

Summary
Control of central nervous system (CNS) pathogens by CD8 T cells is key to avoid fatal neuroinflammation. Yet the modalities of MHC I presentation in the brain are poorly understood. Here we analyze the antigen presentation mechanisms underlying CD8 T cell-mediated control of the Toxoplasma gondii parasite in the CNS. We show that MHC I presentation of an efficiently processed model antigen (GRA6-OVA), even when not expressed in the bradyzoite stage, reduces cyst burden and dampens encephalitis in C57BL/6 mice. Antigen presentation assays with infected primary neurons reveal a correlation between lower MHC I presentation of tachyzoite antigens by neurons and poor parasite control in vivo. Using conditional MHC I-deficient mice, we find that neuronal MHC I presentation is required for robust restriction of T. gondii in the CNS during chronic phase, showing the importance of MHC I presentation by CNS neurons in the control of a prevalent brain pathogen.
Introduction
The brain is endowed with specialized innate and adaptive immune mechanisms that ensure both the detection and mitigation of neurotropic infections (Klein and Hunter, 2017; Russo and McGavern, 2015) but some pathogens can chronically reside within the central nervous system (CNS). Accumulating evidence point to intricate links between neuroinflammation and the development of neurodegenerative diseases (Colonna and Butovsky, 2017; Heneka et al., 2015). By eliciting various degrees of inflammation, chronically persisting CNS pathogens are likely to influence brain processes, including age-related cognitive dysfunctions (Cabral et al., 2017; McManus and Heneka, 2017; Mohle et al., 2016). A better understanding of the mechanisms by which immune components of the CNS, such as CD8 T cells, detect and control persisting microorganisms is needed not only to improve containment of these pathogens but also to potentially alleviate detrimental effects of chronic infections on brain functions.
The protozoan intracellular parasite Toxoplasma gondii is a widespread foodborne pathogen (Guo et al., 2016), which commonly infects humans. Due to the lack of an effective drug targeting its encysted bradyzoite stage, this parasite cannot be cleared from the brain. Hence, with a worldwide seroprevalence of ~30% (Pappas et al., 2009) that can reach up to 50% in certain countries (Wilking et al., 2016), T. gondii is thought to reside in the brains of more than 2 billion individuals. Following dissemination of the rapidly-dividing tachyzoites in the host, the parasite converts into slower-growing bradyzoites, which chronically persist within cysts in muscles and in the CNS (Ferguson and Hutchison, 1987). In the CNS, multiple resident cell types may be in contact with tachyzoites but neurons are the only cells supporting the development of cysts (Cabral et al., 2016; Melzer et al., 2010), which are mostly found within intact host cells (Ferguson and Hutchison, 1987).
The clinical outcome of T. gondii infection critically depends on the host immune status and more specifically on a fully functional T cell compartment. In immunocompetent humans, acute infection remains mildly symptomatic but chronic presence of T. gondii in the brain (referred to as latent toxoplasmosis) has been associated with neuropsychiatric disorders, such as schizophrenia (Torrey and Yolken, 2003) and cognitive changes (Stock et al., 2017), although conflicting data exist on this question (Perry et al., 2016; Wyman et al., 2017). In rodents, which are natural hosts of T. gondii, the parasite has been reported to influence the course of neurodegenerative disorders (Cabral et al., 2017; Mohle et al., 2016) and to cause major behavioral modifications (Vyas, 2015). In case of T cell lymphopenia or sub-optimal function (e.g. due to HIV/AIDS or immune suppressive treatment), individuals become at risk of developing T. gondii encephalitis (TE), a fatal neuroinflammatory disease that is still common among HIV-infected people (Ondounda et al., 2016). TE is characterized by high cyst burden, tachyzoite replication foci in the CNS, massive immune cell influx, activation of recruited and local myeloid cells and cerebral tissue damage (Parlog et al., 2014). TE is also associated with activation and exhaustion of CD4 T cells, leading to functional attrition of CD8 T cells (Hwang et al., 2016).
Mouse studies have highlighted a variety of innate immune mechanisms that can contribute to parasite control in the brain. These include the production of pro-inflammatory mediators by innate mononuclear cells (Biswas et al., 2015; Sa et al., 2015) and neutrophils (Biswas et al., 2017), as well as anti-microbial pathways triggered by IFNγ/STAT1 in astrocytes (Hidano et al., 2016). Yet, these processes are typically not sufficient to prevent TE pathogenesis. In contrast, CD8 T cells and MHC I, in particular the H-2 Ld MHC I allele, are pivotal to drive robust and durable brain parasite control and to dampen encephalitis, a status known as TE resistance (Blanchard et al., 2008; Brown et al., 1995). Consequently, C57BL/6 mice, which are devoid of H-2 Ld, are a good model to study the pathogenesis of T. gondii encephalitis while mice expressing H-2 Ld (e.g. BALB/c or congenic B6.H-2d mice) are TE-resistant (Blanchard et al., 2015). In mice bearing the H-2d MHC haplotype, protection from TE relies on the induction of CD8 T cells specific for an immunodominant Ld-restricted peptide that is efficiently processed from the T. gondii-secreted GRA6 protein in infected macrophages and dendritic cells (DC) (Blanchard et al., 2008; Feliu et al., 2013). This protective response is maintained without a contraction phase, by continuous production of effector CD8 T cells via a proliferative, antigen-dependent population, displaying a memory-effector hybrid phenotype (Chu et al., 2016). While induction of GRA6-specific peripheral CD8 T cell responses in the periphery is a clear prerequisite to enable robust parasite control in the brain (Feliu et al., 2013), the determinants underlying CD8-mediated surveillance of T. gondii in the CNS in the context of TE resistance remain ill-defined.
To address this question without modifying the endogenous GRA6 protein, which plays a role in cystogenesis (Fox et al., 2011), we created transgenic parasites ectopically expressing a model antigen composed of the H-2 Kb-restricted OVA-derived SIINFEKL epitope in fusion with the C-terminus of GRA6, a known immunogenic position (Feliu et al., 2013). Compared to parasites expressing the same SIINFEKL epitope within a different source antigen (vacOVA), C57BL/6 mice infected with the GRA6-OVA-expressing parasites displayed limited brain inflammation. Using a promoter restricting expression of the GRA6-OVA antigen to tachyzoites, we showed that CD8 T cell recognition of this antigen at the tachyzoite stage is enough to ensure parasite restriction in the brain. Measurements of antigen presentation by primary neuronal cultures infected with parasites leading to TE (vacOVA) or to a reduced CNS inflammation (GRA6-OVA) suggested the implication of neuronal MHC I presentation in the control of brain parasite load. To formally address this possibility, we developed a mouse model enabling selective ablation of the H-2 Ld allele (naturally conferring resistance to TE) in neurons. Our results revealed that although neuronal MHC I presentation is dispensable for CNS accumulation of T. gondii-specific CD8 T cells, it is critical for durable parasite control.
Results
A CD8 T cell-dependent TE-resistant model in C57BL/6 mice based on GRA6-OVA expression
To study how CD8 T cell-mediated detection and restriction of T. gondii in the CNS are achieved, we sought to create a C57BL/6-based model in which parasites expressing a tractable CD8 T cell model antigen would be efficiently or poorly controlled at chronic phase, leading to limited vs. active brain inflammation and thereby mimicking T. gondii latency vs. encephalitis. Since CD8 T cell responses specific for the GRA6-derived HPGSVNEFDF (HF10) epitope are pivotal for resistance to T. gondii encephalitis in H-2 Ld+ mice (Blanchard et al., 2008; Feliu et al., 2013) and since GRA6 C-terminus is a preferential location for antigenicity (Buaillon et al., 2017; Feliu et al., 2013), we reasoned that the addition of the H-2 Kb-restricted chicken ovalbumin (OVA)-derived SIINFEKL model epitope to the C-terminus of GRA6 may protect C57BL/6 mice from the CNS inflammation that is typically observed after infection by type II parasites. To test this hypothesis, we generated type II parasites (Tomato+ Pru) expressing the GRA6-OVA model antigen. In order to preserve the final antigen processing steps, the OVA-derived SIINFEKL peptide was flanked by 5 amino acids that are naturally present in OVA (Fig. 1a). In parallel, we took advantage of a previously described (Schaeffer et al., 2009) Tomato+ Pru strain that expresses the SIINFEKL epitope embedded in a different dense granule-secreted antigen: the vacuolar SAG1ΔGPI-OVA protein (abbreviated here as vacOVA) (Fig. 1a). The resulting parasites are designated as Tg.pGRA6/GRA6-OVA and Tg.pTUB/vacOVA, respectively. In line with the known dual-stage activity of the tubulin and GRA6 promoters respectively controlling the expression of vacOVA and GRA6-OVA, these two antigenic constructs were detected both in tachyzoites and bradyzoites (Schaeffer et al., 2009) (Fig. 1a). To ensure that the comparison of these two transgenic parasites was valid, we evaluated the in vivo dissemination, access to the brain and CD8 T cell responses elicited in the course of acute toxoplasmosis. Ten days post-infection, comparable OVA-specific CD8 T cell responses were induced in the spleen by both strains, which disseminated in the spleen and, even more efficiently than the parental strain, in the brain (Sup. Fig. 1a, b, c, d). Yet 3 weeks post-infection, i.e. at the beginning of chronic stage, the Tg.pGRA6/GRA6-OVA parasites were more robustly restricted in the CNS than the Tg.pTUB/vacOVA parasites. Such a difference was observed in H-2b (Kb-positive) C57BL/6, which develop prominent SIINFEKL-specific CD8 T cell responses in the CNS (Fig. 4d, e), but not in H-2k (Kb-negative) CBA mice (Fig. 1b, c, d and Fig S1e, f, g). As CBA mice are unable to present the SIINFEKL peptide, these data show (i) that the in vivo fitness of the Tg.pGRA6/GRA6-OVA is not altered and (ii) that the beneficial impact of GRA6-OVA on CNS parasite control in C57BL/6 mice is likely linked to H-2Kb-restricted CD8 T cell responses.
Figure 1. Expression of GRA6-OVA antigen by T. gondii leads to efficient parasite control and lower CNS inflammation.
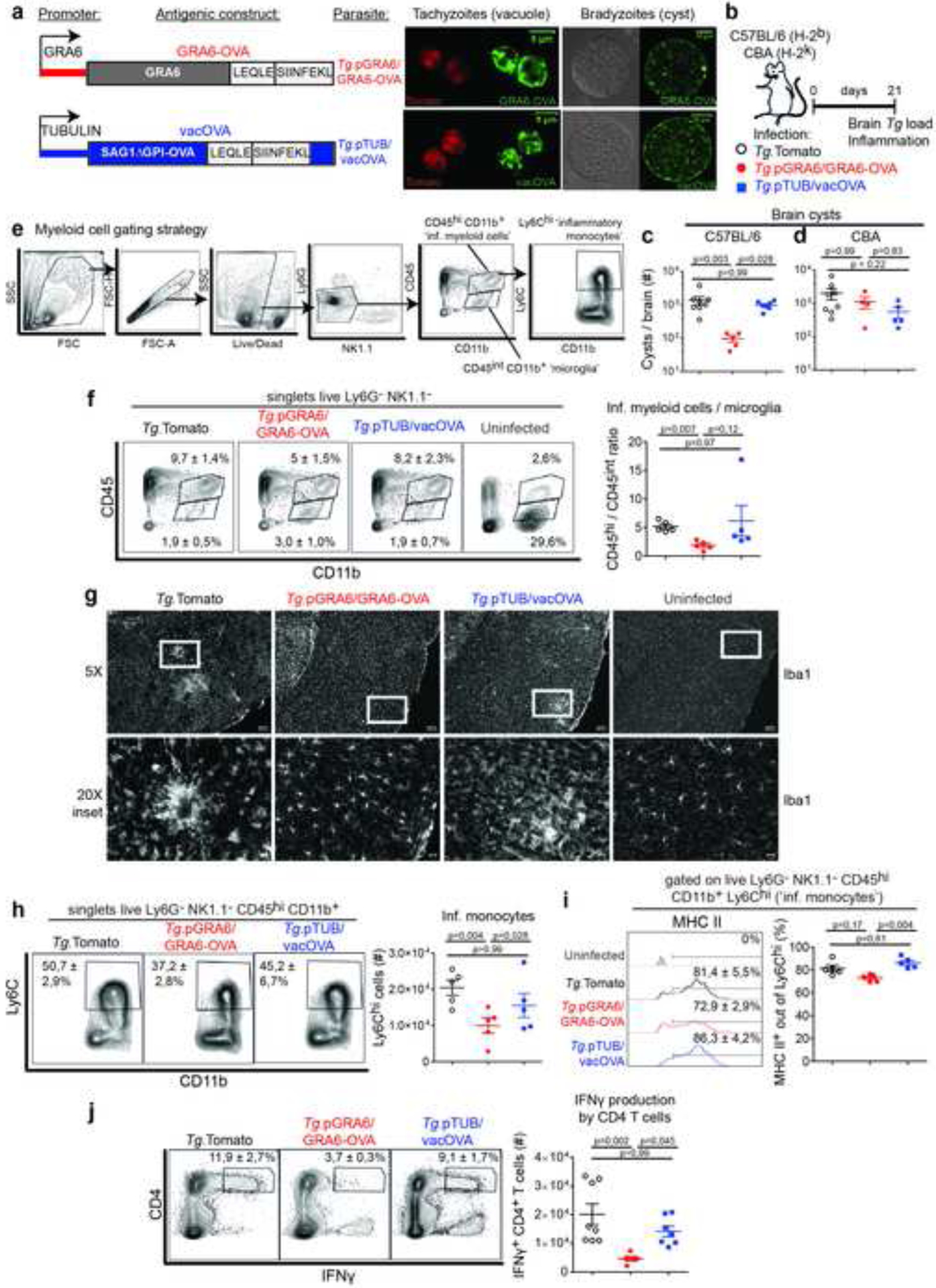
(a) Schematics of GRA6-OVA and vacOVA antigenic constructs expressed in the Tomato+ Prugnaud (Pru) parental strain. GRA6-OVA: fusion protein between GRA6(II) and LEQLE-SIINFEKL sequence, driven by the GRA6 promoter. vacOVA: fusion protein between SAG1ΔGPI and amino acids [140–386] of OVA, which contain the LEQLE-SIINFEKL sequence, driven by the tubulin promoter. Representative immunofluorescent images of a parasitophorous vacuole and an ex vivo cyst to illustrate activity of both promoters in tachyzoites and bradyzoites. Red: intrinsic Tomato fluorescence. Green: anti-SIINFEKL staining for GRA6-OVA, anti-full length OVA staining for vacOVA. (b) Schematics of experimental infections in mice infected i.p. with either of the 3 parasite strains. Analyses of brain parasite load and immunological status 3 weeks post-infection. (c, d) Number of brain cysts enumerated microscopically (mean +/− SEM) in H-2b C57BL/6 mice (c) and H-2k CBA mice (d). (e) Flow cytometry gating strategy to analyze myeloid cells in the CNS. (f) Analysis of inflammatory CD45hi CD11b+ myeloid cells and resident CD45int CD11b+ (‘microglia’) cells. Numbers on the representative contour plots show the mean percentage +/− SD of each subset out of single live Ly6G− NK1.1− cells. Graph shows the ratio (mean +/− SEM) of CD45hi over CD45int cells. (g) Representative brain cortical sections from uninfected and infected mice, stained for Iba1. Scale bar: 100 μm for 5X images, 25 μm for 20X images (h) Analysis of Ly6Chi inflammatory monocytes. Numbers on the representative contour plots show the mean percentage +/− SD of Ly6Chi cells out of single live Ly6G− NK1.1− CD45hi CD11b+ cells. Graph shows absolute numbers (mean +/− SEM). (i) Proportion of MHC II+ cells (DC) among Ly6Chi monocytes (mean percentage +/− SD). (j) IFNγ production by CNS-isolated CD4 T cells after incubation with brefeldin A. Numbers on the representative contour plots show the mean percentage +/− SD of IFNγ+ out of CD4+ T cells. Graph shows absolute numbers (mean +/− SEM). N = 4–8 mice / group. (c, j) Two experiments pooled. (d) Three experiments pooled. (f, h, i) One experiment representative of 3 independent experiments. (g) Representative field of view taken from a brain per condition. See also Figure S1.
