

Abstract
Motivation
Genomic DNA replicates according to a reproducible spatiotemporal program, with some loci replicating early in S phase while others replicate late. Despite being a central cellular process, DNA replication timing studies have been limited in scale due to technical challenges.
Results
We present TIGER (Timing Inferred from Genome Replication), a computational approach for extracting DNA replication timing information from whole genome sequence data obtained from proliferating cell samples. The presence of replicating cells in a biological specimen leads to non-uniform representation of genomic DNA that depends on the timing of replication of different genomic loci. Replication dynamics can hence be observed in genome sequence data by analyzing DNA copy number along chromosomes while accounting for other sources of sequence coverage variation. TIGER is applicable to any species with a contiguous genome assembly and rivals the quality of experimental measurements of DNA replication timing. It provides a straightforward approach for measuring replication timing and can readily be applied at scale.
Availability and implementation
TIGER is available at https://github.com/TheKorenLab/TIGER.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
High-throughput DNA sequencing has become a central technique in biomedicine. It can be applied for whole genome sequencing or targeted sequencing of genomic DNA, as well as coupled to a variety of biochemical techniques, such as ChIP-seq, ATAC-seq and others in order to study the epigenome. Many assays that utilize DNA sequencing, in particular, the genotyping of copy number variations (CNVs) or alterations (CNAs), as well as many epigenomic assays, rely on binning read counts along chromosomes as a measure of genetic or epigenetic spatial heterogeneity. Non-uniform read coverage is thus used to measure variable DNA copy number or the preferential presence of open chromatin, DNA-bound proteins or other epigenetic marks at different locations across chromosomes.
Another, less appreciated factor that influences sequencing read coverage along chromosomes is DNA replication timing. During the S phase of the cell cycle, DNA is replicated according to a defined spatiotemporal program in which replication initiates at specific sites along chromosomes (replication origins) and at specific times (Fragkos et al., 2015). The locations and activation times of origins along chromosomes, together with the rate of replication fork progression, define a non-uniform landscape of genome replication with different chromosomal regions replicating at different times along S phase. DNA replication timing is highly reproducible among cells and samples, and conserved in evolution. Early replication timing is strongly correlated with high gene density, active gene expression, open chromatin and activating histone marks (Aladjem, 2007). On the other hand, late-replication is correlated with a higher mutation rate in both somatic and germline cells, and replication timing in general has been shown to interface with various aspects of genome stability (Gaboriaud and Wu, 2019; Koren, 2014). Taken together, DNA replication timing is a central cellular process that bridges genetic and epigenetic inheritance with important implications to evolution, development and disease.
Genomic measurements of DNA replication timing typically rely on sequencing DNA from a population of cells enriched for cells in S phase, in order to identify replicated DNA or an increase in the copy number of certain genomic regions [reviewed in (Hulke et al., 2020)]. While these experiments have been applied to various species and cell types, they remain relatively difficult to implement in general, and on large scales in particular, limiting progress in the DNA replication field. However, we have previously shown that samples containing a sufficient fraction of cells in S phase demonstrate measurable imbalances in DNA copy number along chromosomes, allowing the inference of replication timing profiles without cell labeling or sorting. Specifically, by computationally generating pseudo-data representing samples with 0-100% cells in S phase, we showed that as little as 10% of cells being in S phase is sufficient in order to infer high-quality DNA replication timing profiles (Koren et al., 2014). DNA replication in these S phase cells leads to an increased DNA copy number in proportion to the replication timing of the respective genomic regions. After parsing these signals from other influences on DNA copy number (in particular, CNVs/CNAs, as well as technical influences such as alignability and GC content effects), high-resolution replication timing profiles can be derived directly from these sequence data. These profiles are of equivalent quality to replication timing profiles measured using sorted cells, while avoiding much of the associated experimental manipulations (Ding et al., 2020; Koren et al., 2014).
