermediate reaction with NO under similar conditions employed in our LC-MS experiments (Figure 4). An anaerobic solution of 40 μM FeII TxtE with 1 mM Trp was prepared in buffer (pH 8.0). This FeII TxtE solution was mixed with air-saturated buffer in the premix circuit for 1 s to generate the ferric-superoxo intermediate. Next, this ferric-superoxo intermediate was mixed with buffered NO. The spectral features of the ferric-superoxo intermediate were not observed in the dead-time spectrum. Instead, the dead-time spectrum exhibited well-resolved absorbance features with maxima at 365, 436, 547, and 578 nm, consistent with the formation of a heme ferric-nitrosyl ({FeNO}6 in Enemark−Feltham notation45) species (Figure S6), within the stopped-flow dead time of 2 ms. The rapid loss of the ferric-superoxo intermediate is best explained by its rapid reaction with NO to form the putative FeIII-OONO, which is followed by 4-NO2-Trp formation and regeneration of FeIII TxtE (Figure 1). The observation of {FeNO}6 instead of high-spin FeIII TxtE is consistent with the high concentration of unreacted NO used for the experiment. The kobs for the ferric-superoxo reaction with NO at 20 °C was estimated to be >1700 s−1, more than 200000-fold faster than the TxtE kautox of 0.008 s−1.
Figure 4.
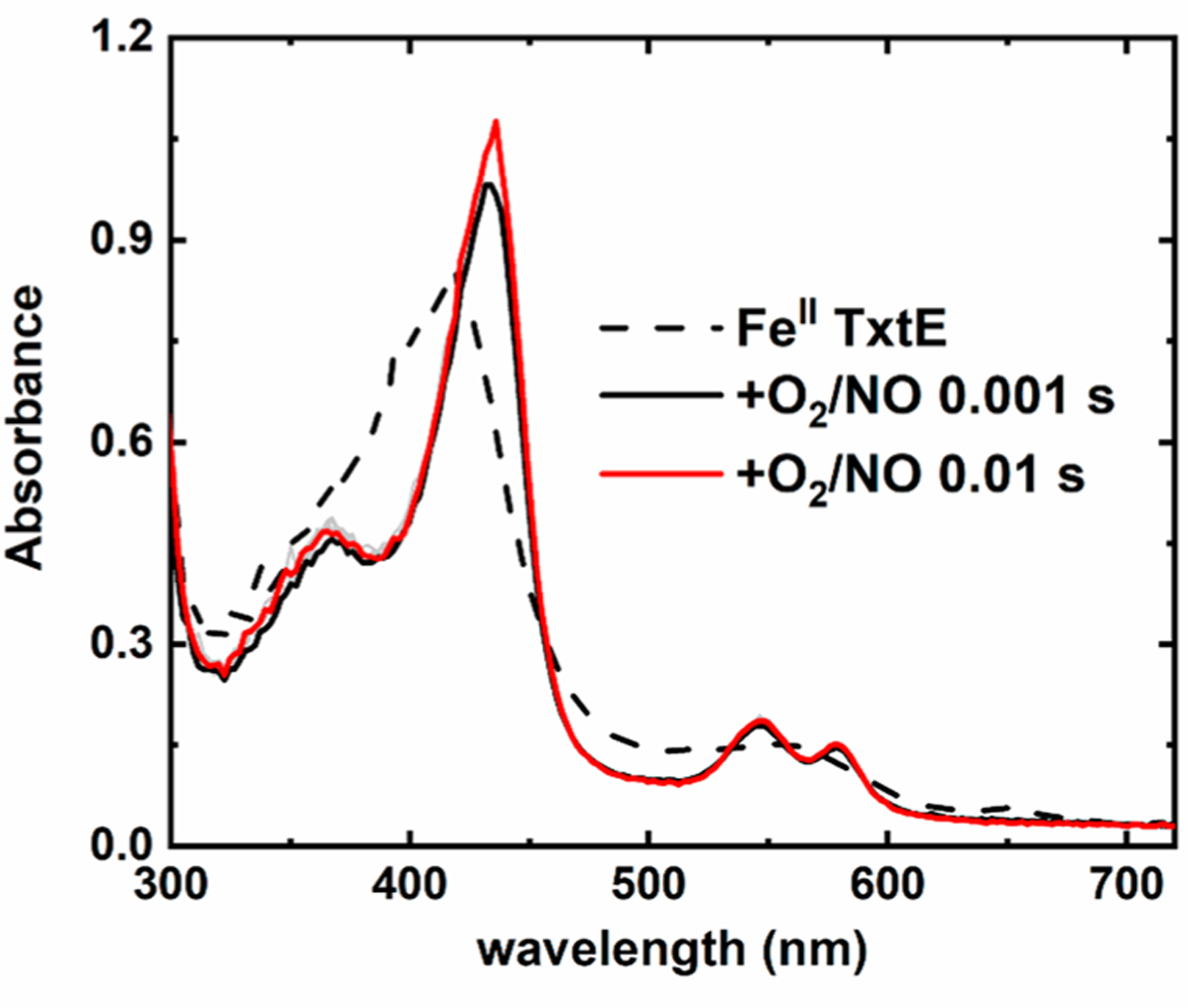
Stopped-flow sequential-mixing spectral time course of the TxtE ferric-superoxo intermediate vs NO over 10 ms at pH 8.0 and 20 °C. The dashed trace is the spectrum of FeII TxtE acquired from mixing FeII TxtE with deoxygenated buffer in both mixing steps. Conditions after stopped-flow mixing: 10 μM FeII TxtE, 250 μM Trp,70 μM O2, and 600 μM NO in 100 mM Tris (pH 8.0) at 20 °C in a 1 cm path length cuvette.
Stopped Flow of the TxtE Ferric-Superoxo Intermediate versus Excess Reducing Equivalents.