Figure 4. Defective control of vacOVA-expressing T. gondii is not due to improper mobilization and effector differentiation of CD8 T cells in CNS.
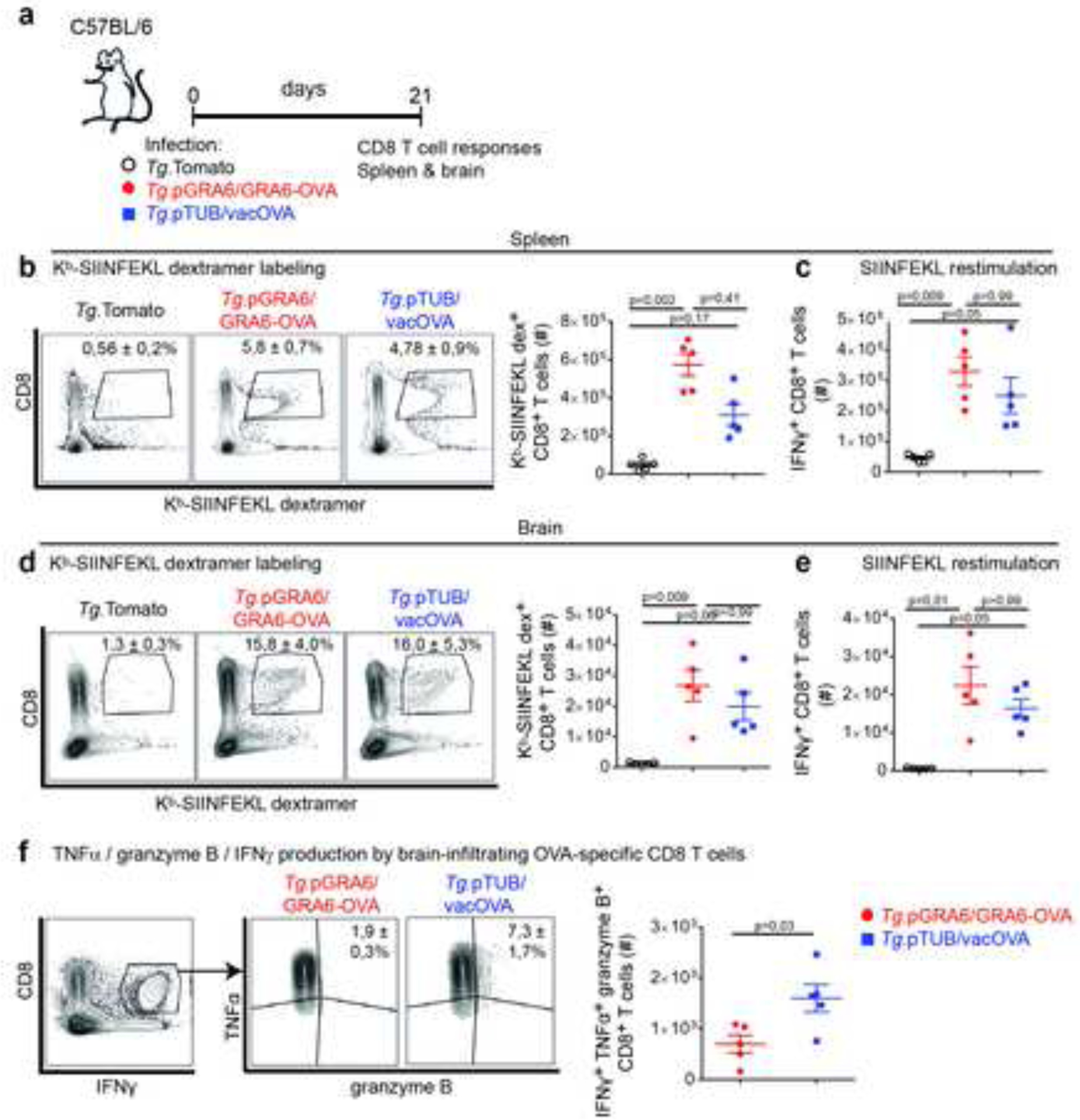
(a) Schematics of experimental infections in C57BL/6 mice infected i.p. with either of the 3 parasite strains. Evaluation of CD8 T cell responses in spleen and brain 3 weeks post-infection. (b, d) Kb-SIINFEKL dextramer labeling of spleen (b) and brain-infiltrating (d) CD8 T cells. Numbers on the representative contour plots show the mean percentage of dextramer+ out of CD8+ T cells +/− SD. Graph shows mean +/− SEM of absolute numbers. (c, e) Absolute numbers of IFNγ-producing CD8 T cells from spleen (c) and brain (e) following in vitro restimulation with OVA-derived SIINFEKL peptide (mean +/− SEM). (f) Analysis of triple-producing IFNγ+ TNF+ granzyme B+ brain CD8 T cells after restimulation with SIINFEKL peptide. Numbers on the representative contour plots show mean percentage of TNF+ granzyme B+ out of IFNγ+ CD8 T cells +/− SD. Graph shows absolute numbers of triple-producing CD8 T cells (mean +/− SEM). N=5 mice/ group. (b, c, d, e) Representative of 4 independent experiments. (f) Representative of 3 independent experiments.
Next we assessed whether the lower load of Tg.pGRA6/GRA6-OVA in the CNS indeed correlated with reduced brain inflammation. Besides a high cyst burden, the encephalitis caused by T. gondii is typically characterized by foci of tachyzoite replication, activation of brain-resident microglia and macrophages, infiltrates of activated lymphocytes and myeloid cells comprising inflammatory monocytes and DC (Biswas et al., 2015; Blanchard et al., 2015; Hwang et al., 2016; John et al., 2011; O’Brien et al., 2019; O’Brien et al., 2017; Zhang et al., 2014). As a reflection of the myeloid cell infiltrate, the relative abundance of brain-infiltrating CD11b+ CD45hi over brain-resident microglia (typically characterized as CD11b+ CD45int at steady-state), was reduced in mice infected with parasites expressing GRA6-OVA (Fig. 1e, f). Although the surface levels of MHC II and CD86 on CD11b+ CD45int microglia did not dramatically differ between the infected groups at this time point (Fig S1h, i, j, k), Iba1+ cells were found accumulated in foci in the cortex of mice infected with the parental or Tg.pTUB/vacOVA parasites (Fig. 1g). There was no substantial difference in the level of CD86 expression on CD11b+ Ly6Chi inflammatory monocytes across the infected groups (Fig S1l) but the infiltration of inflammatory Ly6Chi monocytes was decreased in the brains Tg.pGRA6/GRA6-OVA-infected mice (Fig. 1h), which also displayed the lowest proportion of monocyte-derived MHCII+ DC (Fig. 1i). At last, less activated IFNγ-producing CD4 T cells were found in brains from Tg.pGRA6/GRA6-OVA-infected mice (Fig. 1j).
Together, these results indicate that, in comparison to the parental strain or to vacOVA-expressing parasites, infection with GRA6-OVA-expressing type II T. gondii elicits efficient parasite control in the CNS and reduced CNS inflammation. They establish a useful model to study TE resistance in C57BL/6 mice.
Comparable OVA-specific CD8 T cell responses elicited by GRA6-OVA when expressed by both parasite stages or when restricted to tachyzoites
Depending on the time post-infection (early dissemination vs. chronic stage) and the immunological context examined (effective vs. poor parasite control), the CNS tissue can harbor varying numbers of T. gondii tachyzoites and bradyzoites (Bhadra et al., 2013). In order to examine which parasite stage(s) is/are efficiently held in check by CD8 T cells in the CNS in the context of latent toxoplasmosis, we generated parasites where GRA6-OVA expression was restricted to tachyzoites. To this aim, we drove expression of the GRA6-OVA antigen with the SAG1 promoter, which is active only at the tachyzoite stage, thereby generating the Tg.pSAG1/GRA6-OVA strain (Fig. 2a). Using two complementary assays, we confirmed that GRA6-OVA was indeed shut-down in Tg.pSAG1/GRA6-OVA bradyzoites. First, tachyzoite-infected fibroblasts were treated with apicidin, a histone deacetylase inhibitor which upregulates several genes involved in bradyzoite conversion (Bougdour et al., 2009; Boyle et al., 2006). We observed by Western blot that GRA6-OVA expression was lost in apidicin-treated Tg.pSAG1/GRA6-OVA bradyzoites (Fig. S2a, b). Because only part of the bradyzoite differentiation program is mimicked by apicidin treatment (Bougdour et al., 2009), we also evaluated expression of the respective antigenic constructs in ex vivo isolated bona fide cysts. GRA6-OVA could be visualized in Tg.pGRA6/GRA6-OVA cysts and, confirming the in vitro data, it was not detectable in Tg.pSAG1/GRA6-OVA cysts (Fig. 2b).
Figure 2. CD8 T cell responses against GRA6-OVA antigen are comparable whether or not bradyzoites express the antigen.
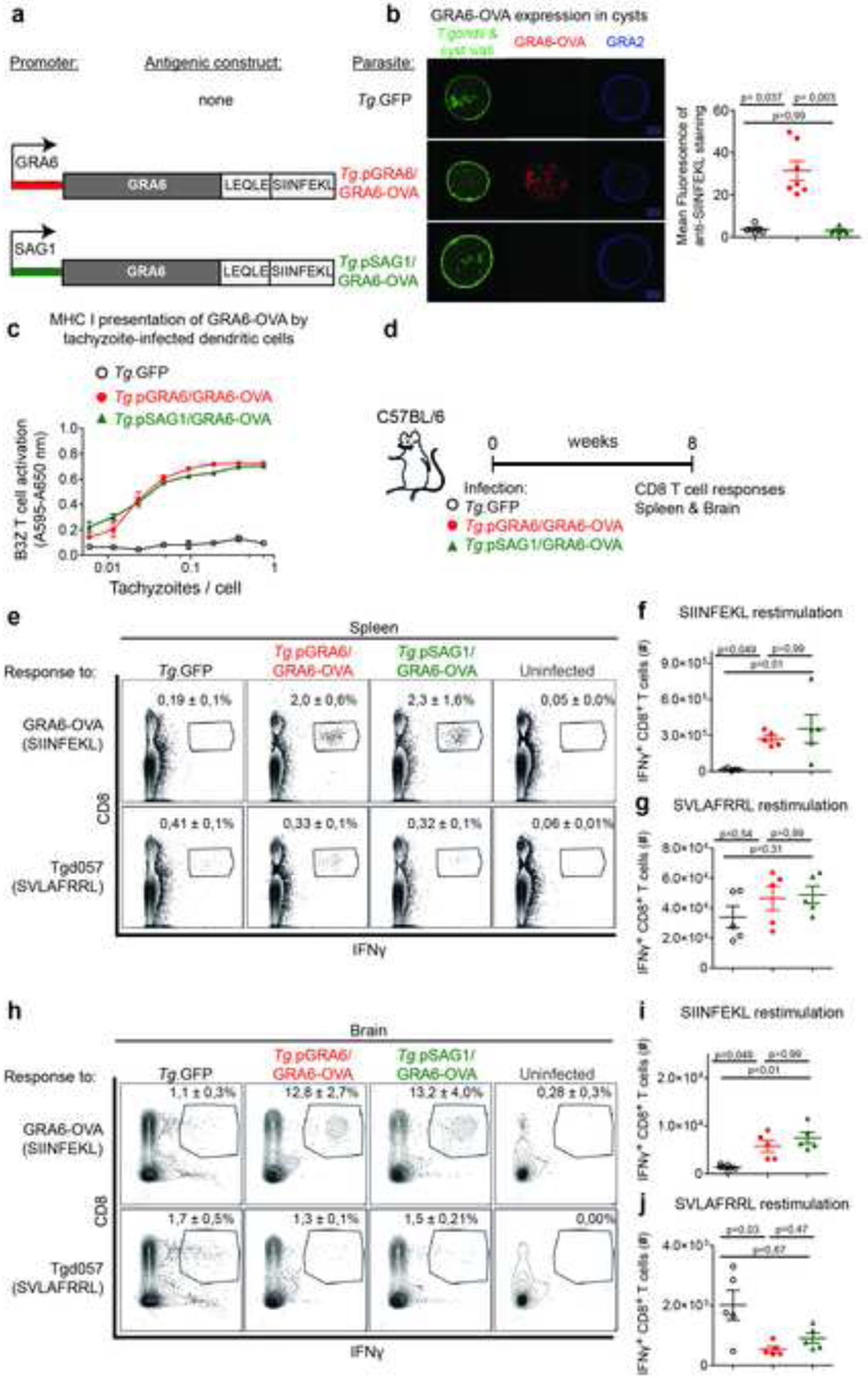
(a) Schematics of the antigenic constructs introduced in GFP+ Prugnaud strain (Tg.GFP). GRA6-OVA is driven either by the GRA6 promoter, active at the tachyzoite and bradyzoite stages, or by the tachyzoite-restricted SAG1 promoter. (b) Immunofluorescence staining of ex vivo-isolated cysts from CBA brains. Green: intrinsic parasite fluorescence and lectin-stained cyst wall. Red: GRA6-OVA detected with anti-SIINFEKL antibody. Blue: GRA2. Scale bar 10 μm. Right panel: quantification of SIINFEKL fluorescence within cyst. Each dot represents one cyst. (c) MHC I Kb presentation of GRA6-OVA-derived SIINFEKL peptide by MutuDC infected with the indicated parasite lines, assessed by absorbance measurements following incubation with LacZ-inducible OVA-specific B3Z CD8 T cell hybridomas. (d) Schematics of experimental infections in C57BL/6 mice infected i.p. with either of the 3 parasite strains. Evaluation of CD8 T cell responses 2 months post-infection. (e-j) IFNγ-producing CD8 T cells from spleen (e, f, g) or brain (h, i, j) following in vitro restimulation with OVA-derived SIINFEKL peptide (f, i) and Tgd057-derived SVLAFRRL peptide (g, j). (e, h) Numbers on the representative contour plots show the mean percentage +/− SD of IFNγ+ cells out of CD8+ T cells. (f, g, i, j) Absolute numbers (mean +/− SEM) of IFNγ+ CD8+ T cells. N = 5 mice / group. Representative of two independent experiments. See also Figure S2.
