

Abstract
Oncogenic gene fusions are hybrid genes that result from structural DNA rearrangements, leading to deregulated activity. Fusions involving the neuregulin-1 gene (NRG1) result in ErbB-mediated pathway activation and therefore present a rational candidate for targeted treatment. The most frequently reported NRG1 fusion is CD74-NRG1, which most commonly occurs in patients with invasive mucinous adenocarcinomas (IMA) of the lung, although several other NRG1 fusion partners have been identified in patients with lung cancer, including ATP1B1, SDC4, and RBPMS. NRG1 fusions are also present in patients with other solid tumors, such as pancreatic ductal adenocarcinoma (PDAC). In general, NRG1 fusions are rare across different types of cancer, with a reported incidence of <1%, with the notable exception of IMA, which represents ≈2‒10% of lung adenocarcinomas and has a reported incidence of ≈10‒30% for NRG1 fusions. A substantial proportion (≈20%) of NRG1 fusion-positive non-small-cell lung cancer (NSCLC) cases are non-mucinous adenocarcinomas. ErbB-targeted treatments, such as afatinib, a pan-ErbB tyrosine kinase inhibitor, are potential therapeutic strategies to address unmet treatment needs in patients harboring NRG1 fusions.
Keywords: gene fusion, NRG1, invasive mucinous adenocarcinoma, non-small-cell lung cancer, ErbB-targeted treatment, afatinib
INTRODUCTION
Gene fusions are hybrid genes that result from structural DNA rearrangements including translocations and insertions, transcription read-through, or splicing.1 Oncogenic gene fusions result in deregulated activity, and are the driver events responsible for initiation and maintenance of various types of cancer.1–3
For some cancers driven by gene fusions, the successful development of targeted therapies has improved clinical outcomes. For example, tyrosine kinase inhibitors (TKI) such as imatinib, dasatinib and nilotinib have markedly improved outcomes in patients with chronic myeloid leukemia (CML), which is driven by the BCR-ABL1 tyrosine kinase, a fusion protein created by a chromosomal translocation.2,4
The last few years have seen the identification of a number of activating gene fusions and the demonstration of efficacy and subsequent approval of a number of TKIs targeting these driver fusions. A prime example is the anaplastic lymphoma kinase (ALK) protein; ALK TKIs have been shown to be highly effective in ALK-rearranged lung adenocarcinomas,3 leading to dramatic improvement of survival rate.5,6 ALK fusions are also found in other tumors, albeit at a lower frequency than in non-small-cell lung cancer (NSCLC).7 Other targetable fusions have been identified across a number of tumor types such as the rearrangements of ROS1 in NSCLC,8 RET in thyroid malignancies and in NSCLC,9,10 and the three neurotrophic receptor tyrosine kinase genes (NTRK1, NTRK2, and NTRK3) that can undergo chromosomal rearrangements leading to functional gene fusions.11–14
In this context, fusions involving the neuregulin-1 gene (NRG1) were first identified by transcriptome sequencing of lung adenocarcinomas, which revealed fusion of CD74 to NRG1.15,16 Subsequently, many other NRG1 fusion partners have been identified.17,18 NRG1 fusions are present at a low incidence in multiple tumor types but are enriched in invasive mucinous adenocarcinomas (IMA) of the lung.18–22
NRG1 fusions result in aberrant expression of the Epidermal Growth Factor (EGF)-like domain of neuregulin-1 (NRG1) on the cell surface, which serves as a ligand for ErbB3 (HER3) and induces the formation of heterodimers, most frequently ErbB2-ErbB3, but also with EGFR (ErbB1) and ErbB4. This leads to pathologic activation of the phosphoinositide 3-kinase-protein kinase B (PI3K-AKT), mitogen-activated protein kinase (MAPK), and other signaling pathways, resulting in abnormal cell proliferation.16 NRG1 fusions and the subsequent ErbB-mediated constitutive pathway activation therefore represent a rational therapeutic target.
The aim of this narrative review is to provide information on the biology and detection of NRG1 fusions, their role in the development of certain types of cancer, and potential intervention modalities that target the ErbB pathway, including afatinib, as it has been used most frequently in a clinical context, for the treatment of NRG1 fusion-driven tumors. To identify relevant published data on NRG1 and NRG1 gene fusions, we conducted literature searches of PubMed using “Neuroregulin-1” and “afatinib” and “gene fusion” and “NRG1 fusion”. We also searched Google Scholar, as well as conference proceedings from recent major congresses, including ASCO, ESMO, AACR, and WCLC. Searches were updated on March 16th, 2020.
Biology of NRG1 and NRG1 fusions
Neuregulins and their normal function
The ErbB family of receptor tyrosine kinases is composed of four members (EGFR, ErbB2 [HER2], ErbB3, and ErbB4).16 There are 13 extracellular ligands for the ErbB family members, which can be divided into those that bind EGFR and ErbB4, and those that bind ErbB3 and ErbB4.16 This latter group is known as the neuregulins and comprises four members (NRG1–4).23 NRG1 and NRG2 bind to ErbB3, and NRG1‒4 bind to ErbB4. Upon ligand binding, these receptors form homo- or heterodimers, which results in phosphorylation of the intrinsic kinase domain, and activation of the PI3K-AKT, MAPK, and other pathways.16
The NRG1 gene is located on chromosome 8 (8p12 region) and encodes NRG1.24 The NRG1 gene is translated to generate six different proteins (I–VI) and at least 31 isoforms.25–28 NRG1 proteins are structurally related to EGF and contain an EGF-like motif that binds and activates ErbB3 and ErbB4.29–34 Most NRG1 isoforms are synthesized as membrane-anchored precursors with their EGF domain positioned outside the cell. Proteolytic processing leads to the release of mature, diffusible NRG1, which can activate ErbB3 and ErbB4 on distant cells.35 An exception is NRG1 type III isoform, which passes through the membrane twice and retains its EGF domain tethered to the membrane after proteolytic processing;36 this restricts type III NRG to signaling to adjacent cells.16
NRG1 binding to ErbB3 can induce heterodimerization, preferentially with the ErbB2 receptor. This allows ErbB2 to phosphorylate the kinase-defective ErbB3 and leads to activation of downstream pathways including the PI3K-AKT and MAPK pathways (Figure 1).34,37,38 NRG1 also has the capacity to bind ErbB4, similarly leading to dimerization as either a homodimer or a heterodimer with ErbB2/ErbB3, and subsequent activation.24,39–41 Different ErbB homo- and heterodimers activate different pathways to different degrees;42 this might explain differences in response to targeted therapies observed in patients.
Figure 1: Mechanism of action figure.
