

Abstract
Although the treatment of myocardial infarction (MI) has improved considerably, it is still a worldwide disease with high morbidity and high mortality. Whilst there is still a long way to go for discovering ideal treatments, therapeutic strategies committed to cardioprotection and cardiac repair following cardiac ischemia are emerging. Evidence of pathological characteristics in MI illustrates cell signaling pathways that participate in the survival, proliferation, apoptosis, autophagy of cardiomyocytes, endothelial cells, fibroblasts, monocytes, and stem cells. These signaling pathways include the key players in inflammation response, e.g., NLRP3/caspase-1 and TLR4/MyD88/NF-κB; the crucial mediators in oxidative stress and apoptosis, for instance, Notch, Hippo/YAP, RhoA/ROCK, Nrf2/HO-1, and Sonic hedgehog; the controller of myocardial fibrosis such as TGF-β/SMADs and Wnt/β-catenin; and the main regulator of angiogenesis, PI3K/Akt, MAPK, JAK/STAT, Sonic hedgehog, etc. Since signaling pathways play an important role in administering the process of MI, aiming at targeting these aberrant signaling pathways and improving the pathological manifestations in MI is indispensable and promising. Hence, drug therapy, gene therapy, protein therapy, cell therapy, and exosome therapy have been emerging and are known as novel therapies. In this review, we summarize the therapeutic strategies for MI by regulating these associated pathways, which contribute to inhibiting cardiomyocytes death, attenuating inflammation, enhancing angiogenesis, etc. so as to repair and re-functionalize damaged hearts.
Subject terms: Cardiovascular diseases, Molecular medicine
Introduction
Cardiovascular diseases are the leading cause of death disease worldwide, of which the death toll due to ischemic heart disease accounted for as much as 49.2% in 20191,2. Acute myocardial infarction (MI) is usually caused by a thrombus blocking an artery or a bypass graft, characterized by an abrupt reduction in blood flow to the myocardium, ultimately leading to heart failure and death2,3. Restoring blood flow to rescue hypoxic-ischemic tissue is considered to be an effective strategy4–6. Thrombolysis, percutaneous coronary intervention (PCI), and coronary artery bypass grafting are the most common methods for the treatment of acute MI in the clinic4–6. Although these methods significantly reduce the patient mortality rate7, complications occur in an unpredictable manner, including hemorrhage, ischemia-reperfusion injury, and coronary restenosis5,8. Therefore, it is necessary to pursue more innovative and effective avenues to preserve myocardial function and avoid heart failure progression.
Post MI, in the injured myocardium, the inflammation, fibrosis, and angiogenesis phases in the injured myocardium overlap9,10 (Fig. 1). Suffering from ischemia-hypoxia, the apoptotic wave of cardiomyocytes within hours to days, and the damaged tissue triggers an inflammatory reaction, which results in the development of granulation tissue with infiltration of immunocytes that release pro-inflammatory cytokines and chemokines9,11. Along with the recruitment of myeloid cells and the transduction of pro-inflammatory signals, including transforming growth factor-β (TGF-β)/SMADs and Wingless (Wnt)/β-catenin, fibroblasts produce collagen and endothelial cells are activated by pro-angiogenic phosphoinositide-3 kinase/protein kinase B (PI3K/Akt), Janus kinase/signal transducer and activator of transcription (JAK/STAT), and angiogenesis commences9–12. The new capillaries not only bring nutrients to the border zone of the infarct but also provide energy for fibroblasts to differentiate into myofibroblasts, which is crucial for sustaining the integrity of the structure and function of the heart through compensation9,10. Simultaneously, myofibroblasts activate TGF-β, and Wnt/β-catenin signaling to escape apoptosis and improve survival13. However, reactive fibrosis and cardiac remodeling lead to cardiac dysfunction9,14.
Fig. 1.

Schematic diagram of the pathophysiology of different cell phenotypes and representative pathways involved in infarct hearts (created with BioRender.com). After myocardial infarction, various cell signaling pathways are activated. Oxidative stress and the death of tissue, particularly apoptotic and necrotic cardiomyocytes, trigger the inflammatory response. Immunocytes infiltrate the infarct area and release inflammatory factors. Meanwhile, cardiac fibroblasts transform into cardiac myofibroblasts and secrete extracellular matrix, and endothelial cells migrate, proliferate and form a network of blood vessels to promote the cardiac repair. However, pathological hypertrophy of the myocardium affected by inflammation, coupled with reactive fibrosis, would eventually lead to cardiac remodeling and heart failure. MAPK, mitogen-activated protein kinase; Hippo/YAP, Hippo/Yes-associated protein; RhoA/ROCK, Ras homolog family member A/Rho associated coiled-coil containing protein kinase; Nrf2/HO-1, nuclear factor erythroid derived 2-related factor 2/heme oxygenase-1; TLR4/MyD88/NF-κB Toll-like receptor 4/MyD88/nuclear factor-κB; NLRP3/caspase-1, the nucleotide-binding domain, leucine-rich-repeat family, pyrin-domain-containing 3/caspase-1; TGF-β/SMADs, transforming growth factor-β/SMADs; Wnt/β-catenin, Wingless/β-catenin; PI3K/Akt, phosphoinositide-3 kinase/protein kinase B; EndoMT. endothelial-to-mesenchymal transition
Notably, cell signaling pathways have critical roles in regulating these pathophysiological conditions. Some cell signaling pathways such as Notch, nuclear factor erythroid-derived 2-related factor 2/heme oxygenase-1 (Nrf2/HO-1), Ras homolog family member A/Rho-associated coiled-coil containing protein kinase (RhoA/ROCK), as well as Sonic hedgehog pathways regulate cardiac regeneration, reactive fibrosis, and cardiac hypertrophy, mediate the survival, proliferation, apoptosis, differentiation and other phenotypes of cells12,15–19. In general, considering cell signaling pathways as a regulating network that participate in a variety of processes after MI, it is pivotal to comprehend the mechanism of pathophysiological processes post MI. And understanding the signal transduction of molecular events eventually contributes to the recognization of the influence of signaling pathways on the progress of MI, and further leads to the discovery of novel therapeutic strategies.
Over the past few decades, enthusiastic attempts have been made to improve post-infarction prognosis in MI by targeting signaling pathways, which are known as emerging therapies, including pharmacotherapy, gene therapy, protein therapy, cell therapy, and exosome therapy12,20,21. These therapies address the essential causes of MI progression by targeting key signaling pathways. For example, inhibition of the Toll-like receptor 4 (TLR4)/MyD88/nuclear factor-κB (NF-κB) and TGF-β pathways alleviate excessive inflammation and cardiac fibrosis22,23. On the other hand, enhancing activation of the PI3K/Akt and mitogen-activated protein kinase (MAPK) pathways promotes the formation of functional vasculatures24. Apart from the anti-fibrosis strategy, the anti-inflammation, and therapeutic angiogenesis strategies targeting molecular mechanisms have also been well confirmed and applied for the treatment of MI9,11,15,25,26. Over the past decade, more advanced studies have shown that promoting the proliferation of pre-existing cardiomyocytes to drive endogenous cardiac regeneration by regulating Hippo/Yes-associated protein (YAP) signaling is viable, as another means of treating cardiac ischemic injury27–29.
To date, increasing numbers of preclinical studies and clinical trials were designed to pursue effective therapeutic strategies for MI. From this perspective, comprehending and summarizing the existing evidence of cell signaling pathways associated with the development and treatment of MI are essential and promising. Therefore, in this review, we explore the roles of several key signaling pathways in MI: PI3K/Akt, Notch, TGF-β/SMADs, Wnt/β-catenin, NLRP3/caspase-1, TLR4/MyD88/NF-κB, Nrf2/HO-1, RhoA/ROCK, MAPK, JAK/STAT, Hippo/YAP, and Sonic hedgehog pathways. Herein, we discuss the crucial functions of these signaling pathways in pathophysiological conditions post ischemia, all of which are promising therapeutic targets in the therapeutic strategies of MI.
PI3K/Akt pathway in MI
The PI3K/Akt pathway has been identified as a key mechanism in the occurrence, progression, and treatment of MI30. An increasing number of studies have found that the components of this pathway are activated in response to cell-external or -internal stimuli31,32, implicated in survival, proliferation, apoptosis, migration, and other physiological or pathological processes30,33–35. When PI3K converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3), Akt is activated as the core molecule in the pathway36,37. PIP3 binds to the Pleckstrin homology (PH) domain of Akt to alter its conformation, exposing Ser473 and Thr308 sites36. Finally, phosphoinositide dependent kinase 1 (PDK1) and PDK2 phosphorylate Thr308 and Ser473 of Akt, regulate cardiac recovery following MI via the downstream signaling pathway36,38 (Fig. 2a).
Fig. 2.

a PI3K/Akt signaling pathway and targeted therapy in Myocardial infarction (MI). PI3K/Akt is involved in the regulation of cardiac remodeling, regeneration, and repair post-ischemia. This pathway responds to the stimulus, likewise growth factor/growth factor receptor signaling and so on. Phosphorylated PI3K and Akt activate the downstream molecules, VEGF, eNOS, while inhibiting mTOR(C1), GSK-3β, FOXO, respectively. GF, growth factor; GFR growth factor receptor, PI3K Phosphoinositide-3 kinase, Akt protein kinase B, PIP2 phosphatidylinositol 4,5-bisphosphate, PIP3 phosphatidylinositol 3,4,5-trisphosphate, PDK phosphoinositide dependent kinase, PH Pleckstrin homology, PTEN phosphatase and tensin homolog, VEGF vascular endothelial growth factor, eNOS endothelial nitric oxide synthase, mTORC1/2 mammalian target of rapamycin complex 1/2, GSK-3β glycogen synthase kinase 3β, FOXO forkhead box subfamily O, AZIN2-sv lncRNA-AZIN2 splice variant, S1P sphingosine-1-phosphate, Ezh2 enhancer of zeste homolog 2. b Notch signaling pathway and targeted therapy in MI. RBP-JК recombination signal-binding protein-JК, NICD notch intracellular domain, CSL CBF1/Rbpj (mammalian), Su(H) (Drosophila), and Lag-1 (Caenorhabditis elegans), CX Chuanxiong, CS Chishao VA velvet antler, YQHX Yiqihuoxue prescription, AGS astragaloside
Downstream molecules of the PI3K/Akt pathway in MI
As downstream effectors of Akt, endothelial nitric oxide synthase (eNOS)39, vascular endothelial growth factor (VEGF)40, mammalian target of rapamycin (mTOR)33, glycogen synthase kinase 3β (GSK-3β)41, and forkhead box subfamily O (FOXO)42 govern cell growth, proliferation, apoptosis, and cardiovascular homeostasis (Fig. 2a).
eNOS is a member of the family of NOS enzymes encoded by Nos2, that catalyzes the conversion of l-arginine into nitric oxide (NO). In the heart, Nos2 is expressed in vascular endothelial and smooth muscle cells, cardiomyocytes, and cardiac fibroblasts. NO has been proven to be a key mediator in cardiac remodeling39. Deletion of eNOS induced the profibrotic effect, resulting in excessive cardiac fibrosis39, which might provide a therapeutic target for myocardial fibrosis through activation of eNOS. In addition, activation of eNOS contributes to myocardial angiogenesis43, similar to the role of VEGF in therapeutic angiogenesis post MI.
Studies have shown that mTOR consists of two complexes, mTOR complex 1 (mTORC1) and mTORC2. They are both essential for cardiac remodeling following MI, because they regulate apoptosis, autophagy44–46, and inflammation47. Upregulation of autophagy is a cardioprotection mechanism response in stress48,49. Autophagy can be inhibited by the activity of mTORC150, leading to reduced survival of cardiomyocytes in an in vitro injury model and aggravating infarction in vivo in myocardial ischemia51. Nevertheless, mTORC2 primarily responds to stimulation of insulin and insulin-like growth factors, which seem to also regulate cell proliferation and polarity52–54, protecting the heart from ischemic damage45. Furthermore, GSK-3β alleviates the inhibition of autophagy mediated by mTORC1 in myocardial cells and aggravates ischemic injury after prolonged myocardial ischemia55.
FOXOs are not only involved in tumorigenesis but are also involved in the deterioration of MI, in particular, FOXO356,57. It has been noted that, following ischemia, constitutively active FOXO3a is associated with poor prognosis, resulting in deficient angiogenesis due to the increase in apoptosis and a reduction in proliferation in vascular smooth muscle cells (VSMCs)42. The signaling stimuli of growth factors phosphorylate Akt1 and FOXO3a, limit FOXO3a transcriptional activity, and enhance cardiomyocyte survival and native angiogenesis in the aftermath of an ischemic event35,58.
The PI3K/Akt pathway as a beneficial signaling mechanism for MI therapy
Drugs
Phosphatase and tensin homolog (PTEN) is widely considered to be a negative regulator of PI3K/Akt by dephosphorylating PIP3 to PIP259,60, participating in pathological processes in ischemic myocardium61,62. In preclinical studies, pharmacological inhibitors of PTEN, including HOpic61 and VO-OHpic63, have shown admirable efficacy in reducing the inhibition of PI3K and promoting angiogenesis61, apoptosis resistance, and survival63. Moreover, emerging evidence confirmed that PTEN is involved in cardiac remodeling post infarction, the decrease of PTEN activity was associated with subsequent reductions in leukocyte infiltration, cardiomyocyte proliferation, and adverse cardiac remodeling62,64.
As mentioned above, mTOR-dependent signal transduction is implicated in cardiac remodeling, and an mTOR inhibitor has been verified to augment autophagy and limit the infarct size of ischemia myocardium44,65. Rapamycin and its derivatives are common therapeutic agents that reinforce autophagy but also limit apoptosis33,66–68. Moreover, sphingosine-1-phosphate and tanshinone IIA have been highlighted as potential therapeutic targets that inhibit mTOR to promote angiogenesis and encourage myocyte autophagy following MI69,70.
Protein therapy and Gene therapy
With the application of recombinant proteins and viral vectors in cardiovascular diseases, increasing studies are attempting to use developing techniques for cardiovascular disease treatment71,72. In response to gene and protein expression of FMS-like tyrosine kinase 3 upregulated by intramyocardial injection of the recombinant FMS-like tyrosine kinase 3 ligand, cardiomyocytes are protected from apoptosis, and cardiac remodeling and function of the infarct heart were improved through Akt-dependent signaling73. Interestingly, gene editing of SERCA2a exerted similar cardioprotective effects74.
Studies have shown that non-coding RNAs, including microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs), represent novel therapeutic tools for MI. A growing number of studies have observed that miRNA-2175,76, miRNA-130a77, miR221-3p78, and miR-30179 are mediated by suppression of PTEN and activation of PI3K-dependent signaling. Moreover, studies on lncRNAs indicated that small nucleolar RNA host gene 1 (Snhg1) directly binds to PTEN to form a positive feedback loop with PTEN/Akt/c-Myc to induce cardiomyocyte proliferation80. Furthermore, miR-99a plays a cardioprotective role in postinfarction cardiac remodeling81.
In recent years, with the advent of the theory of competing for endogenous RNAs (ceRNAs), several studies have attempted to explore their detailed molecular regulatory mechanisms in MI82–85. For example, lncRNA GAS5 competes with miR-21 to inhibit the negative regulation of miR-21 to target PDCD4 and PI3K mRNAs83. Similarly, acts as a ceRNA to sponge miR-93-5p mediates activation of the Rac1/PI3K/Akt pathway, revealing that CircHIPK3 could be a potential target for simultaneously reducing cardiac fibrosis and apoptosis84. In addition, suppression the of lncRNA-AZIN2 splice variant (AZIN2-sv) to the PTEN/Akt pathway was released by absorbing miR-214-induced angiogenesis and myocardial repair85. LncRNA UCA1 relieves cardiomyocytes via declining miR-122 and activating the Akt/mTOR pathway86. Likewise, studies illustrate that lncRNA UCA1 and DANCR are cardioprotective by decreasing miRNA-mediated mTOR signaling86,87.
Cell therapy and exosome therapy
In recent decades, stem cell therapy has gained attention due to its viability and potential use in cardiac repair21,88,89. Stem cells secrete cytokines and extracellular vesicles to modulate the processes following MI21,76,90. Transplanted bone-marrow endothelial progenitor cells (EPCs) in the myocardium trigger PI3K/Akt/FoxO signaling underlying the existence of Period 291. Another study mentioned that bone marrow-derived mesenchymal stem cells (BMMSCs) release paracrine factors that exert a protective effect on cardiomyocytes against hypoxia based on overexpression of Akt192. However, due to the unfavorable survival rate of regenerative cells, it is necessary to explore novel strategies to improve the efficacy of stem cell therapy21. Improving stem cell engraftment and reparative potency in injured cardiac tissue might be an alternative. Human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and thymosin β4 microspheres were simultaneously injected into pigs after MI induction, and the microspheres delivered thymosin β4 to improve the engraftment and reparative properties of stem cells post-transplantation by heightening Akt activity93. In addition, relying on activation of the Akt pathway, nerve growth factor nanoparticles enhanced the therapeutic potency of human umbilical cord mesenchymal stem cells (hUCMSCs)94 and paracrine effects on Akt-modified BMMSC-mediated cardiac protection and functional improvement92,95, similar to the cardioprotective effects of edaravone-treated96, EGb761-treated97 TMSB4-transfected98 or IP6K-inhibited99 BMMSCs and rosuvastatin-supplemented adipose-derived stem cells (ADSCs)100.
As a possible modality that may supplant cell therapy, exosome therapy is an emerging novel approach for the treatment of MI76,90,101. Based on the evidence of in vivo experiments and exosomal miRNA arrays derived from human explant-derived cardiac stem cells (CSCs), exosomes from healthy donors exhibited a scarcity of heart protection compared to exosomes from patients with heart failure, and exhibited an impaired ability by blunting miR-21-5p/PTEN/AKT102. In addition, exosomes secreted from aged mesenchymal stem cells (MSCs) enhanced the angiogenesis and survival of cardiomyocytes via the miR-221-3p/PTEN/Akt pathway78. By switching PI3K signaling, analogously, exosomes excreted from SDF1-overexpressing MSCs displayed an advantageous effect on myocardial cells and cardiac endothelial cells after ischemia103.
Notch signaling pathway in MI
The Notch signaling pathway has been demonstrated to play a critical role in mammalian cardiac development. During embryonic heart development, Notch1 is highly expressed in immature myocardium and expressed at low levels in postnatal myocardium. Notch1, Hes1 and Jagged1 levels in adult hearts are very low at birth. However, their levels in cardiomyocytes are significantly increased 4 days after MI104, suggesting that the Notch signaling pathway is involved in the regulation of myocardial injury. Many studies have found that Notch signaling induces stem cell differentiation105, promotes neovascularization106, and alleviates myocardial fibrosis107 and other multiple effects108, further mediating the repair of myocardial ischemic injury and improving cardiac function109. Other studies have also shown that activation of Notch signaling limits the range of myocardial ischemia and improves myocardial function after MI110. Additionally, there is evidence indicating that the Notch pathway is associated with the improvement of MI by improving angiogenesis108,111,112, improving cardiac regeneration and cardioprotection108,113, and reducing fibrosis107, apoptosis114, and oxidative stress108,115.
The Notch pathway improves angiogenesis
Notch signaling also has physiological effects on the phenotype and functional differentiation of vascular endothelial cells. Notch1, Notch4, Jagged1, DLL-1, and DLL-4 are all expressed in endothelial cells, and only the correct binding of ligands and receptors can induce normal endothelial cell function116 (Fig. 1b). Notch1 acts as a mechanical sensor in adult arteries, where endothelial cells transform mechanical forces into intracellular signals116. Intracellular signals are essential for vascular homeostasis, junction integrity, and endothelial cell elongation116. The Notch pathway is also correlated with VEGFA signaling in regulating the differentiation of endothelial cells, the sprouting of capillary networks, and the branching and fusion of endothelial tubes117.
The Notch pathway reduces myocardial fibrosis
Cardiac fibroblasts proliferate and differentiate into myofibroblasts after myocardial injury, express smooth muscle actin (SMA), secrete collagen, and participate in tissue repair108. However, progressive fibroblast proliferation and differentiation result in the excessive synthetic secretion of collagen, eventually leading to myocardial fibrosis108.
The Notch pathway plays a crucial role in myocardial fibrosis. It directly regulates the expression of α-SMA through activation of the primary effector CSL in endothelial cells and vascular smooth muscle cells118. Many studies have suggested that activation of the Notch1 signaling pathway prevents myocardial fibrosis. For example, Notch1 knockout mice were more likely to develop myocardial fibrosis after myocardial injury than wild type mice119, while enhanced Notch1 activity inhibited the transformation of fibroblasts into myoblast fibroblasts by antagonizing TGF-β1/SMAD3 signaling107. Moreover, some therapies have been developed to explore the application of stem cells or miRNAs to decrease fibrosis110,120. For example, investigators transplanted N1ICD-overexpressing C-MSCs into MI mice and observed decreased myocardial fibrosis after MI110. Another study also used miR-29b to inhibit myocardial fibrosis by activating the Dll4-Notch1-Hes l signaling pathway in MI rats120.
Besides Notch1, Notch3 reportedly inhibits cardiac fibroblast proliferation, promotes apoptosis, and reduces the transition of fibroblasts to myofibroblasts121. They found that Notch3-mediated cardiac fibroblast activity by negatively regulating the RhoA/ROCK/HIF-1α-signaling pathway121. In addition, expression of Notch-4 was also observed in cardiac fibroblasts118.
The Notch pathway reduces cardiomyocyte apoptosis
In vitro and in vivo studies have suggested that the Notch pathway plays a significant role in reducing cardiomyocyte apoptosis114. In an in vitro experiment in a hypoxic cardiomyocyte model, Notch1-regulated apoptosis by down-regulating Bcl-2 and Bax and up-regulating caspase-9 and -3114. At the same time, the Notch signaling pathway exerts an anti-apoptotic effect by regulating the transcription factor RBP-J in MI mice122. Additionally, another study reported that Notch1 inhibits the binding of NF-КB to DNA, thereby playing a negative regulatory role in inhibiting apoptosis and enhancing cell survival123,124.
The Notch pathway reduces oxidative stress in cardiomyocytes
The function of the Notch pathway in antioxidative stress has been reported in several studies105,125,126. For instance, TNF-α inhibitor was demonstrated to suppress oxidative stress in myocardial ischemia/reperfusion (I/R) injury partly through Notch1 signaling125. Considering that the Notch pathway correlates with antioxidative stress, researchers have developed several therapeutic methods and stem cells to upregulate Notch1 signaling to reduce oxidative stress105,126. Overexpression of aldolase A (ALDOA) decreases the hypoxia/reperfusion-triggered oxidative stress and apoptosis in cardiomyocytes by upregulating VEGF/Notch1/Jagged 1 axis126. Another study used EV-C-MSCs carrying N1ICD and found that they decreased the apoptosis of endothelial cells and cardiomyocytes under oxidative stress and ischemic injury in vitro105.
The Notch pathway in the improvement of cardiac regeneration and cardioprotection
During the early postnatal stage, Notch pathway activation is important for regulating cardiomyocyte proliferation127. Notch signaling plays a crucial role in cardiac development, guiding cell fate decisions that underlie myocyte, and vessel differentiation127. In adults, Notch signaling is inhibited in healthy individuals because epigenetic modification of the Notch pathway suppresses cardiac regeneration ability127. However, Notch signaling is activated when injury, hypoxia, and diseases are encountered.
It was reported that reactivation of the Notch pathway is crucial for adult zebrafish to drive cardiac regeneration after injury and in HMGB1-mediated cardiac regeneration128,129. In addition, it also promotes the growth, survival, and differentiation of cardiac progenitor cells into smooth muscle lineages in vitro130. Another study knocked out the Notch1 gene in bone marrow-derived stem cells to treat MI mice, and they observed impaired cardiac repair, suggesting that the Notch signaling pathway plays an important role in the myocardial repair of bone marrow-derived stem cells131.
Besides the role of the Notch pathway in cardiac repair, much preclinical and clinical evidence has also revealed the cardioprotective role of Notch signaling pathways. In a high glucose cell model of hypoxic injury, the Jagged1-Notch signaling pathway exerts a cardioprotective effect113. Another study suggested that upregulation of Notch3 and Notch4 mRNA levels, as well as NICD-3 and -4 in cardiomyocytes induces therapeutic benefits in chronic HF132. Furthermore, clinical evidence is also emerging for the use of Notch1 signaling-activated BMMSCs in patients with ischemic heart disease131.
Correlation between Notch and other signaling pathways in MI
Akt signaling
Notch signaling is reportedly activated by the C-Met/HGF and PI3K/Akt signaling pathways after myocardial injury. Interestingly, Notch also enhances the expression of PI3K/Akt signaling in adult myocardium following myocardial injury110. This mutually supportive crosstalk suggests a positive survival feedback mechanism between Notch and Akt signaling110.
Notch signaling and hypoxia
The imbalance between oxygen supply and oxygen consumption during hypoxia activates oxygen transport and hypoxic cellular metabolism pathways133. Studies have confirmed that Notch signaling is sensitive to hypoxia, and there are multiple direct and indirect interactions between Notch signaling molecules and the hypoxia-inducible factor (HIF) signaling pathway133. First, hypoxia activates the Notch pathway. The gradual accumulation of HIF in tissues stimulates the Notch signaling pathway by activating the expression and synthesis of the exogenous intracellular domain (NICD) promoter to initiate expression of the downstream genes Hes1 and Hey2134. Moreover, inhibition of miR-363 protects cardiomyocytes against hypoxia-induced apoptosis through the promotion of Notch1 expression and the activation of Notch signaling135.
Second, the Notch pathway and hypoxia exert synergistic effects. For example, myocardial ischemia also activates the Notch signaling pathway and induces HIF expression by expressing the target gene Hesl136, alleviating myocardial I/R injury136. Moreover, the HIF-1α-Notch1 pathway is required for the generation of arterial endothelial cells for arteriogenesis and revascularization of ischemic tissue134. This synergistic effect of HIF-1α and the Notch signaling pathway maximizes the rescue of damaged myocardia.
Hypoxia induces expression of Notch ligand Dll 4 and target genes Hey1 and Hey2, activating the Dll 4-Notch-Hey 2 signaling pathway, whose activation is dependent on the activation of HIF-1α and Notch137. Elevated expression of Dll 4 and Hey2 in endothelial progenitor cells inhibits the chicken ovalbumin upstream promoter transcription factor II (Coup-TF II), regulating the production of arteries138. Hey inhibits HIF-1α-induced gene expression136, which suggests that there is negative feedback to prevent hypoxia-induced gene overexpression139.
Application of the Notch pathway in intervention therapy for MI
To date, there is very limited evidence regarding the application of Notch in clinical therapy. Previous studies investigated whether the Notch signaling-induced proangiogenic effect may be the reason for the beneficial effect after the treatment of MI using traditional Chinese medicine and cell therapy62,133,140–145. Many studies have reported that the regulation of non-coding RNAs including miRNAs123,146–154, lncRNAs155,156, and circRNAs147 could exert a therapeutic role in myocardial repair. Moreover, some drugs are reported to correlate with the Notch pathway157–161. For example, it has been reported that Notch signaling participates in the antiapoptotic effects of liraglutide on cardiomyocytes against high glucose-induced myocardial damage157. Oestrogen receptor β activation enhances Notch1 signaling and its downstream mediator-PI3K/Akt signaling to improve myocardial function in MI model158. Although previous studies have suggested that the Notch signaling pathway may be a target of treatment for MI, most are preclinical evidence. Therefore, it is of great significance to further explore the role of Notch signaling in all possible therapies in clinical practice. Up to now, the benefit of melatonin, a regulator of Notch1/Mfn2 pathway145, has been investigated in many clinical trials for coronary heart disease and shows a potential promising clinical application value in reducing infarction size162. However, some evidence suggested melatonin did not improve the myocardial salvage163. It remains to be studied whether melatonin protects the adverse myocardial remodeling in patients with MI.
NLRP3/caspase-1/IL-1β signaling pathway in MI
Some studies suggest that imbalanced inflammation facilitates adverse myocardial remodeling through the activation of one of the most well-known innate inflammatory signaling pathways, the nucleotide-binding domain, leucine-rich-repeat family, pyrin-domain-containing 3 (NLRP3)/caspase-1 inflammasome pathway164,165. It has also been shown that the NLRP3 inflammasome plays an indispensable role in the development and progression of inflammation in MI166 (Fig. 3a).
Fig. 3.

a NLRP3/caspase-1 signaling pathway and its correlated intervention after MI. NLRP3/caspase-1 inflammasome pathway mediated inflammation, pyroptosis, oxidative stress, fibrosis, cardiac remodeling following MI. When NLRP3 is activated by DAMPs and PAMPs, it binds to ASC adaptor molecule and aggregates with pro-caspase-1. Then the NLRP3 inflammasome converts pro-caspase-1 to caspase-1, which catalyzes the conversion of pro-IL-1β and pro-IL-18 to its mature product IL-1β and IL-18. ATP adenosine triphosphate, LPS lipopolysaccharide, PAMPs pathogen-associated molecular patterns, DAMPs Danger-associated molecular patterns, TLR4 toll-like receptor 4, NLRP3 nucleotide-binding domain, leucine-rich-repeat family, pyrin-domain-containing 3, ASC activating signal cointegrator, IL interleukin, NF-κB nuclear factor-κBn, OLT1177 Dapansutrile. b TLR4/MyD88/NF-κB signaling pathway and its correlated intervention after MI. TLR4/MyD88/NF-κB signaling pathway mediated inflammation, pyroptosis, apoptosis, fibrosis, ventricular arrhythmias and lipid metabolism after myocardial infarction. Cardiac injury generates endogenous signals that activate the TLR4/MyD88/NF-κB signaling pathway. The activation of the TLR signaling pathway originates from the cytoplasmic TIR domain that associates with a TIR domain-containing adaptor, MyD88. This signaling pathway activates NF-κB, a transcription factor, and subsequently induce the production of proinflammatory cytokines. TLR4 Toll-like receptor 4, TIRAP TIR (Toll/IL-1 receptor) domain-containing adapter protein, IRAK-4 IL-1 receptor-associated kinase-4, TRAF6 tumor necrosis factor receptor-associated factor 6, IKK IκB kinase, NF-κB Nuclear factor-κB, Lenti shRNA Lentivirus short hairpin RNA, TAK-242 resatorvid, RP-105 radioprotective 105
Activation of the NLRP3/caspase-1 inflammasome pathway in MI
The canonical NLRP3 inflammasome is an intracellular protein complex consisting of the NOD-like receptor (NLR) family member NLRP3, the adaptor protein apoptosis-associated speck-like protein containing a caspase-activating and recruitment domain (ASC), and pro-caspase-1167. PRRs, such as Toll-like receptor 4, recognize a priming signal of infection or tissue damage to activate the inflammatory transcription factor NF-kB, which increases NLRP3, pro-interleukin (IL) -1β, and pro-IL-18168. When NLRP3 is activated, it binds to the activating signal cointegrator (ASC) adaptor molecule and aggregates with pro-caspase-1. Then, the NLRP3 inflammasome converts pro-caspase-1 to caspase-1, which catalyzes the conversion of pro-IL-1β and pro-IL-18 to its mature products IL-1β and IL-18169. IL-1β and IL-18 cause inflammation and tissue damage by regulating immune cell recruitment, cytokine production, and extracellular matrix turnover in the inflammatory response following MI170,171 (Fig. 3a).
Increasing evidence shows that MI is accompanied by inflammatory responses that lead to leukocyte accumulation, the release of inflammatory cytokines/chemokines, myocardial damage, healing, and scar formation172. Therefore, it is important to preserve the heart function and prevent the development of adverse remodeling through timely repression and containment of inflammatory signals173. Several studies focusing on the relationship between NLRP3 inflammasome activation and patients with MI have been reported. Defects in the inflammasome and associated proteins may be involved in promoting ischemic heart disease174.
The NLRP3/caspase-1 inflammasome pathway-mediated inflammation, pyroptosis, oxidative stress, fibrosis, and cardiac remodeling following MI
Many molecules and transcription factors participate in the regulation of the NLRP3/caspase-1 inflammasome pathway in MI. Several studies have shown that nicorandil, isofraxidin, resveratrol (RES, a naturally occurring polyphenol), and short-term aminooxyacetic acid (an inhibitor of aspartate aminotransferase in the aspartate-arginosuccinate shunt) exert cardioprotective effects through inhibition of the NLRP3 inflammasome to reduce MI-induced inflammation175–177. Meanwhile, the inhibition of glycogen synthase kinase-3β or cathepsin B also alleviates activation of the NLRP3 inflammasome in MI178,179. Furthermore, several factors, such as nicorandil180 and growth differentiation factor 11181, exert cardioprotective effects by inhibiting the NLRP3/caspase-1 inflammasome pathway to reduce MI-induced pyroptosis. A recent study investigated whether the NLRP3/caspase-1 pathway also plays a unique role in regulating oxidative stress182. In addition, salvianolate and resveratrol reduce cardiac fibrosis by inhibiting NLRP3 inflammasome signaling and the TGF-β1/SMAD2 signaling pathway in post-MI rats176,183. Moreover, NLRP3 inflammasome activation plays an essential role in cardiac remodeling and malignant ventricular arrhythmia after MI165,179,184–186. Besides the cardiac cells, deficiency of the epigenetic regulator Tet2 in hematopoietic cells is associated with elevated IL-1β-NLRP3 inflammasomes to induce greater cardiac dysfunction185. In addition, a previous study focused on the deterioration of bone vascular function in ischemic heart disease and found that inhibition of NLRP3 partially prevented the loss of type H vasculature after MI in mice187.
Some non-coding RNAs also regulate NLRP3/caspase-1 levels in MI. Recent studies have shown that miR-703188 and miR-133b189 attenuate pyroptosis and hypoxia injury by inhibiting NLRP3/caspase-1 after MI. Moreover, in hypoxic cardiomyocytes, lncRNA H19 overexpression also inhibits NLRP3/caspase-1 to suppress the cell apoptosis rate and promote the cell proliferation rate190.
Furthermore, MSCs exosome treatment reduces white blood cell accumulation and expression of the NLRP3 inflammasome around the infarct area in mouse hearts subjected to left coronary artery (LCA) ligation191. Increased NLRP3 inflammasome activity also plays a role in the pathogenesis of aging-related functional decline in human ADSCs in the aging hosts192. As such, the NLRP3 inflammasome is a key mediator of the post-MI inflammatory response and tissue injury.