Having validated the stage expression profile of these two antigenic constructs, we analyzed MHC I presentation, CD8 T cell responses and dissemination to the brain during acute phase. Using LacZ-inducible reporter CD8 T cell hybridomas that specifically respond to the Kb-SIINFEKL MHC-peptide complexes (B3Z), we observed an equivalent presentation of the SIINFEKL peptide by DC infected with tachyzoites, regardless of the promoter used (Fig. 2c), which was consistent with a similar induction of OVA-specific CD8 T cell responses in the spleen 10 days post infection (Fig. S2c, d). In addition, in this context, initial parasite invasion of the brain was not substantially altered by the presence of GRA6-OVA compared to the parental strain (Fig. S2e).
We then proceeded to analyze CD8 T cell responses, parasite burden and CNS inflammation at chronic phase. We chose to perform the analyses at late chronic phase, i.e. 2 months post-infection, to limit the potential effects of pSAG1/GRA6-OVA residual expression that would be expected in the early days of chronic phase, when bradyzoites have converted only recently. Whether or not GRA6-OVA was expressed in bradyzoites, both strains elicited comparable CD8 T cell responses to the SIINFEKL peptide in the spleen and brain (Fig. 2d, e, f, h, i). CD8 T cells specific for another Kb-restricted peptide, the Tgd057-derived SVLAFRRL epitope, were found in similar proportions in the 3 groups in the spleen and were slightly more abundant in the brains of mice infected with the parental Tg.GFP (Fig. 2e, g, h, j). In conclusion, CD8 T cell responses developing against a T. gondii tachyzoite-restricted antigen are maintained throughout chronic infection, even in the absence of antigen expression by bradyzoites.
Tachyzoite-restricted GRA6-OVA expression is sufficient to provide robust CNS parasite control and to dampen TE
We next assessed the impact of tachyzoite-restricted GRA6-OVA expression on brain parasite control and TE pathogenesis (Fig. 3a). Compared to the parental strain, we observed a major reduction in brain cysts (Fig. 3b) and parasite DNA (Fig. 3c) in C57BL/6 mice infected with T. gondii expressing GRA6-OVA independently from the promoter. Consistent with brain parasite control being primarily mediated by Kb-OVA-specific CD8 responses (see Fig. 1d), parasite burdens were similar in chronically infected TE-susceptible H-2k CBA mice (Fig. 3d). In order to assess TE pathogenesis, we evaluated the CNS immune infiltration and activation of myeloid and CD4 T cells. Regardless of the promoter, mice infected with GRA6-OVA-expressing strains displayed a lower ratio of CD11b+ CD45hi inflammatory myeloid cells over CD11b+ CD45int microglia (Fig. 3e, f) and microglial populations were less activated, as evidenced by reduced CD86 and MHC II surface expression (Fig. 3g, h). We observed a less pronounced accumulation of CD11b+ Ly6Chi inflammatory monocytes (Fig. 3i), which displayed a lower, though not significant, level of CD86 expression (Fig. 3j) and among which there were fewer differentiated MHC II+ DC (Fig. 3k). At last, these brains contained less IFNγ-producing CD4 T cells (Fig. 3l). These results show that C57BL/6 mice infected with Tg.pGRA6/GRA6-OVA and Tg.pSAG1/GRA6-OVA have reduced brain inflammation, in contrast to mice infected with parental Tg.GFP.
Figure 3. Tachyzoite-restricted GRA6-OVA antigen confers sustained parasite control and protection against T. gondii encephalitis.
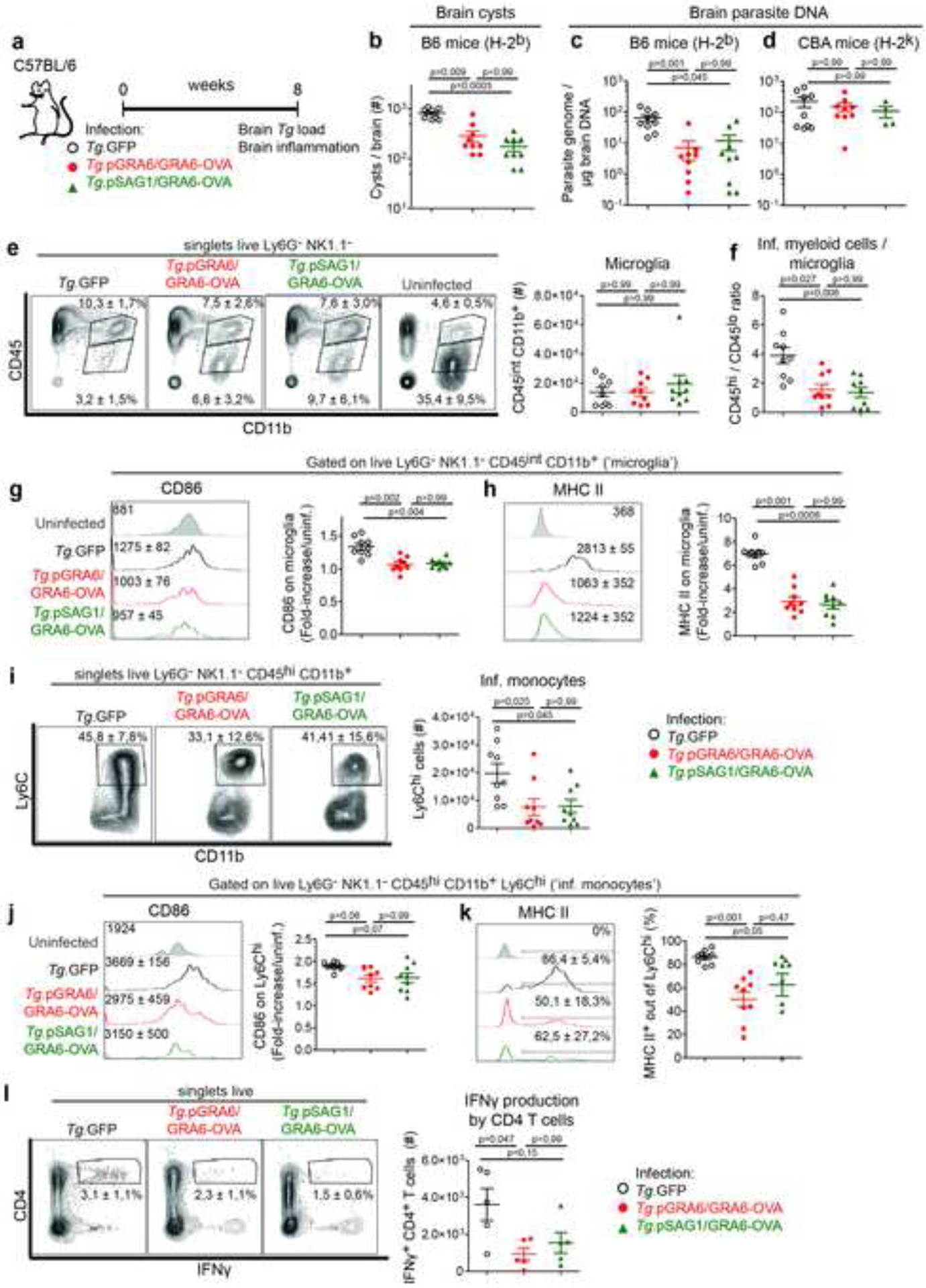
(a) Schematics of experimental infections in C57BL/6 mice infected i.p. with either of the 3 parasite strains. Analyses of brain parasite load and immune infiltrates 2 months post-infection. (b) Brain cysts enumerated microscopically in infected C57BL/6 mice (mean +/− SEM). (c, d) Parasite burden measured by qPCR on genomic DNA from infected C57BL/6 (c) or CBA (d) brains (mean +/− SEM). (e, f) Analysis of inflammatory CD45hi CD11b+ myeloid cells and resident CD45int CD11b+ (‘microglia’) cells. Numbers on the representative contour plots show the mean percentage +/− SEM of each subset out of single live Ly6G− NK1.1− cells. Graphs show the number of CD45int CD11b+ microglia (mean +/− SEM) (e) or the ratio of CD45hi over CD45int cells (mean +/− SEM) (f). (g, h) Expression level of MHC II (g) and CD86 (h) on the surface of microglia. Numbers on the representative histograms show the geomean +/− SD. Graphs display the fold-increase of each marker with respect to microglia from uninfected mice (mean +/− SEM). (i) Analysis of Ly6Chi inflammatory monocytes. Numbers on the representative contour plots show the mean percentage +/− SD of Ly6Chi cells out of single live Ly6G− NK1.1− CD45hi CD11b+ cells. Graph shows absolute numbers (mean +/− SEM). (j) Expression level of CD86 on the surface of Ly6Chi monocytes. Numbers on the representative histograms show the geomean +/− SD. Graph displays the fold-increase over uninfected (mean +/− SEM). (k) Proportion of MHC II+ cells (DC) among Ly6Chi monocytes. Numbers on the representative histograms and the graph show mean percentages +/− SD. (l) IFNγ production by CNS-infiltrating CD4 T cells after incubation with brefeldin A. Numbers on the representative contour plots show the mean percentage of IFNγ+ out of CD4+ T cells +/− SD. Graph shows absolute numbers (mean +/− SEM). For all panels, N = 9 mice / group with 2 experiments pooled, except for (l) where N = 5 mice / group from one experiment.
Altogether, this set of data indicates that CD8 T cell recognition of an efficiently processed tachyzoite-derived antigen is sufficient to provide effective parasite control in the CNS, thereby limiting the development of TE.
Limited TE development conferred by GRA6-OVA expression correlates with efficient neuronal MHC I presentation
We next sought to gain further insights into the modalities of MHC I presentation within the CNS, in particular regarding the nature of the antigen-presenting cells that determine the pathogenesis of TE. To this aim, we took advantage of the Tg.pTUB/vacOVA and Tg.pGRA6/GRA6-OVA parasite strains described in Figure 1, which express the same CD8 T cell epitope embedded in differentially protective source antigens, leading to varying levels of parasite control and CNS inflammation. We first interrogated whether the poor containment of Tg.pTUB/vacOVA parasites compared to Tg.pGRA6/GRA6-OVA could be due to improper recruitment and activation of OVA-specific CD8 T cells in the cerebral tissue. In contrast to the experiments in which the GRA6-OVA antigen was restricted to tachyzoites (see Fig. 2 and 3), there was no concern about residual antigen expression in the early chronic phase. Furthermore, the differences in brain parasite burden and inflammation of Figure 1 were observed at 3 weeks post-infection. Therefore, we assessed the abundance, specificity and effector functions of splenic and brain-infiltrating CD8 T cells 3 weeks post-infection (Fig. 4a). In the spleen, substantial SIINFEKL-specific CD8 T cells were elicited by both GRA6-OVA- and vacOVA-expressing parasites, with a slightly higher magnitude in the context of Tg.pGRA6/GRA6-OVA infection (Fig. 4b, c). In the brain, the numbers of Kb-SIINFEKL dextramer+ CD8 T cells and of IFNγ-producing CD8 T cells after SIINFEKL peptide restimulation were similar between Tg.pTUB/vacOVA and Tg.pGRA6/GRA6-OVA-infected mice (Fig. 4d, e). Analysis of the effector potential of OVA-specific CD8 T cells following in vitro peptide restimulation showed that vacOVA-expressing parasites in fact led to larger quantities of triple-producing IFNγ+ TNF+ granzyme B+ CD8 T cells (Fig. 4f). This may be related to the higher parasite burden observed in this setting. A more abundant antigenic material could, via MHC I presentation by local APC, accelerate CD8 T cell differentiation into effector cells. In conclusion however, poor control of vacOVA-expressing parasites is not due to defective infiltration or impaired effector differentiation of T. gondii-specific CD8 T cells in the infected CNS. On this ground, we hypothesized that what may be subpar in the vacOVA context is the recognition of infected target cells by CD8 T cells.
Neurons being the most predominant host cells for T. gondii in the CNS (Cabral et al., 2016; Ferguson and Hutchison, 1987; Melzer et al., 2010), we speculated that inefficient neuronal MHC I presentation of vacOVA compared to GRA6-OVA may underlie impaired parasite control. To test this possibility, we set out to probe antigen presentation by primary neurons infected with Tg.pTUB/vacOVA and Tg.pGRA6/GRA6-OVA tachyzoites using the SIINFEKL-specific LacZ-inducible B3Z reporter CD8 T cell hybridomas (Fig. 5a). First, we optimized neuronal culture conditions to ensure that the proportion of glial cells was minimal because these are potent antigen-presenting cells that may blur the interpretation of the assay (Fig. S3a, b). We also verified that 24 h post-infection, > 95% of the T. gondii tachyzoite-containing vacuoles were harbored in neurons (Fig. S3c) and that there were no differences in neuronal infection rates between the two parasite strains (Fig. 5b, c). Antigen presentation measurements revealed that the GRA6-OVA antigen was processed and presented by H-2Kb to the reporter CD8 T cells more efficiently than the vacOVA antigen (Fig. 5d).
Figure 5. Poorer Kb-SIINFEKL presentation by neurons infected with T. gondii expressing vacOVA compared to neurons infected with T. gondii expressing GRA6-OVA.