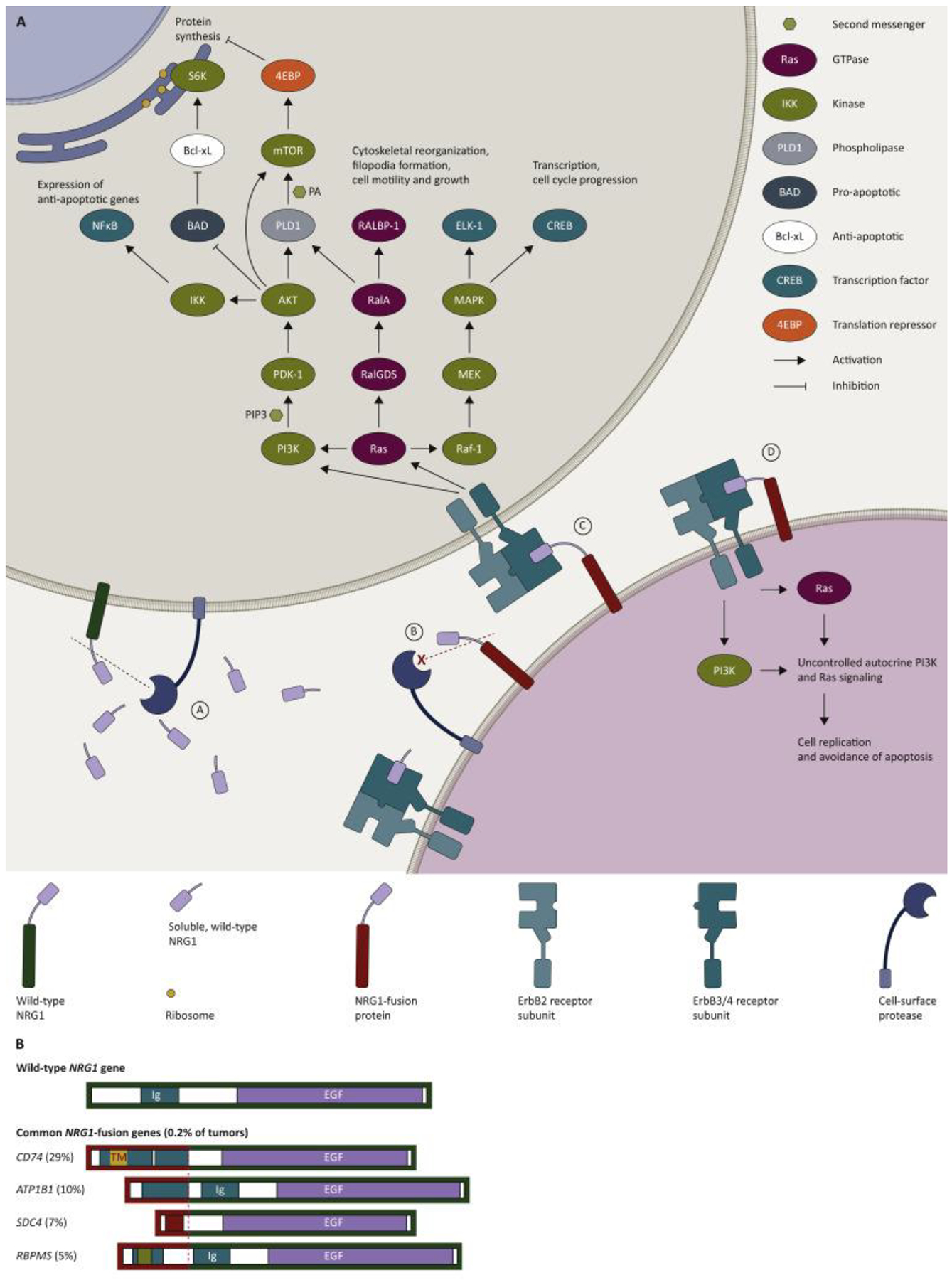
Figure 1A. NRG1-fusion proteins drive tumorigenic signaling pathways (adapted from Fernandez-Cuesta and Thomas, 201516, Ferrer et al 2018,78 Gille et al 1996,79 Populo et al 2012,80 Sakatomoto et al 2009,81 Shi et al 2007,82 Yang et al 1995,83 Manning and Cantley, 200784) and common NRG1-fusions (redrawn from Jonna et al 201948).
A. Wild type NRG1 may be cleaved by proteases to release soluble NRG1 containing the EGF-like region. Soluble NRG1 diffuses to activate distant ErbB3 or ErbB4 receptor subunits (paracrine signaling). Activated receptors dimerize, forming ErbB3- or ErbB4-containing homo- or hetero ErbB dimers, preferentially involving ErbB2.
B. NRG1 fusions by losing, altering or misplacing cleavage sites may become poorer substrates for proteases, e.g. ADAM17, resulting in excess NRG1 tethered to the cell surface.
C. NRG1-fusion proteins hold the EGF-like domain close to the cell surface, causing uncontrolled juxtacrine signaling. Aberrant NRG1 signaling drives cell proliferation and avoidance of apoptosis via PI3K and RAS dependent signaling and activation of downstream signalling molecules, including RALBP-1, CREB, ELK-1, 4EBP, S6K and NFκB.
D. NRG1-fusion proteins may also drive autocrine signaling via the same PI3K/Ras dependent pathways.
Figure 1B. Common NRG1-fusions (redrawn from Jonna et al 201948). Schematics of the wild-type NRG1 gene (green border) and the most common NRG1-fusions (red border, from 21,858 tumours, of more than 26 different types48). The CD74-NRG1 fusions shown are CD74 exon 6; CD74 (exon 8)-NRG1 fusions were also reported. The dotted line indicates the point at which NRG1 and the fusion partner are joined. The EGF-like region is shown in purple.
The role of NRG1 fusions in cancer
Discovery and incidence of NRG1 fusions
The first NRG1 fusion was reported in 2014 by Fernandez-Cuesta and colleagues.15 This team identified a fusion of CD74 to NRG1 while screening a cohort of 25 EGFR and KRAS wild-type lung adenocarcinoma specimens, using a novel computational data analysis strategy. The NRG1 isoform involved in the fusion belonged to type III and contained a β-type EGF domain, which binds with greater affinity to receptors than the α-type.37 Subsequently, they identified an additional four cases of CD74–NRG1 while screening a cohort of 102 pan-negative adenocarcinomas. All five cases were in never-smoker females with IMA of the lung.
Subsequently, CD74–NRG1 has emerged as the most frequently reported NRG1 fusion,24,43 and most CD74–NRG1 fusions have been reported in patients with lung IMA.15,17,19,44–48
Other NRG1 fusion partners identified in patients with lung cancer include SLC3A2,17,19,48 SDC4,43,45,48 RBPMS,45,48,49 WRN,45 VAMP2,50 ATP1B1,18,21,48ROCK1,43,48 RALGAPA1,51 TNC,48 MDK,48 DIP2B,48 MRPL13,48 DPYSL2,48 PARP8,48 and ITGB1.49 In an analysis of 21,858 tumor specimens that underwent anchored multiplex PCR for targeted RNA sequencing, the prevalence of NRG1 fusions was 0.2%. Of these, CD74 was the most common partner (29%), followed by ATP1B1 (10%), SDC4 (7%), and RBPMS (5%).48
Several other NRG1-fusion partners have also been identified across various solid tumor types,43,48 including POMK (colorectal cancer),41,52 APP (pancreatic ductal adenocarcinoma [PDAC]),21,53 CDH6 (PDAC),21 ATP1B1 (cholangiocarcinoma and PDAC),18,21,53 and CLU (ovarian cancer).54
Proposed pro-tumoral mechanism of NRG1 fusions
Characterization of the CD74–NRG1 fusion protein provided insight into the pro-tumoral effects of this aberration. Type III NRG1 proteins have cytosolic N-termini and membrane-tethered EGF-like domains. In CD74–NRG1 fusions, the CD74 portion is predicted to replace the transmembrane domain of NRG1 III-β3, resulting in a membrane-tethered EGF-like domain. Fernandez-Cuesta and colleagues demonstrated that the EGF-like domain of the fusion proteins was overexpressed on the cell surface.15 Maintenance of the extracellular EGF domain at the cellular surface in CD74–NRG1 fusions promotes activation of the ErbB/PI3K/NF-κB pathway. Subsequently, ErbB3 expression was noted at high levels, and the proteins were phosphorylated, in fusion-positive cases.15 In addition, high ErbB3 expression was significantly associated with NRG1 fusions in a cohort of patients with lung cancer.22 It was hypothesized that CD74–NRG1 signals through induction of ErbB2–ErbB3 heterodimers that activate the PI3K-AKT pathway, as has been shown for wild-type NRG1.15 This was confirmed by transduction of lung cancer cell lines with viruses expressing the CD74–NRG1 fusion. Transduced cells showed increased levels of phosphorylated ErbB2 and ErbB3, AKT and S6K compared with the empty vector control. The presence of phosphorylated ErbB3 and AKT both depended on the presence of the EGF-like domain of CD74–NRG1 in the fusion. The AKT pathway was also stimulated in Ba/F3 cells expressing normal levels of ErbB2 and ErbB3 when they were co-cultured with NIH-3T3 cells expressing CD74–NRG1.15
Other groups have confirmed that NRG1 fusions promote signaling through ErbB2/ErbB3 heterodimers.17,19,43,50 Signaling through EGFR was less important than through ErbB2 and ErbB3.43 The role of ErbB4 remains unclear. It has also been consistently reported in different cancer types, and with different NRG1 fusions, that ErbB2/ErbB3 heterodimers activate signaling through both the PI3K-AKT pathway and the MAPK pathway, with phosphorylation of PI3K, AKT and mTOR being observed, as well as ERK.17,19,43,45,50
In another study, the CD74–NRG1 fusion was shown to confer cancer stem cell (CSC)-like properties, including sphericalization, on immature, progenitor-like cells.55 CSCs are a subgroup of cancer cells that have the ability to self-renew and differentiate into other types of mature cells.24 Murayama and colleagues found that expression of CD74–NRG1 induced signaling through ErbB2-ErbB3 heterodimers, which activated the PI3K-AKT pathway, leading to activation of NF-κB and insulin-like growth factor 2 (IGF2), leading to sphericalization of the cells. The CD74–NRG1 fusion not only induced sphere-forming ability in vitro, but also enhanced the tumor-initiating ability in vivo, suggesting that the fusion gene could be involved in tumor development by inducing CSC properties. The CD74–NRG1 fusion did not stimulate rapid growth of CSCs; however, CSCs are thought to be resistant to stressful conditions, hinting at a potential resistance to conventional chemotherapeutics in tumors expressing this fusion.55