Clinical prospects of the NLRP3/caspase-1 inflammasome pathway
As mentioned above, preclinical studies have shown that inhibition of the NLRP3 inflammasome has beneficial effects on preventing infarction injury after MI. Hence, many inhibitors have been developed based on the functional effect of this molecule regarding the treatment of MI. Pharmacological inhibition of the NLRP3 inflammasome via an NLRP3 inflammasome inhibitor (16673-34-0), an intermediate in the synthesis of glyburide, limits cell death and left ventricle systolic dysfunction after ischemia in mice193. Porcine MI models treated with the NLRP3-inflammasome inhibitor MCC950 (6 or 3 mg/kg) markedly preserve the left ventricular ejection fraction194. Moreover, Li, X., et al. noninvasively demonstrated the therapeutic effects of MCC950 in AMI using (18)F-FDG PET imaging195. The covalent NLRP3 inflammasome inhibitor oridonin reduces expression levels of NLRP3, IL-1β, IL-18, and myocardial fibrosis and preserves cardiac function in a mouse MI model196. JC124, a benzenesulfonamide analog used as an NLRP3 inflammasome inhibitor, is now being further studied in mouse models of acute MI, but the results have not yet been published197. OLT1177 (dapansutrile), a β-sulfonyl nitrile molecule and a novel NLRP3 inflammasome inhibitor, preserves myocardial function in I/R or non-reperfused anterior MI mouse models198,199.
Previous studies found that increase of ATP levels following ischemia/reperfusion stimulates P2X7-mediated release of IL-1β, IL-18, and ROS, promoting myocardial damage and declining cardiac function200,201. In contrast, inhibition of P2X7 (brilliant blue G) abrogates the protective ATP-driven effect of short bouts of I/R conditioning and results in increased infarct sizes202. Additionally, colchicine (a drug with broad anti-inflammatory effects, including inhibitory effects on the NLRP3 inflammasome)203 and canakinumab (inhibition of IL-1β)204 have shown efficacy in preventing major adverse cardiovascular events in phase III trials in patients with ischemic heart disease.
There are also several large, randomized placebo-controlled trials. For example, CANTOS205 tested subcutaneous canakinumab 300 mg every 3 months against placebo in patients with a history of MI and serum C-reactive protein (CRP) > 2 mg/L, demonstrating efficacy in preventing major cardiovascular events but increased rates of fatal infections. COLCOT206 (in patients with recent MI) and LoDoCo2207 (in patients with chronic coronary syndromes) tested oral colchicine 0.5 mg daily vs. placebo, demonstrating prevention of major cardiovascular events with a slightly increased risk of pneumonia in COLCOT (0.9% vs. 0.4%) but not in LoDoCo2. Expanding translational research using selective NLRP3 inhibitors is necessary to fully evaluate the potential of NLRP3 inflammasome inhibition in cardiovascular disease.
TLR4/MyD88/NF-κB-signaling pathway in MI
Innate immune cells identify danger signals via engagement of Toll-like receptors (TLRs), a family of transmembrane receptors that activates downstream pro-inflammatory cascades208. TLRs are an important class of protein molecules involved in non-specific immunity that serve as a bridge between non-specific and specific immunity, as well as recognizes invasion and activates the immune response209. To date, more than 10 TLRs have been identified. TLR4 has been the most studied TLR and is widely present on the surface of a variety of cells, such as macrophages210, dendritic cells211, endothelial cells212, and epithelial cells213.
Functional enrichment analyses of 134 genes (gene expression omnibus, GEO database) from patients with different phases of MI identified several hub genes (IL1R1, TLR2, and TLR4) associated with the progression of MI, which can be used as new diagnostic molecules for MI214. Previous cardiac studies have shown that the activation of TLR4 causes increased expression of proinflammatory cytokines, leading to inflammatory responses and additional damage to the already injured myocardium172. Notably, the TLR4-signaling pathways correlate with infarct severity but not with the extent of inflammation. TLR4 and downstream gene expression profiles are upregulated in both infarcted and remote myocardium following MI215,216. In addition, necrotic cardiac myocytes release a wide range of endogenous signals due to MI (S100A1, S100A8/A9, HMGB1, galectin-3, S100β, IL-1α, etc.), associated with significant TLR4 induction217–219. Moreover, platelet activating factor receptor (PTAFR), TLR4, miR-149-5p, miR-6778-3p, and miR-520a-3p were found to be involved in the progression of stable coronary artery disease to AMI in a clinical study220. Conversely, a recent study showed that patients with ST-segment elevation MI have increased expression of a series of genes that implicate NF-κB activity, including HIF-1α, NF-κBIα, IL-18R1/2, MMP9, and IL-8, but reduced expression of TLR4-induced genes, such as TNF-α221. Therefore, further studies focused on the expression of TLR4 and downstream genes in different stages and categories of cardiac disease are needed to confirm these findings (Fig. 2b).
The TLR4/MyD88/NF-κB-signaling pathway mediates inflammation, pyroptosis, apoptosis, fibrosis, ventricular arrhythmias and lipid metabolism after myocardial infarction
Some molecules or transcription factors participate in the regulation of TLR4/MyD88/NF-κB in MI. Gentianella acuta, astaxanthin, astragaloside IV, and danshen (Salvia miltiorrhiza) may ameliorate inflammatory injury via the TLR4/MyD88/NF-κB signaling pathway after acute MI222–225. On the other hand, Li et al. indicated the involvement of the TLR4/MyD88/NF-κB/NLRP3 signaling pathway in attenuating pyroptosis in MI rats treated with nicorandil180. Inhibition of the TLR4/TNF-α signaling pathway in dapsone-mediated cardioprotection also ameliorates apoptosis in rats226. Moreover, the TLR4/MyD88/NF-κB pathway plays a unique role in ameliorating myocardial fibrosis via modified citrus pectin23. Activation of the TLR4/CaMKII signaling pathway is related to vulnerability to ventricular arrhythmias in myeloid differentiation protein 1 (MD1) deletion mice after MI227.
In addition, some metabolism-related factors are also involved in the regulation of the TLR4/MyD88/NF-κB pathway as follows: HIF-1α and apolipoprotein A-I mimetic peptide 4F (4F) may attenuate myocardial injury by minimizing TLR4 upregulation in post-MI rats228,229; cardiac TLR4 is preferentially upregulated by oxidized cholesterol in rats with MI230. Similarly, activation of the TLR4-MyD88 signaling pathway in a hyperlipidemic environment inhibits the lisinopril-mediated cardioprotective effect231. Moreover, electroacupuncture, a physiotherapy factor, may alleviate the excessive inflammatory response after MI by inhibiting the expression of the IL-23/IL-17 axis in MI rats, and TLR4 may be involved during the process232. As such, targeting these factors during different phases of MI may offer an effective therapeutic approach for preserving the function of the ischemic heart.
Some non-coding RNAs are also involved in regulating the TLR4/MyD88/NF-κB signaling pathway in MI. Previous studies have shown that miR-125b-5p, miR-708, and miR-421 attenuate anoxia/reoxygenation injury and the inflammatory response by blocking TLR4 signaling via targeting circRNA nuclear factor IX233, HMGB1234, and JAK2/STAT3235. Furthermore, M1 macrophage-derived extracellular vesicles may promote cardiac dysfunction through TRL4-dependent NF-κB236. Moreover, MSCs exosomes attenuate myocardial ischemia injury in mice by shuttling miR-182/TLR4, which modifies the polarization status of macrophages237. These studies shed new light on potential therapeutic tools for myocardial ischemic injury.
The clinical perspective of TLR4/MyD88/NF-κB inhibition
Sustaining TLR4 activation may lead to deleterious myocardial inflammation; hence, studies have explored several approaches regarding the negative regulation of TLR4. Many preclinical studies focused on inhibiting the TLR4/MyD88/NF-κB signaling pathway have shown beneficial effects in preventing infarction injury after MI. The TLR4 antagonist, ApTOLL238 may be effective in an in vivo pig model of AMI by decreasing inflammatory production of IL-1β and IL-6 and increasing production of IL-10. In addition, radioprotective 105 (RP105), a TLR4 homolog that competitively inhibits TLR4 signaling, confers protective effects on cardiac function after MI239. Moreover, the nanoparticle-mediated administration of TAK-242, a chemical inhibitor of TLR4, attenuates AMI injury by regulating TLR4-dependent monocyte/macrophage-mediated inflammation in a mouse model240. In addition, the clinical drugs metformin and methotrexate, act as TLR4 and NF-κB inhibitors to reduce MI size and improve cardiac function in animal post-MI models241,242. Furthermore, research focusing on gene therapy shows that injection of lentivirus shRNA against TLR4 into the infarcted heart significantly decreases infarct size and improves cardiac function in vivo243. However, the prevention or treatment of cardiac diseases using TLR4 inhibitors or antagonists has not currently been launched in human clinical trials. Further studies are still required to devise methods for protecting the myocardium from additional damage and to contribute to the treatment of MI.
NRF2/HO-1 signaling pathway in MI
NRF2 is the product of the NFE2L2 gene and consists of seven functional domains244. It belongs to the Cap ‘n’ Collar (CNC) subfamily245. NRF2 is extremely unstable and easily degraded in a non-stress state246. NRF2 is an important factor that maintains ROS homeostasis and participates in the regulation of antioxidant genes247. It may sense oxidative signals and transfer signaling molecules to the nucleus, initiating antioxidant gene transcription248. In acute kidney injury, stroke, and other diseases, the use of NRF2-activated compounds effectively reduces ROS, preventing or delaying disease progression249,250.
Heme oxygenase (HO) is a rate-limiting enzyme that catalyzes heme to biliverdin Ixα, carbon monoxide (CO), and iron251. HO-1, HO-2, and HO-3 all belong to the three isoenzymes in the HO system, and all of them show the same catalytic activity252. As a downstream target of NRF2, HO-1 is involved in antioxidant stress and cell protection. For example, HO-1 protects retinal ganglion cells253, liver cells254, and hippocampal neurons255 from I/R injury. In addition, HO-1 can also enter mitochondria to regulate autophagy and inflammation in cells256. Therefore, the protective effect of HO-1 on myocardial cells after MI should not be ignored.
The function of the NRF2/HO-1 signaling pathway in MI
NRF2 plays a crucial role in combating various oxidative stress responses and heart remodeling after MI (Fig. 3a). For example, in the NRF2-KO mouse model, the important role of NRF2 in protecting multiple organs, including the heart, has been widely confirmed17,257,258. Moreover, deletion of NRF2 induces significantly higher mortality of mice after MI is significantly higher than that of mice in the control group, demonstrating that NRF2 plays an important role in MI17. In addition, the important role of HO-1 in the long-term treatment and rehabilitation of MI has also been confirmed. After the modeling of acute MI in rats that received HO-1 pretreatment, in long-term follow-up observations, compared to the control group, the long-term survival rate and myocardial function are significantly increased, and left ventricle remodeling was significantly decreased259,260.
Apoptosis
NRF2/HO-1 is an important pathway that exists in almost all cells types in the body to maintain homeostasis and reduce oxidative stress261. The apoptosis of myocardial cells after MI is one of the important reasons leading to impaired heart function262. Studies have shown that wogonin263, hirudin264, dapsone226, and rosuvastatin combined with low-dose carvedilol265 all act on the NRF2/HO-1 pathway to protect cardiomyocytes from oxidative stress damage after MI and reduce cardiomyocyte apoptosis. The final outcome maintains normal cardiomyocyte function and myocardial tissue structure as well as prevents ventricular remodeling. When HO-1 is successfully activated in rabbit I/R models, it reduces the occurrence of myocardial apoptosis by inhibiting the translocation of NF-κB and AP-1266. In addition, pre-injection of HO-1 or HO-1 activator into the heart significantly reduced MI size and myocardial apoptosis267,268. All this evidence suggests that HO-1 can directly treat MI by reducing oxidative stress-induced damage.
Hypoxia and oxidative stress
Stem cell therapy is one of the most promising therapies in MI269. However, stem cells injected into the border area after MI cause a large number of deaths due to environmental effects such as hypoxia and ischemia, which reduce their therapeutic utility. Overexpression of HO-1 in stem cells effectively solves the tolerance of stem cells to hypoxia and oxidative stress, and simultaneously enhances their paracrine function, thereby increasing the survival rate and enhancing the therapeutic effects270–272. This provides an experimental basis for improving the therapeutic effect of stem cells in the future.
NRF2/HO-1 also protects cardiomyocytes from oxidative stress by regulating ion channels. Excessive Ca2+ influx leads to activation of Ca2+-dependent degradation enzymes, which in turn leads to cellular oxidative stress and dysfunction. Carbon monoxide is the product of HO-1 decomposing heme, which promotes the proliferation of VSMCs and protects cardiomyocytes by inhibiting L-type Ca2+ channels and T-type Ca2+ channels273,274. The proper function of ion channels is closely related to mitochondria. When cardiomyocytes are in an ischemic state, it leads to the deposition of excess ROS and the dysfunction of mitochondrial membrane potential275.
The predictive effect of HO-1 in the blood on MI prognosis
In current clinical studies, it remains controversial whether the levels of HO-1 expression in the blood are correlated with the degree of MI. During the six-month follow-up of AMI discharge, researchers found that increased HO-1 exhibits a significant association with lower severity of coronary artery disease276. However, another two studies suggested the opposite conclusion. SM Chen et al. demonstrated that compared to the control group, expression levels of HO-1 in patients with stable angina pectoris, unstable angina pectoris, and acute MI displayed a rising trend related to disease severity277. Another cohort study of non-cardiac surgery showed that the incidence of adverse cardiac events in elderly patients with high HO-1 expression before surgery was greater than that in elderly patients with low HO-1 expression after non-cardiac surgery278. We think there are three possible reasons for this divergence. The first is that the source of HO-1 is the patients’ blood, and HO-1 in the blood does not fully represent the true condition of HO-1 in the damaged heart tissues. The second is that the damaged myocardium releases high levels of ROS. These increased levels of ROS do not increase the expression of HO-1279. Therefore, whether it is reasonable to use HO-1 in the blood to detect the level of myocardial damage needs further investigation. The last reason is that the total number of samples included was relatively small, and cannot objectively reflect the real situation of HO-1. Therefore, it is necessary to investigate HO-1 expression and MI severity in a larger population in the future.
RhoA/ROCK signaling pathway in MI
RhoA is one of the most important members of the Rho family, and the primary function of the Rho family is widely known for its key role in regulating the cytoskeleton of actin in eukaryotic organisms. The spatiotemporal regulation of RhoA activation is responsible for cellular morphology, attachment, and cell movement280. Under the regulation of guanine nucleotide exchange factor (GEF), GTPase activating proteins (GAPs), and guanine nucleotide dissociation inhibitor (GDI), RhoA switches back and forth between the inactive GDP state and active GTP state to play a biological role281. In addition, mammalian RhoA shares a common post-translational modification region (PTM) at its carboxyl terminus (COOH)282. This region allows RhoA to anchor to the cell membrane, which is necessary for its activation. Only activated RhoA can bind to cell membranes and regulate signaling molecules282. GDI is a negative regulator of RhoA that inactivates RhoA and disconnects it from the membrane to the cytoplasm, and this effect can be reversed by GDF, which allows RhoA to anchor to the cell membrane and restart the cycle again283.
RhoA plays a crucial role in regulating the development and differentiation of the nervous system and cardiovascular system in the embryonic period. For example, during the development of the central nervous system, RhoA regulates neuronal migration mediated by radial glia284. In the cardiovascular system, the primary role of RhoA in its early formation is to promote heart tube fusion, while in the later stage of formation, RhoA plays a role in the construction of the conduction system285,286. In addition, RhoA also mediates the differentiation of coronary artery smooth muscle cells and epicardial cells287. In myocardial cells, RhoA regulates L-type Ca2+ currents and potassium channels288,289. In addition, it is a potential inhibitor of the cardiac fast Na+ current290.
ROCK is the key downstream target of RhoA. It consists of an N-terminal domain and a C-terminal cysteine-rich domain located in the PH motif domain291. ROCK has 2 subtypes: ROCK1 and ROCK2292. They contain 1354 and 1388 amino acids, respectively, and there are 65% and 55% similar homologies in their amino-acid sequence and kinase domains293. Therefore, they are similar in structure and function294. Nevertheless, due to their distinct localization of tissue and subcellular structure295, there are differences in their functions in certain diseases. For example, in diabetic nephropathy, ROCK1 is involved in mitochondrial dynamics and cell differentiation, while ROCK2 is related to inflammation, fibrosis, and cell death296. In airway hyperresponsiveness, although both ROCK1 and ROCK2 can mediate ozone-induced airway hyperresponsiveness, the mechanism is different297.
Is there any difference between the roles of ROCK1 and ROCK2 in the heart? The answer is yes. ROCK1 cardiac-specific knockout mice exhibit myocardial hypertrophy, but cardiac-specific ROCK2 knockout mice do not display signs of myocardial hypertrophy298–300. These results provide evidence for further exploring the mechanism of ROCK1 and ROCK2 in cardiomyocyte hypertrophy after MI (Fig. 4b).
Fig. 4.

a Nrf2/HO-1 signaling pathway and targeted therapy post MI. NRF2/HO-1 plays a crucial role in combating various oxidative stress responses and heart remodeling after MI. It exists in almost all kinds of cells in the body to maintain homeostasis and reduce oxidative stress. In addition, this pathway also plays an important role in stem cell therapy of MI and prognosis prediction of MI. KEAP1 kelch like ECH associated protein 1, NRF2 nuclear factor erythroid-derived 2-related factor 2, HO-1 heme oxygenase-1, ARE antioxidant responsive element, ROS reactive oxygen species. b RhoA/ROCK signaling pathway and targeted therapy post MI. RhoA switches back and forth between inactive GDP state and active GTP state, so as to play its biological role. ROCK is a downstream molecule of Rhoa. They all play a role in fibrosis, ventricular remodeling, and cardiac repair after myocardial infarction. Many drugs, including statins, can play their role in treating myocardial infarction by targeting the RhoA/ROCK pathway. RhoA Ras homolog family member A, ROCK Rho associated coiled-coil containing protein kinase, HIF-1α hypoxia inducible factor-1α, HMG-CoA hydroxymethylglutaryl-CoA, GAP GTPase-activating protein, GDI guanine dissociation inhibitor, GDP guanosine diphosphate, GTP guanosine triphosphate, GEF guanine nucleotide exchange factor
The function of the RhoA/ROCK signaling pathway in MI
There is no doubt that the RhoA/ROCK signaling pathway plays a crucial role in cardiovascular diseases including MI301. However, the direct role of RhoA in the myocardium is rarely studied at present, but it is certain that RhoA directly or indirectly regulates the death and survival of myocardial cells, myocardial hypertrophy, and fibrosis after ischemic injury302,18. These effects may be related to its regulation of actin, cell morphology, and ion channel status303.
Myocardial fibrosis
Myocardial fibrosis is an important pathophysiological process in the border area after MI. It has been confirmed that HIF-1α plays an important role in fibrosis after MI304,305. The RhoA/ROCK signaling pathway is upstream of HIF-1α. The profibrotic effect of HIF-1α is negatively regulated by Notch3 via the RhoA/ROCK/HIF-1α signaling pathway121. It is of great significance to further understand the pathogenesis of cardiac fibrosis. Estradiol and nicorandil are two common clinically utilized drugs. Injecting the above two drugs into the border area of MI significantly reduced the occurrence of fibrosis by inhibiting the RhoA/ROCK signaling pathway306,307. Recently, fasudil, a protein kinase inhibitor based on the structure of isoquinoline sulfonamide, was approved for clinical use as the first ROCK inhibitor308. Although fasudil is mainly used to treat cerebrovascular diseases309,310, its therapeutic effect has been demonstrated in animal models with myocardial fibrosis after MI311,312. Its appearance offers hope for fibrosis after MI. This application prospect is worth investigating in myocardial fibrosis after MI.
Oxidative stress
Oxidative stress is an important pathophysiological process after MI. Numerous studies have shown that the Rho signaling pathway participates in related reactions such as oxidative stress and inflammation313–315. At present, ligation of the left anterior descending coronary artery and isoproterenol injection are the two primary methods for modeling MI. The former is a mechanical blockage of blood flow that leads to MI316. The latter causes oxidative stress in the heart, which leads to progressive mitochondrial damage and changes in cardiac biochemical parameters317. Therefore, the use of isoproterenol injection can be used to further explore the performance of oxidative stress after MI. At present, it has been found that dexmedetomidine318, berberine319, ibuprofen320, and fasudil321 regulate the RhoA/ROCK pathway to protect cardiomyocytes from damage caused by isoproterenol. The ultimate result of these interventions preserves heart function and prevents cardiomyocyte death and ventricular remodeling.
Statins and MSCs in the treatment of MI by regulating the RhoA/ROCK signaling pathway
Statins are clinically important lipid-lowering drugs. Studies have shown that statins can protect the heart after MI. For example, statins such as rosuvastatin322 and fluvastatin323 protect myocardial cells and reduce apoptosis after MI by regulating the RhoA/ROCK pathway. Nevertheless, MI accompanied by an increase in ROS and leakage of cytochrome c and Ca2+ increases the myotoxicity of statins324. Hence, it is significant to explore the most appropriate dose between treatment and poisoning for the application of statins in MI.
Y-27632 is a specific inhibitor of ROCK. When used to iPSCs, it guides the differentiation of iPSCs into cardiac progenitor cells325, and is useful for cell therapy in cardiovascular diseases. Due to differential molecular target binding, another representative statin, atorvastatin inhibits the RhoA/ROCK pathway and its downstream molecules326. This may be due to RhoA non-muscle myosin II taking center stage in cell adhesion and migration327, which provides an important reference for future treatment of MI with drugs combined with MSCs.
MAPK signaling pathway in MI
Mitogen-activated protein kinases (MAPKs) are a class of highly conserved serine/threonine protein kinases in cells that transmit signals through a three-level cascade. To date, four primary branches of the MAPK signaling pathway have been identified, ERK, c-JNK, p38/MAPK, and ERK5328,329. These kinases are sequentially activated and jointly regulate many important physiological and pathological effects, such as proliferation, growth, and differentiation of cardiac resident cells, for example, cardiomyocytes, fibroblasts, endothelial cells, and macrophages330. To date, many attractive inhibitors and antagonists have been developed based on the crucial role of the MAPK/ERK pathway331,332.
Although MAPK signal transduction has been well studied, the clinical efficacy of this pathway inhibitor in MI is not uniform, MAPKs’ functional mechanism and effect in MI remain to be further studied330,333. In this section, we mainly introduce the role of the MAPK signaling pathway in MI from the aspects of drug therapy and molecular and non-coding RNA regulation and discuss the prospects (Fig. 5a).
Fig. 5.

a MAPK signaling pathway and targeted therapy post MI following MI. MAPKs are a class of highly conserved serine/threonine protein kinases in cells that transmit signals through a three-level cascade. There are four main branches of MAPK signaling pathway, namely the ERK, the c-JNK, the p38/MAPK and the ERK5. Hsp90 Heat shock protein 90, α1-AR Alpha1 adrenergic receptor, CXCR7 CXC chemokine receptor 7, Mst1 mammalian sterile 20-like kinase 1, EPO erythropoietin. b JAK/STAT signaling pathway and targeted therapy following MI. JAK/STAT regulates transmembrane receptor and nuclear communication through four steps: (1) Cytokines bind to receptors, leading to dimerization of receptor molecules, and JAKs are activated and phosphorylated; (2) STAT protein is recruited to the docking site formed by these phosphorylated tyrosine sites; (3) STATs are phosphorylated and activated, which enables them to dimerize; and (4) STAT–STAT dimer translocates to the nucleus and regulates gene expression. JAK Janus kinase, STAT signal transduction and activator of transcription, EGCG epigallocatechin-3-gallate, gp130 glycoprotein 130, VEGF vascular endothelial growth factor, Bcl-2 B-cell lymphoma-2, iNOS inductible nitric oxide synthase
Apoptosis
Drugs
Apoptosis is one of the most notable phenotypes mediated by the MAPK signaling pathway. Apoptosis of myocardial cells after MI leads to decreased cardiac function, while apoptosis of non-myocardial cells may aggravate the formation of cardiac scars after MI334. Therefore, effectively avoiding apoptosis through regulation of the MAPK signaling pathway is attractive. Some drugs target this signaling pathway. Kuanxiong aerosol inhibits myocardial injury induced by isoproterenol by inhibiting the MAPK signaling pathway335. The classic lipid-lowering drug atorvastatin significantly improves cardiac function and cardiomyocyte apoptosis in post-MI rats, and its mechanism is related to activation of the ERK1/2 signaling pathway336.
Molecular regulation
Some molecular effectively promote the MAPK signaling pathway and achieve regulate the phenotype of apoptosis. Wang et al. found that overexpression of Mammalian sterile 20-like kinase-1 (MST1) leads to activation of the JNK pathway, which initiates caspase-9-mediated cardiomyocyte apoptosis337. However, the activation of the MAPK signaling pathway is not necessarily negative, and it is widely reported that activating the ERK signaling pathway exerts a protective function in oxidative damage-induced cell death338. For example, ghrelin plays a cardioprotective role in mammals. It significantly reduces apoptosis after MI, and its mechanism is related to the activation of Raf-1-MEk1/2-ERK1/2 signaling pathway339.
Conversely, some molecules also inhibit the MAPK pathway. Erythropoietin is a glycoprotein secreted by perivascular cells in the proximal convoluted tubules of the renal cortex340. Studies have shown that erythropoietin reduces myocardial apoptosis after MI by inhibiting the JNK signaling pathway341. In addition, the regulator of G-protein signaling 5 (RGS5) is an important member of the RGS family that is closely related to cardiovascular diseases342,343. It was found that cardiac function in RGS5 knockout mice was significantly decreased after MI, the infarct area was significantly increased, and obvious apoptosis occurred, which may partially activate the NF-κB and MAPK signaling pathways344. This means that upregulation of RGS5 inhibits the MAPK signaling pathway, reducing myocardial apoptosis. Thus, based on the MAPK signaling pathway, RGS5 is a promising molecular therapeutic target.
Of note, apoptosis of non-cardiomyocytes, including myofibroblasts, after MI may aggravate myocardial remodeling and decrease cardiac function. This is because interstitial non-cardiomyocytes such as granulation tissue form scar tissue through apoptosis and death334,345. Therefore, blocking non-cardiomyocyte apoptosis through the MAPK signaling pathway is also a feasible method for attenuating cardiac dysfunction after MI. Sphingosylphosphorylcholine has this effect. Li et al. found that sphingosylphosphorylcholine inhibits the CaM/p38/STAT3 signaling pathway and attenuates apoptosis of cardiac myofibroblasts induced by hypoxia334,346.
Non-coding RNAs
Genomic studies based on high-throughput sequencing and microarrays also focus on the potential effects of non-coding RNAs on apoptosis after regulating the MAPK pathway347,348. The myocardial tissue of rats with MI infarction was injured in different degrees, and levels of miR-539 were significantly decreased. Further studies demonstrated that apoptosis and autophagy were increased after upregulation of miR-539 expression in H9C2 cells, which may be related to the targeted inhibition of MEK expression by miR-539349. Moreover, p38 is one of the target genes of miR-125b. It can up-regulate the expression of miR-125b, inhibits the expression of p38 and p-p38 to inhibit apoptosis350. In addition, lncRNA MALAT1 downregulation significantly improves myocardial function after MI in rats, which may be related to inhibition of the ERK/MAPK signaling pathway351.
Fibrosis and hypertrophy
Myocardial fibrosis and hypertrophy after MI are key links of pathological ventricular remodeling that are closely related to the MAPK signaling pathway, and targeted regulation of this pathway is of great significance for improving ventricular remodeling352,353.
We mentioned that ANO1 attenuates post-MI myocardial fibrosis through the TGF-β/SMADs pathway354. However, it was been reported that ANO1 also causes fibrosis by activating the MAPK pathway355. We believe that the comprehensive effect of ANO1 in vivo depends on the synergistic effect of multiple pathways, which needs to be further studied. MST1 has also been reported to be associated with fibrosis and activated MST1 induces myocardial fibrosis after MI337. Additionally, Li et al. found that Sprouty3 was predicted to be a potential fibrosis-related target gene of miR-143-3p. MiR-143-3p promotes fibrosis through Sprouty3 degradation and downstream activation of the P38, ERK, and JNK pathways356.
Heat shock protein 90 is a common molecular chaperone that regulates the classic MAPK signaling pathway357. Tamura et al. found that heat shock protein 90 causes myocardial hypertrophy using in vitro and in vivo experiments. The mechanism may be related to increasing the stability of c-Raf in cardiomyocytes and activating the classical Raf/MEK/ERK pathway358. Moreover, knocking out alpha1 adrenergic receptors increased the degree of myocardial hypertrophy after MI, indicating that the deletion of Alpha1 adrenergic receptors may lead to more serious pathological myocardial remodeling in MI mouse hearts359.
Inflammation
Inflammatory injury occurs in the heart after MI, and a variety of inflammatory mediators participate in the process of MI. The severity of the inflammatory reaction also determines the severity of MI, as well as the continuous pro-inflammatory response which leads to ventricular remodeling after MI360. The MAPK signaling pathway is correlated with the inflammatory phenotype, and targeted intervention in this pathway improves the prognosis of AMI by interfering with the occurrence and development of inflammation361. Duan et al. evaluated the cardioprotective effect of Osthole, an active component of Cnidium monnieri extract, in AMI. They found that Osthole improves post-MI symptoms in rats by decreasing the expression of inflammatory cytokines via activations of the MAPK pathway362. Morin is a bioflavonoid that resists isoproterenol-induced myocardial necrosis in rats. Results indicated that levels of proteins related to the MAPK pathway (p-JNK, P38, p-ERK1/2) and related inflammatory indices (TNF-α and IL-6) were changed, indicating that morin reduces inflammatory markers by the regulating MAPK pathway and exerts a protective effect on myocardial injury363. In addition, MST1 knockdown reduces inflammation and protects the heart muscle from damage after chronic infarction337. Erythropoietin also reduces inflammation after heart attacks341. Other studies regarding molecular regulation also demonstrated a correlation between the MAPK signaling pathway and inflammation; for example, low expression of RGS5 leads to activation of part of the MAPK signaling pathway and increases the occurrence of inflammation344. Inhibiting the expression of C-X-C chemokine receptor type 7 prevents the polarization and chemotaxis of M1 macrophages and reduces the occurrence of inflammation, which may be related to the activation of the ERK1/2 pathway364. In addition, miR-26b further inhibits the MAPK signaling pathway by targeting PTGS2, reducing the inflammatory response in mice after MI365.
Angiogenesis
For MI, in theory, blood flow may be richer by increasing the number of blood vessels supplying ischemic tissue366 and targeting this pathway to promote angiogenesis could be a strategy for improving the prognosis of MI. Danhong injection is a type of traditional Chinese medicine for the treatment of cardiovascular diseases367. Li et al. found that after treating MI mice with Danhong injection in vivo and in vitro, the infarct area was significantly decreased, the capillary density increased, and the proliferation and migration ability of HUVECs was significantly improved. This may be related to the drug upregulating miR-126 and indirectly activating the ERK pathway368. Wnt is a secretory glycoprotein that plays a role in autocrine or paracrine signaling369. Wnt11 activates the Wnt/PKC/JNK signaling pathway, promotes angiogenesis and improves cardiac function after MI370. In addition, epiregulin also activates the ERK1/2 pathway and promotes angiogenesis after MI371. IκB Kinase α is also related to angiogenesis. Knockout of IκB Kinase α enhanced the MEK1/2/ERK1/2 pathway and reduced angiogenesis in mice after MI372.
Clinical trials of the MAPK pathway in MI
In addition to the widely used statins which have a good effect on the prognosis of MI, the drugs developed in the clinic are mainly targeted at individual molecules in each branch of the MAPK pathway373,374. As a novel p38 MAPK inhibitor, losmapimod can effectively inhibit the expression of p38 MAPK α and β subtypes. In a phase II clinical trial, the drug effectively improved the prognosis of patients with MI and was well tolerated after oral administration375. But Michelle L O’Donoghue ‘s team found that although the use of losmapimod reduced the inflammatory response in patients after MI compared with placebo, it did not reduce the risk of major ischemic cardiovascular events376,377. Therefore, we think that the selection of Losmapimod as a therapeutic agent for patients with MI remains to be discussed, and the selection of other molecules of this pathway as therapeutic targets may be another treatment idea.
In conclusion, the MAPK signaling pathway is important due to the number of phenotypes involved. Future research should effectively promote the dominant phenotypes caused by this pathway, such as angiogenesis and inflammation reduction, and inhibit the undesirable phenotypes caused by this pathway, such as myocardial fibrosis and cardiac apoptosis. In short, fully understanding the transduction mechanism of the MAPK signaling pathway, taking this signaling pathway as the research target of MI therapy, and developing methods to improve cardiac function after MI are the keys to solving MI challenges.
JAK/STAT signaling pathway in MI
JAK protein is a cytoplasmic tyrosine kinase associated with the intracellular domain of membrane-bound receptors378. Its function is to transduce signals from extracellular ligands (such as cytokines and growth factors) to the nucleus to coordinate cellular responses378. There are 4 members in the JAK family (JAK1, JAK2, JAK3, and TYK2) and 7 members in STAT (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6)379. The JAK/STAT signaling pathway, also known as the IL-6 signaling pathway, is regulated by cytokines and participates in many important biological processes including cell proliferation, differentiation, apoptosis, and immune regulation380, which mainly regulate transmembrane receptors communicating to the nucleus381.
JAK/STAT regulates transmembrane receptors and nuclear communication through four steps: (1) cytokines bind to receptors, leading to dimerization of receptor molecules, and JAKs are activated and phosphorylated; (2) STAT protein is recruited to the docking site formed by these phosphorylated tyrosine sites; (3) STATs are phosphorylated and activated, which enables them to dimerize; and (4) the STAT-STAT dimer translocates to the nucleus and regulates gene expression (Fig. 5b)382. The JAK/STAT pathway is closely related to the occurrence and development of many diseases, such as rheumatoid arthritis383, Parkinson’s disease384, multiple sclerosis385, tumors, and cancer386. Of note, studies have shown that JAK/STAT can be used for the therapeutic intervention of cardiovascular diseases387–391.