The ability to infer DNA replication timing from whole-genome sequence data provides an incredibly powerful approach for advancing the replication timing field. It provides a means of easily measuring replication timing in various samples, and is highly scalable. In addition to the utility of this approach for investigating replication timing, the influences of DNA replication on sequence read coverage is a potential confounder in numerous genetic and epigenetic studies that rely on coverage analysis and/or read counting applications. The ability to extract replication timing signals from these data enables their identification, and hence separation, from the other biological factors being studied.
Here, we introduce TIGER (Timing Inferred from Genome Replication), a unified computational pipeline for extracting DNA replication timing information from whole-genome sequence data. TIGER analyzes DNA copy number, corrects for alignability, GC bias and CNVs/CNAs, filters outliers and smoothes and normalizes the copy number data to obtain high-resolution replication timing profiles. TIGER is applicable to samples containing proliferating cells of any species with a contiguous reference genome assembly.
2 Materials and methods
2.1 Whole genome sequence data
Whole genome sequence data for mouse embryonic fibroblasts (MEFs) were obtained from Yang et al. (2019) (SRA accession number PRJNA554729; 38–42× coverage). Whole genome sequence data for two mouse embryonic stem cells (mESC) and four induced pluripotent stem (iPS) cell lines were obtained from Sugiura et al. (2014) (SRA accession number DRP000548; 14–27× coverage). Data for the human ESC line CHB1 was obtained from Ding et al. (2020) (dbGaP accession number: phs001957; 17× coverage), for the human ESC line HUES63 from Merkle et al. (2020) (30× coverage) and for the human LCL GM12878 from Eberle et al. (2017) (dbGap accession number: phs001224.v1.p1; 50× coverage).
2.2 Whole genome sequencing
The GM12878 lymphoblastoid cell line (Coriell Institute) was grown in RPMI 1640 medium (Corning) supplemented with 15% FBS at 37°C in a 5% CO2 atmosphere. DNA was isolated using the MasterPure™ DNA Purification Kit (Epicentre) and libraries were prepared with the TruSeq DNA PCR-Free Library Prep Kit (Illumina). Paired-end sequencing was performed for 150 cycles with the Illumina HiSeq X Ten.
2.3 Alignability filter
Sequence fastq files were aligned using BWA-MEM to either the human hg19 reference genome or the mouse mm10 reference genome. The reference genomes were used to generate short fragments of 100bps that correspond to each location in that genome. These fragments were then aligned back to the same reference genome, after which fragments that did not align to a single location were flagged and added to a list of non-uniquely alignable loci. This was defined as the alignability filter. Previous research suggested that there is no benefit in using fragments larger than 100bps, even for sequencing libraries with reads longer than 100bps (Handsaker et al., 2015).
2.4 ‘Read number windows’
The alignability filter was used to define genomic windows of equal number of uniquely alignable base pairs, which were used for all subsequent analyses. Since non-uniquely alignable loci are not uniformly distributed across the genome, the resulting windows vary with respect to their physical length. We recommend a window size of 10Kb of uniquely alignable base pairs by default, although shorter or longer windows can also be considered depending on data quality and/or sequence coverage. Windows that span gaps in the reference genome (∼0.1% of the human genome version hg19 and ∼0.2% of the mouse genome version mm10) were removed from further consideration.
2.5 Counting reads in read number windows
The locations of all sequence reads were extracted from BAM files using SAMtools after quality filters for sequence reads that were not primary alignments, were PCR duplicates or had MAPQ scores lower than 10. Only the first reads in read pairs were used. Using the alignability filter, non-uniquely alignable reads were removed from further analysis.
2.6 GC content normalization
We corrected for GC effects at the level of sequencing library fragments by calculating the relationship between read coverage and the GC content of DNA sequences corresponding to typical sequencing fragment lengths (400 bp). This was implicated in four steps.
