

Abstract
Coumarin is an important six-membered aromatic heterocyclic pharmacophore, widely distributed in natural products and synthetic molecules. The versatile and unique features of coumarin nucleus, in combination with privileged sulfonamide moiety, have enhanced the broad spectrum of biological activities. The research and development of coumarin, sulfonamide-based pharmacology, and medicinal chemistry have become active topics, and attracted the attention of medicinal chemists, pharmacists, and synthetic chemists. Coumarin sulfonamide compounds and analogs as clinical drugs have been used to cure various diseases with high therapeutic potency, which have shown their enormous development value. The diversified and wide array of biological activities such as anticancer, antibacterial, anti-fungal, antioxidant and anti-viral, etc. were displayed by diversified coumarin sulfonamides. The present systematic and comprehensive review in the current developments of synthesis and the medicinal chemistry of coumarin sulfonamide-based scaffolds give a whole range of therapeutics, especially in the field of oncology and carbonic anhydrase inhibitors. In the present review, various synthetic approaches, strategies, and methodologies involving effect of catalysts, the change of substrates, and the employment of various synthetic reaction conditions to obtain high yields is cited.
Keywords: coumarin sulfonamide, synthesis, anticancer agents, carbonic anhydrase inhibitors, SAR
1. Introduction
Ever since the first time that coumarin 1 was isolated from natural source tonka bean (Dipteryx odorata), commonly known as cumaru, in 1820 by Vogel [1,2]. Coumarin is an old, important, and diversified oxygen containing six membered heterocyclic classes of 1,2 benzopyrones, which naturally occur in plants and many other species, such as fungi (Armillariella tabescens, Fomitopsis officinalis) and bacteria (Streptomyces niveus, Escherichia coli) [3,4]. More than 1300 coumarins are present in plants, which play vital role in physiology and overall functioning of plants [1]. The general structure of coumarin 1 is given below (Figure 1). In the mid-nineteenth century, the research and development of coumarin-based compounds and hybrid structures began via the famous Perkin condensation reaction between acetic anhydride and salicylaldehyde. The different synthetic classical techniques, such as Knoevenagel, Perkin and Pechmann reactions, are applied to achieve simple coumarins [5,6,7]. The rapid developments in the synthetic chemistry of coumarins have been made, due to their wide therapeutic potential as medicinal drugs. The coumarin scaffolds displayed an array of biological activities, such as coumarin chalcone derivatives, coumarin aryl sulfonamides, and coumarin hydrazine–hydrazone hybrids, etc., which were screened to investigate their anticancer activities [8,9,10,11,12]. Coumarin scaffolds are extensively studied for their antioxidant [13,14,15,16], antibacterial [17,18,19,20], anti-fungal [21,22,23], anti-inflammatory [24,25], anti-diabetic [26], vasorelaxant [27], analgesic [28], anti-HIV [29], antimicrobial [30], anti-coagulation [31], and anti-pyretic [32] activities, etc.
Figure 1.
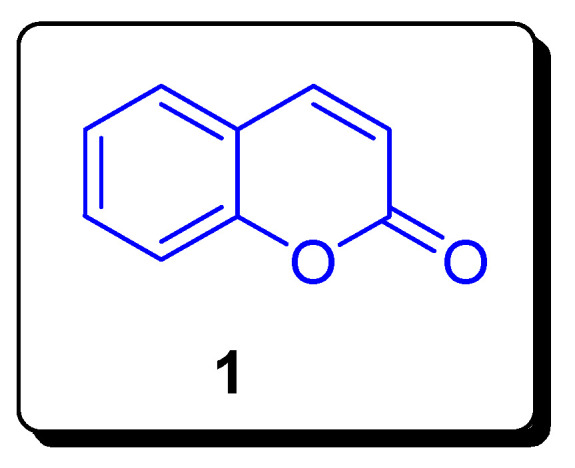
Structure of coumarin 1.
The combination of sulfonamide moieties with coumarin nucleus is attractive, as well as being a versatile platform for the research and development of a novel class of bioactive-targeted therapeutic agents [33]. The coumarin sulfonamides such as sulfocoumarin 2 and chlorophenyl-based coumarin sulfonamide 3 executed significant activity against carbonic anhydrase inhibitors IX and XII, while CAI17 coumarin sulfonamide scaffold 4 displayed remarkable anti-metastatic activity (Figure 2) [34,35,36].
Figure 2.

Structures of biologically active coumarin sulfonamides 2–4.
This review article comprehensively reviews the overall current progress of coumarin sulfonamides in medicinal chemistry as carbonic anhydrase inhibitor and anticancer agents. The successful synthetic approaches, substitution pattern, and structure–activity relationship of different bioactive compounds are discussed.
2. Coumarin Sulfonamides as Anti-Cancer Agents and Carbonic Anhydrase Inhibitors
Cancer is one of the most lethal, notable complex and serious threats to human health, and has attracted attention worldwide. All over the world, about 7.6 million people die due to cancer every year, and around 13 million people will likely die before 2030. In 2020, globally, almost 10 million people died due to cancer [37,38]. Extensive research and development work have been conducted in the field of oncology to develop anticancer therapeutic agents, and large breakthroughs and great strides have been made over past 60 years [39]. Coumarin sulfonamide derivatives and analogs have therapeutic potential against different types of cancer cell lines and CAs (carbonic anhydrases). CAs are also known as carbonate dehydratases [34]. CAs are metalloenzymes which are present in all life forms, and are essential for equilibria between different simple but significant reaction species, such as carbon dioxide, proton, and bicarbonate [40,41,42,43,44]. In 1933, 88 years ago, these enzymes were discovered, and are still an extraordinary example of convergent evolution, and extensively studied and investigated for biomedical inhibitory activities. CAs were found in bacteria, archaea and eukarya; genetically, at least eight (α-, β-, γ-, δ-, ζ-, η-, θ- and ι-CAs) distinct families [40,41,42,43,44,45]. The α-CAs family is present in vertebrates, and the bacteria, algae, and cytoplasm of green plants, while β-CAs are found in bacteria, the chloroplasts of monodicotyledons and dicotyledons, and algae. The γ-CAs are mainly present in archaea and some bacteria, the δ-, ζ- and θ-CAs are present in some marine diatoms, and the η-CAs are present in protozoa. The ι-CAs were discovered in marine phytoplankton, as well as in some bacteria [45,46,47,48,49,50,51,52,53,54,55]. There are five membrane-bound isozymes: CA-IV, CA-IX, CA-XII, CA-XIV, and CA-XV), five cytosolic forms CA-I, CA-II, CA-III, CA-VII, and CA-XIII, a secreted CA isozyme CA-VI, and two mitochondrial forms, CA-VA and CA-VB [56,57,58,59,60]. CAs inhibition mechanism with coumarins was unraveled with kinetic and X-ray crystallographic techniques. The first natural product, coumarin, was bound to human isoform hCA-II, but the formation of the enzyme inhibitor complex is not a rapid process, it takes 6 h for incubation period, while other classes take just 15 min for the incubation period [61,62,63,64,65,66]. The coumarin sulfonamides’ anticancer and CAs inhibition activities are discussed below in more detail.
2.1. Benzenesulfonamide-Based Coumarins as Carbonic Anhydrases II and IX Inhibitors
Wang and coworkers designed a solvent-free green methodology to synthesize substituted coumarin containing sulfonamides derivatives, and screened for carbonic anhydrase inhibitory activities. In this synthetic strategy, Meldrum’s acid was reacted with various substituted phenol 5 to achieve substituted malonic acid-based mono phenol esters 6 in (91–94%) yield, which further cyclized with Eaton’s reagent under mild conditions to yield 4-hydroxycoumarin 7 in (79–91%) yield. In the next step, substituted 3-formyl-4-chlorocoumarin 8 (59–73%) was obtained by Vilsmeiere Haack reactions in dimethylformamide (DMF) and phosphoryl chloride. Derivative 8 was treated with substituted sulfonamides in ethanol at room temperature (rt) to 50 °C, leading to the formation of final coumarin sulfonamide derivative 9 in (45–79%) yield (Scheme 1) [67].
Scheme 1.

Synthesis of coumarin containing sulfonamide derivative 9.
The benzenesulfonamide coumarins’ eighteen derivatives were afforded and screened for their in vitro anticancer activity against mouse melanoma cells (B16–F10) and breast carcinoma cell lines (MCF-7), and two human carbonic anhydrase against hCAs II (cytosolic off target isoform) and hCAs IX (trans-membrane tumor-associated isoform). The IC50 calculations were done by using Origin 8.6 software using an inhibitory model with the sum of squares of the residuals minimized. In this study, the most active derivative was substituted dimethyl pyrimidine-based coumarin benzene sulfonamide 9a (Figure 3), which displayed the highest and remarkable significant anticancer potential against MCF-7 cell lines with IC50 0.0088 µM when compared with the reference drugs doxorubicin IC50 0.072 µM and semaxanib IC50 0.012 µM. Both the virtual screening and anticancer activity results for MCF-7 showed that the over-expressed CA might be the most active therapeutic candidate that coumarin sulfonamides interacted with. The substituted pyrimidine-based coumarin benzene sulfonamide 9b (Figure 3) and di-tert-butyl substituted coumarin benzenesulfonamide containing pyrimidine 9c (Figure 3) displayed strong inhibition against hCAs II and hCAs IX isoforms with IC50 values of 0.063 µM and 0.124 µM (Table 1) respectively, when compared with standard drugs acetazolamide (AAZ) and sulfanilamide (SA). The SAR studies investigated that the introduction of thiazole and methyl pyrimidine substitutions in the benzenesulfonyl ring of the coumarin enhanced the anticancer and carbonic anhydrase inhibition activities of the below-mentioned coumarin derivatives [67].
Figure 3.