The JAK/STAT signaling regulates myocardial apoptosis
It was reported that ischemic myocardium causes cell damage to different degrees and types, and cell apoptosis is one of them392. Previous studies regarding the role of the JAK/STAT pathway in the cardiac tissue have primarily focused on the investigation of STAT1 and STAT3387. For example, the supernatant of necrotic primary cardiomyocytes (Necrotic-S) activates the JAK1-STAT1 pathway and promotes the nuclear translocation of c-Fos and NF-κB p65 after simulating the MI microenvironment, further inducing hypoxia myocardial cell apoptosis, but STAT1 silencing inhibited Necrotic-S-induced cardiomyocyte apoptosis388. Moreover, STAT1 reportedly also induces apoptosis in myocardial I/R by upregulating caspase-1389. Unlike STAT1’s pro-apoptotic effect, STAT3 exhibits an anti-apoptotic effect390. In the rabbit I/R model, the expression of anti-apoptotic genes BCL-2 and p-STAT3 protein significantly decreased. After injection of opioid receptors, the expression of BCL-2 and p-STAT3 increased, and the number of apoptotic cardiomyocytes decreased391. Furthermore, after treatment with the JAK2 inhibitor AG-490, phosphorylation of STAT3 in the myocardium of rats with MI was significantly inhibited, and the activity of caspase-3, Bax expression, and the number of apoptotic cells were significantly increased393. These studies indicate that the JAK/STAT pathway is closely related to the apoptotic response after MI, and STAT1 and STAT3 seem to have opposite effects.
The JAK/STAT in angiogenesis
STAT3 plays an important role in the formation of blood vessels, and this process is essential for controlling compensatory hypertrophy and remodeling394. Not only that, the JAK/STAT signaling pathway also induces polarization of M2 macrophages, promoting myocardial angiogenesis and myocardial functional reconstruction356,395. Specific STAT3 knockout mice displayed no changes in VEGF expression, but these mice exhibited levels of VEGF inhibitors, such as thrombospondin 1 (TSP-1), and increased levels of proteins involved in the formation of interstitial matrix, such as osteopontin (OPN) and plasminogen activator inhibitor-1 (PAI-1)394. This leads to a pro-fibrotic and anti-angiogenic state in the heart after STAT3 is knocked out394. Granulocyte colony stimulating factor (G-CSF) and erythropoietin promote angiogenesis as well as improve cardiac function in MI through the JAK2/STAT pathway in a dose-dependent manner396. These studies demonstrated that the JAK/STAT pathway plays a crucial role in the promotion of heart remodeling via controlling angiogenesis.
The JAK/STAT signaling as a therapeutic target for MI
Continuous activation of STAT transcription factors, especially STAT1, STAT3, and STAT5, has been described in a variety of malignant transformations397. Most studies indicate that STAT3 is an oncogene and that inhibiting STAT3 prevents tumor progression398,399, but activation of STAT3 is essential for protecting the cardiac tissue, such as by promoting angiogenesis or reducing apoptosis390. Therefore, in contrast to cancer treatment, the treatment of cardiovascular disease requires activation of STAT3 signaling390.
STAT3 is mainly activated by the interleukin (IL)-6 cytokine family, which activates glycoprotein 130 (gp130) and further causes the phosphorylation of JAK1 and STAT3400. Nevertheless, gp130 also activates other signal transduction cascades, including PI3k/Akt pathway401, as well as MAPKs (ERK1/2, JNK, p38, and ERK5)402–404. Of note, the differential activation of cell types and cytokines selectively activates many pathways with distinct relative activation intensities400. This activation can select the protective effects of these cytokines in ischemia and hypertrophic myocardium in treatment while diminishing their harmful effects400. In addition, many studies have shown that remote ischemic preconditioning (RIPC) reduces the area of MI and prevents I/R injury by activating JAK/STAT405,406. The JAK/STAT signaling may have multiple target genes, and the upregulation of the target genes may be harmful to the myocardium. For example, the STAT3 target gene iNOS induces the production of nitric oxide through IL-6 and reduces cardiac contractility407. Unlike STAT3 activation, STAT1 inhibitors exhibit cardioprotective effects408. EGCG, a STAT-1 phosphorylation and activation inhibitor, reportedly reduces infarct size and improves hemodynamic recovery and ventricular function in the I/R rat heart408. Hence, activation of JAK/STAT in MI should be carefully controlled, and a clear treatment strategy that balances JAK/STAT signaling should be developed to protect the heart from pathophysiological stress390.
Preclinical studies of the JAK/STAT signaling pathway have shown beneficial effects in preventing infarct injury after MI. Current clinical studies have also found that intracoronary perfusion mobilization of peripheral blood stem cells (PBSCs) and G-CSF in patients with myocardial infarction can improve left ventricular systolic function and remodeling, but the efficacy and safety of this study should be evaluated in a large randomized controlled trial409. However, we have not yet retrieved cases of the use of JAK/STAT inhibitors or agonists in human clinical trials to prevent or treat myocardial infarction. Further research is still needed to devise methods to protect the myocardium from additional damage and to aid in the management of MI.
TGF-β/SMADs signaling pathway in MI
At present, the human TGF-β family includes several members, such as TGF-β, bone morphogenetic protein (BMP), growth and differentiation factor (GDF), activin, inhibin, and so on410. After the TGF-β family binds to TGFβRII, TGFβRI is phosphorylated on specific serine and threonine residues and finally forms a heterocomplex411,412. The receptor complex reacts with the downstream effector SMADs protein and eventually regulates the transcription of the target gene413. At present, there are 8 kinds of SMADs, which are divided into 3 classes according to their functions, namely, receptor-regulated SMAD (R-SMAD), common SMAD (Co-SMAD), and inhibitory SMAD (I-SMAD)414. The TGF-β complex binds to R-SMADs and Co-SMADs to form heteromers, translocates into the nucleus in an R-SMAD-Co-SMAD complex, and transduces specific signals to regulate the transcription of target genes to exert its biological effects415 (Fig. 6a).
Fig. 6.

a TGF-β/SMADs signaling pathway and targeted therapy in MI. After TGF-β family binds to TGFβRII, TGFβRI is phosphorylated on specific serine and threonine residues, and finally forms a heterocomplex. The receptor complex reacts with the downstream effector molecule SMADs protein and eventually regulates the transcription of the target gene. RI receptors type I, RII receptors type II, TF transcriptional factor, R-Smad receptor-regulated Smad, Co-Smad common Smad, I-Smad inhibitory Smad, TMAO trimethylamine N-oxide, KLF5 Kruppel-like factor 5, Cytl1 cytokine-Like 1, ANO1 Anoctamin-1, CTRP9 C1q/tumor necrosis factor-related protein-9. b Wnt/β-catenin signaling pathway and targeted therapy in MI. Wnt signaling is considered as a basic growth regulation pathway. The binding of Wnt to the Frizzled receptor family and low-density lipoprotein receptor-related protein 5 (LRP5) or LRP6 co-receptors stimulates the canonical Wnt/β-catenin signaling pathway, thereby regulating the stability of β-catenin and context-related transcription. Wnt wingless, LRP LDL receptor-related protein, APC adenomatous polyposis coli, GSK-3β glycogen synthase kinase 3β, VEGF vascular endothelial growth factor, SMAD small mother against decapentaplegic, IL interleukin, DKK2 Dickkopf-related protein 2
This signaling pathway is related to the occurrence and development of different diseases, including tumors, tissue fibrosis, cardiovascular diseases as well as rheumatic immune diseases416–418. Researchers have found that this pathway also plays a crucial role in myocardial fibrosis, apoptosis, and other pathological processes after MI419. Therefore, there are an increasing number of studies targeting this pathway for the treatment of MI22,420.
TGF-β/SMADs and myocardial fibrosis after MI
Previous studies have shown that the TGF-β/SMADs signaling pathway plays a critical role in tissue repair421. TGF-β1 is closely related to post-MI and ventricular remodeling and is one of the most important factors promoting myocardial fibrosis422. To date, recombinant TGF-β1 protein is widely used to establish the fibrosis model in vitro423–425.
Cardiac fibroblasts secrete the pro-fibrosis cytokine TGF-β1 and activate the TGF-β1/SMADs signaling pathway after MI426. Moreover, TGF-β1 and downstream SMAD2/3/4 expression are increased to varying degrees in the infarct area and the infarct boundary area427. In addition, TGF-β1 promotes the transformation of cardiac fibroblasts into myofibroblasts. Expression of α-SMA, a hallmark of mature myofibroblasts, was significantly increased in the infarct boundary area428,429. Myofibroblasts further release related inflammatory factors, angiotensin-II, and other cytokines that promote fibrosis, further aggravating cardiac fibrosis429,430. It is worth noting that in the early stage of MI, increased expression of TGF-β1 promotes the recruitment of fibroblasts to the infarction site and secretion of collagen and other substances to promote the recovery of myocardial injury431; however, continued fibrotic responses cause cardiac remodeling and reduced heart function, which eventually lead to heart failure426. Therefore, it is of great significance to explore the positive and negative effects of TGF-β and effectively regulate the expression of key molecules in this pathway for the development of new therapeutic strategies for myocardial fibrosis432,433.
Drugs
Due to the critical role of the TGF-β/SMADs signaling in MI, many inhibitors and antagonists have been developed. Recent studies have found that simvastatin can downregulate TGF-activated kinase 1, reducing TGF-β expression, and improving ventricular remodeling434. Notably, the antihypertensive drug valsartan significantly decreased the expression of TGF-β/SMADs, HIF-1α, and fibrosis-related proteins in rats after MI and significantly improved the cardiac function, infarct size, wall thickness, and myocardial vascularization in ischemic hearts304. Liu’s group showed that the combination of LCZ696 (an angiotensin receptor-neprilysin inhibitor) and benazepril (an angiotensin-converting enzyme inhibitor) exerts a good positive regulatory effect on myocardial fibrosis after MI in mice, and its mechanism was also closely related to the decrease in TGF-β1435. Besides conventional inhibitors and antagonists, additional vitamin D supplementation and aerobic resistance training also regulate the expression of collagen type I and III by downregulating the TGF-β1/SMAD2/3 signaling pathway, further improving cardiac function and alleviating cardiac fibrosis436.
Additionally, several active components of traditional Chinese herbs may also have anti-fibrosis effects through this pathway. Salvianolic acid B, an effective component of Salvia miltiorrhiza, reduces myocardial collagen fibers, decreases the expression of TGF-β1 and SMAD2/3, and increases expression of SMAD7 in vivo and in vitro, which ultimately improves fibrosis437. In addition, it was found that tanshinone IIA reduces the expression levels of collagen type I and III, TGF-β, α-SMA, MP2, and MMP9 in myocardial infarcted rats and angiotensin-induced cardiac fibroblasts438. Yu et al. also found that Ginsenoside Re may improve cardiac dysfunction induced by MI and reduce ventricular remodeling by regulating the AMPK/TGF-β1/SMAD2/3 signaling pathway439.
Molecular regulation
Through the application of gene therapy (gene silencing, gene knockout, gene overexpression, etc.), chemical reagents, and recombinant proteins, key molecules in this pathway can be regulated, thus affecting the occurrence and development of MI440,441. Trimethylamine N-oxide is an intestinal microbial metabolite that is reported to be relevant to the poor prognosis of ischemic heart disease. It activates the TGF-βRI/SMAD2 pathway and aggravates excess cardiac fibrosis and dysfunction after MI442. β-Arrestins are the signaling molecules involved in the desensitization of β-adrenergic receptors. Upregulation of β-Arrestins in cardiac fibroblasts after MI promotes the transformation of fibroblasts to myofibroblasts and collagen synthesis stimulated by TGF-β443. In addition, cytokine-like 1 may aggravate myocardial fibrosis after MI by activating the TGF-β/SMADs signaling pathway444.
Notably, some molecules can also reduce fibrosis by negatively regulating this pathway. For example, ANO1 is a calcium-activated chloride channel protein in human cardiac fibroblasts. In in vivo and in vitro experiments, the degree of cardiac fibrosis was decreased after overexpression of ANO1354. C1q/tumor necrosis factor-related protein-9 has been found to reverse ventricular remodeling and effectively reduce visceral fibrosis via the SMAD2/3 signaling pathway445. Moreover, Nogo-C protein and exogenous BMP-7, which can inhibit this pathway, have also been reported to reduce fibrosis and improve ventricular remodeling446,447. Overexpression of the Notch1 intracellular domain antagonizes TGF-β 1-induced SMAD3 phosphorylation and alleviates the occurrence of fibrosis107. Besides, Notch3 has been found to have similar effects448. Therefore, it may be a promising method for Notch signal activators and TGF-β/SMADs signaling inhibitors to be used for the treatment of fibrosis after MI.
Non-coding RNAs
In recent years, studies regarding non-coding RNAs have emerged and a growing number of findings have demonstrated that the non-coding RNAs play very important roles in regulating the TGF-β/SMADs signaling pathway449–451. Accordingly, it was found that miR-195 promotes fibrosis in MI rats upregulating TGF-β1/SMADs pathway452. Downregulating the expression of miR-130 upregulates the expression of peroxisome proliferator-activated receptor γ and indirectly inhibits TGF-β1, suppressing cardiac fibrosis453. In addition, including but not limited to MALAT1, CircRNA 010567, miR-133a and miR-224 have also been found to affect cardiac remodeling after MI by regulating this pathway454–457.
Of note, some non-coding RNAs directly target key molecules of this pathway to play a regulatory role in MI. For example, SMAD7 is not only the I-SMAD of the TGF-β/SMADs signaling pathway but is also the direct target of Lnc-Ang362. Upregulation of Lnc-Ang362 directly suppressed the expression of SMAD7, promoted the expression of this pathway, and aggravated fibrosis after MI458. In addition, SMAD7 is the direct target of miR-216-5p, and overexpression of miR-216-5p aggravates the occurrence of fibrosis. CircHNRNPH1, a sponge of miR-216-5p, downregulates the expression of miR-216-5p and indirectly upregulates the expression of SMAD7, attenuating reactive fibrosis459.
Cell therapy
Cell therapy is seen as a promising clinical approach, and the application of BMMSCs is a kind of cell therapy that improves cardiac function after MI460. Wei et al. found that ultrasound targeted microbubble destruction-mediated galactose lectin-7-small interfering RNA therapy enhanced the homing ability of BMMSCs, inhibited TGF-β1/SMADs signaling pathway activation and reduced fibrosis after MI461. Hypoxic preconditioned MSCs reduce the activation of fibroblasts by secreting leptin, which may involve inhibition of the TGF-β/SMAD2 signaling pathway462. In addition, MSC transplantation combined with pioglitazone improves myocardial remodeling through the TGF-β1/SMADs signaling pathway463.
The role of the TGF-β/SMADs signaling pathway in apoptosis after MI
The TGF-β/SMADs signaling pathway mediates multiple phenotypes, which not only plays a role in tissue repair but also apoptosis464. After MI, continuous ischemia and hypoxia will lead to activation of TGF-β, which leads to high expression of SMAD2/3, resulting in apoptosis of cardiomyocyte, and further aggravating myocardial injury392,465. The reduction in cardiomyocyte apoptosis during MI is beneficial for the improvement of cardiac function; therefore, targeting this pathway and regulating pericardial apoptosis are particularly important.
There are few studies on the treatment of apoptosis based on the TGF-β/SMADs pathway in the field of MI, and in recent years, it has mainly focused on the regulation of this pathway by ncRNAs. Kruppel-like factor 5 (KLF5) promotes apoptosis in cardiomyocytes and it has been found that KLF5 may activate the TGF-β/SMAD2/3 signaling pathway by downregulating miR-27a, resulting in cardiomyocyte injury after MI466. MiR-808 downregulates the expression of TGF-β1, inhibits the expression of caspase-3 and caspase-9, and inhibits cardiomyocyte apoptosis465. Exocrine bodies derived from ADSCs contain miR-671, which reduces cardiomyocyte apoptosis by inactivating the TGFβRII/SMAD2 axis467. Moreover, LncRNA SOX2-OT aggravates hypoxia-induced cardiomyocyte injury by regulating the miR-27a-3p/TGFβRI axis468. In addition, downregulation of circRNA 010567 expression improves cardiac function and inhibits myocardial apoptosis. The mechanism may be related to inhibition of the TGF-β signaling pathway454.
In most cases, apoptosis is not beneficial in the heart after MI, regardless of whether it occurs in cardiomyocytes or non-cardiomyocytes334. However, TGF-β/SMADs are a double-edged sword, and prematurely targeting inhibition of this pathway to inhibit apoptosis inevitably affects tissue repair in the early stage of MI. Briefly, much more work should be done on the development of new therapeutics targeting the TGF-β/SMADs signaling pathway.
Clinical trials of the TGF-β/SMADs pathway in MI
The statin, angiotensin converting enzyme inhibitor (ACEI) and angiotensin receptor antagonist (ARB) mentioned above that may inhibit this pathway have been widely used in clinical practice and achieved good results in patients with MI469,470. Besides, N-acetylcysteine (NAC) can reduce serum TGF-β levels in Patients with ST-segment elevation MI471. In addition, short-term use of Sodium Tanshinone IIA can also effectively reduce left ventricular remodeling in MI patients472. However, there is still a lack of inhibitors or agonists of the TGF-β/SMADs signaling pathway in large-scale clinical trials, coupled with the two-sidedness of this pathway in the process of cardiac obstruction, further research is needed on the effects of targeted drugs and timing of drug use on patients with MI.
Wnt/β-catenin signaling pathway in MI
The Wnt signaling pathway is related to the development process and affects the cell cycle at various time points473. Simply put, Wnt is a growth stimulating factor that causes cell proliferation474. At the same time, it acts as a directional growth factor in the process of tissue growth475–477. In the field of developmental evolution and cancer therapy, Wnt signaling has been considered as a basic growth regulation pathway473. It is divided into two categories: β-catenin-dependent signaling (canonical pathway) and β-catenin-independent signaling (non-canonical pathway)478. Binding of Wnt to the Frizzled receptor family and low-density lipoprotein receptor-related protein 5 (LRP5) or LRP6 co-receptors stimulates the canonical Wnt/β-catenin signaling pathway, thereby regulating the stability of β-catenin and context-related transcription479. On the other hand, the transmembrane receptor Tyr kinases Ror2 and Ryk and Frizzled receptors that act independently of LRP5 or LRP6, activate the non-canonical Wnt pathway479. This pathway drives cell movement480 and changes in polarity481.
Increasing evidence has shown that Wnt signaling is triggered during the pathological process of MI injury (Fig. 6b). Studies have demonstrated that Wnt activation is related to pathological stages after MI, including inflammation, angiogenesis, and fibrosis482. Analysis of the expression of Wnt proteins indicated that Wnt-2, Wnt-4, Wnt-10b, and Wnt-11 were significantly upregulated 5 days after MI483. The researchers used Axin2-LacZ to express LacZ in cells with active typical Wnt signaling, demonstrating that Wnt signaling is activated in cardiomyocytes located in the border zone of the infarct484. In the TopGAL mouse model expressing the marker β-gal under the control of TCF/LEF1, an increase in Wnt signaling activity was detected 4 days after MI483.
The Wnt/β-catenin signaling pathway and inflammation in MI
The repair of infarct myocardium includes three stages: inflammation, proliferation, and maturity. Inflammation is first activated in MI172. Wnt-5a promotes the release of IL-1, IL-6, and IL-8 from monocytes, indicating that it has a pro-inflammatory effect485. β-catenin-mediated signals are activated in pro-inflammatory macrophages after MI, which is manifested by increased lymphocyte infiltration levels and increased expression of pro-inflammatory cytokines486. In addition, another study reported that the absence of Wnt inhibitory factor 1 (WIF1) causes increased inflammatory monocytes and severe adverse remodeling, while overexpression of WIF1 weakens the monocyte response and improves cardiac function487.
The Wnt/β-catenin signaling pathway and angiogenesis in MI
Angiogenesis manifests as newly formed blood vessels by endothelial cells, which is conducive to heart repair and functional recovery after MI482. A previous study showed that the Wnt signaling pathway is located in the cytoplasm of the vascular endothelium during the neovascularization process after MI, which is reflected by the accumulation of β-catenin488. In fact, many negative Wnt modulators have been shown to promote angiogenesis in the heart after MI482. Overexpression of the FrzA/sFRP-1 gene increases capillary density in MI scars through the inhibition of Wnt signaling489. Likewise, Dickkopf2 (DKK2), another Wnt inhibitor, stimulates endothelial cell angiogenesis after MI via LRP6/APC activation490. Nevertheless, one study also found that the allosteric inhibitor NP12 stabilizes β-catenin and activates the Wnt signaling pathway, which in turn promotes angiogenesis and improves ventricular function after MI491.
The Wnt/β-catenin signaling pathway and cardiac fibrosis in MI
Cardiac remodeling is regarded as a key determinant of the clinical outcome in heart disease and cardiac fibrosis is a major aspect of the remodeling process492. Myocardial fibrosis is an important pathophysiological process observed after MI493. Studies have shown that the Wnt/β-catenin signaling pathway plays a major role in the regulation of cardiac fibrosis494. Interestingly, TGF-β signaling also interacts with the Wnt signaling pathway and plays a key role in the differentiation of myofibroblasts492. Regarding the interaction between the Wnt and TGF-β signaling, studies have demonstrated that Wnt3a can up-regulate TGF-β signaling through the canonical β-catenin-dependent Wnt signaling of SMAD2, inducing myofibroblast differentiation495. In acute ischemic heart injury, the upregulates Wnt1 is initially expressed in the epicardium and then expressed by cardiac fibroblasts in the injured area496. Wnt1 induces cardiac fibroblasts to proliferate and express pro-fibrosis genes496. Except for the role of Wnt, the absence of β-catenin in cardiac fibroblasts alleviates pressure-overload-induced fibrosis in mice, preserves cardiac function, and reduces interstitial fibrosis497. In addition to research on signaling molecules, based on the results of Cui et al.498, miR-145 also reduces heart fibers by directly targeting SOX9 in fibroblasts and regulating the Akt/GSK-3β/β-catenin signaling pathway change. This shows that miRNAs can also inhibit cardiac fibrosis after MI.
The Wnt/β-catenin pathway as a therapeutic target for MI
Since Wnt/β-catenin plays a critical role in MI, the development/use of Wnt/β-catenin inhibitors has been attractive for MI therapy. Pyrvinium, a Wnt inhibitor, was successfully used to stabilize β-catenin and inhibit Axin degradation499. An increase in Ki-67+ cells was observed in the peri-infarct and distal myocardium of animals treated with pyrvinium, which reduced adverse cardiac remodeling499. ICG-001, a β-catenin inhibitor, inhibits the β-catenin signaling pathway and reduces the expression of S100A4, alleviating cardiac fibrosis in mice, indicating that S100A4 may be a therapeutic target for cardiac fibrosis500. Due to differences in target binding, UM206 is a selective frizzled protein antagonist, which inhibits Wnt/Frizzled signaling and was used to reduce the expansion of infarct size and prevent the development of heart failure501. In addition, WNT-974 improves the recovery of heart function after ligation of the left anterior descending coronary artery by reducing the undesirable remodeling of the infarct tissue482. Its mechanism involves preventing the production of collagen in cardiomyocytes by blocking the secretion of Wnt3 (a pro-fibrotic agonist) from cardiac fibroblasts and its signal transmission to cardiomyocytes502. These studies indicate that Wnt pathway inhibitors are a class of potential drugs that treat MI through many mechanisms, including increasing angiogenesis, inhibiting fibrosis, and stimulating heart regeneration.
In recent years, the role of non-coding RNA in MI has emerged. One study found that miR-26a-5p targets WNT5A to inhibit the activity of the Wnt/β-catenin signaling pathway, inhibit H/R-induced cardiomyocyte damage and apoptosis, and restore cell viability467. However, additional studies have observed that miRNAs activate the Wnt pathway to promote the development of MI, and their inhibitors may be more therapeutic. For example, miR-30b-5p promotes myocardial cell apoptosis in rats with MI by activating the Wnt/β-catenin signaling pathway503. MiR-154 has the same effect as miR-30b-5p504. MiR-34a inhibitors505 and miR-423-5p inhibitors506 reduce apoptosis and cardiomyocyte damage after MI in rats by activating the Wnt/β-catenin signaling pathway to improve cardiac function. From the above studies, the roles of non-coding RNA in the Wnt pathway are not entirely the same, and understanding these differences may require further research.
Cell therapy has been extensively tested to restore heart function after MI88,89. Cardiac progenitor cells induced by human induced pluripotent stem cells using cardiogenic small molecules effectively regenerate the infarcted heart and reduce fibrosis, and can target a variety of genes related to cardiac differentiation signaling pathways, including Wnt, cytoskeleton remodeling, and TGF-β induced epithelial mesenchymal transition (EMT) signal, VEGF507. Another study found that the coordinated angiogenesis of cardiac MSCs and direct induction of TGF-β/Wnt signals in MSCs in the myocardium initiate an accelerated healing process and promote heart recovery508. In addition, activation of the Akt/GSK3β/β-catenin signaling axis helps cortical bone-derived stem cells (CBSCs) to play an important protective role in the myocardium by reducing the area of MI, improving cardiac function, and increasing capillary density509. Interestingly, a study found that miR-497 inhibitors activate the Wnt/β-catenin pathway to promote the effects of BMMSCs transplantation in the treatment of MI510.
At present, treatment of MI based on the Wnt/β-catenin signaling pathway has been verified in many animal experiments, but animal models cannot fully replicate all the processes that occur after human MI, so the results of preclinical studies should be carefully explained511. Many current clinical studies have found that Wnt/β-catenin targeted drug therapy or stem cell therapy are more widely used in various cancer patients. Two clinical phase I studies have shown that the Wnt/β-catenin pathway is related to the inhibitors CWP232291512 and OMP-18R5513 can improve the occurrence of adverse events. However, in the research of myocardial infarction, drug therapy and stem cell therapy have been fully explored in preclinical research, and there is still a lack of clinical research to further transform and verify the important role of the Wnt/β-catenin signaling pathway in the prevention and treatment of myocardial infarction. Future research may begin with drugs that have been shown to target Wnt signaling in diseases such as cancer to further test the benefits of intervening in Wnt signaling in cardiovascular disease. These experiments are likely to shed more light on the feasibility and benefits of targeting Wnt signaling in cardiovascular disease.
Hippo signaling pathway in MI
Because the Hippo pathway has been implicated in regulating organ size and tissue homeostasis514,515, there is infinite interest in uncovering the regulatory mechanism of the Hippo pathway in MI29. As an evolutionarily conserved signaling pathway, the key components in mammals include MST1/2, Salvador family protein 1 (SAV1), large tumor suppressors (LATS1/2), Mps one binder kinase activator-like 1A/1B (MOB1A/1B), Yes-associated protein (YAP), and PDZ-binding motif (TAZ), which maintain high consistency with Drosophila516,517. In response to microenvironmental cues, Hippo kinase MST1/2 heterodimerizes with SAV1, and consequently phosphorylates LATS1/2 and the coactivator MOB1, in turn activating the coordinated ubiquitination and 14-3-3 binding of phosphorylated YAP and TAZ, finally suppressing their nuclear localization and degradation29,518 (Fig. 7a).
Fig. 7.

a Hippo/YAP signaling pathway and targeted therapy after MI. Canonical and noncanonical Hippo pathways are vital mechanisms of homeostasis, repair, and regeneration in the heart. In canonical Hippo/YAP, when the pathway turns “on”, the activated MST, SAV, LATS, and MOB leads phosphorylated YAP/TAZ detained in the cytoplasm or degraded, progressively. In contrast, the pathway turns “off” up-regulating the coaction of YAP/TAZ and other transcription factors. MST1/2 mammalian sterile 20-like kinases 1/2, SAV1 salvador family protein 1, LATS1/2 large tumor suppressors 1/2, MOB1A/1B Mps one binder kinase activator-like 1A/1B, YAP Yes-associated protein, TAZ PDZ-binding motif. b Sonic Hedgehog signaling pathway and its correlated intervention after MI. Shh Sonic Hedgehog, Ptc Patched, Smo smoothened, Gli glioma-associated oncogene homolog, TMP tetramethylpyrazine, AGS astragaloside
The Hippo pathway in cardiomyocyte regeneration after MI
Evidence from the latest study showed that MI induces regional patterns of cycling cardiomyocytes519. Since studies have found that the Hippo pathway plays an important role in homeostasis of the cardiovascular system, by controlling cardiomyocyte proliferation and survival29, it has been suggested that there may be tremendous potential for targeting the Hippo pathway for therapeutic intervention in MI520. Moderate loss of function of the Hippo component is a desirable strategy for alleviating cardiac injury in MI521. Among these molecules, MST1 works as a forward modulating regulator in cardiac dysfunction induced by ischemia522. Suppressing the activation of MST1 mainly mitigates adverse cardiac remolding and relieves heart dysfunction523,524. Additionally, Sav was found to be inversely associated with cardiac function and angiogenesis, and positively related to cardiac fibrosis525,526; promotion of LATS2 is deemed to be a negative mediator in cardiomyocyte proliferation527. In the nucleus, activation of transcriptional effector YAP/TAZ, either by inactivation of Hippo kinase cascade components, or by forced activation of YAP/TAZ in a Hippo-independent manner, is desirable for cardiomyocyte renewal therapy. When non-phosphorylated YAP and TAZ enter the nucleus, they bind to transcription cofactors, such as TEA domain transcription factor (TEAD)528 and paired-like homeodomain 2 (PITX2)529, to activate target cardiomyocyte protection genes514. Based on experimental discoveries, YAP activation induces cardiomyocytes to re-enter the myocyte cycle and proliferate in both fetal and adult mouse hearts519, and likewise, overexpression of YAP1 mediated by adeno-associated virus (AAV9) alleviates injury and improves the heart function530.
Besides the canonical Hippo pathway, numerous studies have focused on the molecular mechanisms of Hippo components in cardiac regulation post injury. Epigenetics, Han et al. pointed out the involvement of α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5)-mediated m6A demethylation promotes the translation of YAP, consequently, leading to the promotion of cardiomyocyte proliferation, reduction of infarct size, and marked the restoration of cardiac function531. The beneficial effect of gene therapy with constitutive AAV-gp130 has been demonstrated to promote the proliferation of cardiomyocytes by activating macrophage recruitment via the Hippo-independent Src-Yap pathway532. Heart regeneration following MI is regulated by an intricate network of signaling cascades, and signaling between and within cells is highly complex533. Since research has gradually focused on exploring the mechanism, by means of high-throughput sequencing, the synergistic effect on cardiac recovery has been reflected534,535. Multiple signaling pathways including PI3K/Akt, BMP-SMAD1/5, Hippo/YAP, and MAPK/ERK, are all controlled via lysophosphatidic acid3 (LPA3) mediation to enhance cardiac function and heart regeneration534, and the EMT-like regenerative response is regulated by ERBB2-mediated YAP535. These results revealed that the independent Hippo pathway regulates transcriptomics and proteomics in cardiomyocyte regeneration during injury.
The Hippo pathway mediates inflammation, fibrosis, and angiogenesis following cardiac injury
In addition to the myocardium responding to the occurrence of infarction through the Hippo pathway, the pericardium, inflammatory cells, cardiac fibroblasts, and vascular endothelial cells play an essential role in regulating cardiac function through this pathway during the recovery phase29,536,537. Deleted Yap and Taz in the adult murine epicardium resulted in defective regulatory T cell infiltration following tamoxifen-induced injury, leading to cardiac fibrosis, cardiomyopathy, and a high rate of mortality, along with pro-fibrotic F4/80+ macrophages recruitment538. Moreover, during the progression of MI, the exaggerated fibrotic response, in general, leads to progressive heart failure. Recent studies have investigated whether the Hippo pathway plays a unique role in regulating fibroblast state transitions. LATS1/2 and YAP are required for maintaining cardiac fibroblasts in a resting state and myofibroblast differentiation; hence, deletion of Lats1/2 or inhibition of YAP limits the YAP-dependent inflammation and fibrogenesis response to injury539,540. Admittedly, prolonged fibrogenesis contributes to scar expansion and heart failure9, and effective interventions to prevent or reverse cardiac fibrosis are urgently needed. Although SMAD/TGF-β signaling is commonly regarded as the core regulator in cardiac fibrosis, it has been shown that SKI also triggers the Hippo pathway and deactivates TAZ to inhibit myofibroblast activation541. In contrast, angiogenesis is generally encouraged to prevent heart failure after MI. The heat shock protein (HSPA12B) in endothelial cells cooperates with YAP to regulate the process of vascular remodeling542.
Therapeutic strategies for MI based on the Hippo pathway
As a potential contributor to the regulation of cardiac regeneration, inflammatory, fibrotic, and angiogenic phenotypes, the Hippo signaling pathway is considered a desirable target for treatment. Except for the studies focusing on gene therapy mentioned in the previous section525,530,532,534, administering a high dose of AAV9-Sav-short hairpin RNA (AAV9-Sav-shRNA) directly into border zone cardiomyocytes revealed a mild improvement in the ejection fraction of pig heart526, similar to findings in mouse model from Leach’s group525.
Apart from studies that have demonstrated the feasibility of gene therapy, the newest studies have focused on other interventions, for instance, drug therapy, cell therapy, and therapies based on biomaterials, exosomes, and non-coding RNAs. Drug intervention for suppressing activation of MST1 might represent a promising strategy for cardiac protection522. The cardioprotective effects of oncostatin M523, luteolin543, and melatonin544 have been verified by observing enhanced cardiomyocyte autophagy and mitochondrial biogenesis in MI by targeting MST1. Moreover, to extend the duration of pharmaceutical drug delivery, Chen et al. encapsulated, the fluorine substituent of TAZ-12, TT-10, into polylactic-co-glycolic acid nanoparticles, which effectively activated the cell cycle of hiPSC-CMs and inhibit apoptosis by upregulating YAP545. Feng et al. implanted reduced graphene oxide (rGO)/silk fibroin-modified nanofibrous biomaterials into the heart, showing a direct effect on preventing rat ventricular remodeling via YAP/TAZ546. Due to the regenerative properties of stem cells, stem cell therapy has been engaged to repair injured heart tissues547. Intriguingly, with the aid of exosome biocompatibility, human cardiac explant-derived progenitor cells (CPCs)-derived exosomes carried the extracellular matrix protein periostin to regulate the cardiomyocyte proliferation548. Remarkably, non-coding RNAs including miRNAs have been demonstrated to sufficiently induce cardiomyocyte proliferation and regeneration549,550. In a recent study, high-content miRNA screening of hiPSC-CMs confirmed the core node of the Hippo pathway in controlling cardiomyocyte proliferation as a potential miRNA target551. In particular, miR-93, miR-302, and miR-367 attenuate cardiac remodeling by targeting LATS2527,552,553, Mst1, and Mob1b553 and promote angiogenesis552 after MI. Of interest, although transduction of non-coding RNAs can be achieved by gene therapy using AAVs or small nucleic acids, and delivery of biomaterial nanoparticles or engineering exosomes, enveloping non-coding RNAs could facilitate their delivery to the damaged myocardium with high efficiency and safety554.