To provide further evidence that the ET components used in these studies cannot reduce the TxtE ferric-superoxo intermediate, stopped-flow UV−vis spectrophotometry was used to monitor the decay of the ferric-superoxo intermediate in the presence of PdXred (Figure 5A,B). An anaerobic solution containing FeII TxtE with 4 equiv of PdXred and excess Trp was mixed with deoxygenated buffer (pH 8.0). This resulting spectrum exhibited a shoulder at 408 nm and a peak at 551 nm, consistent with the spectrum for FeII TxtE with additional spectral contributions from PdXred. Mixing this protein solution with air-saturated buffer resulted in the immediate disappearance of the FeII TxtE spectrum and the appearance of features at 423 and 553 nm. These features are consistent with formation of the TxtE ferric-superoxo intermediate, as verified from the 4 ms minus PdXred difference spectrum (Figure 5A, inset). Thereafter, two phases were observed. The first phase exhibited broad absorbance increases across the 300−700 nm region (Figure 5A). A difference spectrum of the 65 s time spectrum minus the 1 s time spectrum exhibited features consistent with oxidized PdX (PdXox);34 therefore, this first phase was assigned as oxidation of PdXred. The second phase, from 65 to 510 s, featured an absorbance increase at 395 and concomitant decreases at 450 and 560 nm (Figure 5B). The 510 s minus PdXox difference spectrum is identical to the spectrum of high-spin FeIII TxtE (Figure 5A, inset). On the basis of these assignments, the time course monitors the following reaction scheme:
| (2) |
| (3) |
| (4) |
Figure 5.
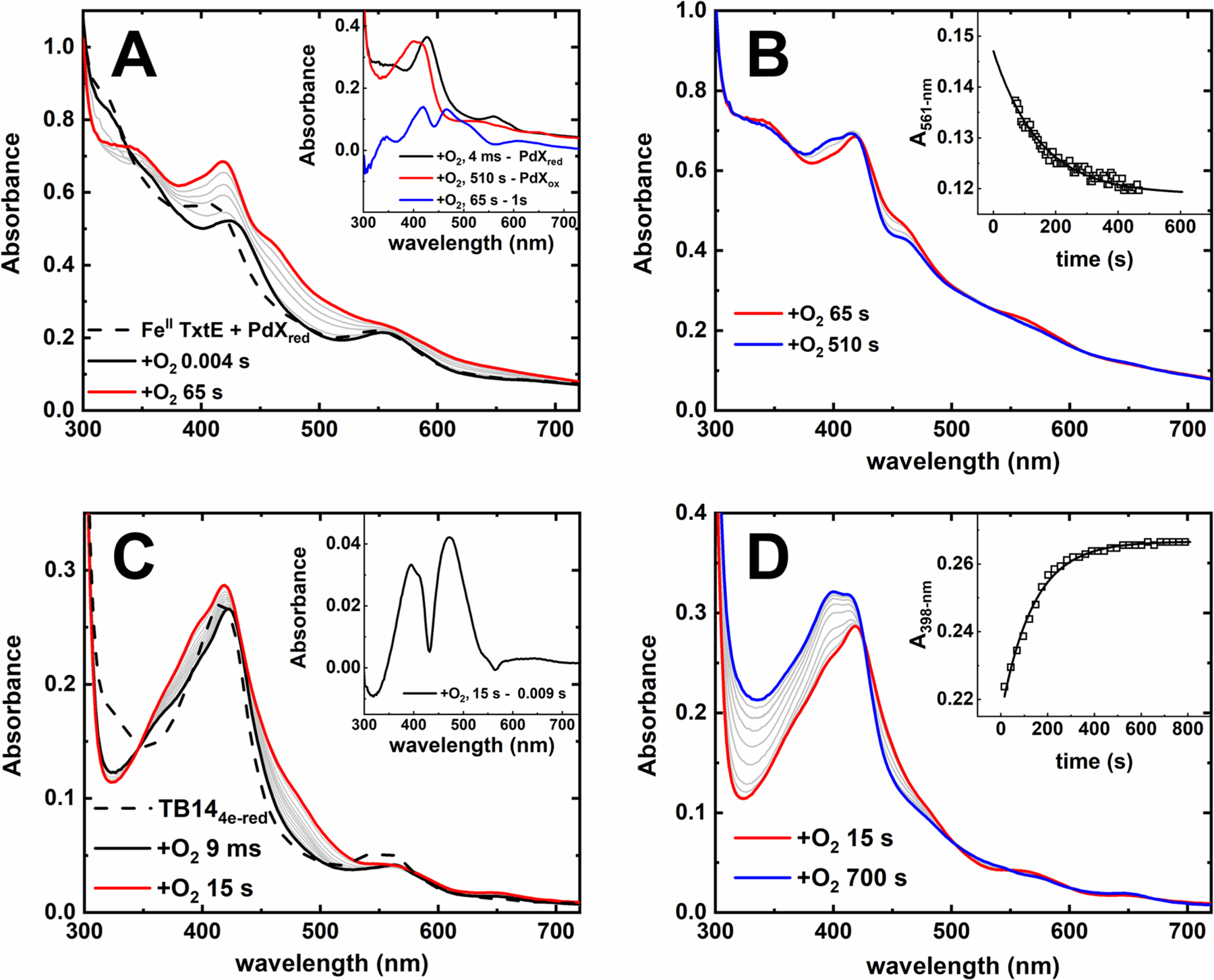
Stopped-flow spectral time courses of (A and B) anaerobic FeII TxtE and PdXred or (C and D) TB144e‑red mixed with O2 in the presence of excess Trp at pH 8.0 and 21 °C. Each time course is divided into (A and C) a fast phase and (B and D) a slow phase. The dashed trace was obtained by mixing the anaerobic solution against deoxygenated buffer. Solid black, red, or blue traces were recorded at times indicated in the figure legends. Gray traces were recorded at intermediate times. The inset shows single-exponent fits to representative A561 or A398 traces. Conditions after mixing: (A and B) 12.5 μM FeII TxtE, 50 μM PdXred, 250 μM Trp, and 130 μM O2 and (C and D) 20 μM TB144e‑red, 250 μM Trp, and 130 μM O2. All solutions were in 100 mM Tris (pH 8.0), and all reactions performed at 20 °C in a 1 cm path length cuvette.