Structure of the most active anticancer and CA inhibitors coumarin sulfonamides 9a–9c.
Table 1.
Anticancer data of compound 9a and CAs inhibition data of compounds 9b and 9c.
| Compound | MCF-7 µM | Compounds | hCAs II µM | hCAs IX µM |
|---|---|---|---|---|
| 9a | 0.0088 | 9b | - | 0.124 |
| Doxorubicin | 0.065 | 9c | 0.063 | - |
| Semaxanib | 0.0031 | AAZ | 0.016 | 0.028 |
| - | - | SA | 0.26 | 0.29 |
2.2. Thiazole-Sulfonamide Coumarin Hybrids as hCA I and hCA II Inhibitors
Kurt and colleagues developed a solvent-free approach to achieve the unsubstituted thiazole-based coumarin sulfonamides 17 by the reaction of 2-hydroxybenzaldehyde 10, L-proline and ethyl 3-oxobutanoate 11, by heating for 0.5 h at a temperature of 80–90 °C to obtain 3-acetylcoumarin 12 in 92% yield, which further refluxed for 15 min in chloroform and bromine solutions to obtain 3-(bromoacetyl) coumarin 13 in 98% yield. Refluxing compound 13 with thiourea 14 in ethanol for 1 h gives 2-amino coumarin thiazolyl derivatives 15 (90% yield) that were further treated with benzenesulfonyl chloride 16 derivatives at 60 °C in pyridine, which led to the synthesis of thiazole-based coumarin sulfonamides 17 in 68–82% yield (Scheme 2) [68].
Scheme 2.

Solvent-free synthesis of coumarin sulfonamide derivatives 17.
In this study, the thiazole ring of acetazolamide was combined with coumarin moiety to afford biologically active, substituted benzenesulfonamide-based coumaryl thiazole hybrids, and was screened for its anticancer activity against hCA I and hCA II (human carbonic anhydrase isoforms). Among all these compounds, the scaffold coumarin-thiazole-based naphthalene-2-sulpho-namide 17a (Figure 4) displayed the strongest inhibition against hCA I and hCA II with the IC50 values 5.63 µM and 8.48 µM (Table 2), respectively. The SAR showed that bulky substituents such as s tert-butyl, naphthalene and iodine increase inhibitory activity, so compound 17a showed the most potent inhibitory activity due to the steric effect of bulky group substitution, such as naphthalene on sulfonyl group against hCA I and hCA II [68].
Figure 4.

Structures of the most active antioxidant and CA inhibitors coumarin sulfonamides 17a.
Table 2.
CAs inhibition data and antioxidant data of compounds 17a–17b.
| Compound | hCA I IC50 (µM) |
hCA II IC50 (µM) |
|---|---|---|
| 17a | 5.63 | 8.48 |
2.3. Sulfonyl Ureido Coumarins Hybrids as Carbonic Anhydrase Inhibitors
Bozdag and collogues described a single step reaction to afford substituted sulfonyl ureido coumarins 20 in 53–88% yield by the treatment of coumarin 18 and sulfonyl ureido isocyanates 19 in acetonitrile (ACN) or dry acetone (Scheme 3) [69].
Scheme 3.

Synthesis of substituted sulfonyl ureido coumarins 20.
The ary lsulfonylureido coumarin derivatives were evaluated for their inhibitory activity against hCA I and II (carbonic anhydrase cytosolic inhibitor) and hCA IX and XII (tumor-associated isoforms). The 4-chloro-substituted coumarin benzenesulfonamide 20a (Figure 5) exhibited the highest inhibitory activity with a KI value 20.2 nM against hCA IX and 6.0 nM against hCA XII (Table 3). Acetazolamide (AAZ) was used as a standard reference drug with KI = 25.0 nM and KI = 5.7 nM (Table 3) against hCA IX and hCA XII, respectively. The SAR showed that analogue 20a was the most potent due to the presence of electron withdrawing Cl atom in the benzene ring of the sulfonyl ureido group [69].
Figure 5.

Structures of the most active coumarin sulfonamide CA inhibitor 20a.
Table 3.
CAs inhibition data compound 20a.
| Compound | hCA IX KI (nM) |
hCA XII KI (nM) |
|---|---|---|
| 20a | 20.2 | 6.0 |
| AAZ | 25.0 | 5.7 |
2.4. Benzene Sulfonamido-Coumarinyl Hydrazones Hybrids as CA Inhibitors
Chandak et al., in 2016, synthesized sulfonamide bearing coumarin derivatives by a Hantzsch thiazole synthetic approach as shown in Scheme 4. In this synthetic strategy, the thiazoyl hydrazine methylidene pyrazole 31 derivatives were achieved from 4-hydrazinobenzenesulfonamide hydrochloride, further converted into pyrazole-based carbaldehyde bearing thiosemicarbazones, and finally reacted to substituted bromoacetyl-based coumarins 30 by condensation reaction. In the second step, different 6-substituted 3-bromoacetylcoumarins 30 and 4-thioureido-benzenesulfonamide achieved 2-amino-substituted-coumarinylthiazoles 32 by condensation reaction. In the next step, heterocyclic series 33 containing three IBTs prepared by treatment of 2-aminobenzothiazole-6-sulfonamide that first obtained from sulfanilamide and 6-substituted-3-bromoacetylcoumarins. On the other hand, the derivatives of series 4, different 3-acetylcoumarins 29 and 4-hydrazinobenzenesulfonamide hydrochloride 34 by refluxing together in aqueous ethanol with anhydrous sodium acetate, give benzenesulfonamido-coumarinyl hydrazones, 35 (Scheme 4) [70].
Scheme 4.

Synthesis of sulfonamide bearing coumarin derivatives 31–35.
The following sulfonamide-bearing coumarin scaffold consisted of twenty-four compounds evaluated for the inhibition of hCA I, II, IX and XII (human carbonic anhydrase isoforms). Among all of these, the 32a compound (Figure 6) exhibited strong potent inhibitory activity with a KI value 2.28 nM (Table 4) against hCA IX, as compared to standard compound AZA with a KI range 25.0 nM. Moreover, analogues 32a and 32b were most potent with KI values 0.54 nM against hCA XII when compared to AZA with a KI value 5.7 nM. The hybrid structure 4-{2-[1-(2-oxo-2H-chromen-3-yl)ethylidene]hydrazino} benzenesulfonamide 35a revealed the highest activity KI = 13.23 nM for hCA II in comparison with reference drug AZA with KI value 12.1 nM. The compound 4-{2-[1-(6-bromo-2-oxo-2H-chromen-3-yl)ethylidene]hydrazino}benzenesulfonamide 35b (Figure 6) screened potent inhibitory activity with a KI value 21.95 nM against hCA I, as compared to standard compound AZA (acetazolamide) with a KI range 250.0 nM (Table 4). The SAR showed that the introduction of bromo and unsubstituted H-atom on coumarin increase the carbonic anhydrase inhibitory activity of derivatives 35a and 35b (Figure 6), while the presence of electron-withdrawing Cl-atom and unsubstituted H-atom on coumarin enhances the inhibitory activity of compounds 32a and 32b [70].
Figure 6.

Structures of the most active coumarin sulfonamide CA inhibitors 32a–32b and 35a–35b.
Table 4.
Coumarin sulfonamide as CA inhibitors 32a–32b and 35a–35b.
| Compound Number | hCA I KI (nM) |
hCA II KI (nM) |
hCA IX KI (nM) |
hCA XII KI (nM) |
|---|---|---|---|---|
| 32a | 263.49 | 21.20 | 2.28 | 0.54 |
| 32b | 349.63 | 17.46 | 2.54 | 0.54 |
| 35a | 220.13 | 13.23 | 58.61 | 4.4 |
| 35b | 21.95 | 1751.72 | 23.59 | 0.62 |
| AZA | 250.0 | 12.1 | 25.0 | 5.7 |
2.5. Pyrazole-Based Coumarin Sulfonamides as CA Inhibitors and Anticancer Agents
Lu et al. in 2016 reported a synthetic approach in which acylhydrazone was converted to substituted pyrazole-based coumarin sulfonamide derivatives by consecutive reactions of various substrates (Scheme 5). The substituted acetophenone 36 was treated with dimethyl oxalate using sodium methoxide in methanol for 6 h under refluxing conditions to achieve different substituted chalcones 37 on treatment with 4-hydrazinyl benzenesulfonamide, in the presence of methanol for 6 h, to afford scaffold 38. In the second step, analogue 38 reacted with hydrazine monohydrate solution at 80 °C for 8 h to give substituted hydrazide derivatives 39. The 4-chloro-coumarin-3-aldehyde 42 was afforded by treating 4-hydroxy coumarin 40 with a mixture of phosphoric trichloride and dimethyl formamide. In the last step, the derivatives 43 were obtained (60–90%) by the combination of derivative 39 and 4-chloro-coumarin-3-aldehyde 42 at room temperature for 12 h in ethanol and AcOH, as presented in Scheme 5 [71].
Scheme 5.

Synthesis of coumarin sulfonamide derivatives 43.
The series of pyrazole moiety containing coumarin sulfonamides was tested for anti-proliferation activities in vitro, against four cancer cell lines (HeLa, HepG2, F10, A549) and two non-cancer cell lines (293T, L02). These synthetic scaffolds were also evaluated for inhibitory activities against various inhibitors, such as COX-2 and COX-1. The derivative 43a (Figure 7) possessing most powerful anti-proliferative activity against HeLa, HepG2, F10, and A549 cell lines with IC50 values of 0.36 ± 0.05 µM, 0.85 ± 0.08 µM, 2.27 ± 0.17 µM and 2.56 ± 0.34 µM, respectively. The celecoxib was used as a standard compound with IC50 values 7.79 ± 0.84 µM for HeLa, 10.03 ± 0.84 µM for HepG2, 14.36 ± 0.96 µM for F10, and 15.64 ± 1.23 µM (Table 5) for A549, respectively. The analogue 43b (Figure 7) exhibited the highest activity against the 293T cell line, with IC50 range 101.24 ± 2.27 µM and derivative 43c (Figure 8) displaying strong activity against the L02 cell line with IC50 104.57 ± 2.73 µM. Celecoxib was used as a reference drug with IC50 values 95.26 ± 2.28 µM against 293T, and 98.15 ± 2.39 µM against L02, respectively. The scaffold 43d (Figure 7) showed the highest inhibitory activity with IC50 value 39.45 ± 1.33 µM against COX-1, while scaffold 43a proved to be good inhibitor against COX-2 with IC50 = 0.09 ± 0.01 µM. Celecoxib was used as standard reference compound with IC50 value 43.37 ± 1.44 against COX-1 and 0.31 ± 0.12 (Table 6) against COX-2, respectively [71].
Figure 7.