Others have observed the induction of cancer-like properties in cells expressing NRG1 fusions. Shin et al. showed that an SLC3A2–NRG1 fusion induced cancer cell migration via increased phosphorylation of FAK and Src.17 They also found that ErbB2/ErbB3 heterodimers were activated by a soluble factor in a juxtacrine and/or autocrine manner. Dhanasekaran and colleagues found that overexpression of CD74–NRG1 induced epithelial to mesenchymal transition (EMT) as shown by increased VIM and SNAIL protein expression and decreased CDH1 levels. Microarray analysis identified overexpression of EMT markers, SRC and ErbB pathways, and under-expression of cell adhesion markers, supporting an oncogenic phenotype.45
It has been suggested that the fusion partner may influence the localization of NRG1 to the plasma membrane,48 causing it to mimic wild-type NRG1 type III. The NRG1 fusion partners SLC3A2, VAMP2, RBPMS, WNR and SDC4 were predicted to retain the membrane-bound EGF-like domain of the wild-type NRG1 III-β3 form, suggesting they would retain the oncogenic potential of CD74–NRG1.22 Jonna and colleagues identified a small proportion of fusions where exon 2 of NRG1 was spliced to upstream non-coding exons of the fusion partner; this could result in an N-terminal truncated version of NRG1, currently of unknown clinical significance.48
Most NRG1 fusions result in the expression of chimeric proteins (e.g. CD74–NRG1), but a WRN–NRG1 gene fusion was found to result in the overexpression of full-length NRG1 regulated by the WRN gene promoter.45 Fusion of the NRG1 gene downstream (3ʹ) of a 5ʹ partner brings the EGF-like domain-containing exons of this gene under the control of a foreign transcription regulator.
A number of studies have shown that the NRG III-β isoform is involved in the fusion.15,21 Isoforms of NRG1 with the β isoform of the EGF-like domain have higher affinity for the receptors than those with the α isoform.37 It could be hypothesized that the combination of overexpression and the higher affinity EGF-like domain result in greater pro-tumoral potential.
Detection of NRG1 fusions
Until recently, fusion proteins and genes in solid tumors were detected primarily using immunohistochemistry and fluorescence in situ hybridization (FISH) techniques; important advances in gene fusion detection include DNA next-generation sequencing (NGS) and the introduction of targeted gene fusion panels on RNA (Table 1).1,56
Table 1:
The advantages and disadvantages of different assays available for the detection of NRG1 and other gene fusions
| Type of assay | Input | Advantages | Disadvantages |
|---|---|---|---|
| FISH56,75 | DNA | Widely available and used routinely for other fusions; laboratories are familiar with this technique Faster than NGS techniques |
Labor intensive and difficult to interpret (requires diagnostic expertise) Lacks scalability for high-volume multitarget testing Does not provide fusion partner breakpoint precision |
| Anchored multiplex PCR for targeted RNA sequencing (AMP)75 | RNA | Clinical sensitivity and specificity better than standard FISH assays Unlike other PCR methods, AMP enables enrichment of a target region with knowledge of only one of its ends and may be used to assess sequence read complexity based on random start sites |
Quality of archived samples may be compromised Costly |
| Next-generation sequencing of DNA (MSK-IMPACT)43,56 | DNA | Next-generation sequencing generally requires a low quantity of tumor material (low DNA inputs) | Results may not be as reliable as targeted RNA sequencing Deep sequencing is required to detect NRG1 |
| Targeted RNA sequencing (MSK Solid Fusion Assay, Archer FusionPlex)43,56 | RNA | May be more reliable than next-generation sequencing of DNA Not impacted by large intronic regions Requires low RNA inputs |
Long-term storage of samples not recommended (quality of sample may be compromised) |
| IHC22,75 | Proteins (antigens) | Widely available Faster than other techniques Detection of phosphorylated ErbB3 by IHC may potentially be a useful screening method to select for NRG1-positive patients (good correlation with FISH results) |
Relies on availability of good-quality antibodies and on qualitative scoring Does not provide fusion partner breakpoint precision |
FISH = fluorescent in situ hybridization; IHC = immunohistochemistry; NGS = next generation sequencing; PCR = polymerase chain reaction
In a large analysis of 17,485 solid tumors, detection of NRG1 fusions was undertaken using DNA NGS (MSK-IMPACT) for all tumors and targeted RNA sequencing (MSK-Solid Fusion Assay, Archer FusionPlex) for certain tumours.43 Targeted RNA sequencing detected NRG1 fusions in several KRAS wild-type IMAs and one pancreatic adenocarcinoma, which were not previously detected by next-generation DNA sequencing alone.43 These findings suggest that NRG1 fusions may be missed due to the complexity and diversity of fusion architecture and the large intronic regions characteristic of NRG1. Overall, RNA sequencing may be more reliable than DNA NGS at detecting NRG1 fusions. The unidirectional targeted approach of RNA sequencing allows the detection of unknown fusion partners, which will be particularly useful in light of the variability of NRG1 fusion partners.43 Also of interest are findings from a whole-exome sequencing and RNA-seq analysis of Japanese patients with lung adenocarcinoma who were negative for the driver oncogenes defined by conventional laboratory testing, which identified CD74-NRG2α as an actionable novel fusion.57
Prevalence and characteristics of patients with NRG1 fusions
In general, NRG1 fusions are rare across different types of cancer, typically occurring in <1% in most reported series (Figure 2).15,19,43,48,58 For example, using prospective DNA NGS, Drilon and colleagues detected NRG1 rearrangements in 3 of 2,079 patients (0.14%) with lung adenocarcinomas, 1 of 791 patients (0.13%) with pancreatic adenocarcinoma, and 1 of 2,703 patients (0.04%) with breast carcinoma.43
Figure 2: Incidence of NRG1 fusions in cancer.