Fitting A561 traces extracted from time courses collected with low-intensity light (Figure S7) provided an estimated kobs for the decay of the ferric-superoxo intermediate in the presence of PdXred of 0.0072 (0.0015) s−1. This value is similar to the kautox for the TxtE ferric-superoxo intermediate in the absence of excess reducing equivalents (Table 2). Therefore, the presence of PdXred has no effect on the lifetime of this intermediate.
Similar results were observed in time courses of TB144e‑red mixed with O2 (Figure 5C,D). An anaerobic sample of TB144e‑red exhibits absorbance features centered at 412 and 549 nm. This spectrum is consistent with the spectrum of FeII TxtE with absorption contributions from the flavin cofactors in the reductase domain of TB14. Mixing the TB144e‑red solution with air-saturated buffer (pH 8.0) resulted in the disappearance of the TB144e‑red spectrum within the dead time and the appearance of features centered at 361, 430, and 561 nm. This spectrum was consistent with formation of the ferric-superoxo intermediate. Thereafter, there are two phases. The first phase occurs over the first 15 s; the corresponding spectral time course exhibits a rise in absorption from 350 to 550 nm (Figure 5C). The 15 s minus 0.009 s difference spectrum shown in the inset exhibits features consistent with oxidized flavin.46 Therefore, this first phase was assigned to oxidation of the TB144e‑red flavin cofactors. The second phase, from 15 to 700 s, exhibits an increase in absorbance at 395 nm and the loss of the resolved Q-band at 561 nm. These spectral changes resemble those assigned to autoxidation at the heme site (Figure 3). However, there is an unexpected simultaneous absorbance increase in the 300−350 nm region not observed in our other autoxidation time courses. We suspected this was a photochemical process unrelated to TxtE autoxidation. Time courses collected with low-intensity light lack the absorbance increase from 300 to 350 nm (Figure S8), verifying this conclusion. Therefore, we assign the second phase as autoxidation of the TB14 ferric-superoxo intermediate. Fitting A398 traces to a single exponent collected with low-intensity light provided an estimated kautox of 0.0064 (0.001) s−1, a value statistically identical to kautox in the absence of excess reducing equivalents (Table 2).
DISCUSSION
Our data are consistent with a mechanism in which FeII TxtE binds O2 to form a ferric-superoxo intermediate, which subsequently reacts with NO to mediate Trp nitration (Figure 6). Ferrous TxtE fully supports Trp nitration with a high electron coupling efficiency (70 ± 8%) and requires no additional reducing equivalents (Figure 2B and Table 1). Therefore, only one exogenous electron is needed for nitration. Stopped-flow spectral time courses support the conclusion that O2 binds to FeII TxtE to form a ferric-superoxo intermediate (Figure 3). This intermediate decays to FeIII TxtE over several minutes by autoxidation (Figure 3; kautox = 0.008 s−1). By contrast, this intermediate decays within 2 ms in the presence of excess NO (Figure 4). The observed product spectrum of this reaction is consistent with a heme {FeNO}6 species, which was attributed to reaction of unreacted NO with the high-spin FeIII TxtE. These observations are consistent with the mechanism of Louka et al.29
Figure 6.
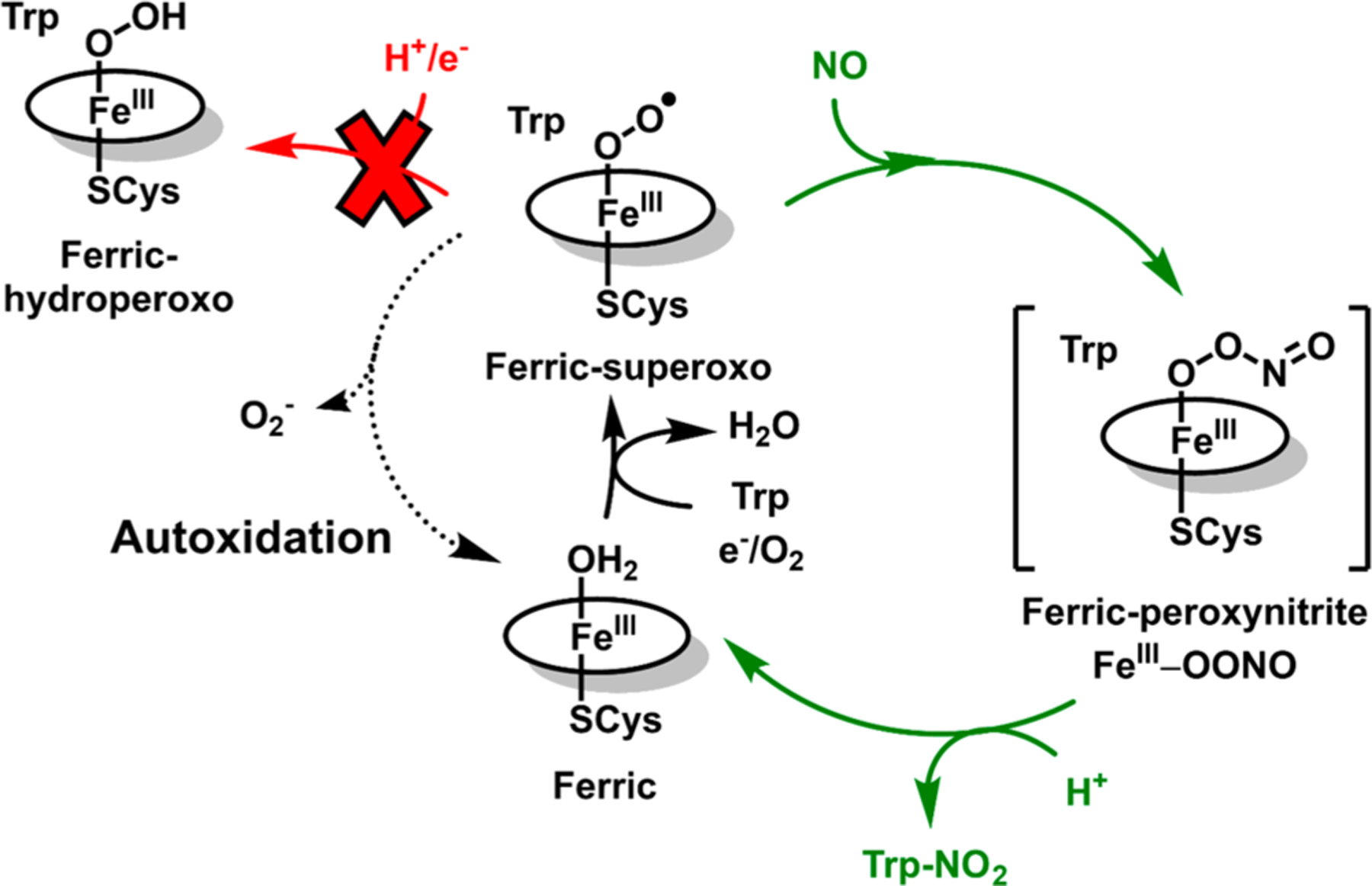
Summary of results from this study. The resistance of the TxtE ferric-superoxo intermediate to reduction avoids formation of Trp-OH and leaves autoxidation as the only uncoupling pathway available to TxtE. Brackets surround proposed intermediates not yet characterized for TxtE.