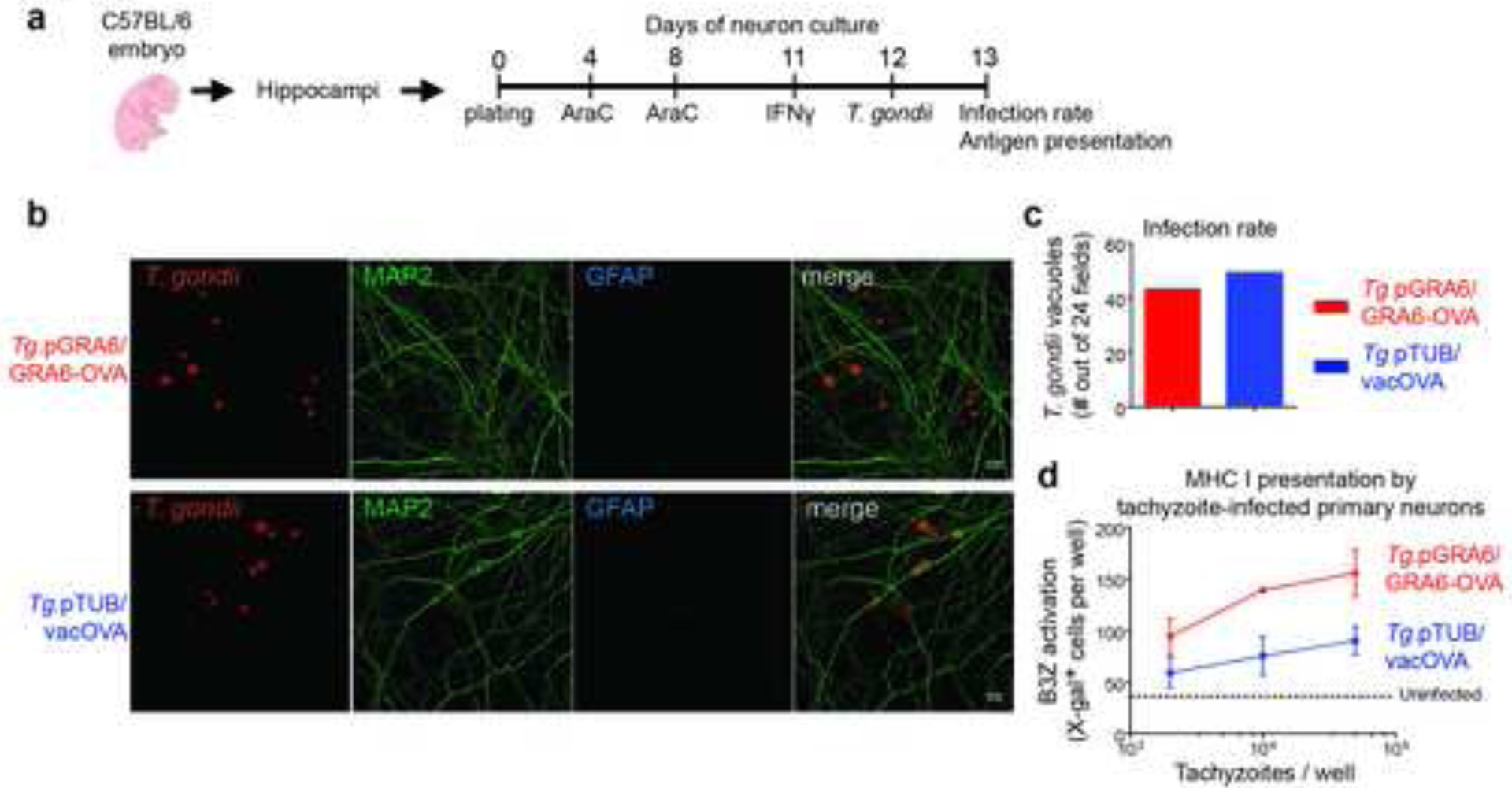
(a) Timeline of antigen presentation assay with Tg-infected primary neuronal cultures. AraC was added to inhibit growth of glial cells. (b, c) Tropism and infection rate of the two Tomato+ parasites following co-staining with MAP2 (green) and GFAP (blue). Pictures represent maximum intensity projections. Out of 24 fields, no vacuole was detected in a glial cell. Scale bar 10 μm. (d) Kb–SIINFEKL presentation by primary neurons infected for 24 h with the indicated tachyzoites, assessed by absorbance measurements following incubation with SIINFEKL-specific LacZ-inducible B3Z CD8 T cell hybridomas. (b, c, d) Representative of 2 independent experiments. See also Figure S3.
These data support the notion that failure to control vacOVA-expressing parasites in the CNS may be related to sub-optimal MHC I presentation of T. gondii tachyzoite antigens by infected neurons.
MHC I Ld selective ablation in neurons hampers control of parasite load in the CNS
To formally address the in vivo requirement for neuronal MHC I antigen presentation in the control of CNS parasites, we designed a Cre-loxP-based conditional deletion approach. Because MHC I molecules play important functions during brain development and plasticity (Corriveau et al., 1998; Elmer and McAllister, 2012; Huh et al., 2000), we reasoned that it would be appropriate to leave the endogenous C57BL/6 MHC I locus intact while introducing a floxed version of H-2 Ld, the allele that was originally associated with TE resistance (Blanchard et al., 2008; Brown et al., 1995). We thus generated transgenic B6.LdLox mice expressing a floxed Ld gene, in which exons 1 to 3 are flanked by LoxP sites (Fig. 6a). As expected, T cells, B cells and DC in the spleen of B6.LdLox mice displayed Kb levels that were analogous to C57BL/6 mice (Fig. 6b). Surface expression of Ld in B6.LdLox mice was slightly reduced compared to B6.H-2d congenic mice, which naturally express H-2 Ld (Fig. 6c) but despite this, B6.LdLox and B6.H-2d mice mounted robust CD8 T cell responses to the Ld-restricted T. gondii GRA6-derived HF10 peptide in the spleen and brain (Fig. 6d, e, f). Accordingly, compared to C57BL/6 mice, B6.LdLox mice were potent in their ability to control brain T. gondii during chronic infection (Fig. 6g).
Figure 6. MHC I presentation by CNS neurons is required for efficient brain control of T. gondii at chronic phase.

(a) Schematic representation of the LdLox DNA cassette introduced in C57BL/6 fertilized eggs to generate the B6.LdLox mice. The cassette is a 12-kb genomic sequence comprising the Ld gene modified with 2 LoxP sites flanking exons 1 to 3 (domains L, N and C1 according to nomenclature from (Evans et al., 1982)) and the endogenous 5’ and 3’ UTR regulatory sequences from BALB/c. (b, c) Flow cytometry labeling of H-2 Kb (b) and H-2 Ld (c) on the surface of CD19− CD3+ T cells, CD3− CD19+ B cells and CD3− CD19− CD11c+ DC in the spleen of the indicated mouse strains. Numbers on the histograms show the geomean +/− SD. (b, c) Representative of two independent experiments. N=2–4 mice / group. (d) Schematics of experimental infections in C57BL/6, B6.LdLox and congenic B6.H-2d mice infected i.p. with Tg.pTUB/vacOVA, which naturally expresses GRA6. Evaluation of CD8 T cell responses and parasite load 3 weeks later. (e, f) Absolute numbers of IFNγ-producing CD8 T cells isolated from spleen (e) and brain (f), following in vitro restimulation with Ld-restricted GRA6-derived HF10 peptide (mean +/− SEM). (g) Number of brain cysts in mice of the indicated genotypes (mean +/− SEM). (e-g) Two experiments pooled with N=3–4 mice / group. (h) Schematics of the breeding strategy and outcome in terms of MHC I molecules expressed by neurons vs. non-neuronal cells in Cre− and Cre+ mice. (i) Schematics of experimental infections in C57BL/6, B6.LdLox.Cre+ or Cre− infected per os with 15 cysts of the 76K strain. Evaluation of CD8 T cell responses and brain parasite load 3 weeks later. (j) Number of brain cysts enumerated microscopically. (k) Brain parasite burden measured by qPCR on genomic DNA. (l) Absolute number of total CD8 T cells isolated from infected brains. (m) Absolute numbers of brain-isolated IFNγ-producing CD8 T cells, following in vitro restimulation with Ld-restricted GRA6-derived HF10 peptide. (j-m) Graphs show the mean +/− SEM. N=5–6 mice / group. Representative of 2 independent experiments. See also Figure S4, S5 and S6.
In order to selectively eliminate Ld from neurons, we bred B6.LdLox with B6.CamKIIα-iCre mice, which express the Cre recombinase in all excitatory glutamatergic neurons of the CNS (forebrain, hippocampus, olfactory lobe and scattered cells in hypothalamus) (Casanova et al., 2001; Wang et al., 2013). In Cre+ mice, CamKIIα+ neurons should express only Kb and Db whereas all other cells should express Kb, Db and Ld (Fig. 6h). The level of Ld on the surface of leukocytes isolated from infected brains was indeed comparable regardless of Cre expression (Fig. S4a). In addition, as shown in primary co-cultures of neuronal and glial cells from CamKIIα-Cre+ vs. CamKIIα-Cre− mice, Ld expression was significantly reduced in Cre+ neurons while it was not changed in Cre+ glial cells (Fig. S4b, c). To monitor parasite-specific CD8 T cell responses, we infected mice with a T. gondii strain that naturally carries the endogenous GRA6 and Tgd057 proteins but is devoid of the OVA antigen. Effector CD8 T cell responses specific for the GRA6-derived Ld-restricted HPGSVNEFDF peptide and the Tgd057-derived Kb-restricted SVLAFRRL peptide were elicited similarly in the periphery of Cre+ and Cre− mice (Fig. S5a, b, c). Parasite dissemination in the spleen, peritoneum and brain throughout acute infection was also comparable in the presence or absence of Ld on neurons (Fig. S5d, e, f). To evaluate the impact of Ld neuronal deletion during chronic infection, we infected mice per os with the 76K unmanipulated strain, akin to what occurs during natural infection. On week 3 post-infection, there was a >10-fold increase in brain parasite burden in mice devoid of Ld in neurons, reflected both by elevated cyst numbers (Fig. 6i, j) and parasite DNA (Fig. 6k). Brain parasite control in Cre+ mice was as poor as that observed in chronically infected TE-susceptible C57BL/6 mice (Fig. 6j, k), indicating that the selective absence of Ld on neurons critically impairs the ability of local CD8 T cells to restrict CNS parasites. To check if Ld deficiency in neurons impeded the development of cerebral CD8 T cell responses, we quantified the numbers of total CD8 T cells and GRA6-specific CD8 T cells in chronically infected brains. Absence of Ld in neurons did not abrogate the infiltration of total CD8 T cells (Fig. 6l), nor did it preclude the accumulation of IFNγ-producing GRA6-specific CD8 T cells (Fig. 6m). Evaluation of the ratio of inflammatory myeloid cells over microglia, of the activation status of microglia and of the IFNγ-producing CD4 T cells in the CNS did not reveal substantial differences between Cre− and Cre+ animals (Fig. S6), suggesting that at 3 weeks post-infection, neuronal presentation of T. gondii antigens by Ld plays a limited role in regulating CNS inflammation. Altogether, our findings establish that efficient MHC I presentation of immunodominant T. gondii antigen by tachyzoite-infected neurons is dispensable for the accumulation of CD8 T cells in the CNS but is a prerequisite for sustained parasite control by CD8 T cells in this organ.
Discussion
Combining transgenic T. gondii parasites, antigen presentation assays with primary neurons and a mouse model of conditional MHC I deletion, our data support a model whereby the efficacy of CD8 T cell-mediated surveillance of tachyzoite-infected neurons determines the severity of CNS inflammation. Notably, C57BL/6 mice infected with GRA6-OVA-expressing parasites durably control the parasite in the CNS and show limited encephalitis throughout late chronic infection.
Our work sheds light on the stage specificity of CD8 T cell-mediated surveillance of T. gondii in the CNS. In the context of TE development, two-photon laser scanning microscopy imaging of infected brains previously revealed that CD8 T cells contact granuloma-like structures containing tachyzoites and that they interact in a cognate manner with individual CD11b+ or CD11c+ antigen-presenting cells (APC) that are not necessarily infected (John et al., 2011; Schaeffer et al., 2009). It was also found that CD8 T cells largely ignore neurons harboring cysts (Schaeffer et al., 2009), in line with the notion that, as ‘dormant’ persisting structures, cysts must be impervious to host immunity. However bradyzoites within tissue cysts are dynamic and replicative entities (Watts et al., 2015) and intra-neuronal cysts contain up to thousands of bradyzoites that continue to express and secrete a number of proteins, some of which are major CD8 T cell targets (e.g. ROP7 (Frickel et al., 2008) and GRA6). Therefore, in the context of latent toxoplasmosis, a different scenario may prevail whereby CD8 T cells could directly recognize and eliminate cysts. In accordance, it was reported in a TE reactivation model in which immunodeficient BALB/c mice are treated with sulfadiazine, that following adoptive transfer, total splenic CD8 T cells isolated from chronically infected mice could reduce cyst count through a perforin-dependent, IFNγ-independent, modus operandi (Suzuki et al., 2010). Here, in the course of primary infection of an immunocompetent host, we report that T. gondii-infected neurons are able to process tachyzoite antigens and to display antigenic fragments via MHC I for CD8 T cell recognition. This is then sufficient to restrict brain cyst load up to 2 months post-infection, stressing the notion that CD8 T cell recognition of tachyzoite-derived antigens is a prominent driver to limit the development of TE. In this model, we hypothesize that antigenic peptides are displayed by neuronal MHC I as soon as a neuron gets infected by a tachyzoite and that this is enough to trigger cytotoxic T lymphocyte (CTL)-mediated containment of the parasite invader. An interesting implication is that in the context of TE resistance, the parasites are controlled ‘from the beginning’ (as tachyzoites) and that bradyzoite conversion only minimally occurs. It will now be essential to determine by which mechanisms bradyzoites actively escape CD8 T cell surveillance, including in the context of the robust CD8 T cell responses leading to reduced encephalitis. Current drugs are ineffective on cysts, hence being able to restore presentation of bradyzoite antigens in order to boost cyst clearance by CD8 T cells could ultimately open new therapeutic avenues for at-risk individuals.
Another asset of our work is the creation of a mouse model to genetically test the importance of neuronal MHC I presentation in the control of T. gondii within the CNS. In vitro validation of the B6.LdLox model using primary neuronal cultures showed a selective reduction of Ld in neurons compared to glial cells, but not a complete loss. The residual expression of Ld observed in Cre+ neurons in this assay may be linked to the late and/or heterogenous expression of the CamKIIα promoter in in vitro cultures as well as to the half-life of the Ld protein following Cremediated excision. Since our attempts to detect Ld directly on brain sections have not been successful, one cannot exclude that a fraction of CamK-Cre+ neurons still retain Ld expression in vivo following infection. Regardless, we found major consequences on parasite burden, indicating that MHC I presentation by CamKα+ excitatory glutamatergic neurons plays an important role for the control of T. gondii in the CNS. Although initially controversial, it is now indisputable that MHC I molecules are expressed by neurons, at the surface of both axons and dendrites (Elmer and McAllister, 2012). Neuronal MHC I plays essential functions during brain development by restricting the activity-dependent plasticity of connections (Huh et al., 2000) and negatively regulating synaptic density (Glynn et al., 2011). In addition, several lines of evidence support the notion that neuronal MHC I can present antigenic peptides and activate CD8 T cells. Expression of a neo-antigen by neuronal subtypes, like the orexinergic neurons (Bernard-Valnet et al., 2016) or Purkinje cells (Yshii et al., 2016), was associated with CTL granule polarization and destruction of the neuron subset expressing the antigen. In viral infections such as with Lymphocytic Choriomeningitis Virus (LCMV) (Kreutzfeldt et al., 2013; Rall et al., 1995), Theiler’s murine encephalomyelitis virus (McDole et al., 2010) and Borna disease virus (Chevalier et al., 2011), CTL have been shown to interact with infected neurons in a MHC I- and antigen-specific manner. Our work now demonstrates the essentiality of MHC I presentation for effective CD8 T cell control of a widespread, non-viral, neurotropic pathogen.
What may be the outcome of neuronal MHC I presentation on neuron function? In certain contexts, contacts of neurons with CTL lead to neuron killing (Bernard-Valnet et al., 2016; Cebrian et al., 2014) but in other situations, they induce more subtle morphological changes, such as an increase in membrane permeability (Chevalier et al., 2011), synaptic stripping (Di Liberto et al., 2018) or a loss of axon integrity (Sauer et al., 2013), without triggering immediate apoptosis. An alteration of electrical properties has also been reported (Meuth et al., 2009). Future investigations should be undertaken to address whether cognate contacts between CTL and T. gondii-infected neurons could result in alterations of the electrical activity of individual neurons or integrated neuronal networks (Casanova et al., 2018). These experiments may ultimately shed light on some mechanisms behind the behavioral alterations induced by T. gondii.
