

Abstract
Host pattern recognition receptors (PRRs) are crucial for sensing pathogenic microorganisms, launching innate responses, and shaping pathogen-specific adaptive immunity during infection. Rickettsia spp., Orientia tsutsugamushi, Anaplasma spp., Ehrlichia spp., and Coxiella burnetii are obligate intracellular bacteria, which can only replicate within host cells and must evade immune detection to successfully propagate. These five bacterial species are zoonotic pathogens of clinical or agricultural importance, yet, uncovering how immune recognition occurs has remained challenging. Recent evidence from in-vitro studies and animal models has offered new insights into the types and kinetics of PRR activation during infection with Rickettsia spp., A. phagocytophilum, E. chaffeensis, and C. burnetii, respectively. However, much less is known in these regards for O. tsutsugamushi infection, until the recent discovery for the role of the C-type lectin receptor Mincle during lethal infection in mice and in primary macrophage cultures. This review gives a brief summary for clinical and epidemiologic features of these five bacterial infections, focuses on fundamental biologic facets of infection, and recent advances in host recognition. In addition, we discuss knowledge gaps for innate recognition of these bacteria in the context of disease pathogenesis.
Keywords: Pattern Recognition Receptor, Innate Immunity, Obligate Intracellular Bacteria, Orientia tsutsugamushi, Rickettsia, Anaplasma, Ehrlichia, Coxiella burnetii
BACKGROUND
Pattern recognition receptors (PRRs) can sense a stunning array of self- or non-self- molecules, serving as sentinels of infection. They can detect pathogen-associated molecular patterns (PAMPs), along with host damage-associated molecular patterns (DAMPs), to initiate immune responses [1–5]. Four major families of PRRs have been identified and each can sense distinct molecular motifs or structures, playing specific or cooperative roles during infection. The cross-talks among different or same receptor family members, as well as their downstream signaling pathways, can lead to pathogen and host context-dependent immunological outcomes [2–5]. Different PRRs collectively shape both the innate and adaptive arms of immunity to provide pathogen-specific responses, which can lead to infection control or promote disease pathogenesis.
Toll-Like Receptors (TLRs) were the first identified family of PRRs [6]. TLRs are transmembrane proteins which occupy both plasma and endosomal membranes [2]. Those located on the cell surface detect bacterial components, including lipoproteins (TLRs 1, 2, and 6) [7–10], lipopolysaccharide (TLR4) [11], and flagellin (TLR5) [12]. In contrast, endosomal TLRs detect nucleic acids of viral or parasitic origin, including double-stranded RNA (TLR3) [13], single-stranded RNA (TLR7 and TLR8) [14–17], and CpG-containing single-stranded DNA (TLR9) [18]. After ligand binding, TLRs interact with an adaptor complex consisting of either 1) myeloid differentiation primary-response protein 88 (MyD88), which is shared by all TLRs, or 2) Toll/IL-1R-domain-containing adapter-inducing interferon-β (TRIF), which is utilized by TLR3 and TLR4 [2]. The result of TLR signaling via MyD88 is transcription of NF-κB- and AP-1-dependent genes, whereas signaling via TRIF results in transcription of NF-κB, AP-1, and IRF3 controlled genes and induction of necroptosis [2]. While TLRs are also implicated in sensing DAMPs, this aspect of signaling is outside the scope of this manuscript and excellently reviewed elsewhere [19, 20].
Nucleotide Binding Oligomerization Domain-Like Receptors (NLRs) are located within the cytoplasm and contain a nucleotide-binding domain and leucine-rich repeat domain, the latter of which is involved in sensing PAMPs and DAMPs [3]. NLRs are grouped into subfamilies based on the presence of additional domains, including caspase activation and recruit domains (NLRC), pyrin domains (NLRP), and others [3]. Within the NLRC subfamily, NOD1 and NOD2 are widely studied and known to recognize the building blocks of peptidoglycan (muropeptides and muramyl dipeptides, respectively) [21, 22]. Activated NOD1 and NOD2 may interact with the receptor-interacting-serine-threonine-kinase2 (RIP2) to stimulate NF-κB mediated transcription or, alternatively, IRF7/3 to induce interferon responses [3]. Another member of the NLRC subfamily, NLRC4, recognizes intracellular flagellin and contributes towards inflammasome assembly, leading to Caspase-1 activation and secretion of the proinflammatory cytokine IL-1β [3, 23–26]. Members of the NLRP subfamily, including NLRP3, respond to potassium efflux, reactive oxygen species, and bacterial lipoproteins to activate the inflammasome and proinflammatory cytokine secretion (namely IL-1β) [3]. NLRs have also been implicated in numerous other cellular processes, including autophagy, and have been shown to crosstalk with TLRs and RIG-I (below) [3].
Like NLRs, Retinoic Acid-Inducible Gene I (RIG-I)-Like Receptors (RLRs) are localized in the cytosol [27]. The RLR family includes RIG-I, melanoma differentiation-associated protein 5, and laboratory of genetics and physiology protein 2 [4]. This family contains a central helicase domain and carboxy-terminal domain, which collectively recognize immunostimulatory RNA harboring 5’-PPP moieties [4]. Upon activation, RIG-I interacts with mitochondrial antiviral-signaling protein to initiate type-1 interferon responses and NF-κB translocation [4]. While activation of RLRs has historically been implicated in sensing viral infection, recent evidence has shown they may also sense mitochondrial RNA [28, 29] and could play a wider role in sensing DAMPs than previously thought.
C-Type Lectin Receptors (CLRs) are a diverse superfamily comprised of over 1,000 proteins that, by definition, contain at least one C-type lectin-like domain [5]. CLRs are expressed predominately in myeloid cells and can be found secreted or anchored to the plasma membrane [5, 30]. This family of receptors recognizes endogenous and exogenous carbohydrate or glycolipid moieties [5]. Activation of CLRs shapes inflammation through the adaptor protein spleen tyrosine kinase (SYK). CLRs interact with SYK via an immunoreceptor tyrosine-based inhibitory or immunoreceptor tyrosine-based activation motif in its own cytoplasmic tail, or through coupling with signaling partners (mainly FcγRs or DAP10/12) [5]. The majority of CLRs studied in the context of bacterial recognition are members of the Group II asialoglycoprotein receptor family, including Dectin-1 and dendritic cell immunoreceptor subfamilies [5]. Mincle (Macrophage inducible C-type lectin; also known as Clec4e), the most well-characterized CLR, is a member of the dendritic cell immunoreceptor subfamily known to recognize bacterial glycolipids as well as host DAMPs [5]. The outcome of Mincle activation is highly varied and context dependent but includes inflammatory macrophage (MΦ) polarization, induction of type 1-skewed T helper responses, and proinflammatory cytokine production [5].
Studies identifying PRR activation during bacterial infection have focused extensively on extracellular or facultative intracellular bacteria, with scant evidence available for obligate intracellular bacteria. However, obligate intracellular bacteria are a group of clinically important organisms that are highly prevalent throughout the world [31, 32]. This unique group includes the Rickettsiales (Rickettsia spp., O. tsutsugamushi, Anaplasma spp., Ehrlichia spp.), as well as C. burnetii. Virtually all these obligate intracellular bacteria have zoonotic infection cycles (Table 1). For example, Rickettsia spp. can be found on all continents except Antarctica and are transmitted to humans via numerous blood-feeding arthropods (including ticks, lice, and fleas) [32, 33], while O. tsutsugamushi is endemic across Southeast Asia and transmitted via mite [34]. In contrast, Anaplasma and Ehrlichia are predominately found within the United States and transmitted via numerous species of tick [35, 36]. C. burnetii, while found globally, is endemic to the Mediterranean region and commonly spread via livestock secretions [37]. However, recent evidence indicates that ticks may serve as a vector of transmission [38–40].
Table 1.
Overview of Obligate Intracellular Bacterium Characteristics and Defined Immune Sensing
| Obligate Intracellular Species | Reservoir | Transmission | Primary Host Cell for Replication | Location in Host Cell | Receptor(s) in Recognition | Adaptor Proteins Implicated | |
|---|---|---|---|---|---|---|---|
| Animal Vector | Arthropod Vector | ||||||
| Orientia tsutsugamushi | Rodents, Leptotromibidium mites | Not Known | Leptotromibidium * | Endothelial Cells, Neutrophils, Monocytes/Macrophages, Dendritic Cells | Cytosol | TLR2 [63] TLR4 [61] RIG-I [64] NLRC5 [65] Mincle [49] |
STING [64] |
| Rickettsia spp. | Various: Rodents, Dogs, Forest animals (opossums, squirrels) | Not Known | Various: Dermancentor, Ixodes, Amblyomma, Bothriocroton, Rhipicephalus, Rhizomucor, more | Endothelial Cells, Macrophages | Cytosol | TLR2 [78] TLR4 [76–78] NLRP3 [80] |
Myd88 [74] ASC [77, 80] |
| Anaplasma phagocytophilum | Forest animals (mice, skunks, deer), Horses, Household pets | Not Known | Ixodes*, Amblyomma, Dermacentor | Neutrophils | Vacuole | NOD2 [98] NLRC4 [100] TLR2 [96] |
RIP2 [98] MyD88 [98] STAT1 [96] |
| Ehrlichia chaffeensis | White-tailed deer*, coyotes, dogs | Not Known | Amblyomma*, Ixodes, Dermacentor | Monocytes/Macrophages | Vacuole | TLR2 [111, 113] TLR7 [111] TLR9 [114] NOD2 [113] |
Myd88 [112] |
| Coxiella burnetii | Animal livestock (goats, cattle, sheep, dairy cows), Birds, Reptiles, Arthropods (Ixodes, Dermacentor, Haemaphysalis) | Fluid secretions (amniotic fluid, feces, vaginal secretions, urine) *, Unpasteurized Milk | Not Known | Monocytes/Macrophages | Vacuole | NOD2 [135] TLR1 [135] TLR2 [137, 138] TLR4 [137] |
MyD88 [135–137] TRIF [137] Myd88, TRIF [137] |
Denotes primary mode of transmission
The biology of obligate intracellular pathogens necessitates immune evasion since replication can only occur within host cells. Thus, these bacteria exhibit many unique characteristics compared with extracellular bacteria, most prominently the lack of immunostimulatory cell wall components and extensive genome reduction [31]. O. tsutsugamushi, for example, lacks biosynthetic pathways for both peptidoglycan and lipopolysaccharide (LPS) [41]. Additionally, the genome of O. tsutsugamushi is comprised of ~1.2 million base pairs [41], which is in stark contrast with the ~5 million base pair genome of Salmonella typhi [42] or the ~4.5 million base pair genome Escherichia coli [43]. Very little is known regarding recognition of obligate intracellular bacteria and how PRRs orchestrate the immune response to such pathogens. Direct comparative analyses among these bacteria are severely lacking. To our knowledge, only one report has used primary human dendritic cells and directly compared innate immune signatures among obligate intracellular bacterial species, including O. tsutsugamushi and C. burnetii [44]. Studies aimed at defining the PRR response have been complicated by challenges associated in working with these agents, including technical difficulties in propagating large-scale cultures and the necessity of biocontainment facilities for O. tsutsugamushi, Rickettsia spp., and C. burnetii [31, 41, 45].
While highly treatable, the Rickettsiales and C. burnetii are often overlooked causes infection and severe disease usually includes immunopathogenic features [32, 46–48]. Thus, thoroughly defining activated PRRs and their impact on immune signatures would yield significant insight into disease pathogenesis and potential treatments for the severely ill. In this review, we discuss critical clinical and epidemiologic features of the Rickettsiales and C. burnetii, along with recent advances in the understanding of PRR sensing during initial infections with these obligate intracellular bacteria. Finally, we highlight key areas for future studies to define the potential links between PRRs and disease pathogenesis.
METHODS
Databases, search strategy, study selection, and RNA sequencing
Articles for obligate intracellular bacteria of interest were identified through scouring relevant publications from electronic sources. Searching was performed via Ovid-Medline and Pubmed-Medline. Studies were identified by combining search terms for bacteria of interest and PRR of interest. For example, studies for O. tsutsugamushi were identified by searching “Orientia and TLR”, “Orientia and NLR”, “Orientia and RLR”, “Orientia and CLR”. JRF and ZDC reviewed abstracts generated by the search for relevance and, unless a seminal publication, only included reports from the recent 10 years. Host gene transcriptional profiles were based on late stages of lethal infection in mice and tissue analyses by using NanoString, RNAseq, or qRT-PCR approaches [49, 50].
ORIENTIA TSUTSUGAMUSHI
Epidemiology and clinical features
O. tsutsugamushi is the causative agent of scrub typhus, a life-threatening disease of globally widening impact. Approximately 1 million cases of scrub typhus occur each year in an endemic region termed the “tsutsugamushi triangle”, which spans throughout southeast Asia and northern Australia [34]. Recent reports, however, have shown serological prevalence of scrub typhus in historically non-endemic regions, including South America [51, 52] and Africa [53, 54]. While rodents may serve as an animal reservoir of this bacterium [55], O. tsutsugamushi is predominately maintained in Leptotrombidium mites (commonly known as chiggers) [56]. The bacterium is transmitted to humans via bite of larval stage mites and disease pathogenesis mainly occurs in highly vascularized organs (lung, liver, brain, etc.) [34]. Scrub typhus may manifest as interstitial pneumonia, liver damage, and meningoencephalitis [34]. If left untreated, disease can progress to multi-organ failure with fatality rates ranging from 0–70% (median of 6%) [34, 57, 58].