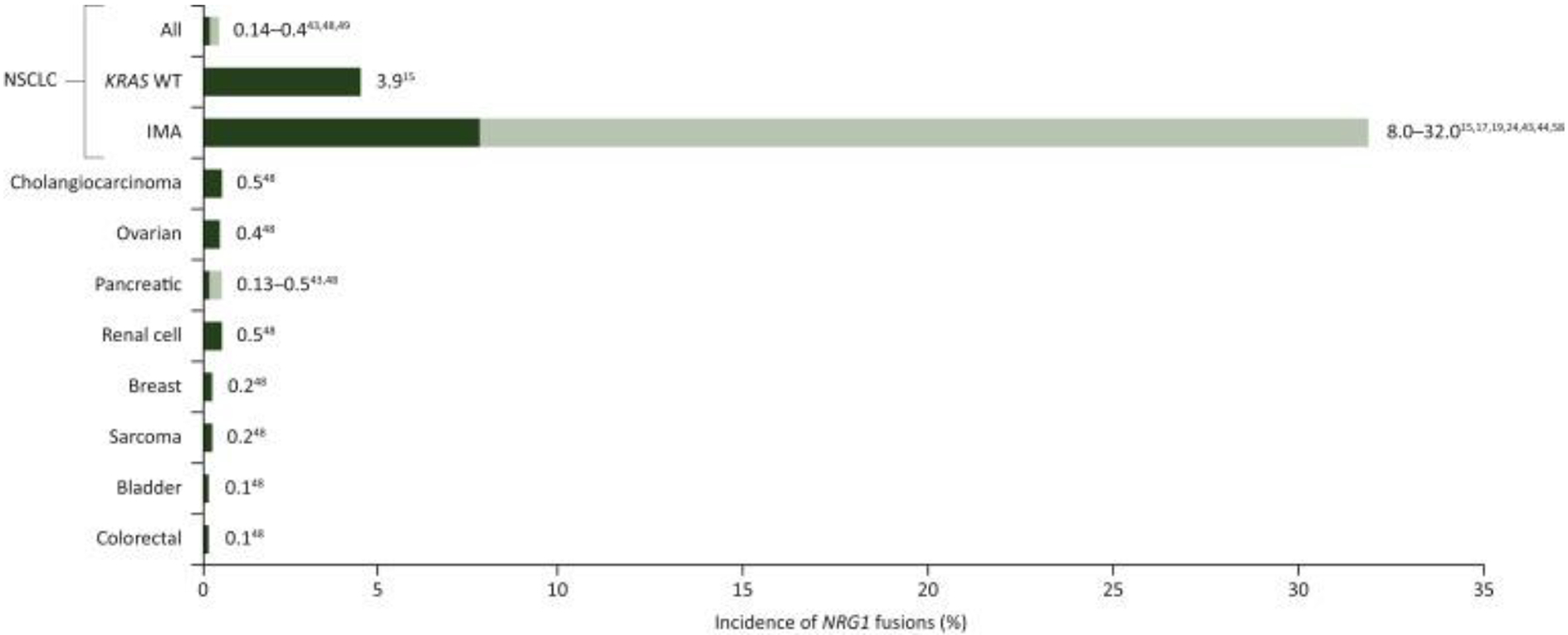
CRC = colorectal cancer; IMA = invasive mucinous adenocarcinoma; KRAS WT = Kirsten rat sarcoma viral oncogene homolog wild-type; NSCLC = non-small cell lung cancer; RCC = renal cell carcinoma
Note, for pancreatic cancer, where reported (Jonna et al. 201948), all cases were KRAS WT
Subsequent analysis of RNA-seq data for 8,984 tumors from The Cancer Genome Atlas identified NRG1 rearrangements across various tumor histologies, as follows: breast (AKAP13-NRG1: 0.01%); head and neck cancer (THBS1-NRG1 and PDE7A-NRG1: 0.49%); lung adenocarcinoma (SDC4-NRG1: 0.22%); renal clear cell carcinoma (PCM1-NRG1: 0.19%); squamous cell lung cancer (THAP7-NRG1 and SMAD4-NRG1: 0.21%); ovarian cancer (RAB3IL1-NRG1: 0.24%); pancreatic cancer (ATP1B1-NRG1: 0.55%); uterine carcinosarcoma (NRG1-PMEPA1: 1.75%), and prostate cancer (NRG1-STMN2: 0.3%).43
Jonna and colleagues48 reported on the incidence of NRG1 fusions in 21,858 solid tumor specimens that underwent RNA sequencing, and found that 0.2% harbored an NRG1 fusion. Although the greatest incidence of NRG1 fusions was in NSCLC (25 of 41 cases in total), this represented only 0.3% of NSCLC cases tested. Other NRG1-postive tumor types included gallbladder cancer, renal cell carcinoma, bladder cancer, ovarian cancer, pancreatic cancer, breast cancer, neuroendocrine tumor, sarcoma, and colorectal cancer. As previously noted, NRG1 fusions do appear to be more common in IMA and have been reported in 5 of 72 IMA cases (7%) using anchored multiplex PCR to perform NGS,20 6 of 90 IMA cases (7%) using whole-transcriptome sequencing,19 16 of 59 IMA cases (27%) using targeted sequencing and RT-PCR,17 and 16 of 51 IMA cases (31%) using FISH.22 In Drilon and colleagues’ study,43 in patients with KRAS wild-type IMA, NRG1 fusions were detected in 4 of 36 patients (11%) using targeted RNA sequencing.
KRAS mutations are the most frequent oncogenic driver mutations in IMA, occurring in 28‒87% of cases.59 It was thought that NRG1 fusions were mutually exclusive with KRAS mutations.19–21 However, a growing number of studies have shown this may not be the case. One series of 16 IMA patients with NRG1 fusions identified KRAS codon 12 mutations in nine cases (56%).17 Trombetta et al. found concurrent NRG1 rearrangements with KRAS mutations in six of 18 cases (33%).22 With respect to PDAC, it appears that almost all patients harboring NRG1 fusions are KRAS wild-type, which provides a useful screening strategy for identifying PDAC patients with NRG1 fusions.21,53,60
Initial reports of NRG1 fusions in patients with IMA indicated that most of the patients were female and never smokers,19 and much of the work was done in an East Asian population.15 However, a recent evaluation of a cohort of 85 Caucasian lung cancer patients found NRG1 rearrangements in 31% of patients with IMA and 3% of those with non-IMA.22 All of the IMA cases also lacked EGFR mutations and ALK rearrangements.22 These findings indicate that NRG1 fusions are important drivers of IMA in both Asian and Caucasian populations. Moreover, recent results from the international NRG1 registry indicate a substantial proportion (≈20%) of NRG1-positive NSCLC are non-mucinous adenocarcinomas, and there were also cases of non-adenocarcinoma NSCLC.61
Prognostic significance of NRG1 fusions
The prognostic value of NRG1 fusions in IMA is unclear. As noted earlier, NRG1 fusions may lead to CSC formation;55 and CSCs are known to be related to tumor recurrence and metastasis, chemo-resistance and poor prognosis.24
In an evaluation of survival in a series of 59 patients with IMA, patients harboring tumors with NRG1 fusions had a significantly shorter overall survival (OS) than those without NRG1 fusions (median 51.9 months vs. not reached; p=0.019). This was also true for patients with stage I disease (median 48.1 months vs not reached; p=0.009).17 In contrast, Shim and colleagues reported that OS was no worse in patients with mucinous compared with non-mucinous lung adenocarcinoma, and that there was no difference in prognosis for patients with gene fusions.20
Potential treatments targeting the NRG1/ErbB pathway
Preclinical evidence of ErbB inhibition activity in NRG1-positive models
As NRG1 proteins are ligands of ErbB receptors,21,62 ErbB-targeted treatments, such as monoclonal antibodies GSK2849330 and zenocutuzumab (MCLA-128) and small molecules afatinib, tarloxotinib, lapatinib, erlotinib and neratinib, are of particular interest. For example, afatinib downregulates ErbB signaling by binding to the kinase domains of EGFR, ErbB2, and ErbB4 and irreversibly inhibiting tyrosine kinase autophosphorylation.63 In a preclinical model in which EFM-19 cells were exposed to conditioned media from human lung cancer cells expressing exogenous CD74-NRG1 fusion protein, afatinib and lapatinib suppressed ErbB2, ErbB3, and ERK phosphorylation, indicating downregulation of ErbB signaling.19 Anti-tumor activity and downstream ErbB signaling inhibition was also demonstrated with the anti-ErbB3 monoclonal antibody GSK2849330, and to a lesser extent with afatinib, in an NRG1-rearranged patient-derived xenograft model.43 Preclinical studies have also demonstrated efficacy with tarloxotinib, a hypoxia-activated pan-ErbB kinase inhibitor in NRG1 fusion-positive cell lines and patient-derived xenografts.64 Initial anti-tumor activity has also been demonstrated with zenocutuzumab (MCLA-128).65 In cell lines with aberrant NRG1-expression, exposure to GSK2849330, afatinib, erlotinib, and neratinib significantly inhibited growth and suppressed ErbB2, ErbB3, and AKT phosphorylation.43
Studies using NRG1 fusions from patient cases have shown that afatinib reduced phosphorylation of downstream pathways when cells were treated with it.19,21 Although, in another study, afatinib was not effective at preventing signaling in cells carrying NRG1 fusions; whereas, an anti-ErbB3 monoclonal antibody, GSK2849330, was.43
As part of the transcriptomic analysis of 5 patients with NRG1 fusions (1 lung cancer, 1 cholangiocarcinoma and 3 PDAC), 4 had extremely high levels of RNA expression ranging between the 93rd and 100th percentile of expression compared to NRG1 levels seen across cancers in the TCGA dataset.18,53 Moderately high RNA expression levels were also observed in ErbB2 and ErbB3 for all 5 patients, and low to normal levels were seen in EGFR and ErbB.