Importantly, our study does not rule out important contributions from other CNS-resident cells in regulating the course of toxoplasmosis via MHC I presentation of pathogen fragments. The main impact of Ld deletion in neurons is to hamper parasite control but the consequences on brain inflammation appear more modest. It does not seem so surprising since regardless of the Cre status, Ld presentation is expected to remain intact in all other brain-resident and - infiltrating populations, which are likely to be critical for T cell/myeloid cell infiltration. Chief among these are microglia, which are potent antigen-presenting cells involved in regulating effector and memory CD8 T cells that have reached the CNS (Colonna and Butovsky, 2017). In chronically LCMV-infected mice, microglia act as a viral reservoir and they can be prompted to present viral antigens that promote viral purge by CD8 T cells (Herz et al., 2015). In T. gondii-infected CNS, microglia are activated in the vicinity of T. gondii replication foci during TE (Zhang et al., 2014). Since Ld elimination from neurons only modestly impacted CD8 T cell accumulation, it is tempting to speculate that Ld deletion from microglia would instead profoundly disrupt CD8 T cell infiltration and/or memory CD8 T cell formation in the brain. Astrocytes is another cell type, which antigen-presenting function would deserve further scrutiny (Wilson and Hunter, 2004). T cells may encounter astrocytes either in the parenchyma or when crossing the blood-brain barrier endothelium that is surrounded by astrocytic endfeet. Studies in viral infections uncovered a possible interplay between astrocytes and CD8 T cells (Xie and Yang, 2015). Thanks to the above-described mouse model and to a recently published floxed Kb system (Malo et al., 2018), it will now be possible to explore the role(s) of MHC I presentation by CNS-resident cells in homeostasis and disease. In the case of chronic T. gondii infection, these models may be useful to better understand how brain function is impacted by this widespread parasite.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nicolas Blanchard (nicolas.blanchard@inserm.fr)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human cell lines
Human Foreskin Fibroblasts (HFF) were purchased from ATCC (see Key Resources Table). They were maintained in DMEM supplemented with 10 % FCS (Gibco). Gender is unknown.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| LEAF™ Purified anti-mouse CD16/32 antibody | BioLegend | Cat# 101321, RRID:AB_1877064 |
| PE Rat Anti-Mouse CD8a | Thermo Fisher Scientific | Clone 53-6.7; Cat# 12-0081-83; RRID:AB_465531 |
| BV421 Rat Anti-Mouse CD8a | BD Biosciences | Clone 53-6.7; Cat# 563898; RRID:AB_2738474 |
| BV510 Rat Anti-Mouse CD4 | BD Biosciences | Clone RM4-5; Cat# 563106; RRID:AB_2687550 |
| PE-Cy™7 Rat Anti-Mouse CD4 | BD Biosciences | Clone RM4-5; Cat# 552775; RRID:AB_394461 |
| APC Rat Anti-Mouse IFN-γ | BD Biosciences | Clone XMG1.2; Cat# 554413; RRID:AB_398551 |
| Alexa Fluor® 700 Rat anti-Mouse TNF | BD Biosciences | Clone MP6-XT22; Cat # 558000; RRID:AB_396980 |
| PE Rat anti-mouse Granzyme B Monoclonal Antibody, eBioscience™ | Thermo Fisher Scientific | Clone NGZB; Cat # 12-8898-82; RRID:AB_10870787 |
| BV421 Hamster Anti-Mouse CD3e | BD Biosciences | Clone 145-2C11; Cat # 562600; RRID:AB_11153670 |
| PE-Cy™7 Rat Anti-Mouse CD19 | BD Biosciences | Clone 1D3; Cat # 552854; RRID:AB_394495 |
| PE Hamster Anti-Mouse CD11c | BD Biosciences | Clone HL3; Cat # 557401; RRID:AB_396684 |
| PE-CF594 Rat Anti-CD11b | BD Biosciences | Clone M1/70; Cat # 562287; RRID:AB_11154216 |
| PerCP-Cy5.5 mouse anti-mouse H-2Kb Antibody | Biolegend | Clone AF6-88.5; Cat # 116516; RRID:AB_1967133 |
| Alexa Fluor® 700 Rat Anti-Mouse CD4 | BD Biosciences | Clone RM4-5; Cat # 557956; RRID:AB_396956 |
| Brilliant Violet 510™ Rat anti-mouse Ly-6G | BioLegend | clone 1A8; Cat # 127633; RRID:AB_2562937 |
| PerCP-Cy™5.5 Rat Anti-Mouse CD45 | BD Biosciences | Clone 30-F11; Cat # 550994; RRID:AB_394003 |
| PE Mouse Anti-Mouse NK-1.1 | BD Biosciences | Clone PK136; Cat# 557391; RRID:AB_396674 |
| Alexa Fluor® 700 rat anti-mouse CCR2 | R & D Systems | Clone # 475301; Cat # FAB5538N; RRID:AB_2725739 |
| APC Rat anti-Mouse CD86 | BD Biosciences | clone GL1; Cat # 558703; RRID:AB_2075114 |
| Brilliant Violet 711™ Rat anti-mouse Ly-6C | BioLegend | clone HK1.4; Cat # 128037; RRID:AB_2562630 |
| PE Rat Anti-CD11b | BD Biosciences | Clone M1/70; Cat # 553311; RRID: AB_394775 |
| Alexa Fluor 700 rat anti-mouse CD86 | BD Biosciences | Clone GL1; Cat # 560581; RRID:AB_1727517 |
| FITC Rat Anti-Mouse I-A/I-E | BD Biosciences | Clone 2G9; Cat # 553623; RRID:AB_394958 |
| FITC Hamster anti-mouse CD3 | BD Biosciences | Clone 145-2C11; Cat # 553062; RRID:AB_394595 |
| Monoclonal Anti-MAP2 (2a+2b) antibody produced in mouse | Sigma-Aldrich | clone AP-20; Cat # M1406; RRID:AB_477171 |
| Anti-Glial Fibrillary Acidic Protein (GFAP) antibody | Millipore | Cat # AB5804; RRID:AB_2109645 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat # A-11001; RRID:AB_2534069 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat # A-21244; RRID:AB_2535812 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat # A-11032; RRID:AB_2534091 |
| F(ab’)2-Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat # A-11070; RRID:AB_2534114 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen | Cat # A21429; RRID:AB_2535850 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat # A-21235; RRID: AB_2535804 |
| Anti-Rabbit IgG (H+L), HRP Conjugate antibody | Promega | Cat # W4011; RRID:AB_430833 |
| Anti-Mouse IgG (H+L) Antibody, HRP Conjugated | Promega | Cat # W4021; RRID:AB_430834 |
| Rabbit anti-HPGSVNEFDF | (Buaillon et al., 2017) | N/A |
| Rabbit anti-SIINFEKL | Biotem, this study | N/A |
| Mouse anti-GRA 1 | Biotem; M.-F. Cesbron-Delauw | clone TG17.43 |
| Mouse anti-GRA2 | Biotem; M.-F. Cesbron-Delauw | clone Tg17-179 |
| H2-Kb-SIINFEKL Dextramer PE | Immudex | Cat # JD2163 |
| Rabbit anti-TgProfilin | D. Soldati-Favre | PRF556 |
| Rabbit anti-chicken OVA | Sigma | Cat # C6534; RRID: AB_258953 |
| Rabbit anti-Iba1 | Wako | Cat # 019-19741; RRID: AB_839504 |
| Rabbit anti-MAP2 | Millipore | Cat# AB5622; RRID: AB_91939 |
| anti-H-2Ld AF647 | Biotem, (Ozato et al., 1980) | clone 30-5-7 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| HPGSVNEFDF (HF10) | Genecust | N/A |
| SIINFEKL | Genecust | N/A |
| SVLAFRRL | Genecust | N/A |
| Mouse IFN-γ premium grade | Miltenyi Biotec | Cat # 130-105-774 |
| Mycophenolic acid | Sigma-Aldrich | Cat # M5255 |
| Xanthine | Sigma-Aldrich | Cat # X-2502 |
| DNAse I from bovine pancreas | Sigma-Aldrich | Cat # DN25 |
| Collagenase D from Clostridium Histolyticum | Roche | Cat # 11088882001 |
| Rhodamine labeled Dolichos Biflorus Agglutinin (DBA) | Vector Laboratories | Cat # RL-1032; RRID: AB_2336396 |
| eBioscience™ Fixable Viability Dye eFIuor™ 450 | Invitrogen | Cat # 65-0863-14 |
| eBioscience™ Fixable Viability Dye eFIuor™ 660 | Invitrogen | Cat # 65-0864-14 |
| LIVE/DEAD™ Fixable Green Dead Cell Stain Kit, for 488 nm excitation | Invitrogen | Cat # L34970 |
| Triton X-100 | Sigma-Aldrich | Cat # X100 |
| Tween®20 | Sigma-Aldrich | Cat # P1379 |
| ProLong Diamond Anti-Fade Mounting medium with DAPI | Invitrogen | Cat # P36962 |
| Paraformaldehyde 20 % aqueous solution | Electron Microscopy Sciences | Cat # 15713 |
| Apicidin | Sigma-Aldrich | Cat # A8851 |
| Laemmli Sample Buffer | BIO-RAD | Cat # 1610737 |
| Percoll | GE Healthcare | Cat # 17-0891-01 |
| eBioscience Brefeldin A solution | Thermo Fisher Scientific | Cat # 00-4506-51 |
| eBioscience Permeabilization Buffer (10X) | Thermo Fisher Scientific | Cat # 00-8333-56 |
| Poly-D-lysine | Merck Millipore | Cat # A-003-E |
| UltraPure™ DNase/RNase-Free Distilled Water | Invitrogen | Cat # 10977035 |
| Laminin Mouse Protein, Natural | Invitrogen | Cat # 23017-015 |
| Papain | Worthington Biochemical Corporation | Cat # LK003176 |
| Bovine Serum Albumin | Dutscher | Cat # SH30574.02 |
| Trypsin Inhibitor from chicken egg white | Roche | Cat # 10109878001 |
| B-27 supplement | Gibco | Cat # 17504044 |
| GlutaMAX Supplement | Gibco | Cat # 35050061 |
| Cytarabine Hydrochloride | Sigma-Aldrich | Cat # C6645 |
| Normal Goat Serum | Vector Laboratories | Cat # S-1000 |
| chlorophenol red-β-D-galactopyranoside CPRG | Roche | Cat # 10884308001 |
| Glutaraldehyde 8% aqueous solution | Electron Microscopy Science | Cat # 16019 |
| X-gal | Sigma-Aldrich | Cat # B4252 |
| Potassium Ferrocyanide | Sigma-Aldrich | Cat # 60279 |
| Potassium Ferricyanide | Sigma-Aldrich | Cat # 60299 |
| Magnesium Chloride Hexahydrate | Sigma-Aldrich | Cat # M2670 |
| Tetrodotoxin | Sigma-Aldrich | Cat # T8024 |
| TBS | Euromedex | Cat # ET220 |
| DAPI | Sigma-Aldrich | Cat # D9542 |
| Fluoromount medium | Electron Microscopy Sciences | Cat # 17984-25 |
| Critical Commercial Assays | ||
| DNEasy Blood and Tissue Kit | Qiagen | Cat # 69504 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| MutuDC | (Fuertes Marraco et al., 2012), H. Acha-Orbea | N/A |
| Human Foreskin Fibroblasts (HFF) | ATCC | Cat# SCRC-1041 |
| SIINFEKL-specific LacZ-inducible CD8 T cell reporter hybridomas (B3Z) | (Karttunen et al., 1992), N. Shastri | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: CBA: CBA/JRj | Janvier | N/A |
| Mouse: B6 or C57BL/6: C57BL/6J | Janvier | N/A |
| Mouse: B6.H-2d: B6.C-H2d/bByJ | Jax | Cat # 000359 |
| Mouse: B6.CamKIIα-iCre | (Casanova et al., 2001), G. Schutz | N/A |
| Mouse: B6.LdLox: | This study | N/A |
| T. gondii: 76K | (Bonnart et al., 2017); D. Buzoni-Gatel | N/A |
| T. gondii: Tg.Tomato: Pru.Δhxgprt.tdTOMATOprom TUB | (Schaeffer et al., 2009) | N/A |
| T. gondii: Tg.pTUB/vacOVA: Pru.Δhxgprt.tdTOMATOprom TUB.SAG1ΔGPI-OVA[140-386]prom TUB/3’utr DHFR +BLE | (Schaeffer et al., 2009) | N/A |
| T. gondii: Tg.pGRA6/GRA6-OVA: Pru.Δhxgprt.tdTOMATOprom TUB.GRA6II-LEQLE-SIINFEKLprom GRA6/3’utr GRA2 +HXGPRT | This study | N/A |
| T. gondii: Tg.GFP: Pru.Δhxgprt.GFPprom GRA1.click beetle LUCprom DHFR | (Kim et al., 2007) | N/A |
| T. gondii: Tg.pGRA6/GRA6-OVA: Pru.Δhxgprt.GFPprom GRA1.click beetle LUCprom DHFR.GRA6II-LEQLE-SIINFEKLprom GRA6/3’utr GRA2 +HXGPRT | This study | N/A |
| T. gondii: Tg.pSAG1/GRA6-OVA: Pru.Δhxgprt.GFPprom GRA1.click beetle LUCprom DHFR.GRA6II-LEQLE-SIINFEKLprom SAG1/3’utr GRA2 +HXGPRT | This study | N/A |
| Oligonucleotides | ||
| TOX9: 5’-AGGAGAGATATCAGGACTGTAG | (Feliu et al., 2013) | N/A |
| TOX11: 5’-GCGTCGTCTCGTCTAGATCG | (Feliu et al., 2013) | N/A |
| pri58-F: 5’-TTCCGAGCAGGTGACCTGGGTC | This study | N/A |
| pri92-R: 5’-CGTACGGGTACCATGGTTACAGTTTTTCAAAGTTGATTATACTCTCAAGCTGCTCAAGAAAATCAAACTCATTCACACTTCCCGGGT | This study | N/A |
| pri28F : 5’-CTAGATACCGTTCGTATAATGTATGCTATACGAAGTTATACTAGTGCTAGCATAACTTCGTATAATGTATGCTATACGAACGGTAT | This study | N/A |
| pri29R : 5’-CTAGATACCGTTCGTATAGCATACATTATACGAAGTTATGCTAGCACTAGTATAACTTCGTATAGCATACATTATACGAACGGTAT | This study | N/A |
| Recombinant DNA | ||
| pLd4 | (Evans et al., 1982), T. Hansen | internal ID: NBpla93 |
| pLd4Lox | This study | internal ID: NBpla100 |
| pGRA6/GRA6-OVA | This study | internal ID: NBpla119 |
| pSAG1/GRA6-OVA | This study | internal ID NBpla190 |
| Software and Algorithms | ||
| ImageJ | NIH | N/A |
| FlowJo | TreeStar | N/A |
| Prism | GraphPad | N/A |
Mouse cell lines and primary cells
MutuDC, a C57BL/6-derived dendritic cell line, were obtained from H. Acha-Orbea (see Key Resource Table) and grown using the recommended protocol, as in (Fuertes Marraco et al., 2012). Bone marrow-derived DC from F1 C57BL/6 X B6.H-2d (H-2bxd) mice were generated as in (Buaillon et al., 2017).
