

Summary
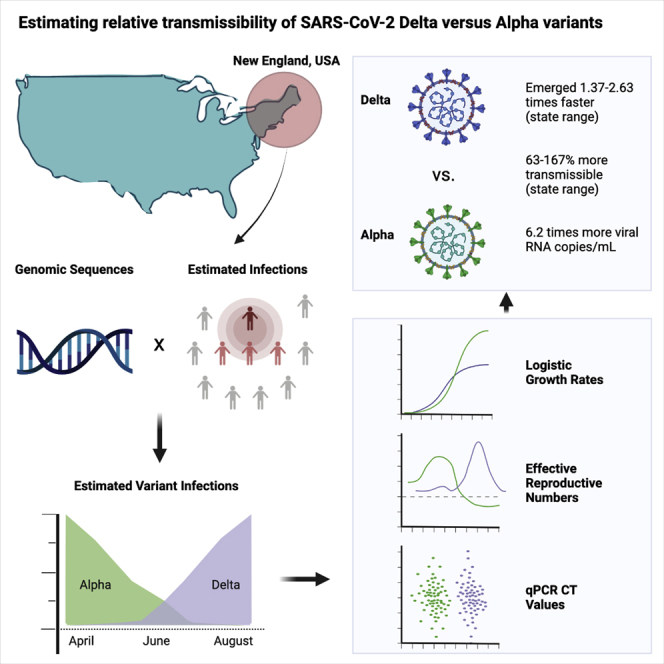
The SARS-CoV-2 Delta variant rose to dominance in mid-2021, likely propelled by an estimated 40%–80% increased transmissibility over Alpha. To investigate if this ostensible difference in transmissibility is uniform across populations, we partner with public health programs from all six states in New England in the United States. We compare logistic growth rates during each variant’s respective emergence period, finding that Delta emerged 1.37–2.63 times faster than Alpha (range across states). We compute variant-specific effective reproductive numbers, estimating that Delta is 63%–167% more transmissible than Alpha (range across states). Finally, we estimate that Delta infections generate on average 6.2 (95% CI 3.1–10.9) times more viral RNA copies per milliliter than Alpha infections during their respective emergence. Overall, our evidence suggests that Delta’s enhanced transmissibility can be attributed to its innate ability to increase infectiousness, but its epidemiological dynamics may vary depending on underlying population attributes and sequencing data availability.
Keywords: SARS-CoV-2, COVID-19, genomic epidemiology, viral sequencing, variant of concern, VOC, transmissibility, viral emergence
Graphical abstract
Highlights
-
•
SARS-CoV-2 Delta variant emerges 1.37–2.63 times faster than Alpha (state range)
-
•
Delta is on average 63%–167% more transmissible than Alpha (state range)
-
•
Delta infections average 6.2 times more viral RNA copies per milliliter than Alpha
-
•
Variant transmissibility estimates depend on innate, population, and data factors
The SARS-CoV-2 Delta variant displaced the previously dominant Alpha variant by mid-2021, ostensibly because of 40%–80% increased transmissibility. To determine whether this difference is uniform across populations, Earnest et al. estimate variant-specific logistic growth rates and effective reproductive numbers for six states in New England finding substantial heterogeneity.
Introduction
The evolution and emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants associated with increased transmissibility, more severe disease, and/or decreased vaccine effectiveness continue to exacerbate the coronavirus disease 2019 (COVID-19) pandemic.1 A SARS-CoV-2 variant is a virus with a defining set of mutations that distinguishes it from viruses belonging to other lineages.2 In particular, two SARS-CoV-2 variants with enhanced transmissibility substantially altered the pandemic’s trajectory: Alpha (B.1.1.7 lineage) and Delta (B.1.617.2 and AY.x sub-lineages). Alpha, defined in part by a N501Y amino acid substitution in the spike gene receptor binding domain that may affect ACE2 binding, was first detected in the United Kingdom in late 2020 and became the dominant global variant by early 2021.3, 4, 5 Delta, containing the spike L452R and P681H mutations (in the receptor binding domain and near the furin cleavage site, respectively) that may affect antibody recognition, was first recognized in India in early 2021 and displaced Alpha as the dominant variant by mid-2021.5 This shift led to a significant resurgence in COVID-19 cases in many countries.6, 7, 8, 9
Transmission rates can be affected by two main factors: innate attributes of the variant itself and the specific population in which it spreads. Variant-associated attributes may lead to innately increased transmissibility (e.g., increased viral loads, longer infection duration, decreased infectious doses).10 The rapid spread of Delta in many locations around the world suggests that it is innately more transmissible than Alpha and other SARS-CoV-2 variants. However, estimates of Delta’s transmissibility may also vary among populations because of differences in underlying immunity, control measures, behaviors, and demographics. For example, a variant that is more likely to cause vaccine breakthroughs may have a larger observed transmissibility advantage in populations with higher vaccination rates because it can spread to more individuals. In addition, the quality and volume of data generated by sequencing programs in different locations can influence estimates. Studies conducted in the United Kingdom estimated that Delta is 40%–80% more transmissible than Alpha, which itself was more transmissible than the SARS-CoV-2 lineages previously in circulation.11 The World Health Organization similarly estimated a 55% increase in Delta transmissibility on the basis of data from India and the United Kingdom.12 To understand whether these estimates are applicable elsewhere, it is critical to compare the relative transmissibility of SARS-CoV-2 variants in different locations to test the sensitivity of estimates to population-specific conditions. Accurate variant transmissibility estimates enable us to begin exploring drivers of transmissibility differences between populations.
In this study, we posed several important questions that arose with Delta: (1) how much more transmissible was Delta than Alpha, (2) what was the range in relative transmissibility estimates across states, and (3) was Delta more transmissible because it caused higher viral loads during infection? To investigate each, we partnered with SARS-CoV-2 genomic surveillance programs from all six New England states: Connecticut, Maine, Massachusetts, New Hampshire, Rhode Island, and Vermont. Using logistic growth rates and estimated effective reproductive numbers,4,13,14 we found that Delta was consistently more transmissible than Alpha, but the relative difference varied across states. Furthermore, we found on average 6.2 (95% confidence interval [CI] 3.1–10.9) times more viral RNA copies per milliliter from samples collected from Delta infections compared with Alpha infections during their respective emergence periods, supporting the hypothesis that Delta may be more transmissible because it generates higher viral loads. Overall, we estimated that Delta is 63%–167% more transmissible than Alpha (range across states). Our data indicated that the overall transmission advantage of Delta may in part be attributed to its innate ability to enhance infections and that the range in estimates between populations may be driven by differences in underlying characteristics and sequencing data availability.
Results
Genomic surveillance revealed similar variant frequency trajectories across New England
In response to emerging SARS-CoV-2 variants, all states within the New England region of the United States (Connecticut, Maine, Massachusetts, New Hampshire, Rhode Island, and Vermont) increased their virus sequencing capacity through local and regional partnerships (Figure 1A). From early April to mid-July 2021, at least 5% of the weekly reported COVID-19 cases were being sequenced from each state (with the exception of 7 days in June for New Hampshire and 10 days for Vermont when daily mean coverage dropped to 4% and 2%, respectively). The maximum daily sequencing coverage ranged from 16% (New Hampshire) to 46% (Maine). From these state-level sequencing data, we tracked the frequencies of the SARS-CoV-2 Alpha variant (B.1.1.7 lineage), the Delta variant (B.1.617.2 and AY.x sub-lineages), and all other lineages (Figure 1B). We observed similar trajectories in variant frequencies, with other lineages declining as Alpha increased in March and April 2021. Beginning in June 2021, Delta rapidly displaced the Alpha and other lineages. We also observed that the emergence of Delta resulted in a “selective sweep” and more fully dominated the variant landscape compared with Alpha. By the final week in July 2021, Delta constituted the vast majority of sequenced samples in all states (range 94%–100%). In contrast, although Alpha was the main variant in early 2021, we still observed other lineages maintained in the population.
Figure 1.

