

Abstract
Environmental microorganisms continue to serve as a major source of bioactive natural products (NPs) and as an inspiration for many other scaffolds in the toolbox of modern medicine. Nearly all microbial NP-inspired therapies can be traced to field expeditions to collect samples from the environment. Despite the importance of these expeditions in the search for new drugs, few studies have attempted to document the extent to which NPs or their corresponding production genes are distributed within a given environment. To gain insight into this, the geographic occurrence of NP ketosynthase (KS) and adenylation (A) domains was documented across 53 and 58 surface sediment samples, respectively, covering 59,590 square kilometers of Lake Huron. Overall, no discernable NP geographic distribution patterns were observed for 90,528 NP classes of nonribosomal peptides and polyketides detected in the survey. While each sampling location harbored a similar number of A domain operational biosynthetic units (OBUs), limited overlap of OBU type was observed, suggesting that at the sequencing depth used in this study, no single location served as a NP ‘hotspot’. These data support the hypothesis that there is ample variation in NP occurrence between sampling sites and suggests that extensive sample collection efforts are required to fully capture the functional chemical diversity of sediment microbial communities on a regional scale.
1. Introduction
The preparation of Pyocyanase in 1899 and the discovery of bioactive natural products (NPs) penicillin and gramicidin in 1928 and 1939, respectively, marked the beginning of modern microbial drug discovery efforts.1–4 Since then, environmental microorganisms have served as a major source of bioactive NPs and as an inspiration for a plethora of therapeutic scaffolds. These small molecules have generated therapies for an array of diseases such as cancer, bacterial infections, immune disorders, and others, as 34% of FDA approved drugs from 2000 to 2014 were NPs or NP-derived.5 Importantly, nearly all of these microbial NP-inspired therapies resulted from field expeditions to collect samples from the environment. In general, these field expeditions have been guided by the hypothesis that environments in diverse geographic locations contain different ecological pressures, and as a result harbor minimally-overlapping populations of NP biosynthetic pathways.6–8
Despite the importance of sample collection expeditions toward the search for new drugs, few studies have attempted to document the extent to which NPs or their corresponding production genes are distributed in any given environment. Charlop-Powers et al. compared the NP biosynthetic potential of soil samples from a diverse array of environmental microbiomes.9 Their analyses of 185 soil microbiomes collected from five continents suggested that geographic distance and local environment contributed to biosynthetic diversity differences observed between samples.9 Additionally, Lemetre et al. found that changes in latitude correlated with changes in biosynthetic domain composition within soil samples on a continent-wide scale.10 Borsetto et al. correlated the observed differences in biosynthetic gene cluster (BGC) diversity in a range of soils within metagenome data, with the microbial community present at each site and with geographic location, and suggested that environmental variables influence the biosynthetic potential at a given site.11 Similarly, Sharrar et al. found that patterns of abundance of BGC types varied by taxonomy in soil bacteria, and that bacteria with higher biosynthetic potential were associated with specific types of soil vegetation.12 These studies demonstrate that biosynthetic domain composition can differ with changing geography and/or variables within the soil. Thus, characterizing the geographic distribution of NP-producing BGCs at a finer geographical resolution will inform front-end discovery practices such as sample collection and microbial library generation, which traditionally have a high degree of uncertainty.13
Due to decreasing sequencing costs and availability of online tools, probing microbial-based chemical diversity in nature has become attainable without relying on cultivation techniques. To gain insight into how specific NP classes are distributed in an environment, the occurrence of NP domains was characterized in up to 58 surface sediment samples covering a 59,590 square kilometer region in Lake Huron. Ketosynthase (KS) domains from polyketide synthases (PKS) and adenylation (A) domains from nonribosomal peptide synthetase (NRPS) were examined, as they represent conserved domains within two common classes of NPs that often encode for the production of antibiotics, siderophores, and other bioactive compounds. The current study provides preliminary evidence that there is substantial variation in NP composition between sampling sites on a regional scale and suggests that extensive sample collection efforts will be required to fully capture the BGC diversity that exists in sediment. Investigating BGC distribution patterns and dynamics in Lake Huron represents an essential initial step toward the design of a more methodical environmental sample collection approach, a critical front-end process that has been largely unchanged since antibiotic discovery efforts began in the early 20th century.
2. Results and Discussion
2.1. Characterization of BGC Domain Sequence Diversity in Sediment
In August and September of 2014, 59 samples were collected from Lake Huron – a geographic region that spans 59,590 square kilometers (Table S1). To confirm the bacterial diversity present represents populations that commonly occur in freshwater systems, the taxonomic diversity of bacteria at each site was assessed using microbial 16S rRNA gene amplicons (Supp. Experimental Procedures). Results were congruent with those of typical lake bacterial populations (Supp. Table S4).14 To assess the composition of NP domains at each collection site, previously designed degenerate primers were used to amplify the KSα domain for PKS II15 and the A domain for NRPS genes from genomic DNA (gDNA) extracted from sediment samples.16 The KSα and A domains were selected because they are among the most conserved catalytic domains of the PKS type II and NRPS gene clusters respectively. Furthermore, this sequence conservation has yielded primer sets for PCR amplification15–17 as well as bioinformatic tools and databases to facilitate the annotation and prediction of NPs.18,19
The selected conserved regions were PCR-amplified from genomic DNA using a two-stage PCR protocol, as described previously.20 Briefly, 613 bp fragments of KSα (β-ketoacyl synthase) and 700 bp fragments of NRPS A domains were amplified using degenerate oligonucleotides, respectively.15,16 All primers were synthesized with a locus-specific sequence as well as a universal 5′ tail.20 Resulting sequences were filtered using profile hidden Markov models (pHMMs) downloaded from antiSMASH’s HMM detection modules to remove non-specific sequences.21 These models are based on known and predicted KSα and A domain architectures.22 Filtered sequences were then clustered at 85% similarity to approximate compound class designations and to avoid overestimation of chemical diversity in sediment.23 Sequences were extracted from the manually curated and annotated BGC database MIBiG19, subjected to different clustering thresholds, and evaluated for their ability to group according to similar biosynthetic origins/molecular products. The optimal clustering threshold fluctuated and was dependent on the specific compound class and ranged from 80% to 90%. Therefore, analysis proceeded using an 85% similarity threshold. At 85% similarity, the sequence groupings – or operational biosynthetic units (OBUs) – represent an estimation of compound classes. To further scrutinize this clustering method, amplicons from a control Streptomyces strain, Streptomyces coelicolor A3(2), were subjected to this process (see Methods section 4.5).24 S. coelicolor A3(2) produces two KSα domain-containing compounds (actinorhodin and a spore pigment) and twelve A domain-containing compounds (CDA1b, CDA2a, CDA2b, CDA3a, CDA3b, CDA4a, CDA4b, coelibactin, coelimycin P1, undecylprodigiosin, SCO-2138, and a putative tris-hydroxamate tetrapeptide iron chelator coelichelin).24,25 Analysis of S. coelicolor A3(2) amplicons at 85% similarity yielded two KSα domain OBUs and fifteen A domain OBUs, and confirmed this as a suitable threshold to organize 300 bp fragments into groups that represent compound classes.