Although current clinical trials of cardiac regenerative therapies have encountered obstacles, revealing limitations and difficulties in translating preclinical experiments into the clinic, there are still several studies aiming to overcome this bottleneck555,556. Pioneer showed the cardioprotective effects of melatonin, acting as a suppressor of MST1544, administered in patients with ST-segment elevation MI (STEMI) after primary percutaneous coronary intervention162.
Undoubtedly, the molecules of Hippo signaling components are potential target spots for cardiac disease treatment. However, extensive experiments focused on these therapeutic strategies converge on the Hippo pathway in large mammal preclinical models and high-quality clinical trials are still required to advance toward clinical application.
Sonic hedgehog signaling pathway in MI
Hedgehog was discovered in 1980 by Nusslein-Volhard and Wieschaus to regulate the polarity of Drosophila segments557. There is only one Hh gene in fruit flies, while mammals have three: Sonic Hedgehog (Shh), Indian Hedgehog (Ihh), and Desert Hedgehog (Dhh)558. All three Hh gene-secreted proteins exhibit catalytic capacity. Shh is the most widely distributed in human tissues and cells, participating in gene transcription, regulating the expression of cytokines and functional proteins558, and playing an extremely important role in regulating embryonic growth and development, angiogenesis, and tumor cell proliferation559.
The Sonic hedgehog signaling pathway is composed of the signaling molecule hedgehog, patch receptor Ptc (Patched), Smoothened (Smo), and Glioma-associated oncogene homolog (Gli)560,561. Unlike other growth and development signaling pathways, the Sonic hedgehog signaling pathway is highly dependent on a single organelle, the primary cilia560,561. The cilia are packed with proteins required for Sonic hedgehog signal transduction, the important signal components are concentrated in a small area on the tip of the cilia to achieve an effective response, and their distribution on the cilia changes according to the presence or absence of Sonic hedgehog signals562. The Sonic hedgehog pathway has a pleiotropic effect in alleviating cardiac ischemic injury by improving angiogenesis and recruiting EPCs560,561, protecting myocardial cells by decreasing apoptosis and oxidative stress19, and reducing the occurrence of reperfusion arrhythmia563 (Fig. 7b). Previous studies used a Sonic hedgehog agonist to activate the Sonic hedgehog pathway564, and they found that coronary artery density was increased and coronary artery function was also improved after MI564. Accordingly, blocking the Sonic hedgehog signal of cardiomyocytes reduced the expression of coronary angiogenic genes and the number of vessels564.
The Sonic hedgehog pathway in promoting angiogenesis after MI
During myocardial ischemia, expression levels of Sonic hedgehog and Ptc increased, which promotes bone marrow-derived EPCs to induce angiogenesis and promotes the expression of angiogenic factors565. Studies have indicated that Sonic hedgehog gene therapy regulates angiogenesis by activating the Sonic hedgehog pathway565, improves the speed and quantity of coronary angiogenesis566,567, and then increases coronary blood supply which further improves cardiac function566. Previous studies also used a Sonic hedgehog agonist to activate the Sonic hedgehog pathway564, and they found that coronary artery density was increased and coronary artery function was also improved after MI564. Accordingly, blocking the Sonic hedgehog signal of cardiomyocytes reduced the expression of coronary angiogenic genes and the number of vessels564.
Many studies have demonstrated the mechanism of Sonic hedgehog signaling in promoting angiogenesis560,561,565,567,568. First, the Sonic hedgehog pathway increases the number of EPCs and promotes their function. The Sonic hedgehog pathway recruits EPCs to the site of myocardial ischemia, promotes neovascularization, inhibits myocardial fibrosis, and prevents myocardial apoptosis in a chronic myocardial ischemia model565. Another study used microparticles to carry Sonic hedgehog morphogen (MPShh+) and EPCs to improve vascular regeneration and found that MPShh+ increased the angiogenesis of EPCs and the production of NO567. Second, overexpression of Sonic hedgehog in endothelial cells increased VEGF to regulate angiogenesis560,561. During ischemia and hypoxia, increased expression of Sonic hedgehog and Ptc not only promotes the induction of angiogenesis by bone marrow-derived EPCs but also promotes expression of angiogenic factors, including angiopoietin and VEGF, in cardiac microvascular endothelial cells560,561. Third, the function of endothelial cells could be promoted by the Sonic hedgehog pathway568. It has also been suggested that the hedgehog signaling pathway, as the target gene of platelet-derived growth factor-BB (PDGF-BB), upregulates ERK1/2 and phosphorylates Akt, playing a role in the migration and recruitment of vascular endothelial cells568.
Considering the differential mechanisms of Sonic hedgehog pathways on angiogenesis, researchers have used multiple methods to improve angiogenesis after MI, such as stem cell therapy and pharmacological compounds569–571. Some studies injected Sonic hedgehog modified-CD34+ cells into the edge of acute MI in mice and found that the infarct size was significantly reduced569,570. Additionally, the activation of BMMSCs in Sonic hedgehog pathways induced angiogenesis and endogenous cardiac regeneration through paracrine effects571,572. Another study used erythropoietin to induce Sonic hedgehog signaling to repair the heart after MI573. Consistent with this, the activation of hedgehog signaling in the adult heart leads to an increase in coronary vessel density574. These studies implicate Sonic hedgehog signaling as an essential regulator of coronary vascular development and as a potential therapeutic target for coronary heart diseases. Further studies should explore whether the Sonic hedgehog signaling-induced angiogenesis has therapeutic value in MI.
Activation of the Sonic hedgehog pathway decreases cardiomyocyte apoptosis
It was reported that increased survival and decreased apoptosis of cardiomyocytes enhance the repair of myocardial function564. Activation of the Sonic hedgehog pathway increases the survival rate of cardiomyocytes and reduces apoptosis caused by myocardial ischemia575. Sonic hedgehog also promotes the recovery of left ventricular function by decreasing programmed myocardial cell death564. This study also demonstrated that downregulating the Sonic hedgehog signaling of cardiomyocytes leads to apoptosis and dysfunction of cardiomyocytes564. Based on the role of the Sonic hedgehog pathway in myocardial apoptosis, previous studies used different compounds including miRNAs576, the agonists and antagonists of Sonic hedgehog2,577, and cell therapy, to explore the mechanism of Sonic hedgehog in apoptosis2. One study suggested that silencing miR-802-5p targets PTCH1 and activates the Sonic hedgehog signaling pathway to inhibit apoptosis and reduce myocardial injury after MI576. Adding the Sonic hedgehog signaling pathway receptor agonist to oxygen glucose deprivation (OGD)-induced myocardial cells downregulated the expression of Bcl-2 and Bax, and decreased the number of apoptotic cells. Nevertheless, the administration of the antagonist SANT-1 had the opposite effect2. In addition, in a diabetic myocardial ischemia model, autologous cell therapy using diabetic EPCs suppressed myocardial apoptosis and improved angiogenesis, thus reducing cardiac fibrosis and finally restoring myocardial function through the Shh/Bmi1/p53 axis578.
The Sonic hedgehog pathway in decreasing oxidative stress
Besides the role of the Sonic hedgehog on cardiomyocytes and apoptosis, activation of the Sonic hedgehog pathway also reduces oxidative stress after MI. Many agonists and antagonists of the Sonic hedgehog have been developed to explore their role in oxidative stress. One study reported that purmorphamine, a Sonic hedgehog agonist, prevents the ovariectomized heart from myocardial injury by attenuating the expression of TNF-α and MPO levels and the release of LDH and CK-MB577. However, silencing the effects of Shh using cyclopamine, a specific inhibitor of Shh, or siRNA, an inhibitor of the Shh receptor Patched, strongly reduced the production of NO579. These studies suggest the potential role of Sonic hedgehog in the decrease in oxidative stress. Furthermore, another study used antioxidative strategies and found that it could reactivate the endogenous Sonic hedgehog pathway and contribute to myocardial healing as well as the improvement of diabetic cardiac function580. Based on its antioxidative role, researchers have applied new methods to decrease injury of oxidase stress. Microparticles (MPs) reportedly carry molecules in the Sonic hedgehog pathway to induce expression of NO and decreases the production of reactive oxygen species579. Injection of MPs also improved endothelial function by decreasing oxidase stress injury579.
The Sonic hedgehog pathway increases autophagy after MI
Studies have shown that autophagy plays an important role after MI and that activating autophagy could represent a new therapeutic method for cardiac protection581. Up to now, few studies have reported the role of the Sonic hedgehog pathway in activating autophagy. The main mechanism was correlated with the AMPK pathway19,579. The Sonic hedgehog pathway promotes the phosphorylation of the AMPK pathway and combines with it to induce autophagy579. One study added SAG to H9C2 cardiomyocytes with OGD and found that SAG stimulated autophagy and promoted H9C2 cardiomyocyte survival, and they suggested that the Sonic hedgehog pathway protected cardiomyocytes through an AMPK-dependent autophagy19. In addition, inhibition of autophagy using AMPK inhibitor also weakened the protective effect of Sonic hedgehog on myocardial cell autophagy after infarction19.
The controversial role of the Sonic hedgehog pathway in myocardial I/R injury
Although most studies have reported the protective role of Sonic hedgehog pathways in MI582, there are only a few studies have reported the opposite results in the model of myocardial I/R injury583,584. One study proposed that Sonic hedgehog had no cardio-protective effect on cardiomyocytes after myocardial I/R injury583. They found that the overexpression of Sonic hedgehog in human stem cell-derived cardiomyocytes did not increase vascularization of the infarct scar583. Another study even suggested that the Sonic hedgehog pathway plays a detrimental role in myocardial repair. They found that simvastatin decreased myocardial I/R injury by inhibiting the Sonic hedgehog pathway584. The opposite results of the Sonic hedgehog pathway may be explained by the different models (MI and I/R models) used in previous studies. The permeant MI model and myocardial I/R injury model may induce slightly different scars and lead to slightly different repair mechanisms which may change how the tissue responds to Sonic hedgehog signaling.
The Sonic hedgehog pathway in clinical applications
Since Sonic the hedgehog pathway has critical roles in promoting myocardial repair, it may serve as a potential cardiac therapeutic target565,585. Sonic hedgehog gene therapy may have considerable therapeutic potential in individuals with acute and chronic myocardial ischemia by triggering the expression of multiple trophic factors and engendering tissue repair in the adult heart565.
The first application is microparticles (MPs), which are small fragments generated from the plasma membrane after cell stimulation. A previous study activated the Sonic hedgehog pathway by N-Shh or shed membrane microparticles harboring Sonic hedgehog ligand (MPs (Shh+)) to protect the heart from I/R injury by preventing the occurrence of arrhythmias563. Secondly, it can be applied in gene targeted therapy578,582,586. There are many methods to activate the expression of Sonic hedgehog in cardiomyocytes, including recombination of Sonic hedgehog proteins, using microparticles loaded with Sonic hedgehog, knocking out Patched genes, injection of Sonic hedgehog mRNA, as well as the Sonic hedgehog receptor agonists585. These methods could improve the motility of smooth muscle cells, induce the migration of smooth muscle cells, recruit parietal cells into neovascularization, upregulate VEGF and angiopoietin, increase the number of capillaries and promote cardiac vascular maturation after MI578, reduce myocardial cell apoptosis578, inhibit left ventricular remodeling578, increase the number of myocardial cells582, and improve cardiac function after MI585. Thirdly, the application in comprehensive therapy. The Sonic hedgehog signaling pathway promotes cardiac function by upregulating angiogenic genes and enhancing the mobilization of bone marrow-derived progenitor cells. Combination therapy using PHSHH and AMD3100 effectively stimulates progenitor cell mobilization, improves capillary density, and reduces myocardial fibrosis to enhance cardiac function recovery575. Lastly, some drugs, including tetramethylpyrazine and astragaloside IV were reported to preserve cardiac function after MI by upregulating Sonic hedgehog, Smo, and Gli-2587. Tempol reduced oxidative stress to restore the endogenous Shh pathway and improve diabetic cardiac function580.
Some clinical trials have explored the potential therapeutic effect of CD34+ cells588, BMMSCs589, and erythropoietin590–592 in coronary heart disease. Although some of these clinical trials show application prospects, they are not widely used in clinical practice. Further studies should explore whether these cell therapy and drugs of activating Sonic hedgehog signaling-induced angiogenesis has therapeutic value and could be safely and effectively applied in patients with MI.
Conclusion and perspectives
Ischemic heart disease has become a serious threat to human life and health, therefore novel therapeutic strategies for the treatment of MI are in urgent need. Over the past decades, the developed therapeutic strategies have taken into consideration the impact of the cellular and molecular levels in MI pathological processes as well as the treatment procedures. Herein, most of the current strategies in MI therapy show promising clinical application prospects in the recovery of MI such as pharmacotherapy, gene therapy, protein therapy, cell therapy, as well as exosome therapy. It is evident that the preclinical experiments and clinical experiments targeting molecular signaling following myocardial ischemia have achieved promising effects. In this review, we comprehensively highlighted and summarized the most relevant signaling pathways involved in MI treatment (Table 1).
Table 1.
Intervention of signaling pathways in MI
| Therapeutic strategy | Diseases (Model) | Target pathway | Intervention | Author/Year |
|---|---|---|---|---|
| Drug | MI | PI3K/Akt/mTOR | Everolimus (mTOR inhibitor) | Buss et al. 200965 |
| TAC | eNOS | L-NAME (eNOS inhibitor) | Kazakov et al. 201339 | |
| I/R | PI3K/Akt/GSK-3β | Kaempferide | Wang et al. 201741 | |
| Langendorff system | PTEN/PI3K/Akt | VO-OHpic | Li et al. 201863 | |
| MI | PI3K/Akt/mTOR | TNP (IP7 inhibitor) | Deng et al. 201946 | |
| MI/H2O2 | mTOR | Tanshinone IIA | Zhang et al. 201970 | |
| MI | PTEN/PI3K/Akt | HOpic (PTEN Inhibitor) | Feng et al. 202061 | |
| MI | mTOR | Rapamycin | Chen et al. 202147 | |
| MI | PI3K/Akt/mTOR | Ivabradine | Dai et al. 202167 | |
| MI/SGD | mTOR | Sphingosine-1-phosphate | Yang et al. 202169 | |
| MI | PI3K/Akt/VEGF | Gastrin | Fu et al. 2021605 | |
| MI | Notch | TNF-α inhibitor | Pei et al. 2015125 | |
| MI | Notch | Melatonin | Pei et al. 2016145 | |
| MI | Notch | Yiqihuoxue prescription | Wu et al. 2017143 | |
| MI | Notch | Astragaloside | Yu et al. 2017144 | |
| MI | Notch |
Ligusticum Chuanxiong Radix Paeonia |
Shi et al. 2019140 | |
| MI | Notch | Pigment epithelium-derived factor | Liu et al. 2019159 | |
| MI | Notch | Oestrogen Receptor β | Du et al. 2020158 | |
| I/R | Notch | ALDOA | Luo et al. 2020126 | |
| MI | TLR4 | Metformin | Soraya et al. 2014241 | |
| MI | NF-κB | Methotrexate | Maranhão et al. 2017242 | |
| I/R | NF-κB, AP-1 | Hemin | Yeh et al. 2009266 | |
| MI | HO-1, connexin-43 | Cobalt protoporphyrin | Kusmic et al. 2014267 | |
| MI | EET/HO-1 | Agonists of EETs | Cao et al. 2015268 | |
| MI | NRF2/HO-1 | Wogonin loaded NPs | Bei et al. 2020263 | |
| MI | KEAP1/NRF2/HO-1 | Hirudin | Zhang et al. 2020264 | |
| MI |
NRF2/HO-1 TLR4/TNF-α |
Dapsone | Abdelzaher et al. 2021226 | |
| MI | PI3K/Akt/Nrf2/HO-1 | Rosuvastatin combined with low-dose carvedilol | Baraka et al. 2021265 | |
| MI | RhoA | Rosuvastatin | Bulhak et al. 2007322 | |
| MI | RhoA/ROCK | Coptisine | Gong et al. 2012319 | |
| MI | RhoA/ROCK | Estradiol | Lee et al. 2014306 | |
| MI | RhoA/ROCK | Ibuprofen | Patel et al. 2016320 | |
| MI | RhoA/ROCK | Nicorandil | Lee et al. 2018307 | |
| MI | RhoA/ROCK | Fasudil | Zhou et al. 2020321 | |
| MI | RhoA/ROCK | Fluvastatin | Yi et al. 2020323 | |
| MI | RhoA/ROCK | Dexmedetomidine | Sun et al. 2021318 | |
| MI | MAPK | Osthole | Yeung et al. 2018361 | |
| MI | MAPK | Atorvastatin | Zeng et al. 2019336 | |
| MI | MAPK | Danhong injection | Li et al. 2019368 | |
| MI | MAPK | KXA | Lu et a. 2021335 | |
| MI | Wnt/β-catenin | Pyrvinium | Saraswati et al. 2010499 | |
| MI | Wnt/β-catenin | Wnt antagonist Dickkopf2 | Min et al. 2011490 | |
| MI | Wnt/β-catenin | Aldehyde dehydrogenase-2 | Zhao et al. 2015494 | |
| MI | Wnt/Frizzled | UM206 | Uitterdijk et al. 2016501 | |
| MI | Wnt/β-catenin | NP12 | Baruah et al. 2017491 | |
| MI | Wnt/β-catenin | ICG-001 | Qian et al. 2018500 | |
| MI | TGF-β2, TGF-β3 | Fasudil | Hattori et al. 2004312 | |
| MI | TGFβ1/TAK1 | Fasudil | Li et al. 2012311 | |
| MI | TGF-β/SMADs | Valsartan | Sui et al. 2015304 | |
| MI | TGF-β/SMADs | Simvastatin | Xiao et al. 2016434 | |
| MI | TGF-β/SMADs | Salvianolic acid B | Gao et al. 2019437 | |
| MI | TGF-β/SMADs | Ginsenoside Re | Yu et al. 2020439 | |
| MI | TGF-β/SMADs | LCZ696 + benazepril | Liu et al. 2021435 | |
| MI | TGF-β/SMADs | Vitamin D + ART | Mehdipoor et al. 2021436 | |
| MI | TGF-β/SMADs | Tanshinone IIA | Chen et al. 2021438 | |
| MI | JAK/STAT | IL-33 | Li et al. 2019395 | |
| MI | JAK/STAT | Hyaluronic acid Oligosaccharides | Lee et al.2019606 | |
| MI | Hippo/YAP | Luteolin | Hu et al. 2016543 | |
| MI | Hippo/YAP | Melatonin | Hu et al. 2017544 | |
| STEMI | Hippo/YAP | Melatonin Adjunct | Dominguez-Rodriguez et al. 2017162 | |
| MI | Hippo/YAP | oncostatin M | Yang et al. 2018522 | |
| MI | YAP | TT-10-delivered NPs | Chen et al. 2021545 | |
| MI | Sonic Hedgehog | Erythropoietin | Ueda et al. 2010573 | |
| MI | Sonic Hedgehog | Tempol | Xiao et al. 2015580 | |
| MI | Sonic Hedgehog |
Tetramethylpyrazine Astragaloside IV |
Wang et al. 2017587 | |
| MI | Sonic Hedgehog | Purmorphamine | Sharma et al. 2018577 | |
| MI | Sonic Hedgehog | Simvastatin | Feng et al. 2020584 | |
| Gene therapy | MI | PI3K/Akt/FOXO3a |
Ab-NGF Ad-human NGF |
Meloni et al. 201058 |
| MI | PTEN/PI3K/Akt | Ad-PTEN | Parajuli et al. 201264 | |
| MI | mTOR | lenti-miR-99a | Li et al. 201481 | |
| MI | PI3K/Akt/FOXO | Period 2 KO EPC | Sun et al. 201491 | |
| MI | PTEN/PI3K/Akt | miR-130a | Lu et al. 201577 | |
| MI | Akt |
Ad-GHSR-1a GHSR-1a siRNA |
Yuan et al. 201632 | |
| MI | PTEN/PI3K/Akt |
miR-21 miR-146a |
Huang et al. 201675 | |
| MI | PTEN/PI3K/Akt | EnMSCs-Exo-miR-21 | Wang et al. 201776 | |
| MI | Akt/PDGF‐D |
Akt‐hucMSCs-Exo PDGF‐D siRNA |
Ma et al. 201790 | |
| MI | PI3K/p-Akt | AAV9-activated PDGFR-β | Yue et al. 201931 | |
| MI | PTEN/PI3K/Akt |
sh-GAS5 miR-142-5p inhibitor |
Du et al. 201982 | |
| MI | PTEN/PI3K/Akt | sh-AZIN2-sv | Li et al. 201985 | |
| OGD | PI3K/Akt/mTOR | pEX-UCA1 | Zhang et al. 201986 | |
| Chronic MI | Akt/FoxO3a | AAV9-SERCA2a | Kumarswamy et al. 201274 | |
| MI | PTEN/PI3K/Akt | PTEN cKO mice | Liang et al. 202062 | |
| MI | PTEN/PI3K/Akt |
Senescent MSCs-Exo miR-221-3p |
Sun et al. 202078 | |
| MI | PTEN/PI3K/Akt | miR-301a | Zhen et al. 202079 | |
| MI | PTEN/PI3K/Akt |
sh-GAS5 OE-GAS5 miR-21 mimics |
Zhou et al. 202083 | |
| Hypoxia | PI3K/Akt/mTOR | pcDNA3.1-DANCR | Qiu et al. 202087 | |
| MI | PTEN/PI3K/Akt |
lncRNA Snhg1 Snhg1 cKO mice |
Li et al. 202180 | |
| MI | PI3K/Akt |
sh-CircHIPK3 miR-93-5p agomiR |
Wu et al. 202184 | |
| MI | Notch | miR-199b | Chen et al.148 | |
| MI | Notch | miR-429 | Xu et al. 2016154 | |
| MI | Notch | miR-363 | Meng et al. 2017135 | |
| MI | Notch | miR-208a | Zhang et al. 2018123 | |
| MI | Notch | miR-1 | Chen et al. 2018153 | |
| MI | Notch | CYP2J2 | Zhao et al. 2018161 | |
| MI | Notch | miR-29b | Liu et al. 2019120 | |
| MI | Notch | miR-374 | Zhao et al. 2019152 | |
| MI | Notch | KCNQ1OT1 | Wang et al. 2019155 | |
| MI | Notch | lncRNA XIST | Zhang et al. 2019156 | |
| MI | Notch | KRT1 | Fang et al. 2019160 | |
| MI | Notch | miR-384-5p | Fan et al. 2020112 | |
| MI | Notch | CircRNA Hipk3 | Si et al. 2020147 | |
| MI | Notch | miR-29b-3p | Yang et al. 2020149 | |
| MI | Notch | miR-124a | Xu et al. 2021146 | |
| MI | Notch | miR-133 | Zhang et al. 2021150 | |
| MI | Notch | miR-106a-363 | Jung et al. 2021151 | |
| MI | P2X7/NLRP3 | Brilliant blue G | Vessey et al. 2010202 | |
| MI | NLRP3 | 16673-34-0 | Marchetti et al. 2015193 | |
| MI | NLRP3 | MCC950 | van Hout et al. 2017194 | |
| MI | NLRP3 | JC124 | Fulp et al. 2018197 | |
| MI | NLRP3 | OLT1177 | Toldo et al. 2019198 | |
| MI | NLRP3 | Oridonin | Gao et al. 2021196 | |
| MI | TLR4 | TAK-242 | Fujiwara et al. 2019240 | |
| MI | TLR4 | ApTOLL | Ramirez-Carracedo et al. 2020238 | |
| I/R | HO-1 | AAV-HO-1 | Melo et al. 2002260 | |
| I/R | HO-1 | AAV2-HO-1 | Liu et al. 2007259 | |
| MI | TLR4 | Radioprotective 105 (RP105) | Louwe et al. 2014239 | |
| MI | TLR4 | lenti-shRNA | Liu et al. 2015243 | |
| MI | β-MHC, ANF, BNP | NRF2 KO | Strom et al. 201717 | |
| MI | RhoA/ROCK/HIF-1α | Notch3 siRNA | Shi et al. 2020121 | |
| MI | MAPK | α1-AR KO | Yeh et al. 2017359 | |
| MI | MAPK | miR-539 | Hui et al. 2017349 | |
| MI | MAPK | Mst1KO | Wang et al. 2018337 | |
| MI | MAPK | RGS5 KO | Ding et al. 2018344 | |
| MI | MAPK | ghrelin | Eid et al. 2019339 | |
| MI | MAPK | EPO | Li et al. 2019341 | |
| MI | MAPK | 17-AAG (Hsp90 inhibitor) | Tamura et al. 2019358 | |
| MI | MAPK | Morin | Verma et al. 2019363 | |
| MI | MAPK | Epiregulin siRNA | Cai et al. 2019371 | |
| MI | MAPK | IκB Kinase α KO | Cao et al. 2019372 | |
| MI | MAPK | miR-26b | Ge et al. 2019365 | |
| MI | MAPK | miR-143-3p | Li et al. 2019356 | |
| MI | MAPK | MALAT1 | Fan et al. 2019351 | |
| MI | MAPK | SPC | Li et al. 2019346 | |
| MI | MAPK | lenti-ANO1-RNAi | Tian et al. 2020355 | |
| MI | MAPK | CXCR7 shRNA | Zhang et al. 2020364 | |
| MI | MAPK | pMSCV-EGFP-Wnt11 | Wang et al. 2020370 | |
| MI | MAPK | miR-125b | Qiao et al. 2020350 | |
| MI | Wnt/β-catenin | Transgenic Mice OE FrzA | Barandon et al. 2003489 | |
| MI | Wnt/β-catenin | miR-34a antagomir | Li et al. 2019505 | |
| I/R | Wnt/β-catenin | miR-423-5p inhibitor | Zhu et al. 2019506 | |
| MI | Wnt/β-catenin | miR-30b-5p inhibitor | Chi et al. 2020503 | |
| MI | Akt/GSK‐3β/β‐catenin | Ad‐miR‐145 | Cui et al. 2021498 | |
| I/R | Wnt/β-catenin | miR-26a-5p | Yan et al. 2021467 | |
| MI | TGF-β/SMADs | Cytl1 KO | Kim et al. 2016444 | |
| MI | TGF-β/SMADs |
Notch3 siRNA Notch3 cDNA |
Zhang et al. 2016448 | |
| MI | TGF-β/SMADs | Ad-ANO1-GFP | Gao et al. 2017354 | |
| MI | TGF-β/SMADs | Exogenous BMP-7 | Jin et al. 2018447 | |
| MI | TGF-β/SMADs |
Nogo-C KO Nogo-C-shRNA |
Weng et al. 2018446 | |
| MI | TGF-β/SMADs | miR-130 | Chu et al. 2018453 | |
| MI | TGF-β/SMADs | TMAO | Yang et al. 2019442 | |
| MI | TGF-β/SMADs | β-arrestins siRNA | Philip et al. 2019443 | |
| MI | TGF-β/SMADs |
Ad‐CTRP9 shCTRP9 |
Liu et al. 2019445 | |
| MI | TGF-β/SMADs |
Ad‐N1ICD/Smad3 Ad‐shN1ICD/Smad3 |
Zhou et al. 2019107 | |
| MI | TGF-β/SMADs | miR-133a | Yu et al. 2019456 | |
| MI | TGF-β/SMADs | miR-224 | Xu et al. 2019457 | |
| MI | TGF-β/SMADs | MALAT1 | Huang et al. 2019455 | |
| MI | TGF-β/SMADs | miR-195 | Wang et al. 2020452 | |
| MI | TGF-β/SMADs | miR-808 | Zhang et al. 2020465 | |
| MI | TGF-β/SMADs | Lnc-Ang362 | Chen et al. 2020458 | |
| MI | TGF-β/SMADs | LncRNA SOX2-OT | Yang et al. 2020468 | |
| MI | TGF-β/SMADs | CircRNA 010567 | Bai et al. 2020454 | |
| MI | TGF-β/SMADs | KLF5-specific inhibitor ML264 | Tian et al. 2021466 | |
| MI | TGF-β/SMADs | miR-671 | Wang et al. 2021467 | |
| MI | TGF-β/SMADs | CircHNRNPH1 | Li et al. 2021459 | |
| I/R | JAK/STAT | EGR-1 | Mudaliar et al. 2017406 | |
| MI | JAK/STAT/c-Fos |
miR-181 miR-150 |
Zhu et al. 2017388 | |
| I/R | JAK/STAT | Unacylated ghrelin (UAG) | Sawashita et al. 2020405 | |
| MI | Hippo/YAP | Tg-DN-Mst1 | Odashima et al. 2007524 | |
| MI | Hippo/YAP |
lenti-miR302-367 OE pLKO.1-Yap shRNA |
Tian et al. 2015553 | |
| MI | Hippo/YAP |
AAV9-SAV-shRNA SAV cKO mice |
Leach et al. 2017525 | |
| hiPSCs | Hippo/YAP | miR-302d OE | Xu et al. 2019607 | |
| MI | Hippo/YAP |
LPA3-KO mice AAV9-LPA3-OE |
Wang et al. 2020534 | |
| MI | Hippo/YAP | lenti-miR-93 OE | Ma et al. 2020552 | |
| MI | Hippo/YAP | AAV9-SAV-shRNA | Liu et al. 2021526 | |
| MI | Hippo/TAZ | Ad-SKI | Landry et al. 2021541 | |
| MI | YAP/TAZ | YAP cKO mice | Ramjee et al. 2017538 | |
| MI | YAP | AAV9-human YAP | Lin et al. 2014530 | |
| MI | YAP | AAV-gp130 | Li et al. 2020532 | |
| MI (HF) | YAP |
caERBB2-OE mice pAAV-CMV-YAP |
Aharonov et al. 2020535 | |
| MI | YAP | YAP cKO mice | Francisco et al. 2020540 | |
| MI | YAP |
HSPA12B cKO mice YAP cKO mice |
Fan et al. 2020542 | |
| MI | YAP | AAV9-ALKBH5-KO | Han et al. 2021531 | |
| MI | YAP | hCPC-Exo carried periostin | Balbi et al. 2021548 | |
| MI | Sonic Hedgehog | Shh-AMD3100 | Roncalli et al. 2011575 | |
| MI | Sonic Hedgehog | MSCs | Tang et al. 2013571 | |
| MI | Sonic Hedgehog | PEG hydrogel | Johnson et al. 2015586 | |
| MI | Sonic Hedgehog | MPsSHH+ | Paulis et al. 2015582 | |
| MI | Sonic Hedgehog | DM EPCShh | Xiao et al. 2019578 | |
| MI | Sonic Hedgehog | MPsSHH+ | Bueno-Betí et al. 2019567 | |
| MI | Sonic Hedgehog | MPsSHH+ | Ghaleh et al. 2020563 | |
| MI | Sonic Hedgehog | miR-802-5p | Li et al. 2021576 | |
| Cell therapy | Hypoxia | Akt | Akt-MSCs | Gnecchi et al. 200592 |
| MI | Akt | Akt-MSCs | Gnecchi et al. 200695 | |
| MI | Notch | Embryonic stem cell | Tsang et al. 2017134 | |
| MI | Notch |
MSCs CSCs |
Shevchenko et al. 2019141 | |
| MI | Wnt/β-catenin |
hiPSCs CPCs ISX-9 |
Xuan et al. 2018507 | |
| I/R | Akt/GSK3β/β-Catenin |
Cortical bone-derived stem Cell Trop2 |
Li et al. 2018509 | |
| MI | TGF-β/SMADs | MSCs+pioglitazone | Chen et al. 2014462 | |
| MI | TGF-β/SMADs | Hypoxic preconditioned MSCs | Hou et al. 2015463 | |
| MI | TGF-β/SMADs | BMMSCs | Wei et al. 2021461 | |
| MI | JAK/STAT | G-CSF- and erythropoietin-based cell therapy | Kang et al. 2008396 | |
| MI | Sonic Hedgehog | MSCs (Shh) | Ahmed et al. 2010572 | |
| MI | Sonic Hedgehog | CD34 (Shh) | Mackie et al. 2012569 | |
| MI | Sonic Hedgehog | hiPSCs | Munarin et al. 2020608 | |
| Exosome therapy | MI | PTEN/PI3K/Akt | Explant-derived cardiac stromal cells-Exo | Qiao et al. 2019102 |
| MI | Sonic Hedgehog | PAMsHGF | Riaud et al. 2021609 | |
| Protein therapy | MI | PI3K/Akt | Recombinant FLT3 Ligation | Pfister et al. 201473 |
| MI | YAP/TAZ | rGO/Silk Fibroin-Modified Nanofibrous Patches | Feng et al. 2021546 | |
| Combination therapy | ||||
|
Drug Cell therapy |
H/SD | PI3K/Akt |
EGb761 BMMSCs |
Li et al. 201197 |
|
Drug Gene therapy |
HFD MI |
mTORC1 |
Rapamycin Ad-Rheb |
Sciarretta et al. 201251 |
|
Drug Gene therapy |
MI | PI3K/Akt/mTOR |
PI3Kγ KO mice AAV9-PRAS40 Insulin |
Völkers et al. 201345 |
|
Drug Gene therapy |
MI | eNOS |
L-NAME (eNOS inhibitor) human HSPA12B OE mice |
Li et al. 201343 |
|
Drug Cell therapy |
MI | PI3K/Akt/ FOXO3a | Rosuvastatin; AD-MSCs | Zhang et al. 2013100 |
|
Drug Cell therapy |
MI | Akt |
TNP (IP6Ks inhibitor) MSCs |
Zhang et al. 201499 |
|
Drug Cell therapy |
MI | Akt |
Edaravone BMMSCs CSCs |
Zhang et al. 201696 |
|
Cell therapy Gene therapy Exosome therapy |
MI | PI3K/Akt/mTOR |
MSCs SDF1 OE Exo-SDF1 |
Gong et al. 2019103 |
|
Protein therapy Cell therapy |
MI | Akt |
Thymosin β4 hiPSC-CMs |
Tan et al. 202193 |
|
Protein therapy Cell therapy |
MI | PI3K/Akt |
NGF hucMSCs |
Luo et al. 202194 |
|
Gene therapy Cell therapy |
MI | Akt |
lenti-TMSB4 BMMSCs |
Tang et al. 202198 |
|
Gene therapy Cell therapy |
MI | HO-1 | MSCs OE HO-1 | Zeng et al. 2008272 |
|
Gene therapy Cell therapy |
MI | HO-1 | MSCs OE HO-1 | Zeng et al. 2010270 |
|
Gene therapy Cell therapy |
MI | HO-1 | MSCs OE HO-1 | Jiang et al. 2011271 |
|
Drug Cell therapy |
MI | RhoA/ROCK/ERK | Atorvastatin | Zhang et al. 2014326 |
|
Drug Cell therapy |
MI | TGF-β/Wnt |
Epicardial erythropoietin Stem cells |
Klopsch et al. 2018508 |
|
Gene Cell therapy |
MI | Wnt/β-catenin |
miR-497 antagomir BMMSCs |
Tang et al. 2019510 |
AAV adeno-associated virus, Ab-NGF nerve growth factor-neutralizing antibody, Ad adenoviral virus, AD-MSCs adipose-derived mesenchymal stem cells, AZIN2-sv lncRNA-AZIN2 splice variant, CD34(Shh) Sonic hedgehog-modified human CD34+ cells, cKO conditional knockout, CSCs cardiac stem cells, DM EPCShh adenovirus Shh-modified diabetic EPCs, EETs epoxyeicosatrienoic acids, EnMSCs human endometrium-derived mesenchymal stem cells, EPC endothelial progenitor cell, Exo exosome, FLT3 FMS-like tyrosine kinase 3, G-CSF granulocyte colony stimulating factor, GAS5 growth arrest-specific transcript 5, GHSR-1a growth hormone secretagogue receptor1a, gp130 glycoprotein 130, H/SD hypoxia/serum deprivation, H2O2 hydrogen peroxide, hCPCC human cardiac explant-derived progenitor cells, HF heart failure, HFD high fat diet-induced obesity and metabolic syndrome, hiPSCs human-induced pluripotent stem cells, hiPSC-CMs human-induced pluripotent stem cell-derived cardiomyocytes, HSPA12B heat shock protein A12B, hucMSCs human umbilical cord mesenchymal stem cells, I/R ischemia and reperfusion, IP6Ks inositol hexakisphosphate kinases, IP7 inositol pyrophosphates, lenti lentivirus, lncRNA long non-coding RNA, L-NAME L-N(G)-nitroarginine methyl ester, MI myocardial infarction, miRNA microRNA, MPsSHH+ shed membrane microparticles harboring SHH ligand, MSCs mesenchymal stem cells, NPs nanoparticles, OE overexpression, OGD oxygen-glucose deprivation, PAMsHGF pharmacology active microcarriers encapsulated hepatocyte growth factor, PDGF-D platelet‐derived growth factor D, PEG biodegradable polyethylene glycol, Rheb Ras homology enriched in brain, SDF1 stromal-derived factor 1, SGD serum-and glucose-deficient, shRNA short hairpin RNA, Shh Sonic hedgehog, siRNA small interfering RNA, Snhg1 small nucleolar RNA host gene 1, STEM ST-segment elevation myocardial infarction, TAC transverse aortic constriction, Tg-DN-Mst1 overexpression of dominant negative Mst1, TMSB4 thymosin β4 gene, TNP N6-(p-nitrobenzyl) purine, UCA1 urothelial carcinoma associated 1
It is well-established that the damage of cardiac tissue caused by ischemia-hypoxia is a composite result of the cellular change in response to stimuli, in addition, these cells also participate in cardiac repair and regeneration following MI10,360,392. It follows from the above that cardiac protection and functional restoration can be achieved through a multi-targeted approach, which modulates the flow of cellular signals in different indigenous or migrated cells. Herein, in order to describe the pivotal role of signaling pathways in the biological process of MI vividly, we diagramed the fundamental signaling pathways in cardiomyocytes, endothelial cells, fibroblasts, monocytes, as well as (myeloid or transplanted) stem cells, in the pathological changes and the treatment of MI (Figs. 8, 9). Principal signaling pathways mentioned here include the PI3K/Akt, Notch, TGF-β/SMADs, Wnt/β-catenin, NLRP3/caspase-1, TLR4/MyD88/NF-κB, Nrf2/HO-1, RhoA/ROCK, MAPK, JAK/STAT, Hippo/YAP, and Sonic hedgehog pathways, which mainly centered on various pathological states such as inflammation, oxidative stress, fibrosis, hypertrophy, apoptosis, survival, angiogenesis, and regeneration post MI (Fig. 9). Remarkably, these pathways form a complex and homeostatic regulating network, rather than act in isolation. In this context, it should be emphasized that the novel therapies which mediate crosstalk pathways may exert more beneficial effects in cardiac repair and secondary prevention of MI.