TLR/RIG-I/NLR-mediated immune recognition
O. tsutsugamushi is an LPS-negative, Gram-negative coccobacillus which primarily infects endothelial cells and phagocytes (MΦs, neutrophils, and dendritic cells) [41]. Compared with the other four bacterial species of interest, O. tsutsugamushi has unique biology, as well as host recognition mechanisms (Fig. 1). After the bacterium is internalized via endocytosis or phagocytosis, it rapidly escapes the endosome to freely inhabit the cytosol [41]. The bacteria can utilize microtubules to traffic to the perinuclear region where replication occurs. O. tsutsugamushi replicates slowly, with peak rates occurring over 1 – 5 days post-infection (dpi), and then exits host cells via a budding-like mechanism [41, 59]. A recent report has shown O. tsutsugamushi actively inhibits NF-κB activation to evade host responses during its replication process [60]. However, very few reports have examined innate recognition of O. tsutsugamushi and mechanisms of PRR sensing have remained obscure.
Figure 1. Orientia tsutsugamushi intracellular life cycle and host innate responses.
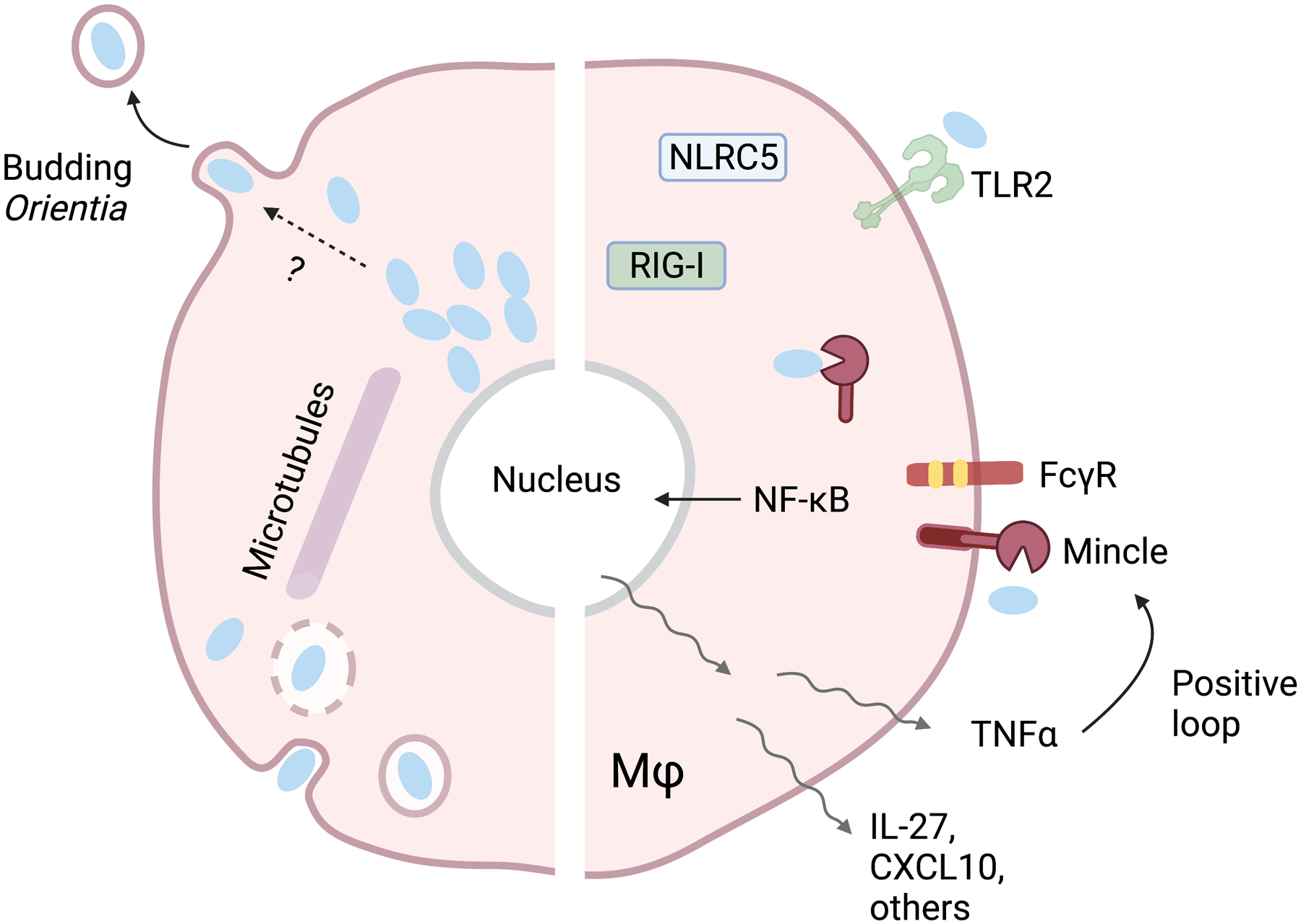
After entering the host cell through endocytosis, Orientia bacteria rapidly escape the endosome, move to the perinuclear region via microtubules, and replicate freely (and slowly) in the cytoplasm. O. tsutsugamushi then exits the cell via a poorly defined budding mechanism. Host immune recognition is mediated by Mincle/Fcγ receptor-regulated mechanisms, although the involvement of other sensors (TLR2, RIG-I, NLRC5) are reported. Mincle signaling promotes expression of NF-κB-induced proinflammatory genes, including TNFα, IL-27, CXCL10, among others. TNFα, in turn, can promote Mincle expression via a positive feedback loop, enhancing proinflammatory responses.
While TLR activation during O. tsutsugamushi infection has been shown to occur, the evidence is still debatable. One study in humans suggests a TLR4 mutation (D299G) linked to increased scrub typhus susceptibility [61]. This mutation is within the ligand binding pocket of TLR4 and has been implicated in susceptibility to tuberculosis [62]. Since O. tsutsugamushi lacks LPS, the observed link between TLR4 and susceptibility to scrub typhus is likely due to yet-to-be-defined or indirect mechanisms. This speculation was supported by another study utilizing human TLR4- or TLR2-transfected HEK293 cells, showing that TLR4 does not directly recognize O. tsutsugamushi, while TLR2 may sense bacterial components and promote IL-6 secretion [63]. The role of TLR2 in O. tsutsugamushi infection is supported in bone marrow-derived dendritic cells (BMDC), as infected TLR2−/− BMDCs secrete less IL-6 and TNFα than wild-type (WT) controls [63]. When TLR2−/− and WT C57BL/6 mice were infected intradermally (producing a self-limiting infection), no differences in survival or bacterial loads were observed [63]. However, when mice were infected via the intraperitoneal route (producing lethal infection), TLR2−/− mice had milder disease scores and pathology, but greater bacterial loads in the lung, spleen, and peritoneum, than WT mice. Since O. tsutsugamushi-infected TLR2−/− and WT mice had comparable levels of IL-6 and TNFα transcripts, the biological function of TLR2 in this infection is unclear.
Like TLRs, the role of the cytosolic sensor RIG-I in sensing O. tsutsugamushi is debatable. Min et al have shown that infected mouse embryonic fibroblasts lacking functional MAVS, RIG-I, or STING express less IFN-β and TNFα transcripts than their WT comparators early in infection [64]. However, data for other in-vitro time points or in mice are lacking.
Controversial evidence for NLR activation during O. tsutsugamushi infection has also been reported. For in vitro studies in nonprofessional phagocytes (HeLa and primary human aortic endothelial cells), O. tsutsugamushi can reduce NLRC5 protein levels at late infection (72 hpi) to downregulate major histocompatibility complex-1 expression [65]. In THP-1 monocyte-like cells, however, NLRC5 expression was only temporarily reduced early in infection and rebounded by 72 hpi [65]. Additionally, Cho et al have shown infection-associated NOD1 protein expression, as well as reduced proinflammatory cytokine protein expression in NOD1-knock-down cells [66]. However, NOD1/NOD2 involvement in O. tsutsugamushi infection could not be validated by a separate team, who utilized mouse BMMΦ lacking RIP2, NLRP3, NLRP4, or AIM2, respectively [67].
CLR-mediated immune recognition
The first evidence for Mincle, a unique member in the CLR family, in response to O. tsutsugamushi was reported in 2021 via comprehensive molecular and immunological approaches [49]. Firstly, differential host gene expression profiling analyses of tissues collected from lethally infected C57BL/6 mice (via intravenous route) have revealed 36-fold increase of Mincle (also known as Clec4e) in the lungs, as well as ~400 – 14,000-fold increase in the brains at 10 dpi (prior to host death) (Table 2). Simultaneously, a low degree of TLR, NLR, and RLR expression has been observed. Secondly, multiple approaches (NanoString, qRT-PCR, Western blot, and immunofluorescent staining) consistently confirmed Mincle activation in conjunction with the upregulation of Mincle signaling partner (FcγRs) and proinflammatory cytokines/chemokines (CXCL9-11, TNFα, IL-27) in inflamed or damaged lungs. Thirdly, our in-vitro studies using BMMΦ revealed upregulated Mincle RNA and protein levels in response to live or inactivated O. tsutsugamushi, which positively correlated to upregulated type 1-promoting markers (CXCL9-11, TNFα, IL-27), MΦ chemotactic markers (CCL2-7), and the neutrophil chemotactic marker CXCL1. In contrast, infected Mincle−/− BMMΦ exhibited abrogated transcription of CCL2 and CXCL1, implicating the receptor in propagating inflammation. Finally, given that both bacterium-carrying and uninfected MΦs express Mincle, and that tissue Mincle levels reach peaks at late stages of disease, we speculated the contribution of host factors in driving Mincle expression. Indeed, we have confirmed a positive, synergetic role of TNFα in regulating Mincle expression, since BMMΦ pre-treated with TNFα prior to infection can greatly enhance Mincle, IL-27, and CXCL10 expression; some of these effects are markedly reduced infected Mincle−/− cells. Together, our studies confirmed, for the first time, an important role of Mincle in sensing live versus inactivated O. tsutsugamushi. We have proposed that Mincle/FcγR activation via bacterial glycoprotein/glycolipid motifs and innate host factors (e.g. TNFα) is one of the major mechanisms that program MΦs to a M1-like phenotype, - contributing to Th1/M1-skewed inflammatory responses in O. tsutsugamushi-infected mice and human patients (Fig. 1) [49, 50, 68–71].
Table 2.
PRR Gene Expression in O. tsutsugamushi-Infected C57BL/6 Mice
| Fold Change (D10 vs. D0) | ||||
|---|---|---|---|---|
| Gene/Alias (Encoded Protein) | Lung NanoString (Ref [49]) | Brain NanoString (Ref [49]) |
Brain RNAseq |
|
| CLR | Clec4e (Macrophage Inducible C-type Lectin; Mincle) | 36.00 | 441.21 | 14082.01 |
| Clec4d (Macrophage C-type Lectin; MCL) | - | - | 118.47 | |
| Clec5a (Myeloid DAP12-Associating Lectin 1; MDL) | 6.96 | 6.34 | 4.15 | |
| CLR Partner | Fcgr4 (Fcγ Receptor 4) | 18.77 | 517.89 | 138.43 |
| Fcgr1 (Fcγ Receptor 1) | 7.11 | 12.79 | 7.21 | |
| Fcgr3 (Fcγ Receptor 3) | 3.81 | 7.93 | 5.54 | |
| Fcgr2b (Fcγ Receptor 2b) | 3.20 | 14.81 | 5.37 | |
| TLR | Toll-like receptor 1 | 3.13 | 9.19 | 9.88 |
| Tlr2 | 1.97 | ns | 8.46 | |
| Tlr4 | 0.98 | 5.46 | 4.64 | |
| Tlr6 | 4.28 | - | 2.84 | |
| MyD88 | 1.85 | 3.54 | 2.86 | |
|
NLR and
RLR |
Nlrc5 (NLR Family CARD Domain Containing 5) | - | - | 79.53 |
| Nod1 (Nucleotide-Binding Oligomerization Domain-Containing Protein 1) | 1.38 | - | 4.35 | |
| Nod2 (Nucleotide-Binding Oligomerization Domain-Containing Protein 2) | 2.60 | ns | 4.48 | |
| Nlrp3 (NLR Family Pyrin Domain Containing 3) | 3.29 | - | 3.46 | |
| Ddx58 (RIG-I, retinoic acid-inducible gene I) | 0.80 | 6.53 | 4.61 | |
All values presented are statistically significant changes (p < 0.05) unless denoted not significant (ns).
Knowledge gaps and future studies
The evidence for PRR involvement during O. tsutsugamushi infection has been enigmatic. Research has been placed on examining classical PRRs such as TLRs, RIG-I, and NLRs; however, none of these receptors play significant roles during infection. The studies for TLR2/4 are inconclusive [63] and challenging to link to the biology of O. tsutsugamushi since this bacterium lacks LPS and conventional peptidoglycan. The RIG-I- or NLR-related studies have intrinsic limitations due to the use of mouse embryonic fibroblasts or other cell lines [64, 66]. Mincle-mediated pathways can not only sense and differentiate live versus inactivated O. tsutsugamushi, but also enhance the inflammatory responses in MΦ [49], which provokes many questions. Is Mincle a key sensor during infection in experimental animals and in human patients? If so, what is the bacterium- and/or host-derived ligands for Mincle activation at early versus late stages of infection? How does Mincle interact with other PPRs for infection control and/or immunopathogenesis? Studies aimed at assessing the biological functions of Mincle on in-vivo infection will yield insights into immune recognition of this severely neglected bacterium.