Structures of coumarin sulfonamide as anticancer agents and COX inhibitors 43a–43d.
Table 5.
Coumarin sulfonamide as anticancer agents 53a–53c.
| Compound Number | HeLa IC50 (µM) |
HepG2 IC50 (µM) |
F10 IC50 (µM) |
A549 IC50 (µM) |
293T IC50 (µM) |
L02 IC50 (µM) |
|---|---|---|---|---|---|---|
| 43a | 0.36 ± 0.05 | 0.85 ± 0.08 | 2.27 ± 0.17 | 2.56 ± 0.34 | 234.46 ± 4.52 | 267.28 ± 4.87 |
| 43b | 16.19 ± 1.26 | 20.04 ± 1.29 | 26.24 ± 1.57 | 26.14 ± 1.13 | 101.24 ± 2.27 | 116.35 ± 2.69 |
| 43c | 7.58 ± 0.63 | 15.66 ± 1.34 | 9.58 ± 0.87 | 22.41 ± 1.07 | 106.62 ± 2.43 | 104.57 ± 2.73 |
| Celecoxib | 7.79 ± 0.84 | 10.03 ± 0.84 | 14.36 ± 0.96 | 15.64 ± 1.23 | 95.26 ± 2.28 | 98.15 ± 2.39 |
Figure 8.
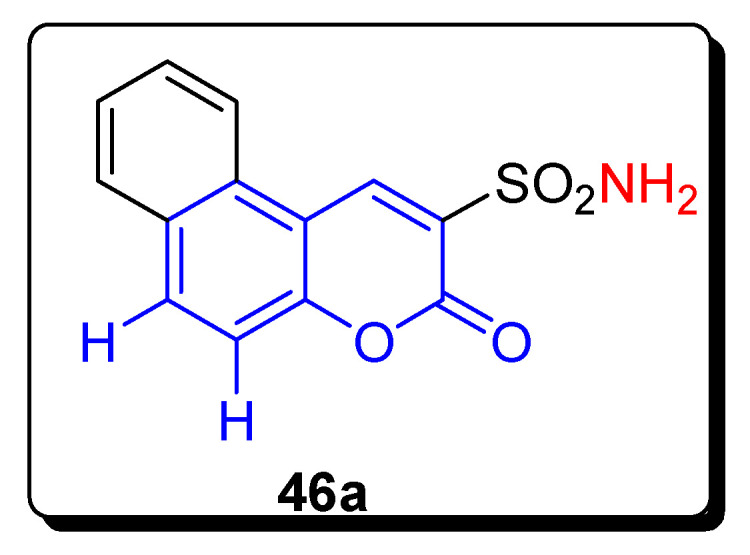
Structures of 3-sulfonamide-coumarin derivative 46a as anticancer agents and hCAs inhibitors.
Table 6.
Coumarin sulfonamide as COX inhibitors 43d and 43a.
| Compound Number | COX-1 IC50 (µM) |
COX-2 IC50 (µM) |
|---|---|---|
| 43d | 39.45 ± 1.33 | 7.13 ± 0.81 |
| 43a | 48.20 ± 1.30 | 0.09 ± 0.01 |
| Celecoxib | 43.37 ± 1.44 | 0.31 ± 0.12 |
2.6. 3-Sulfamoyl Coumarins against Cancer-Related IX and XII Isoforms of hCAs
Dar’in et al. in 2021 synthesized series of seventeen 3-sulfonamide substituted coumarin scaffolds via the reaction of ethyl 2-sulfamoylacetate 45 and various substituted salicylaldehydes 44 in n-butanol at 110 °C for 2–6 h under basic conditions. The reaction mixture was stirred to obtain 3-sulfamoyl coumarines 46 in 30–84% yield (Scheme 6) [72].
Scheme 6.

Synthesis of 3-sulfonamide coumarin derivatives 43.
The 3-sulfonamide substituted coumarin derivatives screened for their in vitro metalloenzyme human carbonic anhydrase inhibitor, such as hCA I, II, IX, and XII. The 3-sulfonamide-substituted naphthalene coumaryl derivative 46a (Figure 8) showed that the incorporation of extra aromatic rings may be detrimental for the anti-hCA activity of 3-sulfamoyl coumarins. The derivatives 46a only showed micromolar KI values against all studies enzyme isoforms, as shown in Table 7, as compared with the reference drug AAZ (acetazolamide). The analogue 46a also displayed good inhibition and retained its selectivity for the A431 cell line over non-tumorigenic normal human fibroblast cell line WI-26 AV4. The IC50 value of compound 46a grows with an increasing incubation period (24 h to 72 h), from 9.73 ± 3.13 μM to 70.14 ± 26.06 μM, in comparison with standard reference drug Gefitinib (32.17 ± 3.44 μM, 16.02 ± 2.87 μM), as shown in Table 7. The promising compound 46a was not the strongest CA IX/XII inhibitor, and apoptosis was induced by activating caspases in a dose-dependent manner. The DNA intercalation mechanism was followed by this compound 46a for anti-proliferative effects, and so this compound has the potential to become a viable lead motif for the further development of next-generation anticancer agents [72].
Table 7.
Coumarin-3-sulfonamide 46a as anticancer agent and hCAs inhibitors.
| Compound | A431 IC50 (µM) 24 h–72 h |
CA I KI (nM) |
CA II KI (nM) |
CA IX KI (nM) |
CA XII KI (nM) |
|---|---|---|---|---|---|
| 46a | 9.73–70.14 | 75,100 | >100 μM | 6371 | 7990 |
| Gefitinib | 32.17–16.02 | - | - | = | - |
| AAZ | - | 250 | 12.5 | 25 | 5.7 |
2.7. Triazole-Bridged Coumarin Sulfonamides as CAs Inhibitors and Anticancer Agents
Kurt and coworkers in 2019 developed a synthetic approach to achieve the target coumarin sulfonamde motifs 53a–i by consecutive reactions from various substrates for the evaluation of carbonic anhydrases (CA I, II, IX, and XII) inhibition and cytoxicities potential against human colorectal adenocarcinoma cell line HT-29 and healthy HEK293T embryonic kidney cells lines. In the first step, 7-hydroxy coumarin 47 was treated with 1,3-dibromopropane to furnish scaffold 48, which was further treated with sodium azide to afford propylazide bearing coumarin derivative 49. On the other hand, various aldehydes 50 hydroxy groups were combined with the propargyl group of propargyl bromide in DMF at rt to yield propargyl substituted aldehydes 51, which were further treated with derivative 49 via a click reaction to obtain aldehyde-based triazole bridged coumarin derivative 52, by using CuSO4·5H2O as a catalyst. The 4-(aminomethyl)benzenesulfonamide was reacted with the formyl group of compound 52 in alcoholic basic conditions, to achieve target triazole moiety containing coumarin sulfonamides 53, as depicted in Scheme 7 [73].
Scheme 7.

Synthesis of triazole-based coumarin sulfonamide derivatives 53.
Kurt et al. in 2019 evaluated the triazole-bridged coumarin sulfonamide scaffolds against four physiologically significant isoforms CA I, II, IX, and XII of carbonic anhydrases by utilizing stopped flow carbon dioxide hydrase assay and anticancer activities against colorectal cancers HT-29 and healthy HEK293T embryonic kidney cells lines by MTT assay. CA IX is a prominent target for, in particular, colorectal cancers HT-29 cell line, which is overexpressed; this resulted in poor prognosis. The triazole-bridged coumrin sulfonamide 53a (Figure 9) displayed significant and the highest hCA IX inhibition with the KI of 45.5 nM in comparison with the standard reference drug AAZ (25.8 nM) as displayed in Table 8. The scaffold 53a exhibited excellent anti-proliferative activity (IC50 17.01 ± 1.35 μM) as compared with the reference drug doxorubicin (IC5.38 ± 1.40 μM), while this compound 53a showed the least cytotoxicity (118.73 ± 1.19 μM) against healthy HEK293T embryonic kidney cells lines. On the basis of these findings, this novel compound 53a cellular proliferates in human colon cancer cells by specifically targeting the CA IX and CA XII expression. The data indicated that the lead compound 53a may be a promising drug candidate [73].
Figure 9.

Structure of triazole-based soumarinsulfonamide derivative 53a as anticancer agents and hCAs inhibitors.
Table 8.
CAs inhibition data compound 53a.
| Compound | hCA IX KI (nM) |
HT-29 IC50 (μM) |
HEK293T IC50 (μM) |
|---|---|---|---|
| 53a | 45.5 | 17.01 ± 1.35 | 118.73 ± 1.19 |
| AAZ | 25.8 | 53.78 ± 1.75 | - |
| Doxirubicin | - | 5.38 ± 1.40 | 1.051 ± 0.57 |
2.8. Substituted Coumarins Sulfonamide as Selective Human CA IX and XII Inhibitors
Abdelrahman and his colleagues in 2021 designed synthetic strategies to achieve different, substituted coumarin sulfonamide derivatives and screened for different biological activities and molecular dynamics. In the first synthetic approach, 3-acetylcoumarins 54 were condensed with 4-sulfamoylbenzoic acid hydrazide 55 and 4-hydrazino benzenesulfonamide 57 in boiling glacial acetic acid, to achieve 3-substituted coumarin sulfonamide derivatives 56a,b and 58a,b, as depicted in Scheme 8 [74].
Scheme 8.

Synthesis of 3-substituted coumarin sulfonamide 56a,b and 58a,b.
The coumarin sulfonamide hydrazones 56a, 56b and hydrazides 58a and 58b (Figure 10) displayed the potent hCA I inhibitory activity with inhibition constant KI 98.8, 159.7, 77.6 and 92.5 nM in comparison with standard reference drug AAZ (KI 250, 12, 25 and 5.7 nM), as described in Table 9. Sulfonamide moiety is essential in the hydrazone derivatives 56a,b and hydrazide 58a,b for the inhibition of both hCA I and II isoforms. The replacement of hydrazone moiety of scaffolds 58a,b with hydrazide in scaffolds 56a,b slightly enhanced inhibitory activity against hCA I and hCA II isoforms [74].
Figure 10.