SARS-CoV-2 sequencing coverage and variant frequency tracking
(A) Confirmed cases per 100,000 population (bars) and percentage of cases sequenced (lines) by state (7 day rolling average), January to August 2021. The variability in percentage of cases sequenced represents changing sample availability and suitability for sequencing. The drop in percentage sequenced at the end of August does not reflect real decreases in sequencing coverage but instead (1) the 1 to 3 week delays between sample collection and sequence availability and (2) how the data are plotted using 7 day rolling averages.
(B) Weekly proportion of sequenced genomes belonging to each variant category with 95% confidence intervals, January to August 2021. A breakdown of the number of genomes (n = 33,408) by state and lineage is included in Tables S1–S3.
Delta emerged faster than Alpha and dominated the variant landscape
Although Delta rose to dominance within several months of its emergence (Figure 1), it was unknown how quickly it emerged relative to Alpha in different populations. We addressed this knowledge gap by comparing the initial growth rates of Delta and Alpha across New England. As Alpha and Delta emerged at different times, we defined their emergence periods as the 90 days following their initial detection in each state (Figure 2A). We then estimated the logistic growth rate of Alpha and Delta during their respective state-specific emergence periods (Figure 2B). We found that Delta emerged faster than Alpha despite higher vaccination rates during mid-2021.
Figure 2.

Variant logistic growth rates during their respective emergence periods in the context of infections and vaccination
(A) Estimated infections per 100,000 population (gray bars, left axis) and percentage of the population fully vaccinated (black lines, right axis) (7 day rolling average), with the colored rectangles indicating the 90 day emergence periods for each variant.
(B) Predicted probability of a given sequence belonging to each variant category over time determined by a binomial logistic regression for the variant category as the outcome and the number of days since the first detection as the predictor, estimating the logistic growth rate for Alpha versus Delta. Shown with 95% confidence intervals. The analysis is restricted to the first 90 days of emergence in each state as shown in (A). During the Alpha emergence period, we had the following number of Alpha genomes for each state: Connecticut, n = 1;221; Maine, n = 508; Massachusetts, n = 2;062; New Hampshire, n = 298; Rhode Island, n = 641; and Vermont, n = 466. During the Delta emergence period, we had the following number of Delta genomes for each state: Connecticut, n = 301; Maine, n = 108; Massachusetts, n = 268; New Hampshire, n = 30; Rhode Island, n = 136; and Vermont, n = 82.
(C) The regression coefficients (slopes) of the logistic growth rate from (B) with 95% confidence intervals. A sensitivity analysis varying the emergence periods by ±30 days is presented in Figure S1 and Table S4.
Although Alpha appeared to initially outpace Delta, as indicated by its steeper growth curve during the early emergence period (Figure 2B), we hypothesized that this was due to gaps in surveillance programs that impeded detection of Alpha but were addressed before Delta emerged. In some states, the predicted probability of a given sequence belonging to Alpha is non-zero at the time of first phylogenetic detection, providing further support for this hypothesis. As noted previously (Figure 1A), sequencing coverage improved over time in all states as incident cases declined. The probability of a given sequenced sample belonging to Alpha at the start of its emergence period was 11% in Vermont, 8% in Connecticut, and between 1% and 2% in the remaining states, indicating that Alpha likely was circulating for some time before its first detection (Figure 2B). In contrast, the probability of a given sequenced sample belonging to Delta was 0% at the start of the emergence period in all states. We estimated that the logistic growth rate for Delta was 2.63 times greater than Alpha in Vermont, 2.51 times greater in Connecticut, 1.98 times greater in Rhode Island, 1.95 times greater in Maine, 1.75 times greater in New Hampshire, and 1.37 times greater in Massachusetts (Figure 2C). From the first sequenced detection, it took Delta on average 71 days (range across states 54–92 days) to become dominant (to surpass 50% predicted frequency; Figure 2B). Given that the Alpha and Delta variants circulating across New England were intermixed, the differences in the growth rates between states are likely due in part to population-specific factors. As an initial exploration, we compared the increase in the logistic growth rate for Delta versus Alpha with the vaccination rates or estimated infections (Figure 2A) per state at the start of the Delta emergence period. We noted an association between the relative emergence speed of Delta with state vaccination rates; however, states with earlier Delta detection dates, such as Massachusetts, necessarily have lower vaccination rates during the Delta emergence period. We did not note an association between the relative emergence speed of Delta and estimated infections per 100,000 population in each state. Finally, we performed a sensitivity analysis varying the emergence period by ±30 days from the selected 90 day emergence period (Figure S1; Table S4). Using a 60 day emergence period, Delta emerged 1.30–1.57 times faster (range across states) in all states except Maine and New Hampshire, where Alpha emerged 2.33 and 4.55 times faster than Delta, respectively. Using a 120 day emergence period, Delta emerged 1.97–3.30 times faster (range across states).
Delta was more transmissible than Alpha in all New England states
We showed that Delta emerged faster in New England than Alpha had previously (Figure 2) and rose to higher levels of dominance, almost completely displacing Alpha and other lineages (Figure 1). However, we still do not know how much more transmissible Delta was than Alpha when they were co-circulating and the extent to which our estimates varied across. To answer this question, we adapted our previously developed framework to estimate the variant-specific effective reproductive number (Rt) from inferred SARS-CoV-2 infections.13,15 Our Rt estimates for each variant approximate the time-varying average number of secondary cases from a primary infection within a population. Rt estimates greater than 1 imply that COVID-19 cases associated with variants will increase in the future. We report the length of the Alpha/Delta co-circulation period (STAR Methods) and estimated mean Alpha and Delta infections per 100,000 population during the co-circulation period for each state (Figure S2A). We computed Rt for each variant category during January to August 2021 (Figure 3A) by combining the frequency estimates from our genomic surveillance data (Figure 1B) with daily estimated SARS-CoV-2 infections (Figure 2A). Specifically, we used a multi-step bootstrapped sampling approach to generate 1,000 samples containing the estimated number of variant-specific infections. Our approach is further detailed in STAR Methods. Delta had Rt > 1 for the majority of the time period following its emergence, which exceeded the Rt estimates for Alpha and other lineages (Figure 3). Our mean Rt estimates for other lineages was <1 for all states, ranging from 0.87 to 0.91. Prior to the emergence of Delta, our mean Rt for Alpha was 1.20 across states, dropping to a mean of 0.78 as vaccination increased during the period following Delta’s emergence. Our mean Rt estimate for Delta was 1.40, ranging from 1.27 (New Hampshire) to 1.65 (Vermont). We found that the Rt for Delta exceeded that of Alpha in all states for the majority of the time following its initial detection (Figure 3B). We then estimated that the mean Rt ratio of Delta to Alpha during their co-circulation period was 2.67 in Vermont, 2.04 in Maine, 1.92 in New Hampshire, 1.83 in Rhode Island, 1.78 in Connecticut, and 1.63 in Massachusetts, suggesting that Delta was 63%–167% more transmissible on average than Alpha (range across states).
Figure 3.

Comparison of variant effective reproductive numbers to estimate relative transmissibility
(A) Estimated effective reproductive number (Rt) over time for each variant category calculated from the inferred number of infections using EpiEstim.15,16 We used a multi-step bootstrap sampling approach to generate 1,000 samples containing the estimated number of variant-specific infections. We obtained mean Rt estimates for each of the 1,000 bootstrapped samples and plotted the overall mean and 95% confidence intervals (2.5% and 97.5% quantiles across the bootstrapped samples). Note that the y axis differs from that in (B).
(B) Daily mean ratios of Rt values for Delta compared with Alpha from (A). For each bootstrapped sample described in (A), we calculated the daily ratio of the Delta to Alpha Rt estimates. We plotted the mean across the 1,000 bootstrapped samples and the 95% confidence intervals, calculated the same as in (A). For (A) and (B), the upper limit of Delta’s confidence intervals in Maine, New Hampshire, and Vermont are not plotted but are displayed in Figures S2B and S2C.