RICKETTSIA SPP.
Epidemiology and clinical features
Rickettsia spp. can be found on all continents except Antarctica, causing a wide range of human diseases [33]. Bacteria in this genus are classified into four groups based on taxonomy and associated epidemiologic features. The spotted fever group (SFG) includes R. rickettsii (Rocky Mountain Spotted Fever), R. conorii (Boutonneuse fever), R. africae (African tick-bite fever), and R. parkeri (Maculatum disease), which are transmitted to humans via ticks. The transitional group includes R. akari (Rickettsialpox), R. australis (Queensland fever), and R. felis (flea borne spotted fever), which are transmitted to humans via fleas, ticks, or mites. The typhus group (TG) consists of R. prowazekii (epidemic typhus) and R. typhi (murine typhus), which are transmitted to humans via fleas, lice, or flying squirrels. Finally, the ancestral group is composed of R. canadensis and R. bellii and is not associated with any human diseases [31, 72]. The rickettsioses display a diverse array of clinical symptoms and severity. Most infections begin with constitutional symptoms accompanied by rash [32]. However, disease can progress to multiorgan failure and other life-threatening syndromes if not promptly treated [32]. Case fatality rates differ greatly among rickettsioses, with Rocky Mountain Spotted Fever and epidemic typhus ranking among the highest (15–65%) [72, 73]. As such, R. rickettsii and R. prowazekii have garnered significant research interest due to high infectivity, significant mortality, as well as the potential for use as bioterrorism agents [72]. Interestingly, although no fatal cases have been reported, recrudescence of epidemic typhus, known as Brill-Zinsser disease, can occur years after initial infection [32].
TLR/MyD88-mediated immune recognition
Rickettsia are Gram-negative, LPS-positive bacilli which primarily infect host endothelial cells and MΦs [72]. After entering a host cell via endocytosis, Rickettsia will escape the endolysosome to replicate freely within the cytoplasm in a manner like O. tsutsugamushi [31]. The ability to subvert autophagy plays a major role in bacterial survival, but the underlying mechanisms remain largely unexplored [74, 75]. Replication is followed by direct invasion of neighboring cells (spotted fever group) or host cell lysis (typhus group) (Fig. 2), which is in sharp contrast to the budding mechanism employed by O. tsutsugamushi [31].
Figure 2. Rickettsia spp. intracellular life cycle and host innate responses.
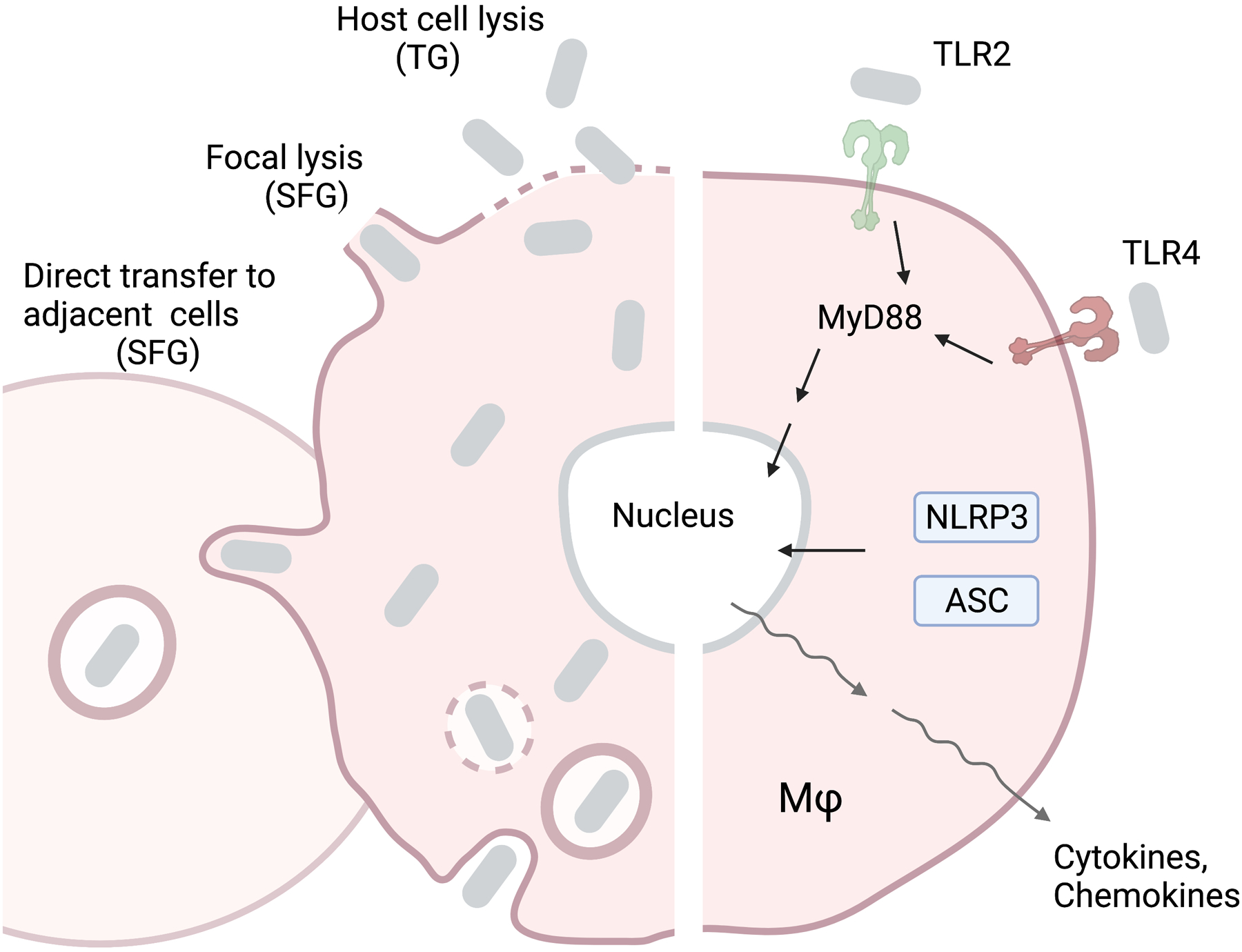
After entering the host cell through endocytosis, Rickettsia bacteria undergo endolysosome escape and cytoplasmic replication, similar to O. tsutsugamushi. However, these bacteria exit from the host cell either by direct transfer to the adjacent cell (Spotted Fever Group), focal lysis (Spotted Fever Group), or cell lysis (Typhus Group). Host immune recognition is mediated by TLR2/4-regulated mechanisms, which activate the MyD88 pathway, or NLRP3/ASC, which activate the inflammasome. Recognition of Rickettsia is followed by rapid transcription of proinflammatory cytokines and chemokines.
TLR2/4- and MyD88-mediated mechanisms are the most well characterized pathways for innate recognition of Rickettsia. Early evidence indicated that C3H/HeJ mice (naturally deficient in TLR4 function) were more susceptible to R. conorii challenge and harbored greater bacterial loads in the brain and lungs than C3H/HeN mice (TLR4 competent) [76]. Infected C3H/HeJ mice also displayed reduced splenic natural killer cell activation that could be rescued by adoptive transfer of TLR4 competent DCs, implicating TLR4 as a driver for this process [76]. Additionally, the secretion of proinflammatory cytokines (IL-1α and TNFα) was much lower in infected C3H/HeJ primary brain microvascular endothelial cells than C3H/HeN cells [76]. TLR4 has also been shown to recognize R. australis [77]. Infected TLR4−/− C57BL/6 BMMΦ produced less pro-IL-1β transcripts than WT controls and harbored greater bacterial loads [77]. Additionally, WT cells, but not TLR4−/− BMMΦ produced pro-IL-1β transcripts in response to purified R. australis LPS in TLR4−/− BMMΦ indicating that rickettsial LPS is the likely stimulus for TLR4. Thus, TLR4 recognizes R. conorii and R. australis and contributes to generating the proinflammatory response.
TLR2 also plays a role in sensing Rickettsia. Quevedo-Diaz et al used in-vitro systems to examine whether R. akari can activate TLR2/4 [78]. Heat-killed R. akari was added to HEK293T cells stably transfected to express human TLR2 or TLR4. After addition of heat-killed R. akari, both TLR2- and TLR4-expressing HEK293T cells exhibited NF-κB activation, whereas TLR2/4 negative cells did not. Using a luciferase-based assay, the authors then showed that engineering specific amino acid residue mutations within TLR2 (R753Q) or TLR4 (D299G) abrogated NF-κB activation in HEK293T cells, further implicating the receptors in directly recognizing the bacterium. When anti-TLR2 and anti-TLR4 antibodies were used simultaneously in human monocytes exposed to heat-killed R. akari, there was a 60% reduction in TNFα expression compared to control cells. However, the effects of these antibodies were modest in cells exposed to live R. akari. It is possible that live bacteria can stimulate TNFα expression in TLR2/4-dependent and -independent manners, and that TLR2 may play a minor role in immune responses to rickettsia. This speculation was supported by a recent study, which revealed no differences in survival or bacterial loads between R. conorii infected TLR2−/− and WT C57BL/6 mice [74].
Nevertheless, MyD88 is essential for host sensing and protection against Rickettsia. MyD88−/− C57BL/6 mice are highly susceptible to high-dose R. conorii (0% survival), compared to WT mice (100% survival), due to uncontrolled bacterial growth in the liver, spleen, and lungs [74] R. australis-infected MyD88−/− mice also displayed low type 1 and proinflammatory cytokines in the lungs (IFNγ, TNFα, IL-6, and IL-1β transcripts) and sera (IFNγ, IL-12-p40, IL-12-p70, IL-6, G-CSF proteins). Histologic analysis revealed reduced MΦ numbers and reduced frequency/size of inflammatory infiltrations in the liver, concomitant with reduced neutrophils in the lung of infected MyD88−/− animals. In-vitro studies of infected MyD88−/− BMDCs revealed reduced MHC-II expression and no increase in IL-12-p40 expression compared with WT cells, indicating impaired DC maturation. Together, these findings indicate that MyD88 is responsible for host protection against R australis via DC maturation and the generation of type 1-skewed responses.
NLR- and ASC-mediated immune recognition
New evidence on how NLRs shape the inflammasome during rickettsial infection has recently emerged. Inflammasome activation can lead to different infection outcomes, depending on the Rickettsia species examined. R. parkeri-induced inflammasome activation has been shown to antagonize type-1 interferon responses in vitro and in vivo, allowing bacterial growth [79]. This phenomenon was shown via infected Caspase 1/11−/− C57BL/6 BMMΦs, which exhibited reduced bacterial loads and increased type 1 interferon compared with WT [79]. Yet, no specific NLR was implicated in driving these findings. In contrast, the inflammasome has been shown to inhibit R. australis growth. R. australis-infected human peripheral blood mononuclear cell (PBMC)-derived MΦs and C57BL/6 BMMΦs can rapidly secrete IL-1β as early as 3 hours post-infection (hpi) [80]. Concomitantly, NLRP3 transcripts were significantly increased in infected cells by 4 hpi, leading the authors to examine the role of NLRP3 and ASC (a key NLR-inflammasome adaptor protein) in recognizing R. australis infection. NLRP3−/− BMMΦs exhibited reduced IL-1β secretion across different infectious doses, whereas cleaved Caspase-1 (indicating inflammasome activation) was evident only at a high dose. For in-vivo studies, infected NLRP3−/− mice harbored high bacterial loads in the spleen, but not within the liver or lung, when compared with WT mice. The lack of impact on survival or histopathology in NLRP3−/− mice indicates that while NLRP3 may contribute to tissue specific responses, it is not essential for controlling infection in-vivo. Examining the role of ASC, the authors observed ASC−/− MΦs infected with R. australis produced virtually no IL-1β, IL-18, or activated Caspase-1 protein, but did not follow up with in-vivo characterization. A subsequent study analyzing the role of inflammasome activation during R. australis infection revealed a powerful role for ASC and potential crosstalk with TLR4 [77]. ASC−/− C57BL/6 mice infected with a sublethal dose of R. australis were highly susceptible to infection, evidenced by 90% of these animals succumbing to disease. Bacterial loads within the liver, lung, and spleen were significantly greater in ASC−/− animals, indicating the role of ASC in host resistance against R. australis infection. Additionally, serum levels of IL-1β, IL-18, and IFNγ were significantly reduced in infected ASC−/− mice when compared with controls in the terminal phase of disease. Interestingly, infected ASC−/− BMMΦs harbored greater bacterial loads and produced more pro-IL-1β than WT. Purified R. australis LPS also stimulated pro-IL-1β transcription in ASC−/− MΦs. Considering that pro-IL-1β was not induced upon treatment with R. australis LPS in TLR4−/− MΦs, the authors posit that ASC-driven inflammation is triggered by TLR4-mediated IL-1β production.
Knowledge gaps and future studies
While TLR4 is likely a key innate sensor for Rickettsia (Fig. 2), defining its molecular interactions is not straightforward for several reasons. Rickettsia spp. have very low levels of LPS (1–2% of total biomass) [81]. Considering the inherent challenges associated in cultivating this obligate intracellular bacterium at a large scale, extraction and purification of rickettsial LPS in sufficient quantities for in-vitro and in-vivo analysis remains exceedingly difficult. Perhaps not surprisingly then, very few comparative studies have been performed analyzing structural or immunologic differences between rickettsial LPS and LPS from other Gram-negative bacteria such as E. coli or Salmonella [81, 82]. Therefore, bioinformatic and modeling studies are needed to predict structural interactions between rickettsial ligands and TLRs. Despite NLRP3 and ASC involvement during Rickettsial infection, upstream mediators of the inflammasome, including ROS production and ion imbalance, have not been explored [80]. There is also a need to address whether inflammasome activation through NLRP3 and ASC occurs via host DAMP molecules, bacterial components/pathways, or other indirect signals. There is a stark need for more studies to examine innate recognition of Rickettsia, as most have focused on relatively few species in the context of a few sensing receptors. Furthermore, studies examining other classes of PRRs, including the CLRs and RLRs, in recognizing the rickettsiae are lacking. Whether modes of innate recognition are universal across SFG, transitional group, and typhus group rickettsia remains to be determined.