Of the 59 sediment samples, 6 from the KSα dataset and 1 from the A domain dataset did not return sufficient quality data to be included in the analysis. In total, 1,818 KSα OBUs (5,815 total sequences) throughout 53 sediment samples, and 171,527 A domain OBUs (1,730,091 total sequences) throughout 58 sediment samples were observed. This represents approximately 34 KSα and 2,957 A domain OBUs per sediment sample (Table 1). These original numbers were then adjusted to account for suspected overestimation of chemical diversity, as described in the following section. The large disparity in KSα and A domain OBU counts may be attributed to (1) primer biases and accuracy, (2) depth of sequencing, and (3) the size of the family to which these domains belong. A domains belong to a large superfamily of adenylate-forming enzymes,26 in contrast to the smaller KSα (α-ketoacyl synthases) domain family, which are known to produce aromatic polyketides and polyenes, and whose primers were designed specifically for strains within the Streptomyces genus.27,28 The number and putative identity of OBUs for each compound class is listed in Supporting Tables S6A–B. As previously reported, the KSα primers are highly degenerate, with substantial off-target amplification.29 Due to this limitation, KSα data, including distribution analysis and maps, can be found in the Supplemental Information.
Table 1.
A and KSα domain abundances in sediment.
| KSα | A | |
|---|---|---|
| Total # of OBUs detected | 1,818 | 171,527 |
|
| ||
| Total # of OBUs after adjustment by the split correction factor | 1,198 | 90,528 |
|
| ||
| Average # of OBUs per sample after adjustment by the split correction factor | 23 (±18) | 1,561 (±798) |
2.2. Analysis of Characterized NP BGC Distribution in Lake Sediment
In order to assess the occurrence of known NP BGC classes across Lake Huron sediment, the identity of each OBU was verified. Sequence representatives from each OBU were aligned against domain sequences extracted from the MIBiG database using the DIAMOND alignment tool via its default settings.30,31 MIBiG associates BGCs with known NP structures, allowing prediction of the product of each matching OBU and as a result, estimation of the chemical diversity at each sample site. To ensure that a 300 bp amplicon is sufficient for structural annotation, sequences from control strain S. coelicolor A3(2) were amplified, sequenced, and aligned (Supplementary Experimental Procedures).24 Amplified KSα and A domain sequences from S. coelicolor A3(2) aligned appropriately against coelichelin, coelibactin, and select calcium-dependent antibiotic (CDA) sequences from S. coelicolor in MIBiG at a maximum e-value of 3.90 e−43. In general, an e-value smaller than 0.01 is considered a reliable hit for homology matches, while an e-value in the range of 1e−50 is considered a match of high reliability.32 These results were used as a guide to select a list of annotated OBUs to map across lake sediment. Based on empirical tests and comparison to e-values obtained from the S. coelicolor A3(2) control, a maximum e-value threshold of 1.2 e−15 was selected for KSα domain OBUs and 1.3 e−11 for A domain OBUs. These stringent cutoffs allowed only high-confidence OBU assignments to be used in the study.
Once OBU sequence representatives were aligned against sequences from the MIBiG database, the majority of these could not be assigned to known chemical compound classes. In total, of the 1,818 KSα domain OBUs that were observed across 53 samples, 32 (1.7%) were assigned to known compound classes. Similarly, of 171,527 total A domain OBUs observed across 58 samples, 108 (0.06%) were assigned to known compound classes. Of particular note is that some distinct OBU sequence representatives were assigned to the same compound class (for example, five separate OBU sequence representatives aligned to rifamycin), which resulted in an overestimation of compound classes present in sediment. To correct for this, it was necessary to estimate the average number of times a compound class was divided into separate OBUs in the dataset; this average was deemed a “split correction factor” (see Supplementary Table S6 for discussion). The total number of observed OBUs was then divided by that factor, resulting in a more accurate estimation of the compound classes present in sediment: a total of 1,198 KSα domain OBUs, of which 21 (1.8%) were known compound classes, and a total of 90,528 A domain OBUs, of which 57 (0.06%) were known compound classes. Further details are listed in Supplementary Tables S6A–B.
Of the 78 OBUs matched to known classes of PKS (21) and NRPS (57) NPs in MIBiG, distribution maps of compounds that occurred in at least two distinct locations were generated after rarefaction analysis to the lowest sample count (15 sequences for KSα domain OBUs and 3,487 for A domain OBUs). A total of 30 OBUs met these criteria.