The first step (implicated in the script ‘TIGER_generate_processing_files’ and performed once for a given genome and window size) calculates the GC content of all 401 bp (200 bp on each side of each considered base pair) fragments in the genome. It saves all the genomic positions belonging to each GC content bin (401 total bins), excluding positions falling within the alignability filter. Subsequently, for each read number window (defined above), the number of base pairs belonging to each GC content bin are counted.
The second step (implicated in the script ‘TIGER_generate_replication_profiles’) was performed separately for each sample. It assigns each autosomal sequence read in the sample to a GC content bin and then calculates the relationship between GC content and read coverage (as fraction of autosomal reads divided by fraction of autosomal base pairs) in that sample.
Only genomic regions with a copy number consistent with the sample’s ploidy were considered for calculating the GC content bias. To implement this, a segmentation algorithm (implicated in ‘TIGER_segment_filt’; see section ‘Removal of copy number outliers’ below) was used to identify genomic regions with outlier copy numbers. This process is particularly important for samples that harbor aneuploidies or segmental copy number alterations; not removing these from the GC content correction may introduce biases affecting the entire genome.
In a third step, the GC content distribution of each read number window was used to calculate the expected number of reads in each window given the GC effects, the total coverage of the library and the ploidy of the sample (assumed here to be 2 by default). Specifically, in each window, the number of bps that fall into each GC bin was multiplied by the GC bias factor for that bin. This was applied only to bins of 20–80% GC (i.e. bins 81 to 321 of the 401 bins). These numbers were then summed, multiplied by the mean number of reads per window (which effectively normalizes for the overall sequencing coverage of the sample), and divided by 2 (to make the genome diploid after normalization).
Finally, the actual read counts per window (second step) were divided by the expected read count (third step) to derive a ‘normalized’ DNA copy number profile.
2.7 Removal of copy number outliers
DNA replication timing leads to continuous, low-amplitude changes in DNA copy number rather than larger, stepwise changes characteristic of CNVs/CNAs and other outlier copy number measurements (e.g. segmental duplications or other regions with problematic mapping). To separate replication timing from these other factors, we use a segmentation algorithm. Specifically, we used the Matlab function segment with an ARX model with parameters [0 1 1] and a default R2 (assumed variance of the innovations in the model) value of 0.04. Segmentation was applied on contiguous genomic regions between gaps in the reference genome. Data points within segments that have copy number values more than a given number of standard deviations (set by default to 1.5) from the autosomal data point mean values were removed. Subsequently, the same standard deviation threshold was applied within each individual chromosome. The former removes chromosomes or large chromosomal regions (e.g. chromosome arms) that have an abnormal copy number compared to the remainder of the genome (e.g. trisomies), while the latter more effectively removes short segments with outlier copy number compared to their chromosomal vicinity. Segmentation-based filtering is optimal for replication timing data because it minimizes the removal of real replication timing peaks and valleys (the segments corresponding to them receive values close to the genome average despite data points close to peaks and valleys being relatively diverged from the average); and because it removes data points that are not copy number outliers by themselves but belong to longer segments that are outliers.
2.8 Filtering, smoothing and normalization
To generate the final replication timing profiles, the raw data (read number windows after GC correction) was subjected to several additional steps. First, CNVs/CNAs and copy number outliers were removed using TIGER_segment_filt. This is similar to the GC filtering, applied once again on the GC-corrected data.
Second, the profiles were smoothed with a cubic smoothing spline using the Matlab function csaps with a default parameter of 10−17. Smoothing of MEF data was repeated because the raw data was more noisy (consistent with MEFs being less proliferative). We independently smoothed contiguous chromosome regions, defined as segments without a reference genome gap greater than 50Kb and without a data gap greater than 100Kb. Smoothing is the fundamental step that generates continuous replication timing profiles.
Last, the data were normalized to units of standard deviation.