ANAPLASMA PHAGOCYTOPHILUM
Epidemiology and clinical features
A. phagocytophilum is the etiologic agent of human granulocytic anaplasmosis, a potentially lethal febrile illness endemic to the Northeast and North Central United States [35]. Anaplasma spp. were once considered Ehrlichia and are closely related to the genus Rickettsia [36, 83, 84]. Epidemiological surveys have revealed Anaplasma spp. are maintained in a large pool of hosts, ranging from small mammals and birds to various species of deer and even horses [85]. Transmission occurs through the bite of Ixodes ticks and humans are the accidental dead-end host [86]. Co-infection is common since Ixodes ticks may also transmit Babesia microti, Borrelia burgdorferii, and encephalitic viruses [87]. Anaplasma genetic material has also been detected in sequenced saliva from Amblyomma and Dermacentor ticks, however, transmission from these vectors has not been well-studied [88]. Following infection, early clinical symptoms are nonspecific and include fever, chills, headache, and myalgias. In a small percentage of cases that do not receive proper treatment (< 1%), HGA can manifest into hematological issues, along with respiratory distress, renal failure, septic shock, and more [89]. While the clinical course of this disease has been characterized, host immune recognition remains less clear. Considering the incidence rate of human granulocytic anaplasmosis in the United States has shown a growing trend since 2008 [90, 91], careful examination into the host immunological response is warranted.
Immune recognition
A. phagocytophilum is a Gram-negative bacterium that preferentially infects neutrophils [92]. After invading a neutrophil, intracytoplasmic replication occurs in clusters of bacteria known as morulae, which can be identified through blood smear approaches [93]. A slew of cellular events initiated by the bacteria prevents its detection and elimination. Previous reports have shown that inhibition of apoptosis occurs via stimulation of the PI3K/Akt and p38 MAPK pathways, which prolongs survival of an infected cell [94, 95]. A. phagocytophilum also lacks genes necessary for LPS and peptidoglycan synthesis, which facilitates passive immune evasion [83].
Early reports suggest a possible role for TLR2 and MyD88 in sensing A. phagocytophilum in-vitro [96, 97]. Recently, the link between MyD88- or TRIF-dependent TLRs and inflammation was explored via in-vitro and in-vivo approaches. Infected MyD88−/−, MyD88/TRIF−/−, and TLR2/3/4/7/9−/− murine Hoxb8 neutrophils showed reduced proinflammatory responses (Nos2 transcripts; TNFα, CCL4, and CCL5 secretion) when compared with WT cells [98]. Despite differences in inflammation in-vitro, no phenotypic differences were observed in-vivo between infected WT C57BL/6 or TRIF−/− mice. Collectively, these studies imply that while both MyD88- and TRIF-dependent TLRs may sense A. phagocytophilum, they do not influence the outcome of infection.
Several studies have suggested NLR activation in response to A. phagocytophilum. First, human primary neutrophils were shown to upregulate RIP2 transcripts within 4 hpi [99]. When the effect of RIP2 was tested in C57BL/6 mice, infected RIP2−/− animals were shown to exhibit higher bacterial loads in blood and delayed clearance of infection when compared with WT mice [99]. Additionally, when serum cytokine/chemokine responses were measured, infected RIP2−/− mice displayed two-fold less IFNγ levels. This led the authors to speculate that NLRs signaling through RIP2 may contribute to mounting the Th1 response to Anaplasma [99]. A separate study by Müller and colleagues built upon this by analyzing the contribution of specific NLRs during infection [98]. They observed significantly increased A. phagocytophilum loads in the blood and lungs of NOD2−/− C57BL/6 mice throughout the course of infection, but both NOD2−/− and WT mice were able eventually clear the bacterium at a similar rate [98]. Using Hoxb8 murine neutrophils, no differences in bacterial load or proinflammatory markers were observed between infected WT, NOD1−/−, NOD2−/−, and NLRP3−/− cells [98]. However, A. phagocytophilum has been shown to activate NLRC4 via a unique mechanism. A. phagocytophilum-infected BMMΦ produced increased amounts of prostaglandins (PGE2, PGD2, TXA2), paralleled with increased activity of cyclooxygenase and phospholipase enzymes [100]. A unique feature of this pathway was shown downstream, where increased levels of PGE2 led to the initiation and activation of the NLRC4 inflammasome complex [100]. The known activators of this inflammasome pathway are flagellin and T3SS, both of which are absent in Anaplasma. How Anaplasma infection activates the NLRC4 inflammasome remains unclear.
The contribution of other PRRs in sensing A. phagocytophilum is less understood, with no studies examining RLRs and a single study revealing no role for CLRs. To examine the contribution of CLRs, DAP12−/−, FcRγ−/−, SYKdel/del C57BL6 mice were infected with A. phagocytophilum and followed for duration of disease [98]. No differences in survival or bacterial loads in blood, spleen, or lung were observed between WT and DAP12−/−, FcRγ−/−, or SYKdel/del mice. Additionally, infected DAP12−/− and FcRγ−/− Hoxb8 neutrophils exhibited no differences in bacterial loads or proinflammatory markers compared with WT counterparts. Notably, Sykdel/del Hoxb8 neutrophils were not studied, as these cells could not be cultivated in sufficient quantities. Thus, CLRs do not contribute significantly to controlling A. phagocytophilum infection in-vivo or generating neutrophil inflammation in-vitro.
Knowledge gaps and future studies
Despite advances in understanding innate responses to A. phagocytophilum, many challenges remain. One major hurdle in defining immune signatures associated with severe disease is that lethal models of Anaplasmosis have not yet been developed. While murine models of disease, including C57BL/6 and BALB/c, accurately mimic pathologic features associated with human disease, infection is cleared generally within 20 days of infection [101]. Even infection of immunodeficient models, including SCID−/− mice, are non-lethal [102]. Thus, immunologic differences between mild and severe infection may be blurred. While TLRs were the first PRR family studied, they were found to play a small role in-vivo and in-vitro [98, 103, 104]. Since Anaplasma spp. lack both peptidoglycan and LPS, the limited role for TLRs may not be surprising. Additionally, the strongest evidence for NLR involvement largely hinges on findings from knocking out the adaptor protein RIP2 [99]. Specific NLRs contributing to the innate response remain undefined and future studies are needed to identify subclasses of NLRs which may recognize A. phagocytophilum. Finally, the studies of host innate responses in the absence of Ixodes ticks is a concern, as tick saliva can modulate or dampen initial immune responses to A. phagocytophilum infection. While one study with BMMΦs treated with saliva from Ixodes scapularis has reported reduced TLR and NLR signaling [87], this aspect of infection has not yet been widely addressed.
EHRLICHIA CHAFFEENSIS
Epidemiology and clinical features
E. chaffeensis is the causative agent of human monocytic ehrlichiosis, an emerging tick-borne illness found predominantly in the Southeastern and South-Central United States [36]. E. chaffeensis is maintained in white-tailed deer [105] and transmitted to humans via bite of the Lonestar tick (Amblyomma americanum) [106]. Early symptoms of the disease are nonspecific (fever, headache, myalgia, anorexia, and chills); however, approximately 40% of identified cases have required hospitalization due to multiorgan failure, resulting in case fatality rates of 2–3% [107]. Clinical findings during E. chaffeensis infection include leukopenia, thrombocytopenia, anemia, and elevated liver aminotransferases, which are often confused with A. phagocytophilum, resulting in under-reporting and misdiagnosis [107].
Immune recognition
E. chaffeensis shares many microbiological features with A. phagocytophilum and other Rickettsiales. It is a Gram-negative, small cocci, with primary tropism for monocytes and macrophages, but may also infect hepatocytes and endothelial cells [108]. After entering the host cell via endocytosis, the bacterium differentiates from the infectious (dense core) form to the replicative (reticulate) form (Fig. 3) [36, 107]. E. chaffeensis is well-adapted to subvert immune detection in both host and vector, as it lacks genes for LPS or peptidoglycan biosynthesis, like O. tsutsugamushi and A. phagocytophilum [109]. Thus, understanding immune recognition of this important pathogen has remained elusive.
Figure 3. Ehrlichia chaffeensis intracellular life cycle and host innate response.
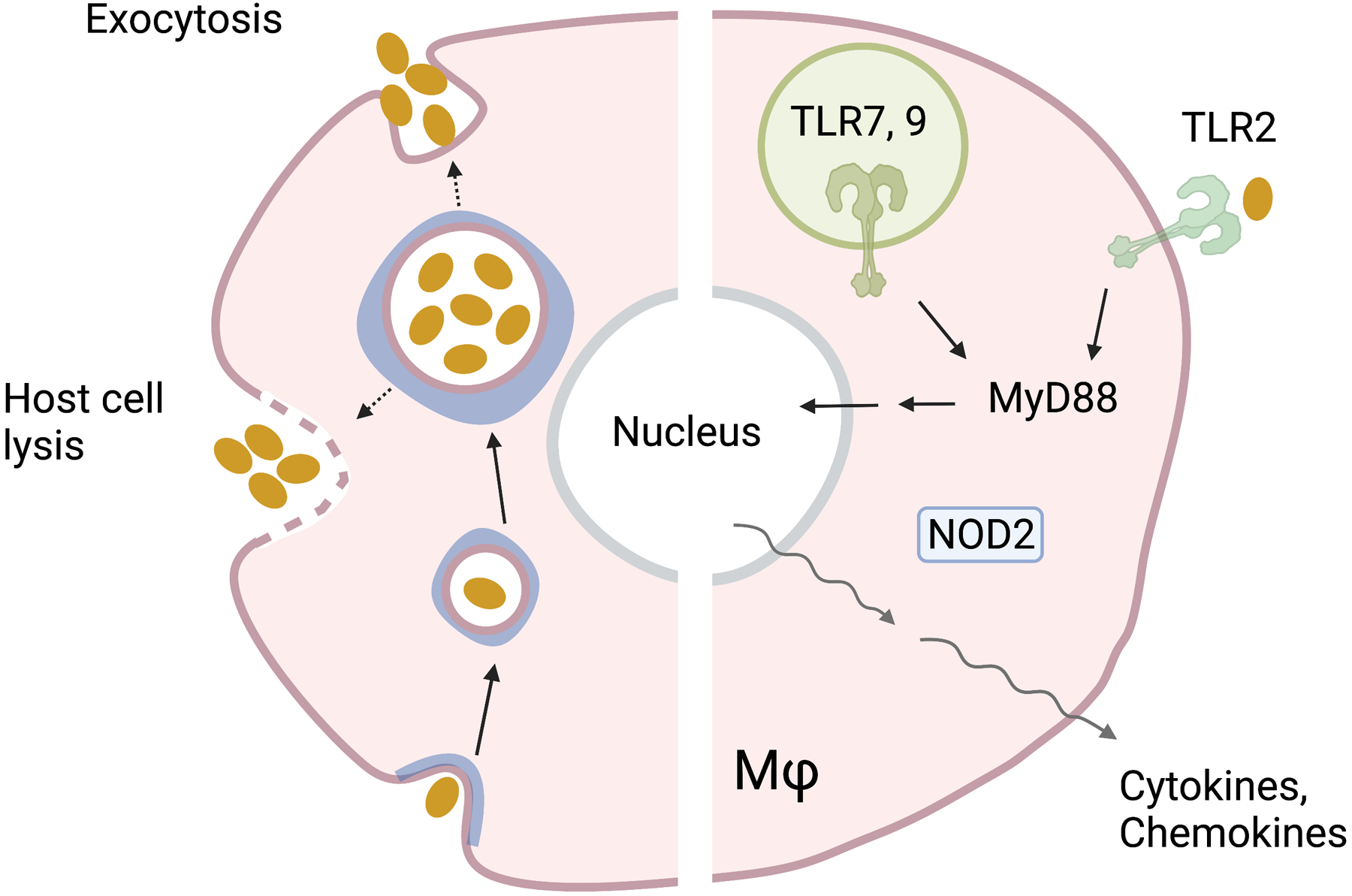
Ehrlichia enters the cell via Caveolae (in blue)-mediated endocytosis. Initial entry is characterized by an infectious dense core which later differentiates to a replicative reticulate form. Bacteria can exit from the cell by host cell lysis or exocytosis. Host immune recognition is mediated by TLR2 on the cell surface, or TLR7/TLR9 within the endosome, leading to MyD88 pathway activation. NOD2 may also sense Ehrlichia within the cytoplasm. The phylogenetically related organism A. phagocytophilum induces a PRR activation signature like Ehrlichia, for innate recognition by TLR2 and NOD2. Together, TLR and NOD2 recognition can activate the transcription of proinflammatory cytokines and chemokines.