However, neither these experiments nor those previously reported were performed in the presence of excess reducing equivalents and ET partners. If the TxtE ferric-superoxo intermediate could be reduced to form a ferric-(hydro)peroxo intermediate, then evidence for either Trp hydroxylation or dissociation of H2O2 (via peroxide shunt) should be observed (Figure 1). Indeed, reduction of the ferric-superoxo intermediate could account for the high level of uncoupling observed for TxtE and RufO.5,14 Unexpectedly, we observed that TB14 could produce Trp-OH under O2-turnover conditions; however, we were able to attribute its formation to an off-pathway process dependent on H2O2 (Figure 2A). Therefore, we can confidently rule out TxtE uncoupling by formation of Trp-OH. Electron coupling efficiencies of Trp nitration by TxtE in the presence and absence of excess reducing equivalents were used as a probe for other potential uncoupling pathways (Table 1). Single-turnover nitration by FeII TxtE exhibits a coupling efficiency 25−50-fold higher than that of TxtE turnover (Table 1). This higher electron coupling efficiency rules out an uncoupling pathway originating from the putative FeIII-OONO intermediate to, for example, form NO3−. Finally, TB141e‑red exhibited an electron coupling efficiency lower than those of TB142e‑red and TB144e‑red. The simplest explanation for this observation is that the ferric-superoxo intermediate of TxtE cannot be reduced to the ferric-(hydro)peroxo species, a prerequisite for electron uncoupling by the peroxide shunt.
Supporting this conclusion, the time courses monitoring the decay of the ferric-superoxo intermediate in the presence of reduced CYP ET partners (Figure 5) lacked any spectroscopic or kinetic evidence for reduction of the ferric-superoxo intermediate. Cryoreduction of the CYP101A1 ferric-superoxo to the ferric-peroxo species shifts the Soret band from 417 to 440 nm.47 In addition, prior studies with CYP101A1 show rapid decay of the ferric-superoxo intermediate to the FeIII species with a kobs from 85 to 140 s−1 (3−4 °C) or 390 s−1 (25 °C) upon addition of PdXred.48–51 This faster decay was attributed to reduction of the ferric-superoxo intermediate, leading to substrate hydroxylation, and ultimately faster formation of FeIII CYP101A1. Neither the FeII TxtE with PdXred nor the TB144e‑red experiments shown in Figure 5 showed evidence for accumulation of spectral features that can be attributed to two-electron reduced intermediates. In fact, other than accumulation of spectroscopic features related to oxidation of the ET partners, the time courses closely resembled that of autoxidation of TxtE. In addition, the kobs for decay of the ferric-superoxo intermediate to FeIII TxtE is statistically identical to kautox regardless of the presence of excess reducing equivalents (Table 2). The lack of spectral evidence for intermediate reduction along with the lack of a change in intermediate decay kinetics strongly suggests that the TxtE ferric-superoxo intermediate is resistant to reduction.
The apparent recalcitrance of the TxtE ferric-superoxo intermediate to reduction enables efficient nitration activity. If the TxtE ferric-superoxo intermediate could be either reduced or reacted with NO, the two pathways would compete for the fate of Trp to form the hydroxylated product, Trp-OH, and the nitrated product 4-NO2-Trp (Figure 6). As an example of how this competition could affect product distributions, we can use reduction rate constants from CYP101A1. For this enzyme, the reduction of ferric-superoxo is rapid with a rate constant of 85−140 s−1 at 3−4 °C.49–51 The reaction of NO with the TxtE ferric-superoxo intermediate reported by Louka et al. was also performed at 4 °C and was complete within 100 ms. Therefore, the kobs of the reaction of NO with TxtE ferric-superoxo has a lower limit of 35 s−1 at 4 °C. Under these conditions, hydroxylation of Trp would outcompete its nitration by a 2:1 ratio. Alternatively, we can also use a rate constant of 3 s−1 for cytochrome b5 reduction of the cytochrome P450 2B4 ferric-superoxo intermediate. In this case, Trp hydroxylation would still moderately compete with its nitration by a 1:12 ratio.52–54 Therefore, the ability of TxtE to resist the reduction of the ferric-superoxo intermediate appears to improve the efficiency of nitration activity by avoiding access to intermediates that lead to substrate hydroxylation.
Further enhancing the TxtE nitration efficiency is the remarkably slow autoxidation of Trp-bound TxtE (kautox = 0.008 s−1) at 20 °C. The observed TxtE kautox is slower than that for CYP102A1 (0.140 s−1 at 15 °C) and CYP2B4 (0.09 s−1 at 15 °C), orders of magnitude slower than that for testosterone-bound CYP3A4 (3.6 s−1 at 24 °C), and similar to that of CYP101A1.55,56 The slow kautox further improves the efficiency of TxtE nitration by increasing the lifetime of the ferric-superoxo intermediate, which promotes its reaction with NO and subsequent Trp nitration.