Structures of coumarin sulfonamide derivative 56a,b and 58a,b hCAs inhibitors.
Table 9.
CAs inhibition data compounds 56a,b and 58a,b.
| Compound | hCA I KI (nM) |
hCA II KI (nM) |
hCA IX KI (nM) |
hCA XII KI (nM) |
|---|---|---|---|---|
| 56a | 98.8 | 29.1 | 43.8 | 10.1 |
| 56b | 159.7 | 40.0 | 24.2 | 55.8 |
| 58a | 77.6 | 26.3 | 31.6 | 19.7 |
| 58b | 92.5 | 12.1 | 19.8 | 34.6 |
| AAZ | 250.0 | 12.0 | 25.0 | 5.7 |
In the second synthetic approach, Abdelrahman and coworkers carried out the bromination of 3-acetylcoumarins 59a,b with bromine in the presence of glacial acetic acid to furnish 3-(bromoacetyl)coumarins 60a,b, which subsequently refluxed in ethanol with sodium benzenesulfinates 61a,b to achieve phenylsulfonyl-based acetyl coumarin motifs 62a,b. The coumarin arylsulfonehydrazones 63a–d and hydrazide 64a,b scaffolds were furnished in 74–82% yield via the treatment of compound 62a,b with phenyl hydrazine and benzoic acid hydrazide in ethanolic glacial acetic acid solution and refluxed for 2 h, as shown in Scheme 9 [74].
Scheme 9.

Synthesis of 3-substituted coumarin sulfonamide 63a–d and 64a,b motifs.
The coumarins, sulfolanamide 63a,b and 64a–d displayed excellent selectivity profiles, but these scaffolds have very little potential of inhibition; these derivatives barely inhibited hCA IX/XII carbonic anhydrases. These motifs inhibit the hCA II isoform up to 100 μM concentration.
In the final dual tail synthetic strategy (Scheme 10), 4-sulfamoylbenzoic acid hydrazide, and 4-hydrazinobenzenesulfonamide were refluxed with 3-(2-(phenylsulfonyl) acetyl) coumarins 62a,b in ethanol, to achieve coumarin sulfonamide arylsulfone hydrazide 65a,b and coumarin sulfonamide arylsulfonehydrazones 66a–d respectively, in the 74–78% yield as depicted in Scheme 10 [74].
Scheme 10.

Synthesis of 3-substituted coumarin sulfonamide 65a,b and 65a–d motifs.
The incorporation of the arylsulfone moiety in the dual tail coumarin sulfonamide hydrazide 65a,b and hydrazone 66a–d (Figure 11)derivatives lowered the inhibition efficacy toward hCA I and hCA II isoforms in comparison to their counterpart derivatives 56 and 58. The most selective dual tail coumarin sulfonamide derivatives were compounds 65a and 66d, as shown in Table 10. In the present study, hydrazides 58b and 65a proved the best and excellent CA IX inhibitors with low nanomolar potencies (9.8 and 18.6 nM, respectively) [75].
Figure 11.

Structures of coumarin sulfonamide derivative 65a and 66d hCAs inhibitors.
Table 10.
CAs inhibition data compounds 65a,b and 66a–d.
| Compound | hCA I KI (nM) |
hCA II KI (nM) |
hCA IX KI (nM) |
hCA XII KI (nM) |
|---|---|---|---|---|
| 65a | 1537 | 224.8 | 18.6 | 32.3 |
| 65b | 1352 | 365.2 | 42.1 | 76.8 |
| 66a | 824.2 | 442.4 | 89.1 | 74.4 |
| 66b | 2596 | 559.7 | 76.0 | 91.1 |
| 66c | 3455 | 276.0 | 63.6 | 36.2 |
| 66d | 3962 | 538.3 | 29.2 | 12.0 |
| AAZ | 250.0 | 12.0 | 25.0 | 5.7 |
2.9. Coumarin Sulfonamide as RAF/MEK Inhibitors and Anticancer Agents
Aoki and coworkers synthesized compound 67 by the benzylation reaction of ethyl acetoacetate, which was further treated with 4-chlororesorcinol 68 by the Pechmann reaction to furnish substituted coumarin derivative 69 (Scheme 11). On the other hand, the substituted coumarin aniline derivative 57 was obtained by the carbamoylation of the phenoic hydroxyl group with N,N-dimethyl carbamoyl chloride after the reduction of a nitro group was carried out by using SnCl2. The sulfamoyl chloride or sulfamoyl oxazolizinone was treated with scaffold 70, which led to the formation of sulfamide 71, as described in Scheme 11 [75].
Scheme 11.

Synthesis of amino-based coumarin sulfonamide derivatives 71.
The series of newly reported coumarin containing sulfonamides was evaluated for their enzyme inhibition activities and tested for in vivo antitumor activity against HCT 116 (human colon cancer), MEK1, C-Raf, and HT-29 xenograft. The scaffold 71a (Figure 12) showed the highest antitumor inhibition activities against MEK1, HCT116 and HT-29 with IC50 values 7 nM, 4 nM, and 1 nM, respectively. Moreover, 71b (Figure 12) was the most potent against C-Raf, with an IC50 value of 5 nM. The derivatives 71c and 72d (Figure 12) displayed the highest antitumor activity with IC50 value 4 nM against C-Raf. The analogue 71e (Figure 12) was the most active MEK1 inhibitor, with IC50 value 5 nM, while the derivative 71c demonstrated the strongest anticancer activity with IC50 23 nM (Table 11) against HCT1169, respectively [75].
Figure 12.

Structures of coumarin sulfonamide as anticancer agents and COX inhibitors 71a–71e.
Table 11.
Coumarin sulfonamide as RAF/MEK inhibitors and anticancer agents 71a–71e.
| Compound Number |
C-Raf IC50 (nM) |
MEK1 IC50 (nM) |
HCT116 IC50 (nM) |
HT-29 IC50 (nM) |
|---|---|---|---|---|
| 71a | 8 | 7 | 4 | 1 |
| 71b | 5 | 11 | 12 | 9 |
| 71c | 4 | 36 | 23 | - |
| 71d | 4 | 47 | 36 | - |
| 71e | 40 | 5 | 93 | - |
3. Pyrazoline-Based Coumarin Sulfonamide Hybrids as Anticancer Agents
Amin and the team reported that synthetic methodology to furnish pyrazoline-based coumarin sulfonamide hybrids by refluxing acetic anhydride with 7-hydroxy substituted chromene 72 for 5 h, to produce substituted acetate chromenyl 73, which was further heated with AlCl3 at 145 °C for 1 h to afford hydroxy acetyl chromen-2-one derivative 74. By refluxing compound 74 and scaffold 75 in methyl iodide and dry acetone for 1 day to synthesize scaffold 76, which reacted with aryl aldehyde and 10% NaOH in ethanol at rt for 1 day to yield substituted methoxy containing aryl acryloyl chromen 77, which further proceeds in two steps. At the first step, derivatives 77 reacted with 4-substituted phenyl sulphonyl hydrazines 78 in absolute ethanol by refluxing at 8–12 h to afford pyrazoline-based coumarin sulfonamide hybrids 79 in 35–68% yield (Scheme 12) [76].
Scheme 12.


Synthesis of pyrazoline-based coumarin sulfonamide derivatives 79.
The coumarin-pyrazoline containing phenyl sulfonyl moiety was screened for anticancer therapeutic efficacy against nine different tumor cell lines, including colon cancer, melanoma cancer, and breast cancer. The pyrazoline-based coumarin sulfonamide derivative 79a (Figure 13) showed the best anticancer activities against tumor cell lines. The compound 79a exhibited the highest antitumor activity against colon cancer (HCT-116), melanoma (LOX IMVI), and breast cancer (MCF7). Scaffold 79a possessed the most powerful activity displaying the GI50 value 0.94 µM against HCT-116, 0.87 µM against LOX IMVI, and 0.49 µM (Table 12) against MCF7. The values were measured by different parameters for the three analogues GI50, TGI, and LC50 against every cell line. GI50 is the cytotoxicity parameter for the molar concentration of the scaffold that decreases 50% of the cell growth, and is observed as a growth-inhibitory level of effect; TGI (cytostatic activity) is the molar concentration of the derivative, leading to total growth inhibition; LC50 is the molar concentration of the compound that causes 50% net cell death. The SAR showed that the presence of electron-withdrawing chlorine atom on the fourth position of the phenyl sulfonyl structure and the phenyl group on the 5th position of the pyrazoline ring was responsible for the most potent antitumor activity of the scaffold 79a [76].
Figure 13.
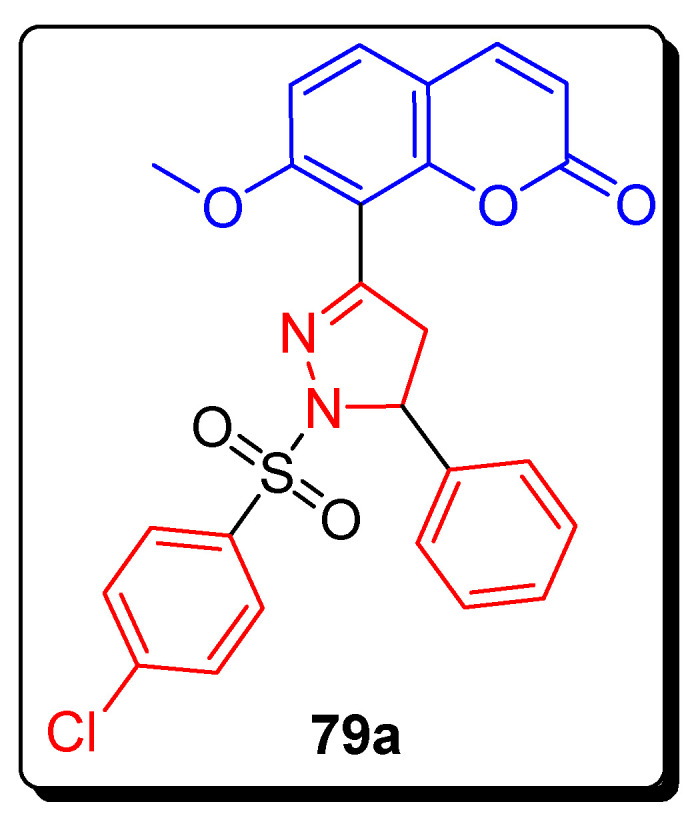
Structure of the most active coumarin sulfonamide derivative 79a.
Table 12.
Anti-cancer activities of compound 79a.
| Subpanel/Tumor Cell Lines | Compound 79a GI50 (µM) |
|---|---|
| Colon Cancer | |
| HCT-116 | 0.94 |
| Melanoma Cancer | |
| LOX IMVI | 0.87 |
| Breast Cancer | |
| MCF7 | 0.49 |
3.1. Pyrazoline-Based Coumarin Sulfonamide Hybrids as Anticancer Agents
Zhang et al. in 2021 reported the synthesis of the coumarin sulfonamides derivatives 84 by the consecutive reactions of various substrates. In the first step, the substituted aromatic amines 80 were combined with methyl 2-chlorosulfonylacetate 81 rt by refluxing in TEA for 3 h to afford derivative N-phenyl sulfomyl acetate 82. The scaffold sulfomyl acetae 82 further reacted with 4-(diethylamino) salicylaldehyde 83 in the presence of piperidine at 87 °C under reflux for 30 min to furnish substituted N-phenyl sulfomyl moiety containing diethyl amini coumarin motifs 84, as depicted in Scheme 13 [77].
Scheme 13.