In addition, separately for Delta and Alpha, we calculated the multiplicative increase in Rt, another measure of relative transmissibility (Figure S3).14 For this estimate, we exponentiated the coefficients from the binomial logistic regression and multiplied them by the mean generation interval to estimate the change in the probability of a given sequence belonging to a lineage over a generation interval. The multiplicative increase in Rt for Delta was greater than that for Alpha in all states. Across states, we observed a mean 1.30 increase in the probability of a sample belonging to Alpha over a generation interval, compared with 1.69 for Delta. Our multiplicative increase in Rt estimates suggests that Delta had the greatest advantage in Maine (1.99-fold increase) and the lowest advantage in Massachusetts (1.45-fold) (Figure 3C). Differences in transmissibility estimates between states may be due to a combination of viral, underlying population, and data factors that are further described in the Discussion.
Delta infections on average had a larger number of viral RNA copies than Alpha infections during their respective emergence periods
One potential mechanism for Delta’s increased transmissibility relative to Alpha (Figures 2 and 3) is that infections with the Delta variant could lead to higher virus titers than those with Alpha. To test this hypothesis, we compared the qRT-PCR cycle threshold (CT) values of sequence-confirmed Alpha and Delta infections (anterior nares or nasopharyngeal swabs) available from four institutes in New England: Yale University (Connecticut), Jackson Laboratory (Connecticut), Mass General Brigham (Massachusetts), and the Health and Environmental Testing Laboratory (Maine) (Figure 4). PCR CT values are a metric of virus RNA copies, and lower CT values indicate that there are more copies. We consistently found lower CT values for Delta infections across all institutes, but some comparisons did not yield significant differences.
Figure 4.

Cross-sectional PCR data from Alpha and Delta samples
PCR CT values (inverted y axis) plotted by institute and variant category, limiting Alpha samples to January to March 2021 and Delta samples to June to August 2021 to account for their respective emergence periods. Monthly CT values for Alpha and Delta for Yale University are shown in Figures S4A and S4B. For each institute, the means of the two variant categories were compared using a t test, with statistical significance symbols corresponding to the following values: ns (p > 0.05), ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001. Data from the full month of August were not available for most of the institutes at the time of analysis. The Yale University (Connecticut) Alpha (n = 541) and Delta (n = 250) data are from the N1 primer/probe set (originally from the “CDC assay”) of a research use-only RT-PCR assay to discriminate among variants (data shown as virus RNA copies per milliliter in Figure S4C).17 The Jackson Laboratory (Connecticut) Alpha (n = 204) and Delta (n = 60) data are from the N gene primer/probe set of the TaqPath COVID-19 Combo Kit (Thermo Fisher Scientific). The Mass General Brigham (Massachusetts) Alpha (n = 41) and Delta (n = 153) data are from the E gene of the Roche Cobas SARS-CoV-2 test (ORF1a data shown in Figure S4D). The Health and Environmental Testing Laboratory (Maine) Alpha (n = 16) and Delta (n = 88) data are from the N1 primer/probe set (originally from the “CDC assay”) of the OPTI SARS-CoV-2 RT-PCR Test (OPTI Medical Systems).
Importantly, PCR CT values from cross-sectional tests can be biased by the epidemic period because viral loads are dynamic and tend to decrease with time.18 During the emergence phase of an epidemic, most PCR tests come from recent infections, whereas the opposite is true when the epidemic is declining. The result is that PCR CT values could be higher (meaning less virus detected) during the declining phase even though the infection dynamics are the same throughout the epidemic. We first investigated if variant-specific PCR CT values change during different epidemic phases by running a one-way ANOVA for Alpha and Delta samples (separately) generated by one of the institutes (Yale University) to test for significant differences between monthly mean CT values (Figures S4A and S4B). We found a significant difference for only the Alpha samples. Using a post hoc Tukey’s honestly significant difference (HSD) test to investigate pairwise differences in monthly CT values while controlling for the experiment-wise error rate, we found that for the Alpha samples, there was a significant difference only for March versus April. If the epidemic phase were affecting our mean monthly CT values, we would expect higher CT values (corresponding to lower viral loads) for April versus March, as April is further in the declining phase of Alpha. We observe the opposite, providing further support for our previous finding that we did not observe evidence of an epidemic phase effect on our monthly CT values. Still, to account for any effects that the epidemic period may have on our comparisons, we limited our analysis to the approximate emergence phase of each variant: January to March 2021 for Alpha and June to August for Delta. Furthermore, the PCR CT data that we used from the four institutes are from different assays and some target different genes (though most target the nucleocapsid [N] gene). The Yale University data are from the N1 primer/probe set (originally from the “Centers for Disease Control and Prevention [CDC] assay”) of a “research use only” assay17; the Health and Environmental Testing Laboratory data are from the same N1 primer/probe set at Yale but from the OPTI SARS-CoV-2 RT-PCR Test; the Jackson Laboratory data are from the N gene primer/probe set of the TaqPath COVID-19 Combo Kit; and the Mass General Brigham data are from the envelope (E) and open reading frame 1a (ORF1a) gene primer/probe set of the Roche Cobas SARS-CoV-2 test. Therefore, we analyzed the PCR CT values independently for each institute and gene target.
Assessing cross-sectional PCR data from the four institutes, we consistently found lower mean CT values (more viral RNA copies) from Delta compared with Alpha nasal swab samples (Figure 4). The differences were significant from the Yale University (p ≤ 0.0001) and Jackson Laboratory (p ≤ 0.001) data, but not from Mass General Brigham and the Health and Environmental Testing Laboratory (p > 0.05 for each). In addition, for the Yale University samples, we used a standardized PCR curve to translate the CT values into viral RNA copies per milliliter.19 We found 6.2 (95% CI 3.1–10.9) times more RNA copies per milliliter (non-log scale) on average in Delta anterior nares swab samples compared with Alpha samples (Figure S4C). Thus, during their respective emergence periods, upper respiratory tract samples collected from individuals infected with Delta on average had higher viral copies than from Alpha infections, possibly contributing to enhanced transmissibility. An important limitation of this analysis is that viral load differences may change later in each variant’s epidemic trajectory as PCR tests increasingly come from less recent infections, and thus our findings are restricted to the respective emergence periods. We discuss our CT analysis limitations further in the Discussion.
Discussion
The SARS-CoV-2 variant Delta emerged across the United States in mid-2021, displacing previous variants, including Alpha. Assessing the variability in relative emergence growth rates and transmissibility estimates of Delta versus Alpha across different settings remains an important public health question. With more than 3 million SARS-CoV-2 genomes available in public repositories, large-scale and globally diverse assessments can be conducted. However, there is always a risk when analyzing diverse sources of data without input from the data submitters on possible sampling biases (e.g., targeted sequencing in certain settings or sub-state locations) that may not be apparent in the repositories. We directly partnered with SARS-CoV-2 genomic surveillance programs from all six states in the New England region of the United States (Figure 1) to confidently assess emergence growth rates and relative transmissibility at the state level. We found that the logistic growth rates (Figure 2) and effective reproductive numbers (Figure 3) of Delta were consistently greater than Alpha across all New England states, although there was variation among states.
The estimated initial growth rates for Alpha and Delta could have been influenced by population factors that differed between each variant’s respective emergence period. At the time of Alpha’s emergence in January and February 2021, 0% of the population across New England was reported as being fully vaccinated (Figure 2A). In comparison, when Delta first emerged in March and April 2021, 18%–37% of the population was fully vaccinated. Estimated infections per 100,000 population were also substantially lower during Delta’s emergence period (Figure 2A). However, with rising vaccination rates, all of the states began relaxing capacity constraints from late February to late March 2021. States continued rolling back COVID-19 mitigation measures with the majority lifted by the end of May 2021, although some states maintained indoor masking for unvaccinated individuals.20 The emergence of Delta also occurred within the background of other variants, including Alpha (Figure 1B). Thus, the fitness landscape for SARS-CoV-2 variants may have changed dramatically during 2021, potentially playing out differently across the states and explaining the large range in relative growth rates between Delta and Alpha (Figure 2C). Finally, the initial growth rates are sensitive to the length of the emergence period (Figure S1). We found consistently faster relative growth rates for Delta versus Alpha when using a longer emergence period (120 days instead of 90 days) but had mixed findings when using a shorter emergence period (60 days instead of 90 days) (Table S4). Shorter emergence periods may exacerbate the potential aforementioned biases in surveillance between Alpha and Delta, leading to more variable results. This underscores the importance of understanding the sequencing context in the early days of a variant’s emergence when making these estimates.