Comparison of the TxtE crystal structure to that of CYP101A1 may provide some structure−function clues as to how the TxtE ferric-superoxo intermediate resists reduction.11,39,57 Two key residues in CYP101A1, D251 and T252, sometimes termed the acid-alcohol pair, are critical for its hydroxylation activity (Figure 7, left panel). A key role of the T252 residue is to promote the protonation of the FeIII-OOH to form compound I. A T252A variant of CYP101A1 hinders electron coupling as evidenced by both a lower ratio of product formation per mole of NADH consumed and a marked increase in H2O2 production.58,59 This has been attributed to uncoupling of H2O2 from the FeIII-OOH intermediate due to slower proton transfer. The role of D251 is to promote a critical conformational change in the CYP101A1 I helix that allows for orienting catalytic waters near the active site upon O2 binding.57,60,61 A D251N CYP101A1 variant exhibited a greatly decreased rate of NADH consumption. This was attributed to the absence of the catalytic water residues that inhibited reduction of the ferric-superoxo intermediate. The TxtE crystal structure shown in the right panel of Figure 7 is of Trp-bound FeIII TxtE and does not show O2 bound. Nevertheless, this structure clearly shows that in the place of the acid-alcohol pair of CYP101A1 are the P249 and T250 residues (Figure 7, right panel). Therefore, TxtE lacks D251 as well as other residues, such as N255, needed to support the change in conformation of helix I. With P249 in this position, it is likely that the ordered waters will be different compared to that of CYP101A1, which may account for the observed resistance of the TxtE ferric-superoxo intermediate to reduction.
Figure 7.
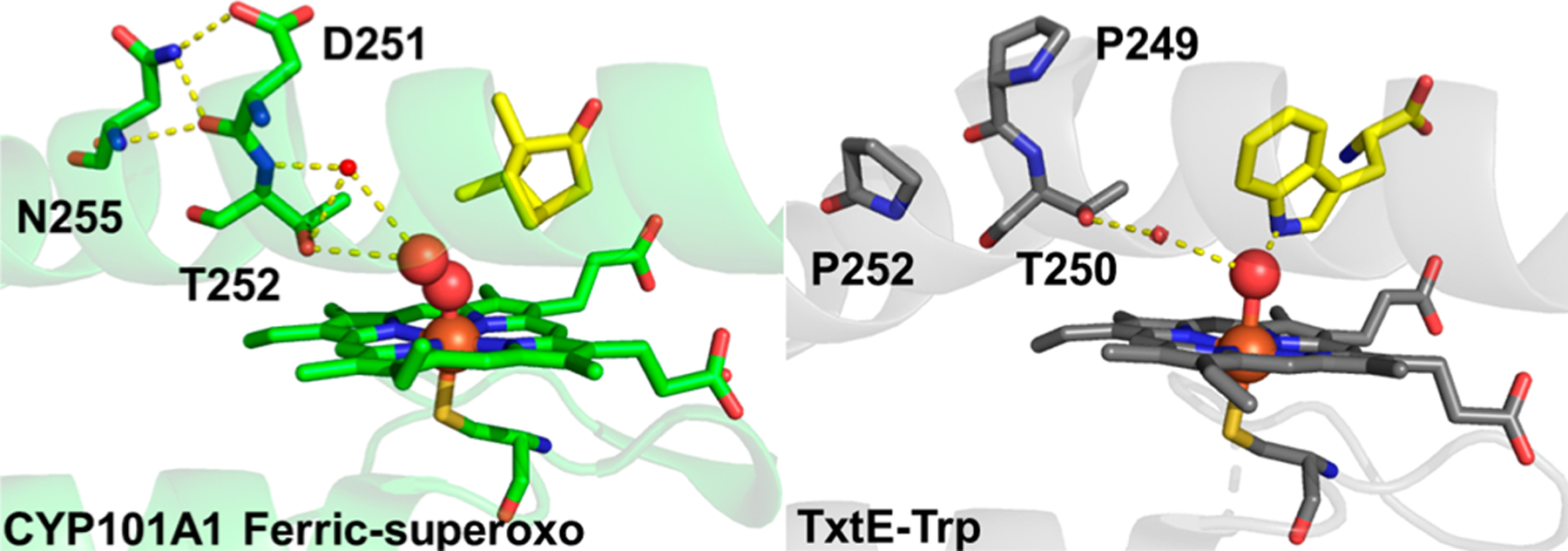
Crystal structures of the CYP101A1 ferric-superoxo intermediate (Protein Data Bank entry 1DZ8) and Trp-bound FeIII TxtE (Protein Data Bank entry 4TPO). Carbon atoms are colored green for CYP101A1 and gray for TxtE, oxygen atoms red, nitrogen atoms dark blue, and sulfur atoms yellow, and the iron atom is colored orange. H-Bonds are represented by dashed yellow lines.
Finally, we can narrow down the possible reasons for the poor electron coupling observed for steady-state nitration by RufO and TxtE. One possibility for poor coupling is autoxidation of the ET cofactors, such as the flavin cofactors of the TB14 reductase domain.62 This possibility could also account for evidence of H2O2 formation in TB14-turnover samples (Figure 2A). However, poor electron coupling is also observed for the FNR/Fd/TxtE system (Table 1) and, therefore, cannot be attributed to the TB14 reductase domain. Nevertheless, a more suitable reductase that is also resistant to autoxidation could increase the coupling efficiency. To date, a native TxtE reductase has yet to be found.63
An alternative possibility is the insufficient NO delivery rates by the NO generators used for in vitro experiments. In the presence of high NO concentrations, similar to those used in our limited-turnover experiments, we estimated the kobs for the reaction of the ferric-superoxo intermediate with NO (>1700 s−1) to be more than 200000-fold faster than the rate of TxtE autoxidation (0.008 s−1). These rate constants suggest that uncoupling by autoxidation will occur only when the level of NO in the reaction mixture is low or nearly depleted. Improvement in the in vitro efficiency of TxtE or its variants is expected in the presence of a NO donor that aligns better with the lifetime of the ferric-superoxo intermediate.
For in vivo considerations, the delivery of NO by bNOS is critical to balance efficient nitration with overproduction of NO that can (1) scavenge O2, a necessary co-substrate for nitration, and (2) be lethal to the cell at high concentrations.