Fig. 8.

Novel targeted therapeutic strategies and mechanisms in MI treatment (Created with BioRender.com). In terms of the signaling pathways, potential therapeutic strategies for MI that have been proposed to include drug, gene therapy, protein therapy, cell therapy, and exosome therapy. According to the pathological process of MI, the specific targeted mechanism of these therapies could be classified into four categories: (1) anti-inflammation, (2) anti-fibrosis, (3) cardioprotection and cardiac regeneration, and (4) pro-angiogenesis. Dot-labeled subtitles refer to the representative targeting drugs or bioactive molecules. ACEI angiotensin converting enzyme inhibitor, ARB angiotensin receptor antagonist, EPCs endothelial progenitor cells, lncRNA long non-coding RNA, miRNA microRNA, mTOR mammalian target of rapamycin, NAC N-acetylcysteine, NLRP3 nucleotide-binding domain, leucine-rich-repeat family, pyrin-domain-containing 3, PTEN phosphatase and tensin homolog, TGF-β1 transforming growth factor-β1, IL-1β interleukin-1β, TLR4 Toll-like receptor 4, NF-κB nuclear factor-κB, MSCs mesenchymal stem cells
Fig. 9.
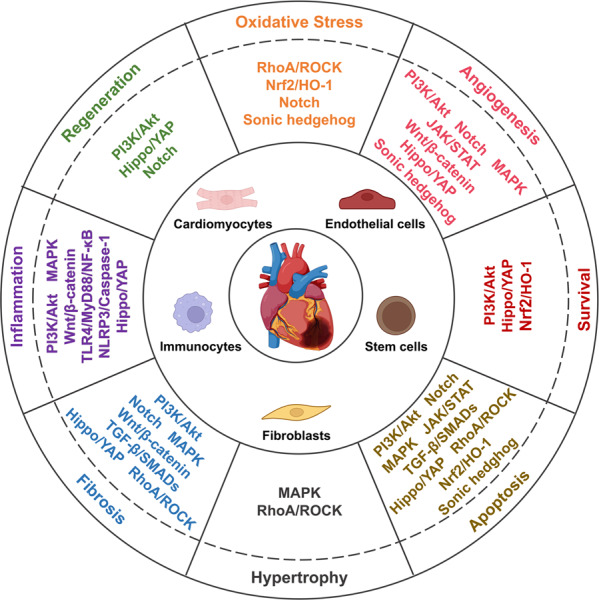
Cell signaling pathways participant in regulating pathological processes and phenotypes after MI (Created with BioRender.com). PI3K/Akt phosphoinositide-3 kinase/protein kinase B, TGF-β/SMADs transforming growth factor-β/SMADs, Wnt/β-catenin wingless/β-catenin, NLRP3/caspase-1 nucleotide-binding domain, leucine-rich-repeat family, pyrin-domain-containing 3/caspase-1, TLR4/MyD88/NF-κB toll-like receptor 4/MyD88/Nuclear factor-κB, Nrf2/HO-1 nuclear factor erythroid derived 2-related factor 2/heme oxygenase-1, MAPK mitogen-activated protein kinase, JAK/STAT Janus kinase/signal transducer and activator of transcription, Hippo/YAP Hippo/Yes-associated protein
In the preclinical studies for MI treatment, the potential effect of drug, gene therapy, and cell therapy on MI point out the promising direction of clinical research. The drugs, such as Ivabradine, colchicinef, canakinumab, rapamycin, and melatonin have been investigated in clinical trials (Table 2). Incorporating the findings of preclinical studies, some of the drugs could target the important molecules of signaling pathways in cardiac repair and recovery of cardiac function. It is remarkable that most of the drugs listed in this review are working through a multi-targeted approach, which directs to multiple molecular targets in different intracellular signaling pathways. For instance, melatonin possesses antioxidant and anti-inflammatory activities post MI593; rosuvastatin resists the inflammatory response and excessive fibrosis594,595. Besides the usage of drugs, there is also a great possibility to combine drug therapy and classical therapeutic strategies properly. For example, as an adjunct to primary PCI for acute STEMI, the administration of melatonin showed a significant reduction in the infarct size162. However, further studies are still needed to explore the intended population, side effects, and optimal dose of drugs596.
Table 2.
Some clinical trials of novel therapeutic strategies for MI
| Strategy (drug) | Molecular markers/Signal pathways | Register number | Phase | Estimated/actual enrollment | Status |
|---|---|---|---|---|---|
| Ivabradine | PI3K/Akt/mTOR | NCT02446990 | III | 19102 | Completed |
| Rapamycin/Sirolimus | mTOR | NCT00552669 | IV | 200 | Completed |
| NCT04951050 | NA | 200 | Not yet recruiting | ||
| Losmapimod | MAPK | NCT02145468 | III | 3503 | Completed |
| Canakinumab | NLRP3/IL-1β | NCT01327846 | III | 10066 | Completed |
| NCT01900600 | NA | 15 | Completed | ||
| Colchicine | NLRP3/IL-1β | NCT02551094 | III | 4745 | Completed |
| ACTRN12614000093684 | III | 5522 | Completed | ||
| NCT01709981, NCT02594111 | IV | 280 | Completed | ||
| NCT01906749 | IV | 500 | Unknown | ||
| NCT00754819 | II & III | 80 | Completed | ||
| NCT05130892 | IV | 132 | Recruiting | ||
| Erythropoietin | MAPK, TGF-β, Wnt, Sonic Hedgehog | NCT00390832 | III | 138 | Completed |
| UMIN000005721 | NA | 600 | Completed | ||
| NCT00423020 | IV | 72 | Completed | ||
| NCT00149058 | II | 124 | Unknown | ||
| NCT00524901 | II | 10 | Completed | ||
| Estradiol | RhoA/ROCK | NCT00402636 | IVIV | 502 | Completed |
| Estrogen | RhoA/ROCK | NCT00123539 | NA | 334 | Terminated |
| Nicorandil | RhoA/ROCK | NCT01396395 | IV | 402 | Completed |
| Dexmedetomidine | RhoA/ROCK | NCT03095469 | I | 200 | Unknown |
| Valsartan | TGF-β/SMADs | NCT03309618 | NA | 36 | Completed |
| Sildenafil | JAK2/STAT3, RhoA/ROCK | NCT01046838 | IV | 70 | Completed |
| G-CSF | JAK2/STAT3, NF-κB | NCT00596479 | NA | 50 | Completed |
| NCT00886509 | NA | 50 | Completed | ||
| NCT00307879 | II | 20 | Terminated | ||
| NCT00043628 | II | 35 | Completed | ||
| Methotrexate | NF-κB | NCT01741558 | II | 80 | Completed |
| Metformin | TLR4 | NCT01217307 | II & III | 380 | Completed |
| Melatonin | Notch, Hippo/YAP | NCT00640094 | II | 272 | Terminated |
| NCT03966235 | IV | 74 | Unknown | ||
| NCT00640094 | II | 272 | Terminated | ||
| NCT01172171 | II | 41 | Completed | ||
| Fasudil | TGFβ1/TAK1, TGF-β2, TGF-β3, RhoA/ROCK | NCT03753269 | IV | 600 | Not yet recruiting |
| Statin | PI3K/Akt/ FOXO3a, TGF-β/SMADs, Sonic Hedgehog, RhoA/ROCK/ERK, PI3K/Akt/Nrf2/HO-1 | NCT00128024 | IV | 460 | Completed |
| Hirudin | KEAP1/Nrf2/HO-1 | NCT03664180 | IV | 2856 | Recruiting |
| Strategy (gene therapy) | |||||
| VEGF-A165 plasmid | VEGF/PI3K/Akt | NCT00135850 | I& II | 48 | Completed |
| AdGVVEGF121cDNA | VEGF/PI3K/Akt | NCT01174095 | I | 31 | Completed |
| Endocardial adenovirus VEGF-D gene transfer | VEGF/PI3K/Akt | NCT01002430 | I | 30 | Completed |
| Bicistronic VEGF-A165/bFGF plasmid | VEGF(bFGF)/PI3K/Akt | NCT00620217 | II | 52 | Completed |
| Strategy (Cell therapy) | |||||
| CD34+ cell | PI3K/Akt, Sonic Hedgehog | NCT00300053 | II | 321 | Completed |
| NCT03471611 | I | 20 | Active, not recruiting | ||
| NCT01508910 | III | 291 | Completed | ||
| CD133+ cell | PI3K/Akt, Wnt | NCT00694642 | I& II | 28 | Completed |
| NCT02870933 | IV | 30 | Completed | ||
| EPCs | VEGF/PI3K/Akt/eNOS, Dll4/Notch/Hey2, Sonic Hedgehog, Akt/HO-1 | NCT00494247 | IV | 60 | Completed |
| MSCs | PI3K/Akt/mTOR, Wnt/β-catenin, TGF-β/SMADs, Notch, Sonic Hedgehog | NCT00418418 | II | 60 | Unknown |
PI3K/Akt phosphoinositide-3 kinase/protein kinase B, MAPK mitogen-activated protein kinase, NLRP3/IL-1β, mTOR mammalian target of rapamycin, NLRP3/IL-1β nucleotide-binding domain, leucine-rich-repeat family, pyrin-domain-containing 3/ interleukin-1β, TGF-β/SMADs transforming growth factor-β/SMADs, Wnt Wingless, RhoA/ROCK/ERK Ras homolog family member A/Rho-associated coiled-coil containing protein kinase/extracellular regulated protein kinases, JAK/STAT Janus kinase/signal transducer and activator of transcription, NF-κB nuclear factor-κB, TLR4 toll-like receptor 4, Hippo/YAP, Hippo/Yes-associated protein, FOXO3a forkhead box subfamily O3a, KEAP1 kelch like ECH-associated protein 1, Nrf2/HO-1 nuclear factor erythroid derived 2-related factor 2/heme oxygenase-1, G-CSF granulocyte colony stimulating factor, VEGF vascular endothelial growth factor, bFGF basic fibroblast growth factor, eNOS endothelial nitric oxide synthase, EPCs endothelial progenitor cells, MSCs mesenchymal stem cells
Up to now, with the clinical application of gene and cell therapies in MI, some of the current results are encouraging: For example, as the pro-angiogenic growth factor, VEGF binds to VEGF receptors and activates the downstream signaling pathway to promote angiogenesis40. Since D. W. Losordo et al. directed myocardial gene transfer of VEGF to treat MI and improved myocardial perfusion in patients in 1998597, VEGF gene therapy has been considered an effective therapy for myocardial ischemia, and the long-term safety of gene strategy has been confirmed over a 10 years follow-up in cardiovascular disease598; additionally, stem cell therapy for MI is being carried out in many studies, and these adequately powered results promote the development of clinical translation in this field. Further studies also indicated that stem cell therapy might be a potential cardioprotective technique to complement PCI or thrombolytic therapy after AMI599,600. Although results of clinical studies on stem cell therapy for myocardial infarction have a certain degree of inconsistency601,602, the low immunogenicity, differentiation potential, paracrine action of stem cells could facilitate further studies to demonstrate their clinical efficacy in MI603,604. Remarkably, in this review, we list a lot of registered clinical trials (Table 2) which aim to assess the therapeutic potential of gene and stem cell therapy in clinical application, integrated with some basic research findings regarding the influences of therapeutic strategies on cell signaling molecule expression. It is undeniable that, with the gradual development of clinical research, the treatment of coronary heart disease targeting these signaling pathways may be advanced from molecular mechanisms to therapeutic potentials, from bench to bed eventually.
In conclusion, the importance of therapeutic strategies targeting cell signaling molecule expression is emerging which we can not ignore, because it provides us with new evolutional solutions for MI treatment that show potential efficacy in preclinical studies and clinical trials. Moreover, characterization of signaling pathway transduction and regulation in MI development is critical for the determination of targeted therapeutic protocols. Since we have fully combed the roles of signaling pathways in the pathological development and treatment of MI, and the future research direction of myocardial infarction treatment, this information will contribute to the exploration and application of novel therapeutic strategies for MI.
Acknowledgements
The study was funded by National Natural Science Foundation of China (Grant No. 82172534) and National Key R&D Program of China (Grant No. 2020YFC2008502) and 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University.
Author contributions
Q.W. and C.Y.F. designed and wrote the manuscript. Q.Z., L.W., S.Q.W., H.X.C., L.X., G.Q.P., and Y.W. did literature search and wrote the manuscript and drafted figures. Y.F.J. and C.Q.H. revised manuscript. All authors listed have made a substantial contribution to the work. All authors have read and approved the article.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Qing Zhang, Lu Wang
Contributor Information
Chenying Fu, Email: fcying_2004@163.com.
Quan Wei, Email: weiquan@scu.edu.cn.
References
- 1.Roth GA, Mensah GA, Fuster V. The global burden of cardiovascular diseases and risks: a compass for global action. J. Am. Coll. Cardiol. 2020;76:2980–2981. doi: 10.1016/j.jacc.2020.11.021. [DOI] [PubMed] [Google Scholar]
- 2.Roth GA, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J. Am. Coll. Cardiol. 2020;76:2982–3021. doi: 10.1016/j.jacc.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Golforoush P, Yellon DM, Davidson SM. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic Res. Cardiol. 2020;115:73. doi: 10.1007/s00395-020-00829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackman N, Bergmeier W, Stouffer GA, Weitz JI. Therapeutic strategies for thrombosis: new targets and approaches. Nat. Rev. Drug Discov. 2020;19:333–352. doi: 10.1038/s41573-020-0061-0. [DOI] [PubMed] [Google Scholar]
- 5.Doenst T, et al. PCI and CABG for treating stable coronary artery disease: JACC review topic of the week. J. Am. Coll. Cardiol. 2019;73:964–976. doi: 10.1016/j.jacc.2018.11.053. [DOI] [PubMed] [Google Scholar]
- 6.Sabatine MS, Braunwald E. Thrombolysis In Myocardial Infarction (TIMI) Study Group: JACCFocus Seminar 2/8. J. Am. Coll. Cardiol. 2021;77:2822–2845. doi: 10.1016/j.jacc.2021.01.060. [DOI] [PubMed] [Google Scholar]
- 7.Sabatine MS, et al. Percutaneous coronary intervention with drug-eluting stents versus coronary artery bypass grafting in left main coronary artery disease: an individual patient data meta-analysis. Lancet. 2021;398:2247–2257. doi: 10.1016/S0140-6736(21)02334-5. [DOI] [PubMed] [Google Scholar]
- 8.McCarthy CP, et al. Left ventricular thrombus after acute myocardial infarction: screening, prevention, and treatment. JAMA Cardiol. 2018;3:642–649. doi: 10.1001/jamacardio.2018.1086. [DOI] [PubMed] [Google Scholar]
- 9.Fraccarollo D, Galuppo P, Bauersachs J. Novel therapeutic approaches to post-infarction remodelling. Cardiovasc. Res. 2012;94:293–303. doi: 10.1093/cvr/cvs109. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Reboll MR, Korf-Klingebiel M, Wollert KC. Angiogenesis after acute myocardial infarction. Cardiovasc. Res. 2021;117:1257–1273. doi: 10.1093/cvr/cvaa287. [DOI] [PubMed] [Google Scholar]
- 11.Viola M, de Jager SCA, Sluijter JPG. Targeting inflammation after myocardial infarction: a therapeutic opportunity for extracellular vesicles? Int. J. Mol. Sci. 2021;22:7831. doi: 10.3390/ijms22157831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung M, Dodsworth M, Thum T. Inflammatory cells and their non-coding RNAs as targets for treating myocardial infarction. Basic Res. Cardiol. 2018;114:4. doi: 10.1007/s00395-018-0712-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan D, Kassiri Z. Modulation of cardiac fibrosis in and beyond cells. Front. Mol. Biosci. 2021;8:750626. doi: 10.3389/fmolb.2021.750626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang W, Xiong Y, Li X, Yang Y. Cardiac fibrosis: cellular effectors, molecular pathways, and exosomal roles. Front. Cardiovasc. Med. 2021;8:715258. doi: 10.3389/fcvm.2021.715258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Contessotto P, Pandit A. Therapies to prevent post-infarction remodelling: from repair to regeneration. Biomaterials. 2021;275:120906. doi: 10.1016/j.biomaterials.2021.120906. [DOI] [PubMed] [Google Scholar]
- 16.Cannatà A, Ali H, Sinagra G, Giacca M. Gene therapy for the heart lessons learned and future perspectives. Circ. Res. 2020;126:1394–1414. doi: 10.1161/CIRCRESAHA.120.315855. [DOI] [PubMed] [Google Scholar]
- 17.Strom J, Chen QM. Loss of Nrf2 promotes rapid progression to heart failure following myocardial infarction. Toxicol. Appl Pharm. 2017;327:52–58. doi: 10.1016/j.taap.2017.03.025. [DOI] [PubMed] [Google Scholar]
- 18.Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-kinase in the cardiovascular system. Circ. Res. 2016;118:352–366. doi: 10.1161/CIRCRESAHA.115.306532. [DOI] [PubMed] [Google Scholar]
- 19.Xiao Q, et al. AMP-activated protein kinase-dependent autophagy mediated the protective effect of sonic hedgehog pathway on oxygen glucose deprivation-induced injury of cardiomyocytes. Biochem. Biophys. Res. Commun. 2015;457:419–425. doi: 10.1016/j.bbrc.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Chen J, Cowan DB, Wang D-Z. Non-coding RNAs in cardiac regeneration: mechanism of action and therapeutic potential. Semin. Cell Dev. Biol. 2021;118:150–162. doi: 10.1016/j.semcdb.2021.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beliën H, Evens L, Hendrikx M, Bito V, Bronckaers A. Combining stem cells in myocardial infarction: the road to superior repair? Med. Res. Rev. 2022;42:343–373. doi: 10.1002/med.21839. [DOI] [PubMed] [Google Scholar]
- 22.Goumans MJ, Ten Dijke P. TGF-β signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 2018;10:a022210. doi: 10.1101/cshperspect.a022210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu GR, et al. Modified citrus pectin ameliorates myocardial fibrosis and inflammation via suppressing galectin-3 and TLR4/MyD88/NF-κB signaling pathway. Biomed. Pharmacother. 2020;126:110071. doi: 10.1016/j.biopha.2020.110071. [DOI] [PubMed] [Google Scholar]
- 24.Liang X, et al. Overexpression of ERBB4 rejuvenates aged mesenchymal stem cells and enhances angiogenesis via PI3K/AKT and MAPK/ERK pathways. FASEB J. 2019;33:4559–4570. doi: 10.1096/fj.201801690R. [DOI] [PubMed] [Google Scholar]
- 25.Martinez M, Andriantsitohaina RJCR. Microparticles in angiogenesis: therapeutic potential. Circ. Res. 2011;109:110–119. doi: 10.1161/CIRCRESAHA.110.233049. [DOI] [PubMed] [Google Scholar]
- 26.Sahoo S, Losordo DJCR. Exosomes and cardiac repair after myocardial infarction. Circ. Res. 2014;114:333–344. doi: 10.1161/CIRCRESAHA.114.300639. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen N, et al. A calcineurin–Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature. 2020;582:271–276. doi: 10.1038/s41586-020-2228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senyo S, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flinn MA, Link BA, O’Meara CC. Upstream regulation of the Hippo-Yap pathway in cardiomyocyte regeneration. Semin. Cell Dev. Biol. 2020;100:11–19. doi: 10.1016/j.semcdb.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghigo A, Laffargue M, Li M, Hirsch E. PI3K and calcium signaling in cardiovascular disease. Circ. Res. 2017;121:282–292. doi: 10.1161/CIRCRESAHA.117.310183. [DOI] [PubMed] [Google Scholar]
- 31.Yue Z, et al. PDGFR-β signaling regulates cardiomyocyte proliferation and myocardial regeneration. Cell Rep. 2019;28:966–978e964. doi: 10.1016/j.celrep.2019.06.065. [DOI] [PubMed] [Google Scholar]
- 32.Yuan MJ, et al. GHSR-1a is a novel pro-angiogenic and anti-remodeling target in rats after myocardial infarction. Eur. J. Pharm. 2016;788:218–225. doi: 10.1016/j.ejphar.2016.06.032. [DOI] [PubMed] [Google Scholar]
- 33.Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ. Res. 2018;122:489–505. doi: 10.1161/CIRCRESAHA.117.311147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Razavi H, Hamilton J, Feng QJP, therapeutics Modulation of apoptosis by nitric oxide: implications in myocardial ischemia and heart failure. Pharm. Ther. 2005;106:147–162. doi: 10.1016/j.pharmthera.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Ronnebaum S, Patterson C. The FoxO family in cardiac function and dysfunction. Annu. Rev. Physiol. 2010;72:81–94. doi: 10.1146/annurev-physiol-021909-135931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hennessy B, Smith D, Ram P, Lu Y, Mills GJNRDD. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 37.Hua H, et al. Targeting Akt in cancer for precision therapy. J. Hematol. Oncol. 2021;14:128. doi: 10.1186/s13045-021-01137-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eisenreich A, Rauch U. PI3K inhibitors in cardiovascular disease. Cardiovasc. Ther. 2011;29:29–36. doi: 10.1111/j.1755-5922.2010.00206.x. [DOI] [PubMed] [Google Scholar]
- 39.Kazakov A, et al. Inhibition of endothelial nitric oxide synthase induces and enhances myocardial fibrosis. Cardiovasc. Res. 2013;100:211–221. doi: 10.1093/cvr/cvt181. [DOI] [PubMed] [Google Scholar]
- 40.Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. 2019;176:1248–1264. doi: 10.1016/j.cell.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D, et al. Kaempferide protects against myocardial ischemia/reperfusion injury through activation of the PI3K/Akt/GSK-3β pathway. Mediators Inflamm. 2017;2017:5278218. doi: 10.1155/2017/5278218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tucka J, et al. Akt1 regulates vascular smooth muscle cell apoptosis through FoxO3a and Apaf1 and protects against arterial remodeling and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014;34:2421–2428. doi: 10.1161/ATVBAHA.114.304284. [DOI] [PubMed] [Google Scholar]
- 43.Li J, et al. HSPA12B attenuates cardiac dysfunction and remodelling after myocardial infarction through an eNOS-dependent mechanism. Cardiovasc. Res. 2013;99:674–684. doi: 10.1093/cvr/cvt139. [DOI] [PubMed] [Google Scholar]
- 44.Buss SJ, Riffel JH, Katus HA, Hardt SE. Augmentation of autophagy by mTOR-inhibition in myocardial infarction: when size matters. Autophagy. 2010;6:304–306. doi: 10.4161/auto.6.2.11135. [DOI] [PubMed] [Google Scholar]
- 45.Völkers M, et al. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation. 2013;128:2132–2144. doi: 10.1161/CIRCULATIONAHA.113.003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng J, et al. Inositol pyrophosphates mediated the apoptosis induced by hypoxic injury in bone marrow-derived mesenchymal stem cells by autophagy. Stem Cell Res. Ther. 2019;10:159. doi: 10.1186/s13287-019-1256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen G, Phan V, Luo X, Cao DJ. The mechanistic target of rapamycin complex 1 critically regulates the function of mononuclear phagocytes and promotes cardiac remodeling in acute ischemia. J. Mol. Cell. Cardiol. 2021;159:62–79. doi: 10.1016/j.yjmcc.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009;16:31–38. doi: 10.1038/cdd.2008.163. [DOI] [PubMed] [Google Scholar]
- 49.Nakai A, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 50.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sciarretta S, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samidurai A, Kukreja RC, Das A. Emerging role of mTOR signaling-related miRNAs in cardiovascular diseases. Oxid. Med. Cell. Longev. 2018;2018:6141902. doi: 10.1155/2018/6141902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014;114:549–564. doi: 10.1161/CIRCRESAHA.114.302022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hua H, Kong Q, Yin J, Zhang J, Jiang Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: a challenge for cancer therapy. J. Hematol. Oncol. 2020;13:64. doi: 10.1186/s13045-020-00904-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J. Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circ. Res. 2011;109:502–511. doi: 10.1161/CIRCRESAHA.111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Calissi G, Lam EW, Link W. Therapeutic strategies targeting FOXO transcription factors. Nat. Rev. Drug Discov. 2021;20:21–38. doi: 10.1038/s41573-020-0088-2. [DOI] [PubMed] [Google Scholar]
- 57.Fender AC, Dobrev D. Nitric oxide as a fragile switch between cardioprotection and cardiac injury. Int. J. Cardiol. 2021;343:102–103. doi: 10.1016/j.ijcard.2021.09.001. [DOI] [PubMed] [Google Scholar]
- 58.Meloni M, et al. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ. Res. 2010;106:1275–1284. doi: 10.1161/CIRCRESAHA.109.210088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee Y-R, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 2018;19:547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 60.Worby C, Dixon J. PTEN. Annu. Rev. Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 61.Feng Q, et al. PTEN inhibitor improves vascular remodeling and cardiac function after myocardial infarction through PI3k/Akt/VEGF signaling pathway. Mol. Med. 2020;26:111. doi: 10.1186/s10020-020-00241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang T, et al. Loss of phosphatase and tensin homolog promotes cardiomyocyte proliferation and cardiac repair after myocardial infarction. Circulation. 2020;142:2196–2199. doi: 10.1161/CIRCULATIONAHA.120.046372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Z, Cheng Z, Haifeng Y, Chen M, Li L. PTEN signaling inhibitor VO-OHpic improves cardiac myocyte survival by mediating apoptosis resistance in vitro. Biomed. Pharmacother. 2018;103:1217–1222. doi: 10.1016/j.biopha.2018.04.141. [DOI] [PubMed] [Google Scholar]
- 64.Parajuli N, Yuan Y, Zheng X, Bedja D, Cai ZP. Phosphatase PTEN is critically involved in post-myocardial infarction remodeling through the Akt/interleukin-10 signaling pathway. Basic Res. Cardiol. 2012;107:248. doi: 10.1007/s00395-012-0248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buss SJ, et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2009;54:2435–2446. doi: 10.1016/j.jacc.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 66.Sanches-Silva A, et al. Therapeutic potential of polyphenols in cardiovascular diseases: Regulation of mTOR signaling pathway. Pharm. Res. 2020;152:104626. doi: 10.1016/j.phrs.2019.104626. [DOI] [PubMed] [Google Scholar]
- 67.Dai Y, Chen Y, Wei G, Zha L, Li X. Ivabradine protects rats against myocardial infarction through reinforcing autophagy via inhibiting PI3K/AKT/mTOR/p70S6K pathway. Bioengineered. 2021;12:1826–1837. doi: 10.1080/21655979.2021.1925008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao G, et al. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int. J. Mol. Med. 2020;45:195–209. doi: 10.3892/ijmm.2019.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang LG, et al. Sphingosine-1-phosphate induces myocyte autophagy after myocardial infarction through mTOR inhibition. Eur. J. Pharm. 2021;907:174260. doi: 10.1016/j.ejphar.2021.174260. [DOI] [PubMed] [Google Scholar]
- 70.Zhang X, et al. Tanshinone IIA protects against heart failure post-myocardial infarction via AMPKs/mTOR-dependent autophagy pathway. Biomed. Pharmacother. 2019;112:108599. doi: 10.1016/j.biopha.2019.108599. [DOI] [PubMed] [Google Scholar]
- 71.Segers VFM, Lee RT. Protein therapeutics for cardiac regeneration after myocardial infarction. J. Cardiovasc. Transl. Res. 2010;3:469–477. doi: 10.1007/s12265-010-9207-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cao G, Xuan X, Zhang R, Hu J, Dong H. Gene therapy for cardiovascular disease: basic research and clinical prospects. Front. Cardiovasc. Med. 2021;8:760140. doi: 10.3389/fcvm.2021.760140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pfister O, et al. FLT3 activation improves post-myocardial infarction remodeling involving a cytoprotective effect on cardiomyocytes. J. Am. Coll. Cardiol. 2014;63:1011–1019. doi: 10.1016/j.jacc.2013.08.1647. [DOI] [PubMed] [Google Scholar]
- 74.Kumarswamy R, et al. SERCA2a gene therapy restores microRNA-1 expression in heart failure via an Akt/FoxO3A-dependent pathway. Eur. Heart J. 2012;33:1067–1075. doi: 10.1093/eurheartj/ehs043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang W, et al. Combination of microRNA-21 and microRNA-146a attenuates cardiac dysfunction and apoptosis during acute myocardial infarction in mice. Mol. Ther. Nucleic Acids. 2016;5:e296. doi: 10.1038/mtna.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang K, et al. Enhanced cardioprotection by human endometrium mesenchymal stem cells driven by exosomal microRNA-21. Stem Cells Transl. Med. 2017;6:209–222. doi: 10.5966/sctm.2015-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu C, et al. Attenuation of cardiac dysfunction and remodeling of myocardial infarction by microRNA-130a are mediated by suppression of PTEN and activation of PI3K dependent signaling. J. Mol. Cell. Cardiol. 2015;89:87–97. doi: 10.1016/j.yjmcc.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun L, et al. Down-regulated exosomal MicroRNA-221-3p derived from senescent mesenchymal stem cells impairs heart repair. Front. Cell Dev. Biol. 2020;8:263. doi: 10.3389/fcell.2020.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhen L, et al. miR-301a-PTEN-AKT signaling induces cardiomyocyte proliferation and promotes cardiac repair post-MI. Mol. Ther. Nucleic Acids. 2020;22:251–262. doi: 10.1016/j.omtn.2020.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li M, et al. LncRNA Snhg1-driven self-reinforcing regulatory network promoted cardiac regeneration and repair after myocardial infarction. Theranostics. 2021;11:9397–9414. doi: 10.7150/thno.57037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Q, et al. Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J. Cell. Mol. Med. 2014;18:919–928. doi: 10.1111/jcmm.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Du J, Yang ST, Liu J, Zhang KX, Leng JY. Silence of LncRNA GAS5 protects cardiomyocytes H9c2 against hypoxic injury via sponging miR-142-5p. Mol. Cells. 2019;42:397–405. doi: 10.14348/molcells.2018.0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou XH, Chai HX, Bai M, Zhang Z. LncRNA-GAS5 regulates PDCD4 expression and mediates myocardial infarction-induced cardiomyocytes apoptosis via targeting MiR-21. Cell Cycle. 2020;19:1363–1377. doi: 10.1080/15384101.2020.1750257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu Y, et al. Silencing CircHIPK3 sponges miR-93-5p to inhibit the activation of Rac1/PI3K/AKT pathway and improves myocardial infarction-induced cardiac dysfunction. Front. Cardiovasc. Med. 2021;8:645378. doi: 10.3389/fcvm.2021.645378. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Li X, et al. Inhibition of AZIN2-sv induces neovascularization and improves prognosis after myocardial infarction by blocking ubiquitin-dependent talin1 degradation and activating the Akt pathway. EBioMedicine. 2019;39:69–82. doi: 10.1016/j.ebiom.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Z, et al. Long non-coding RNA UCA1 relieves cardiomyocytes H9c2 injury aroused by oxygen-glucose deprivation via declining miR-122. Artif. Cells Nanomed. Biotechnol. 2019;47:3492–3499. doi: 10.1080/21691401.2019.1652630. [DOI] [PubMed] [Google Scholar]
- 87.Qiu L, et al. Long non-coding RNA DANCR alleviates hypoxia-caused H9c2 cells damage through up regulation of HIF-1α. Artif. Cells Nanomed. Biotechnol. 2020;48:533–541. doi: 10.1080/21691401.2020.1725026. [DOI] [PubMed] [Google Scholar]
- 88.Arjmand B, et al. Regenerative medicine for the treatment of ischemic heart disease; status and future perspectives. Front. Cell Dev. Biol. 2021;9:704903. doi: 10.3389/fcell.2021.704903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carvalho E, Verma P, Hourigan K, Banerjee R. Myocardial infarction: stem cell transplantation for cardiac regeneration. Regen. Med. 2015;10:1025–1043. doi: 10.2217/rme.15.63. [DOI] [PubMed] [Google Scholar]
- 90.Ma J, et al. Exosomes derived from Akt-modified human umbilical cord mesenchymal stem cells improve cardiac regeneration and promote angiogenesis via activating platelet-derived growth factor D. Stem Cells Transl. Med. 2017;6:51–59. doi: 10.5966/sctm.2016-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sun YY, et al. Period 2 is essential to maintain early endothelial progenitor cell function in vitro and angiogenesis after myocardial infarction in mice. J. Cell. Mol. Med. 2014;18:907–918. doi: 10.1111/jcmm.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gnecchi M, et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat. Med. 2005;11:367–368. doi: 10.1038/nm0405-367. [DOI] [PubMed] [Google Scholar]
- 93.Tan SH, et al. Thymosin β4 increases cardiac cell proliferation, cell engraftment, and the reparative potency of human induced-pluripotent stem cell-derived cardiomyocytes in a porcine model of acute myocardial infarction. Theranostics. 2021;11:7879–7895. doi: 10.7150/thno.56757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luo W, et al. NGF nanoparticles enhance the potency of transplanted human umbilical cord mesenchymal stem cells for myocardial repair. Am. J. Physiol. Heart Circ. Physiol. 2021;320:H1959–H1974. doi: 10.1152/ajpheart.00855.2020. [DOI] [PubMed] [Google Scholar]
- 95.Gnecchi M, et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J. 2006;20:661–669. doi: 10.1096/fj.05-5211com. [DOI] [PubMed] [Google Scholar]
- 96.Zhang GW, et al. Edaravone promotes activation of resident cardiac stem cells by transplanted mesenchymal stem cells in a rat myocardial infarction model. J. Thorac. Cardiovasc. Surg. 2016;152:570–582. doi: 10.1016/j.jtcvs.2016.02.071. [DOI] [PubMed] [Google Scholar]
- 97.Li S, et al. Biphasic effect of EGb761 on simulated ischemia-induced rat BMSC survival in vitro and in vivo. Life Sci. 2011;88:853–863. doi: 10.1016/j.lfs.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 98.Tang S, et al. TMSB4 Overexpression Enhances the Potency of Marrow Mesenchymal Stromal Cells for Myocardial Repair. Front Cell Dev Biol. 2021;9:670913. doi: 10.3389/fcell.2021.670913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Z, et al. Selective inhibition of inositol hexakisphosphate kinases (IP6Ks) enhances mesenchymal stem cell engraftment and improves therapeutic efficacy for myocardial infarction. Basic Res. Cardiol. 2014;109:417. doi: 10.1007/s00395-014-0417-x. [DOI] [PubMed] [Google Scholar]