Toxoplasma gondii
For all experiments, type II 76K or Prugnaud (Pru) derivatives (see Key Resource Table for details) were used. Pru tachyzoites were maintained in vitro by serial passages on confluent monolayers of HFF using DMEM supplemented with 1 % FCS (Gibco). 76K parasites were passaged in vivo in CBA mice by per os inoculation of 100 cysts every 2-month. For intra-peritoneal infections, infected HFF were scraped, tachyzoites were released through a 23G needle, filtered through a 3 μm polycarbonate hydrophilic filter (it4ip S.A.) and 2–5×10^2 tachyzoites were injected in 200 μl PBS. For per os infections, 200 μl of brain homogenate containing 15 cysts was administered by oral gavage.
Mice
Animal care and use protocols were carried out under the control of the National Veterinary Services and in accordance with the European regulations (EEC Council Directive, 2010/63/EU, September 2010). Protocols inducing pain (CE no. 2015–02) were approved by the local Ethical Committee for Animal Experimentation registered by the ‘Comité National de Réflexion Ethique sur l’Expérimentation Animale’ under no. CEEA122. CBA/JRj and C57BL/6J (B6) mice were purchased from Janvier (France). B6.C-H2d/bByJ, abbreviated as B6.H-2d, were purchased from Jackson Laboratories (Bar Harbor, ME, USA). F1 C57BL/6 X B6.H-2d (H-2bxd) were bred for this study. B6.CamKIIα-iCre were a gift from G. Schutz (Casanova et al., 2001). B6.LdLox were generated by additive transgenesis following microinjection of a linearized DNA cassette containing a floxed H-2Ld allele into the pronucleus of C57BL/6J fertilized eggs. The floxed Ld gene with two LoxP sites flanking exons 1 to 3 was obtained after modification of the pLd4 plasmid that harbors a 12-kb BALB/c-derived genomic sequence (Evans et al., 1982). More details on the pLd4Lox plasmid construction are shown below. All mice were housed and bred under specific pathogen-free conditions at the ‘Centre Régional d’Exploration Fonctionnelle et de Ressources Expérimentales’ (CREFRE-Inserm UMS006). Age of mice used in experiments was 8–20 weeks. Mice used in experiments were males. Mice in B6.LdLox.CamK-Cre+/− experiments were littermates. Number of mice and experimental replicates are indicated in the respective figure legends.
METHOD DETAILS
Generation of transgenic parasites
Hxgprt-deficient GFP+ (Kim et al., 2007) and tdTomato+ Pru (Schaeffer et al., 2009) were used as parental strains. Tachyzoites were transfected with plasmids encoding the GRA6II open-reading frame in frame with nucleotides encoding LEQLE-SIINFEKL, yielding the GRA6-OVA antigenic construct. GRA6-OVA was flanked by gra2 3’ UTR and by either the endogenous gra6 promoter and 5’ UTR (pGRA6/GRA6-OVA) or the by the sag1 promoter and 5’UTR (pSAG1/GRA6-OVA). Plasmids were generated by a combination of DNA fragment synthesis (GeneArt, Thermo Fisher) and standard cloning procedures and verified by sequencing. Parasite transfections were performed as previously described (Feliu et al., 2013). In brief, 2×10^7 freshly egressed tachyzoites were electroporated with 50 μg of NotI (New England Biolabs)-linearized plasmid DNA. After selection in culture medium supplemented with mycophenolic acid (Sigma, 25 μg/ml) and xanthine (Sigma, 50 μg/ml), resistant tachyzoites were cloned by limiting dilution in flat-bottom 96-well plates and presence of the transgene was verified by PCR and sequencing.
Bradyzoite differentiation and Western blot
For in vitro bradyzoite differentiation assays, HFF were seeded in 6-W plates and infected with 10^6 tachyzoites. After 24 h, the medium was replaced and supplemented with 40 nM of the histone deacetylase inhibitor apicidin (Sigma-Aldrich) in order to promote bradyzoite differentiation(Bougdour et al., 2009; Boyle et al., 2006). After 24 h for the untreated or 72 h for the apicidin-treated, parasites were released with a 23-G needle, lyzed in 1x Laemmli buffer (Bio-Rad) containing 10 % 2-mercaptoethanol (Bio-Rad) and heated for 5 min at 70°C. Total cell lysates were forced through a 29-G needle, separated by electrophoresis on 15 % polyacrylamide gels and transferred to nitrocellulose membranes. Immunologic detection was performed using purified rabbit anti-SIINFEKL antisera (custom-made, Biotem, Grenoble) and mouse anti-GRA1 (Biotem) followed by horseradish peroxidase-conjugated antibodies (Promega). Peroxidase activity was visualized by chemiluminescence and quantified using a ChemiDoc XRS+ system (Bio-Rad).
Isolation of spleen and brain leukocytes
Spleens and brains were dissociated in complete RPMI (Gibco) supplemented with 10 % vol/vol FCS (Gibco). Splenocytes were mashed through a 100 μm cell strainer (Falcon). Brains were homogenized using a Potter, minced and digested for 45 min in RPMI containing 1 mg/ml collagenase (Roche) and 0.1 mg/ml DNAse (Sigma-Aldrich). Samples were then centrifuged at 600 g and suspended in 60 % (vol/vol) Percoll (GE Healthcare). A 30 % (vol/vol) Percoll solution was overlaid and the tubes were centrifuged at 1000 g for 20 min. Brain leukocytes were recovered from the interface. In both cases, erythrocytes were lyzed using ACK buffer (100 μM EDTA, 160 mM NH4Cl and 10 mM NaHCO3).
Ex vivo T cell stimulation
One-fifth of Percoll-isolated brain leukocytes or 10^6 splenocytes were incubated for 4 h 15 min at 37°C in the presence of brefeldin A (eBioscience) with 10^5 bone marrow-derived DC from F1 C57BL/6 X B6.H-2d (H-2bxd) mice or with C57BL/6-derived MutuDC (Fuertes Marraco et al., 2012), plus 200 nM of the following peptides as indicated in the legends: GRA6-derived HPGSVNEFDF (HF10) presented by H-2Ld, OVA-derived SIINFEKL presented by H-2Kb and Tgd057-derived SVLAFRRL presented by H-2Kb.
Flow cytometry
Following Fc receptor saturation (Biolegend) and dead cell detection with AF488 Live Dead/Cell marker (Invitrogen) in PBS, cell suspensions were surface labelled with CD8α BV421 (53–6.7, 1/300, BD Pharmingen) or CD8α PE (53–6.7, 1/300, eBioscience), CD4 BV510 (RM4–5, 1/200, BD Horizon) or CD4 Pe-Cy7 (RM4–5, 1/300, BD Pharmingen). Intracellular IFNγ (IFNγ-APC or AF700, XMG1.2 1/300, BD Pharmingen), TNFα (TNFα AF700, MP6-XT22, 1/300, BD Pharmingen), Granzyme B (Granzyme B PE, NGZB, 1/200, eBioscience) were detected after fixation in 4 % paraformaldehyde solution (PFA, Electron Microscopy Sciences) and permeabilization with the Permeabilization Buffer kit (eBioscience).
For ex vivo MHC I labeling, 10^6 splenocytes were stained with FcR block and AF488 Live/Dead cell marker in PBS. They were labelled with CD3 BV421 (145–2C1, 1/300, BD Horizon) or CD3 FITC (145–2C1, 1/300, BD Pharmingen), CD19 Pe-Cy7 (1D3,1/400, BD Pharmingen), CD11c PE (N418, 1/300, eBioscience), CD11b PE-CF594 (M1/70, 1/3000, BD Horizon) or CD11b PE (M1/70, 1/400, BD Pharmingen), anti-H-2Ld AF647 (30-5-7, 1/100, Biotem) or anti-H-2Kb PerCP-Cy5.5 (AF6–88.5, 1/300, Biolegend). Samples were fixed using 4% PFA.
For ex vivo Kb-SIINFEKL dextramer analysis, splenocytes and brain leukocytes were incubated 1 h at 37 °C with a solution of dextramer H-2 Kb-SL8 PE (1/50, Immudex). Cells were stained with FcR block and eFluor 660 Fixable Viability Dye (1/1000, eBioscience) in PBS and then labelled with CD8α BV421 (53–6.7, 1/300, BD Horizon) and CD4 AF700 (RM4–5, 1/200, BD Pharmingen) or CD4 BV510 (RM4–5, 1/200, BD Horizon).
To analyze the phenotype and activation of CNS-isolated myeloid cells, brain leukocytes were stained with FcR block and eFluor 450 Fixable Viability Dye (1/1000, eBioscience) in PBS and then labelled using the following antibodies: Ly6G BV510 (1/200, 1A8, Biolegend), CD45 PerCP-Cy5.5 (30-f 11, 1/300, BD Pharmingen), CD11b PE-CF594 (M1/70, 1/3000, BD Horizon), NK1.1 PE (PK 136, 1/400, BD), CCR2 AF700 (FAB5538N, 1/200, R&D Systems), CD86 APC (GL1,1/300, BD Pharmingen) or CD86 Alexa Fluor 700 (GL1, 1/500, BD Pharmingen), Ly6C BV711 (HK1.4, 1/1800, Biolegend), MHC II I-A/I-E FITC (2G9, 1/300, BD Pharmingen). In all cases, samples were ultimately fixed in 4 % PFA before acquisition on a BD Fortessa and analyzed using FlowJo (Tree Star).
Parasite load analysis
For cyst enumeration, 5 % of total brain homogenate was labeled with rhodamine or fluorescein-conjugated Dolichos Biflorus Agglutinin (Vector Laboratories). Cysts were counted using an inverted fluorescence microscope. Quantification of parasite DNA by qPCR was performed on genomic DNA extracted from 5 % of each brain homogenate, 10^6 splenocytes or 5×10^5 peritoneal exudate cells using the DNEasy Blood & Tissue Kit (Qiagen). As described earlier (Feliu et al., 2013), a 529-bp repeat element in the T. gondii genome was amplified using the TOX9 and TOX11 primers (sequences shown in Key Resource Table). The number of parasite genome per μg of tissue DNA was estimated by comparison with a standard curve, established with a known number of Pru tachyzoites.
IF labeling of ex vivo cysts
Five percent of the brain homogenate was stained with rhodamine-conjugated DBA lectin, fixed in 4% PFA for 10 min at room temperature (RT) and permeabilized in PBS - 0.2% Triton™ X100 (Sigma-Aldrich). Samples were then incubated with a custom-made rabbit anti-SIINFEKL (1/300, Biotem) or anti-OVA (1/500, Sigma) and a mouse anti-GRA2 (1/3000, Biotem) diluted in PBS BSA 3 % (Dutscher), followed by incubation with AF555-coupled anti-rabbit Immunoglobulin G (IgG) or AF488-coupled anti-rabbit (1/500) and AF647-coupled anti-mouse IgG (1/500, Life Technologies, Thermo Fisher) diluted in PBS-BSA 3 %. Samples were mounted using ProLong™ Diamond Anti-Fade containing DAPI (Life Technologies, Thermo Fisher scientific) and imaged using a Zeiss LSM710 confocal microscope. Quantifications of the GRA6-OVA signal were performed using ImageJ software (NIH). Briefly, a filled mask encompassing the entire area of the cyst was created based on the DBA lectin staining and the parasite fluorescence. The mean intensity of the GRA6-OVA signal of masked pixels was measured and averaged from 3 equatorial planes for every individual cyst.
IF labeling of HFF
HFF were infected with Tg.pGRA6/GRA6-OVA or Tg.pTUB/vacOVA parasites during 24 h. After 2 washes in PBS, cells were fixed in PFA 4% for 20 min at RT and quenched in PBS glycine 100mM (Sigma) for 10 min at RT. Cells were then washed in PBS and incubated with primary polyclonal rabbit OVA (1/500, Sigma) or rabbit anti-SIINFEKL (1/300, Biotem) in PBS-BSA 0,2%-saponin 0,05 %. After washing in the same buffer, cells were incubated with AF488-coupled anti-rabbit IgG (1/500, Life Technologies). Coverslips were mounted using ProLong Diamondᵀᴹ Anti-Fade containing DAPI (Life Technologies, Thermo Fisher scientific). Images were acquired with a 63X objective on a Zeiss LSM710 confocal microscope.
Primary hippocampal neuronal cultures
Primary neuronal cultures were derived from C57BL/6 embryonic day 17 hippocampi. One day before dissection, flat-bottom 24-well plates with glass coverslips and flat-bottom 96-well plates were coated with Poly-D-lysine (Merck) dissolved in Ultra-Pure™ Distilled Water (Gibco) to the final concentration of 38,5 μg/ml. Coated plates were stored overnight at 37°C. The day of the dissection, plates were washed twice with Ultra-Pure™ Distilled Water (Gibco) and coated with laminin (mouse, 1/500, Invitrogen) during 3 h at 37°C. After dissection, hippocampi were digested 8 min at 37°C with Papain solution (Worthington Biochemical Corp.) at 1 U/ml final activity. Digestion was stopped using a solution composed of 1.5 mg/ml BSA (Dutscher), 1.5 mg/ml Trypsin inhibitor from chicken egg white (Sigma-Aldrich) and 66.7 μg/ml DNAse (Sigma-Aldrich) in PBS. Tissues were mechanically dissociated by trituration with a plastic pipette, filtered through a 70 μm strainer (Falcon) and centrifuged at 210 g for 7 min. Cells were suspended in Neurobasal®-A medium (Gibco) containing 2% B27® Supplement (vol/vol) (Gibco), 1% GlutaMAX™-I (vol/vol) (Gibco), 120 U/ml Penicillin, 120 μg/ml Streptomycin (Gibco) and seeded at 2×10^5 cells per well for the 24-well plate and 4×10^4 cells per well for the 96-well plate. Cells were then incubated at 37°C, 5% CO2. At day 4 and 8, primary cultures were treated with 5 μM of cytarabine hydrochloride (AraC) (Sigma-Aldrich) in order to inhibit growth of glial cells. At day 11, 100 U/ml of mouse IFNγ (Miltenyi Biotec) was added. Twelve days after the beginning of the culture, neurons were infected with Tg.pGRA6/GRA6-OVA or Tg.pTUB/vacOVA strains. Respectively, 5×10^4 or a serial dilution of 5×10^4 to 2×10^3 tachyzoites were added in each well of the 24-well or 96-well plate. To assess Ld expression, hippocampal cultures were established with B6.LdLox.Cre+/− pups at postnatal day 1 using a similar procedure as above, with the following modifications: 1,5×10^5 cells were seeded per well in a 24 - well plate and were not treated with AraC. At day 13, primary cultures were treated with mouse IFNγ (0,5 μg/ml) and 1 μM Tetrodotoxin (TTX) (Sigma-Aldrich) as in (Chevalier et al., 2011).