2.9 DNA replication timing data
For comparison of TIGER results to reference replication timing data, we used S/G1 or Repli-seq data for the same cell types. S/G1 replication profiles for the human cell line GM12878 were obtained from Massey et al. (2019) (SRA accession number PRJNA419407) and re-aligned to the human reference genome hg19, while Repli-seq data for mESCs (D3, 46 C and TT2), iPS and MEF cell lines, aligned to the mouse reference genome mm10, were obtained from ReplicationDomain.com (Weddington et al., 2008). Mouse Repli-seq data were further smoothed (Matlab csaps function with parameter 10−17) in order to match the smoothing scales with the TIGER data.
3 Results
DNA replication timing has previously been measured on a genomic scale either by labeling (e.g. using BrdU), isolating and sequencing replicated DNA, or by sorting replicating (S phase) cells and sequencing their genome in comparison to the sequences of non-replicating (G1 phase) cell DNA. The need to enrich for replicating cells (as well as the labeling of cells in the former approach) is a limiting factor for the routine and large-scale application of these techniques. An alternative is to avoid cell sorting, and instead, rely on proliferating cell samples in order to detect the low-amplitude fluctuations in DNA copy number along chromosomes that occur as a result of DNA replication in a subset of cells. Given the highly quantitative nature of next-generation DNA sequencing, even small changes in DNA copy number caused by DNA replication in a subset of cells could potentially be detected. There are two main challenges in inferring replication timing from unsorted rather than sorted cells. First, the replication timing signal is weaker: instead of a 2-fold difference in DNA copy number between replicated and non-replicated genomic regions in pure S phase cell samples (or an even larger fold-difference in labeled DNA), in unsorted samples the fold-difference would theoretically equal the fraction of S phase cells in the sample (for example, if 20% of the cells are in S phase, a 1.2-fold difference in copy number along chromosomes is expected). Second, lack of sorting also means that control, G1 cells, are not sorted. Typically, concomitantly sorted G1 cells serve as ideal controls for CNVs/CNAs, alignability and GC content effects on sequencing read coverage. These factors represent biological and technical influences on DNA copy number measurements independently of DNA replication.
We previously showed that accurate DNA replication timing data can be inferred from the whole-genome DNA sequences of proliferating cell cultures (Ding et al., 2020; Koren et al., 2014). To achieve this, broad-scale DNA copy number (i.e. sequence read depth) fluctuations across chromosomes are calculated from the sequence data, while the reference genome and the sequence data itself (analyzed at a narrower spatial scale) are used to calculate alignability and GC content effects. These, in turn, are used as the equivalent of in silico generated G1 cell DNA sequence data with which the read depth data (approximating S phase sequence data) is normalized. Following additional steps of outlier filtering, smoothing and normalization, DNA replication timing profiles are obtained. These replication timing profiles are highly reproducible and highly consistent with replication timing profiles measured by sorting and sequencing S and G1 phase cells (or following BrdU labeling). Moreover, the replication profiles obtained directly from sequence data typically have sharper peaks and valleys than those obtained using other methods (Ding et al., 2020; Koren et al., 2014) (also see Fig. 3 below). This may be related to the avoidance of technical manipulations of cells and DNA. The approach of inferring replication timing from sequence data provides the most effective and scalable way so far to study DNA replication timing.
Fig. 3.