- 100.Zhang Z, et al. Rosuvastatin enhances the therapeutic efficacy of adipose-derived mesenchymal stem cells for myocardial infarction via PI3K/Akt and MEK/ERK pathways. Basic Res. Cardiol. 2013;108:333. doi: 10.1007/s00395-013-0333-5. [DOI] [PubMed] [Google Scholar]
- 101.Wang X, et al. The application potential and advance of mesenchymal stem cell-derived exosomes in myocardial infarction. Stem Cells Int. 2021;2021:5579904. doi: 10.1155/2021/5579904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qiao L, et al. microRNA-21-5p dysregulation in exosomes derived from heart failure patients impairs regenerative potential. J. Clin. Investig. 2019;129:2237–2250. doi: 10.1172/JCI123135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gong XH, Liu H, Wang SJ, Liang SW, Wang GG. Exosomes derived from SDF1-overexpressing mesenchymal stem cells inhibit ischemic myocardial cell apoptosis and promote cardiac endothelial microvascular regeneration in mice with myocardial infarction. J. Cell. Physiol. 2019;234:13878–13893. doi: 10.1002/jcp.28070. [DOI] [PubMed] [Google Scholar]
- 104.de la Pompa JL. Notch signaling in cardiac development and disease. Pediatr. Cardiol. 2009;30:643–650. doi: 10.1007/s00246-008-9368-z. [DOI] [PubMed] [Google Scholar]
- 105.Xuan W, Khan M, Ashraf M. Extracellular vesicles from notch activated cardiac mesenchymal stem cells promote myocyte proliferation and neovasculogenesis. Front. Cell Dev. Biol. 2020;8:11. doi: 10.3389/fcell.2020.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huang H, Huang F, Huang JP. Transplantation of bone marrow-derived endothelial progenitor cells overexpressing Delta-like-4 enhances functional neovascularization in ischemic myocardium. Mol. Med. Rep. 2013;8:1556–1562. doi: 10.3892/mmr.2013.1657. [DOI] [PubMed] [Google Scholar]
- 107.Zhou XL, et al. Notch signaling inhibits cardiac fibroblast to myofibroblast transformation by antagonizing TGF-β1/Smad3 signaling. J. Cell. Physiol. 2019;234:8834–8845. doi: 10.1002/jcp.27543. [DOI] [PubMed] [Google Scholar]
- 108.Li Y, Hiroi Y, Liao JK. Notch signaling as an important mediator of cardiac repair and regeneration after myocardial infarction. Trends Cardiovasc. Med. 2010;20:228–231. doi: 10.1016/j.tcm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Luxán G, D’Amato G, MacGrogan D, de la Pompa JL. Endocardial notch signaling in cardiac development and disease. Circ. Res. 2016;118:e1–e18. doi: 10.1161/CIRCRESAHA.115.305350. [DOI] [PubMed] [Google Scholar]
- 110.Gude NA, et al. Activation of Notch-mediated protective signaling in the myocardium. Circ. Res. 2008;102:1025–1035. doi: 10.1161/CIRCRESAHA.107.164749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang F, et al. Mesenchymal stem cells modified with miR-126 release angiogenic factors and activate Notch ligand Delta-like-4, enhancing ischemic angiogenesis and cell survival. Int. J. Mol. Med. 2013;31:484–492. doi: 10.3892/ijmm.2012.1200. [DOI] [PubMed] [Google Scholar]
- 112.Fan J, Xu W, Nan S, Chang M, Zhang Y. MicroRNA-384-5p promotes endothelial progenitor cell proliferation and angiogenesis in cerebral ischemic stroke through the delta-likeligand 4-mediated notch signaling pathway. Cerebrovasc. Dis. 2020;49:39–54. doi: 10.1159/000503950. [DOI] [PubMed] [Google Scholar]
- 113.Wu F, Yu B, Zhang X, Zhang Y. Cardioprotective effect of Notch signaling on the development of myocardial infarction complicated by diabetes mellitus. Exp. Ther. Med. 2017;14:3447–3454. doi: 10.3892/etm.2017.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yu B, Song B. Notch 1 signalling inhibits cardiomyocyte apoptosis in ischaemic postconditioning. Heart Lung Circ. 2014;23:152–158. doi: 10.1016/j.hlc.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 115.Boopathy AV, Pendergrass KD, Che PL, Yoon YS, Davis ME. Oxidative stress-induced Notch1 signaling promotes cardiogenic gene expression in mesenchymal stem cells. Stem Cell Res. Ther. 2013;4:43. doi: 10.1186/scrt190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mack JJ, et al. NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 2017;8:1620. doi: 10.1038/s41467-017-01741-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shoeibi S, Mozdziak P, Mohammadi S. Important signals regulating coronary artery angiogenesis. Microvasc. Res. 2018;117:1–9. doi: 10.1016/j.mvr.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 118.Noseda M, et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004;94:910–917. doi: 10.1161/01.RES.0000124300.76171.C9. [DOI] [PubMed] [Google Scholar]
- 119.Croquelois A, et al. Control of the adaptive response of the heart to stress via the Notch1 receptor pathway. J. Exp. Med. 2008;205:3173–3185. doi: 10.1084/jem.20081427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu Y, Wang H, Wang X, Xie G. MiR-29b inhibits ventricular remodeling by activating notch signaling pathway in the rat myocardial infarction model. Heart Surg. Forum. 2019;22:E019–E023. doi: 10.1532/hsf.2079. [DOI] [PubMed] [Google Scholar]
- 121.Shi J, et al. Notch3 modulates cardiac fibroblast proliferation, apoptosis, and fibroblast to myofibroblast transition via negative regulation of the RhoA/ROCK/Hif1α axis. Front. Physiol. 2020;11:669. doi: 10.3389/fphys.2020.00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.He Y, et al. Blockade of RBP-J-mediated notch signaling pathway exacerbates cardiac remodeling after infarction by increasing apoptosis in mice. Biomed. Res. Int. 2018;2018:5207031. doi: 10.1155/2018/5207031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang S, Zhang R, Wu F, Li X. MicroRNA-208a regulates H9c2 cells simulated ischemia-reperfusion myocardial injury via targeting CHD9 through Notch/NF-kappa B signal pathways. Int. Heart J. 2018;59:580–588. doi: 10.1536/ihj.17-147. [DOI] [PubMed] [Google Scholar]
- 124.Cheng P, et al. Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. J. Immunol. 2001;167:4458–4467. doi: 10.4049/jimmunol.167.8.4458. [DOI] [PubMed] [Google Scholar]
- 125.Pei H, et al. TNF-α inhibitor protects against myocardial ischemia/reperfusion injury via Notch1-mediated suppression of oxidative/nitrative stress. Free Radic. Biol. Med. 2015;82:114–121. doi: 10.1016/j.freeradbiomed.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 126.Luo G, Wang R, Zhou H, Liu X. ALDOA protects cardiomyocytes against H/R-induced apoptosis and oxidative stress by regulating the VEGF/Notch 1/Jagged 1 pathway. Mol. Cell. Biochem. 2021;476:775–783. doi: 10.1007/s11010-020-03943-z. [DOI] [PubMed] [Google Scholar]
- 127.Felician G, et al. Epigenetic modification at Notch responsive promoters blunts efficacy of inducing notch pathway reactivation after myocardial infarction. Circ. Res. 2014;115:636–649. doi: 10.1161/CIRCRESAHA.115.304517. [DOI] [PubMed] [Google Scholar]
- 128.Zhao L, et al. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl Acad. Sci. USA. 2014;111:1403–1408. doi: 10.1073/pnas.1311705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Limana F, et al. Transcriptional profiling of HMGB1-induced myocardial repair identifies a key role for Notch signaling. Mol. Ther. 2013;21:1841–1851. doi: 10.1038/mt.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gude N, et al. Notch activation enhances lineage commitment and protective signaling in cardiac progenitor cells. Basic Res. Cardiol. 2015;110:29. doi: 10.1007/s00395-015-0488-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li Y, et al. Notch1 in bone marrow-derived cells mediates cardiac repair after myocardial infarction. Circulation. 2011;123:866–876. doi: 10.1161/CIRCULATIONAHA.110.947531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Øie E, et al. Activation of Notch signaling in cardiomyocytes during post-infarction remodeling. Scand. Cardiovasc. J. 2010;44:359–366. doi: 10.3109/14017431.2010.511256. [DOI] [PubMed] [Google Scholar]
- 133.Wang K, et al. Hypoxia-stressed cardiomyocytes promote early cardiac differentiation of cardiac stem cells through HIF-1α/Jagged1/Notch1 signaling. Acta Pharm. Sin. B. 2018;8:795–804. doi: 10.1016/j.apsb.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tsang KM, et al. Embryonic stem cell differentiation to functional arterial endothelial cells through sequential activation of ETV2 and NOTCH1 signaling by HIF1α. Stem Cell Rep. 2017;9:796–806. doi: 10.1016/j.stemcr.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Meng X, et al. Inhibition of miR-363 protects cardiomyocytes against hypoxia-induced apoptosis through regulation of Notch signaling. Biomed. Pharmacother. 2017;90:509–516. doi: 10.1016/j.biopha.2017.03.080. [DOI] [PubMed] [Google Scholar]
- 136.Sainson RC, Harris AL. Hypoxia-regulated differentiation: let’s step it up a Notch. Trends Mol. Med. 2006;12:141–143. doi: 10.1016/j.molmed.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 137.Diez H, et al. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp. Cell Res. 2007;313:1–9. doi: 10.1016/j.yexcr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 138.Anbara T, Sharifi M, Aboutaleb N. Endothelial to mesenchymal transition in the cardiogenesis and cardiovascular diseases. Curr. Cardiol. Rev. 2020;16:306–314. doi: 10.2174/1573403X15666190808100336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gustafsson MV, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 140.Shi WL, et al. Combination of Ligusticum chuanxiong and Radix paeonia promotes angiogenesis in ischemic myocardium through notch signalling and mobilization of stem cells. Evid. Based Complement. Altern. Med. 2019;2019:7912402. doi: 10.1155/2019/7912402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shevchenko EK, et al. Combination of mesenchymal stromal cells and cardiac stem cells in a multilayer cell construct promotes activation of notch signaling and initiation of endothelial differentiation. Bull. Exp. Biol. Med. 2019;166:548–552. doi: 10.1007/s10517-019-04390-7. [DOI] [PubMed] [Google Scholar]
- 142.Li Y, et al. Velvet antler mobilizes endothelial progenitor cells to promote angiogenesis and repair vascular endothelial injury in rats following myocardial infarction. Front. Physiol. 2018;9:1940. doi: 10.3389/fphys.2018.01940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wu J, et al. Effect of Yiqihuoxue prescription on myocardial energy metabolism after myocardial infarction via cross talk of liver kinase B1-dependent Notch1 and adenosine 5’-monophosphate-activated protein kinase. J. Tradit. Chin. Med. 2017;37:378–386. [PubMed] [Google Scholar]
- 144.Yu J, Zhang X, Zhang Y. Astragaloside attenuates myocardial injury in a rat model of acute myocardial infarction by upregulating hypoxia inducible factor-1α and Notch1/Jagged1 signaling. Mol. Med. Rep. 2017;15:4015–4020. doi: 10.3892/mmr.2017.6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pei H, et al. Melatonin prevents adverse myocardial infarction remodeling via Notch1/Mfn2 pathway. Free Radic. Biol. Med. 2016;97:408–417. doi: 10.1016/j.freeradbiomed.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 146.Xu, W., Jiang, S. & Liu, Q. MicroRNA-124a protects the myocardium against ischemia reperfusion injury through regulation of the notch signaling pathway. Braz. J. Cardiovasc. Surg. (2021). [DOI] [PMC free article] [PubMed]
- 147.Si X, et al. circRNA Hipk3 induces cardiac regeneration after myocardial infarction in mice by binding to notch1 and miR-133a. Mol. Ther. Nucleic Acids. 2020;21:636–655. doi: 10.1016/j.omtn.2020.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen T, et al. MicroRNA-199b modulates vascular cell fate during ips cell differentiation by targeting the notch ligand Jagged1 and enhancing VEGF signaling. Stem Cells. 2015;33:1405–1418. doi: 10.1002/stem.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang Q, et al. Aberrant expression of miR-29b-3p influences heart development and cardiomyocyte proliferation by targeting NOTCH2. Cell Prolif. 2020;53:e12764. doi: 10.1111/cpr.12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang B, et al. Effect of MiR-133 on myocardial cell apoptosis in rats with myocardial infarction through the Notch1 signaling pathway. Minerva Med. 2021;112:303–305. doi: 10.23736/S0026-4806.19.06226-8. [DOI] [PubMed] [Google Scholar]
- 151.Jung JH, et al. miR-106a-363 cluster in extracellular vesicles promotes endogenous myocardial repair via Notch3 pathway in ischemic heart injury. Basic Res. Cardiol. 2021;116:19. doi: 10.1007/s00395-021-00858-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhao Z, et al. Protective role of microRNA-374 against myocardial ischemia-reperfusion injury in mice following thoracic epidural anesthesia by downregulating dystrobrevin alpha-mediated Notch1 axis. J. Cell. Physiol. 2019;234:10726–10740. doi: 10.1002/jcp.27745. [DOI] [PubMed] [Google Scholar]
- 153.Chen C, et al. MicroRNA-1 regulates the differentiation of adipose-derived stem cells into cardiomyocyte-like cells. Stem Cells Int. 2018;2018:7494530. doi: 10.1155/2018/7494530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Xu H, Jin L, Chen Y, Li J. Downregulation of microRNA-429 protects cardiomyocytes against hypoxia-induced apoptosis by increasing Notch1 expression. Int. J. Mol. Med. 2016;37:1677–1685. doi: 10.3892/ijmm.2016.2558. [DOI] [PubMed] [Google Scholar]
- 155.Wang Y, et al. Methylation-dependent transcriptional repression of RUNX3 by KCNQ1OT1 regulates mouse cardiac microvascular endothelial cell viability and inflammatory response following myocardial infarction. FASEB J. 2019;33:13145–13160. doi: 10.1096/fj.201900310R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang M, et al. Silence of lncRNA XIST represses myocardial cell apoptosis in rats with acute myocardial infarction through regulating miR-449. Eur. Rev. Med. Pharm. Sci. 2019;23:8566–8572. doi: 10.26355/eurrev_201910_19172. [DOI] [PubMed] [Google Scholar]
- 157.Wu J, Xie F, Qin Y, Liu J, Yang Z. Notch signaling is involved in the antiapoptotic effects of liraglutide on rat H9c2 cardiomyocytes exposed to hypoxia followed by reoxygenation. J. Int. Med. Res. 2020;48:300060520948394. doi: 10.1177/0300060520948394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Du M, et al. Oestrogen receptor β activation protects against myocardial infarction via Notch1 signalling. Cardiovasc. Drugs Ther. 2020;34:165–178. doi: 10.1007/s10557-020-06949-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu X, et al. Pigment epithelium-derived factor increases native collateral blood flow to improve cardiac function and induce ventricular remodeling after acute myocardial infarction. J. Am. Heart Assoc. 2019;8:e013323. doi: 10.1161/JAHA.119.013323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Fang HC, et al. KRT1 gene silencing ameliorates myocardial ischemia-reperfusion injury via the activation of the Notch signaling pathway in mouse models. J. Cell. Physiol. 2019;234:3634–3646. doi: 10.1002/jcp.27133. [DOI] [PubMed] [Google Scholar]
- 161.Zhao Q, et al. Endothelium-specific CYP2J2 overexpression improves cardiac dysfunction by promoting angiogenesis via Jagged1/Notch1 signaling. J. Mol. Cell. Cardiol. 2018;123:118–127. doi: 10.1016/j.yjmcc.2018.08.027. [DOI] [PubMed] [Google Scholar]
- 162.Dominguez-Rodriguez A, et al. Usefulness of early treatment with melatonin to reduce infarct size in patients with ST-segment elevation myocardial infarction receiving percutaneous coronary intervention (from the melatonin adjunct in the acute myocardial infarction treated with angioplasty trial) Am. J. Cardiol. 2017;120:522–526. doi: 10.1016/j.amjcard.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 163.Ekeloef S, et al. Effect of intracoronary and intravenous melatonin on myocardial salvage index in patients with ST-elevation myocardial infarction: a Randomized Placebo Controlled trial. J. Cardiovasc. Transl. Res. 2017;10:470–479. doi: 10.1007/s12265-017-9768-7. [DOI] [PubMed] [Google Scholar]
- 164.Takahashi M. Role of the inflammasome in myocardial infarction. Trends Cardiovasc. Med. 2011;21:37–41. doi: 10.1016/j.tcm.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 165.Wei Z, et al. Loss of Camk2n1 aggravates cardiac remodeling and malignant ventricular arrhythmia after myocardial infarction in mice via NLRP3 inflammasome activation. Free Radic. Biol. Med. 2021;167:243–257. doi: 10.1016/j.freeradbiomed.2021.03.014. [DOI] [PubMed] [Google Scholar]
- 166.Swanson KV, Deng M. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015;265:35–52. doi: 10.1111/imr.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bauernfeind FG, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019;20:3328. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kaplanski G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol. Rev. 2018;281:138–153. doi: 10.1111/imr.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Abbate A, et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 2020;126:1260–1280. doi: 10.1161/CIRCRESAHA.120.315937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Libby P. Targeting inflammatory pathways in cardiovascular disease: the inflammasome, interleukin-1, interleukin-6 and beyond. Cells. 2021;10:951. doi: 10.3390/cells10040951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hermansson C, et al. Reduced expression of NLRP3 and MEFV in human ischemic heart tissue. Biochem. Biophys. Res. Commun. 2013;430:425–428. doi: 10.1016/j.bbrc.2012.11.070. [DOI] [PubMed] [Google Scholar]
- 175.Chen G, Song X, Lin D, Xu P. Isofraxidin alleviates myocardial infarction through NLRP3 inflammasome inhibition. Inflammation. 2020;43:712–721. doi: 10.1007/s10753-019-01158-z. [DOI] [PubMed] [Google Scholar]
- 176.Jiang J, Gu X, Wang H, Ding S. Resveratrol improves cardiac function and left ventricular fibrosis after myocardial infarction in rats by inhibiting NLRP3 inflammasome activity and the TGF-β1/SMAD2 signaling pathway. J. Am. Heart Assoc. 2021;9:e11501. doi: 10.7717/peerj.11501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhao P, et al. Aminooxyacetic acid attenuates post-infarct cardiac dysfunction by balancing macrophage polarization through modulating macrophage metabolism in mice. J. Cell. Mol. Med. 2020;24:2593–2609. doi: 10.1111/jcmm.14972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang S, et al. Glycogen synthase kinase-3β inhibition alleviates activation of the NLRP3 inflammasome in myocardial infarction. J. Mol. Cell. Cardiol. 2020;149:82–94. doi: 10.1016/j.yjmcc.2020.09.009. [DOI] [PubMed] [Google Scholar]
- 179.Liu A, Gao X, Zhang Q, Cui L. Cathepsin B inhibition attenuates cardiac dysfunction and remodeling following myocardial infarction by inhibiting the NLRP3 pathway. Mol. Med. Rep. 2013;8:361–366. doi: 10.3892/mmr.2013.1507. [DOI] [PubMed] [Google Scholar]
- 180.Zhu N, Chen F, Chen ZQ, Zhong GL, Zhu JJ. Nicorandil inhibits TLR4/MyD88/NF-κB/NLRP3 signaling pathway to reduce pyroptosis in rats with myocardial infarction. Inflammation. 2021;246:1938–1947. doi: 10.1177/15353702211013444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Li Z, et al. GDF11 inhibits cardiomyocyte pyroptosis and exerts cardioprotection in acute myocardial infarction mice by upregulation of transcription factor HOXA3. Cell Death Dis. 2020;11:917. doi: 10.1038/s41419-020-03120-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nagoor Meeran MF, et al. α-Bisabolol protects against β-adrenergic agonist-induced myocardial infarction in rats by attenuating inflammation, lysosomal dysfunction, NLRP3 inflammasome activation and modulating autophagic flux. Food Funct. 2020;11:965–976. doi: 10.1039/c9fo00530g. [DOI] [PubMed] [Google Scholar]
- 183.Katzenberger DR, et al. Salvianolate reduces atrial fibrillation through suppressing atrial interstitial fibrosis by inhibiting TGF-β1/Smad2/3 and TXNIP/NLRP3 inflammasome signaling pathways in post-MI rats. Ann. Pharmacother. 2018;51:255–265. doi: 10.1016/j.phymed.2018.09.238. [DOI] [PubMed] [Google Scholar]
- 184.Louwe MC, et al. Absence of NLRP3 inflammasome in hematopoietic cells reduces adverse remodeling after experimental myocardial infarction. J. Cell. Mol. Med. 2020;5:1210–1224. doi: 10.1016/j.jacbts.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sano S, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol. 2018;71:875–886. doi: 10.1016/j.jacc.2017.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liu W, et al. Activation in M1 but not M2 macrophages contributes to cardiac remodeling after myocardial infarction in rats: a critical role of the calcium sensing receptor/NRLP3 inflammasome. Cell. Physiol. Biochem. 2015;35:2483–2500. doi: 10.1159/000374048. [DOI] [PubMed] [Google Scholar]
- 187.Hoffmann J, Luxán G. Post-myocardial infarction heart failure dysregulates the bone vascular niche. Nat. Commun. 2021;12:3964. doi: 10.1038/s41467-021-24045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wei X, et al. MiR-703 protects against hypoxia/reoxygenation-induced cardiomyocyte injury via inhibiting the NLRP3/caspase-1-mediated pyroptosis. J. Bioenerg. Biomembr. 2020;52:155–164. doi: 10.1007/s10863-020-09832-w. [DOI] [PubMed] [Google Scholar]
- 189.Zhou Y, Huang H, Hou X. MicroRNA-133b alleviates hypoxia injury by direct targeting on NOD-like receptor protein 3 in rat H9c2 cardiomyocyte. Cardiol. Res. Pract. 2019;2019:8092461. doi: 10.1155/2019/8092461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li M, Ge Q, Zhang J, Han Y. LncRNA H19 suppresses pyroptosis of cardiomyocytes to attenuate myocardial infarction in a PBX3/CYP1B1-dependent manner. J. Inflamm. Res. 2021;476:1387–1400. doi: 10.1007/s11010-020-03998-y. [DOI] [PubMed] [Google Scholar]
- 191.Kore RA, et al. MSC exosome-mediated cardioprotection in ischemic mouse heart comparative proteomics of infarct and peri-infarct areas. Mol. Cell. Biochem. 2021;476:1691–1704. doi: 10.1007/s11010-020-04029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lee TM, et al. Host pre-conditioning improves human adipose-derived stem cell transplantation in ageing rats after myocardial infarction: role of NLRP3 inflammasome. J. Cell. Mol. Med. 2020;24:12272–12284. doi: 10.1111/jcmm.15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Marchetti C, et al. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. Biomed. Res. Int. 2015;66:1–8. doi: 10.1097/FJC.0000000000000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.van Hout GP, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017;38:828–836. doi: 10.1093/eurheartj/ehw247. [DOI] [PubMed] [Google Scholar]
- 195.Li X, et al. (18)F-FDG PET imaging-monitored anti-inflammatory therapy for acute myocardial infarction: exploring the role of MCC950 in murine model. J. Nucl. Cardiol. 2020;28:2346–2357. doi: 10.1007/s12350-020-02044-0. [DOI] [PubMed] [Google Scholar]
- 196.Gao RF, et al. The covalent NLRP3-inflammasome inhibitor Oridonin relieves myocardial infarction induced myocardial fibrosis and cardiac remodeling in mice. Int. Immunopharmacol. 2021;90:107133. doi: 10.1016/j.intimp.2020.107133. [DOI] [PubMed] [Google Scholar]
- 197.Fulp J, et al. Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: design, synthesis, and biological characterization. J. Med. Chem. 2018;61:5412–5423. doi: 10.1021/acs.jmedchem.8b00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Toldo S, et al. The NLRP3 inflammasome inhibitor, OLT1177 (Dapansutrile), reduces infarct size and preserves contractile function after ischemia reperfusion injury in the mouse. J. Cardiovasc. Pharm. 2019;73:215–222. doi: 10.1097/FJC.0000000000000658. [DOI] [PubMed] [Google Scholar]
- 199.Aliaga J, Bonaventura A. Preservation of contractile reserve and diastolic function by inhibiting the NLRP3 inflammasome with OLT1177(®) (Dapansutrile) in a mouse model of severe ischemic cardiomyopathy due to non-reperfused anterior wall myocardial infarction. Molecules. 2021;26:3534. doi: 10.3390/molecules26123534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mezzaroma E, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl Acad. Sci. USA. 2011;108:19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hesse J, et al. CD73-derived adenosine and tenascin-C control cytokine production by epicardium-derived cells formed after myocardial infarction. FASEB J. 2017;31:3040–3053. doi: 10.1096/fj.201601307R. [DOI] [PubMed] [Google Scholar]
- 202.Vessey DA, Li L, Kelley M. Pannexin-I/P2X 7 purinergic receptor channels mediate the release of cardioprotectants induced by ischemic pre- and postconditioning. J. Cardiovasc. Pharm. Ther. 2010;15:190–195. doi: 10.1177/1074248409360356. [DOI] [PubMed] [Google Scholar]
- 203.Bouabdallaoui N, et al. Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT) Eur. Heart J. 2020;41:4092–4099. doi: 10.1093/eurheartj/ehaa659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ridker PM, et al. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- 205.Ridker PM, et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 206.Tardif JC, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 2019;381:2497–2505. doi: 10.1056/NEJMoa1912388. [DOI] [PubMed] [Google Scholar]
- 207.Nidorf SM, et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 2020;383:1838–1847. doi: 10.1056/NEJMoa2021372. [DOI] [PubMed] [Google Scholar]
- 208.Dolasia K, Bisht MK, Pradhan G, Udgata A, Mukhopadhyay S. TLRs/NLRs: shaping the landscape of host immunity. Int. Rev. Immunol. 2018;37:3–19. doi: 10.1080/08830185.2017.1397656. [DOI] [PubMed] [Google Scholar]
- 209.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 210.Rayees S, et al. Macrophage TLR4 and PAR2 signaling: role in regulating vascular inflammatory injury and repair. Front. Immunol. 2020;11:2091. doi: 10.3389/fimmu.2020.02091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Park HJ, et al. A novel TLR4 binding protein, 40S ribosomal protein S3, has potential utility as an adjuvant in a dendritic cell-based vaccine. J. Immunother. Cancer. 2019;7:60. doi: 10.1186/s40425-019-0539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Qu D, et al. Focal TLR4 activation mediates disturbed flow-induced endothelial inflammation. Cardiovasc. Res. 2020;116:226–236. doi: 10.1093/cvr/cvz046. [DOI] [PubMed] [Google Scholar]
- 213.Shang J, Liu W, Yin C, Chu H, Zhang M. Cucurbitacin E ameliorates lipopolysaccharide-evoked injury, inflammation and MUC5AC expression in bronchial epithelial cells by restraining the HMGB1-TLR4-NF-κB signaling. Mol. Immunol. 2019;114:571–577. doi: 10.1016/j.molimm.2019.09.008. [DOI] [PubMed] [Google Scholar]
- 214.Li M, et al. Identification of post-myocardial infarction blood expression signatures using multiple feature selection strategies. Front. Physiol. 2020;11:483. doi: 10.3389/fphys.2020.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Haas B, et al. Adenosine reduces cell surface expression of toll-like receptor 4 and inflammation in response to lipopolysaccharide and matrix products. J. Cardiovasc. Transl. Res. 2011;4:790–800. doi: 10.1007/s12265-011-9279-x. [DOI] [PubMed] [Google Scholar]
- 216.van Hout GP, Arslan F, Pasterkamp G, Hoefer IE. Targeting danger-associated molecular patterns after myocardial infarction. Expert Opin. Ther. Targets. 2016;20:223–239. doi: 10.1517/14728222.2016.1088005. [DOI] [PubMed] [Google Scholar]
- 217.Rohde D, et al. S100A1 is released from ischemic cardiomyocytes and signals myocardial damage via Toll-like receptor 4. EMBO Mol. Med. 2014;6:778–794. doi: 10.15252/emmm.201303498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. 2013;2013:828354. doi: 10.1155/2013/828354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhang W, et al. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J. Am. Heart Assoc. 2015;4:e001993. doi: 10.1161/JAHA.115.001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Xiao SJ, et al. Uncovering the differentially expressed genes and pathways involved in the progression of stable coronary artery disease to acute myocardial infarction using bioinformatics analysis. Nat. Genet. 2021;25:301–312. doi: 10.26355/eurrev_202101_24396. [DOI] [PubMed] [Google Scholar]
- 221.van der Pouw Kraan TC, et al. Systemic toll-like receptor and interleukin-18 pathway activation in patients with acute ST elevation myocardial infarction. J. Mol. Cell. Cardiol. 2014;67:94–102. doi: 10.1016/j.yjmcc.2013.12.021. [DOI] [PubMed] [Google Scholar]
- 222.Sun JH, et al. Gentianella acuta prevents acute myocardial infarction induced by isoproterenol in rats via inhibition of galectin-3/TLR4/MyD88/NF-?B inflammatory signalling. Inflammopharmacology. 2021;29:205–219. doi: 10.1007/s10787-020-00708-4. [DOI] [PubMed] [Google Scholar]
- 223.Zaafan MA, Abdelhamid AM. The cardioprotective effect of astaxanthin against isoprenaline-induced myocardial injury in rats: involvement of TLR4/NF-κB signaling pathway. Eur. Rev. Med. Pharm. Sci. 2021;25:4099–4105. doi: 10.26355/eurrev_202106_26052. [DOI] [PubMed] [Google Scholar]
- 224.Shi H, Zhou P. Astragaloside IV prevents acute myocardial infarction by inhibiting the TLR4/MyD88/NF-κB signaling pathway. J. Food Biochem. 2021;45:e13757. doi: 10.1111/jfbc.13757. [DOI] [PubMed] [Google Scholar]
- 225.Wang X, et al. Danshen (Salvia miltiorrhiza) restricts MD2/TLR4-MyD88 complex formation and signalling in acute myocardial infarction-induced heart failure. J. Cell. Mol. Med. 2020;24:10677–10692. doi: 10.1111/jcmm.15688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Abdelzaher WY, et al. Dapsone ameliorates isoproterenol-induced myocardial infarction via Nrf2/ HO-1; TLR4/ TNF-α signaling pathways and the suppression of oxidative stress, inflammation, and apoptosis in rats. Front. Pharm. 2021;12:669679. doi: 10.3389/fphar.2021.669679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Fu H, Shuai W, Kong B, Jiang X, Huang H. MD1 depletion predisposes to ventricular arrhythmias in the setting of myocardial infarction. Heart Lung Circ. 2021;30:869–881. doi: 10.1016/j.hlc.2020.09.938. [DOI] [PubMed] [Google Scholar]
- 228.Chen QF, Wang W, Huang Z, Huang DL. Hypoxia-inducible factor-1α attenuates myocardial inflammatory injury in rats induced by coronary microembolization. Acad. Bras. Cienc. 2020;92:e20190658. doi: 10.1590/0001-3765202020190658. [DOI] [PubMed] [Google Scholar]
- 229.Moreira RS, et al. Synthetic apolipoprotein A-I mimetic peptide 4F protects hearts and kidneys after myocardial infarction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020;318:R529–R544. doi: 10.1152/ajpregu.00185.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Khorrami A. Oxidized cholesterol exacerbates toll-like receptor 4 expression and activity in the hearts of rats with myocardial infarction. Acta Cir. Bras. 2020;12:43–50. doi: 10.34172/jcvtr.2020.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ma Y, et al. Hyperlipidemia inhibits the protective effect of lisinopril after myocardial infarction via activation of dendritic cells. J. Cell. Mol. Med. 2020;24:4082–4091. doi: 10.1111/jcmm.15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sun K, et al. [Effect of electroacupuncture on expression of IL-23/IL-17 axis and TLR4 in infarcted tissue in rats with myocardial infarction] Zhongguo Zhen Jiu. 2021;41:1023–1028. doi: 10.13703/j.0255-2930.20200824-k0001. [DOI] [PubMed] [Google Scholar]
- 233.Wang X, Sun Q, Hu W. Carvedilol protects against the H2O2-induced cell damages in rat myoblasts by regulating the Circ_NFIX/miR-125b-5p/TLR4 signal axis. Oxid. Med. Cell. Longev. 2021;78:604–614. doi: 10.1097/FJC.0000000000001095. [DOI] [PubMed] [Google Scholar]
- 234.Zhang P, et al. miR-708 affords protective efficacy in anoxia/reoxygenation-stimulated cardiomyocytes by blocking the TLR4 signaling via targeting HMGB1. Sci. Rep. 2020;54:101653. doi: 10.1016/j.mcp.2020.101653. [DOI] [PubMed] [Google Scholar]