Synthesis of diethylamini-based substituted coumarin sulfonamides 84.
The following coumarin-based sulfonamide derivatives were afforded and screened for their in vitro antitumor activity against MDA-MB-231 (human breast cancer cell line), HCT-116 (human colon cancer cell line), and KB (human oral epidermoid carcinoma cell line). The substituted N-phenyl sulfomyl moiety containing diethyl amini coumarin motifs 84a (Figure 14) displayed the highest inhibitory anti-cancer efficacy against MDA-MB-231 cell line with IC50 value 9.33 ± 1.81 µM in comparison with the reference compounds 5-fluorouracil with IC50 8.59 ± 0.52 µM (Table 13). The SAR investigated that the introduction of methoxy group on the benzene ring and diethylamino substituent at C-7 position and sulfonamide moiety at C-3 position of the coumarin enhanced antitumor activity. The compound 84a had unique and versatile therapeutic efficacy, and properties against tumors, such as invasions, inducing apoptosis and the inhibition of cell migration. Scaffold 84a upregulated the expression of caspase-3 and enhanced cancer cell apoptosis by increasing reactive oxygen species (ROS) level in MDA-MB-231 cells, as indicated by ROS assay and Western blotting analysis. The results of this study suggested that scaffold 84a could be a promising lead anti-cancer agent which requires further exploration and research to become an antitumor drug [77].
Figure 14.

Structure of the most active coumarin sulfonamide derivative 84a.
Table 13.
Anti-cancer activities of compound 84a.
| Tumor Cell Lines | Compound 84a GI50 (µM) |
|---|---|
| Human breast cancer cell line | |
| MDA-MB-231 | 9.33 ± 1.81 |
| 5-Fluorouracil | 8.59 ± 0.52 |
3.2. Pyrazole Sulfonyl Coumarins Hybrids as Anticancer Agents and Anti-Migratory Activity
El-Sawy synthesized substituted sulfonyl coumarin derivatives via the treatment of sulfonyl chloride chromene 85 were refluxed with 2-acetyl-2-cyanoacetohydrazide using drops of triethylamine containing absolute ethanol to obtain substituted amino acetyl dihydropyrazol derivative 86 in 85% yield. Moreover, compound 85 and amino-based pyrazolinone reacted in the presence of dry dioxane in ethanol to afford sulfonic acid-substituted chromene-based pyrazolyl amide 87 in a 77% yield. In another approach, scaffold sulfonyl-based chromene malononitrile 88 in 76% yield was afforded by refluxing scaffold 85 with malononitrile. The derivative 88 further cyclized with hydrazine hydrate using triethylamine, along with dry ethanol, to produce pyrazole 89 in an 82% yield (Scheme 14) [78].
Scheme 14.

Synthesis of coumarin sulfonamide derivatives 86–87, 89.
The non-cytotoxic tested compounds were screened against HepG2 (hepatocellular carcinoma cells). The compounds significantly inhibited MMP-2 activity at p value ˂ 0.001 as a percentage of control. The results of this study demonstrated that the derivatives 86, 87, and 89 (Figure 15) are non-toxic angiogenesis inhibitors, and the compound 86 proved to be a promising anti-angiogenic agent. The coumarin sulfonamide derivative 89 displayed the highest MMP dependent (protease-dependent) anti-migratory activity (Table 14), as compared to all other tested compounds. The SAR showed that the presence of electron donating di-amino moiety on the pyrazole ring of scaffold 28 was responsible for the highest anti-migratory effect [78].
Figure 15.

Structures of the most active coumarin sulfonamide 86–87, 89.
Table 14.
Anti-migratory activity of compound 89.
| Compound Number | Anti-Migratory Effect |
|---|---|
| 89 | 4.7 |
3.3. Coumarin Benzomidazole Sulfonamide Hybrids as Anticancer Agents
Holiyachi and coworkers in 2016 reported synthetic protocols to afford a series of substituted sulfonamide-based coumarin benzimidazole hybrids 93 with different substituents. The substituted 4-formylcoumarins 90 and ortho-phenylenediamine (OPD) 91 in DMF in the presence of p-Toluenesulphonic acid (p-TsOH) to achieve benzimidazole moiety contained substituted coumarin derivative 92 with an excellent yield 89–98%. The N-sulphonation of benzimidazole moiety of coumarin derivatives 92 was carried with p-toluenesulfonyl chloride (TsCl) in the presence of TEA to furnish the coumarin-benzimidazole sulfonamide hybrids 93 in a good to excellent yield—75–90% (Scheme 15) [79].
Scheme 15.

Synthesis of coumarin benzimidazole-sulfonamide derivatives 93.
The novel series of coumarin-benzimidazole sulfonamide hybrids (93a–c) was tested against different cancerous cell lines, such as HeLa (human cervix cancer) cell lines and human colon HT29 cancer cell lines. Among all these, the scaffolds 93a (methyl coumarin) and 93b (methoxy coumarin) exhibited the highest anticancer activity towards HeLa line with GI50 36.2 and 35.3 respectively, while the analogues 93a and 93d (bromo coumarin) displayed the most powerful anticancer activity against HT when compared with the standard drug Adriamycin (ADR), respectively. The SAR highlighted that the presence of methyl, methoxy, and bromine substituent on the 6-position of coumarin ring enhanced the anticancer activity, as depicted in Figure 16 [79].
Figure 16.

Structures of the most active coumarin benzimidazole sulfonamides 93a–c.
3.4. Coumarin-Proline Sulfonamide Motifs as Anticancer Agents
Durgapal and Soman in 2019 report the synthetic approach to achieve coumarin-proline sulfonamide derivatives 100, and screened for their anticancer and antidiabetic therapeutic potential. In this synthetic strategy (Scheme 16), the carbamate-protected 3-aminophenol 95 was afforded by the reaction of ethyl chloroformate with 3-aminophenol 94. The 7-carboethoxy amino coumarin 96 was obtained by the Pechmann reaction of 3-aminophenol 94 with ethyl acetoacetate in 70% ethanolic H2SO4. The amino-substituted 4-methyl coumarin motifs 97 were afforded by the deprotection of derivative 96 in H2SO4/CH3COOH (1:1). The derivative 98 was achieved by the reaction of (L)-N-Boc-proline, first at 0–5 °C by stirring with ethyl chloroformate in tetrahydrofuran (THF) by adding derivative 97 solution and TEA dropwise, and then refluxed the mixture for 8 h in THF. The compound 98 contained the protection of the Boc group which was deprotected through trifluoroacetic acid (TFA) to obtain derivative 99, which was further treated with different substituted benzene sulfonyl chloride derivatives by using sodium hydrogen carbonate in CH2Cl2:H2O (1:1) at rt to furnish coumarin-proline sulfonamide derivatives 100 (Scheme 16) [80].
Scheme 16.

Synthesis of coumarin-proline sulfonamide derivatives 100.
The series of coumarin-proline sulfonamide derivatives were evaluated as anticancer against different cancer cell lines, such as the breast cancer cell line (MCF7), and a lung cancer cell line (A549), using the MTT assay and DPP-IV inhibition assay. The coumarin-proline sulfonamide scaffold 100a (Figure 17) was the most potent against MCF-7 with an IC50 value of 1.07 mM, as compared to reference standard drug fluorouracil with an IC50 value of 45.04 mM (Table 15). The SAR showed that the activity of compound 100a was significantly increased, due to the presence of methyl substituent at a parallel position of benzene ring to sulfonamide group [80].
Figure 17.

Structures of the most active coumarin-proline sulfonamide hybrid 100a.
Table 15.
Anti-cancer activity of compound 100a.
| Compound Number | MCF-7 IC50 (mM) |
|---|---|
| 100a | 1.07 |
| Fluorouracil | 45.04 |
3.5. Coumarin-6-Sulfonamide Derivatives as Anticancer Agents
Sabt and colleagues reported the synthesis of coumarin moiety containing sulfonamides and evaluated for apoptotic anti-proliferative activity. The chromene-based sulfonyl chloride 101 and 4-aminoacetophenone 102 reacted in pyridine in the presence of dichloromethane for 24 h at rt to yield acetyl phenyl-bearing chromene-based sulfonamide 103 that further reacted in two different steps. In this step, scaffold 103 refluxed with substituted phenyl hydrazine in the presence of absolute ethanol for 8 h to obtain coumarin-6-sulfonamide derivatives 104 in the 58–66% yield (Scheme 17) [81].
Scheme 17.

Synthesis of coumarin-6-sulfonamide derivatives 104.
The Sabt and coworkers synthesized coumarin containing substituted sulfonamide structural hybrids and were tested for apoptotic proliferation inhibition potential against the colon cancer cell line (Caco-2), hepatocellular carcinoma cell line (HepG2), and breast cancer cell line (MCF-7). The scaffold phenyl hydrazono containing coumarin-6-sulfonamide 104a (Figure 18) showed the excellent and remarkable proliferation inhibition activity with IC50 value 8.53 ± 0.72 µM, in comparison with standard drug doxorubicin having IC50 = 4.10 ± 1.37 µM (Table 16) [81].
Figure 18.

Structure of anticancer coumarin sulfonamide derivative 104a.
Table 16.
Coumarin sulfonamide as anticancer agents.
| Compound Number | HepG2 IC50 (µM) |
MCF7 IC50 (µM) |
Caco-2 IC50 (µM) |
|---|---|---|---|
| 104a | 26.99 ± 2.01 | 14.30 ± 1.18 | 8.53 ± 0.72 |
| Doxorubicin | 5.43 ± 0.24 | 3.18 ± 0.32 | 4.10 ± 1.37 |
Sabt and colleagues prepared coumarin-6-sulfonamides (107a,b and 109a,b) via the treatment of compound 103 with thiosemicarbazide using a catalytic amount of acetic acid in ethanol by refluxing for 6 h to furnish corresponding coumarin sulfonamide hydrazine carbothioamide 105 in a 34% yield. The intermediate 105 further refluxing with phenacyl bromide 106a and coumarin-3-acetylbromide 106b for 8 h using dioxane to achieve thiazoles moiety containing coumarin sulfonamides 107a in an 80% and 107b in a 63% yield. They further treated the intermediate 105 with bromoacetic acid 108a and 2-bromopropanoic acid 108b using anhydrous sodium acetate in acetic acid to afford the target thiazolidinone derivatives 109a,b with a 40–64% yield (Scheme 18) [81].
Scheme 18.