Clinical evidence of ErbB inhibition activity in NRG1-positive patients
Clinical evidence of response to ErbB inhibition in patients with NRG1 fusion-positive tumors has been seen with several different agents (Table 2). To date, the most frequently reported agent showing responses in this setting has been afatinib, although reports of its efficacy in NRG1 fusion-positive tumors is currently limited to case reports in patients with IMA, PDAC, lung adenocarcinoma, ovarian cancer and cholangiocarcinoma (Table 2). Partial responses were achieved for up to 10 months in patients with IMA, and up to 5.5 months in patients with PDAC (Table 2). As well as the case reports discussed here, a global, multicenter registry has been set up to identify patients with NRG1 fusion-positive NSCLCs.61 Eighty patients have been included on the registry so far, including 12 treated with afatinib. Four of the 12 patients had an objective response [partial response (PR) or stable disease (SD)] to afatinib treatment, with a median progression-free survival of 3.5 months (range 0.6–16.5 months).61
Table 2:
Reported cases of ErbB-targeted treatment in patients with NRG1 fusion-positive tumors
| Patient | Treatment | Tumor type | NRG1 fusion partner | Best response | Duration of response (months) | Reference |
|---|---|---|---|---|---|---|
| 1 | Afatiniba | Lung adenocarcinoma | SLC3A2 | PR | 12 | Gay et al. 201776 |
| 2 | Afatiniba | Lung adenocarcinoma | SDC4 | PR | 12 | Jones et al. 201718 |
| 3 | Afatiniba | IMA | CD74 | PR | 10 | Gay et al. 201776 |
| 4 | Afatiniba | IMA | CD74 | PR | 6.5 | Cheema et al. 201777 |
| 5 | Afatiniba | IMA | CD74 | SD | 3 | Drilon et al. 201843 |
| 6 | Afatiniba | IMA | CD74 | PD | - | Drilon et al. 201843 |
| 7 | Afatiniba | IMA | SDC4 | PD | - | Drilon et al. 201843 |
| 8 | Afatiniba (dose reduced to 30 mg/day due to toxicity) | PDAC | ATP1B1 | PR | 5.5 | Jones et al. 201953 |
| 9 | Afatiniba 30 mg/day | PDAC | APP | PR | 5 (ongoing) | Jones et al. 201953 |
| 10 | Afatiniba (dose reduced due to cutaneous AEs) | PDAC | ATP1B1 | PR | 3 | Heining et al. 201821 |
| 11 | Afatiniba monotherapy followed by trastuzumab and pertuzumab | Ovarian cancer | CLU | SD | >36 | Murumagi et al. 201954 |
| 12 | Afatiniba (dose reduced to 30 mg/day due to rash/diarrhea) | Cholangiocarcinoma | ATP1B1 | PR | 8 | Jones et al. 201718 |
| 13 | GSK2849330b 30 mg/kg IV q2wk | IMA | CD74 | PR | 19 | Drilon et al. 201843 |
| 14 | Lumretuzumabc 800 mg IV q2wk plus erlotinib | IMA | SLC3A2 | SD | 4 | Han et al. 201766 |
| 15 | Lumretuzumabc 800 mg IV q2wk plus erlotinib | IMA | SLC3A2 | SD | 4 | Han et al. 201766 |
| 16 | Zenocutuzumabd (MCLA-128) 750 mg IV q2wk | PDAC | ATP1B1 | PR | 7+ (ongoing) | Schram et al. 201965 |
| 17 | Zenocutuzumabd (MCLA-128) 750 mg IV q2wk | PDAC | ATP1B1 | SD | 7+ (ongoing) | Schram et al. 201965 |
| 18 | Zenocutuzumabd (MCLA-128) 750 mg IV q2wk | NSCLC | CD74 | PR | 4.5+ (ongoing) | Schram et al. 201965 |
| 19 | Pertuzumab and erlotinib | PDAC | SARAF, CDH6 | PR | 3 | Heining et al. 201821 |
Oral afatinib, 40 mg/day unless stated otherwise;
GSK2849330 (anti-ErbB3 monoclonal antibody) has undergone a phase 1 trial (NCT01966445) but does not appear to be undergoing further development (clinicaltrials.gov);
Lumretuzumab is an anti-ErbB3 monoclonal antibody;
Zenocutuzumab (MCLA-128) is a bispecific humanized monoclonal antibody.
AEs = adverse events; APP = amyloid beta precursor protein; ATP1B1 = ATPase Na+/K+ Transporting Subunit Beta 1; CDH6 = Cadherin6; CD74 = CD74 molecule (Major Histocompatibility Complex, Class II Invariant Chain); CLU = clusterin; IMA = invasive mucinous adenocarcinoma; IV = intravenous; NSCLC = non-small-cell lung cancer; PD = progressive disease; PDAC = pancreatic ductal adenocarcinoma; PR = partial response; q2wk = every 2 weeks; SD = stable disease; SARAF = Store-Operated Calcium Entry Associated Regulatory Factor; SDC4 = syndecan 4; SLC3A2 = Solute Carrier Family 3 Member 2
By contrast, some case reports have shown primary resistance to afatinib in patients with NRG1 fusions. Drilon and colleagues investigated afatinib treatment in four patients with NRG1-rearranged lung cancers, and found progressive disease (PD) shortly after starting treatment. Three of the four patients had CD74–NRG1 fusions and one had an SDC4–NRG1 fusion. One of the patients had previously been treated with the anti-ErbB3 molecule GSK2849330 (no longer in development), and had developed PD after a PR lasting 19 months.43
These response discrepancies highlight the need for a global data and treatment sharing platform to gather as much data as possible on these uncommon drivers. In this context, a global multicenter registry of thoracic oncologists was launched to improve our understanding of clinicopathologic characteristics and response to treatment of patients with NRG1 fusion-driven NSCLC.61
Other case reports or basket studies in patients with NRG1 fusion-driven solid tumors include the anti-ErbB3 agent lumretuzumab (two patients with SD for 4 months)66 and the bispecific humanized monoclonal antibody zenocutuzumab (MCLA-128) (three patients with ongoing responses: 7 months PR, 4.5 months PR, and 7 months SD; currently in clinical phase 1/2)65,67 (Table 2). Other potential targeted treatments against NRG1 fusions include the ErbB2 inhibitors lapatinib, trastuzumab, and pertuzumab, the pan-HER inhibitor tarloxotinib, as well as the ErbB3 inhibitors patritumab (U3–1287; AMG-888), seribantumab (MM-121), AV-203, 9F7-F11, and LJM716. However, only preclinical or phase 1 (pharmacokinetic/pharmacodynamic) data are currently available for these agents.68–74
Association of NRG1 fusions with resistance to ALK inhibitors
Activation of NRG1 fusion-driven pathways may also act as a mechanism of resistance following treatment with certain TKIs, including ALK inhibitors. A novel NRG1 fusion (RALGAPA1–NRG1) was detected in an ALK+ patient who had progressed on alectinib. Cells engineered to express this fusion were resistant to the ALK inhibitor crizotinib, but sensitive to afatinib.51 In addition, increased phosphorylated ErbB3 was observed in these engineered cells. The induction of the NRG1 fusion in these cells led to the loss of phosphorylation of SHP2, which is known to be a signaling adaptor for ALK. Analysis of samples from the patient found that signaling via both the ALK and NRG1 fusions was functional. Further analysis of earlier tumor specimens from this patient showed that the RALGAPA1–NRG1 fusion had coexisted with the ALK fusion, and so was not a de novo mechanism of resistance but may have contributed to the short response to crizotinib.