Our estimates of the transmission advantage (measured as the mean Rt ratio of Delta to Alpha) for Massachusetts (63%) and Connecticut (78%) were within the 40%–80% estimate range provided by the United Kingdom.11 We estimated a greater transmission advantage for Delta in Rhode Island (83%), New Hampshire (92%), Maine (104%), and Vermont (167%). This variation may be driven by differences in the underlying state populations, such as population density, vaccination rates, travel patterns, control measures, behaviors, and competing variants in circulation. In addition, the differences could reflect the noisier Delta Rt estimates we observed for states with fewer infections and, as a result, fewer genomic sequences. In particular, we noted wide confidence intervals around the Delta Rt estimates for Maine, New Hampshire, and Vermont, the states with the greatest Delta to Alpha transmission advantage (Figures S2B and S2C). It is possible that additional sequencing data from these states would bring their estimates of relative transmissibility more in line with those from other locations.
Our study adds to the growing evidence that Delta may be more transmissible in part by causing higher viral loads during acute infections.21, 22, 23, 24, 25 We found significantly lower mean PCR CT values (corresponding to more viral RNA copies) for Delta versus Alpha infections from nasal swab samples tested by Yale University and Jackson Laboratory in Connecticut (Figure 4). The overall pattern of lower CT values for Delta versus Alpha held for the Massachusetts and Maine data, although the differences in mean values were not significant. This may be due to relatively small sample sizes for Alpha in those locations, specimen type used for analysis (anterior nares vs. nasopharyngeal swab), and/or changing CT cutoff criteria for sequencing (e.g., the Alpha CT values from Mass General Brigham do not extend above 31, suggesting a stringent cutoff). For the Yale data, we used a standardized curve to translate the CT values into virus copies and found that Delta samples had on average 6.2 (95% CI 3.1–10.9) times more viral RNA copies per milliliter than Alpha samples during their respective emergence periods (Figure S4C).
In conclusion, although we determined that the Delta SARS-CoV-2 variant emerged faster and was more transmissible than Alpha in New England, there was heterogeneity in our estimates across states. Our analysis demonstrated that, in addition to possible innate differences between variants, there may be other factors at play such as vaccination levels, transmission levels, demographics, behavior, control measures, and sequencing data availability that affected each variant’s estimated transmissibility within a given population. Finally, state sequencing volumes and coverage variability may be partly behind the range of estimates that we observed. The exact mechanisms driving differences in variant transmissibility among states are unclear, but these factors remain important when considering how another variant might rise to dominance in the future and the sequencing data required to accurately assess its ascent. It is impossible to predict when and where the next variant of concern will emerge, as the explosive recent emergence of Omicron has shown, and thus it is important to enhance SARS-CoV-2 genomic surveillance and further our understanding of how different population characteristics and sequencing data availability affect variant transmissibility estimates.
Limitations of the study
An important limitation to our comparative analyses was that sequencing coverage improved in states over time (Figure 1A), and thus there may have been a greater delay in the time to the first sequenced detection for Alpha versus Delta. In addition, because of the uncertainty around the serial interval, we selected an uncertain serial interval approach to explore various possible distributions when calculating Rt (STAR Methods). Our state-specific results should be considered within the context of their variability in sequencing coverage and the volume of sequences. Overall, the states with the greatest Delta to Alpha transmission advantage (Vermont, Maine, and New Hampshire) also had the widest confidence intervals around their Rt estimates (Figures S2B and S2C), reflecting the challenges of estimating variant-specific Rt values when relatively few genomic sequences are available. In particular, Vermont had more variable sequencing coverage because of low case counts, and its results should be interpreted cautiously (Figure 1A). Thus, variant frequencies based on a relatively small number of sequences are likely driving some of the uncertainty around Rt estimates and variability across states (Figures 3A, S2B, and S2C). In addition, we did not account for the lag between the time of estimated infections and when the sequenced samples were collected, but we do not believe that this would have a substantial impact on our Rt estimates, particularly given the 21 day sliding window we used. We conducted our analysis at the state level, however there could be within-state heterogeneity in our estimates. Drivers of heterogeneity could include a lack of even sequencing coverage across each state. We attempted to assess the within-state location of each sequence used in our analysis, but only the state location was typically available among publicly available data. In addition, there could be differences in population demographics, behavior, immunity, and/or control measures at more local levels that could lead to more heterogeneous estimates.
For the PCR CT analysis, we necessarily used only confirmed variant sequence data, which represents a fraction of the total PCR CT data and is biased against higher CT values that lack sufficient virus RNA for sequencing. It is also important to be cautious in interpreting CT values among institutes because of differences in the diagnostic PCR test platforms, specimen types included, and subsequent CT cut-off values. For this reason, we did not aim to directly compare institutes but rather sought to establish whether an overall pattern of lower CT values for Delta versus Alpha samples held. Mean CT values can also be influenced by the epidemic trajectory, as discussed previously. To assess whether we observe this in our data, we plotted Alpha and Delta CT values (separately) by month for the Yale data (Figures S4A and S4B) and did not find a pattern in monthly mean CT values indicative of a bias due to the epidemic trajectory. To further control for possible differences in CT values driven by the epidemic trajectory, we also limited our comparison of mean viral loads between variants to only the initial 3 months following their respective emergence. Additional patient metadata such as estimated time since infection would enable a more extensive analysis in differences in variant viral loads.
Consortia
The members of the New England Variant Investigation Team are Ahmad Altajar, Alexandra DeJesus, Anderson Brito, Anne E. Watkins, Anthony Muyombwe, Brendan S. Blumenstiel, Caleb Neal, Chaney C. Kalinich, Chen Liu, Christine Loreth, Christopher Castaldi, Claire Pearson, Clare Bernard, Corey M. Nolet, David Ferguson, Erika Buzby, Eva Laszlo, Faye L. Reagan, Gina Vicente, Heather M. Rooke, Heidi Munger, Hillary Johnson, Irina R. Tikhonova, Isabel M. Ott, Jafar Razeq, James C. Meldrim, Jessica Brown, Jianhui Wang, Johanna Vostok, John P. Beauchamp, Jonna L. Grimsby, Joshua Hall, Katelyn S. Messer, Katie L. Larkin, Kyle Vernest, Lawrence C. Madoff, Lisa M. Green, Lori Webber, Luc Gagne, Maesha A. Ulcena, Marianne C. Ray, Marissa E. Fisher, Mary Barter, Matthew D. Lee, Matthew T. DeFelice, Michelle C. Cipicchio, Natasha L. Smith, Niall J. Lennon, Nicholas A. Fitzgerald, Nicholas Kerantzas, Pei Hui, Rachel Harrington, Randy Downing, Rashida Haye, Ryan Lynch, Scott E. Anderson, Scott Hennigan, Sean English, Seana Cofsky, Selina Clancy, Shrikant Mane, Stephanie Ash, Stephanie Baez, Steve Fleming, Steven Murphy, Sushma Chaluvadi, Tara Alpert, Trevor Rivard, Wade Schulz, and Zoe M. Mandese.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| SARS-CoV-2 genomes | GISAID | https://www.gisaid.org/ |
| Confirmed COVID-19 cases | Johns Hopkins Center for Systems Science and Engineering | https://github.com/CSSEGISandData/COVID-19 |
| Estimated COVID-19 infections | Covidestim | https://covidestim.org/ |
| 2019 U.S. state populations | United States Census Bureau | https://www.census.gov/ |
| State population vaccination percentages | Centers for Disease Control and Prevention | https://data.cdc.gov/ |
| Software and algorithms | ||
| R | RStudio: Integrated Development for R | Version 1.4.1106 |
| EpiEstim: Estimate Time Varying Reproduction Numbers from Epidemic Curves (R Package) | Cori et al., 2013 | Version 2.2-4 |
| Other | ||
| Connecticut SARS-CoV-2 PCR CT values | Yale University, Jackson Laboratory | https://github.com/grubaughlab/2021_paper_Delta-v-Alpha |
| Massachusetts SARS-CoV-2 PCR CT values | Mass General Brigham | https://github.com/grubaughlab/2021_paper_Delta-v-Alpha |
| Maine SARS-CoV-2 PCR CT values | Health and Environmental Testing Laboratory | https://github.com/grubaughlab/2021_paper_Delta-v-Alpha |
Resource availability
Lead contact
Further information and requests for resources and data should be directed to and will be fulfilled by the lead contact, Dr. Nathan Grubaugh (nathan.grubaugh@yale.edu).