These 30 OBUs were further categorized into antibiotics, siderophores, and other bioactive NP classes such as anticancer and antiviral compounds. OBUs from each of the 30 classes were mapped and patterns of occurrence were assessed (representative OBUs per category are shown in Figure 2, while maps for the remaining OBUs are shown in Supplementary Figures S3–5). The size of the colored circles are proportional to the number of sequences detected at each sampling site, after rarefaction. Figures 2A–D show the distribution of cyclomarin, surugamide, pyoverdin, and coelichelin classes. For example, sequence reads for cyclomarin class antibiotics (Figure 2A) were detected in five distinct geographic locations across the lake, while sequence reads for pyoverdin-type siderophores (Figure 2C) were detected in 38 distinct geographic locations across the lake. Four of these locations contained both compounds. Overall, the distribution profiles among the compound classes analyzed were non-overlapping in lake sediment. In general, siderophores were the most frequently detected compound class in lake sediment, exceeding that of antibiotics and other bioactive NPs.
Figure 2.
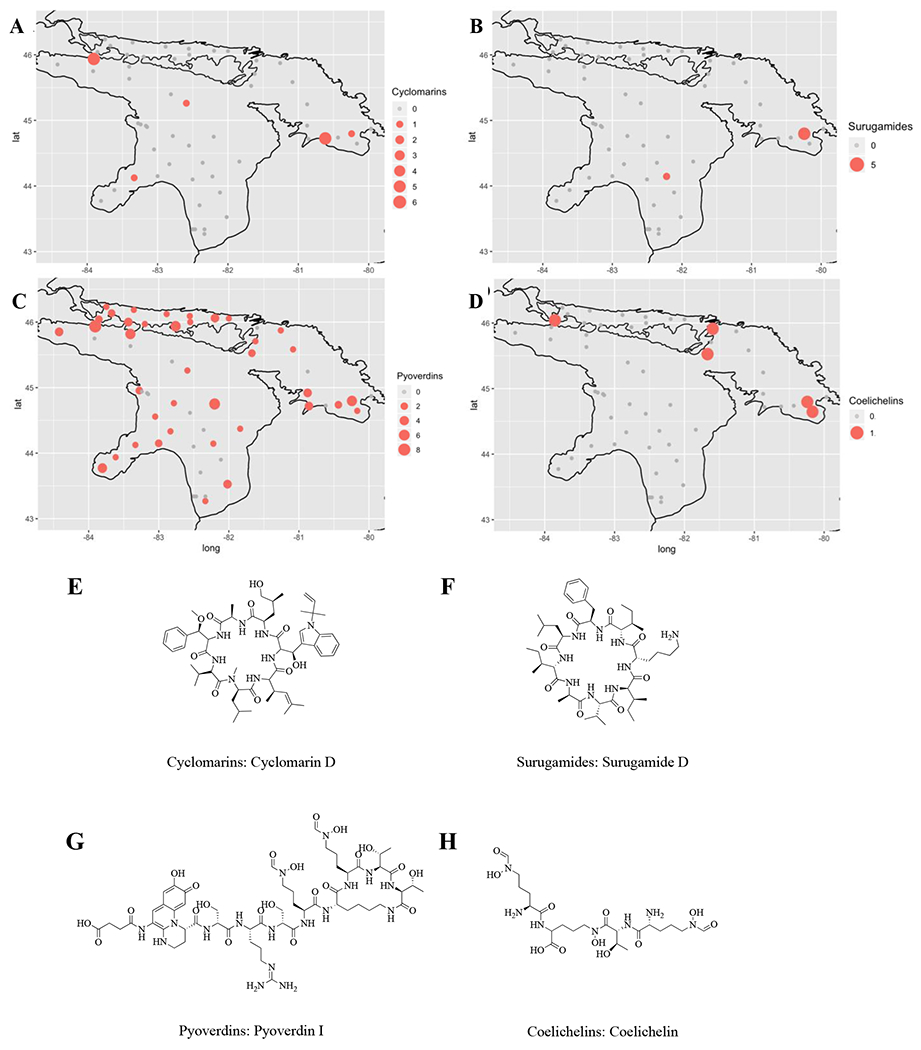
Detection of domain sequences of select NP classes in Lake Huron sediment. Figures 2A–D show the detection and relative read abundance of cyclomarin, surugamide, pyoverdin, and coelichelin classes, respectively. Figures S3–5 depict the distribution of additional NP classes. Different sized circles represent sequence read abundance at a rarefaction depth of 13 sequences per sample for KSα domain sequences and of 3,487 sequences per sample for A domain sequences at each collection site in Lake Huron. Representative structures from each of the four compound classes are shown in Figures 1E–H.
2.3. Analysis of Uncharacterized NP BGC Distribution in Lake Sediment
The majority of OBUs detected in Lake Huron sediment were not assigned to known compound classes (98.3% KSα domain OBUs and 99.9% of A domain OBUs, respectively). Instead of constructing maps for all 90,528 uncharacterized A domain OBUs, the number of locations at which a given OBU was detected was plotted (Figure 3). This allowed determination of the frequency of occurrence of OBUs across lake sediment. Figure 3 demonstrates that the vast majority of A domain OBUs (96.5%) occurred in fewer than 10 samples (in varying occurrence patterns, data not shown), across the 58 locations. For example, 40,003 OBUs (83.7%) were detected in only a single sediment location, and 2,524 OBUs (5.3%) were detected in only two locations (in varying occurrence patterns). However, no more than 1,042 OBUs were detected at any single sampling site (Figure 4); thus, the genetic diversity detected is broadly distributed. Figures 3 and 4 together demonstrate that there is little overlap among occurrence patterns of these OBUs, indicating that there are not select NP ‘hotspots’ among our 58 sampling sites and that NP occurrence varies considerably across Lake Huron sediment.
Figure 3.