IF labeling of primary neuronal cultures
Twenty-four hours after infection, cells were fixed with 4 % PFA for 20 min at RT. After 2 × 5 min washes with PBS, cells were permeabilized with PBS - 0,05 % Triton™ X100 (Sigma-Aldrich) for 5 min at RT. Following 3 × 5 min washes in PBS, non-specific binding sites were blocked with PBS - 5 % Normal Goat Serum (NGS) (Vector Laboratories) - 0,05 % Tween®20 (Sigma-Aldrich) for 1 h at RT. Cells were incubated overnight at 4°C with primary monoclonal mouse anti-microtubule-associated protein 2 (MAP2, AP-20, 1/500, Sigma-Aldrich) and polyclonal rabbit anti-glial fibrillary acidic protein (GFAP, 1/500, Merck) diluted in PBS - 3 % NGS - 0,05 % Tween®20. Cells were washed 1 × 5 min with PBS, 3 × 5 min with PBS - 0,05 % Tween®20 and 5 min with PBS. Cells were incubated for 2 h at RT in presence of secondary antibodies AF488-coupled anti-mouse immunoglobulin G (IgG) and AF647-coupled anti-rabbit IgG (Life Technologies, Thermo Fisher) diluted in PBS - 3 % NGS - 0,05 % Tween®20 (1/500) and protected from light. Samples were mounted using ProLong™ Diamond Anti-Fade with DAPI (Life Technologies, Thermo Fisher). Z-stacks were acquired with a 63X objective on a Zeiss LSM710 confocal microscope and analyzed using ImageJ software. Extracellular H-2 Ld was stained with 1,2 μg/ml of mouse anti Ld (clone 30-5-7, Biotem) diluted in PBS-3 % NGS for 30 min on ice followed by two washes with cold PBS. Steps of intracellular staining were the same as described above except for the antibodies used. Cells were stained with a polyclonal rabbit anti-microtubule-associated protein 2 (MAP2, 1/1000, Merck) or with a polyclonal rabbit anti-glial fibrillary acidic protein (GFAP, 1/500, Merck), followed by both AF488-coupled anti-rabbit IgG (1/500, Life Technologies, Thermo Fisher) and AF594-coupled anti-mouse IgG (1/500, Life Technologies, Thermo Fisher). Quantification of Ld labeling was done with ImageJ. The intensity of Ld labeling was recorded on multiple sections after applying a mask corresponding to the MAP2+ or GFAP+ pixels.
IF labeling of brain cortical floating sections
After transcardial perfusion with NaCl 0,9 %, hemibrains were removed, fixed in 4 % PFA for 48 h at 4°C and washed in PBS for 5 min and 24 h at 4°C. Brains were transferred in PBS - 30 % sucrose for 48 h at 4°C, sectioned coronally at 25 μm-thickness and stored in PBS - 30 % ethylene glycol - 30 % glycerol at −20°C until processed further. For this, sections were washed 4 × 5 min in TBS (Euromedex), permeabilized with TBS - 0,1 % Triton™ X100 (Sigma-Aldrich) for 10 min at RT and rinsed once with TBS for 5 min. After antigen retrieval with TBS - 100 mM Tris - 12 mM EDTA - 0,05 % Tween®20 (Sigma-Aldrich) at pH 9 during 20 min at 100°C, brain sections were washed 3 × 5 min in TBS. Non-specific binding sites were blocked with TBS - 5 % Normal Goat Serum (NGS) (Vector Laboratories) - 5 % BSA (Dutscher) - 0,1 % Tween®20 for 1 h at RT. Floating sections were incubated overnight at 4°C with rabbit anti-Iba1 (1/1000, Wako) in TBS - 3 % NGS - 0,1 % Tween®20. Following several washes in TBS - 0,1 % Tween®20 and TBS, sections were incubated for 2 h at RT with secondary antibody AF647-coupled anti-rabbit IgG (Thermo Fisher) diluted in TBS - 3 % NGS - 0,1 % Tween®20 (1/500) and protected from light. Samples were washed alternatively in TBS - 0,1 % Tween®20 and TBS twice and incubated with 1 μg/ml of DAPI (Sigma-Aldrich) in TBS for 30 min at RT. Sections were mounted in Fluoromount medium (Electron Microscopy Sciences) after 2 washes in TBS. Images were acquired with 5 × and 20 × objectives on Apotome Zeiss microscope and analyzed using ImageJ software.
In vitro antigen presentation assays
Antigen presentation measurements with MutuDC were performed as described in(Buaillon et al., 2017). In brief, 5×10^4 MutuDC were seeded into flat-bottom 96-well plates and infected for 24 h with serially diluted tachyzoites. The proportion of infected cells (i.e. GFP+ or Tomato+) was controlled by flow cytometry. Presentation of the SIINFEKL peptide by Kb was assessed following 16 h incubation at 37°C with 10^5 LacZ-inducible B3Z reporter hybridomas per well. Production of β-galactosidase by the hybridomas was quantified using the chromogenic substrate chlorophenol red-β-D-galactopyranoside (CPRG, Roche). Absorbance was read at 595 nm with a reference at 655 nm with a spectrophotometer (VersaMax, Molecular Devices).
For antigen presentation with primary neurons, 24 h after neuron infection, 10^5 B3Z reporter hybridomas were seeded per well of the 96-well plate and incubated for 16 h at 37°C. After fixation with 2 % PFA - 0,2 % glutaraldehyde (Electron Microscopy Sciences) for 20 min at RT and 2 × 5 min washes with PBS, cells were incubated with a solution containing 1 mg/ml 5-bromo-4chloro-3-indolyl-beta-D-galactopyranoside (X-gal, Sigma-Aldrich), 5 mM potassium ferrocyanide (Sigma-Aldrich), 5 mM potassium ferricyanide (Sigma-Aldrich), 2 mM MgCl2 (Sigma-Aldrich) in PBS. X-gal-positive (blue) cells were counted microscopically with a 20X magnification. With neurons, the X-gal assay was preferred over the bulk CPRG read-out because of its higher sensitivity.
Plasmids
pGRA6/GRA6-OVA
The pGRA6/GRA6-OVA plasmid was constructed by In-fusion (Takara) cloning of a GRA6(II)-LEQLE-SIINFEKL insert (abbreviated as GRA6-OVA) into the 5’ BstEII / 3’ NcoI-linearized and gel purified pGRA.HPT.GRA6(II) vector, a T. gondii expression vector containing a HXGPRT resistance cassette (Feliu et al., 2013). The GRA6-OVA sequence was PCR-amplified from type II Pru genomic DNA using a forward (pri58-F) and a reverse primer (pri92-R) (sequences shown in Key Resource Table) in order to introduce the LEQLE-SIINFEKL coding sequence at the C-terminus of GRA6(II) and to add extremities annealing with the ends of the linearized vector for the fusion cloning. The resulting pGRA6/GRA6-OVA plasmid (internal ID NBpla119) contains the GRA6-OVA coding sequence flanked by the endogenous GRA6(II) promoter/5’UTR and the 3’UTR from GRA2(I) as well as the HXGPRT resistance cassette.
pSAG1/GRA6-OVA
To obtain the pSAG1/GRA6-OVA plasmid, the pGRA6/GRA6-OVA plasmid was modified to replace the GRA6(II) promoter/5’UTR and GRA6-OVA coding sequence by a synthetic DNA fragment containing the SAG1 promoter/5’UTR and GRA6-OVA coding sequence using 5’ HindIII/3’ NcoI restriction cloning. The sequence of the introduced synthetic fragment is shown below, with HindIII/NcoI sites underlined and GRA6-OVA ORF in bold:
AAGCTTTTACATCCGTTGCCTTTTCCACGGTCCGTGATTTCATGTGCGTGCAGCTTCAAAGACTGGTCGTTGCGACTAATAAGACTGCAGTGACAGGTCGAATGGTGGGCACCTTGCTGATGACTATCTACTGCAAAGTCTGAGACAACGAACGAAACTTCCCACACGAGGCATTTGAAACTGACGGTGTCTAGGTAATATGCACTGCAAGACACGGTACTGGGGCCTCGCTGAATTAGGGGCCGATCTCGTTGCCCTATCAGTGCTCACAGTGCCGCAACGTAACACCAGGGCAGGTTCTTGACAGTGGCAACAATGTGCGACGGGCGTGTGAACGTTTCGTAGTCATAGCGCTAGCACGTACCTAGCCACATGGTCGTGAGGAGCTTTACCATGCGTCTAGAAGGTGGATGCGGGACACGCCTTCCTGGCCTTTGGCTCCCGAGACGCGTGTTCTAACCACAAACCTTGAGACGCGTGTTCCAACCACGCACCCTGACACGCGTGTTCCAACCACGCACCCTGAGACGCGTGTTCTAACCACGCACCCTGAGACGCGTGTTCTAACCACGCACCCTGAGACGCGTGTTCTGCCGCACAATGTGCACCTGTAGGAAGCTGTAGTCACTGCTGATTCTCACTGTTCTCGGCAAGGGCCGACGACCGGAGTACAGTTTTTGTGGGCAGAGCCGTTGTGCAGCTTTCCGTTCTTCTCGGTTGTGTCACATGTGTCATTGTCGTGTAAACACACGGTTGTATGGCACACGGTGGCATCTATCTGAGGCAGAAGCGTAACTTCTGTCCTTTAACTGTCTCCACAGTTGCTGTGGTCTTTGTAGTCTTCATGGGTGTACTCGTCAATTCGTTGGGTGGAGTCGCTGTCGCAGCAGACAGCGGTGGTGTTAGGCAGACCCCTTCGGAAACCGGTTCGAGCGGTGGACAGCAAGAAGCAGTGGGGACCACTGAAGACTATGTCAACTCTTCGGCGATGGGCGGTGGCCAAGGCGACTCGTTAGCTGAAGATGATACAACCTCCGATGCGGCGGAGGGCGACGTTGACCCTTTTCCCGCGCTGGCGAATGAGGGGAAGTCGGAGGCGCGTGGCCCGTCGCTCGAGGAAAGAATCGAAGAACAGGGCACAAGACGACGTTACTCCTCTGTTCAAGAACCACAAGCGAAGGTGCCTAGCAAACGAACACAGAAACGCCACAGACTCATTGGTGCTGTGGTGTTGGCAGTATCTGTGGCAATGCTTACCGCTTTCTTTCTTCGAAGGACTGGACGACGCTCTCCCCAAGAACCATCTGGGGGTGGTGGTGGAAATGATGCAGGCAATAATGCTGGGAACGGTGGGAATGAAGGCAGAGGTGAAGGAGGCGAGGATGACAGGCGCCCGTTGCACCCGGGAAGTGTGAATGAGTTTGATTTTCTTGAGCAGCTTGAGAGTATAATCAACTTTGAAAAACTGTAACCATGG
The resulting pSAG1/GRA6-OVA plasmid (internal ID NBpla 190) contains the GRA6-OVA coding sequence flanked by the SAG1 promoter/5’UTR and the 3’UTR from GRA2(I) as well as a HXGPRT resistance cassette.
pLd4Lox plasmid
The pLd4 plasmid (internal ID NBpla93, kind gift of T. Hansen) contains 12-kb of genomic sequence of BALB/c H-2 Ld gene inserted into pBR327 backbone between EcoRI and HindIII (Evans et al., 1982).
A pair of reverse complement primers (pri28F and pri29R, see sequences in Key Resource Table) were annealed in vitro in order to create two distinct LoxP sites (to make sure that recombination is irreversible) flanked by cohesive ends that permit cloning into XbaI site. SpeI and NheI restriction sites, which are compatible for religation with XbaI, were included between the LoxP sites:
CTAGATACCGTTCGTATAATGTATGCTATACGAAGTTATACTAGTGCTAGCATAACTTCGTATAATGTATGCTATACGAACGGTAT
The XbaI-digested annealed fragment was cloned into XbaI-linearized pUC19 to obtain pUC19 | LoxP |SpeI |NheI | LoxP (internal ID NBpla95). A 1.6kb XbaI-excised fragment from pLd4 containing the first 3 exons (L,N,C1 (Ozato et al., 1983)) was cloned into NheI-linearized pUC19 | LoxP |SpeI |NheI | LoxP to obtain pUC19 | LoxP | SpeI | Ld exons 1–3 | LoxP (internal ID NBpla99).
The LoxP | SpeI | Ld exons 1–3 | LoxP fragment was then excised with XbaI and cloned into XbaI-opened pLd4 to obtain pLd4Lox (internal ID Nbpla100). Clones were confirmed by restriction digest and sequencing.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses for all experiments were performed with Prism software v7 (GraphPad). In experiments comparing only two groups, non-parametric Mann-Whitney tests were used to compare the experimental group with the control group. For other experiments including 3 groups, non-parametric ANOVA tests (Kruskal-Wallis with Dunn’s correction for multiple tests) were used. Individual P values are indicated on the figures.
Supplementary Material
Acknowledgments
F. L’Faqihi-Olive, V. Duplan-Eche, A.-L. Iscache, L. de la Fuente for technical assistance at the CPTP-Inserm U1043 flow cytometry facility; S. Allart, A. Canivet-Laffitte, D. Daviaud for technical assistance at the CPTP-Inserm U1043 imaging facility; G. Tavernier and Y. Barreira at INSERM UMS006-CREFRE for the generation of the B6.LdLox mice, R. Balouzat and the zootechnicians at INSERM UMS006-CREFRE mouse facility; the Blanchard and Robey teams for help and discussions; M.-F. Cesbron-Delauw for the anti-GRA1 and anti-GRA2 antibodies, H. Acha-Orbea for the MutuDC, D. Buzoni-Gatel for the 76K parasites, S.-K. Kim, J. Boyle and J. Boothroyd for the GFP+ Pru tachyzoites; T. Hansen for the pLd4 plasmid; G. Schutz for the B6.CamKIIα-iCre mice; I. Cebrian and D. Dunia for critical reading of the manuscript.
This work was supported by ‘Institut National de la Santé et de la Recherche Médicale’, Idex Toulouse ‘Attractivity Chair’ Program (to ER and NB), Human Frontier Science Program Organization (CDA00047/2011 to NB), PIA PARAFRAP Consortium (ANR-11-LABX0024 to NB), PIA ANINFIMIP equipment (ANR-11-EQPX-0003 to NB), Agence Nationale pour la Recherche (ANR-18-CE15-0015 to NB and ES), ‘Fondation pour la Recherche Médicale’ to AS (FDT20170436953).
Footnotes
Declaration of interests
The authors declare no competing interests.