Materials availability
This study did not generate any unique reagents, but raw data and code for this study can be found in the Supplemental Materials and via the resources specified in Data and Code Availability.
Experimental model and subject details
Ethics statement
Yale University (Connecticut)
The Institutional Review Board from the Yale University Human Research Protection Program determined that the RT-qPCR testing and sequencing of de-identified remnant COVID-19 clinical samples obtained from clinical partners conducted in this study is not research involving human subjects (IRB Protocol ID: 2000028599).
Jackson Laboratory (Connecticut)
The Institutional Review Board of The Jackson Laboratory determined that use of de-identified residual COVID-19 clinical samples obtained from the Clinical Genomics Laboratory for RT-qPCR testing and sequencing for this study is not research involving human subjects (IRB Determination: 2020-NHSR-021).
Mass General Brigham (Massachusetts)
The Institutional Review Board of Partners Human Research determined that the use of excess, de-identified COVID-19 clinical specimens obtained within the Partners Healthcare network for RT-qPCR testing and genomic sequencing for this study is not research involving human subjects (IRB Protocol ID: 2019P003305). In addition, the Institutional Review Board of the Massachusetts Department of Public Health has reviewed and approved this study to perform genomic sequencing of coronaviruses (IRB Protocol ID: 1603078).
Health and Environmental Testing Laboratory (Maine)
A Memorandum of Understanding between The State of Maine Department of Health and Human Services and The Jackson Laboratory determined that extracted viral RNA from human respiratory specimens which have tested positive for SARS-CoV-2 and are used for sequencing will not be used in human subjects, in clinical trials, or for diagnostic purposes involving human subjects.
Method details
Growth rates and transmissibility estimation
Genomic surveillance data
We obtained SARS-CoV-2 genomic sequence data from GISAID as of August 13, 2021.26, 27, 28 We restricted the dataset to our states of interest (Connecticut, Maine, Massachusetts, New Hampshire, Rhode Island, and Vermont) and removed sequences that lacked a lineage assignment (N = 4977), had mismatched metadata for the sample collection location (N = 81), or were outside of our January 1, 2021 - August 1, 2021 time period of interest (N = 4260). This yielded a final dataset of 33,408 genomes. We categorized the GISAID SARS-CoV-2 genomes into 3 mutually exclusive categories: Alpha (B.1.1.7), Delta (B.1.617.2 and all AY.x sub-lineages existing as of August 13, 2021), and Other (any sequences not included in the prior categories that had a lineage assignment). A breakdown of the number of genomes by state and lineage is included in Tables S1–S3.
Confirmed cases per 100K population
We obtained confirmed case data from the Johns Hopkins Center for Systems Science and Engineering.29 We used state population estimates for 2019 from the United States Census Bureau to calculate confirmed cases per 100K population (Figure 1A).30
Percent of the population fully vaccinated
We obtained the percent of the state populations that were fully vaccinated from the Centers for Disease Control and Prevention (dataset downloaded August 11, 2021) (Figure 2A).31
Infections per 100K population
We obtained estimated infections for each state from Covidestim, a Bayesian nowcasting approach that accounts for differences in diagnosis and reporting by anchoring its estimates to death data, which are generally more reliable than case data (dataset downloaded November 11, 2021).15 We used state population estimates for 2019 from the United States Census Bureau to calculate estimated infections per 100K population (Figure 2A).30
RT-qPCR and lineage identification
Yale University (Connecticut)
Clinical samples (anterior nares in viral transport media) were received from confirmed SARS-CoV-2 positive individuals from routine testing provided by Yale New Haven Hospital. The samples were primarily from Connecticut and were collected for a variety of inpatient and outpatient testing programs. Nucleic acid was extracted from 300 μL of the original sample using the MagMAX viral/pathogen nucleic acid isolation kit, eluting in 75 μl of elution buffer. The extracted nucleic acid was tested for SARS-CoV-2 RNA using a “research use only” (RUO) RT-qPCR assay.17 For this analysis, only the CT values from the CDC N1 primer/probe set were used (Figure 4). N1 CT values were also converted into SARS-CoV-2 RNA copies using a standard curve, as previously described (Figure S4C).19
To determine the SARS-CoV-2 lineage, samples with CT values ≤ 35 were sequenced using the Illumina COVIDSeq Test RUO version. Amplicons were pooled and cleaned before quantification with Qubit High Sensitivity dsDNA kit. The resulting libraries were sequenced using a 2x150 approach on an Illumina NovaSeq at the Yale Center for Genomic Analysis. Each sample was given at least 1 million reads. Samples were typically processed in sets of 93 or 94 with negative controls incorporated during the RNA extraction, cDNA synthesis, and amplicon generation steps. The reads were aligned to the Wuhan-Hu-1 reference genomes (GenBank MN908937.3) using BWA-MEM v.0.7.15.32 Adaptor sequences were trimmed, primer sequences were masked, and consensus genomes were called (simple majority >60% frequency) using iVar v1.3.133 and SAMtools.34 An ambiguous ‘N’ was used when fewer than 20 reads were present at a site. In all cases, negative controls were analyzed and confirmed to consist of at least 99% Ns. Pangolin v.2.4.235 was used to assign lineages.36
Jackson Laboratory (Connecticut)
Clinical samples were received in The Jackson Laboratory Clinical Genomics Laboratory (CGL) as part of a statewide (Connecticut) COVID-19 surveillance program, with the majority of samples representing asymptomatic screening of nursing home and assisted living facility residents and staff. Total nucleic acids were extracted from anterior nares swabs in viral transport media or saline (200 μL) using the MagMAX Viral RNA Isolation kit (ThermoFisher) on a KingFisher Flex purification system. Samples were tested for the presence of SARS-CoV-2 RNA using the TaqPath COVID-19 Combo Kit (ThermoFisher). For this analysis, only the CT values from the N gene primer/probe set were used (Figure 4).
Samples with CT values ≤ 30 for the N gene target were prepared for sequencing using the Illumina COVIDSeq Test kit. Sequencing was performed on an Illumina NovaSeq or NextSeq in the CGL. Data analysis was performed using the DRAGEN COVID Lineage App in BaseSpace Sequence Hub. Sequences with >80% of bases with non-N basecalls and ≥1500-fold median coverage were considered successful and were submitted to GISAID. Lineages were assigned using pangolin v.2.4.235 and the most current version of the pangoLEARN assignment algorithm.
Mass General Brigham (Massachusetts)
Clinical samples were received from confirmed SARS-CoV-2 positive tests collected at Massachusetts General Brigham testing facilities during routine testing, via nasopharyngeal swabs in viral or molecular transport media. Clinical samples included in the analysis were tested for the presence of SARS-CoV-2 RNA using the Roche Cobas SARS-CoV-2test on the 6800 system, targeting both the E and ORF1a genes. CT values were analyzed independently for both the E (Figure 4) and ORF1a (Figure S4D) gene primer/probe sets.