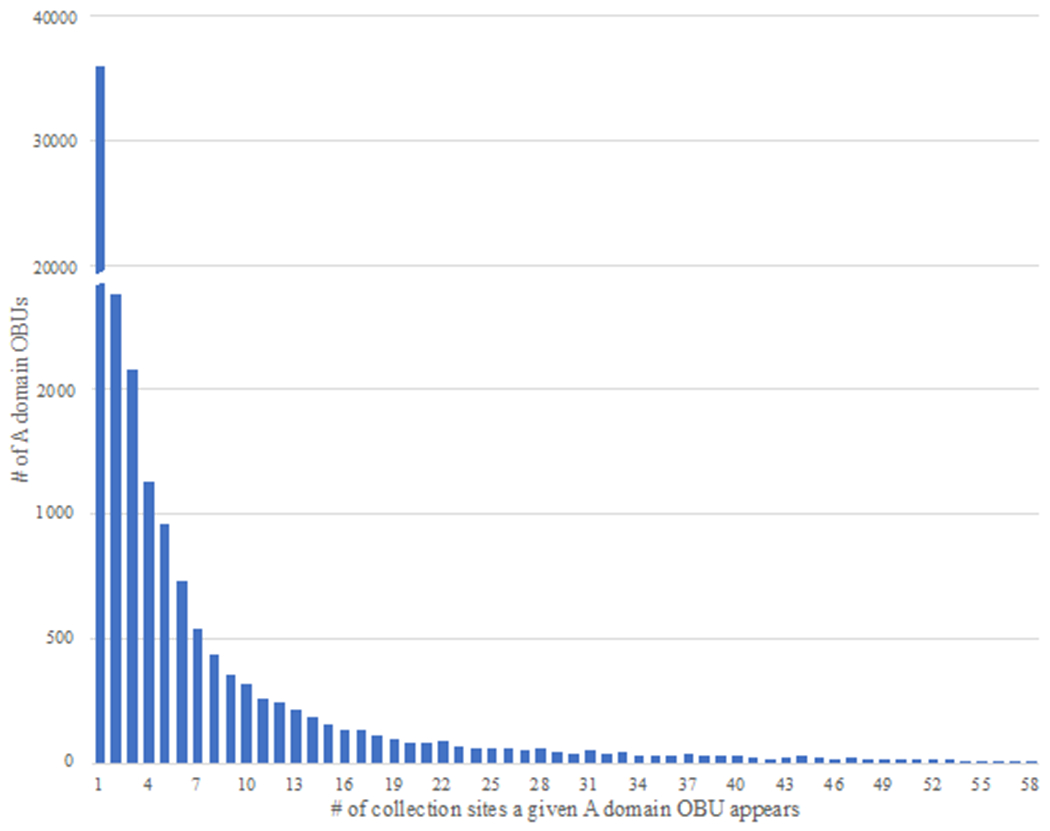
The number of locations from which an A domain OBU occurs. The majority of A domain OBUs occur in fewer than ten locations.
Figure 4.
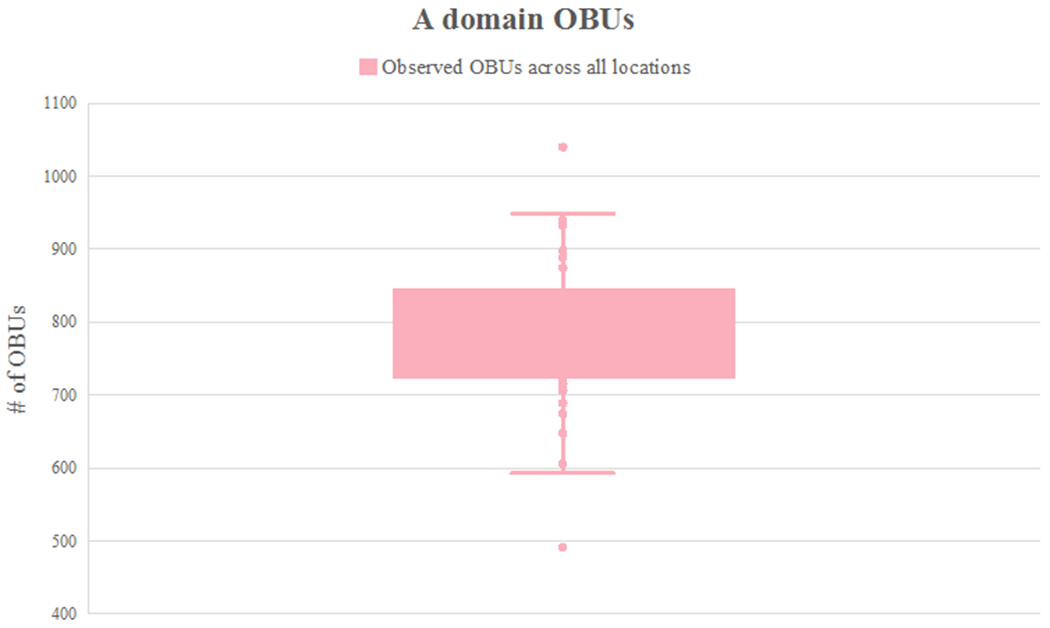
Box plot indicating observed number of A domain OBUs across all locations at a rarefaction depth of 3,487; each point represents the number of A domain OBUs at that location. The number of A domain OBUs that appears in each location ranges from 491 to 1,042.
We sought to determine whether A domain OBUs were likely to co-occur in the environment. Correlation coefficients based on presence/absence and abundance were calculated for each OBU pair for the 1,000 most abundant OBUs from rarified BIOM tables,33 based on the formula in Supp. Table S7. Among these, 0.16% of OBUs displayed a strong positive correlation with each other (a correlation score of 0.9 or above). This analysis further supports the lack of co-occurrence of dominant OBU classes in these sediments. Similarly, correlation analyses were undertaken to assess whether A domain OBUs correlated with the presence of specific Actinobacteria or Proteobacteria OTUs at each location (see Supplemental Information Table S7). No significant correlations were observed. One possible cause of this may be that the detected OBUs are associated with mobile genetic elements and therefore are associated with multiple taxa.34 Alternatively, primer biases (OBU versus OTU) coupled with insufficient OTU sequencing depth prevented sufficient detection of the necessary sequences needed to observe such correlations. Further experiments using shotgun metagenome sequencing will be required to confirm this result.
This study aimed to generate a preliminary assessment of how NP OBUs are distributed across Lake Huron sediment. As shown in Figure 2 (and Supp. Figures S3–S5), among the select 30 characterized OBUs that were analyzed, no discernable patterns of occurrence in Lake Huron surface sediment were observed. Some NP OBUs exhibited frequent occurrences in sediment across the geographic locations sampled, while others were confined to select sample sites.