Our electron coupling efficiency data for TxtE in the absence and presence of excess reducing equivalents point to autoxidation as the only TxtE-centered uncoupling pathway. We conclude that the common CYP ET partners used for these experiments are unable to reduce the TxtE ferric-superoxo intermediate. The resistance of the intermediate to reduction contrasts with typical CYP chemistry and highlights the evolutionary versatility of CYPs.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Matthew Rex and Bhavini Goswami for their assistance with LC-MS.
Funding
J.D.C. thanks the University of Central Florida (UCF) College of Sciences and Department of Chemistry for the startup funding that supported this project and the UCF Academic Advancement Programs for supporting the training and research of M.F.M. by the RAMP-T program. The work by Y.D. and M.C. was supported by National Institutes of Health Grant R35GM128742.
ABBREVIATIONS
- NO
nitric oxide
- ET
electron transfer
- Trp
l-tryptophan
- bNOS
bacterial nitric oxide synthase
- Trp-OH
hydroxy-l-tryptophan
- 4-NO2-Trp
4-nitro-l-tryptophan
- 5-ALA
5-aminolevulinic acid
- PMSF
phenylmethanesulfonyl fluoride
- LC-MS
liquid chromatography-mass spectrometry
- HPLC
high-performance liquid chromatography
- PROLI-NONOate
1-(hydroxy-NNO-azoxy)-l-proline
- DEA-NONOate
diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate
- NADPH
nicotinamide adenine dinucleotide phosphate
- NADH
nicotinamide adenine dinucleotide
- PdX
putidaredoxin
Footnotes
Accession Codes
SCAB_31831, C9ZDC6; camB, P00259.
The authors declare no competing financial interest.
Contributor Information
Christopher P. Martin, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States
Manyun Chen, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development, University of Florida, Gainesville, Florida 32610, United States.
Maria F. Martinez, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States
Yousong Ding, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development, University of Florida, Gainesville, Florida 32610, United States.
Jonathan D. Caranto, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States.
REFERENCES
- (1).Nelson DR (2018) Cytochrome P450 diversity in the tree of life. Biochim. Biophys. Acta, Proteins Proteomics 1866, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Guengerich FP (2018) Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catal 8, 10964–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Guengerich FP, and Munro AW (2013) Unusual cytochrome P450 enzymes and reactions. J. Biol. Chem 288, 17065–17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Barry SM, Kers JA, Johnson EG, Song L, Aston PR, Patel B, Krasnoff SB, Crane BR, Gibson DM, Loria R, and Challis GL (2012) Cytochrome P450-catalyzed L-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat. Chem. Biol 8, 814–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tomita H, Katsuyama Y, Minami H, and Ohnishi Y (2017) Identification and characterization of a bacterial cytochrome P450 monooxygenase catalyzing the 3-nitration of tyrosine in rufomycin biosynthesis. J. Biol. Chem 292, 15859–15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tsutsumi H, Katsuyama Y, Izumikawa M, Takagi M, Fujie M, Satoh N, Shin-Ya K, and Ohnishi Y (2018) Unprecedented cyclization catalyzed by a cytochrome P450 in benzastatin biosynthesis. J. Am. Chem. Soc 140, 6631–6639. [DOI] [PubMed] [Google Scholar]
- (7).Rylott EL, Jackson RG, Sabbadin F, Seth-Smith HM, Edwards J, Chong CS, Strand SE, Grogan G, and Bruce NC (2011) The explosive-degrading cytochrome P450 XplA: biochemistry, structural features and prospects for bioremediation. Biochim. Biophys. Acta, Proteins Proteomics 1814, 230–236. [DOI] [PubMed] [Google Scholar]
- (8).Brandenberg OF, Fasan R, and Arnold FH (2017) Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Steck V, Kolev JN, Ren X, and Fasan R (2020) Mechanism-Guided Design and Discovery of Efficient Cytochrome P450-Derived C-H Amination Biocatalysts. J. Am. Chem. Soc 142, 10343–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huang X, and Groves JT (2018) Oxygen activation and radical transformations in heme proteins and metalloporphyrins. Chem. Rev 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dodani SC, Cahn JK, Heinisch T, Brinkmann-Chen S, McIntosh JA, and Arnold FH (2014) Structural, functional, and spectroscopic characterization of the substrate scope of the novel nitrating cytochrome P450 TxtE. ChemBioChem 15, 2259–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dodani SC, Kiss G, Cahn JK, Su Y, Pande VS, and Arnold FH (2016) Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models. Nat. Chem 8, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zuo R, Zhang Y, Huguet-Tapia JC, Mehta M, Dedic E, Bruner SD, Loria R, and Ding Y (2016) An artificial self-sufficient cytochrome P450 directly nitrates fluorinated tryptophan analogs with a different regio-selectivity. Biotechnol. J 11, 624–632. [DOI] [PubMed] [Google Scholar]
- (14).Zuo R, Zhang Y, Jiang C, Hackett JC, Loria R, Bruner SD, and Ding Y (2017) Engineered P450 biocatalysts show improved activity and regio-promiscuity in aromatic nitration. Sci. Rep 7, 842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jiang G, Zhang Y, Powell MM, Zhang P, Zuo R, Zhang Y, Kallifidas D, Tieu AM, Luesch H, Loria R, and Ding Y (2018) High-yield production of herbicidal thaxtomins and thaxtomin analogs in a nonpathogenic Streptomyces strain. Appl. Environ. Microbiol 84, No. e00164–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Loria R, Bignell DR, Moll S, Huguet-Tapia JC, Joshi MV, Johnson EG, Seipke RF, and Gibson DM (2008) Thaxtomin biosynthesis: the path to plant pathogenicity in the genus Streptomyces. Antonie van Leeuwenhoek 94, 3–10. [DOI] [PubMed] [Google Scholar]