Genomic sequencing was conducted on clinical samples with CT values ≤30 using the Illumina COVIDSeq Test protocol. The resulting libraries were pooled, cleaned, and quantified using the Qubit High Sensitivity dsDNA kit. Sequencing was performed at Massachusetts General Hospital or at the Broad Institute of MIT and Harvard using a 2x150 approach on an Illumina NovaSeq SP, an Illumina NextSeq 550, or an Illumina NextSeq 2000. Sequences were analyzed through the Broad Institute Data Analysis Platform using the viral-ngs 2.1.28 on the Terra platform (app.terra.bio). All of the workflows named below are publicly available via the Dockstore Tool Registry Service (dockstore.org/organizations/BroadInstitute/collections/pgs). Sequences with an assembly length >24000 non-N bases were considered complete genomes. Lineages were assigned using the most up-to-date version of the pangoLEARN assignment algorithm.
Health and Environmental Testing Laboratory (Maine)
Clinical samples (anterior nares swabs in viral transport media) were received from confirmed SARS-CoV-2 positive tests collected at the State of Maine Department of Health and Human Services testing facilities (Health and Environmental Testing Laboratory (HETL)) during routine testing. Viral RNA were extracted on a Thermofisher KingFisher Flex purification using the MagMAX Viral/Pathogen II Nucleic Acid Isolation Kit. Extracted samples were tested for the presence of SARS-CoV-2 RNA using the OPTI SARS-CoV-2 RT-PCR Test (OPTI Medical Systems) with the ABI 7500fast DX thermocycler. For this analysis, only the CT values from the N1 gene primer/probe set were used (Figure 4).
Prior to sequencing, samples were not reanalyzed for the presence of SARS-CoV-2 RNA. HETL utilized the QIAseq DIRECT SARS-CoV-2 Kit from Qiagen for targeted whole genome library preparation. Samples were sequenced on an Illumina MiSeq using a MiSeq® Reagent Kit v2 (300 cycle) kit. QIAGEN CLC Genomics Workbench was used to assemble viral genomes. Consensus sequences were uploaded to Nexstrain and Pango for quality control, strain and clade identification.
Additional SARS-CoV-2 positive samples from HETL were sequenced by the Jackson Laboratory (Maine) using the Illumina COVID-Seq protocol modified to include 6e2 copies/μL of a unique SDSI control spiked into each cDNA sample. Samples were sequenced on a NextSeq500 using paired 75 bp reads by the Genome Technology group on Jackson Laboratory’s Bar Harbor campus. Sequence reads were analyzed using an in-house pipeline (https://github.com/tewhey-lab/SARS-CoV-2-Consensus) that leverages minimap2, samtools and iVar for read mapping and variant calling. Sequences with >80% non-N bases were considered complete genomes for analysis.
Quantification and statistical analysis
We used RStudio (v 1.4.1106) for all analyses and figures.37 Statistical details are available in figure legends and in the below STAR Methods section.
Growth rates and transmissibility estimation
Sequencing coverage
We calculated sequencing coverage per state as the number of curated sequenced genomes made publicly available on GISAID divided by the number of confirmed cases, and plotted the data based on the sample collection date using a 7-day rolling average (Figure 1A).
Variant frequencies among sequenced samples
We divided the number of sequences belonging to each variant category (Delta, Alpha, or Other) by the total number of sequences with an assigned lineage to estimate variant frequencies. We calculated a Jeffreys' interval, using the resulting 0.025 and 0.975 quantiles to form a 95% confidence interval (Figure 1B).
Logistic growth rates
We first defined the emergence period for Alpha versus Delta in each state as the time from each variant category’s first phylogenetic detection in the GISAID data to 90 days afterward. We ran a binomial logistic regression for Alpha and Delta, separately, with the variant category as the outcome and the number of days since the first detection as the predictor. We plotted the smoothed fitted curves for the emergence periods with their 95% confidence intervals (Figure 2B), which shows the probability of a given sequence belonging to a specific variant category over time. We also reported the logistic regression coefficients as the log odds of a given sequence belonging to a specific variant category (Figure 2C). Finally, we ran a sensitivity analysis varying the emergence period +/− 30 days from our selected 90-day period (Figure S1 and Table S4). In Table S4, we report the ratio of the regression coefficients (slopes) of the logistic growth rate shown in Figure S1 for Delta versus Alpha for each state and emergence period.
Effective reproductive number Rt estimates and Rt ratios
To reflect the uncertainty around whether our sampled variant frequencies were representative of the full unknown population of infections, we took a multi-step bootstrapping approach. First, we drew the number of variant-specific genomes Yiv from a multinomial distribution where the number of independent trials was equal to the total number of sequences on day i and the probability of a sequence on day i belonging to variant v being equal to its frequency on day i. We then calculated the sample probability Piv of variant v on day i. We repeat this sampling process 1,000 times.
Next, using our sampled variant proportions, we sampled again from a multinomial distribution 1,000 times. In this case, the number of independent trials was equal to the number of estimated infections on day i and the probability of an infection on day i belonging to variant v being equal to its sample probability Piv. The result for each sample was a time series of the estimated number of Alpha, Delta, and Other infections. We applied a 7-day rolling average to these data to account for day-of-the-week differences in sequence reporting.
Finally, following a previous approach used to estimate the comparative transmissibility of SARS-CoV-2 variants,13 we ran each time series through the EpiEstim R package16 to obtain our mean Rt estimates. Within EpiEstim, we used an uncertain serial interval to estimate Rt over 21-day sliding windows, with the mean serial interval of 5.2 days (allowed to vary between 2.2 and 8.2 days) and a standard deviation of 4 days (allowed to vary between 2.5 and 5.5 days) based on estimates available in the literature.38, 39, 40 For Vermont, we removed a single ‘Other’ variant category sequence on July 21, 2021 (after the ‘Other’ variant category had died out) that was leading to highly inflated Rt estimates. We began Rt estimation when there were at least 12 cumulative estimated infections to achieve a posterior coefficient of variation of 0.325 and restricted estimation to before August 1, 2021 as variant frequencies became less certain after that time due to delays in sequencing and reporting. We further truncated the variant-specific Rt estimates in states where estimated variant-specific infections went consistently to 0 (i.e. died out). EpiEstim assumes an uninformative prior for mean Rt of 5, ensuring that when there are few infections (e.g., zero variant-specific infections for the majority of the 21-day estimation window) the prior becomes disproportionately weighted relative to the data. As a result, in the case of a lineage die-out, Rt estimates erroneously seem to increase. Therefore, we truncated the Rt estimates at the date where the lineage died out. For Alpha Rt estimates, we truncated the following states: Connecticut (July 24, 2021), Maine (July 13, 2021), New Hampshire (July 23, 2021), Rhode Island (July 17, 2021), and Vermont (July 11, 2021). For Other Rt estimates, we truncated the following states: New Hampshire (July 20, 2021), Rhode Island (July 25, 2021), and Vermont (June 25, 2021). We did not truncate any of the Delta Rt estimates as Delta infections grew in all states during our studied time period. We used the same serial interval parameters for all variant categories due to the lack of consensus regarding differences.41,42 We reported the resulting mean Rt estimates across the 1,000 samples and the 95% confidence intervals, calculated as the 2.5 and 97.5% quantiles across the 1,000 mean Rt estimates (Figure 3A). We plotted the daily mean Rt ratio and 95% confidence intervals for Delta versus Alpha over time (Figure 3B). The mean Alpha/Delta co-circulation period across bootstrapped samples was 94 days for Connecticut, 76 for Maine, 121 for Massachusetts, 97 for New Hampshire, 87 for Rhode Island, and 70 for Vermont. We plotted the mean estimated Alpha and Delta infections per 100K during this time period in Figure S2A.
Multiplicative reproductive number estimates
Separately for Alpha and Delta, we calculated the multiplicative increase in Rt, a measure of relative transmissibility by multiplying the coefficients from the previously described binomial logistic regression by the mean generation interval, 5.2 days, and exponentiating to get an estimate of the increase in the probability of a given sequence belonging to a lineage over a generation interval.14 We plotted this against the log-transformed p value associated with each coefficient (Figure S3) to provide the level of support for the multiplicative value estimate.