Synthesis of coumarin-6-sulfonamide derivatives 107a,b and 109a,b.
The thiazole moiety containing coumarin sulfonamide structural hybrids was evaluated against the MCF-7 cell line, HepG2 and Caco-2 cell lines for proliferation inhibition activities. The thiazole moiety containing coumarin sulfonamide analogue 107a (Figure 19) displayed the best anti-proliferative activity against hepatocellular carcinoma cell line with a IC50 value 3.48 ± 0.28 µM (Table 17) in comparison with the standard reference drug doxorubicin of IC50 = 5.43 ± 0.24 µM among all other screened derivatives of this series. The methyl-substituted thiazole-based coumarin sulfonamide 109b (Figure 19) was the most active derivative, which exhibited highly significant anticancer therapeutic potential against MCF7 cell line with IC50 value 10.62 ± 1.35 (Table 17), by comparing it with standard compound doxorubicin drug having an IC50 value 3.18 ± 0.32 µM [81].
Figure 19.

Structure of anticancer thiazole-based coumarin sulfonamide derivative 107a.
Table 17.
Thiazole-based coumarin sulfonamide 107a and 109a as anticancer agents.
| Compound Number | HepG2 IC50 (µM) |
MCF7 IC50 (µM) |
Caco-2 IC50 (µM) |
|---|---|---|---|
| 107a | 3.48 ± 0.28 | 83.23 ± 6.85 | 83.43 ± 7.04 |
| 109b | 25.07 ± 2.08 | 10.62 ± 1.35 | 174.91 ± 12.30 |
| Doxorubicin | 5.43 ± 0.24 | 3.18 ± 0.32 | 4.10 ± 1.37 |
3.6. Coumarin-Based Substituted Benzenesulfonamide Derivatives as Anticancer Agents
Debbabi and coworkers reported the series of substituted cyanoacetohydrazonoethyl methyl-based benzenesulfonamide derivatives (Scheme 19) and evaluated for anticancer and antimicrobial therapeutic potential. The different coumarin-based benzenesulfonamides scaffolds 112 and 114 were achieved in dioxane under refluxing conditions via the reaction of cyanoacetyl containing hydrazonoethyl methyl-based benzenesulfonamide 110 with salicyldehyde 111 and 2-hydroxy naphthalene-1-carbaldehyde 113, respectively (Scheme 19) [82].
Scheme 19.

Synthesis of coumarin-based benzenesulfonamide derivatives 112 and 114.
The series of all newly synthesized cyanoacetohydrazonoethyl-N-ethyl-N-methyl benzenesulfonamide derivatives were evaluated for anti-cancer therapeutic potential against the MCF cell line. The coumarin moiety containing a benzenesulfonamide 112 scaffold (Figure 20) displayed significant and remarkably high anticancer activity against the MCF-7 cell line, with the IC50 value 1.08 µg/mL (Table 18) among all synthesized scaffolds. The MTX (methotrexate) was used as a standard compound with an IC50 value of 12.3 µg/mL (Table 18) against MCF-7 [82].
Figure 20.