Key points/conclusions
NRG1 fusions are present in multiple cancer types, and in a high proportion of lung IMA. Although these gene fusions are relatively uncommon in most cancer types, they are detectable and targetable vulnerabilities, and recent NRG1 registry data indicate that a substantial proportion (≈20%) of NRG1 fusion-positive NSCLC cases are non-mucinous adenocarcinomas. Other NRG1-positive tumor types include pancreatic cancer (e.g. KRAS wild-type PDAC), gallbladder cancer, renal cell carcinoma, bladder cancer, ovarian cancer, breast cancer, neuroendocrine tumor, sarcoma, and colorectal cancer.
Development of new drugs for rare diseases is challenging, but evaluation of drugs already approved for other indications is a pragmatic option. In the case of NRG1 fusion-driven tumors, existing ErbB-targeted treatments, have potential as targeted therapies in tumors harboring NRG1 fusions. Responses have been reported with afatinib in both IMA and gastrointestinal tumors harboring NRG1 fusions. Future studies to evaluate the efficacy of various ErbB-targeted treatments in a larger number of patients with NRG1 fusion-driven tumors may be best suited to a tumor agnostic real-world analysis or basket trials where feasible.
This approach is ongoing in two prospective studies investigating already licensed drugs in patients with potentially actionable genetic alterations. The Drug Rediscovery Protocol trial (DRUP; NCT02925234) is ongoing in the Netherlands and will enroll around 400 participants with any of more than ten targetable tumor profiles. Similarly, the Targeted Agent and Profiling Utilization Registry study (TAPUR; NCT02693535) is ongoing in the US and will enroll over 3000 patients with any of 13 targetable tumor profiles. Other prospective studies exploring novel agents are also underway, including a phase 2 trial evaluating the clinical activity of tarloxotinib in patients with NSCLC harboring an EGFR Exon 20 insertion or HER2-activating mutation and other advanced solid tumors with NRG1/ERBB family gene fusions (NCT03805841), and a phase 1/2 trial of zenocutuzumab (MCLA-128) in patients with solid tumors harboring an NRG1 fusion (NCT02912949).
As the implementation of more broad spectrum genomic and transcriptomic sequencing approaches becomes more common, the number of niche opportunities for targeting cancers expands. The growing number of targetable cancer-driving fusions is an example of the changing landscape of cancer therapy. While tumors with NRG1 fusions do not represent a substantial proportion of cancers overall, the emerging evidence supports the use of existing anticancer agents effectively repurposed to target these cancer drivers.
Highlights:
NRG1 fusions result in ErbB-mediated pathway activation and present a rational therapeutic target
ErbB-targeted treatments, such as afatinib, can be effective therapeutic options in tumors harboring NRG1 fusions
The incidence of NRG1 fusion-driven tumors is low, occurring in <1% of cancers, but is highest (≈10‒30%) in IMA
Acknowledgments
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Christina Jennings and Greg Plosker of GeoMed, an Ashfield company, part of UDG Healthcare plc during the preparation of this article.
Funding:
This work was supported by Boehringer Ingelheim. No grant number is applicable.
Disclosures
JL reports receipt of honoraria for academic/accredited talks from Roche and AstraZeneca; membership of an advisory board and consultancy for AstraZeneca, Boehringer Ingelheim, Roche, Pfizer, and Takeda; and receipt of research grants (funds to institution) from AstraZeneca, Roche, and Eli Lilly. SVL reports membership of an advisory board and consultancy for AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Catalyst, Celgene, G1 Therapeutics, Genentech/Roche, Guardant Health, Janssen, Eli Lilly, Loxo, MSD, Pfizer, PharmaMar, Regeneron, and Takeda; and receipt of research grants (funds to institution) from Alkermes, AstraZeneca, Bayer, Blueprint, Bristol-Myers Squibb, Corvus, Genentech, Eli Lilly, Lycera, Merck, Merus, Molecular Partners, Pfizer, Rain, RAPT, Spectrum, and Turning Point Therapeutics. KT reports employment with Foundation Medicine; receipt of honoraria from BMS and Merck; and receipt of research grants from AstraZeneca. PC reports membership of an advisory board and consultancy for Roche, Pfizer, AstraZeneca, Novartis, Merck, Takeda, and Bristol-Myers Squibb; and receipt of honoraria from Boehringer Ingelheim, Roche, Pfizer, Novartis, Merck, Takeda, and AstraZeneca. JC reports consulting/advisory relationship with AstraZeneca, Boehringer Ingelheim, BMS, Eli Lilly, Novartis, MSD, Pfizer, Roche, and Takeda; and receipt of research funding from AstraZeneca, Boehringer Ingelheim, Pfizer, and Novartis. MRJ reports employment with QIAGEN Inc. AD reports receipt of honoraria from and membership of an advisory board for Ignyta/Genentech/Roche, Loxo/Bayer/Eli Lilly, Takeda, Ariad, Millenium, TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Helsinn, Beigene, BergenBio, Hengrui Therapeutics, Exelixis, Tyra Biosciences, Verastem, MORE Health, and Abbvie; associated research grants (funds to institution) from Pfizer, Exelixis, GlaxoSmithKline, Teva, Taiho, and PharmaMar; research grants from Foundation Medicine; royalties from Wolters Kluwer; CME honoraria from Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, and Research to Practice; and other fees from Merck (food/beverage), Puma (food/beverage), Merus, and Boehringer Ingelheim. AC reports employment with Boehringer Ingelheim. SG reports employment with AstraZeneca as of May 4, 2020. FS reports employment with Boehringer Ingelheim RCV GmbH & Co KG. MD reports membership of an advisory council or committee for Roche, BMS, Nanostring, MSD, Astra Zeneca, Abbvie, Takeda, Boehringer Ingelheim, Blueprint, Merus, and Pfizer; consulting fees from Roche, BMS, MSD, AstraZeneca, Abbvie, Takeda, Boehringer Ingelheim, and Pfizer; and receipt of research grants from Novartis, Nanostring, and Blueprint. CH and RFS have declared no conflicts of interest.