RT-qPCR CT value and virus RNA comparisons
We binned the RT-qPCR CT and virus RNA data (separately) for Alpha (January-March 2021) and Delta (June-August 2021) samples and compared the group means for each institute using a t-test, with statistical significance symbols corresponding to the following values: ns (p > 0.05), ∗ (p ≤ 0.05), ∗∗ (p ≤ 0.01), ∗∗∗ (p ≤ 0.001), ∗∗∗∗ (p ≤ 0.0001) (Figures 4, S4C, and S4D). For the monthly analysis, we ran a one-way ANOVA test for Alpha and Delta samples (separately) to test for significant differences between monthly mean CT values (Figures S4A and S4B). For the Alpha samples, which returned a significant result, we ran post hoc Tukey’s HSD test to investigate pairwise differences in monthly CT errors while controlling for the experiment-wise error rate. To calculate the ratio of mean Alpha to mean Delta virus RNA copies per mL during their respective emergence periods, we sampled with replacement (with size equal to each variant’s sample size), took the mean for Alpha and Delta separately, and calculated the ratio. We repeated this 1,000 times and took the mean, 2.5%, and 97.5% quantiles across the samples. We reported the resulting mean and 95% confidence intervals.
Acknowledgments
We thank the Broad Genomics Platform, the Broad Clinical Research Sequencing Program, and the Broad Data Sciences Platform for their tremendous work making data available for surveillance and research; members of the New England “variant” group who meet weekly to discuss and share data regarding emerging SARS-CoV-2 variants; the groups that continuously make their sequencing data available to the public; the frontline and essential workers for their service during the pandemic; D. MacCannell for his ongoing support and permission to use the data generated by the CDC; Y. Grad for technical advice; and our friends and family, particularly V. Parsons, P. Jack, and S. Taylor, for their support. The graphical abstract was created using BioRender.com. We thank the Yale Center for Research Computing for the computation run on the Farnam cluster. This work was funded by CTSA grant TL1 TR001864 (R.E. and M.E.P.), a Fast Grant from Emergent Ventures at the Mercatus Center at George Mason University (N.D.G.), the Centers for Disease Control and Prevention Broad Agency Announcement 75D30120C09570 (N.D.G.) and 75D30120C09605 (B.L.M.), and CDC Baseline Surveillance Contract 75D30121C10501 (Clinical Research Sequencing Platform, LLC).
Author contributions
Conceptualization, R.E., J.E.L., and N.D.G.; Genomic Surveillance, all authors; Provision of PCR Data, R.U., N.M., N.R., S.E.T., M.I.B., R.T.K., K.Billig., C.B.F.V., M.L.L., D.R.P., K.K., G.O., H.G., S.M., J.M., R.T., M.D.A., J.E.L., N.D.G., and New England Variant Investigation Team; Methods Development, M.E.P., J.E.R., R.T.K., V.E.P., J.E.L., and N.D.G; Data Analysis and Interpretation, R.E., J.E.L., and N.D.G.; Supervision, R.T., M.D.A., D.J.P., J.E.L., and N.D.G.; Writing – Original Draft, R.E. and N.D.G; Writing – Review & Editing, all authors.
Declaration of interests
N.D.G. is a paid consultant for Tempus Labs and the National Basketball Association and has received speaking fees from Goldman Sachs. P.C.S. is a co-founder of, shareholder in, and scientific advisor to Sherlock Biosciences, Inc., as well as a board member of and shareholder in Danaher Corporation. The remaining authors declare no competing interests.
Published: March 11, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100583.
Contributor Information
Rebecca Earnest, Email: rebecca.earnest@yale.edu.
Nathan D. Grubaugh, Email: nathan.grubaugh@yale.edu.
New England Variant Investigation Team:
Ahmad Altajar, Alexandra DeJesus, Anderson Brito, Anne E. Watkins, Anthony Muyombwe, Brendan S. Blumenstiel, Caleb Neal, Chaney C. Kalinich, Chen Liu, Christine Loreth, Christopher Castaldi, Claire Pearson, Clare Bernard, Corey M. Nolet, David Ferguson, Erika Buzby, Eva Laszlo, Faye L. Reagan, Gina Vicente, Heather M. Rooke, Heidi Munger, Hillary Johnson, Irina R. Tikhonova, Isabel M. Ott, Jafar Razeq, James C. Meldrim, Jessica Brown, Jianhui Wang, Johanna Vostok, John P. Beauchamp, Jonna L. Grimsby, Joshua Hall, Katelyn S. Messer, Katie L. Larkin, Kyle Vernest, Lawrence C. Madoff, Lisa M. Green, Lori Webber, Luc Gagne, Maesha A. Ulcena, Marianne C. Ray, Marissa E. Fisher, Mary Barter, Matthew D. Lee, Matthew T. DeFelice, Michelle C. Cipicchio, Natasha L. Smith, Niall J. Lennon, Nicholas A. Fitzgerald, Nicholas Kerantzas, Pei Hui, Rachel Harrington, Randy Downing, Rashida Haye, Ryan Lynch, Scott E. Anderson, Scott Hennigan, Sean English, Seana Cofsky, Selina Clancy, Shrikant Mane, Stephanie Ash, Stephanie Baez, Steve Fleming, Steven Murphy, Sushma Chaluvadi, Tara Alpert, Trevor Rivard, Wade Schulz, and Zoe M. Mandese
Supplemental information
Data and code availability
A summary of the SARS-CoV-2 lineages used from GISAID are available in Tables S1–S3. A complete list of GISAID acknowledgements and all original code and data have been deposited at Github and are publicly available as of the date of publication (https://github.com/grubaughlab/2021_paper_Delta-v-Alpha). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.CDC SARS-CoV-2 variant classifications and definitions. 2021. https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html
- 2.Lauring A.S., Hodcroft E.B. Genetic variants of SARS-CoV-2—what do they mean? JAMA. 2021;325:529–531. doi: 10.1001/jama.2020.27124. [DOI] [PubMed] [Google Scholar]
- 3.Rambaut A., Loman N., Pybus O., Barclay W., Barrett J., Carabelli A., Connor T., Peacock T., Robertson D.L., Volz E., et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. 2020. https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563
- 4.Alpert T., Brito A.F., Lasek-Nesselquist E., Rothman J., Valesano A.L., MacKay M.J., Petrone M.E., Breban M.I., Watkins A.E., Vogels C.B.F., et al. Early introductions and transmission of SARS-CoV-2 variant B.1.1.7 in the United States. Cell. 2021;184:2595–2604.e13. doi: 10.1016/j.cell.2021.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tao K., Tzou P.L., Nouhin J., Gupta R.K., de Oliveira T., Kosakovsky Pond S.L., Fera D., Shafer R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021;22:757–773. doi: 10.1038/s41576-021-00408-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.CDC Delta variant: what we know about the science. 2021. https://www.cdc.gov/coronavirus/2019-ncov/variants/delta-variant.html
- 7.WHO . WHO; 2021. Weekly Epidemiological Update on COVID-19 - 20 July 2021. [Google Scholar]
- 8.Bolze A., Cirulli E.T., Luo S., White S., Cassens T., Jacobs S., Ngyuyen J., Ramirez J.M., Sandoval E., Wang X., et al. Rapid displacement of SARS-CoV-2 variant B.1.1.7 by B.1.617.2 and P.1 in the United States [Internet] medRxiv. 2021 http://medrxiv.org/lookup/doi/10.1101/2021.06.20.21259195 Preprint at bioRxiv. [Google Scholar]
- 9.Challen R., Dyson L., Overton C.E., Guzman-Rincon L.M., Hill E.M., Stage H.B., Brooks-Pollok E., Pellis L., Scarabel F., Pascall D.J., et al. Early epidemiological signatures of novel SARS-CoV-2 variants: establishment of B.1.617.2 in England [Internet] medRxiv. 2021 http://medrxiv.org/lookup/doi/10.1101/2021.06.05.21258365 Preprint at bioRxiv. [Google Scholar]
- 10.Grubaugh N.D., Hodcroft E.B., Fauver J.R., Phelan A.L., Cevik M. Public health actions to control new SARS-CoV-2 variants. Cell. 2021 doi: 10.1016/j.cell.2021.01.044. https://www.cell.com/cell/fulltext/S0092-8674(21)00087-8?dgcid=raven_jbs_aip_email [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.SAGE . GOV.UK; 2021. SPI-M-O: Summary of Further Modelling of Easing Restrictions – Roadmap Step 4 on 19 July 2021, 7 July. [Google Scholar]