TLRs are the most studied class of PRRs in the context of Ehrlichial disease. One unique aspect observed from in vitro studies is that E. chaffeensis can modulate TLR expression to survive within host cells. For example, E. chaffeensis actively downregulates TLR2/TLR4 expression in human monocytes and monocyte-like THP-1 cells, partially due to the action of E. chaffeensis secreted tandem repeat effector protein 120 [110]. Also, the direct interaction of tandem repeat effector protein 120 with members of the Notch signaling pathway can lead to inhibition of PU.1, p38 MAPK, and ERK1/2, as well as increased bacterial survival [110]. While E. chaffeensis modulates TLR2/TLR4 for its replication, the effect of this process on cytokine and chemokine signaling remains controversial. Miura et al. have shown that following infection with E. chaffeensis, BMMΦs from TLR2−/− or TLR4−/− C57BL/6 mice produced significantly higher levels of CXCL2 transcripts (but not IL-1β and TNFα) than WT controls [111]. Surprisingly, when MyD88−/− or MyD88/TRIF−/− MΦs, HEK293 cells (lacking all known TLRs), or specific inhibitors of TLR3-, TLR7-, and TLR9-mediated activation were used for E. chaffeensis infection, there were no major effects on cytokine/chemokine levels in comparison to control counterparts [111]. Collectively, these in vitro studies suggest a limited role for TLRs in the expression of proinflammatory cytokines during infection.
The contribution of TLRs and NLRs in Ehrlichial disease severity and pathogenesis have been examined by several groups [112–114]. A protective role of the MyD88-mediated pathway in a murine model of mild Ehrlichial disease was first reported by Koh et al [112], as MyD88−/− mice harbored greater bacterial loads in the blood and spleen, which coincided with significantly less serum IL-12 levels. Additionally, fewer apoptotic cells, lymphoblasts, MΦs, and neutrophils were observed in the spleens of infected MyD88−/− animals. Examining the cytokine response to E. muris at the cellular level, infected MyD88−/− BMDCs produced significantly less proinflammatory cytokines (IL-12-p40, TNFα, IL-6) than WT comparators. Surprisingly, when the authors attempted to define which TLR could drive such findings, no differences in cytokine production were observed from infected TLR2−/−, TLR3−/−, TLR4−/−, TLR5−/−, TLR7−/−, TLR9−/−, or TLR11−/− DCs. Infection of RIP2−/−, NLRP3−/−, and NLRC4−/− BMDCs also yielded no significant differences for bacterial loads or cytokine secretion. Therefore, each tested TLRs and NLRs, by itself, is insufficient in generating inflammatory cytokines in mild disease caused by E. muris infection.
The model of mild Ehrlichial disease, however, may not represent the immune signatures of severe infection. One study addressed this by comparing the role of TLR2 and NOD2 in both mild and severe forms of Ehrlichial disease. Using infection with Ixodes ovatus Ehrlichia (IOE) to instigate lethal disease and E. muris as model for mild disease, Chattoraj et al discovered contributions for TLR2 and NOD2 in ameliorating or worsening pathogenesis [113]. At 3 dpi in both models, transcripts of TLR2, TLR3, TLR4, and TLR9 were all reduced in the liver compared with mock controls, while IOE-infected group had increased NOD1 transcripts. By the terminal phase of IOE infection, however, TLR2 and MyD88 transcripts were significantly increased compared with mock controls and E. muris counterparts. To evaluate the function of TLR2 in lethal disease, TLR2−/− mice were infected with IOE. TLR2−/− mice succumbed to disease more quickly than WT, with increased hepatic bacterial loads, necrosis, and inflammatory foci by terminality [113]. In stark contrast, IOE-infected NOD2−/− mice exhibited improved survival, enhanced hepatic bacterial clearance, along with fewer hepatic necrotic foci and apoptotic cells. These animals also displayed reduced splenic CD8+ T cells, but increased Natural Killer T cells, CD4 T cells, Th1 signatures, and anti-inflammatory responses compared with WT and TLR2−/− mice [113]. Thus, TLR2 may contribute to controlling infection, while NOD2 may enhance IOE-associated immunopathology.
While significant emphasis has been placed on the contribution of TLR2, immunopathologic analysis of IOE-infected C57BL/6 mice revealed a powerful role for TLR9 in pathogenesis, inflammasome activation, and autophagy [114]. Firstly, IOE-infected MyD88−/− mice displayed improved survival and higher serum IL-10 levels than WT animals, despite harboring greater hepatic bacterial loads. MyD88−/− mice also exhibited dampened hepatic injury and inflammasome activation. Examining the effect of TLR signaling on inflammasome activation at the cellular level, infected MyD88−/− BMMΦ secreted significantly less proinflammatory markers (IL-1β, IL-1α, and TNF) compared with WT cells, while caspase-1 cleavage and lactate dehydrogenase release was impaired. Markers indicating autophagy induction (Beclin-1, Atg5) were also increased in infected MyD88−/− MΦs, implicating TLRs in blunting autophagosome formation. TLR7 and TLR9 were found to drive these findings, as IOE-infected TLR7−/− MΦs produced less IL-1β than WT, with infected TLR9−/− MΦs following the same trend. However, Caspase-1 and Caspase-11 activation were markedly hindered in infected TLR9−/− cells, heavily implicating this receptor in inflammasome activation. Surprisingly, IOE-infected TLR9−/− mice were fully resistant to lethal ehrlichiosis (100% survival) and displayed reduced hepatic injury compared with WT controls [114]. The authors then demonstrated that accumulated mitochondrial DNA is the major TLR9 stimulus and provided robust evidence that TLR9 is the key upstream mediator of MyD88-dependent effects [114].
Knowledge gaps and future studies
How do TLR2, TLR9, or NOD2 sense Ehrlichia during infection (Fig. 3)? The rapid cytokine/chemokine responses to infection reported in multiple studies [111] suggest that bacterium-derived components are likely activating these receptors directly. Proposed ligands for TLR2 include endogenous DAMPs or Ehrlichial lipopeptides, while NOD2 may be stimulated by a low-molecular weight peptidoglycan homolog [113]. However, direct evidence to support these interactions is still lacking. Despite considerable progress in understanding the TLR responses to Ehrlichia in-vitro and in-vivo, no published reports have examined other PRRs, including CLRs and RIG-I. Defining whether or how these additional receptors contribute to generating the immune response is necessary and could provide valuable insight into disease pathogenesis.
COXIELLA BURNETII
Epidemiology and clinical features
C. burnetii is the causative agent of “Q fever”, first identified in Australia in 1937 [40]. Although C. burnetii is distributed across all continents except Antarctica (like Rickettsia spp.), cases of Q fever are most heavily clustered in regions containing livestock and farm animal processing centers [115]. While the reservoir for this bacterium is expansive and composed of various animals, livestock are the most common source implicated in transmission to humans [37]. Inhalation of aerosolized bacteria from livestock birthing or still birth fluid, vaginal mucus, feces, and other secretions is the most common form of spread [116–118]. Ingestion of unpasteurized milk and cheese containing this bacterium is another mode of transmission but poses lower risk of infection [118, 119]. Arthropods may play a role in transmission, as C. burnetii have been identified in various tick species, including Ixodes, Dermacentor, Rhipicephalus and Haemaphysalis [38–40]. However, the role of ticks in human infection and pathogenesis is unknown. Most acute Q fever cases are asymptomatic, but patients may also present with constitutional signs and symptoms, making accurate diagnosis difficult [120]. Furthermore, though rare (1 – 5 % of cases), chronic complications after primary infection may occur and include interstitial fibrosis, hepatitis, encephalitis, as well as endocarditis and valvular pathology [121, 122].
Immune recognition
C. burnetii is a Gram-negative, LPS-positive, pleomorphic, spore-like forming bacterium [123]. While classically considered a strict intracellular pathogen, this categorization may be revisited due to the successful cultivation C. burnetii in cell-free conditions [124, 125]. For in vivo infection, alveolar macrophages or monocytes are the primary target cells for replication following inhalation into the host [126]. C. burnetii then replicates within phagolysosomes, forming a Coxiella containing vacuole (Fig. 4). In contrast to other intracellular bacteria, which either escape the endosome or thwart phagolysosome acidification, Coxiella grows best in the acidified vacuole [127]. The life cycle of C. burnetii takes two forms: a stable small cell variant, capable of penetrating host cells and surviving in the environment, and a large cell variant, which is metabolically active and replicates in host cells [128]. After 5 days, the Coxiella containing vacuole reaches a size that dominates most of the cell volume, and the large cell variant population begins to transition back into the small cell variant, which is then ready to infect another cell [129]. While Coxiella stays within the cell, release of pathogenic factors (AnkG, CaeA, CaeB, IcaA, etc.) into the cytoplasm promotes an anti-apoptotic environment which lengthens the life of an infected cell [130]. Additionally, it is worth noting that C. burnetii strains are divided into two Phase variants for laboratory study, based on virulence and LPS structures. Phase I variants are virulent, synthesize LPS containing highly branched O-chains, and generally are isolated from infected individuals or animals [131]. Phase II variants, in contrast, are avirulent, synthesize truncated O-antigen, and generated via laboratory passage [132]. Phase II variants are immunostimulatory and capable of activating innate immune cells, whereas Phase I variants are better able to evade innate recognition [133]. Differences in LPS structure may explain these findings, with Phase I variant LPS acting to mask other PAMPs residing on the surface of C. burnetii [133]. The ability of Phase I variants to avoid immune detection has been hypothesized as a link to chronic Q fever infection [133], highlighting the importance of efforts to thoroughly understand innate recognition of this bacterium.
Figure 4. Coxiella burnetii intracellular life cycle and host innate responses.
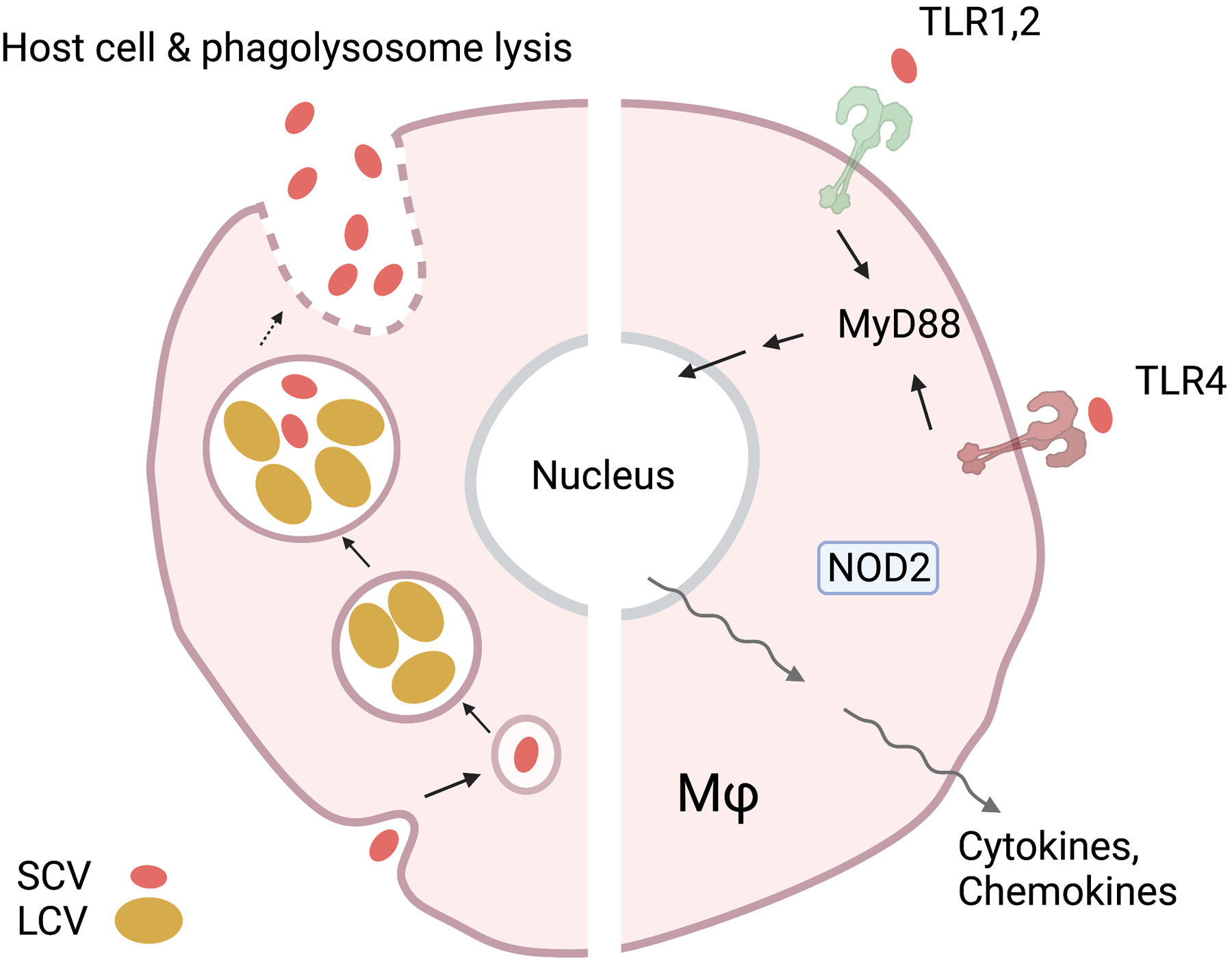
Coxiella exhibits two different morphologic variants during its life cycle. A stable small cell variant (SCV), that can penetrate the cell via endocytosis, and a large cell variant (LCV), which performs metabolic processes and replication. Only the SCV exits the cell via the host-cell-phagolysosome lysis. Host immune recognition is mediated by NOD2, as well as by TLR1/2/4-regulated activation of MyD88 or TRIF. Immune recognition of Coxiella then leads to the transcription of target proinflammatory cytokines and chemokines.