References
- 1.Latysheva NS, Babu MM. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016;44:4487–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soverini S, Mancini M, Bavaro L, et al. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer. 2018;17:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14:735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh VK, Coumar MS. Chronic myeloid leukemia: existing therapeutic options and strategies to overcome drug resistance. Mini Rev Med Chem. 2019;19:333–345. [DOI] [PubMed] [Google Scholar]
- 5.Duruisseaux M, Besse B, Cadranel J, et al. Overall survival with crizotinib and next-generation ALK inhibitors in ALK-positive non-small-cell lung cancer (IFCT-1302 CLINALK): a French nationwide cohort retrospective study. Oncotarget. 2017;8:21903–21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solomon BJ, Kim DW, Wu YL, et al. Final overall survival analysis from a study comparing first-line crizotinib versus chemotherapy in ALK-mutation-positive non-small-cell lung cancer. J Clin Oncol. 2018;36:2251–2258. [DOI] [PubMed] [Google Scholar]
- 7.Ross JS, Ali SM, Fasan O, et al. ALK fusions in a wide variety of tumor types respond to anti-ALK targeted therapy. Oncologist. 2017;22:1444–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossi G, Jocolle G, Conti A, et al. Detection of ROS1 rearrangement in non-small cell lung cancer: current and future perspectives. Lung Canc Targ Ther. 2017;8:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bronte G, Ulivi P, Verlicchi A, et al. Targeting RET-rearranged non-small-cell lung cancer: future prospects. Lung Cancer (Auckl). 2019;10:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subbiah V, Velcheti V, Tuch BB, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018;29:1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. 2016;1:e000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsiao SJ, Zehir A, Sireci AN, Aisner DL. Detection of tumor NTRK gene fusions to identify patients who may benefit from tyrosine kinase (TRK) inhibitor therapy. J Mol Diagn. 2019;21:553–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.FDA. FDA approves entrectinib for NTRK solid tumors and ROS-1 NSCLC. Available at https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc. 2019.
- 14.Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15:731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Cuesta L, Plenker D, Osada H, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014;4:415–422. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Cuesta L, Thomas RK. Molecular pathways: targeting NRG1 fusions in lung cancer. Clin Cancer Res. 2015;21:1989–1994. [DOI] [PubMed] [Google Scholar]
- 17.Shin DH, Lee D, Hong DW, et al. Oncogenic function and clinical implications of SLC3A2-NRG1 fusion in invasive mucinous adenocarcinoma of the lung. Oncotarget. 2016;7:69450–69465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones MR, Lim H, Shen Y, et al. Successful targeting of the NRG1 pathway indicates novel treatment strategy for metastatic cancer. Ann Oncol. 2017;28:3092–3097. [DOI] [PubMed] [Google Scholar]
- 19.Nakaoku T, Tsuta K, Ichikawa H, et al. Druggable oncogene fusions in invasive mucinous lung adenocarcinoma. Clin Cancer Res. 2014;20:3087–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shim HS, Kenudson M, Zheng Z, et al. Unique genetic and survival characteristics of invasive mucinous adenocarcinoma of the lung. J Thorac Oncol. 2015;10:1156–1162. [DOI] [PubMed] [Google Scholar]
- 21.Heining C, Horak P, Uhrig S, et al. NRG1 fusions in KRAS wild-type pancreatic cancer. Cancer Discov. 2018;8:1087–1095. [DOI] [PubMed] [Google Scholar]
- 22.Trombetta D, Graziano P, Scarpa A, et al. Frequent NRG1 fusions in Caucasian pulmonary mucinous adenocarcinoma predicted by Phospho-ErbB3 expression. Oncotarget. 2018;9:9661–9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. [DOI] [PubMed] [Google Scholar]
- 24.Trombetta D, Rossi A, Fabrizio FP, et al. NRG1-ErbB lost in translation: a new paradigm for lung cancer? Curr Med Chem. 2017;24:4213–4228. [DOI] [PubMed] [Google Scholar]
- 25.Tan W, Wang Y, Gold B, et al. Molecular cloning of a brain-specific, developmentally regulated neuregulin 1 (NRG1) isoform and identification of a functional promoter variant associated with schizophrenia. J Biol Chem. 2007;282:24343–24351. [DOI] [PubMed] [Google Scholar]
- 26.Steinthorsdottir V, Stefansson H, Ghosh S, et al. Multiple novel transcription initiation sites for NRG1. Gene. 2004;342:97–105. [DOI] [PubMed] [Google Scholar]
- 27.Meyer D, Yamaai T, Garratt A, et al. Isoform-specific expression and function of neuregulin. Development. 1997;124:3575–3586. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Bates R, Yin DM, et al. Specific regulation of NRG1 isoform expression by neuronal activity. J Neurosci. 2011;31:8491–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Falls DL, Rosen KM, Corfas G, et al. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell. 1993;72:801–815. [DOI] [PubMed] [Google Scholar]
- 30.Peles E, Bacus SS, Koski RA, et al. Isolation of the neu/HER-2 stimulatory ligand: a 44 kd glycoprotein that induces differentiation of mammary tumor cells. Cell. 1992;69:205–216. [DOI] [PubMed] [Google Scholar]
- 31.Holmes WE, Sliwkowski MX, Akita RW, et al. Identification of heregulin, a specific activator of p185erbB2. Science. 1992;256:1205–1210. [DOI] [PubMed] [Google Scholar]
- 32.Lemke GE, Brockes JP. Identification and purification of glial growth factor. J Neurosci. 1984;4:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchionni MA, Goodearl AD, Chen MS, et al. Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature. 1993;362:312–318. [DOI] [PubMed] [Google Scholar]
- 34.Kim HG, Lee CK, Cho SM, et al. Neuregulin 1 up-regulates the expression of nicotinic acetylcholine receptors through the ErbB2/ErbB3-PI3K-MAPK signaling cascade in adult autonomic ganglion neurons. J Neurochem. 2013;124:502–513. [DOI] [PubMed] [Google Scholar]
- 35.Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JY, Miller SJ, Falls DL. The N-terminal region of neuregulin isoforms determines the accumulation of cell surface and released neuregulin ectodomain. J Biol Chem. 2001;276:2841–2851. [DOI] [PubMed] [Google Scholar]
- 37.Tzahar E, Levkowitz G, Karunagaran D, et al. ErbB-3 and ErbB-4 function as the respective low and high affinity receptors of all Neu differentiation factor/heregulin isoforms. J Biol Chem. 1994;269:25226–25233. [PubMed] [Google Scholar]
- 38.Sliwkowski MX, Schaefer G, Akita RW, et al. Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J Biol Chem. 1994;269:14661–14665. [PubMed] [Google Scholar]
- 39.Shin DH, Jo JY, Han JY. Dual targeting of ERBB2/ERBB3 for the treatment of SLC3A2-NRG1-mediated lung cancer. Mol Cancer Ther. 2018;17:2024–2033. [DOI] [PubMed] [Google Scholar]
- 40.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. Embo J. 1997;16:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karunagaran D, Tzahar E, Beerli RR, et al. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. EMBO J. 1996;15:254–264. [PMC free article] [PubMed] [Google Scholar]
- 42.Muthuswamy SK, Gilman M, Brugge JS. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol Cell Biol. 1999;19:6845–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drilon A, Somwar R, Mangatt BP, et al. Response to ERBB3-directed targeted therapy in NRG1-rearranged cancers. Cancer Discov. 2018;8:686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gow CH, Wu SG, Chang YL, Shih JY. Multidriver mutation analysis in pulmonary mucinous adenocarcinoma in Taiwan: identification of a rare CD74-NRG1 translocation case. Med Oncol. 2014;31:34. [DOI] [PubMed] [Google Scholar]