This study is one of the few attempts to document the distribution of specific classes of NPs at a regional scale in an environment representative of a collection expedition.35 The observed NP distribution profiles lend experimental evidence to a few predictable phenomena, that to the best of our knowledge have seldom been demonstrated on a large scale. First, individual compound profiles, particularly those that represent bioactive NPs (antibiotics, anticancer, etc), exhibit sparse occurrence across Lake Huron sediment. Second, some profiles occur more frequently across the collection sites, such as the pyoverdins and griseorhodins (Figure 2C and Supp. Figure S5H). This may suggest that the NP is highly functional in its environment or is located on a mobile genetic element that is commonly transferred between species, among other possibilities. Regardless, of the greater than 90,000 known and uncharacterized NP OBUs analyzed, there is little evidence for discernable patterns of NP occurrence across Lake Huron sediment. This suggests that robust sampling is required to survey an environment of this magnitude, and that oversampling leading to redundant NP recovery is not a major concern (though cultivation methods will be a significant factor in recovering those NP populations from sediment).23 Further experiments should be performed to assess whether the OBU distribution trend observed in this study is also detected in the culturable bacterial population, a metric more appropriate to evaluate the efficiency of most microbial drug discovery programs. An attempt to document OBU recovery from culturable bacterial populations was addressed in other complementary studies.23,36 A similar study that analyzes sequences within an area of higher geographic resolution, at multiple time points, and with consideration toward environmental pressures specific to the benthic lake environment, would provide more detailed information on the available NP chemical space in Lake Huron sediment. Events such as algal blooms or other localized environmental phenomena at the time of collection can influence results in any of the sampled locations.
The need for novel approaches to improve detection of NP BGCs from eDNA
There are a few experimental limitations to the current study (see SI for more detailed explanation). First, the low abundance of sequence reads belonging to NPs can be attributed to undersampling, limited eDNA extracted from sediment, or biases generated from PCR amplification using highly degenerate primers. In addition, the resulting amplicons are only partially representative of the BGC population present in sediment. The design of new primers with a broader detection range can improve discovery of non-traditional BGCs. However, alternative, non-PCR-based approaches such as deep shotgun metagenome sequencing coupled with long-read sequence data (e.g. data generated using Oxford Nanopore and Pacific Biosciences sequencing platforms), or enrichment strategies followed by deep sequencing (e.g., Oxford Nanopore selective sequencing,37 hybridization capture+shotgun metagenome sequencing38) will be necessary for further discovery. Finally, the MIBiG database was used to assess compound classes.19 The number of existing NPs greatly outnumbers the entries in MIBiG, underlining the need for the community to contribute to and expand this valuable resource.
3. Conclusion
Despite decades of collecting soil microorganisms for use in drug discovery, few attempts have been made to measure the extent to which NP production genes are distributed in the environment. In this study, KSα and A domain amplicon sequencing was used to document distribution profiles of NPs across Lake Huron surface sediment. Overall, no discernable NP geographic distribution patterns were observed when comparing OBUs from greater than 90,000 NP classes (NRPS and PKS). We observed that the distribution profiles of the majority of A domain OBUs were non-overlapping across the 58 locations, while each location harbored relatively equal number of OBUs, suggesting that at the sequencing depth used in this study, no single location served as a NP ‘hotspot’. Finally, analysis of the top 1,000 most abundant OBUs detected in Lake Huron sediment, which belong to unknown/uncharacterized NPs, indicate that co-occurrence patterns are rare, but do exist. This preliminary evidence supports that there is ample variation in NP occurrence between sampling sites and suggests that extensive sample collection efforts will be required to fully capture the diversity that exists in sediment on a regional scale. Overall, investigating BGC distribution patterns and dynamics in Lake Huron has highlighted the need for a more methodical environmental sample collection approach, a great unmet need in NP drug discovery.
4. Methods
4.1. Collection of Sediment Samples, Cultivation of Sediment Bacteria on Nutrient Agar
Sediment samples were collected using a PONAR grab in the summer of 2012 from Lake Huron, the Georgian Bay, and the Northern Channel during a research expedition aboard the EPA’s Lake Guardian Research Vessel. Surface depths of sediment are listed in Supp. Table S1. Approximately 1 cm3 of sediment was homogenized, and an aliquot was placed into a 2 mL cryovial containing 20% glycerol. These were stored in cryogenic vials in a Dewar until transported back to the laboratory where they were stored in a −20°C freezer.
4.2. Genomic DNA Isolation from Sediment and Nutrient Agar
Cryogenic vials were thawed at room temperature, and gDNA was extracted from approximately 0.25 g of sediment, using a DNeasy PowerSoil Kit (Qiagen, Netherlands) according to the manufacturer’s instructions.
4.3. KSα and A Domain Amplification and Sequencing
KSα and A domain amplicon sequencing was performed using the same two-step PCR strategy described in the Supporting Information. Briefly, a 613 bp fragment of the KSα (β-ketoacyl synthase) was amplified using degenerate primers (5′-TSGCSTGCTTCGAYGCSATC-3′) and (5′-TGGAANCCGCCGAABCCGCT-3′).15 700-bp NRPS A domain gene fragments were amplified using degenerate oligonucleotides A3F (5′-GCSTACSYSATSTACACSTCSGG-3′) and A7R (5′SASGTCVCCSGTSCGGTAS-3′).16 All primers were synthesized with a locus-specific sequence as well as a universal 5′ tail (i.e., CS1 and CS2 linkers). 20 μL of PCR reaction mixture consisted of 1 μL of DNA at 10 ng/μL, 1 μL of a 10 μM solution of each primer, 10 μL KAPA Taq 2X ReadyMix (Kapa Biosystems), 0.8 μL of DMSO, 3.2 μL of 100 mg mL−1 Bovine Albumin Serum, and 3 μL of DI water. The thermal cycling conditions were set to an initial denaturation step at 95 °C for 5 min; 7 cycles of 1 min at 95 °C, 1 min at 65 °C (annealing temperature was lowered 1 °C per cycle), and 1 min at 72 °C; and 40 cycles of 1 min at 95 °C, 90 s at 58 °C and 1 min at 72 °C; and a final elongation step at 72 °C for 5 min. Amplification products were verified by agarose gel electrophoresis and purified using a QIAquick PCR cleanup kit according to the manufacturer’s protocol (Qiagen). The resulting PCR amplicons were used as templates for the second PCR step, as described above, to incorporate sequencing adapters and sample-specific barcodes. Pooled and purified amplicon libraries, with a 20% phiX spike-in, were loaded onto a MiSeq V3 flow cell, and sequenced using paired-end 2 × 300 reads. Sequencing was performed at the Genome Research Core at the University of Illinois at Chicago.