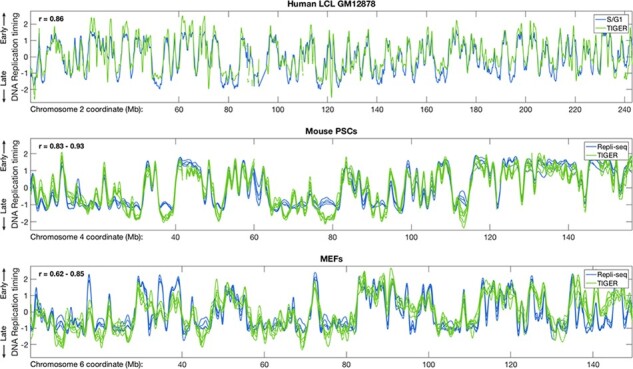
Comparison of TIGER with other experimental approaches for measuring DNA replication timing. TIGER-generated replication timing profiles were compared with S/G1 sequencing for a human LCL or Repli-seq for two mouse cell types. Replication timing profiles were highly concordant between the methods. When compared to Repli-seq, TIGER-generated profiles may even have a higher dynamic range at the earliest and latest replicating regions. The TIGER-generated profiles for MEFs, however, appear relatively noisier, likely due to lower cell proliferation compared to LCLs or PSCs. Four Repli-seq profiles are shown for mouse PSCs and for MEFs, while six TIGER profiles are shown for mouse PSCs and five for MEFs. PSCs include both mESCs and iPS cell lines (for both TIGER and Repli-seq; the differences between mESCs and iPS cells were insignificant). The indicated correlations refer to comparisons between Repli-seq and TIGER profiles
Previously, we inferred replication timing from sequence data by using the pre-processing step of Genome STRiP (software to infer DNA copy number from population-scale sequence data) followed by several custom steps of filtering and smoothing. Here, we introduce TIGER (Timing Inferred from Genome Replication), a dedicated pipeline for inference of replication timing from sequence data that performs all required steps in one package, is optimized for replication timing analysis, and can be applied to any genome for which a contiguous reference sequence is available.
Extracting DNA replication timing information from sequence data involves the analysis of subtle DNA copy number fluctuations along chromosomes. Fundamentally, this is achieved by counting the number of reads in genomic intervals, or windows, across chromosomes. The two main factors that confound the estimation of DNA copy number based on sequence read counts are the alignability of short sequences, which are not uniform across many genomes, with some sequences (e.g. repeats) aligning to more than one genomic location; and GC content, which is also not uniform across the genome and is well-known to influence the efficacy of sequencing and hence the inference of DNA copy number (Aird et al., 2011; Benjamini and Speed, 2012; Chen et al., 2013; Ekblom et al., 2014).
To measure DNA copy number while minimizing the effects of these confounding factors, TIGER defines variable-size genomic windows with uniform alignability based on a reference genome, and counts filtered sequence reads from a BAM file in those windows. It then corrects for GC content effects on read coverage in each particular sequencing library. TIGER subsequently filters CNVs/CNAs and other regions with outlier copy number measurements (e.g. repetitive regions in the genome, reference sequence gaps and other technical artifacts). Last, it smoothes and normalizes the data to produce final replication timing profiles. TIGER is implemented in two scripts. The first, ‘TIGER_generate_processing_files’, is run once per reference genome, read length and desired read window size. This script generates an alignability filter, read number windows and files used for GC correction. The second script, ‘TIGER_generate_replication_profiles’, is run on each individual sequenced sample (or group of samples) and generates DNA replication profiles from the sequence data (Fig. 1).
Fig. 1.
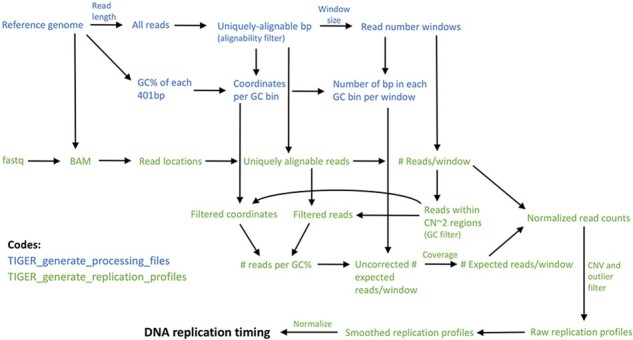
TIGER pipeline overview. TIGER consists of two scripts, the first run once for a given reference genome and window size, and the second applied per sequenced sample. TIGER extracts read locations from a sequencing library, filters reads for alignability, counts reads in genomic windows of uniform alignability, calculates a GC bias factor and uses it to correct the window read counts, filters for CNVs and outliers, and smoothes and normalizes the data to derive the final DNA replication timing profiles
TIGER consists of two scripts, the first run once for a given reference genome and window size, and the second applied per sequenced sample. TIGER extracts read locations from a sequencing library, filters reads for alignability, counts reads in genomic windows of uniform alignability, calculates a GC bias factor and uses it to correct the window read counts, filters for CNVs and outliers and smoothes and normalizes the data to derive the final DNA replication timing profiles.