- 45.Dhanasekaran SM, Balbin OA, Chen G, et al. Transcriptome meta-analysis of lung cancer reveals recurrent aberrations in NRG1 and Hippo pathway genes. Nat Commun. 2014;5:5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang R, Zhang Y, Pan Y, et al. Comprehensive investigation of oncogenic driver mutations in Chinese non-small cell lung cancer patients. Oncotarget. 2015;6:34300–34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duruisseaux M, McLeer-Florin A, Antoine M, et al. NRG1 fusion in a French cohort of invasive mucinous lung adenocarcinoma. Cancer Med. 2016;5:3579–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jonna S, Feldman RA, Swensen J, et al. Detection of NRG1 gene fusions in solid tumors. Clin Cancer Res. 2019;25:4966–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan Y, Zhang Y, Ye T, et al. Detection of Novel NRG1, EGFR, and MET fusions in lung adenocarcinomas in the Chinese population. J Thorac Oncol. 2019;14:2003–2008. [DOI] [PubMed] [Google Scholar]
- 50.Jung Y, Yong S, Kim P, et al. VAMP2-NRG1 fusion gene is a novel oncogenic driver of non-small-cell lung adenocarcinoma. J Thorac Oncol. 2015;10:1107–1111. [DOI] [PubMed] [Google Scholar]
- 51.McCoach CE, Le AT, Gowan K, et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non-small cell lung cancer. Clin Cancer Res. 2018;24:3334–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weinberg B, Renouf DJ, Lim H, et al. NRG1-fusion positive gastrointestinal tumours: afatinib as a novel potential treatment option. Ann Oncol. 2019;30(Suppl4):iv80. [Google Scholar]
- 53.Jones MR, Williamson LM, Topham JT, et al. NRG1 gene fusions are recurrent, clinically actionable gene rearrangements in KRAS wild-type pancreatic ductal adenocarcinoma. Clin Cancer Res. 2019;25:4674–4681. [DOI] [PubMed] [Google Scholar]
- 54.Murumagi A, Ungureanu D, Khan S, et al. Clinical implementation of precision systems oncology in the treatment of ovarian cancer based on ex-vivo drug testing and molecular profiling. Cancer Res. 2019;79(Suppl13):Abstract 2945. [Google Scholar]
- 55.Murayama T, Nakaoku T, Enari M, et al. Oncogenic fusion gene CD74-NRG1 confers cancer stem cell-like properties in lung cancer through a IGF2 autocrine/paracrine circuit. Cancer Res. 2016;76:974–983. [DOI] [PubMed] [Google Scholar]
- 56.Garinet S, Laurent-Puig P, Blons H, Oudart JB. Current and future molecular testing in NSCLC, what can we expect from new sequencing technologies? J Clin Med. 2018;7:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohsaka S, Hayashi T, Nagano M, et al. Identification of novel CD74-NRG2alpha fusion from comprehensive profiling of lung adenocarcinoma in Japanese never or light smokers. J Thorac Oncol. 2020. [DOI] [PubMed] [Google Scholar]
- 58.Trombetta D, Graziano P, Sparaneo A, et al. Recurrent NRG1 rearrangements in Caucasian pulmonary mucinous adenocarcinoma: results from an Italian multi-center cohort. Proceedings: AACR Annual Meeting 2019; March 29-April 3, 2019; Atlanta, GA. Cancer Res. 2019;79(Suppl13):Abstract 4887. [Google Scholar]
- 59.Nakagomi T, Goto T, Hirotsu Y, et al. Genomic characteristics of invasive mucinous adenocarcinomas of the lung and potential therapeutic targets of B7-H3. Cancers (Basel). 2018;10:E478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aguirre AJ. Oncogenic NRG1 fusions: a new hope for targeted therapy in pancreatic cancer. Clin Cancer Res. 2019;25:4589–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duruisseaux M, Liu SV, Han JY, et al. NRG1 fusion-positive lung cancers: Clinicopathologic profile and treatment outcomes from a global multicenter registry. J Clin Oncol. 2019;37:Abstract 9081. [Google Scholar]
- 62.Wilson FH, Politi K. ERBB signaling interrupted: targeting ligand-induced pathway activation. Cancer Discov. 2018;8:676–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ingelheim B. Afatinib (Gilotrif®) US prescribing information. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/201292s014lbl.pdf. 2018.
- 64.Tirunagaru VG, Estrada-Bernal A, Yu H, et al. Tarloxotinib exhibits potent activity in NRG1 fusion and rearranged cancers. Cancer Res. 2019;79(Suppl13):Abstract 2202. [Google Scholar]
- 65.Schram AM. Clinical proof of concept for MCLA-128, a bispecific HER2/3 antibody therapy, in NRG1 fusion-positive cancers. Mol Canc Ther. 2019;18:Abstract PR02. [Google Scholar]
- 66.Han JY, Lim KY, Kim JY, et al. EGFR and HER3 inhibition - a novel therapy for invasive mucinous non-small cell lung cancer harboring an NRG1 fusion gene. J Thorac Oncol. 2017;12:S1274–S1275. [Google Scholar]
- 67.Schram AM, Drilon A, Macarulla Mercade T, et al. A phase 2 basket study of MCLA-128, a bispecific antibody targeting the HER3 pathway, in NRG1 fusion-positive advanced solid tumors. Ann Oncol. 2019;30(Suppl5):Abstract 685TiP. [Google Scholar]
- 68.Le Clorennec C, Bazin H, Dubreuil O, et al. Neuregulin 1 allosterically enhances the antitumor effects of the noncompeting anti-HER3 antibody 9F7-F11 by increasing its binding to HER3. Mol Cancer Ther. 2017;16:1312–1323. [DOI] [PubMed] [Google Scholar]
- 69.Meetze K, Vincent S, Tyler S, et al. Neuregulin 1 expression is a predictive biomarker for response to AV-203, an ERBB3 inhibitory antibody, in human tumor models. Clin Cancer Res. 2015;21:1106–1114. [DOI] [PubMed] [Google Scholar]
- 70.Garner AP, Bialucha CU, Sprague ER, et al. An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or neuregulin. Cancer Res. 2013;73:6024–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang J, Wang S, Lyu H, et al. The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol Cancer. 2013;12:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meulendijks D, Jacob W, Martinez-Garcia M, et al. First-in-human phase i study of lumretuzumab, a glycoengineered humanized anti-HER3 monoclonal antibody, in patients with metastatic or advanced HER3-positive solid tumors. Clin Cancer Res. 2016;22:877–885. [DOI] [PubMed] [Google Scholar]
- 73.Wilson TR, Lee DY, Berry L, et al. Neuregulin-1-mediated autocrine signaling underlies sensitivity to HER2 kinase inhibitors in a subset of human cancers. Cancer Cell. 2011;20:158–172. [DOI] [PubMed] [Google Scholar]
- 74.Tirunagaru VG, Estrada-Bernal A, Yu H, et al. Tarloxotinib exhibits potent activity in NRG1 fusion and rearranged cancers. Cancer Res. 2019;79(Suppl13):Abstract 2202. [Google Scholar]
- 75.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. [DOI] [PubMed] [Google Scholar]
- 76.Gay ND, Wang Y, Beadling C, et al. Durable response to afatinib in lung adenocarcinoma harboring NRG1 gene fusions. J Thorac Oncol. 2017;12:e107–e110. [DOI] [PubMed] [Google Scholar]
- 77.Cheema PK, Doherty M, Tsao MS. A case of invasive mucinous pulmonary adenocarcinoma with a CD74-NRG1 fusion protein targeted with afatinib. J Thorac Oncol. 2017;12:e200–e202. [DOI] [PubMed] [Google Scholar]
- 78.Ferrer I, Zugazagoitia J, Herbertz S, et al. KRAS-mutant non-small cell lung cancer: from biology to therapy. Lung Cancer. 2018;124:53–64. [DOI] [PubMed] [Google Scholar]
- 79.Gille H, Kortenjann M, Strahl T, Shaw PE. Phosphorylation-dependent formation of a quaternary complex at the c-fos SRE. Mol Cell Biol. 1996;16:1094–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci. 2012;13:1886–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sakamoto KM, Frank DA. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin Cancer Res. 2009;15:2583–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi M, Zheng Y, Garcia A, et al. Phospholipase D provides a survival signal in human cancer cells with activated H-Ras or K-Ras. Cancer Lett. 2007;258:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang E, Zha J, Jockel J, et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. [DOI] [PubMed] [Google Scholar]
- 84.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]