- (17).Loria R, Bukhalid RA, Fry BA, and King RR (1997) Plant pathogenicity in the genus Streptomyces. Plant Dis 81, 836–846. [DOI] [PubMed] [Google Scholar]
- (18).King RR, and Calhoun LA (2009) The thaxtomin phytotoxins: sources, synthesis, biosynthesis, biotransformation and biological activity. Phytochemistry 70, 833–841. [DOI] [PubMed] [Google Scholar]
- (19).King RR, Lawrence CH, and Calhoun LA (1992) Chemistry of phytotoxins associated with Streptomyces scabies the causal organism of potato common scab. J. Agric. Food Chem 40, 834–837. [Google Scholar]
- (20).King RR, Lawrence CH, Clark MC, and Calhoun LA (1989) Isolation and characterization of phytotoxins associated with Streptomyces scabies. J. Chem. Soc., Chem. Commun 0, 849–850. [Google Scholar]
- (21).Scheible W-R, Fry B, Kochevenko A, Schindelasch D, Zimmerli L, Somerville S, Loria R, and Somerville CR (2003) An Arabidopsis mutant resistant to thaxtomin A, a cellulose synthesis inhibitor from Streptomyces species. Plant Cell 15, 1781–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Healy FG, Wach M, Krasnoff SB, Gibson DM, and Loria R (2000) The txtAB genes of the plant pathogen Streptomyces acidiscabies encode a peptide synthetase required for phytotoxin thaxtomin A production and pathogenicity. Mol. Microbiol 38, 794–804. [DOI] [PubMed] [Google Scholar]
- (23).Kers JA, Wach MJ, Krasnoff SB, Widom J, Cameron KD, Bukhalid RA, Gibson DM, Crane BR, and Loria R (2004) Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature 429, 79–82. [DOI] [PubMed] [Google Scholar]
- (24).Crane BR, Sudhamsu J, and Patel BA (2010) Bacterial nitric oxide synthases. Annu. Rev. Biochem 79, 445–470. [DOI] [PubMed] [Google Scholar]
- (25).Wach MJ, Kers JA, Krasnoff SB, Loria R, and Gibson DM (2005) Nitric oxide synthase inhibitors and nitric oxide donors modulate the biosynthesis of thaxtomin A, a nitrated phytotoxin produced by Streptomyces spp. Nitric Oxide 12, 46–53. [DOI] [PubMed] [Google Scholar]
- (26).Tomita H, Katsuyama Y, Minami H, and Ohnishi Y (2017) Identification and characterization of a bacterial cytochrome P450 monooxygenase catalyzing the 3-nitration of tyrosine in rufomycin biosynthesis. J. Biol. Chem 292, 15859–15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Caranto JD (2019) The emergence of nitric oxide in the biosynthesis of bacterial natural products. Curr. Opin. Chem. Biol 49, 130–138. [DOI] [PubMed] [Google Scholar]
- (28).Waldman AJ, Ng TL, Wang P, and Balskus EP (2017) Heteroatom-heteroatom bond formation in natural product biosynthesis. Chem. Rev 117, 5784–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Louka S, Barry SM, Heyes DJ, Mubarak MQE, Ali HS, Alkhalaf L, Munro AW, Scrutton NS, Challis GL, and de Visser SP (2020) The catalytic mechanism of aromatic nitration by cytochrome P450 TxtE: Involvement of a ferric-peroxynitrite intermediate. J. Am. Chem. Soc 142, 15764–15779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yosca TH, Ledray AP, Ngo J, and Green MT (2017) A new look at the role of thiolate ligation in cytochrome P450. JBIC, J. Biol. Inorg. Chem 22, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Denisov IG, Makris TM, Sligar SG, and Schlichting I (2005) Structure and chemistry of cytochrome P450. Chem. Rev 105, 2253–2278. [DOI] [PubMed] [Google Scholar]
- (32).Sligar SG, Makris TM, and Denisov IG (2005) Thirty years of microbial P450 monooxygenase research: peroxo-heme intermediates the central bus station in heme oxygenase catalysis. Biochem. Biophys. Res. Commun 338, 346–354. [DOI] [PubMed] [Google Scholar]
- (33).Tan CY, Hirakawa H, Suzuki R, Haga T, Iwata F, and Nagamune T (2016) Immobilization of a bacterial cytochrome P450 monooxygenase system on a solid support. Angew. Chem 128, 15226–15230. [DOI] [PubMed] [Google Scholar]
- (34).Reipa V, Holden M, Mayhew MP, and Vilker VL (2004) Temperature-induced structural changes in putidaredoxin: a circular dichroism and UV-vis absorption study. Biochim. Biophys. Acta, Proteins Proteom 1699, 229–234. [DOI] [PubMed] [Google Scholar]
- (35).Sligar S, Debrunner P, Lipscomb J, Namtvedt M, and Gunsalus I (1974) A role of the putidaredoxin COOH-terminus in P-450cam (cytochrome m) hydroxylations. Proc. Natl. Acad. Sci. U. S. A 71, 3906–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lipscomb JD, Sligar SG, Namtvedt MJ, and Gunsalus IC (1976) Autooxidation and hydroxylation reactions of oxygenated cytochrome P-450cam. J. Biol. Chem 251, 1116–1124. [PubMed] [Google Scholar]
- (37).Guengerich FP (1983) Oxidation-reduction properties of rat liver cytochromes P-450 and NADPH-cytochrome P-450 reductase related to catalysis in reconstituted systems. Biochemistry 22, 2811–2820. [DOI] [PubMed] [Google Scholar]
- (38).Avila L, Wirtz M, Bunce RA, and Rivera M (1999) An electrochemical study of the factors responsible for modulating the reduction potential of putidaredoxin. J. Biol. Inorg. Chem 4, 664–674. [DOI] [PubMed] [Google Scholar]