4.4. Bioinformatic Analyses of BGC Data
Only forward reads were used in further analysis due to the low quality of reverse reads. All sequences generated from the Illumina MiSeq sequencer were trimmed on the ends of the read according to Phred quality scores, then denoised using the DADA2 implemented in Qiime2, and finally chimeras were removed using uchime-denovo as implemented in Qiime2.39,40 The degenerate primer sequences were removed. Filtered and trimmed reads were then 6-frame translated into amino acid sequences using TranslatorX.41 Only frames with no internal stop codons were kept using TranslatorX’s “guess most likely reading frame” option. Amino acid sequences were then filtered via HMMER42 using HMM prebuilt generic detection models downloaded from antiSMASH v5.0.0.21 The following models were used: AMP-binding and A-OX for A domain, and t2ks and t2pks2 for PKS type II. Only sequences that passed the default e-value thresholds were kept, resulting in a much lower number of sequences per sample (Supp. Table S3). Sequences were then clustered at 80%, 85%, 90%, and 95% using USEARCH v11’s UCLUST cluster_fast greedy algorithm via the cluster_fast command. Singletons were kept for the clustering.43 A feature-by-sample abundance matrix (a feature table or biological observation matrix, BIOM)33 file was then created. A representative sequence from each cluster—labeled an OBU—was extracted to a separate file, using the USEARCH v11’s makeudb_usearch command, and the file was aligned against the MIBiG database using DIAMOND.31 Sequence reads belonging to the same molecular class clustered best at 85%. Therefore, the 85% sequence similarity threshold was used for subsequent analyses. An OBU representative sequence was annotated with its BLAST identity only if the pairwise identity was at least 85% and coverage over at least 84 amino acids.44 An OBU-by-sample BIOM file was then created and rarefied to the minimum number of sequences within samples. Singletons were retained during OBU clustering. Since OBU clustering occurred at 85% (as opposed to the single nucleotide/ASV level), changes in a single nucleotide OBU diversity are not expected to change the richness of the sample. In addition, to ensure that singletons were real sequences and not PCR error, 10 singletons from each domain were blasted against the NCBI’s protein database, and all singletons mapped to the correct group (A and KSα domains).43
4.5. Bioinformatic Method Validation Using Reference Strains
Control strain S. coelicolor A3(2) was included in wet lab and bioinformatics analysis to ensure clustering methods and compound identities were valid. S. coelicolor A3(2) was subjected to the same amplification procedure using the degenerate primers that amplify a fragment of the KSα (β-ketoacyl synthase) and a fragment of the A domain. KSα and A domain amplicons were sequenced and analyzed using the strategy described in sections 4.3 and 4.4. Resulting sequence data were then filtered using the same HMM prebuild generic detection models described above. Sequences that passed the default e-value thresholds were kept. Sequences were then clustered at 80% 85%, 90%, and 95%. At 85%, KSα amplicons grouped into two OBUs. A representative sequence from each OBU was mapped against MIBIG for compound identification. Indeed, the representative sequence from the first OBU mapped to actinorhodin and the representative sequence from the second OBU mapped to a spore pigment, as expected. Similarly, at 85%, A domain amplicons grouped into 15 OBUs. After mapping against MIBIG, a representative sequence from 10 OBUs mapped to the CDA family of compounds (CDA1b, CDA2a, CDA2b, CDA3a, CDA3b, CDA4a, CDA4b), one representative sequence mapped to coelimycin P1, one representative sequence mapped to coelibactin, and three representative sequences mapped to coelichelin. There were no OBU representative sequences mapped to the remaining A domain containing compounds undecylprodigiosin and SCO-2138.
Supplementary Material
Figure 1.

Boxplots depicting the variability of KSα and A domain OBU abundance in each sediment sample. A and B represent boxplots of OBU counts after adjustment by the split correction factor, which corrects for overestimations of compound classes present in sediment.
4.6. Acknowledgments
The authors wish to acknowledge the following contributors: A. Li (UIC), K. Rockne (UIC), S. Carlson (formerly UIC), and crew of EPA RV Lake Guardian for assistance with sediment collection; G. Chlipala of UIC’s Core for Research Informatics and A. Naqib of UIC’s DNA Sequencing Core for assistance processing data. This publication was supported by Vahlteich Scholar research funds, the Illinois–Indiana Sea Grant, the Office of Technology Management at UIC, UIC startup funds, and funding from National Institutes of Health grant R01 GM125943.
Footnotes
Supporting Information Available: This material includes additional experimental procedures, accession codes, and sample collection details such as sample names and coordinates. The document also contains detailed sequencing information before and after data filtration steps, analyses of bacterial phyla present in sediment, and occurrence maps of NP OBUs across Lake Huron sediment. This material is available free of charge via the internet at http://pubs.acs.org.