TLR/MyD88-mediated recognition
Multiple studies have examined the role of TLR1/2/4- and MyD88-mediated pathways in recognizing C. burnetii, including reports with human subjects or samples. Evidence from a case-control study revealed a positive association between a single nucleotide polymorphism (SNP) in MyD88 (−938C>A) and the development of chronic Q fever [134]. However, individuals with the TLR1 R80T genotype were found to be less likely to develop chronic disease. The functional consequence of these SNPs was then examined by stimulating whole blood with C. burnetii. Interestingly, infected whole blood from MyD88 (−938C>A) subjects exhibited no differences in cytokine (IL-1β, TNF, IL-2, IL-6, IL-10) production, whereas TLR1 R80T whole blood displayed decreased IL-10 responses. Considering that high serum IL-10 is a marker of poor prognosis for Q fever, the authors were able to show functional relevance of this TLR1 mutation to chronic disease. Additional evidence for the contribution of TLRs stems from Ammerdorffer and colleagues, who investigated the role of TLR polymorphisms in human PBMCs [135]. First, PBMCs infected with C. burnetii Nine Mile (NM) or 3262 strains (Phase I variants) were found to secrete high levels of proinflammatory cytokines (IL-1β, TNFα, and IL-6). For both strains, blocking TLR4 prior to infection did not produce reductions in any cytokine measured, whereas blocking of TLR2 resulted in abrogated IL-1β and IL-6 secretion. PBMCs were then divided into groups based on the presence of TLR SNPs for infection with C. burnetii NM and 3262. Polymorphisms of TLR4 were not associated with reduced cytokine production after infection, whereas PBMCs homozygous for TLR2 P631H displayed decreased IL-1β responses after infection with C. burnetii 3262 only [135]. Furthermore, PBMCs containing homozygous TLR1 variants showed significantly decreased production of both IL-1β and TNFα upon C. burnetii NM and 3262 infections. A similar trend was observed for PBMCs containing homozygous TLR6 P249S SNP, but only in the context of C. burnetii 3262. These findings were then evaluated by using C57BL/6 BMMΦs. Infected TLR2−/− and TLR6−/− cells produced less IL-6 in response to both strains of C. burnetii tested, whereas TLR1−/− cells produced less IL-6 only in response to C. burnetii NM. Collectively, these results show virulent C. burnetii strain specific differences in immune sensing, with shared contributions between TLR1/2 in human PBMCs.
Studies utilizing animal models of Q fever have shed light on the role of TLRs in pathogenesis of infection. Multiple reports have shown that TLR2/4 and MyD88-mediated signaling is essential in controlling the replication of C. burnetii Nine Mile Phase II (NMII) infection in-vitro and in-vivo [136–138]. An early report using CHO cells transfected with functional TLR2 or TLR4 revealed that C. burnetii NMII activated TLR2+ cells only [138]. Expanding upon these findings, they showed that TLR2−/− C57BL/6 BMMΦ displayed significantly increased NMII load at 8 dpi compared to WT cells, along with virtually abolished TNFα and IL-12 secretion, even after treatment with doses as high as 500 bacteria per cell. Notably, TLR4-impaired (C3H/HeJ) MΦ did not exhibit any significant differences compared with control (C3H/HePas) cells. Since C. burnetii contains LPS, they then asked why TLR4 activation was not occurring. To test this, human PBMCs were treated with purified LPS from Phase I and Phase II C. burnetii prior to addition of E. coli endotoxin. From these experiments, they determined that PBMCs treated with C. burnetii Phase I and Phase II LPS exhibited blunted cytokine profiles in response to E. coli endotoxin, indicating that C. burnetii LPS may act antagonistically towards TLR4 [138]. These results suggest that TLR2, but not TLR4, is necessary for regulation and modulation of pro-inflammatory responses in C. burnetii NMII infections.
A subsequent study by Bradley et al revealed that infected MyD88−/− or TRIF−/− C57BL/6 BMMΦ secrete significantly lower, but still substantial, levels of TNFα and IL-6 compared with WT [137]. In contrast, TNFα and IL-6 secretion was virtually abolished in infected MyD88/TRIF−/− MΦ, indicating that TLR responses to NMII rely on both signaling adaptors. Similarly, MyD88/TRIF−/− cells were most permissive to infection when compared against other groups, evidenced by a greater number of intracellular vacuoles. This led the authors to focus on the effects of TLR2 and TLR4. Infected TLR2−/− BMMΦ secreted significantly reduced levels of TNFα and IL-6 following infection and harbor increased bacterial loads, whereas no significant changes were observed for TLR4−/− cells. However, infected TLR2/TLR4−/− cells did not secrete detectable amounts of TNFα and IL-6, suggesting that both receptors crosstalk to produce inflammation in response to NMII. Notably, TLR2/TLR4−/− cells did not exhibit increased bacterial loads when compared with WT controls. Together, these results imply a unique role for TLR2 in controlling bacterial infection and show the importance of TLR2-TLR4 crosstalk for generating inflammatory profiles [137]. A separate study revealed similar findings, showing that MyD88−/− MΦ harbored significantly higher bacterial loads at 72 hpi than WT controls, accompanied by reduced production of IL-6 and IL-10. Together, these results pointed to a role of MyD88 in producing both a cytokine response and regulating the bacterial load in MΦs [136]. After intratracheal infection, the bacterial load of C. burnetii NMII was consistently higher in the lung, spleen, heart, and liver tissues from 7 to 120 dpi, as compared to the MyD88+/− mice which showed complete clearance at 27 dpi [136]. Despite harboring a greater bacterial burden, MyD88−/− mice did not show signs of disease or weight loss. When chemokine/cytokine expression and histopathologic analysis was performed, infected MyD88−/− mice displayed reduced splenic CCL2 and IFNγ responses that correlated with smaller granulomatous foci in the liver. Therefore, MyD88 is fundamental to the control of Coxiella NMII infection.
NLR-mediated immune recognition
Few studies have examined the contribution of NLRs during C. burnetii infection. While one SNP in NOD2 (L1007fsX1) has been associated with development of chronic Q fever, infection of human PBMCs revealed no functional consequence of this variant [134]. However, a separate study analyzing multiple human NOD2 polymorphisms did reveal a functional impact on cytokine secretion in response to infection with two different Phase I strains [135]. Human PBMCs homozygous for NOD2 3020insC secreted significantly less IL-1β and IL-6 in response to C. burnetii 3262, while nine other NOD2 SNPs were found to have no impact on inflammatory responses [135]. Infected PBMCs harboring NOD1 polymorphisms were also found to exhibit cytokine/chemokine profiles like control PBMCs. When these findings were expanded upon using C57BL/6 MΦ, C. burnetii NM stimulation of NOD1−/− cells resulted in 35% reduction of IL-6 secretion and 50% reduction in NOD2−/− cells [135]. Together, these studies suggest a role for NOD2 in sensing C. burnetii.
Knowledge gaps and future studies
The consensus of available data reveals a powerful role of TLR2 in sensing C. burnetii (Fig. 4). Studies utilizing human samples and animal models of infection agree that TLR2 contributes to generating inflammation and controlling bacterial replication [134–138]. The cytokine response also has been shown to involve crosstalk between TLR2 and TLR4, where the effect of TLR4 depends on TLR2 [137]. Reports have shown that TLR1 may also play a role in infection, and thus TLR2 homodimers and TLR1/TLR2 heterodimers could recognize Coxiella [135]. Although no study has yet shown the natural ligands involved, components of this bacterium’s rich lipoprotein cell wall are most likely the culprit. The observation that C. burnetii LPS may be immunosuppressive in a manner like Bartonella is also in line with studies revealing no role for TLR4 alone in contributing to inflammation and controlling infection [138]. While NOD2 is likely involved in sensing this bacterium once it has been engulfed, studies examining pathogenesis of NOD2−/− mice have yet to be performed. Furthermore, evaluation and identification of the natural ligand for NOD2 are needed. Mechanistic studies to identify such ligands may be aided by newly established axenic culturing techniques for C. burnetii, which allow for large-scale bacterial propagation [124, 125]. Finally, the impact of RLRs and CLRs has not yet been evaluated for C. burnetii. Careful assessment of the potential role of both receptor families may yield valuable insight into facets of innate recognition.
CONCLUDING REMARKS
Rickettsia, O. tsutsugamushi, Anaplasma, Ehrlichia, and C. burnetii cause significant human disease across the globe, yet, understanding mechanisms of innate recognition remain challenging. Despite sharing aspects of basic biology, each of these five bacteria exhibit unique characteristics, tropisms, and natural reservoirs (Table 1) which may influence immune recognition. To-date, research efforts have heavily emphasized more well-known PRRs, particularly TLRs and NLRs, while neglecting RLRs or CLRs. This may be due, in part, to the wide availability of TLR- and NLR- deficient mouse strains and reagents, which is severely lacking for CLRs. However, studies examining the impact of CLRs may be very insightful. Considering that CLRs sense both PAMPs and DAMPs, defining the role of these receptors in recognition of obligate intracellular bacteria, as in the case of O. tsutsugamushi with Mincle, may lead to new fields investigation. Continued research into recognition of obligate intracellular bacteria would allow a better understanding of disease pathogenesis and could lead to new therapeutic strategies for patients with severe disease. Additionally, since the United States does not have licensed vaccines for any of these five bacteria discussed herein, evaluating how PRRs shape the adaptive response to infection could yield valuable information for future vaccine design.