Structure of anticancer coumarin-based benzene sulfonamide derivative 112.
Table 18.
Coumarin-based benzenesulfonamides as anticancer agents.
| Compound Number | Human Breast Cell Line (MCF-7) IC50 (µg/mL) |
|---|---|
| 112 | 1.08 |
| MTX | 12.3 |
4. Conclusions
The conducted review of coumarin sulfonamide derivatives and their structural hybrids with various substitution patterns displayed the anticancer and carbonic anhydrases’ different isoforms inhibition potential against a wide variety of cancer cell lines. Cancer is considered a leading cause of high mortality and morbidity. A great number of different anticancer and carbonic anhydrase inhibitors were developed, but due to rapidly evolving drug resistance, reduced the efficacy of curing various types of cancers. Most of the coumarin sulfonamides and benzene sulfonamides containing coumarin moiety derivatives cited in this review are the most active or nearly equal in the efficacy and therapeutic potential, in comparison with marketed anticancer drug and enzyme inhibitors. The SAR analysis provides information to develop novel coumarin sulfonamide derivatives and structural hybrids with improved efficacy, better therapeutic potential, and lower side effects.
Author Contributions
Conceptualization: A.I.; resources: S.A. (Sumbel Afroz), S.H. and I.N.; writing original draft: L.R., A.I. and S.A. (Sajjad Ahmad), F.B.; writing—review and editing: K.K.-M. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.De Souza L.G., Rennã M.N., Figueroa-Villar J.D. Coumarins as cholinesterase inhibitors. A review. Chem. Biol. Interact. 2016;254:11–23. doi: 10.1016/j.cbi.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Peng X.M., Damu L.V., Zhou C.H. Current developments of coumarin compounds in medicinal chemistry. Curr. Pharm. Des. 2013;19:3884–3930. doi: 10.2174/1381612811319210013. [DOI] [PubMed] [Google Scholar]
- 3.Kostova I. Synthetic and natural coumarins as antioxidants. Mini Rev. Med. Chem. 2006;6:365–374. doi: 10.2174/138955706776361457. [DOI] [PubMed] [Google Scholar]
- 4.Pereira M.T., Franco P.D., Vitorio F., Kümmerle E.A. Coumarin compounds in medicinal chemistry: Some important examples from the last years. Curr. Top. Med. Chem. 2018;18:124–148. doi: 10.2174/1568026618666180329115523. [DOI] [PubMed] [Google Scholar]
- 5.Murray R.D.H. Coumarins. Nat. Prod. Rep. 1995;12:477–505. doi: 10.1039/np9951200477. [DOI] [Google Scholar]
- 6.Pereira T.M., Vitório F., Amaral R.C., Zanoni K.P.S., Iha N.Y.M., Kümmerle A.E. Microwave-assisted synthesis and photophysical studies of novel fluorescent N-acylhydrazone and semicarbazone-7-OH-coumarin dyes. New J. Chem. 2016;40:8846–8854. doi: 10.1039/C6NJ01532H. [DOI] [Google Scholar]
- 7.Symeonidis T., Chamilos M., Litina D.J.H., Kallitsakis M., Litinas K.E. Synthesis of hydroxycoumarins and hydroxybenzo[f]- or [h]coumarins as lipid peroxidation inhibitors. Bioorg. Med. Chem. Lett. 2009;19:1139–1142. doi: 10.1016/j.bmcl.2008.12.098. [DOI] [PubMed] [Google Scholar]
- 8.Sashidhara K.V., Kumar A., Kumar J.M., Sinha S.S. Synthesis and in vitro evaluation of novel coumarinechalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010;20:7205–7721. doi: 10.1016/j.bmcl.2010.10.116. [DOI] [PubMed] [Google Scholar]
- 9.Reddy N.S., Mallireddigari M.R., Cosenza S., Gumireddy K., Bell S.C., Reddy E.P., Reddy M.R. Synthesis of new coumarin 3-(N-aryl) sulfonamides and their anticancer activity. Bioorg. Med. Chem. Lett. 2004;14:4093–4097. doi: 10.1016/j.bmcl.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Nasr S.T., Bondock M. Youns, Anticancer activity of new coumarin substituted hydrazide-hydrazone derivatives. Eur. J. Med. Chem. 2014;76:539–548. doi: 10.1016/j.ejmech.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 11.Thakur A., Singla R., Jaitak V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015;101:476–495. doi: 10.1016/j.ejmech.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 12.Devji T., Reddy C., Woo C., Awale S., Kadota S., Carrico-Moniz D. Pancreatic anticancer activity of a novel geranylgeranylatedcoumarin derivative. Bioorg. Med. Chem. Lett. 2011;21:5770–5773. doi: 10.1016/j.bmcl.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Alshibl H.M., Al-Abdullah E.S., Haiba M.E., Alkahtani H.M., Awad G.E.A., Mahmoud A.H., Ibrahim B.M.M., Bari A., Villinger A. Synthesis and Evaluation of New Coumarin Derivatives as Antioxidant, Antimicrobial, and Anti-Inflammatory Agents. Molecules. 2020;25:3251. doi: 10.3390/molecules25143251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melagraki G., Afantitis A., Igglessi-Markopoulou O., Detsi A., Koufaki M., Kontogiorgis C., Hadjipavlou-Litina D.J. Synthesis and evaluation of the antioxidant and anti-inflammatory activity of novel coumarin-3-aminoamides and their alpha-lipoic acid adducts. Eur. J. Med. Chem. 2009;44:3020–3026. doi: 10.1016/j.ejmech.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 15.Fylaktakidou K.C., Hadjipavlou-Litina D.J., Litinas K.E., Nicolaides D.N. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr. Pharm. Des. 2004;10:3813–3833. doi: 10.2174/1381612043382710. [DOI] [PubMed] [Google Scholar]
- 16.Kontogiorgis C., Hadjipavlou-Litina D. Biological evaluation of several coumarins derivatives designed as possible anti-inflammatory/antioxidant agents. J. Enzym. Inhib. Med. Chem. 2003;18:63–69. doi: 10.1080/1475636031000069291. [DOI] [PubMed] [Google Scholar]
- 17.Rehman S.U., Chohan Z.H., Gulnaz F., Supuran C.T. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J. Enzym. Inhib. Med. Chem. 2005;20:333–340. doi: 10.1080/14756360500141911. [DOI] [PubMed] [Google Scholar]
- 18.Kalluraya B., Vishwanatha P., Isloor A.M., Rai G., Kotian M. Synthesis and biological activity of 6-substituted-3-[2-(5-substituted-2-furfurylidenehydrazino)-4-thiazolyl] coumarins. Boll. Chim. Farm. 2000;139:263–266. doi: 10.3390/80200275. [DOI] [PubMed] [Google Scholar]
- 19.Musiciki B., Periers A.M., Laurin P., Ferroud D., Benedetti Y., Lachaud S., Chatreaux F., Haesslein J.L., LLtis A., Pierre C., et al. Improved antibacterial activities of coumarin antibiotics bearing 5′,5′-dialkylnoviose: Biological activity of RU79115. Bioorg. Med. Chem. Lett. 2000;10:1695. doi: 10.1016/S0960-894X(00)00304-8. [DOI] [PubMed] [Google Scholar]
- 20.De Souza S.M., Monache F.D., Smânia A. Antibacterial activity of coumarins. Z. Nat. C. 2005;60:693–700. doi: 10.1515/znc-2005-9-1006. [DOI] [PubMed] [Google Scholar]
- 21.Guerra F.Q.S., de Araújo R.S.A., de Sousa J.P., de Pereira F.O., Mendonça-Junior F.J.B., Barbosa-Filho J.M., de Oliveira Lima E. Evaluation of antifungal activity and mode of action of new coumarin derivative, 7-hydroxy-6-nitro-2h-1-benzopyran-2-one, against Aspergillus spp. Evid. Based Complement. Altern. Med. 2015;2015:925096. doi: 10.1155/2015/925096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montagner C., de Souza S.M., Groposo C., DelleMonache F., Smânia E.F.A., Smânia A., Jr. Antifungal activity of coumarins. Z. Nat. C J. Biosci. 2008;63:21–28. doi: 10.1515/znc-2008-1-205. [DOI] [PubMed] [Google Scholar]
- 23.Sardari S., Mori Y., Horita K., Micetich R.G., Nishibe S., Daneshtalab M. Synthesis and antifungal activity of coumarins and angular furanocoumarins. Bioorg. Med. Chem. 1999;7:1933–1940. doi: 10.1016/S0968-0896(99)00138-8. [DOI] [PubMed] [Google Scholar]
- 24.Ghate M., Manohar D., Kulkarni V., Shobha R., Kattimani S.Y. Synthesis of vanillin ethers from 4-(bromomethyl) coumarins as anti-inflammatory agents. Eur. J. Med. Chem. 2003;38:297–302. doi: 10.1016/S0223-5234(03)00016-3. [DOI] [PubMed] [Google Scholar]
- 25.Christos A.K., Dimitra J.H. Synthesis and anti-inflammatory activity of coumarin derivatives. J. Med. Chem. 2005;48:6400–6408. doi: 10.1021/jm0580149. [DOI] [PubMed] [Google Scholar]
- 26.Li H., Yao Y., Li L. Coumarins as potential antidiabetic agents. J. Pharm. Pharmacol. 2017;69:1253–1264. doi: 10.1111/jphp.12774. [DOI] [PubMed] [Google Scholar]
- 27.Campos-Toimil M., Orallo F., Santana L., Uriarte E. Synthesis and vasorelaxant activity of new coumarin and furocoumarin derivatives. Bioorg. Med. Chem. Lett. 2002;12:783–786. doi: 10.1016/S0960-894X(02)00015-X. [DOI] [PubMed] [Google Scholar]
- 28.Ghate M., Kusanur R.A., Kulkarni M.V. Synthesis and in vivo analgesic and anti-inflammatory activity of some bi heterocyclic coumarin derivatives. Eur. J. Med. Chem. 2005;40:882–887. doi: 10.1016/j.ejmech.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 29.Kostova I., Raleva S., Genova P., Argirova R. Structure-activity relationships of synthetic coumarins as hiv-1 inhibitors. Bioinorg. Chem. Appl. 2006;2006:68274. doi: 10.1155/BCA/2006/68274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ojala T., Remes S., Haansuu P., Vuorela H., Hiltunen R., Haatela K., Vuorela P. Antimicrobial activity of some coumarin containing herbal plants growing in Finland. J. Ethnopharmacol. 2000;73:299–305. doi: 10.1016/S0378-8741(00)00279-8. [DOI] [PubMed] [Google Scholar]
- 31.Abdelhafez M.O., Amin M.K., Batran Z.R., Maher J.T., Nada A.S., Sethumadhavan S. Synthesis, anticoagulant and PIVKA-II induced by new 4-hydroxycoumarin derivatives. Bioorg. Med. Chem. 2010;18:3371–3378. doi: 10.1016/j.bmc.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Ahmad R., Asad M., Siddiqui N.Z., Kumar A. Evaluation of antipyretic and antinociceptive potential of new heterocyclic derivatives of 3-formyl-4-hydroxycoumarin in rats. J. Pharm. Appl. Sci. 2013;3:253–259. [Google Scholar]
- 33.Mostajeran N., Arshad F.A., Aliyan H., Massah A.R. Solvent-free synthesis and antibacterial evaluation of novel coumarin sulfonamides. Pharm. Chem. J. 2018;52:1–7. doi: 10.1007/s11094-018-1756-y. [DOI] [Google Scholar]
- 34.Irfan A., Rubab L., Rehman U.M., Anjum R., Ullah S., Marjana M., Qadeer S., Sana S. Coumarin sulfonamide derivatives: An emerging class of therapeutic agents. Heterocycl. Commun. 2020;26:46–59. doi: 10.1515/hc-2020-0008. [DOI] [Google Scholar]
- 35.Alterio V., Vitale R.M., Monti S.M., Pedone C., Scozzafava A., Cecchi A., de Simone G., Supuran C.T. Carbonic anhydrase inhibitors: X-ray and molecular modeling study for the interaction of a fluorescent antitumor sulfonamide with isozyme II and IX. J. Am. Chem. Soc. 2006;128:8329–8335. doi: 10.1021/ja061574s. [DOI] [PubMed] [Google Scholar]
- 36.Grandane A., Tanc M., Mannelli D.C., Carta F., Ghelardini C., Zalubovskis R., Supuran T.C. Substituted sulfocoumarins are selectivecarbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J. Med. Chem. 2015;58:3975–3983. doi: 10.1021/acs.jmedchem.5b00523. [DOI] [PubMed] [Google Scholar]
- 37.Hassanpour S.H., Dehghani M. Review of cancer from perspective of molecular. J. Cancer Res. Pract. 2017;4:127–129. doi: 10.1016/j.jcrpr.2017.07.001. [DOI] [Google Scholar]
- 38.Arruebo M., Vilaboa N., Sáez-Gutierrez B., Lambea J., Tres A., Valladares M., González-Fernández Á. Assessment of the evolution of cancer treatment therapies. Cancers. 2011;3:3279–3330. doi: 10.3390/cancers3033279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grasso C.S., Wu Y.M., Robinson D.R., Cao X., Dhanasekaran S.M., Khan A.P., Quist M.J., Jing X.J., Lonigro R.J., Brenner J.C., et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Supuran C.T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2012;27:759–772. doi: 10.3109/14756366.2012.672983. [DOI] [PubMed] [Google Scholar]
- 41.Wagner J., Avvaru S.B., Robbins H.A., Scozzafava A., Supuran T.C., McKenna R. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: Solution and crystallographic investigations. Bioorg. Med. Chem. 2010;18:4873–4878. doi: 10.1016/j.bmc.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Supuran C.T. Bacterial carbonic anhydrases as drug targets: Toward novel antibiotics. Front. Pharmacol. 2011;2:34. doi: 10.3389/fphar.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neri D., Supuran C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011;10:767–777. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 44.Supuran C.T. Carbonic anhydrase inhibitors: An editorial. Expert Opin. Ther. Pat. 2013;23:677–679. doi: 10.1517/13543776.2013.778246. [DOI] [PubMed] [Google Scholar]