- 12.Campbell F., Archer B., Laurenson-Schafer H., Jinnai Y., Konings F., Batra N., Pavlin B., Vandemaele K., Van Kerkhove M.D., Jombart T., et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Euro Surveill. 2021 doi: 10.2807/1560-7917.ES.2021.26.24.2100509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petrone M.E., Rothman J.E., Breban M.I., Ott I.M., Russell A., Lasek-Nesselquist E., Kelly K., Omerza G., Renzette N., Watkins A.E., et al. Combining genomic and epidemiological data to compare the transmissibility of SARS-CoV-2 lineages. medRxiv. 2021 doi: 10.1101/2021.07.01.21259859. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J.D., Pearson C.A.B., Russell T.W., Tully D.C., Washburne A.D., et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021:372. doi: 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chitwood M.H., Russi M., Gunasekera K., Havumaki J., Klaassen F., Pitzer V.E., Salomon J.A., Smartwood N.A., Warren J.L., Weinberger D.M., et al. Reconstructing the course of the COVID-19 epidemic over 2020 for US states and counties: results of a Bayesian evidence synthesis model. medRxiv. 2021 doi: 10.1101/2020.06.17.20133983. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cori A., Ferguson N.M., Fraser C., Cauchemez S. A new framework and software to estimate time-varying reproduction numbers during epidemics. Am. J. Epidemiol. 2013;178:1505–1512. doi: 10.1093/aje/kwt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogels C.B.F., Breban M.I., Ott I.M., Alpert T., Petrone M.E., Watkins A.E., Kalinich C.C., Earnest R., Rothman J.E., deJesus J.G., et al. Multiplex qPCR discriminates variants of concern to enhance global surveillance of SARS-CoV-2. PLoS Biol. 2021;19:e3001236. doi: 10.1371/journal.pbio.3001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hay J.A., Kennedy-Shaffer L., Kanjilal S., Lennon N.J., Gabriel S.B., Lipsitch M., Mina M.J. Estimating epidemiologic dynamics from cross-sectional viral load distributions. Science. 2021;373:eabh0635. doi: 10.1126/science.abh0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogels C.B.F., Brito A.F., Wyllie A.L., Fauver J.R., Ott I.M., Kalinich C.C., Petrone M.E., Casanovas-Massana A., Catherine Muenker M., Moore A.J., et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 2020;5:1299–1305. doi: 10.1038/s41564-020-0761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballotpedia Documenting America’s path to recovery. 2021. https://ballotpedia.org/Documenting_America%27s_Path_to_Recovery
- 21.Teyssou E., Delagrèverie H., Visseaux B., Lambert-Niclot S., Brichler S., Ferre V., Marot S., Jary A., Todesco E., Schnurigere A., et al. The Delta SARS-CoV-2 variant has a higher viral load than the Beta and the historical variants in nasopharyngeal samples from newly diagnosed COVID-19 patients. J. Infect. 2021 doi: 10.1016/j.jinf.2021.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kissler S.M., Fauver J.R., Mack C., Tai C.G., Breban M.I., Watkins A.E., Samant R.M., Anderson D.J., Metti J., Khullar G., et al. Viral dynamics of SARS-CoV-2 variants in vaccinated and unvaccinated persons. N. Engl. J. Med. 2021;385:2489–2491. doi: 10.1056/NEJMc2102507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li B., Deng A., Li K., Hu Y., Li Z., Shi Y., Xiong Q., Liu Z., Guo Q., Zou L., et al. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. Nat. Commun. 2022;13:1–9. doi: 10.1038/s41467-022-28089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kislaya I., Rodrigues E.F., Borges V., Gomes J.P., Sousa C., Almeida J.P., Peralta-Santos A., Nunes B. Comparative effectiveness of coronavirus vaccine in preventing breakthrough infections among vaccinated persons infected with Delta and Alpha variants. Emerg. Infect. Dis. 2022;28:331–337. doi: 10.3201/eid2802.211789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Despres H.W., Mills M.G., Shirley D.J., Schmidt M.M., Huang M.-L., Roychoudhury P., Jerome K.R., Greninger A.L., Bruce E.A. Measuring infectious SARS-CoV-2 in clinical samples reveals a higher viral titer:RNA ratio for Delta and Epsilon vs. Alpha variants. Proc. Natl. Acad. Sci. U S A. 2022;119 doi: 10.1073/pnas.2116518119. https://www.pnas.org/content/119/5/e2116518119.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khare S., Gurry C., Freitas L., Schultz M.B., Bach G., Diallo A., Akite N., Ho J., Lee R.T., Yeo W., et al. GISAID’s role in pandemic response. CCDCW. 2021;3:1049–1051. doi: 10.46234/ccdcw2021.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Challenges. 2017;1 doi: 10.1002/gch2.1018. https://pubmed.ncbi.nlm.nih.gov/31565258/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shu Y., McCauley J. GISAID: global initiative on sharing all influenza data – from vision to reality. Eurosurveillance. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.US Census Bureau State population totals: 2010-2019 [internet] 2021. https://www.census.gov/data/datasets/time-series/demo/popest/2010s-state-total.html
- 31.CDC COVID-19 vaccinations in the United States, jurisdiction [internet] 2021. https://data.cdc.gov/Vaccinations/COVID-19-Vaccinations-in-the-United-States-Jurisdi/unsk-b7fc
- 32.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 http://arxiv.org/abs/1303.3997 Preprint at. [q-bio.GN] [Google Scholar]
- 33.Grubaugh N.D., Gangavarapu K., Quick J., Matteson N.L., De Jesus J.G., Main B.J., Tan A.L., Paul L.M., Brackney D.E., Grewal S., et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwam A., Keane T., McCarthy S.A., Davies R.M., et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Toole Á., Scher E., Underwood A., Jackson B., Hill V., McCrone J.T., Colquhoun R., Ruis C., Abu-Dahab K., Taylor B., et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7:veab064. doi: 10.1093/ve/veab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rambaut A., Holmes E.C., O’Toole Á., Hill V., McCrone J.T., Ruis C., Plessis L., Pybus O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.RStudio Team RStudio: integrated development for R [internet] 2020. http://www.rstudio.com/
- 38.Alene M., Yismaw L., Assemie M.A., Ketema D.B., Gietaneh W., Birhan T.Y. Serial interval and incubation period of COVID-19: a systematic review and meta-analysis. BMC Infect. Dis. 2021;21:257. doi: 10.1186/s12879-021-05950-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li M., Liu K., Song Y., Wang M., Wu J. Serial interval and generation interval for imported and local infectors, respectively, estimated using reported contact-tracing data of COVID-19 in China. Front. Public Health. 2020;8:577431. doi: 10.3389/fpubh.2020.577431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du Z., Xu X., Wu Y., Wang L., Cowling B.J., Meyers L.A. Serial interval of COVID-19 among publicly reported confirmed cases. Emerging Infect. Dis. 2020;26:1341–1343. doi: 10.3201/eid2606.200357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pung R., Mak T.M., CMMID COVID-19 working group. Kucharski A.J., Lee V.J. Serial intervals in SARS-CoV-2 B.1.617.2 variant cases. Lancet. 2021;398:837–838. doi: 10.1016/S0140-6736(21)01697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryu S., Kim D., Lim J.S., Ali S.T., Cowling B.J. Serial interval and transmission dynamics during SARS-CoV-2 Delta variant predominance, South Korea. Emerg. Infect. Dis. 2022;28 doi: 10.3201/eid2802.211774. https://pubmed.ncbi.nlm.nih.gov/34906289/ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A summary of the SARS-CoV-2 lineages used from GISAID are available in Tables S1–S3. A complete list of GISAID acknowledgements and all original code and data have been deposited at Github and are publicly available as of the date of publication (https://github.com/grubaughlab/2021_paper_Delta-v-Alpha). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.