Acknowledgements
All figures were generated by using the BioRender platform. This work was supported partially by the National Institute of Allergy and Infectious Diseases grants (R01 AI132674 to LS, T32 AI007526-20 to LS, and R21 AI156536-01 to LS, https://www.niaid.nih.gov), a UTMB Center for Biodefense and Emerging Infectious Diseases Pilot grant (to LS, https://www.utmb.edu/cbeid), and Department of Defense Threat Reduction Agency grant HDTRA1-19-1-0043 (subaward to LS, https://www.dtra.mil/). JF was the recipient of a NIAID Emerging and Tropical Infectious Diseases T32 fellowship. ZC was the recipient of the NIAID Infectious Diseases and Inflammatory Disorders Training Program T35 fellowship (AI078878). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
References:
- 1.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. [DOI] [PubMed] [Google Scholar]
- 2.Fitzgerald KA, Kagan JC. Toll-like Receptors and the Control of Immunity. Cell. 2020;180(6):1044–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity. 2011;34(5):665–79. [DOI] [PubMed] [Google Scholar]
- 4.Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020;20(9):537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown GD, Willment JA, Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol. 2018;18(6):374–89. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Preston-Hurlburt P, Janeway CA Jr., A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–7. [DOI] [PubMed] [Google Scholar]
- 7.Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, et al. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity. 2009;31(6):873–84. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Kawai T, Sanjo H, Copeland NG, Gilbert DJ, Jenkins NA, et al. TLR6: A novel member of an expanding toll-like receptor family. Gene. 1999;231(1–2):59–65. [DOI] [PubMed] [Google Scholar]
- 9.Campos MA, Almeida IC, Takeuchi O, Akira S, Valente EP, Procopio DO, et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. 2001;167(1):416–23. [DOI] [PubMed] [Google Scholar]
- 10.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169(1):10–4. [DOI] [PubMed] [Google Scholar]
- 11.Poltorak A, Smirnova I, He X, Liu MY, Van Huffel C, McNally O, et al. Genetic and physical mapping of the Lps locus: identification of the toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol Dis. 1998;24(3):340–55. [DOI] [PubMed] [Google Scholar]
- 12.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167(4):1882–5. [DOI] [PubMed] [Google Scholar]
- 13.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–8. [DOI] [PubMed] [Google Scholar]
- 14.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–31. [DOI] [PubMed] [Google Scholar]
- 15.Greulich W, Wagner M, Gaidt MM, Stafford C, Cheng Y, Linder A, et al. TLR8 Is a Sensor of RNase T2 Degradation Products. Cell 2019;179(6):1264–75 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–9. [DOI] [PubMed] [Google Scholar]
- 17.Ito T, Amakawa R, Kaisho T, Hemmi H, Tajima K, Uehira K, et al. Interferon-alpha and interleukin-12 are induced differentially by Toll-like receptor 7 ligands in human blood dendritic cell subsets. J Exp Med. 2002;195(11):1507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–5. [DOI] [PubMed] [Google Scholar]
- 19.Mohammad Hosseini A, Majidi J, Baradaran B, Yousefi M. Toll-Like Receptors in the Pathogenesis of Autoimmune Diseases. Adv Pharm Bull. 2015;5(Suppl 1):605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farrugia M, Baron B. The Role of Toll-Like Receptors in Autoimmune Diseases through Failure of the Self-Recognition Mechanism. Int J Inflam. 2017;2017:8391230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7(12):1250–7. [DOI] [PubMed] [Google Scholar]
- 22.Coulombe F, Divangahi M, Veyrier F, de Leseleuc L, Gleason JL, Yang Y, et al. Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J Exp Med. 2009;206(8):1709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7(6):576–82. [DOI] [PubMed] [Google Scholar]
- 24.Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Nunez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol. 2007;37(11):3030–9. [DOI] [PubMed] [Google Scholar]
- 25.Miao EA, Andersen-Nissen E, Warren SE, Aderem A. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol. 2007;29(3):275–88. [DOI] [PubMed] [Google Scholar]
- 26.Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci U S A. 2008;105(7):2562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38(5):855–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Ye X, Dunker W, Song Y, Karijolich J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat Commun. 2018;9(1):4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiang JJ, Sparrer KMJ, van Gent M, Lassig C, Huang T, Osterrieder N, et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat Immunol. 2018;19(1):53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9(7):465–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salje J Cells within cells: Rickettsiales and the obligate intracellular bacterial lifestyle. Nat Rev Microbiol. 2021;19(6):375–90. [DOI] [PubMed] [Google Scholar]
- 32.Blanton LS. The Rickettsioses: A Practical Update. Infect Dis Clin North Am. 2019;33(1):213–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdad MY, Abou Abdallah R, Fournier PE, Stenos J, Vasoo S. A Concise Review of the Epidemiology and Diagnostics of Rickettsioses: Rickettsia and Orientia spp. J Clin Microbiol. 2018;56(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu G, Walker DH, Jupiter D, Melby PC, Arcari CM. A review of the global epidemiology of scrub typhus. PLoS Negl Trop Dis. 2017;11(11):e0006062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bakken JS, Dumler JS. Human granulocytic anaplasmosis. Infect Dis Clin North Am. 2015;29(2):341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paddock CD, Childs JE. Ehrlichia chaffeensis: a prototypical emerging pathogen. Clin Microbiol Rev. 2003;16(1):37–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalaitzakis E, Fancello T, Simons X, Chaligiannis I, Tomaiuolo S, Andreopoulou M, et al. Coxiella burnetii Shedding in Milk and Molecular Typing of Strains Infecting Dairy Cows in Greece. Pathogens. 2021;10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spitalska E, Sparagano O, Stanko M, Schwarzova K, Spitalsky Z, Skultety L, et al. Diversity of Coxiella-like and Francisella-like endosymbionts, and Rickettsia spp., Coxiella burnetii as pathogens in the tick populations of Slovakia, Central Europe . Ticks Tick Borne Dis 2018;9(5):1207–11. [DOI] [PubMed] [Google Scholar]
- 39.Truong AT, Yun BR, Lim J, Min S, Yoo MS, Yoon SS, et al. Real-time PCR biochip for on-site detection of Coxiella burnetii in ticks. Parasit Vectors. 2021;14(1):239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eldin C, Melenotte C, Mediannikov O, Ghigo E, Million M, Edouard S, et al. From Q Fever to Coxiella burnetii Infection: a Paradigm Change. Clin Microbiol Rev. 2017;30(1):115–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salje J Orientia tsutsugamushi: A neglected but fascinating obligate intracellular bacterial pathogen. PLoS Pathog. 2017;13(12):e1006657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baker S, Dougan G. The genome of Salmonella enterica serovar Typhi. Clin Infect Dis. 2007;45 Suppl 1:S29–33. [DOI] [PubMed] [Google Scholar]
- 43.Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–62. [DOI] [PubMed] [Google Scholar]
- 44.Gorvel L, Textoris J, Banchereau R, Ben Amara A, Tantibhedhyangkul W, von Bargen K, et al. Intracellular bacteria interfere with dendritic cell functions: role of the type I interferon pathway. PLoS One. 2014;9(6):e99420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samuel JE, Hendrix LR. Laboratory maintenance of Coxiella burnetii. Curr Protoc Microbiol. 2009;Chapter 6:Unit 6C 1. [DOI] [PubMed] [Google Scholar]
- 46.Mansueto P, Vitale G, Cascio A, Seidita A, Pepe I, Carroccio A, et al. New insight into immunity and immunopathology of Rickettsial diseases. Clin Dev Immunol. 2012;2012:967852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brouqui P, Dumler JS, Raoult D. Immunohistologic demonstration of Coxiella burnetii in the valves of patients with Q fever endocarditis. Am J Med. 1994;97(5):451–8. [DOI] [PubMed] [Google Scholar]
- 48.Melenotte C, Protopopescu C, Million M, Edouard S, Carrieri MP, Eldin C, et al. Clinical Features and Complications of Coxiella burnetii Infections From the French National Reference Center for Q Fever. JAMA Netw Open. 2018;1(4):e181580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fisher J, Card G, Liang Y, Trent B, Rosenzweig H, Soong L. Orientia tsutsugamushi selectively stimulates the C-type lectin receptor Mincle and type 1-skewed proinflammatory immune responses. PLoS Pathog. 2021;17(7):e1009782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soong L, Wang H, Shelite TR, Liang Y, Mendell NL, Sun J, et al. Strong type 1, but impaired type 2, immune responses contribute to Orientia tsutsugamushi-induced pathology in mice. PLoS Negl Trop Dis. 2014;8(9):e3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weitzel T, Jiang J, Acosta-Jamett G, Martinez-Valdebenito C, Lopez J, Richards AL, et al. Canine seroprevalence to Orientia species in southern Chile: A cross-sectional survey on the Chiloe Island. PLoS One. 2018;13(7):e0200362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weitzel T, Acosta-Jamett G, Jiang J, Martinez-Valdebenito C, Farris CM, Richards AL, et al. Human seroepidemiology of Rickettsia and Orientia species in Chile - A cross-sectional study in five regions. Ticks Tick Borne Dis. 2020;11(6):101503. [DOI] [PubMed] [Google Scholar]
- 53.Ghorbani RP, Ghorbani AJ, Jain MK, Walker DH. A case of scrub typhus probably acquired in Africa. Clin Infect Dis. 1997;25(6):1473–4. [DOI] [PubMed] [Google Scholar]
- 54.Osuga K, Kimura M, Goto H, Shimada K, Suto T. A case of Tsutsugamushi disease probably contracted in Africa. Eur J Clin Microbiol Infect Dis. 1991;10(2):95–6. [DOI] [PubMed] [Google Scholar]
- 55.Walker JS, Gan E, Chan Teik C, Muul I. Involvement of small mammals in the transmission of scrub typhus in Malaysia: isolation and serological evidence. Trans R Soc Trop Med Hyg. 1973;67(6):838–45. [DOI] [PubMed] [Google Scholar]
- 56.Phasomkusolsil S, Tanskul P, Ratanatham S, Watcharapichat P, Phulsuksombati D, Frances SP, et al. Transstadial and transovarial transmission of Orientia tsutsugamushi in Leptotrombidium imphalum and Leptotrombidium chiangraiensis (Acari: Trombiculidae). J Med Entomol. 2009;46(6):1442–5. [DOI] [PubMed] [Google Scholar]
- 57.Taylor AJ, Paris DH, Newton PN. A Systematic Review of Mortality from Untreated Scrub Typhus (Orientia tsutsugamushi). PLoS Negl Trop Dis. 2015;9(8):e0003971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonell A, Lubell Y, Newton PN, Crump JA, Paris DH. Estimating the burden of scrub typhus: A systematic review. PLoS Negl Trop Dis. 2017;11(9):e0005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dittrich S, Card E, Phuklia W, Rudgard WE, Silousok J, Phoumin P, et al. Survival and Growth of Orientia tsutsugamushi in Conventional Hemocultures. Emerg Infect Dis. 2016;22(8):1460–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evans SM, Rodino KG, Adcox HE, Carlyon JA. Orientia tsutsugamushi uses two Ank effectors to modulate NF-kappaB p65 nuclear transport and inhibit NF-kappaB transcriptional activation. PLoS Pathog. 2018;14(5):e1007023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Janardhanan J, Joseph Martin S, Astrup E, Veeramanikandan R, Aukrust P, Abraham OC, et al. Single-nucleotide polymorphisms in Toll-like receptor (TLR)-2, TLR4 and heat shock protein 70 genes and susceptibility to scrub typhus. J Hum Genet. 2013;58(11):707–10. [DOI] [PubMed] [Google Scholar]
- 62.Najmi N, Kaur G, Sharma SK, Mehra NK. Human Toll-like receptor 4 polymorphisms TLR4 Asp299Gly and Thr399Ile influence susceptibility and severity of pulmonary tuberculosis in the Asian Indian population. Tissue Antigens. 2010;76(2):102–9. [DOI] [PubMed] [Google Scholar]
- 63.Gharaibeh M, Hagedorn M, Lilla S, Hauptmann M, Heine H, Fleischer B, et al. Toll-Like Receptor 2 Recognizes Orientia tsutsugamushi and Increases Susceptibility to Murine Experimental Scrub Typhus. Infect Immun. 2016;84(12):3379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Min CK, Kim HI, Ha NY, Kim Y, Kwon EK, Yen NTH, et al. A Type I Interferon and IL-10 Induced by Orientia tsutsugamushi Infection Suppresses Antigen-Specific T Cells and Their Memory Responses. Front Immunol. 2018;9:2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodino KG, Adcox HE, Martin RK, Patel V, Conrad DH, Carlyon JA. The Obligate Intracellular Bacterium Orientia tsutsugamushi Targets NLRC5 To Modulate the Major Histocompatibility Complex Class I Pathway. Infect Immun. 2019;87(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho KA, Jun YH, Suh JW, Kang JS, Choi HJ, Woo SY. Orientia tsutsugamushi induced endothelial cell activation via the NOD1-IL-32 pathway. Microb Pathog. 2010;49(3):95–104. [DOI] [PubMed] [Google Scholar]
- 67.Koo JE, Hong HJ, Dearth A, Kobayashi KS, Koh YS. Intracellular invasion of Orientia tsutsugamushi activates inflammasome in asc-dependent manner. PLoS One. 2012;7(6):e39042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soong L Dysregulated Th1 Immune and Vascular Responses in Scrub Typhus Pathogenesis. J Immunol. 2018;200(4):1233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trent B, Liang Y, Xing Y, Esqueda M, Wei Y, Cho NH, et al. Polarized lung inflammation and Tie2/angiopoietin-mediated endothelial dysfunction during severe Orientia tsutsugamushi infection. PLoS Negl Trop Dis. 2020;14(3):e0007675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Astrup E, Janardhanan J, Otterdal K, Ueland T, Prakash JA, Lekva T, et al. Cytokine network in scrub typhus: high levels of interleukin-8 are associated with disease severity and mortality. PLoS Negl Trop Dis. 2014;8(2):e2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bora T, Khan SA. Evaluation of Th1 and Th2 immune response in clinical and sub-clinical scrub typhus infection. Hum Immunol. 2019;80(7):503–9. [DOI] [PubMed] [Google Scholar]
- 72.Walker DH, Ismail N. Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nat Rev Microbiol. 2008;6(5):375–86. [DOI] [PubMed] [Google Scholar]
- 73.Dumler JS, Walker DH. Rocky Mountain spotted fever--changing ecology and persisting virulence. N Engl J Med. 2005;353(6):551–3. [DOI] [PubMed] [Google Scholar]