We first demonstrated the TIGER pipeline on a published 17× coverage whole-genome sequence dataset from the human embryonic stem cell (hESC) line CHB1 (Ding et al., 2020). Following alignment, the locations of sequence reads were extracted, counted in 10Kb windows of uniquely alignable base pairs, corrected for GC content effects, filtered for copy number outliers and smoothed and normalized. While read depth fluctuations are evident from the very first step of visualizing reads along chromosomes, each step further distills DNA replication timing patterns from other factors. The final profiles show the expected ‘wave’ patterns of DNA replication timing gradually alternating between early and late along chromosomes (Fig. 2) and are consistent with replication profiles measured with more traditional methods [(Ding et al., 2020) and see further below].
Fig. 2.
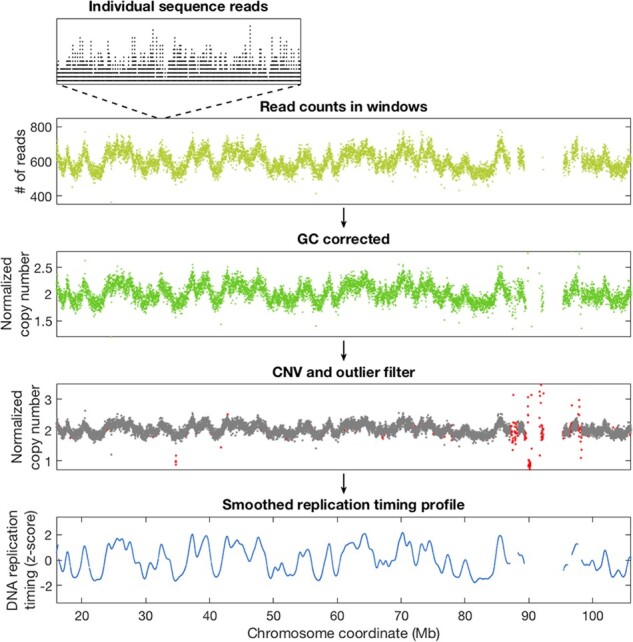
Overview of the TIGER pipeline from sequence reads to replication profiles. Whole-genome sequence data from the CHB1 hESC cell line (Ding et al., 2020) were used. While the number of individual reads mapping to specific locations along chromosomes is not uniform (top inset), counting reads in larger genomic windows (here, 10Kb of uniquely alignable sequence) already reveals the characteristic replication timing wave patterns often observed in sequence data (yellow). GC correction (green) is an important step for removing potential technical influences on sequencing read depth (which are minimal when using PCR-free library preparation as in this example), while removing copy number outliers (red) is necessary to prevent them from confounding the analysis of replication timing. Smoothing then reveals the final DNA replication timing profiles (blue), in which height represents replication timing from early to late, and peaks correspond to the locations at which replication initiates (Color version of this figure is available at Bioinformatics online.)