- 45.Angeli A., Carta F., Supuran C.T. Carbonic Anhydrases: Versatile and Useful Biocatalysts in Chemistry and Biochemistry. Catalysts. 2020;10:1008. doi: 10.3390/catal10091008. [DOI] [Google Scholar]
- 46.Capasso C., Supuran C.T. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: Can bacterial carbonic anhydrases shed new light on evolution of bacteria. J. Enzym. Inhib. Med. Chem. 2015;30:325–332. doi: 10.3109/14756366.2014.910202. [DOI] [PubMed] [Google Scholar]
- 47.Supuran C.T., Capasso C. The η-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin. Ther. Targets. 2015;19:551–563. doi: 10.1517/14728222.2014.991312. [DOI] [PubMed] [Google Scholar]
- 48.Del Prete S., Vullo D., De Luca V., Supuran C.T., Capasso C. Biochemical characterization of the δ-carbonic anhydrase from the marine diatom Thalassiosiraweissflogii, TweCA. J. Enzym. Inhib. Med. Chem. 2014;29:906–911. doi: 10.3109/14756366.2013.868599. [DOI] [PubMed] [Google Scholar]
- 49.Stefanucci A., Angeli A., Dimmito M.P., Luisi G., Del Prete S., Capasso C., Donald W.A., Mollica A., Supuran C.T. Activation of β- and γ-carbonic anhydrases from pathogenic bacteria with tripeptides. J. Enzym. Inhib. Med. Chem. 2018;33:945–950. doi: 10.1080/14756366.2018.1468530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Angeli A., Del Prete S., Alasmary F.A.S., Alqahtani L.S., AlOthman Z., Donald W.A., Capasso C., Supuran C.T. The first activation studies of the η-carbonic anhydrase from the malaria parasite Plasmodium falciparum with amines and amino acids. Bioorg. Chem. 2018;80:94–98. doi: 10.1016/j.bioorg.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 51.Angeli A., Buonanno M., Donald W.A., Monti S.M., Supuran C.T. The zinc—But not cadmium—Containing ζ-carbonic from the diatom Thalassiosiraweissflogii is potently activated by amines and amino acids. Bioorg. Chem. 2018;80:261–265. doi: 10.1016/j.bioorg.2018.05.027. [DOI] [PubMed] [Google Scholar]
- 52.Angeli A., Kuuslahti M., Parkkila S., Supuran C.T. Activation studies with amines and amino acids of the α-carbonic anhydrase from the pathogenic protozoan Trypanosoma cruzi. Bioorg. Med. Chem. 2018;26:4187–4190. doi: 10.1016/j.bmc.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 53.Angeli A., Del Prete S., Osman S.M., Alasmary F.A.S., AlOthman Z., Donald W.A., Capasso C., Supuran C.T. Activation studies with amines and amino acids of the β-carbonic anhydrase encoded by the Rv3273 gene from the pathogenic bacterium Mycobacterium tuberculosis. J. Enzym. Inhib. Med. Chem. 2018;33:364–369. doi: 10.1080/14756366.2017.1422250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Angeli A., Del Prete S., Donald W.A., Capasso C., Supuran C.T. The γ-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae is potently activated by amines and amino acids. Bioorg. Chem. 2018;77:1–5. doi: 10.1016/j.bioorg.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Jensen E.L., Clement R., Kosta A., Maberly S.C., Gontero B. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J. 2019;13:2094–2106. doi: 10.1038/s41396-019-0426-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishimori I., Minakuchi T., Onishi S., Vullo D., Cecchi A., Scozzafava A., Supuran C.T. Carbonic anhydrase inhibitors: Cloning, characterization, and inhibition studies of the cytosolic isozyme III with sulfonamides. Bioorg. Med. Chem. 2007;15:7229–7236. doi: 10.1016/j.bmc.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 57.Köhler K., Hillebrecht A., Schulze W.J., Innocenti A., Heine A., Supuran C.T., Klebe G. Saccharin inhibits carbonicanhydrases: Possible explanation for its unpleasant metallic aftertaste. Angew. Chem. 2007;46:7697–7699. doi: 10.1002/anie.200701189. [DOI] [PubMed] [Google Scholar]
- 58.Supuran C.T., Scozzafava A., Ilies M.A., Briganti F. Carbonic anhydrase inhibitors. Synthesis of sulfonamides incorporating 2,4,6-trisubstituted-pyridinium-ethylcarboxamido moieties possessing membrane-impermeability and in vivo selectivity for the membrane-bound (CA IV) versus the cytosolic (CA I and CA II) isozymes. J. Enzym. Inhib. 2000;15:381–401. doi: 10.1080/14756360009040695. [DOI] [PubMed] [Google Scholar]
- 59.Scozzafava A., Briganti F., Ilies M.A., Supuran C.T. Carbonic anhydrase inhibitors. Synthesis of membrane-impermeant low molecular weight sulfonamides possessing in vivo selectivity for the membrane-bound versus the cytosolic isozymes. J. Med. Chem. 2000;43:292–300. doi: 10.1021/jm990479+. [DOI] [PubMed] [Google Scholar]
- 60.De Simone G., Vitale R.M., Di Fiore A., Pedone C., Scozzafava A., Montero J.L., Winum J.Y., Supuran C.T. Carbonic anhydrase inhibitors:hypoxia-activatable sulfonamides incorporatingdisulfide bonds that target the tumor-associatedisoform IX. J. Med. Chem. 2006;49:5544–5551. doi: 10.1021/jm060531j. [DOI] [PubMed] [Google Scholar]
- 61.Maresca A., Temperini C., Vu H., Pham N.B., Poulsen S.A., Scozzafava A., Quinn R.J., Supuran C.T. Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 2009;131:3057–3062. doi: 10.1021/ja809683v. [DOI] [PubMed] [Google Scholar]
- 62.Maresca A., Temperini C., Pochet L., Masereel B., Scozzafava A., Supuran C.T. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J. Med. Chem. 2010;53:335–344. doi: 10.1021/jm901287j. [DOI] [PubMed] [Google Scholar]
- 63.Supuran C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug. Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 64.Supuran C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016;473:2023–2032. doi: 10.1042/BCJ20160115. [DOI] [PubMed] [Google Scholar]
- 65.Nocentini A., Supuran C.T. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin. Drug Discov. 2019;14:1175–1197. doi: 10.1080/17460441.2019.1651289. [DOI] [PubMed] [Google Scholar]
- 66.Supuran C.T. Coumarin carbonic anhydrase inhibitors from natural sources. J. Enzym. Inhib. Med. Chem. 2020;35:1462–1470. doi: 10.1080/14756366.2020.1788009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Z.C., Qin Y.J., Wang P.F., Yang Y.A., Wen Q., Zhang X., Qiu H.Y., Duan Y.T., Wang Y.T., Sang Y.L., et al. Sulfonamides containing coumarin moieties selectively and potently inhibit carbonic anhydrases II and IX: Design, synthesis, inhibitory activity and 3D-QSAR analysis. Eur. J. Med. Chem. 2013;66:1–11. doi: 10.1016/j.ejmech.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 68.Kurt B.Z., Sonmez F., Bilen C., Ergun A., Gencer N., Arslan O., Kucukislamoglu M. Synthesis, antioxidant and carbonic anhydrase I and II inhibitory activities of novel sulphonamide-substituted coumarylthiazole derivatives. J. Enzym. Inhib. Med. Chem. 2016;31:78–89. doi: 10.3109/14756366.2015.1077823. [DOI] [PubMed] [Google Scholar]
- 69.Bozdag M., Ferraroni M., Carta F., Vullo D., Lucarini L., Orlandini E., Rossello A., Nuti E., Scozzafava A., Masini E., et al. Structural insights on carbonic anhydrase inhibitory action, isoform selectivity, and potency of sulfonamides and coumarins incorporating arylsulfonylureido groups. J. Med. Chem. 2014;57:9152–9167. doi: 10.1021/jm501314c. [DOI] [PubMed] [Google Scholar]
- 70.Chandak N., Ceruso M., Supuran C.T., Sharma P.K. Novel sulfonamide bearing coumarin scaffolds as selective inhibitors of tumor associated carbonic anhydrase isoforms IX and XII. Bioorg. Med. Chem. 2016;24:2882–2886. doi: 10.1016/j.bmc.2016.04.052. [DOI] [PubMed] [Google Scholar]
- 71.Lu X.Y., Wang Z.C., Ren S.Z., Shen F.Q., Man R.J., Zhu H.L. Coumarin sulfonamides derivatives as potent and selective COX-2 inhibitors with efficacy in suppressing cancer proliferation and metastasis. Bioorg. Med. Chem. Lett. 2016;26:3491–3498. doi: 10.1016/j.bmcl.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 72.Dar’in D., Kantin G., Kalinin S., Sharonova T., Bunev A., Ostapenko G.I., Nocentini A., Sharoyko V., Supuran C.T., Krasavin M. Investigation of 3-sulfamoyl coumarins against cancer-related IX and XII isoforms of human carbonic anhydrase as well as cancer cells leads to the discovery of 2-oxo-2H-benzo[h]chromene-3-sulfonamide. A new caspase-activating proapoptotic agent. Eur. J. Med. Chem. 2021;222:113589. doi: 10.1016/j.ejmech.2021.113589. [DOI] [PubMed] [Google Scholar]
- 73.Zengin K.B., Sonmez F., Ozturk D., Akdemir A., Angeli A., Supuran C.T. Synthesis of coumarin-sulfonamide derivatives and determination of their cytotoxicity, carbonic anhydrase inhibitory and molecular docking studies. Eur. J. Med. Chem. 2019;183:111702. doi: 10.1016/j.ejmech.2019.111702. [DOI] [PubMed] [Google Scholar]
- 74.Abdelrahman M.A., Ibrahim H.S., Nocentini A., Eldehna W.M., Bonardi A., Abdel-Aziz H.A., Gratteri P., Abou-Seri S.M., Supuran C.T. Novel 3-substituted coumarins as selective human carbonic anhydrase IX and XII inhibitors: Synthesis, biological and molecular dynamics analysis. Eur. J. Med. Chem. 2021;209:112897. doi: 10.1016/j.ejmech.2020.112897. [DOI] [PubMed] [Google Scholar]
- 75.Aoki T., Hyohdoh I., Furuichi N., Ozawa S., Watanabe F., Matsushita M., Sakaitani M., Ori K., Takanashi K., Harada N., et al. The sulfamide moiety affords higher inhibitory activity and oral bioavailability to a series of coumarin dual selective RAF/MEK inhibitors. Bioorg. Med. Chem. Lett. 2013;23:6223–6227. doi: 10.1016/j.bmcl.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Amin M.K., Eissa A.M., Abou-Seri M.S., Awadallah M.F., Hassan S.G. Synthesis and biological evaluation of novel coumarin-pyrazoline hybrids endowed with phenylsulfonyl moiety as antitumor agents. Eur. J. Med. Chem. 2013;60:187–198. doi: 10.1016/j.ejmech.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 77.Zhang J., Tan Y., Li G., Chen L., Nie M., Wang Z., Ji H. Coumarin Sulfonamides and Amides Derivatives: Design, Synthesis, and Antitumor Activity In Vitro. Molecules. 2021;26:786. doi: 10.3390/molecules26040786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.El-Sawy R.E., Ebaid S.M., Rady M.H., Shalby B.A., Ahmed M.K., Abo-Salem M.H. Synthesis and molecular docking of novel non-cytotoxic anti-angiogenic sulfonyl coumarin derivatives against hepatocellular carcinoma cells in vitro. J. Appl. Pharm. Sci. 2017;7:49–66. doi: 10.7324/JAPS.2017.70207. [DOI] [Google Scholar]
- 79.Holiyachi M., Shastri S.L., Chougala B.M., Shastri L.A., Joshi S.D., Dixit S.R., Nagarajaiah H., Sunagar V.A. Design, Synthesis and Structure-Activity Relationship Study of Coumarin Benzimidazole Hybrid as Potent Antibacterial and Anticancer Agents. ChemistrySelect. 2016;1:4638–4644. doi: 10.1002/slct.201600665. [DOI] [Google Scholar]
- 80.Durgapal S.D., Soman S.S. Evaluation of novel coumarin-proline sulfonamide hybrids as anticancer and antidiabetic agents. Synth. Commun. 2019;49:2869–2883. doi: 10.1080/00397911.2019.1647439. [DOI] [Google Scholar]
- 81.Sabt A., Abdelhafez O.M., Haggar R.S.E., Madkour H.M.F., Eldehna W.M., El-Khrisy E.E.D.A., Abdel-Rahman M.A., Rashed L. ANovel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: Synthesis, in vitro biological evaluation, and QSAR studies. J. Enzym. Inhib. Med. Chem. 2018;33:1095–1107. doi: 10.1080/14756366.2018.1477137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Debbabi K.F., Harbi S.A.A., Saidi H.M.A., Aljuhani E.H., Gl S.M.A.E., Bashandy M.S. Study of reactivity of cyanoacetohydrazonoethyl-N-ethyl-N-methyl benzenesulfonamide: Preparation of novel anticancer and antimicrobial active heterocyclic benzenesulfonamide derivatives and their molecular docking against dihydrofolate reductase. J. Enzym. Inhib. Med. Chem. 2016;31:7–19. doi: 10.1080/14756366.2016.1217851. [DOI] [PubMed] [Google Scholar]