- 74.Bechelli J, Smalley C, Zhao X, Judy B, Valdes P, Walker DH, et al. MyD88 Mediates Instructive Signaling in Dendritic Cells and Protective Inflammatory Response during Rickettsial Infection. Infect Immun. 2016;84(4):883–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voss OH, Gillespie JJ, Lehman SS, Rennoll SA, Beier-Sexton M, Rahman MS, et al. Risk1, a Phosphatidylinositol 3-Kinase Effector, Promotes Rickettsia typhi Intracellular Survival. mBio. 2020;11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jordan JM, Woods ME, Soong L, Walker DH. Rickettsiae stimulate dendritic cells through toll-like receptor 4, leading to enhanced NK cell activation in vivo. J Infect Dis. 2009;199(2):236–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rumfield C, Hyseni I, McBride JW, Walker DH, Fang R. Activation of ASC Inflammasome Driven by Toll-Like Receptor 4 Contributes to Host Immunity against Rickettsial Infection. Infect Immun. 2020;88(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quevedo-Diaz MA, Song C, Xiong Y, Chen H, Wahl LM, Radulovic S, et al. Involvement of TLR2 and TLR4 in cell responses to Rickettsia akari. J Leukoc Biol. 2010;88(4):675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burke TP, Engstrom P, Chavez RA, Fonbuena JA, Vance RE, Welch MD. Inflammasome-mediated antagonism of type I interferon enhances Rickettsia pathogenesis. Nat Microbiol. 2020;5(5):688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smalley C, Bechelli J, Rockx-Brouwer D, Saito T, Azar SR, Ismail N, et al. Rickettsia australis Activates Inflammasome in Human and Murine Macrophages. PLoS One. 2016;11(6):e0157231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fodorova M, Vadovic P, Skultety L, Slaba K, Toman R. Structural features of lipopolysaccharide from Rickettsia typhi: the causative agent of endemic typhus. Ann N Y Acad Sci. 2005;1063:259–60. [DOI] [PubMed] [Google Scholar]
- 82.Fodorova M, Vadovic P, Toman R. Structural features of lipid A of Rickettsia typhi. Acta Virol. 2011;55(1):31–44. [DOI] [PubMed] [Google Scholar]
- 83.Dumler JS, Asanovich KM, Bakken JS. Analysis of genetic identity of North American Anaplasma phagocytophilum strains by pulsed-field gel electrophoresis. J Clin Microbiol. 2003;41(7):3392–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dumler JS, Barbet AF, Bekker CP, Dasch GA, Palmer GH, Ray SC, et al. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent’ as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol. 2001;51(Pt 6):2145–65. [DOI] [PubMed] [Google Scholar]
- 85.Matei IA, D’Amico G, Ionica AM, Kalmar Z, Corduneanu A, Sandor AD, et al. New records for Anaplasma phagocytophilum infection in small mammal species. Parasit Vectors. 2018;11(1):193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Atif FA. Anaplasma marginale and Anaplasma phagocytophilum: Rickettsiales pathogens of veterinary and public health significance. Parasitol Res. 2015;114(11):3941–57. [DOI] [PubMed] [Google Scholar]
- 87.Chen G, Severo MS, Sohail M, Sakhon OS, Wikel SK, Kotsyfakis M, et al. Ixodes scapularis saliva mitigates inflammatory cytokine secretion during Anaplasma phagocytophilum stimulation of immune cells. Parasit Vectors. 2012;5:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Battilani M, De Arcangeli S, Balboni A, Dondi F. Genetic diversity and molecular epidemiology of Anaplasma. Infect Genet Evol. 2017;49:195–211. [DOI] [PubMed] [Google Scholar]
- 89.Ismail N, Bloch KC, McBride JW. Human ehrlichiosis and anaplasmosis. Clin Lab Med. 2010;30(1):261–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dahlgren FS, Heitman KN, Drexler NA, Massung RF, Behravesh CB. Human granulocytic anaplasmosis in the United States from 2008 to 2012: a summary of national surveillance data. Am J Trop Med Hyg. 2015;93(1):66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baker A, Wang HH, Mogg M, Derouen Z, Borski J, Grant WE. Increasing Incidence of Anaplasmosis in the United States, 2012 Through 2016. Vector Borne Zoonotic Dis. 2020;20(11):855–9. [DOI] [PubMed] [Google Scholar]
- 92.Gussmann K, Kirschnek S, von Loewenich FD. Interferon-gamma-dependent control of Anaplasma phagocytophilum by murine neutrophil granulocytes. Parasit Vectors. 2017;10(1):329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Park HS, Shin KS, Son BR, Kim DM, Kim HS, Jeong HW. Human Granulocytic Anaplasmosis Diagnosed Based on a Peripheral Blood Smear Test in South Korea: a Case Report. Jpn J Infect Dis. 2020;73(6):469–72. [DOI] [PubMed] [Google Scholar]
- 94.Sarkar A, Hellberg L, Bhattacharyya A, Behnen M, Wang K, Lord JM, et al. Infection with Anaplasma phagocytophilum activates the phosphatidylinositol 3-Kinase/Akt and NF-kappaB survival pathways in neutrophil granulocytes. Infect Immun. 2012;80(4):1615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Choi KS, Park JT, Dumler JS. Anaplasma phagocytophilum delay of neutrophil apoptosis through the p38 mitogen-activated protein kinase signal pathway. Infect Immun. 2005;73(12):8209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Choi KS, Scorpio DG, Dumler JS. Anaplasma phagocytophilum ligation to toll-like receptor (TLR) 2, but not to TLR4, activates macrophages for nuclear factor-kappa B nuclear translocation. J Infect Dis. 2004;189(10):1921–5. [DOI] [PubMed] [Google Scholar]
- 97.Scorpio DG, von Loewenich FD, Gobel H, Bogdan C, Dumler JS. Innate immune response to Anaplasma phagocytophilum contributes to hepatic injury. Clin Vaccine Immunol. 2006;13(7):806–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muller BJ, Westheider A, Birkner K, Seelig B, Kirschnek S, Bogdan C, et al. Anaplasma phagocytophilum Induces TLR- and MyD88-Dependent Signaling in In Vitro Generated Murine Neutrophils. Front Cell Infect Microbiol. 2021;11:627630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sukumaran B, Ogura Y, Pedra JH, Kobayashi KS, Flavell RA, Fikrig E. Receptor interacting protein-2 contributes to host defense against Anaplasma phagocytophilum infection. FEMS Immunol Med Microbiol. 2012;66(2):211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang X, Shaw DK, Hammond HL, Sutterwala FS, Rayamajhi M, Shirey KA, et al. The Prostaglandin E2-EP3 Receptor Axis Regulates Anaplasma phagocytophilum-Mediated NLRC4 Inflammasome Activation. PLoS Pathog. 2016;12(8):e1005803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Naimi WA, Green RS, Cockburn CL, Carlyon JA. Correction: Naimi WA, et al. Differential Susceptibility of Male versus Female Laboratory Mice to Anaplasma phagocytophilum Infection. Trop. Med. Infect. Dis 2018, 3, 78. Trop Med Infect Dis. 2019;4(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bunnell JE, Trigiani ER, Srinivas SR, Dumler JS. Development and distribution of pathologic lesions are related to immune status and tissue deposition of human granulocytic ehrlichiosis agent-infected cells in a murine model system. J Infect Dis. 1999;180(2):546–50. [DOI] [PubMed] [Google Scholar]
- 103.von Loewenich FD, Scorpio DG, Reischl U, Dumler JS, Bogdan C. Frontline: control of Anaplasma phagocytophilum, an obligate intracellular pathogen, in the absence of inducible nitric oxide synthase, phagocyte NADPH oxidase, tumor necrosis factor, Toll-like receptor (TLR)2 and TLR4, or the TLR adaptor molecule MyD88. Eur J Immunol. 2004;34(7):1789–97. [DOI] [PubMed] [Google Scholar]
- 104.Pedra JH, Sutterwala FS, Sukumaran B, Ogura Y, Qian F, Montgomery RR, et al. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-gamma axis during Anaplasma phagocytophilum infection. J Immunol. 2007;179(7):4783–91. [DOI] [PubMed] [Google Scholar]
- 105.Ewing SA, Dawson JE, Kocan AA, Barker RW, Warner CK, Panciera RJ, et al. Experimental transmission of Ehrlichia chaffeensis (Rickettsiales: Ehrlichieae) among white-tailed deer by Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 1995;32(3):368–74. [DOI] [PubMed] [Google Scholar]
- 106.Anderson BE, Dawson JE, Jones DC, Wilson KH. Ehrlichia chaffeensis, a new species associated with human ehrlichiosis. J Clin Microbiol. 1991;29(12):2838–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stone JH, Dierberg K, Aram G, Dumler JS. Human monocytic ehrlichiosis. JAMA. 2004;292(18):2263–70. [DOI] [PubMed] [Google Scholar]
- 108.Lin M, Rikihisa Y. Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid A biosynthesis and incorporate cholesterol for their survival. Infect Immun. 2003;71(9):5324–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lin M, Rikihisa Y. Ehrlichia chaffeensis downregulates surface Toll-like receptors 2/4, CD14 and transcription factors PU.1 and inhibits lipopolysaccharide activation of NF-kappa B, ERK 1/2 and p38 MAPK in host monocytes. Cell Microbiol. 2004;6(2):175–86. [DOI] [PubMed] [Google Scholar]
- 110.Lina TT, Dunphy PS, Luo T, McBride JW. Ehrlichia chaffeensis TRP120 Activates Canonical Notch Signaling To Downregulate TLR2/4 Expression and Promote Intracellular Survival. mBio. 2016;7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miura K, Matsuo J, Rahman MA, Kumagai Y, Li X, Rikihisa Y. Ehrlichia chaffeensis induces monocyte inflammatory responses through MyD88, ERK, and NF-kappaB but not through TRIF, interleukin-1 receptor 1 (IL-1R1)/IL-18R1, or toll-like receptors. Infect Immun. 2011;79(12):4947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koh YS, Koo JE, Biswas A, Kobayashi KS. MyD88-dependent signaling contributes to host defense against ehrlichial infection. PLoS One. 2010;5(7):e11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chattoraj P, Yang Q, Khandai A, Al-Hendy O, Ismail N. TLR2 and Nod2 mediate resistance or susceptibility to fatal intracellular Ehrlichia infection in murine models of ehrlichiosis. PLoS One. 2013;8(3):e58514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kader M, Alaoui-El-Azher M, Vorhauer J, Kode BB, Wells JZ, Stolz D, et al. MyD88-dependent inflammasome activation and autophagy inhibition contributes to Ehrlichia-induced liver injury and toxic shock. PLoS Pathog. 2017;13(10):e1006644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wardrop NA, Thomas LF, Cook EA, de Glanville WA, Atkinson PM, Wamae CN, et al. The Sero-epidemiology of Coxiella burnetii in Humans and Cattle, Western Kenya: Evidence from a Cross-Sectional Study. PLoS Negl Trop Dis. 2016;10(10):e0005032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Georgiev M, Afonso A, Neubauer H, Needham H, Thiery R, Rodolakis A, et al. Q fever in humans and farm animals in four European countries, 1982 to 2010. Euro Surveill. 2013;18(8). [PubMed] [Google Scholar]
- 117.Alvarez-Alonso R, Basterretxea M, Barandika JF, Hurtado A, Idiazabal J, Jado I, et al. A Q Fever Outbreak with a High Rate of Abortions at a Dairy Goat Farm: Coxiella burnetii Shedding, Environmental Contamination, and Viability. Appl Environ Microbiol. 2018;84(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mioni MSR, Costa FB, Ribeiro BLD, Teixeira WSR, Pelicia VC, Labruna MB, et al. Coxiella burnetii in slaughterhouses in Brazil: A public health concern. PLoS One. 2020;15(10):e0241246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dobos A, Kreizinger Z, Kovacs AB, Gyuranecz M. Prevalence of Coxiella burnetii in Central and Eastern European dairy herds. Comp Immunol Microbiol Infect Dis. 2020;72:101489. [DOI] [PubMed] [Google Scholar]
- 120.Million M, Raoult D. Recent advances in the study of Q fever epidemiology, diagnosis and management. J Infect. 2015;71 Suppl 1:S2–9. [DOI] [PubMed] [Google Scholar]
- 121.Borawski K, Dunaj J, Pancewicz S, Krol M, Czupryna P, Moniuszko-Malinowska A. Coxiella burnetii and Q fever - a review. Przegl Epidemiol. 2020;74(1):43–8. [DOI] [PubMed] [Google Scholar]
- 122.Budgin AM, Abidi MZ, Bajrovic V, Miller MA, Johnson SC. Severe acute Q fever pneumonia complicated by presumed persistent localized Q fever endocarditis in a renal transplant recipient: A case report and review of the literature. Transpl Infect Dis. 2020;22(1):e13230. [DOI] [PubMed] [Google Scholar]
- 123.Leroy Q, Lebrigand K, Armougom F, Barbry P, Thiery R, Raoult D. Coxiella burnetii transcriptional analysis reveals serendipity clusters of regulation in intracellular bacteria. PLoS One. 2010;5(12):e15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sandoz KM, Sturdevant DE, Hansen B, Heinzen RA. Developmental transitions of Coxiella burnetii grown in axenic media. J Microbiol Methods. 2014;96:104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A. 2009;106(11):4430–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat Rev Microbiol. 2013;11(8):561–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pechstein J, Schulze-Luehrmann J, Bisle S, Cantet F, Beare PA, Olke M, et al. The Coxiella burnetii T4SS Effector AnkF Is Important for Intracellular Replication. Front Cell Infect Microbiol. 2020;10:559915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Morgan JK, Luedtke BE, Shaw EI. Polar localization of the Coxiella burnetii type IVB secretion system. FEMS Microbiol Lett. 2010;305(2):177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Moormeier DE, Sandoz KM, Beare PA, Sturdevant DE, Nair V, Cockrell DC, et al. Coxiella burnetii RpoS Regulates Genes Involved in Morphological Differentiation and Intracellular Growth. J Bacteriol. 2019;201(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cordsmeier A, Wagner N, Luhrmann A, Berens C. Defying Death - How Coxiella burnetii Copes with Intentional Host Cell Suicide. Yale J Biol Med. 2019;92(4):619–28. [PMC free article] [PubMed] [Google Scholar]
- 131.Hackstadt T Steric hindrance of antibody binding to surface proteins of Coxiella burnetti by phase I lipopolysaccharide. Infect Immun. 1988;56(4):802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moos A, Hackstadt T. Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immun. 1987;55(5):1144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shannon JG, Howe D, Heinzen RA. Virulent Coxiella burnetii does not activate human dendritic cells: role of lipopolysaccharide as a shielding molecule. Proc Natl Acad Sci U S A. 2005;102(24):8722–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schoffelen T, Ammerdorffer A, Hagenaars JC, Bleeker-Rovers CP, Wegdam-Blans MC, Wever PC, et al. Genetic Variation in Pattern Recognition Receptors and Adaptor Proteins Associated With Development of Chronic Q Fever. J Infect Dis. 2015;212(5):818–29. [DOI] [PubMed] [Google Scholar]
- 135.Ammerdorffer A, Schoffelen T, Gresnigt MS, Oosting M, den Brok MH, Abdollahi-Roodsaz S, et al. Recognition of Coxiella burnetii by toll-like receptors and nucleotide-binding oligomerization domain-like receptors. J Infect Dis. 2015;211(6):978–87. [DOI] [PubMed] [Google Scholar]
- 136.Kohl L, Hayek I, Daniel C, Schulze-Luhrmann J, Bodendorfer B, Luhrmann A, et al. MyD88 Is Required for Efficient Control of Coxiella burnetii Infection and Dissemination. Front Immunol. 2019;10:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bradley WP, Boyer MA, Nguyen HT, Birdwell LD, Yu J, Ribeiro JM, et al. Primary Role for Toll-Like Receptor-Driven Tumor Necrosis Factor Rather than Cytosolic Immune Detection in Restricting Coxiella burnetii Phase II Replication within Mouse Macrophages. Infect Immun. 2016;84(4):998–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zamboni DS, Campos MA, Torrecilhas AC, Kiss K, Samuel JE, Golenbock DT, et al. Stimulation of toll-like receptor 2 by Coxiella burnetii is required for macrophage production of pro-inflammatory cytokines and resistance to infection. J Biol Chem. 2004;279(52):54405–15. [DOI] [PubMed] [Google Scholar]