References
- (1).Aldrich S Alexander Fleming Discovery and Development of Penicillin - Landmark - American Chemical Society. American Chemical Society International Historic Chemical Landmarks. 1999. 10.2307/3561468. [DOI] [Google Scholar]
- (2).Fleming A On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to Their Use in the Isolation of B. Influenzae. British J. Exper. Pathol 1929, 10, 226–236. [Google Scholar]
- (3).Gause GF; Brazhnikova MG Gramicidin S and Its Use in the Treatment of Infected Wounds. Nature 1944, 154, 703. 10.1038/154703a0. [DOI] [Google Scholar]
- (4).Emmerich R; Löw O Bakteriolytische Enzyme Als Ursache Der Erworbenen Immunität Und Die Heilung von Infectionskrankheiten Durch Dieselben. Zeitschrift für Hygiene und Infectionskrankheiten 1899, 31, 1–65. 10.1007/BF02206499. [DOI] [Google Scholar]
- (5).Newman DJ; Cragg GM Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod 2020, 83, 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- (6).Clardy J; Fischbach MA; Currie CR The Natural History of Antibiotics. Curr. Biol 2009, 19, R437–R441. 10.1016/j.cub.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fischbach MA; Walsh CT Antibiotics for Emerging Pathogens. Science 2009, 325 (5944), 1089–1093. 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cheng K; Rong X; Pinto-Tomás AA; Fernández-Villalobos M; Murillo-Cruz C; Huang Y Population Genetic Analysis of Streptomyces Albidoflavus Reveals Habitat Barriers to Homologous Recombination in the Diversification of Streptomycetes. Appl. Environ. Microbiol 2015, 81 (3), 966–975. 10.1128/AEM.02925-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Charlop-Powers Z; Owen JG; Reddy BVB; Ternei M; Guimaraes DO; De Frias UA; Pupo MT; Seepe P; Feng Z; Brady SF Global Biogeographic Sampling of Bacterial Secondary Metabolism. eLife 2015, 2015 (4), e05048. 10.7554/eLife.05048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lemetre C; Maniko J; Charlop-Powers Z; Sparrow B; Lowe AJ; Brady SF Bacterial Natural Product Biosynthetic Domain Composition in Soil Correlates with Changes in Latitude on a Continent-Wide Scale. Proc. Natl. Acad. Sci 2017, 114 (44), 11615–11620. 10.1073/pnas.1710262114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Borsetto C; Amos GCA; da Rocha UN; Mitchell AL; Finn RD; Laidi RF; Vallin C; Pearce DA; Newsham KK; Wellington EMH Microbial Community Drivers of PK/NRP Gene Diversity in Selected Global Soils. Microbiome 2019, 7 (1), 78. 10.1186/s40168-019-0692-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sharrar AM; Crits-Christoph A; Méheust R; Diamond S; Starr EP; Banfield JF Bacterial Secondary Metabolite Biosynthetic Potential in Soil Varies with Phylum, Depth, and Vegetation Type. mBio 2020, 11 (3). 10.1128/mBio.00416-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hernandez A; T. Nguyen L; Dhakal R; T. Murphy B The Need to Innovate Sample Collection and Library Generation in Microbial Drug Discovery: A Focus on Academia. Nat. Prod. Rep 2021, 38, 292–300. 10.1039/D0NP00029A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Newton RJ; Jones SE; Eiler A; McMahon KD; Bertilsson S A Guide to the Natural History of Freshwater Lake Bacteria. Microbiol. Mol. Biol. Rev 2011, 75 (1), 14–49. 10.1128/MMBR.00028-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Metsä-Ketelä M; Salo V; Halo L; Hautala A; Hakala J; Mäntsälä P; Ylihonko K An Efficient Approach for Screening Minimal PKS Genes from Streptomyces. FEMS Microbiol. Lett 1999, 180 (1), 1–6. 10.1016/S0378-1097(99)00453-X. [DOI] [PubMed] [Google Scholar]
- (16).Ayuso-Sacido A; Genilloud O New PCR Primers for the Screening of NRPS and PKS-I Systems in Actinomycetes: Detection and Distribution of These Biosynthetic Gene Sequences in Major Taxonomic Groups. Microbial Ecol. 2005, 49 (1), 10–24. 10.1007/s00248-004-0249-6. [DOI] [PubMed] [Google Scholar]
- (17).Ginolhac A; Jarrin C; Gillet B; Robe P; Pujic P; Tuphile K; Bertrand H; Vogel TM; Perrière G; Simonet P; et al. Phylogenetic Analysis of Polyketide Synthase I Domains from Soil Metagenomic Libraries Allows Selection of Promising Clones. Appl. and Envir. Microbiol 2004, 70 (9), 5522–5527. 10.1128/AEM.70.9.5522-5527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Weber T; Kim HU The Secondary Metabolite Bioinformatics Portal: Computational Tools to Facilitate Synthetic Biology of Secondary Metabolite Production. Synth. Syst. Biotechnol 2016, 1 (2), 69–79. 10.1016/j.synbio.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kautsar SA; Blin K; Shaw S; Navarro-Muñoz JC; Terlouw BR; van der Hooft JJJ; van Santen JA; Tracanna V; Suarez Duran HG; Pascal Andreu V; et al. MIBiG 2.0: A Repository for Biosynthetic Gene Clusters of Known Function. Nucleic Acids Res. 2020, 48 (D1), D454–D458. 10.1093/nar/gkz882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Naqib A; Poggi S; Wang W; Hyde M; Kunstman K; Green SJ Making and Sequencing Heavily Multiplexed, High-Throughput 16S Ribosomal RNA Gene Amplicon Libraries Using a Flexible, Two-Stage PCR Protocol. In: Raghavachari N, Garcia-Reyero N (eds) Gene Expression Analysis. Methods in Molecular Biology. Humana Press, New York, NY. 2018, 1783, 149–169. 10.1007/978-1-4939-7834-2_7. [DOI] [PubMed] [Google Scholar]
- (21).Blin K; Shaw S; Steinke K; Villebro R; Ziemert N; Lee SY; Medema MH; Weber T AntiSMASH 5.0: Updates to the Secondary Metabolite Genome Mining Pipeline. Nucleic Acids Res. 2019, 47 (W1), W81–W87. 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Adamek M; Alanjary M; Ziemert N Applied Evolution: Phylogeny-Based Approaches in Natural Products Research. Nat. Prod. Rep 2019, 36, 1295–1312. 10.1039/C9NP00027E. [DOI] [PubMed] [Google Scholar]