References
- Bernard-Valnet R, Yshii L, Queriault C, Nguyen XH, Arthaud S, Rodrigues M, Canivet A, Morel AL, Matthys A, Bauer J, et al. (2016). CD8 T cell-mediated killing of orexinergic neurons induces a narcolepsy-like phenotype in mice. Proc Natl Acad Sci U S A 113, 10956–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra R, Cobb DA, and Khan IA (2013). Donor CD8+ T cells prevent Toxoplasma gondii de-encystation but fail to rescue the exhausted endogenous CD8+ T cell population. Infect Immun 81, 3414–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas A, Bruder D, Wolf SA, Jeron A, Mack M, Heimesaat MM, and Dunay IR (2015). Ly6C(high) monocytes control cerebral toxoplasmosis. J Immunol 194, 3223–3235. [DOI] [PubMed] [Google Scholar]
- Biswas A, French T, Dusedau HP, Mueller N, Riek-Burchardt M, Dudeck A, Bank U, Schuler T, and Dunay IR (2017). Behavior of Neutrophil Granulocytes during Toxoplasma gondii Infection in the Central Nervous System. Frontiers in cellular and infection microbiology 7, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard N, Dunay IR, and Schluter D (2015). Persistence of Toxoplasma gondii in the central nervous system: a fine-tuned balance between the parasite, the brain and the immune system. Parasite Immunol 37, 150–158. [DOI] [PubMed] [Google Scholar]
- Blanchard N, Gonzalez F, Schaeffer M, Joncker NT, Cheng T, Shastri AJ, Robey EA, and Shastri N (2008). Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat Immunol 9, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnart C, Feuillet G, Vasseur V, Cenac N, Vergnolle N, and Blanchard N (2017). Protease-activated receptor 2 contributes to Toxoplasma gondii-mediated gut inflammation. Parasite Immunol 39. [DOI] [PubMed] [Google Scholar]
- Bougdour A, Maubon D, Baldacci P, Ortet P, Bastien O, Bouillon A, Barale JC, Pelloux H, Menard R, and Hakimi MA (2009). Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J Exp Med 206, 953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle JP, Saeij JP, Cleary MD, and Boothroyd JC (2006). Analysis of gene expression during development: lessons from the Apicomplexa. Microbes Infect 8, 1623–1630. [DOI] [PubMed] [Google Scholar]
- Brown CR, Hunter CA, Estes RG, Beckmann E, Forman J, David C, Remington JS, and McLeod R (1995). Definitive identification of a gene that confers resistance against Toxoplasma cyst burden and encephalitis. Immunology 85, 419–428. [PMC free article] [PubMed] [Google Scholar]
- Buaillon C, Guerrero NA, Cebrian I, Blanie S, Lopez J, Bassot E, Vasseur V, Santi-Rocca J, and Blanchard N (2017). MHC I presentation of Toxoplasma gondii immunodominant antigen does not require Sec22b and is regulated by antigen orientation at the vacuole membrane. Eur J Immunol 47, 1160–1170. [DOI] [PubMed] [Google Scholar]
- Cabral CM, McGovern KE, MacDonald WR, Franco J, and Koshy AA (2017). Dissecting Amyloid Beta Deposition Using Distinct Strains of the Neurotropic Parasite Toxoplasma gondii as a Novel Tool. ASN Neuro 9, 1759091417724915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral CM, Tuladhar S, Dietrich HK, Nguyen E, MacDonald WR, Trivedi T, Devineni A, and Koshy AA (2016). Neurons are the Primary Target Cell for the Brain-Tropic Intracellular Parasite Toxoplasma gondii. PLoS pathogens 12, e1005447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova A, Blatche MC, Ferre CA, Martin H, Gonzalez-Dunia D, Nicu L, and Larrieu G (2018). Self-Aligned Functionalization Approach to Order Neuronal Networks at the Single-Cell Level. Langmuir : the ACS journal of surfaces and colloids 34, 6612–6620. [DOI] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, and Schutz G (2001). A CamKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis 31, 37–42. [DOI] [PubMed] [Google Scholar]
- Cebrian C, Zucca FA, Mauri P, Steinbeck JA, Studer L, Scherzer CR, Kanter E, Budhu S, Mandelbaum J, Vonsattel JP, et al. (2014). MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat Commun 5, 3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier G, Suberbielle E, Monnet C, Duplan V, Martin-Blondel G, Farrugia F, Le Masson G, Liblau R, and Gonzalez-Dunia D (2011). Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLoS pathogens 7, e1002393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HH, Chan SW, Gosling JP, Blanchard N, Tsitsiklis A, Lythe G, Shastri N, Molina-Paris C, and Robey EA (2016). Continuous Effector CD8(+) T Cell Production in a Controlled Persistent Infection Is Sustained by a Proliferative Intermediate Population. Immunity 45, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, and Butovsky O (2017). Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol 35, 441–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, and Shatz CJ (1998). Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21, 505–520. [DOI] [PubMed] [Google Scholar]
- Di Liberto G, Pantelyushin S, Kreutzfeldt M, Page N, Musardo S, Coras R, Steinbach K, Vincenti I, Klimek B, Lingner T, et al. (2018). Neurons under T Cell Attack Coordinate Phagocyte-Mediated Synaptic Stripping. Cell 175, 458–471 e419. [DOI] [PubMed] [Google Scholar]
- Elmer BM, and McAllister AK (2012). Major histocompatibility complex class I proteins in brain development and plasticity. Trends in neurosciences 35, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans GA, Margulies DH, Shykind B, Seidman JG, and Ozato K (1982). Exon shuffling: mapping polymorphic determinants on hybrid mouse transplantation antigens. Nature 300, 755–757. [DOI] [PubMed] [Google Scholar]
- Feliu V, Vasseur V, Grover HS, Chu HH, Brown MJ, Wang J, Boyle JP, Robey EA, Shastri N, and Blanchard N (2013). Location of the CD8 T Cell Epitope within the Antigenic Precursor Determines Immunogenicity and Protection against the Toxoplasma gondii Parasite. PLoS pathogens 9, e1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DJ, and Hutchison WM (1987). The host-parasite relationship of Toxoplasma gondii in the brains of chronically infected mice. Virchows Archiv. A, Pathological anatomy and histopathology 411, 39–43. [DOI] [PubMed] [Google Scholar]
- Fox BA, Falla A, Rommereim LM, Tomita T, Gigley JP, Mercier C, Cesbron-Delauw MF, Weiss LM, and Bzik DJ (2011). Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot Cell 10, 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frickel EM, Sahoo N, Hopp J, Gubbels MJ, Craver MP, Knoll LJ, Ploegh HL, and Grotenbreg GM (2008). Parasite stage-specific recognition of endogenous Toxoplasma gondii-derived CD8+ T cell epitopes. The Journal of infectious diseases 198, 1625–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes Marraco SA, Grosjean F, Duval A, Rosa M, Lavanchy C, Ashok D, Haller S, Otten LA, Steiner QG, Descombes P, et al. (2012). Novel murine dendritic cell lines: a powerful auxiliary tool for dendritic cell research. Front Immunol 3, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn MW, Elmer BM, Garay PA, Liu XB, Needleman LA, El-Sabeawy F, and McAllister AK (2011). MHCI negatively regulates synapse density during the establishment of cortical connections. Nature neuroscience 14, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Mishra A, Buchanan RL, Dubey JP, Hill DE, Gamble HR, Jones JL, and Pradhan AK (2016). A Systematic Meta-Analysis of Toxoplasma gondii Prevalence in Food Animals in the United States. Foodborne pathogens and disease 13, 109–118. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Johnson KR, and McGavern DB (2015). Therapeutic antiviral T cells noncytopathically clear persistently infected microglia after conversion into antigen-presenting cells. Journal of Experimental Medicine 212, 1153–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidano S, Randall LM, Dawson L, Dietrich HK, Konradt C, Klover PJ, John B, Harris TH, Fang Q, Turek B, et al. (2016). STAT1 Signaling in Astrocytes Is Essential for Control of Infection in the Central Nervous System. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, and Shatz CJ (2000). Functional requirement for class I MHC in CNS development and plasticity. Science 290, 2155–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Cobb DA, Bhadra R, Youngblood B, and Khan IA (2016). Blimp-1-mediated CD4 T cell exhaustion causes CD8 T cell dysfunction during chronic toxoplasmosis. J Exp Med 213, 1799–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Ricart B, Tait Wojno ED, Harris TH, Randall LM, Christian DA, Gregg B, De Almeida DM, Weninger W, Hammer DA, and Hunter CA (2011). Analysis of behavior and trafficking of dendritic cells within the brain during toxoplasmic encephalitis. PLoS pathogens 7, e1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karttunen J, Sanderson S, and Shastri N (1992). Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci U S A 89, 6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Karasov A, and Boothroyd JC (2007). Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infect Immun 75, 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, and Hunter CA (2017). Protective and Pathological Immunity during Central Nervous System Infections. Immunity 46, 891–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzfeldt M, Bergthaler A, Fernandez M, Bruck W, Steinbach K, Vorm M, Coras R, Blumcke I, Bonilla WV, Fleige A, et al. (2013). Neuroprotective intervention by interferon-gamma blockade prevents CD8+ T cell-mediated dendrite and synapse loss. J Exp Med 210, 2087–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malo CS, Huggins MA, Goddery EN, Tolcher HMA, Renner DN, Jin F, Hansen MJ, Pease LR, Pavelko KD, and Johnson AJ (2018). Non-equivalent antigen presenting capabilities of dendritic cells and macrophages in generating brain-infiltrating CD8 (+) T cell responses. Nat Commun 9, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDole JR, Danzer SC, Pun RY, Chen Y, Johnson HL, Pirko I, and Johnson AJ (2010). Rapid formation of extended processes and engagement of Theiler’s virus-infected neurons by CNS-infiltrating CD8 T cells. Am J Pathol 177, 1823–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus RM, and Heneka MT (2017). Role of neuroinflammation in neurodegeneration: new insights. Alzheimer’s research & therapy 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer TC, Cranston HJ, Weiss LM, and Halonen SK (2010). Host Cell Preference of Toxoplasma gondii Cysts in Murine Brain: A Confocal Study. J Neuroparasitology 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuth SG, Herrmann AM, Simon OJ, Siffrin V, Melzer N, Bittner S, Meuth P, Langer HF, Hallermann S, Boldakowa N, et al. (2009). Cytotoxic CD8+ T cell-neuron interactions: perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 15397–15409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohle L, Israel N, Paarmann K, Krohn M, Pietkiewicz S, Muller A, Lavrik IN, Buguliskis JS, Schott BH, Schluter D, et al. (2016). Chronic Toxoplasma gondii infection enhances beta-amyloid phagocytosis and clearance by recruited monocytes. Acta neuropathologica communications 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien CA, Batista SJ, Still KM, and Harris TH (2019). IL-10 and ICOS Differentially Regulate T Cell Responses in the Brain during Chronic Toxoplasma gondii Infection. J Immunol 202, 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien CA, Overall C, Konradt C, O’Hara Hall AC, Hayes NW, Wagage S, John B, Christian DA, Hunter CA, and Harris TH (2017). CD11c-Expressing Cells Affect Regulatory T Cell Behavior in the Meninges during Central Nervous System Infection. J Immunol 198, 4054–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondounda M, Ilozue C, and Magne C (2016). Cerebro-meningeal infections in HIV-infected patients: a study of 116 cases in Libreville, Gabon. African health sciences 16, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozato K, Evans GA, Shykind B, Margulies DH, and Seidman JG (1983). Hybrid H-2 histocompatibility gene products assign domains recognized by alloreactive T cells. Proc Natl Acad Sci U S A 80, 2040–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozato K, Hansen TH, and Sachs DH (1980). Monoclonal antibodies to mouse MHC antigens. II. Antibodies to the H-2Ld antigen, the products of a third polymorphic locus of the mouse major histocompatibility complex. J Immunol 125, 2473–2477. [PubMed] [Google Scholar]
- Pappas G, Roussos N, and Falagas ME (2009). Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. International journal for parasitology 39, 1385–1394. [DOI] [PubMed] [Google Scholar]
- Parlog A, Harsan LA, Zagrebelsky M, Weller M, von Elverfeldt D, Mawrin C, Korte M, and Dunay IR (2014). Chronic murine toxoplasmosis is defined by subtle changes in neuronal connectivity. Disease models & mechanisms 7, 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry CE, Gale SD, Erickson L, Wilson E, Nielsen B, Kauwe J, and Hedges DW (2016). Seroprevalence and Serointensity of Latent Toxoplasma gondii in a Sample of Elderly Adults With and Without Alzheimer Disease. Alzheimer disease and associated disorders 30, 123–126. [DOI] [PubMed] [Google Scholar]
- Rall GF, Mucke L, and Oldstone MB (1995). Consequences of cytotoxic T lymphocyte interaction with major histocompatibility complex class I-expressing neurons in vivo. J Exp Med 182, 1201–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MV, and McGavern DB (2015). Immune Surveillance of the CNS following Infection and Injury. Trends in immunology 36, 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa Q, Ochiai E, Tiwari A, Perkins S, Mullins J, Gehman M, Huckle W, Eyestone WH, Saunders TL, Shelton BJ, and Suzuki Y (2015). Cutting Edge: IFN-gamma Produced by Brain-Resident Cells Is Crucial To Control Cerebral Infection with Toxoplasma gondii. J Immunol 195, 796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer BM, Schmalstieg WF, and Howe CL (2013). Axons are injured by antigen-specific CD8(+) T cells through a MHC class I- and granzyme B-dependent mechanism. Neurobiology of disease 59, 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer M, Han SJ, Chtanova T, van Dooren GG, Herzmark P, Chen Y, Roysam B, Striepen B, and Robey EA (2009). Dynamic imaging of T cell-parasite interactions in the brains of mice chronically infected with Toxoplasma gondii. J Immunol 182, 6379–6393. [DOI] [PubMed] [Google Scholar]
- Stock AK, Dajkic D, Kohling HL, von Heinegg EH, Fiedler M, and Beste C (2017). Humans with latent toxoplasmosis display altered reward modulation of cognitive control. Sci Rep 7, 10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Wang X, Jortner BS, Payne L, Ni Y, Michie SA, Xu B, Kudo T, and Perkins S (2010). Removal of Toxoplasma gondii cysts from the brain by perforin-mediated activity of CD8+ T cells. Am J Pathol 176, 1607–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey EF, and Yolken RH (2003). Toxoplasma gondii and schizophrenia. Emerg Infect Dis 9, 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A (2015). Mechanisms of Host Behavioral Change in Toxoplasma gondii Rodent Association. PLoS pathogens 11, e1004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang C, Szabo G, and Sun QQ (2013). Distribution of CaMKIIalpha expression in the brain in vivo, studied by CaMKIIalpha-GFP mice. Brain research 1518, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts E, Zhao Y, Dhara A, Eller B, Patwardhan A, and Sinai AP (2015). Novel Approaches Reveal that Toxoplasma gondii Bradyzoites within Tissue Cysts Are Dynamic and Replicating Entities In Vivo. MBio 6, e01155–01115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilking H, Thamm M, Stark K, Aebischer T, and Seeber F (2016). Prevalence, incidence estimations, and risk factors of Toxoplasma gondii infection in Germany: a representative, cross-sectional, serological study. Sci Rep 6, 22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EH, and Hunter CA (2004). The role of astrocytes in the immunopathogenesis of toxoplasmic encephalitis. Int J Parasitol 34, 543–548. [DOI] [PubMed] [Google Scholar]
- Wyman CP, Gale SD, Hedges-Muncy A, Erickson LD, Wilson E, and Hedges DW (2017). Association between Toxoplasma gondii seropositivity and memory function in nondemented older adults. Neurobiology of aging 53, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, and Yang SH (2015). Interaction of astrocytes and T cells in physiological and pathological conditions. Brain research 1623, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yshii LM, Gebauer CM, Pignolet B, Maure E, Queriault C, Pierau M, Saito H, Suzuki N, Brunner-Weinzierl M, Bauer J, and Liblau R (2016). CTLA4 blockade elicits paraneoplastic neurological disease in a mouse model. Brain : a journal of neurology 139, 2923–2934. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Chen H, Chen Y, Wang L, Cai YH, Li M, Wen HQ, Du J, An R, Luo QL, et al. (2014). Activated microglia contribute to neuronal apoptosis in Toxoplasmic encephalitis. Parasit Vectors 7, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.