Whole-genome sequence data from the CHB1 hESC cell line (Ding et al., 2020) were used. While the number of individual reads mapping to specific locations along chromosomes is not uniform (top inset), counting reads in larger genomic windows (here, 10Kb of uniquely alignable sequence) already reveals the characteristic replication timing wave patterns often observed in sequence data (yellow). GC correction (green) is an important step for removing potential technical influences on sequencing read depth (which are minimal when using PCR-free library preparation as in this example), while removing copy number outliers (red) is necessary to prevent them from confounding the analysis of replication timing. Smoothing then reveals the final DNA replication timing profiles (blue), in which height represents replication timing from early to late, and peaks correspond to the locations at which replication initiates.
We then evaluated the performance of TIGER by comparing its output with previously generated replication timing profiles. We whole-genome-sequenced (without any cell sorting; 16.2× coverage) the human lymphoblastoid cell line (LCL) GM12878, applied TIGER to infer replication timing profiles, and compared them to replication profiles we previously generated for the same cell line by sorting and sequencing S and G1-phase cells (Massey et al., 2019). We further applied TIGER to published whole-genome sequence data of mouse pluripotent stem cell lines (PSCs, including both mouse embryonic stem cells lines and induced pluripotent stem cells; Sugiura et al., 2014) and of mouse embryonic fibroblasts (MEFs; Yang et al., 2019) and compared them to replication timing profiles obtained by Repli-seq (using BrdU-labeling, sorting and sequencing of late-versus-early S phase cells) for the same cell types (Weddington et al., 2008). In all cases, TIGER achieved results comparable (or superior) to previous experimental methods (Fig. 3). TIGER profiles also appear to have comparable resolution to 16-fraction, ‘high resolution’ Repli-seq for identification of replication initiation sites (Supplementary Fig. S1), although the latter approach carries the advantage of identifying allelically asynchronous replication regions (Zhao et al., 2020).
The above results, together with our previous analyses using similar approaches (Ding et al., 2020; Koren et al., 2014), show that TIGER is a proven approach for inferring high-quality replication timing profiles from proliferating cell samples. Furthermore, these results show that TIGER can be effectively applied to different species and cell types. We note, however, that TIGER is currently not applicable to highly rearranged genomes with numerous DNA copy number alterations, such as the genomes of many solid tumors. It can be effectively applied to euploid samples with no or limited aneuploidy and with copy number variations spanning <20% of the genome.
TIGER-generated replication timing profiles were compared with S/G1 sequencing for a human LCL or Repli-seq for two mouse cell types. Replication timing profiles were highly concordant between the methods. When compared to Repli-seq, TIGER-generated profiles may even have a higher dynamic range at the earliest and latest replicating regions. The TIGER-generated profiles for MEFs, however, appear relatively noisier, likely due to lower cell proliferation compared to LCLs or PSCs. Four Repli-seq profiles are shown for mouse PSCs and for MEFs, while six TIGER profiles are shown for mouse PSCs and five for MEFs. PSCs include both mESCs and iPS cell lines (for both TIGER and Repli-seq; the differences between mESCs and iPS cells were insignificant). The indicated correlations refer to comparisons between Repli-seq and TIGER profiles.
4 Discussion
We present TIGER, a computational pipeline that infers DNA replication timing profiles from whole-genome sequence data. TIGER can be applied to sequence data derived from proliferating biological samples of various cell types and species, and provides replication timing data of quality rivaling or exceeding previous experimental approaches. TIGER provides an attractive approach for studies of DNA replication timing, as it can generate replication profiles with minimal experimental manipulations and resources. It can also be applied to genome or epigenome sequence data that were generated for reasons unrelated to DNA replication timing research; in these cases, TIGER can reveal whether replication timing signals are present in these data, which may both confound the original purpose of the experiment, but also provides opportunities for deriving additional replication timing information.