- (23).Elfeki M; Alanjary M; Green SJ; Ziemert N; Murphy BT Assessing the Efficiency of Cultivation Techniques To Recover Natural Product Biosynthetic Gene Populations from Sediment. ACS Chem. Biol 2018, 13 (8), 2074–2081. 10.1021/acschembio.8b00254. [DOI] [PubMed] [Google Scholar]
- (24).Bentley SD; Chater KF; Cerdeño-Tárraga AM; Challis GL; Thomson NR; James KD; Harris DE; Quail MA; Kieser H; Harper D; et al. Complete Genome Sequence of the Model Actinomycete Streptomyces Coelicolor A3(2). Nature 2002, 417 (6885), 141–147. 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- (25).Lautru S; Deeth RJ; Bailey LM; Challis GL Discovery of a New Peptide Natural Product by Streptomyces Coelicolor Genome Mining. Nat. Chem. Biol 2005, 1, 265–269. 10.1038/nchembio731. [DOI] [PubMed] [Google Scholar]
- (26).Schmelz S; Naismith JH Adenylate-Forming Enzymes. Curr. Opin. Struct. Biol 2009, 19 (6), 666–671. 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chen A; Re RN; Burkart MD Type II Fatty Acid and Polyketide Synthases: Deciphering Protein-Protein and Protein-Substrate Interactions. Nat. Prod. Rep 2018, 35, 1029–1045. 10.1039/c8np00040a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Du D; Katsuyama Y; Shin-ya K; Ohnishi Y Reconstitution of a Type II Polyketide Synthase That Catalyzes Polyene Formation. Angew. Chem. Int. Ed 2018, 130 (7), 1972–1975. 10.1002/ange.201709636. [DOI] [PubMed] [Google Scholar]
- (29).Liu L; Salam N; Jiao JY; Jiang HC; Zhou EM; Yin YR; Ming H; Li WJ Diversity of Culturable Thermophilic Actinobacteria in Hot Springs in Tengchong, China and Studies of Their Biosynthetic Gene Profiles. Microb. Ecol 2016, 72, 150–162. 10.1007/s00248-016-0756-2. [DOI] [PubMed] [Google Scholar]
- (30).Kautsar SA; Blin K; Shaw S; Navarro-Muñoz JC; Terlouw BR; van der Hooft JJJ; van Santen JA; Tracanna V; Suarez Duran HG; Pascal Andreu V; et al. MIBiG 2.0: A Repository for Biosynthetic Gene Clusters of Known Function. Nucleic Acids Res. 2019, 48, D454–D458. 10.1093/nar/gkz882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Buchfink B; Xie C; Huson DH Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12 (1), 59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- (32).Scholz M; Ward DV; Pasolli E; Tolio T; Zolfo M; Asnicar F; Truong DT; Tett A; Morrow AL; Segata N Strain-Level Microbial Epidemiology and Population Genomics from Shotgun Metagenomics. Nat. Methods 2016, 13 (5), 435–438. 10.1038/nmeth.3802. [DOI] [PubMed] [Google Scholar]
- (33).McDonald D; Clemente JC; Kuczynski J; Rideout JR; Stombaugh J; Wendel D; Wilke A; Huse S; Hufnagle J; Meyer F; et al. The Biological Observation Matrix (BIOM) Format or: How I Learned to Stop Worrying and Love the Ome-Ome. GigaScience 2012, 464 (1), 1–6. 10.1186/2047-217X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Penn K; Jenkins C; Nett M; Udwary DW; Gontang EA; McGlinchey RP; Foster B; Lapidus A; Podell S; Allen EE; et al. Genomic Islands Link Secondary Metabolism to Functional Adaptation in Marine Actinobacteria. ISME J. 2009, 3 (10), 1193–1203. 10.1038/ismej.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Charlop-Powers Z; Pregitzer CC; Lemetre C; Ternei MA; Maniko J; Hover BM; Calle PY; McGuire KL; Garbarino J; Forgione HM; et al. Urban Park Soil Microbiomes Are a Rich Reservoir of Natural Product Biosynthetic Diversity. Proc. Natl. Acad. Sci 2016, 113 (51), 14811–14816. 10.1073/pnas.1615581113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bech PK; Lysdal KL; Gram L; Bentzon-Tilia M; Strube ML Marine Sediments Hold an Untapped Potential for Novel Taxonomic and Bioactive Bacterial Diversity. mSystems 2020, 5 (5). 10.1128/mSystems.00782-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Edwards HS; Krishnakumar R; Sinha A; Bird SW; Patel KD; Bartsch MS Real-Time Selective Sequencing with RUBRIC: Read Until with Basecall and Reference-Informed Criteria. Sci. Rep 2019, 9 (1), 11475. 10.1038/s41598-019-47857-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhou J; He Z; Yang Y; Deng Y; Tringe SG; Alvarez-Cohen L High-Throughput Metagenomic Technologies for Complex Microbial Community Analysis: Open and Closed Formats. mBio 2015, 6 (1). 10.1128/mBio.02288-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Callahan BJ; McMurdie PJ; Rosen MJ; Han AW; Johnson AJA; Holmes SP DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13 (7), 581–583. 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bolyen E; Rideout JR; Dillon MR; Bokulich NA; Abnet CC; Al-Ghalith GA; Alexander H; Alm EJ; Arumugam M; Asnicar F; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol 2019, 37 (8), 852–857. 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Abascal F; Zardoya R; Telford MJ TranslatorX: Multiple Alignment of Nucleotide Sequences Guided by Amino Acid Translations. Nucleic Acids Res. 2010, 38, 7–13. 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Johnson LS; Eddy SR; Portugaly E Hidden Markov Model Speed Heuristic and Iterative HMM Search Procedure. BMC Bioinformatics 2010, 11 (431), 1471–2105. 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Edgar RC Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26 (19), 2460–2461. 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- (44).Camacho C; Coulouris G; Avagyan V; Ma N; Papadopoulos J; Bealer K; Madden TL BLAST+: Architecture and Applications. BMC Bioinformatics 2009, 10, 1–9. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.