- (39).Yu F, Li M, Xu C, Wang Z, Zhou H, Yang M, Chen Y, Tang L, and He J (2013) Structural insights into the mechanism for recognizing substrate of the cytochrome P450 enzyme TxtE. PLoS One 8, No. e81526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Su J, and Groves JT (2009) Direct detection of the oxygen rebound intermediates, ferryl Mb and NO2, in the reaction of metmyoglobin with peroxynitrite. J. Am. Chem. Soc 131, 12979–12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Su J, and Groves JT (2010) Mechanisms of peroxynitrite interactions with heme proteins. Inorg. Chem 49, 6317–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gardner PR (2005) Nitric oxide dioxygenase function and mechanism of flavohemoglobin, hemoglobin, myoglobin and their associated reductases. J. Inorg. Biochem 99, 247–266. [DOI] [PubMed] [Google Scholar]
- (43).Lipscomb JD, Sligar SG, Namtvedt M, and Gunsalus I (1976) Autooxidation and hydroxylation reactions of oxygenated cytochrome P-450cam. J. Biol. Chem 251, 1116–1124. [PubMed] [Google Scholar]
- (44).Denisov IG, Grinkova YV, Baas BJ, and Sligar SG (2006) The ferrous-dioxygen intermediate in human cytochrome P450 3A4 substrate dependence of formation and decay kinetics. J. Biol. Chem 281, 23313–23318. [DOI] [PubMed] [Google Scholar]
- (45).Enemark J, and Feltham R (1974) Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord. Chem. Rev 13, 339–406. [Google Scholar]
- (46).Hanley SC, Ost TW, and Daff S (2004) The unusual redox properties of flavocytochrome P450 BM3 flavodoxin domain. Biochem. Biophys. Res. Commun 325, 1418–1423. [DOI] [PubMed] [Google Scholar]
- (47).Denisov IG, Makris TM, and Sligar SG (2001) Cryotrapped reaction intermediates of cytochrome P450 studied by radiolytic reduction with phosphorus-32. J. Biol. Chem 276, 11648–11652. [DOI] [PubMed] [Google Scholar]
- (48).Purdy MM, Koo LS, Ortiz de Montellano PR, and Klinman JP (2006) Mechanism of O2 activation by cytochrome P450cam studied by isotope effects and transient state kinetics. Biochemistry 45, 15793–15806. [DOI] [PubMed] [Google Scholar]
- (49).Glascock MC, Ballou DP, and Dawson JH (2005) Direct Observation of a Novel Perturbed Oxyferrous Catalytic Intermediate during Reduced Putidaredoxin-initiated Turnover of Cytochrome P-450-CAM Probing the Effector Rrole of Putidaredoxin in Catalysis. J. Biol. Chem 280, 42134–42141. [DOI] [PubMed] [Google Scholar]
- (50).Brewer CB, and Peterson JA (1988) Single turnover kinetics of the reaction between oxycytochrome P-450cam and reduced putidaredoxin. J. Biol. Chem 263, 791–798. [PubMed] [Google Scholar]
- (51).Tosha T, Yoshioka S, Hori H, Takahashi S, Ishimori K, and Morishima I (2002) Molecular mechanism of the electron transfer reaction in cytochrome P450cam- putidaredoxin: Roles of glutamine 360 at the heme proximal site. Biochemistry 41, 13883–13893. [DOI] [PubMed] [Google Scholar]
- (52).Pompon D, and Coon MJ (1984) On the mechanism of action of cytochrome P-450. Oxidation and reduction of the ferrous dioxygen complex of liver microsomal cytochrome P-450 by cytochrome b5. J. Biol. Chem 259, 15377–15385. [PubMed] [Google Scholar]
- (53).Zhang H, Gruenke L, Arscott D, Shen A, Kasper C, Harris DL, Glavanovich M, Johnson R, and Waskell L (2003) Determination of the rate of reduction of oxyferrous cytochrome P450 2B4 by 5-deazariboflavin adenine dinucleotide T491V cytochrome P450 reductase. Biochemistry 42, 11594–11603. [DOI] [PubMed] [Google Scholar]
- (54).Bonfils C, Balny C, and Maurel P (1981) Direct evidence for electron transfer from ferrous cytochrome b5 to the oxyferrous intermediate of liver microsomal cytochrome P-450 LM2. J. Biol. Chem 256, 9457–9465. [PubMed] [Google Scholar]
- (55).Denisov IG, Grinkova YV, McLean MA, and Sligar SG (2007) The one-electron autoxidation of human cytochrome P450 3A4. J. Biol. Chem 282, 26865–26873. [DOI] [PubMed] [Google Scholar]
- (56).Ost TW, Clark J, Mowat CG, Miles CS, Walkinshaw MD, Reid GA, Chapman SK, and Daff S (2003) Oxygen activation and electron transfer in flavocytochrome P450 BM3. J. Am. Chem. Soc 125, 15010–15020. [DOI] [PubMed] [Google Scholar]
- (57).Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, and Sligar SG (2000) The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287, 1615–1622. [DOI] [PubMed] [Google Scholar]
- (58).Imai M, Shimada H, Watanabe Y, Matsushima-Hibiya Y, Makino R, Koga H, Horiuchi T, and Ishimura Y (1989) Uncoupling of the cytochrome P-450cam monooxygenase reaction by a single mutation, threonine-252 to alanine or valine: possible role of the hydroxy amino acid in oxygen activation. Proc. Natl. Acad. Sci. U. S. A 86, 7823–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Martinis SA, Atkins WM, Stayton PS, and Sligar SG (1989) A conserved residue of cytochrome P-450 is involved in heme-oxygen stability and activation. J. Am. Chem. Soc 111, 9252–9253. [Google Scholar]
- (60).Gerber NC, and Sligar SG (1994) A role for Asp-251 in cytochrome P-450cam oxygen activation. J. Biol. Chem 269, 4260–4266. [PubMed] [Google Scholar]
- (61).Nagano S, and Poulos TL (2005) Crystallographic Study on the Dioxygen Complex of Wild-type and Mutant Cytochrome P450cam Implications for the Dioxygen Activation Mechanism. J. Biol. Chem 280, 31659–31663. [DOI] [PubMed] [Google Scholar]
- (62).Munro AW, Lindsay JG, Coggins JR, Kelly SM, and Price NC (1995) NADPH oxidase activity of cytochrome P-450 BM3 and its constituent reductase domain. Biochim. Biophys. Acta, Bioenerg 1231, 255–264. [DOI] [PubMed] [Google Scholar]
- (63).Healy FG, Krasnoff SB, Wach M, Gibson DM, and Loria R (2002) Involvement of a cytochrome P450 monooxygenase in thaxtomin A biosynthesis by Streptomyces acidiscabies. J. Bacteriol 184, 2019–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.