While TIGER is applicable (and likely optimal) for measuring replication timing in highly proliferating cell samples, it is more limited for biological samples with a low fraction of replicating cells. For the latter, more traditional approaches of labeling and/or isolating replicating cells would perform better. Another consideration when using TIGER is the overall sequencing coverage (or read depth). In contrast to enrichment approaches, for which the signal quality saturates at relatively modest sequencing depths, inferring replication timing from whole genome sequence data benefits from deeper sequence coverage (e.g. 10–30×). Thus, deriving optimal data quality may require deeper sequencing compared to previous approaches, although it also provides the opportunity to increase data resolution and study genomic DNA replication at finer scales than possible before. As a general guideline, for optimal results we recommend applying TIGER to samples with at least 10% S phase cells and at least 10× sequence coverage (assuming 150 bp paired-end reads), although sequence coverage as low as 1× is acceptable for some applications (Supplementary Fig. S2). Last, we note that other technical factors related to DNA extraction, sequencing library preparation, sequencing platform and other potential variables could also affect the data and should be kept as constant as possible.
Further modifications to TIGER would enable its application to allele-specific replication profiling using phased SNP alleles (Koren and McCarroll, 2014), aneuploid and highly rearranged genomes such as cancer genomes, single cells, non-canonical replication events such as re-replication, and more.
Funding
This work was supported by the National Institutes of Health [DP2-GM123495 to A.K.] and the National Science Foundation [MCB-1921341 to A.K.].
Conflict of Interest: none declared.
Data availability
Sequence data generated in this study are available from the NCBI Sequence Read Archive with accession number PRJNA419407.
Supplementary Material
Contributor Information
Amnon Koren, Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853, USA.
Dashiell J Massey, Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853, USA.
Alexa N Bracci, Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853, USA.
References
- Aird D. et al. (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol., 12, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aladjem M.I. (2007) Replication in context: dynamic regulation of DNA replication patterns in metazoans. Nat. Rev. Genet., 8, 588–600. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Speed T.P. (2012) Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res., 40, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.C. et al. (2013) Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS One, 8, e62856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q. et al. (2020) The genetic architecture of DNA replication timing in human pluripotent stem cells. BioRxiv., doi:10.1101/2020.05.08.085324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle M.A. et al. (2017) A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res., 27, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekblom R. et al. (2014) Patterns of sequencing coverage bias revealed by ultra-deep sequencing of vertebrate mitochondria. BMC Genomics, 15, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M. et al. (2015) DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol., 16, 360–374. [DOI] [PubMed] [Google Scholar]
- Gaboriaud J., Wu P.J. (2019) Insights into the link between the organization of DNA replication and the mutational landscape. Genes (Basel), 10, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker R.E. et al. (2015) Large multiallelic copy number variations in humans. Nat. Genet., 47, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulke M.L. et al. (2020) Genomic methods for measuring DNA replication dynamics. Chromosome Res., 28, 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A. (2014) DNA replication timing: coordinating genome stability with genome regulation on the X chromosome and beyond. Bioessays, 36, 997–1004. [DOI] [PubMed] [Google Scholar]
- Koren A. et al. (2014) Genetic variation in human DNA replication timing. Cell, 159, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A., McCarroll S.A. (2014) Random replication of the inactive X chromosome. Genome Res., 24, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey D.J. et al. (2019) Next-generation sequencing enables spatiotemporal resolution of human centromere replication timing. Genes (Basel), 10, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle F.T. et al. (2020) Biological insights from the whole genome analysis of human embryonic stem cells. BioRxiv. [Google Scholar]
- Sugiura M. et al. (2014) Induced pluripotent stem cell generation-associated point mutations arise during the initial stages of the conversion of these cells. Stem Cell Rep., 2, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weddington N. et al. (2008) ReplicationDomain: a visualization tool and comparative database for genome-wide replication timing data. BMC Bioinformatics, 9, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.-H. et al. (2019) Erosion of the epigenetic landscape and loss of cellular identity as a cause of aging in mammals. BioRxiv, doi: 10.1101/808642v1. [DOI] [Google Scholar]
- Zhao P.A. et al. (2020) High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol., 21, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data generated in this study are available from the NCBI Sequence Read Archive with accession number PRJNA419407.