- 235.Guo LL, Guo ML. MicroRNA-421 improves ischemia/reperfusion injury via regulation toll-like receptor 4 pathway. J. Int. Med. Res. 2020;48:300060519871863. doi: 10.1177/0300060519871863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Biemmi V, et al. Inflammatory extracellular vesicles prompt heart dysfunction via TRL4-dependent NF-κB activation. Theranostics. 2020;10:2773–2790. doi: 10.7150/thno.39072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhao J, et al. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc. Res. 2019;115:1205–1216. doi: 10.1093/cvr/cvz040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ramirez-Carracedo R, et al. Targeting TLR4 with ApTOLL improves heart function in response to coronary ischemia reperfusion in pigs undergoing acute myocardial infarction. Biomolecules. 2020;10:1167. doi: 10.3390/biom10081167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Louwe MC, et al. RP105 deficiency aggravates cardiac dysfunction after myocardial infarction in mice. Int. J. Cardiol. 2014;176:788–793. doi: 10.1016/j.ijcard.2014.07.086. [DOI] [PubMed] [Google Scholar]
- 240.Fujiwara M, et al. Nanoparticle incorporating Toll-like receptor 4 inhibitor attenuates myocardial ischaemia-reperfusion injury by inhibiting monocyte-mediated inflammation in mice. Cardiovasc. Res. 2019;115:1244–1255. doi: 10.1093/cvr/cvz066. [DOI] [PubMed] [Google Scholar]
- 241.Soraya H, et al. Chronic treatment with metformin suppresses toll-like receptor 4 signaling and attenuates left ventricular dysfunction following myocardial infarction. Eur. J. Pharm. 2014;737:77–84. doi: 10.1016/j.ejphar.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 242.Maranhão RC, et al. Methotrexate carried in lipid core nanoparticles reduces myocardial infarction size and improves cardiac function in rats. Int. J. Nanomed. 2017;12:3767–3784. doi: 10.2147/IJN.S129324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Liu L, et al. Up-regulated TLR4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction. J. Cell. Mol. Med. 2015;19:2728–2740. doi: 10.1111/jcmm.12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hayes J, Dinkova-Kostova A. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 245.Oyake T, et al. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 1996;16:6083–6095. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Nguyen T, Nioi P, Pickett C. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ray P, Huang B, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kensler T, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1–Nrf2–ARE pathway. Annu. Rev. Pharm. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 249.Wei W, Ma N, Fan X, Yu Q, Ci X. The role of Nrf2 in acute kidney injury: novel molecular mechanisms and therapeutic approaches. Free Radic. Biol. Med. 2020;158:1–12. doi: 10.1016/j.freeradbiomed.2020.06.025. [DOI] [PubMed] [Google Scholar]
- 250.Zhang R, et al. Nrf2-a promising therapeutic target for defensing against oxidative stress in stroke. Mol. Neurobiol. 2017;54:6006–6017. doi: 10.1007/s12035-016-0111-0. [DOI] [PubMed] [Google Scholar]
- 251.Wu J, et al. The non-canonical effects of heme oxygenase-1, a classical fighter against oxidative stress. Redox Biol. 2021;47:102170. doi: 10.1016/j.redox.2021.102170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fernández-Fierro A, et al. Immune modulation by inhibitors of the HO system. Int. J. Mol. Sci. 2020;22:294. doi: 10.3390/ijms22010294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Peng P, Ko M, Chen C, Juan S. Haem oxygenase-1 gene transfer protects retinal ganglion cells from ischaemia/reperfusion injury. Clin. Sci. 2008;115:335–342. doi: 10.1042/CS20070384. [DOI] [PubMed] [Google Scholar]
- 254.Qiao Y, et al. Hepatocellular HO-1 mediated iNOS-induced hepatoprotection against liver ischemia reperfusion injury. Biochem. Biophys. Res. Commun. 2020;521:1095–1100. doi: 10.1016/j.bbrc.2019.11.053. [DOI] [PubMed] [Google Scholar]
- 255.Qi D, et al. HO-1 attenuates hippocampal neurons injury via the activation of BDNF-TrkB-PI3K/Akt signaling pathway in stroke. Brain Res. 2014;1577:69–76. doi: 10.1016/j.brainres.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 256.Ryter S. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019;678:108186. doi: 10.1016/j.abb.2019.108186. [DOI] [PubMed] [Google Scholar]
- 257.Jiang Z, et al. MicroRNA-200a improves diabetic endothelial dysfunction by targeting KEAP1/NRF2. J. Endocrinol. 2020;245:129–140. doi: 10.1530/JOE-19-0414. [DOI] [PubMed] [Google Scholar]
- 258.Durbin R. Letter: acid secretion by gastric mucous membrane. Am. J. Physiol. 1975;229:1726. doi: 10.1152/ajplegacy.1975.229.6.1726. [DOI] [PubMed] [Google Scholar]
- 259.Liu X, et al. Preemptive heme oxygenase-1 gene delivery reveals reduced mortality and preservation of left ventricular function 1 yr after acute myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H48–H59. doi: 10.1152/ajpheart.00741.2006. [DOI] [PubMed] [Google Scholar]
- 260.Melo LG, et al. Gene therapy strategy for long-term myocardial protection using adeno-associated virus-mediated delivery of heme oxygenase gene. Circulation. 2002;105:602–607. doi: 10.1161/hc0502.103363. [DOI] [PubMed] [Google Scholar]
- 261.Tu W, Wang H, Li S, Liu Q, Sha H. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019;10:637–651. doi: 10.14336/AD.2018.0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Liu F, et al. TLR9 is essential for HMGB1-mediated post-myocardial infarction tissue repair through affecting apoptosis, cardiac healing, and angiogenesis. Cell Death Dis. 2019;10:480. doi: 10.1038/s41419-019-1718-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Bei W, Jing L, Chen N. Cardio protective role of wogonin loaded nanoparticle against isoproterenol induced myocardial infarction by moderating oxidative stress and inflammation. Colloids Surf. B Biointerfaces. 2020;185:110635. doi: 10.1016/j.colsurfb.2019.110635. [DOI] [PubMed] [Google Scholar]
- 264.Zhang H, et al. Hirudin protects against isoproternol-induced myocardial infraction by alleviating oxidative via an Nrf2 dependent manner. Int. J. Biol. Macromol. 2020;162:425–435. doi: 10.1016/j.ijbiomac.2020.06.097. [DOI] [PubMed] [Google Scholar]
- 265.Baraka SA, et al. Rosuvastatin and low-dose carvedilol combination protects against isoprenaline-induced myocardial infarction in rats: Role of PI3K/Akt/Nrf2/HO-1 signalling. Clin. Exp. Pharm. Physiol. 2021;48:1358–1370. doi: 10.1111/1440-1681.13535. [DOI] [PubMed] [Google Scholar]
- 266.Yeh C, Chen T, Wang Y, Lin Y, Lin P. HO-1 activation can attenuate cardiomyocytic apoptosis via inhibition of NF-kappaB and AP-1 translocation following cardiac global ischemia and reperfusion. J. Surg. Res. 2009;155:147–156. doi: 10.1016/j.jss.2008.07.044. [DOI] [PubMed] [Google Scholar]
- 267.Kusmic C, et al. Up-regulation of heme oxygenase-1 after infarct initiation reduces mortality, infarct size and left ventricular remodeling: experimental evidence and proof of concept. J. Transl. Med. 2014;12:89. doi: 10.1186/1479-5876-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Cao J, et al. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostagland. Other Lipid Mediat. 2015;116-117:76–86. doi: 10.1016/j.prostaglandins.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Shafei, A. et al. Mesenchymal stem cell therapy: a promising cell-based therapy for treatment of myocardial infarction. J. Gene Med.19, (2017). [DOI] [PubMed]
- 270.Zeng B, Lin G, Ren X, Zhang Y, Chen H. Over-expression of HO-1 on mesenchymal stem cells promotes angiogenesis and improves myocardial function in infarcted myocardium. J. Biomed. Sci. 2010;17:80. doi: 10.1186/1423-0127-17-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Jiang YB, et al. Effects of heme oxygenase-1 gene modulated mesenchymal stem cells on vasculogenesis in ischemic swine hearts. Chin. Med. J. 2011;124:401–407. [PubMed] [Google Scholar]
- 272.Zeng B, et al. Paracrine action of HO-1-modified mesenchymal stem cells mediates cardiac protection and functional improvement. Cell Biol. Int. 2008;32:1256–1264. doi: 10.1016/j.cellbi.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 273.Scragg J, Dallas M, Wilkinson J, Varadi G, Peers C. Carbon monoxide inhibits L-type Ca2+ channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J. Biol. Chem. 2008;283:24412–24419. doi: 10.1074/jbc.M803037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Duckles H, et al. Heme oxygenase-1 regulates cell proliferation via carbon monoxide-mediated inhibition of T-type Ca2+ channels. Pflug. Arch. 2015;467:415–427. doi: 10.1007/s00424-014-1503-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Zhang X, et al. The Nrf-2/HO-1 signaling axis: a ray of hope in cardiovascular diseases. Cardiol. Res Pr. 2020;2020:5695723. doi: 10.1155/2020/5695723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Novo G, et al. Hsp60 and heme oxygenase-1 (Hsp32) in acute myocardial infarction. Transl. Res. 2011;157:285–292. doi: 10.1016/j.trsl.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 277.Chen SM, Li YG, Wang DM. Study on changes of heme oxygenase-1 expression in patients with coronary heart disease. Clin. Cardiol. 2005;28:197–201. doi: 10.1002/clc.4960280410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zheng H, Ma HP, Wang J, Ma M. Preoperative HO-1 levels as prognostic factor for adverse cardiac events in elder patients undergoing non-cardiac surgery. PLoS ONE. 2013;8:e58567. doi: 10.1371/journal.pone.0058567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kumar N, et al. Regulation of adipogenesis by natural and synthetic REV-ERB ligands. Endocrinology. 2010;151:3015–3025. doi: 10.1210/en.2009-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 281.Jaffe A, Hall A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 282.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol. Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 283.Dirac-Svejstrup A, Sumizawa T, Pfeffer S. Identification of a GDI displacement factor that releases endosomal Rab GTPases from Rab-GDI. EMBO J. 1997;16:465–472. doi: 10.1093/emboj/16.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Cappello S, et al. A radial glia-specific role of RhoA in double cortex formation. Neuron. 2012;73:911–924. doi: 10.1016/j.neuron.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 285.Kaarbø M, Crane D, Murrell W. RhoA is highly up-regulated in the process of early heart development of the chick and important for normal embryogenesis. Dev. Dyn. 2003;227:35–47. doi: 10.1002/dvdy.10283. [DOI] [PubMed] [Google Scholar]
- 286.Vicente-Steijn R, et al. RHOA-ROCK signalling is necessary for lateralization and differentiation of the developing sinoatrial node. Cardiovasc. Res. 2017;113:1186–1197. doi: 10.1093/cvr/cvx104. [DOI] [PubMed] [Google Scholar]
- 287.Lu J, et al. Coronary smooth muscle differentiation from proepicardial cells requires rhoA-mediated actin reorganization and p160 rho-kinase activity. Dev. Biol. 2001;240:404–418. doi: 10.1006/dbio.2001.0403. [DOI] [PubMed] [Google Scholar]
- 288.Cachero T, Morielli A, Peralta E. The small GTP-binding protein RhoA regulates a delayed rectifier potassium channel. Cell. 1998;93:1077–1085. doi: 10.1016/s0092-8674(00)81212-x. [DOI] [PubMed] [Google Scholar]
- 289.Olgar Y, et al. Rho-kinase inhibition reverses impaired Ca handling and associated left ventricular dysfunction in pressure overload-induced cardiac hypertrophy. Cell Calcium. 2017;67:81–90. doi: 10.1016/j.ceca.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 290.Abramochkin D, Filatova T, Pustovit K, Dzhumaniiazova I, Karpushev A. Small G-protein RhoA is a potential inhibitor of cardiac fast sodium current. J. Physiol. Biochem. 2021;77:13–23. doi: 10.1007/s13105-020-00774-w. [DOI] [PubMed] [Google Scholar]
- 291.Julian L, Olson M. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5:e29846. doi: 10.4161/sgtp.29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Shimizu T, Liao J. Rho kinases and cardiac remodeling. Circ. J. 2016;80:1491–1498. doi: 10.1253/circj.CJ-16-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Liao J, Seto M, Noma K. Rho kinase (ROCK) inhibitors. J. Cardiovasc. Pharm. 2007;50:17–24. doi: 10.1097/FJC.0b013e318070d1bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Shahbazi R, et al. Targeting ROCK signaling in health, malignant and non-malignant diseases. Immunol. Lett. 2020;219:15–26. doi: 10.1016/j.imlet.2019.12.012. [DOI] [PubMed] [Google Scholar]
- 295.Nakagawa O, et al. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 296.Matoba K, et al. The physiology, pathology, and therapeutic interventions for ROCK isoforms in diabetic kidney disease. Front. Pharm. 2020;11:585633. doi: 10.3389/fphar.2020.585633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Lambert J, Song W. Ozone-induced airway hyperresponsiveness: roles of ROCK isoforms. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015;309:L1394–L1397. doi: 10.1152/ajplung.00353.2015. [DOI] [PubMed] [Google Scholar]
- 298.Okamoto R, et al. FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J. 2013;27:1439–1449. doi: 10.1096/fj.12-217018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zhang Y, et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. FASEB J. 2006;20:916–925. doi: 10.1096/fj.05-5129com. [DOI] [PubMed] [Google Scholar]
- 300.Rikitake Y, et al. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation. 2005;112:2959–2965. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Surma M, Wei L, Shi J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol. 2011;7:657–671. doi: 10.2217/fca.11.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Miyamoto S, et al. Revisited and revised: is RhoA always a villain in cardiac pathophysiology? J. Cardiovasc. Transl. Res. 2010;3:330–343. doi: 10.1007/s12265-010-9192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Hartmann S, Ridley A, Lutz S. The function of Rho-associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front. Pharm. 2015;6:276. doi: 10.3389/fphar.2015.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Sui X, Wei H, Wang D. Novel mechanism of cardiac protection by valsartan: synergetic roles of TGF-β1 and HIF-1α in Ang II-mediated fibrosis after myocardial infarction. J. Cell. Mol. Med. 2015;19:1773–1782. doi: 10.1111/jcmm.12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Janbandhu V, et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell. 2021;29:281–297.e12. doi: 10.1016/j.stem.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Lee TM, Lin SZ, Chang NC. Membrane ERα attenuates myocardial fibrosis via RhoA/ROCK-mediated actin remodeling in ovariectomized female infarcted rats. J. Mol. Med. 2014;92:43–51. doi: 10.1007/s00109-013-1103-4. [DOI] [PubMed] [Google Scholar]
- 307.Lee TM, Lin SZ, Chang NC. Nicorandil regulates the macrophage skewing and ameliorates myofibroblasts by inhibition of RhoA/Rho-kinase signalling in infarcted rats. J. Cell. Mol. Med. 2018;22:1056–1069. doi: 10.1111/jcmm.13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Lorenz R, Wu J, Herberg F, Taylor S, Engh R. Drugging the undruggable: how isoquinolines and PKA initiated the era of designed protein kinase inhibitor therapeutics. Biochemistry. 2021;60:3470–3484. doi: 10.1021/acs.biochem.1c00359. [DOI] [PubMed] [Google Scholar]
- 309.Zhang J, Liu J, Li D, Zhang C, Liu M. Calcium antagonists for acute ischemic stroke. Cochrane Database Syst. Rev. 2019;2:CD001928. doi: 10.1002/14651858.CD001928.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Satoh S, Ikegaki I, Kawasaki K, Asano T, Shibuya M. Pleiotropic effects of the rho-kinase inhibitor fasudil after subarachnoid hemorrhage: a review of preclinical and clinical studies. Curr. Vasc. Pharm. 2014;12:758–765. doi: 10.2174/1570161112666140613115813. [DOI] [PubMed] [Google Scholar]
- 311.Li Q, Xu Y, Li X, Guo Y, Liu G. Inhibition of Rho-kinase ameliorates myocardial remodeling and fibrosis in pressure overload and myocardial infarction: role of TGF-β1-TAK1. Toxicol. Lett. 2012;211:91–97. doi: 10.1016/j.toxlet.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 312.Hattori T, et al. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation. 2004;109:2234–2239. doi: 10.1161/01.CIR.0000127939.16111.58. [DOI] [PubMed] [Google Scholar]
- 313.Kidder G, Montgomery C. Oxygenation of frog gastric mucosa in vitro. Am. J. Physiol. 1975;229:1510–1513. doi: 10.1152/ajplegacy.1975.229.6.1510. [DOI] [PubMed] [Google Scholar]
- 314.Zhao Y, Xu J. Sanggenon C ameliorates cerebral ischemia-reperfusion injury by inhibiting inflammation and oxidative stress through regulating RhoA-ROCK signaling. Inflammation. 2020;43:1476–1487. doi: 10.1007/s10753-020-01225-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Xiang S, et al. PLCε, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal. 2013;6:ra108. doi: 10.1126/scisignal.2004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Samsamshariat S, Movahed M. High rate of right ventricular infarction after ligation of mid left anterior descending artery in rats. Cardiovasc. Revasc. Med. 2005;6:21–23. doi: 10.1016/j.carrev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 317.Allawadhi P, Khurana A, Sayed N, Kumari P, Godugu C. Isoproterenol-induced cardiac ischemia and fibrosis: Plant-based approaches for intervention. Phytother. Res. 2018;32:1908–1932. doi: 10.1002/ptr.6152. [DOI] [PubMed] [Google Scholar]
- 318.Sun T, et al. Dexmedetomidine alleviates cardiomyocyte apoptosis and cardiac dysfunction may be associated with inhibition of RhoA/ROCK pathway in mice with myocardial infarction. Naunyn Schmiedebergs Arch. Pharm. 2021;394:1569–1577. doi: 10.1007/s00210-021-02082-6. [DOI] [PubMed] [Google Scholar]
- 319.Gong LL, et al. Coptisine exert cardioprotective effect through anti-oxidative and inhibition of RhoA/Rho kinase pathway on isoproterenol-induced myocardial infarction in rats. Atherosclerosis. 2012;222:50–58. doi: 10.1016/j.atherosclerosis.2012.01.046. [DOI] [PubMed] [Google Scholar]
- 320.Patel P, Parikh M, Shah H, Gandhi T. Inhibition of RhoA/Rho kinase by ibuprofen exerts cardioprotective effect on isoproterenol induced myocardial infarction in rats. Eur. J. Pharm. 2016;791:91–98. doi: 10.1016/j.ejphar.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 321.Zhou F, Ma K. Fasudil protects against isoproterenol-induced myocardial infarction in mice via inhibiting Rho/ROCK signaling pathway. Eur. Rev. Med. Pharm. Sci. 2020;24:5659–5667. doi: 10.26355/eurrev_202005_21357. [DOI] [PubMed] [Google Scholar]
- 322.Bulhak A, Roy J, Hedin U, Sjöquist PO, Pernow J. Cardioprotective effect of rosuvastatin in vivo is dependent on inhibition of geranylgeranyl pyrophosphate and altered RhoA membrane translocation. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H3158–H3163. doi: 10.1152/ajpheart.01354.2006. [DOI] [PubMed] [Google Scholar]
- 323.Yi Z, Ke J, Wang Y, Cai K. Fluvastatin protects myocardial cells in mice with acute myocardial infarction through inhibiting RhoA/ROCK pathway. Exp. Ther. Med. 2020;19:2095–2102. doi: 10.3892/etm.2020.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.du Souich P, Roederer G, Dufour R. Myotoxicity of statins: mechanism of action. Pharm. Ther. 2017;175:1–16. doi: 10.1016/j.pharmthera.2017.02.029. [DOI] [PubMed] [Google Scholar]
- 325.Cheng Y, et al. Rho-associated kinase inhibitors promote the cardiac differentiation of embryonic and induced pluripotent stem cells. Int. J. Cardiol. 2015;201:441–448. doi: 10.1016/j.ijcard.2015.08.118. [DOI] [PubMed] [Google Scholar]
- 326.Zhang Q, et al. Atorvastatin treatment improves the effects of mesenchymal stem cell transplantation on acute myocardial infarction: the role of the RhoA/ROCK/ERK pathway. Int. J. Cardiol. 2014;176:670–679. doi: 10.1016/j.ijcard.2014.07.071. [DOI] [PubMed] [Google Scholar]
- 327.Vicente-Manzanares M, Ma X, Adelstein R, Horwitz A. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 330.Muslin AJ. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin. Sci. 2008;115:203–218. doi: 10.1042/CS20070430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Perreault S, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway: TRAM-01. BMC Cancer. 2019;19:1250. doi: 10.1186/s12885-019-6442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Diamond EL, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567:521–524. doi: 10.1038/s41586-019-1012-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Ogut O, Brozovich FV. The potential role of MLC phosphatase and MAPK signalling in the pathogenesis of vascular dysfunction in heart failure. J. Cell. Mol. Med. 2008;12:2158–2164. doi: 10.1111/j.1582-4934.2008.00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Hayakawa K, et al. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation. 2003;108:104–109. doi: 10.1161/01.CIR.0000074225.62168.68. [DOI] [PubMed] [Google Scholar]
- 335.Lu Y, et al. Kuanxiong aerosol inhibits apoptosis and attenuates isoproterenol-induced myocardial injury through the mitogen-activated protein kinase pathway. J. Ethnopharmacol. 2021;269:113757. doi: 10.1016/j.jep.2020.113757. [DOI] [PubMed] [Google Scholar]
- 336.Zeng HT, Zhao M, Zhang ZX, Liu ZL, Zhong SM. Atorvastatin improves the cardiac function of rats after acute myocardial infarction through ERK1/2 pathway. Eur. Rev. Med. Pharm. Sci. 2019;23:7120–7127. doi: 10.26355/eurrev_201908_18757. [DOI] [PubMed] [Google Scholar]
- 337.Wang X, Song Q. Mst1 regulates post-infarction cardiac injury through the JNK–Drp1–mitochondrial fission pathway. Cell. Mol. Biol. Lett. 2018;23:21. doi: 10.1186/s11658-018-0085-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol. Rev. 2010;90:1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Eid RA, et al. Ghrelin prevents cardiac cell apoptosis during cardiac remodelling post experimentally induced myocardial infarction in rats via activation of Raf-MEK1/2-ERK1/2 signalling. Arch. Physiol. Biochem. 2019;125:93–103. doi: 10.1080/13813455.2018.1437751. [DOI] [PubMed] [Google Scholar]
- 340.Spivak JL. Erythropoietin. Blood Rev. 1989;3:130–135. doi: 10.1016/0268-960x(89)90008-8. [DOI] [PubMed] [Google Scholar]
- 341.Li GQ, Chen M. Cardioprotective effect of erythropoietin in rats with acute myocardial infarction through JNK pathway. Eur. Rev. Med. Pharm. Sci. 2019;23:153–160. doi: 10.26355/eurrev_201901_18642. [DOI] [PubMed] [Google Scholar]
- 342.Holobotovskyy V, et al. Regulator of G-protein signaling 5 controls blood pressure homeostasis and vessel wall remodeling. Circ. Res. 2013;112:781–791. doi: 10.1161/CIRCRESAHA.111.300142. [DOI] [PubMed] [Google Scholar]
- 343.Hong K, Li M, Nourian Z, Meininger GA, Hill MA. Angiotensin II Type 1 receptor mechanoactivation involves RGS5 (regulator of G protein signaling 5) in skeletal muscle arteries: impaired trafficking of RGS5 in Hypertension. Hypertension. 2017;70:1264–1272. doi: 10.1161/HYPERTENSIONAHA.117.09757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Ding HS, et al. Regulator of G-protein signalling 5 deficiency impairs ventricular remodelling after myocardial infarction by promoting NF-κB and MAPK signalling in mice. Biochem. Biophys. Res. Commun. 2018;499:143–149. doi: 10.1016/j.bbrc.2018.03.082. [DOI] [PubMed] [Google Scholar]
- 345.Humeres C, Frangogiannis NG. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl. Sci. 2019;4:449–467. doi: 10.1016/j.jacbts.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Li Y, et al. Sphingosylphosphorylcholine alleviates hypoxia-caused apoptosis in cardiac myofibroblasts via CaM/p38/STAT3 pathway. Apoptosis. 2020;25:853–863. doi: 10.1007/s10495-020-01639-9. [DOI] [PubMed] [Google Scholar]
- 347.Wang Y, Sun X. The functions of LncRNA in the heart. Diabetes Res. Clin. Pract. 2020;168:108249. doi: 10.1016/j.diabres.2020.108249. [DOI] [PubMed] [Google Scholar]
- 348.Song R, Hu XQ, Zhang L. Mitochondrial MiRNA in cardiovascular function and disease. Cells. 2019;8:1475. doi: 10.3390/cells8121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Hui J, et al. miR-539 as a key negative regulator of the MEK pathway in myocardial infarction. Herz. 2017;42:781–789. doi: 10.1007/s00059-016-4517-2. [DOI] [PubMed] [Google Scholar]
- 350.Qiao GH, Zhu P, Yue L, Yue S. MiR-125b improves acute myocardial infarction in rats by regulating P38/Sirtl/P53 signaling pathway. J. Biol. Regul. Homeost. Agents. 2020;34:1297–1306. doi: 10.23812/20-177-A. [DOI] [PubMed] [Google Scholar]
- 351.Fan YZ, Huang H, Wang S, Tan GJ, Zhang QZ. Effect of lncRNA MALAT1 on rats with myocardial infarction through regulating ERK/MAPK signaling pathway. Eur. Rev. Med. Pharm. Sci. 2019;23:9041–9049. doi: 10.26355/eurrev_201910_19306. [DOI] [PubMed] [Google Scholar]
- 352.Bostan MM, et al. Post-myocardial infarction ventricular remodeling biomarkers-the key link between pathophysiology and clinic. Biomolecules. 2020;10:1587. doi: 10.3390/biom10111587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Paul S. Ventricular remodeling. Crit. Care Nurs. Clin. North Am. 2003;15:407–411. doi: 10.1016/s0899-5885(02)00089-8. [DOI] [PubMed] [Google Scholar]
- 354.Gao Y, et al. ANO1 inhibits cardiac fibrosis after myocardial infraction via TGF-β/smad3 pathway. Sci. Rep. 2017;7:2355. doi: 10.1038/s41598-017-02585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Tian X, et al. ANO1 regulates cardiac fibrosis via ATI-mediated MAPK pathway. Cell Calcium. 2020;92:102306. doi: 10.1016/j.ceca.2020.102306. [DOI] [PubMed] [Google Scholar]
- 356.Li C, et al. MicroRNA-143-3p promotes human cardiac fibrosis via targeting sprouty3 after myocardial infarction. J. Mol. Cell. Cardiol. 2019;129:281–292. doi: 10.1016/j.yjmcc.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 357.Prodromou C. Mechanisms of Hsp90 regulation. Biochem. J. 2016;473:2439–2452. doi: 10.1042/BCJ20160005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Tamura S, Marunouchi T, Tanonaka K. Heat-shock protein 90 modulates cardiac ventricular hypertrophy via activation of MAPK pathway. J. Mol. Cell. Cardiol. 2019;127:134–142. doi: 10.1016/j.yjmcc.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 359.Yeh CC, et al. Shift toward greater pathologic post-myocardial infarction remodeling with loss of the adaptive hypertrophic signaling of alpha1 adrenergic receptors in mice. PLoS ONE. 2017;12:e0188471. doi: 10.1371/journal.pone.0188471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Ong SB, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharm. Ther. 2018;186:73–87. doi: 10.1016/j.pharmthera.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Yeung YT, Aziz F, Guerrero-Castilla A, Arguelles S. Signaling pathways in inflammation and anti-inflammatory therapies. Curr. Pharm. Des. 2018;24:1449–1484. doi: 10.2174/1381612824666180327165604. [DOI] [PubMed] [Google Scholar]
- 362.Duan J, Yang Y, Liu H, Dou PC, Tan SY. Osthole ameliorates acute myocardial infarction in rats by decreasing the expression of inflammatory-related cytokines, diminishing MMP-2 expression and activating p-ERK. Int. J. Mol. Med. 2016;37:207–216. doi: 10.3892/ijmm.2015.2402. [DOI] [PubMed] [Google Scholar]
- 363.Verma VK, et al. Role of MAPK/NF-κB pathway in cardioprotective effect of Morin in isoproterenol induced myocardial injury in rats. Mol. Biol. Rep. 2019;46:1139–1148. doi: 10.1007/s11033-018-04575-9. [DOI] [PubMed] [Google Scholar]
- 364.Zhang J, et al. CXCR7 suppression modulates macrophage phenotype and function to ameliorate post-myocardial infarction injury. Inflamm. Res. 2020;69:523–532. doi: 10.1007/s00011-020-01335-z. [DOI] [PubMed] [Google Scholar]
- 365.Ge ZW, et al. MicroRNA-26b relieves inflammatory response and myocardial remodeling of mice with myocardial infarction by suppression of MAPK pathway through binding to PTGS2. Int. J. Cardiol. 2019;280:152–159. doi: 10.1016/j.ijcard.2018.12.077. [DOI] [PubMed] [Google Scholar]
- 366.Peng L, et al. The effect of pulsed electromagnetic fields on angiogenesis. Bioelectromagnetics. 2021;42:250–258. doi: 10.1002/bem.22330. [DOI] [PubMed] [Google Scholar]
- 367.Feng X, et al. Danhong injection in cardiovascular and cerebrovascular diseases: pharmacological actions, molecular mechanisms, and therapeutic potential. Pharm. Res. 2019;139:62–75. doi: 10.1016/j.phrs.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 368.Li SN, et al. Danhong injection enhances angiogenesis after myocardial infarction by activating MiR-126/ERK/VEGF pathway. Biomed. Pharmacother. 2019;120:109538. doi: 10.1016/j.biopha.2019.109538. [DOI] [PubMed] [Google Scholar]
- 369.Routledge D, Scholpp S. Mechanisms of intercellular Wnt transport. Development. 2019;146:dev176073. doi: 10.1242/dev.176073. [DOI] [PubMed] [Google Scholar]
- 370.Wang J, et al. WNT11-conditioned medium promotes angiogenesis through the activation of non-canonical WNT–PKC–JNK signaling pathway. Genes. 2020;11:1277. doi: 10.3390/genes11111277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Cai Y, Xie KL, Wu HL, Wu K. Functional suppression of Epiregulin impairs angiogenesis and aggravates left ventricular remodeling by disrupting the extracellular-signal-regulated kinase1/2 signaling pathway in rats after acute myocardial infarction. J. Cell. Physiol. 2019;234:18653–18665. doi: 10.1002/jcp.28503. [DOI] [PubMed] [Google Scholar]
- 372.Cao Y, et al. Macrophage-specific IκB kinase α contributes to ventricular remodelling and dysfunction after myocardial infarction. Can. J. Cardiol. 2019;35:490–500. doi: 10.1016/j.cjca.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 373.Wang D, Bai L, Cui XR, Yang XH, Zhang JD. Effectiveness of atorvastatin in the treatment of asymptomatic heart failure after myocardial infarction: a Clinical Study. Adv. Ther. 2020;37:4649–4659. doi: 10.1007/s12325-020-01441-8. [DOI] [PubMed] [Google Scholar]
- 374.Teshima Y, et al. Early atorvastatin therapy improves cardiac function in patients with acute myocardial infarction. J. Cardiol. 2009;53:58–64. doi: 10.1016/j.jjcc.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 375.Newby LK, et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet. 2014;384:1187–1195. doi: 10.1016/S0140-6736(14)60417-7. [DOI] [PubMed] [Google Scholar]
- 376.O’Donoghue ML, et al. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: a Randomized Clinical Trial. JAMA. 2016;315:1591–1599. doi: 10.1001/jama.2016.3609. [DOI] [PubMed] [Google Scholar]
- 377.Cavender MA, et al. Inhibition of p38 MAP kinase in patients with ST-elevation myocardial infarction—findings from the LATITUDE-TIMI 60 trial. Am. Heart J. 2022;243:147–157. doi: 10.1016/j.ahj.2021.08.022. [DOI] [PubMed] [Google Scholar]
- 378.O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–S131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 379.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 380.Bolli R, Dawn B, Xuan YT. Role of the JAK–STAT pathway in protection against myocardial ischemia/reperfusion injury. Trends Cardiovasc. Med. 2003;13:72–79. doi: 10.1016/s1050-1738(02)00230-x. [DOI] [PubMed] [Google Scholar]
- 381.O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–550. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Xin P, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020;80:106210. doi: 10.1016/j.intimp.2020.106210. [DOI] [PubMed] [Google Scholar]
- 383.Crispino N, Ciccia F. JAK/STAT pathway and nociceptive cytokine signalling in rheumatoid arthritis and psoriatic arthritis. Clin. Exp. Rheumatol. 2021;39:668–675. [PubMed] [Google Scholar]
- 384.Qin H, et al. Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J. Neurosci. 2016;36:5144–5159. doi: 10.1523/JNEUROSCI.4658-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Hatami M, et al. STAT5a and STAT6 gene expression levels in multiple sclerosis patients. Cytokine. 2018;106:108–113. doi: 10.1016/j.cyto.2017.10.022. [DOI] [PubMed] [Google Scholar]
- 386.Kowshik J, et al. Astaxanthin inhibits JAK/STAT-3 signaling to abrogate cell proliferation, invasion and angiogenesis in a hamster model of oral cancer. PLoS ONE. 2014;9:e109114. doi: 10.1371/journal.pone.0109114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Barry SP, Townsend PA, Latchman DS, Stephanou A. Role of the JAK–STAT pathway in myocardial injury. Trends Mol. Med. 2007;13:82–89. doi: 10.1016/j.molmed.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 388.Zhu J, et al. miR-181a and miR-150 regulate dendritic cell immune inflammatory responses and cardiomyocyte apoptosis via targeting JAK1-STAT1/c-Fos pathway. J. Cell. Mol. Med. 2017;21:2884–2895. doi: 10.1111/jcmm.13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Stephanou A, et al. Ischemia-induced STAT-1 expression and activation play a critical role in cardiomyocyte apoptosis. J. Biol. Chem. 2000;275:10002–10008. doi: 10.1074/jbc.275.14.10002. [DOI] [PubMed] [Google Scholar]
- 390.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharm. Ther. 2008;120:172–185. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 391.You L, Li L, Xu Q, Ren J, Zhang F. Postconditioning reduces infarct size and cardiac myocyte apoptosis via the opioid receptor and JAK–STAT signaling pathway. Mol. Biol. Rep. 2011;38:437–443. doi: 10.1007/s11033-010-0126-y. [DOI] [PubMed] [Google Scholar]
- 392.Frangogiannis NG. Pathophysiology of myocardial infarction. Compr. Physiol. 2015;5:1841–1875. doi: 10.1002/cphy.c150006. [DOI] [PubMed] [Google Scholar]
- 393.Negoro S, et al. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc. Res. 2000;47:797–805. doi: 10.1016/s0008-6363(00)00138-3. [DOI] [PubMed] [Google Scholar]
- 394.Hilfiker-Kleiner D, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004;95:187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 395.Li J, et al. IL33 attenuates ventricular remodeling after myocardial infarction through inducing alternatively activated macrophages ethical standards statement. Eur. J. Pharm. 2019;854:307–319. doi: 10.1016/j.ejphar.2019.04.046. [DOI] [PubMed] [Google Scholar]
- 396.Kang HJ, Kim HS. G-CSF- and erythropoietin-based cell therapy: a promising strategy for angiomyogenesis in myocardial infarction. Expert Rev. Cardiovasc. Ther. 2008;6:703–713. doi: 10.1586/14779072.6.5.703. [DOI] [PubMed] [Google Scholar]
- 397.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]
- 398.Verhoeven Y, et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020;60:41–56. doi: 10.1016/j.semcancer.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 399.Groner B, von Manstein V. Jak Stat signaling and cancer: opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017;451:1–14. doi: 10.1016/j.mce.2017.05.033. [DOI] [PubMed] [Google Scholar]
- 400.Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J. Cardiovasc. Pharm. 2007;50:126–141. doi: 10.1097/FJC.0b013e318068dd49. [DOI] [PubMed] [Google Scholar]
- 401.Lentzsch S, et al. PI3-K/AKT/FKHR and MAPK signaling cascades are redundantly stimulated by a variety of cytokines and contribute independently to proliferation and survival of multiple myeloma cells. Leukemia. 2004;18:1883–1890. doi: 10.1038/sj.leu.2403486. [DOI] [PubMed] [Google Scholar]
- 402.Wang Y, et al. Receptor subunit-specific action of oncostatin M in hepatic cells and its modulation by leukemia inhibitory factor. J. Biol. Chem. 2000;275:25273–25285. doi: 10.1074/jbc.M002296200. [DOI] [PubMed] [Google Scholar]
- 403.Nakaoka Y, et al. Activation of gp130 transduces hypertrophic signal through interaction of scaffolding/docking protein Gab1 with tyrosine phosphatase SHP2 in cardiomyocytes. Circ. Res. 2003;93:221–229. doi: 10.1161/01.RES.0000085562.48906.4A. [DOI] [PubMed] [Google Scholar]
- 404.Savitz SI, Kessler JA. Leukemia inhibitory factor requires concurrent p75LNTR signaling to induce apoptosis of cultured sympathetic neurons. J. Neurosci. 2000;20:4198–4205. doi: 10.1523/JNEUROSCI.20-11-04198.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Sawashita Y, et al. Remote ischemic preconditioning reduces myocardial ischemia-reperfusion injury through unacylated ghrelin-induced activation of the JAK/STAT pathway. Basic Res. Cardiol. 2020;115:50. doi: 10.1007/s00395-020-0809-z. [DOI] [PubMed] [Google Scholar]
- 406.Mudaliar H, et al. Remote ischemic preconditioning attenuates EGR-1 expression following myocardial ischemia reperfusion injury through activation of the JAK-STAT pathway. Int. J. Cardiol. 2017;228:729–741. doi: 10.1016/j.ijcard.2016.11.198. [DOI] [PubMed] [Google Scholar]
- 407.Yu X, Kennedy RH, Liu SJ. JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J. Biol. Chem. 2003;278:16304–16309. doi: 10.1074/jbc.M212321200. [DOI] [PubMed] [Google Scholar]
- 408.McCormick J, et al. Free radical scavenging inhibits STAT phosphorylation following in vivo ischemia/reperfusion injury. FASEB J. 2006;20:2115–2117. doi: 10.1096/fj.06-6188fje. [DOI] [PubMed] [Google Scholar]
- 409.Kang HJ, et al. Intracoronary infusion of the mobilized peripheral blood stem cell by G-CSF is better than mobilization alone by G-CSF for improvement of cardiac function and remodeling: 2-year follow-up results of the Myocardial Regeneration and Angiogenesis in Myocardial Infarction with G-CSF and Intra-Coronary Stem Cell Infusion (MAGIC Cell) 1 trial. Am. Heart J. 2007;153:237.e231–238. doi: 10.1016/j.ahj.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 410.Hinck AP, Mueller TD, Springer TA. Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2016;8:a022103. doi: 10.1101/cshperspect.a022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Zhao H, Wei J, Sun J. Roles of TGF-β signaling pathway in tumor microenvirionment and cancer therapy. Int. Immunopharmacol. 2020;89:107101. doi: 10.1016/j.intimp.2020.107101. [DOI] [PubMed] [Google Scholar]
- 412.Syed V. TGF-β signaling in cancer. J. Cell. Biochem. 2016;117:1279–1287. doi: 10.1002/jcb.25496. [DOI] [PubMed] [Google Scholar]
- 413.Hata A, Chen YG. TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016;8:a022061. doi: 10.1101/cshperspect.a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 415.Morikawa M, Derynck R. & Miyazono, K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016;8:a021873. doi: 10.1101/cshperspect.a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Colak S, Ten Dijke P. Targeting TGF-β signaling in Cancer. Trends Cancer. 2017;3:56–71. doi: 10.1016/j.trecan.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 417.Zhang X, et al. TGF-β pathway in salivary gland fibrosis. Int. J. Mol. Sci. 2020;21:9138. doi: 10.3390/ijms21239138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Rico MC, Rough JJ, Del Carpio-Cano FE, Kunapuli SP, DeLa Cadena RA. The axis of thrombospondin-1, transforming growth factor beta and connective tissue growth factor: an emerging therapeutic target in rheumatoid arthritis. Curr. Vasc. Pharm. 2010;8:338–343. doi: 10.2174/157016110791112296. [DOI] [PubMed] [Google Scholar]
- 419.Euler G. Good and bad sides of TGFβ-signaling in myocardial infarction. Front. Physiol. 2015;6:66. doi: 10.3389/fphys.2015.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Györfi AH, Matei AE, Distler JHW. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018;68-69:8–27. doi: 10.1016/j.matbio.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 421.Hu HH, et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018;292:76–83. doi: 10.1016/j.cbi.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 422.Khalil H, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017;127:3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Li W, et al. CGRP derived from cardiac fibroblasts is an endogenous suppressor of cardiac fibrosis. Cardiovasc. Res. 2020;116:1335–1348. doi: 10.1093/cvr/cvz234. [DOI] [PubMed] [Google Scholar]
- 424.Jiang C, et al. Xanthohumol inhibits TGF-β1-induced cardiac fibroblasts activation via mediating PTEN/Akt/mTOR signaling pathway. Drug Des. Dev. Ther. 2020;14:5431–5439. doi: 10.2147/DDDT.S282206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Zong J, et al. NLRP1 promotes TGF-β1-induced myofibroblast differentiation in neonatal rat cardiac fibroblasts. J. Mol. Histol. 2018;49:509–518. doi: 10.1007/s10735-018-9789-9. [DOI] [PubMed] [Google Scholar]
- 426.Piek A, de Boer RA, Silljé HH. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016;21:199–211. doi: 10.1007/s10741-016-9536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Hanna A, Frangogiannis NG. The role of the TGF-β superfamily in myocardial infarction. Front. Cardiovasc. Med. 2019;6:140. doi: 10.3389/fcvm.2019.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Fu X, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018;128:2127–2143. doi: 10.1172/JCI98215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Shinde AV, Humeres C, Frangogiannis NG. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:298–309. doi: 10.1016/j.bbadis.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Wang M, et al. GHSR deficiency exacerbates cardiac fibrosis: role in macrophage inflammasome activation and myofibroblast differentiation. Cardiovasc. Res. 2020;116:2091–2102. doi: 10.1093/cvr/cvz318. [DOI] [PubMed] [Google Scholar]
- 431.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Yao Y, et al. ADAMTS16 activates latent TGF-β, accentuating fibrosis and dysfunction of the pressure-overloaded heart. Cardiovasc. Res. 2020;116:956–969. doi: 10.1093/cvr/cvz187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Friebel J, et al. Protease-activated receptor 2 deficiency mediates cardiac fibrosis and diastolic dysfunction. Eur. Heart J. 2019;40:3318–3332. doi: 10.1093/eurheartj/ehz117. [DOI] [PubMed] [Google Scholar]
- 434.Xiao X, et al. Simvastatin ameliorates ventricular remodeling via the TGF-β1 signaling pathway in rats following myocardial infarction. Mol. Med. Rep. 2016;13:5093–5101. doi: 10.3892/mmr.2016.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Liu Y, et al. Combination of LCZ696 and ACEI further improves heart failure and myocardial fibrosis after acute myocardial infarction in mice. Biomed. Pharmacother. 2021;133:110824. doi: 10.1016/j.biopha.2020.110824. [DOI] [PubMed] [Google Scholar]
- 436.Mehdipoor M, Damirchi A, Razavi Tousi SMT, Babaei P. Concurrent vitamin D supplementation and exercise training improve cardiac fibrosis via TGF-β/Smad signaling in myocardial infarction model of rats. J. Physiol. Biochem. 2021;77:75–84. doi: 10.1007/s13105-020-00778-6. [DOI] [PubMed] [Google Scholar]
- 437.Gao H, et al. Salvanic acid B inhibits myocardial fibrosis through regulating TGF-β1/Smad signaling pathway. Biomed. Pharmacother. 2019;110:685–691. doi: 10.1016/j.biopha.2018.11.098. [DOI] [PubMed] [Google Scholar]
- 438.Chen R, Chen W, Huang X, Rui Q. Tanshinone IIA attenuates heart failure via inhibiting oxidative stress in myocardial infarction rats. Mol. Med. Rep. 2021;23:404. doi: 10.3892/mmr.2021.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Yu Y, Sun J, Liu J, Wang P, Wang C. Ginsenoside re preserves cardiac function and ameliorates left ventricular remodeling in a rat model of myocardial infarction. J. Cardiovasc. Pharm. 2020;75:91–97. doi: 10.1097/FJC.0000000000000752. [DOI] [PubMed] [Google Scholar]
- 440.Scimia MC, Gumpert AM, Koch WJ. Cardiovascular gene therapy for myocardial infarction. Expert Opin. Biol. Ther. 2014;14:183–195. doi: 10.1517/14712598.2014.866085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Wang S, et al. rLj-RGD3, a novel recombinant toxin protein from Lampetra japonica, prevents coronary thrombosis-induced acute myocardial infarction by inhibiting platelet functions in rats. Biochem. Biophys. Res. Commun. 2018;498:240–245. doi: 10.1016/j.bbrc.2018.02.021. [DOI] [PubMed] [Google Scholar]
- 442.Yang W, et al. Gut microbe-derived metabolite trimethylamine N-oxide accelerates fibroblast-myofibroblast differentiation and induces cardiac fibrosis. J. Mol. Cell. Cardiol. 2019;134:119–130. doi: 10.1016/j.yjmcc.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 443.Philip JL, Xu X, Han M, Akhter SA, Razzaque MA. Regulation of cardiac fibroblast-mediated maladaptive ventricular remodeling by β-arrestins. PLoS ONE. 2019;14:e0219011. doi: 10.1371/journal.pone.0219011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Kim J, et al. Cytokine-Like 1 regulates cardiac fibrosis via modulation of TGF-β signaling. PLoS ONE. 2016;11:e0166480. doi: 10.1371/journal.pone.0166480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Liu M, et al. CTRP9 ameliorates atrial inflammation, fibrosis, and vulnerability to atrial fibrillation in post-myocardial infarction rats. J. Am. Heart Assoc. 2019;8:e013133. doi: 10.1161/JAHA.119.013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Weng L, et al. Nogo-C regulates post myocardial infarction fibrosis through the interaction with ER Ca(2+) leakage channel Sec61α in mouse hearts. Cell Death Dis. 2018;9:612. doi: 10.1038/s41419-018-0598-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Jin Y, Cheng X, Lu J, Li X. Exogenous BMP-7 facilitates the recovery of cardiac function after acute myocardial infarction through counteracting TGF-β1 signaling pathway. Tohoku J. Exp. Med. 2018;244:1–6. doi: 10.1620/tjem.244.1. [DOI] [PubMed] [Google Scholar]
- 448.Zhang M, et al. Notch3 ameliorates cardiac fibrosis after myocardial infarction by inhibiting the TGF-β1/Smad3 pathway. Cardiovasc. Toxicol. 2016;16:316–324. doi: 10.1007/s12012-015-9341-z. [DOI] [PubMed] [Google Scholar]
- 449.Saadat S, et al. Pivotal role of TGF-β/Smad signaling in cardiac fibrosis: non-coding RNAs as effectual players. Front. Cardiovasc. Med. 2020;7:588347. doi: 10.3389/fcvm.2020.588347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Wang P, et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-β/Smad3 pathway. Sci. Transl. Med. 2018;10:eaat2039. doi: 10.1126/scitranslmed.aat2039. [DOI] [PubMed] [Google Scholar]
- 451.Suzuki HI. MicroRNA control of TGF-β signaling. Int. J. Mol. Sci. 2018;19:1901. doi: 10.3390/ijms19071901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Wang DM, Jin JJ, Tian LM, Zhang Z. MiR-195 promotes myocardial fibrosis in MI rats via targeting TGF-β1/Smad. J. Biol. Regul. Homeost. Agents. 2020;34:1325–1332. doi: 10.23812/20-201-A. [DOI] [PubMed] [Google Scholar]
- 453.Chu X, et al. miR-130 aggravates acute myocardial infarction-induced myocardial injury by targeting PPAR-γ. J. Cell. Biochem. 2018;119:7235–7244. doi: 10.1002/jcb.26903. [DOI] [PubMed] [Google Scholar]
- 454.Bai M, Pan CL, Jiang GX, Zhang YM. CircRNA 010567 improves myocardial infarction rats through inhibiting TGF-β1. Eur. Rev. Med. Pharm. Sci. 2020;24:369–375. doi: 10.26355/eurrev_202001_19935. [DOI] [PubMed] [Google Scholar]
- 455.Huang S, et al. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J. Cell. Physiol. 2019;234:2997–3006. doi: 10.1002/jcp.27117. [DOI] [PubMed] [Google Scholar]
- 456.Yu BT, et al. Role of miR-133a in regulating TGF-β1 signaling pathway in myocardial fibrosis after acute myocardial infarction in rats. Eur. Rev. Med. Pharm. Sci. 2019;23:8588–8597. doi: 10.26355/eurrev_201910_19175. [DOI] [PubMed] [Google Scholar]
- 457.Xu HM, Sui FH, Sun MH, Guo GL. Downregulated microRNA-224 aggravates vulnerable atherosclerotic plaques and vascular remodeling in acute coronary syndrome through activation of the TGF-β/Smad pathway. J. Cell. Physiol. 2019;234:2537–2551. doi: 10.1002/jcp.26945. [DOI] [PubMed] [Google Scholar]
- 458.Chen G, Huang S, Song F, Zhou Y, He X. Lnc-Ang362 is a pro-fibrotic long non-coding RNA promoting cardiac fibrosis after myocardial infarction by suppressing Smad7. Arch. Biochem. Biophys. 2020;685:108354. doi: 10.1016/j.abb.2020.108354. [DOI] [PubMed] [Google Scholar]
- 459.Li W, et al. Epigenetic control of circHNRNPH1 in postischemic myocardial fibrosis through targeting of TGF-β receptor type I. Mol. Ther. Nucleic Acids. 2021;25:93–104. doi: 10.1016/j.omtn.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Trounson A, McDonald C. Stem cell therapies in clinical trials: progress and challenges. Cell Stem Cell. 2015;17:11–22. doi: 10.1016/j.stem.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 461.Wei X, Zheng Y, Zhang W, Tan J, Zheng H. Ultrasound-targeted microbubble destruction-mediated Galectin-7-siRNA promotes the homing of bone marrow mesenchymal stem cells to alleviate acute myocardial infarction in rats. Int. J. Mol. Med. 2021;47:677–687. doi: 10.3892/ijmm.2020.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Chen P, et al. Hypoxia preconditioned mesenchymal stem cells prevent cardiac fibroblast activation and collagen production via leptin. PLoS ONE. 2014;9:e103587. doi: 10.1371/journal.pone.0103587. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 463.Hou J, et al. Peroxisome proliferator-activated receptor gamma promotes mesenchymal stem cells to express Connexin43 via the inhibition of TGF-β1/Smads signaling in a rat model of myocardial infarction. Stem Cell Rev. Rep. 2015;11:885–899. doi: 10.1007/s12015-015-9615-7. [DOI] [PubMed] [Google Scholar]
- 464.Wu Y, et al. PGAM1 deficiency ameliorates myocardial infarction remodeling by targeting TGF-β via the suppression of inflammation, apoptosis and fibrosis. Biochem. Biophys. Res. Commun. 2021;534:933–940. doi: 10.1016/j.bbrc.2020.10.070. [DOI] [PubMed] [Google Scholar]
- 465.Zhang JW, et al. MiR-808 inhibits cardiomyocyte apoptosis and expressions of caspase-3 and caspase-9 in rats with myocardial infarction by regulating TGF-β1 signaling pathway. Eur. Rev. Med. Pharm. Sci. 2020;24:6955–6960. doi: 10.26355/eurrev_202006_21687. [DOI] [PubMed] [Google Scholar]
- 466.Tian Z, Zhang Y, Lyu X. Promoting roles of KLF5 in myocardial infarction in mice involving microRNA-27a suppression and the following GFPT2/TGF-β/Smad2/3 axis activation. Cell Cycle. 2021;20:874–893. doi: 10.1080/15384101.2021.1907512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Wang X, et al. Adipose-derived mesenchymal stem cells-derived exosomes carry MicroRNA-671 to alleviate myocardial infarction through inactivating the TGFBR2/Smad2 Axis. Inflammation. 2021;44:1815–1830. doi: 10.1007/s10753-021-01460-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Yang G, Lin C. Long noncoding RNA SOX2-OT exacerbates hypoxia-induced cardiomyocytes injury by regulating miR-27a-3p/TGFβR1 axis. Cardiovasc. Ther. 2020;2020:2016259. doi: 10.1155/2020/2016259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Latini R, et al. Beneficial effects of angiotensin-converting enzyme inhibitor and nitrate association on left ventricular remodeling in patients with large acute myocardial infarction: the Delapril Remodeling after Acute Myocardial Infarction (DRAMI) trial. Am. Heart J. 2003;146:133. doi: 10.1016/S0002-8703(02)94777-0. [DOI] [PubMed] [Google Scholar]
- 470.Wang H, Fu X. Effects of sacubitril/valsartan on ventricular remodeling in patents with left ventricular systolic dysfunction following acute anterior wall myocardial infarction. Coron. Artery Dis. 2021;32:418–426. doi: 10.1097/MCA.0000000000000932. [DOI] [PubMed] [Google Scholar]
- 471.Talasaz AH, et al. N-Acetylcysteine effects on transforming growth factor-β and tumor necrosis factor-α serum levels as pro-fibrotic and inflammatory biomarkers in patients following ST-segment elevation myocardial infarction. Drugs R. D. 2013;13:199–205. doi: 10.1007/s40268-013-0025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Mao S, Taylor S, Chen Q, Zhang M, Hinek A. Sodium tanshinone IIA sulfonate prevents the adverse left ventricular remodelling: Focus on polymorphonuclear neutrophil-derived granule components. J. Cell. Mol. Med. 2019;23:4592–4600. doi: 10.1111/jcmm.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 474.Niehrs C, Acebron SP. Mitotic and mitogenic Wnt signalling. EMBO J. 2012;31:2705–2713. doi: 10.1038/emboj.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Loh KM, van Amerongen R, Nusse R. Generating cellular diversity and spatial form: Wnt signaling and the evolution of multicellular animals. Dev. Cell. 2016;38:643–655. doi: 10.1016/j.devcel.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 476.Kitajima K, Oki S, Ohkawa Y, Sumi T, Meno C. Wnt signaling regulates left-right axis formation in the node of mouse embryos. Dev. Biol. 2013;380:222–232. doi: 10.1016/j.ydbio.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 477.Schneider J, Arraf AA, Grinstein M, Yelin R, Schultheiss TM. Wnt signaling orients the proximal-distal axis of chick kidney nephrons. Development. 2015;142:2686–2695. doi: 10.1242/dev.123968. [DOI] [PubMed] [Google Scholar]
- 478.Gajos-Michniewicz A, Czyz M. WNT signaling in melanoma. Int. J. Mol. Sci. 2020;21:4852. doi: 10.3390/ijms21144852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009;10:468–477. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- 480.Heisenberg CP, et al. Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature. 2000;405:76–81. doi: 10.1038/35011068. [DOI] [PubMed] [Google Scholar]
- 481.Dabdoub A, et al. Wnt signaling mediates reorientation of outer hair cell stereociliary bundles in the mammalian cochlea. Development. 2003;130:2375–2384. doi: 10.1242/dev.00448. [DOI] [PubMed] [Google Scholar]
- 482.Fu WB, Wang WE, Zeng CY. Wnt signaling pathways in myocardial infarction and the therapeutic effects of Wnt pathway inhibitors. Acta Pharm. Sin. 2019;40:9–12. doi: 10.1038/s41401-018-0060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Aisagbonhi O, et al. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model. Mech. 2011;4:469–483. doi: 10.1242/dmm.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Oerlemans MI, et al. Active Wnt signaling in response to cardiac injury. Basic Res. Cardiol. 2010;105:631–641. doi: 10.1007/s00395-010-0100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Blumenthal A, et al. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood. 2006;108:965–973. doi: 10.1182/blood-2005-12-5046. [DOI] [PubMed] [Google Scholar]
- 486.Huang L, Xiang M, Ye P, Zhou W, Chen M. Beta-catenin promotes macrophage-mediated acute inflammatory response after myocardial infarction. Immunol. Cell Biol. 2018;96:100–113. doi: 10.1111/imcb.1019. [DOI] [PubMed] [Google Scholar]
- 487.Meyer IS, et al. The cardiac microenvironment uses non-canonical WNT signaling to activate monocytes after myocardial infarction. EMBO Mol. Med. 2017;9:1279–1293. doi: 10.15252/emmm.201707565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Blankesteijn WM, van Gijn ME, Essers-Janssen YP, Daemen MJ, Smits JF. Beta-catenin, an inducer of uncontrolled cell proliferation and migration in malignancies, is localized in the cytoplasm of vascular endothelium during neovascularization after myocardial infarction. Am. J. Pathol. 2000;157:877–883. doi: 10.1016/s0002-9440(10)64601-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Barandon L, et al. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation. 2003;108:2282–2289. doi: 10.1161/01.CIR.0000093186.22847.4C. [DOI] [PubMed] [Google Scholar]
- 490.Min JK, et al. The WNT antagonist Dickkopf2 promotes angiogenesis in rodent and human endothelial cells. J. Clin. Investig. 2011;121:1882–1893. doi: 10.1172/JCI42556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Baruah J, et al. The allosteric glycogen synthase kinase-3 inhibitor NP12 limits myocardial remodeling and promotes angiogenesis in an acute myocardial infarction model. J. Biol. Chem. 2017;292:20785–20798. doi: 10.1074/jbc.M117.814376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Hermans KC, Daskalopoulos EP, Blankesteijn WM. The Janus face of myofibroblasts in the remodeling heart. J. Mol. Cell. Cardiol. 2016;91:35–41. doi: 10.1016/j.yjmcc.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 493.Zile MR, et al. Effects of sacubitril/valsartan on biomarkers of extracellular matrix regulation in patients with HFrEF. J. Am. Coll. Cardiol. 2019;73:795–806. doi: 10.1016/j.jacc.2018.11.042. [DOI] [PubMed] [Google Scholar]
- 494.Zhao X, et al. Aldehyde dehydrogenase-2 protects against myocardial infarction-related cardiac fibrosis through modulation of the Wnt/β-catenin signaling pathway. Ther. Clin. Risk Manag. 2015;11:1371–1381. doi: 10.2147/TCRM.S88297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Carthy JM, Garmaroudi FS, Luo Z, McManus BM. Wnt3a induces myofibroblast differentiation by upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PLoS ONE. 2011;6:e19809. doi: 10.1371/journal.pone.0019809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Duan J, et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012;31:429–442. doi: 10.1038/emboj.2011.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Xiang FL, Fang M, Yutzey KE. Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017;8:712. doi: 10.1038/s41467-017-00840-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Cui S, et al. miR-145 attenuates cardiac fibrosis through the AKT/GSK-3β/β-catenin signaling pathway by directly targeting SOX9 in fibroblasts. J. Cell. Biochem. 2021;122:209–221. doi: 10.1002/jcb.29843. [DOI] [PubMed] [Google Scholar]
- 499.Saraswati S, et al. Pyrvinium, a potent small molecule Wnt inhibitor, promotes wound repair and post-MI cardiac remodeling. PLoS ONE. 2010;5:e15521. doi: 10.1371/journal.pone.0015521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Qian L, et al. Downregulation of S100A4 alleviates cardiac fibrosis via Wnt/β -catenin pathway in mice. Cell. Physiol. Biochem. 2018;46:2551–2560. doi: 10.1159/000489683. [DOI] [PubMed] [Google Scholar]
- 501.Uitterdijk A, et al. UM206, a selective Frizzled antagonist, attenuates adverse remodeling after myocardial infarction in swine. Lab. Investig. 2016;96:168–176. doi: 10.1038/labinvest.2015.139. [DOI] [PubMed] [Google Scholar]
- 502.Moon J, et al. Blockade to pathological remodeling of infarcted heart tissue using a porcupine antagonist. Proc. Natl Acad. Sci. USA. 2017;114:1649–1654. doi: 10.1073/pnas.1621346114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Chi, F. et al. MiR-30b-5p promotes myocardial cell apoptosis in rats with myocardial infarction through regulating Wnt/β-catenin signaling pathway. Minerva Med. (2020). [DOI] [PubMed]
- 504.Sun HY, et al. Influence of MiR-154 on myocardial apoptosis in rats with acute myocardial infarction through Wnt/β-catenin signaling pathway. Eur. Rev. Med. Pharm. Sci. 2019;23:818–825. doi: 10.26355/eurrev_201901_16896. [DOI] [PubMed] [Google Scholar]
- 505.Li JH, Dai J, Han B, Wu GH, Wang CH. MiR-34a regulates cell apoptosis after myocardial infarction in rats through the Wnt/β-catenin signaling pathway. Eur. Rev. Med. Pharm. Sci. 2019;23:2555–2562. doi: 10.26355/eurrev_201903_17404. [DOI] [PubMed] [Google Scholar]
- 506.Zhu X, Lu X. MiR-423-5p inhibition alleviates cardiomyocyte apoptosis and mitochondrial dysfunction caused by hypoxia/reoxygenation through activation of the wnt/β-catenin signaling pathway via targeting MYBL2. J. Cell. Physiol. 2019;234:22034–22043. doi: 10.1002/jcp.28766. [DOI] [PubMed] [Google Scholar]
- 507.Xuan W, et al. Cardiac progenitors induced from human induced pluripotent stem cells with cardiogenic small molecule effectively regenerate infarcted hearts and attenuate fibrosis. Shock. 2018;50:627–639. doi: 10.1097/SHK.0000000000001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Klopsch C, et al. Intramyocardial angiogenetic stem cells and epicardial erythropoietin save the acute ischemic heart. Dis. Model. Mech. 2018;11:dmm033282. doi: 10.1242/dmm.033282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Li T, et al. Trop2 guarantees cardioprotective effects of cortical bone-derived stem cells on myocardial ischemia/reperfusion injury. Cell Transpl. 2018;27:1256–1268. doi: 10.1177/0963689718786882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Tang Y, et al. microRNA-497 inhibition mitigates myocardial infarction via enhancing wingless/integrated signal pathway in bone marrow mesenchymal stem cells. J. Cell. Biochem. 2019;120:13403–13412. doi: 10.1002/jcb.28615. [DOI] [PubMed] [Google Scholar]
- 511.Raziyeva K, et al. Preconditioned and genetically modified stem cells for myocardial infarction treatment. Int. J. Mol. Sci. 2020;21:7301. doi: 10.3390/ijms21197301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Lee JH, et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020;4:2032–2043. doi: 10.1182/bloodadvances.2019000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Diamond JR, et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020;184:53–62. doi: 10.1007/s10549-020-05817-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Wang J, Liu S, Heallen T, Martin JF. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018;15:672–684. doi: 10.1038/s41569-018-0063-3. [DOI] [PubMed] [Google Scholar]
- 515.Zheng M, Jacob J, Hung SH, Wang J. The Hippo pathway in cardiac regeneration and homeostasis: new perspectives for cell-free therapy in the injured heart. Biomolecules. 2020;10:1024. doi: 10.3390/biom10071024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Xie, J., Wang, Y., Ai, D., Yao, L. & Jiang, H. The role of the Hippo pathway in heart disease. FEBS J. (2021). [DOI] [PubMed]
- 517.Hilman D, Gat U. The evolutionary history of YAP and the hippo/YAP pathway. Mol. Biol. Evol. 2011;28:2403–2417. doi: 10.1093/molbev/msr065. [DOI] [PubMed] [Google Scholar]
- 518.Heng BC, et al. An overview of signaling pathways regulating YAP/TAZ activity. Cell. Mol. Life Sci. 2021;78:497–512. doi: 10.1007/s00018-020-03579-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Liu X, et al. Cell proliferation fate mapping reveals regional cardiomyocyte cell-cycle activity in subendocardial muscle of left ventricle. Nat. Commun. 2021;12:5784. doi: 10.1038/s41467-021-25933-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Castellan RFP, Meloni M. Mechanisms and therapeutic targets of cardiac regeneration: closing the age gap. Front. Cardiovasc. Med. 2018;5:7. doi: 10.3389/fcvm.2018.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Ma S, Meng Z, Chen R, Guan K-L. The Hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 2019;88:577–604. doi: 10.1146/annurev-biochem-013118-111829. [DOI] [PubMed] [Google Scholar]
- 522.Yang Y, Wang H, Ma Z, Hu W, Sun D. Understanding the role of mammalian sterile 20-like kinase 1 (MST1) in cardiovascular disorders. J. Mol. Cell. Cardiol. 2018;114:141–149. doi: 10.1016/j.yjmcc.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 523.Hu J, et al. OSM mitigates post-infarction cardiac remodeling and dysfunction by up-regulating autophagy through Mst1 suppression. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:1951–1961. doi: 10.1016/j.bbadis.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 524.Odashima M, et al. Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ. Res. 2007;100:1344–1352. doi: 10.1161/01.RES.0000265846.23485.7a. [DOI] [PubMed] [Google Scholar]
- 525.Leach JP, et al. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature. 2017;550:260–264. doi: 10.1038/nature24045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Liu S, et al. Gene therapy knockdown of Hippo signaling induces cardiomyocyte renewal in pigs after myocardial infarction. Sci. Transl. Med. 2021;13:eabd6892. doi: 10.1126/scitranslmed.abd6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Xu F, et al. MicroRNA-302d promotes the proliferation of human pluripotent stem cell-derived cardiomyocytes by inhibiting LATS2 in the Hippo pathway. Clin. Sci. 2019;133:1387–1399. doi: 10.1042/CS20190099. [DOI] [PubMed] [Google Scholar]
- 528.Lin KC, Park HW, Guan K-L. Regulation of the Hippo pathway transcription factor TEAD. Trends Biochem. Sci. 2017;42:862–872. doi: 10.1016/j.tibs.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Tao G, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–123. doi: 10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Lin Z, et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res. 2014;115:354–363. doi: 10.1161/CIRCRESAHA.115.303632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Han Z, et al. ALKBH5 regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics. 2021;11:3000–3016. doi: 10.7150/thno.47354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Li Y, et al. gp130 controls cardiomyocyte proliferation and heart regeneration. Circulation. 2020;142:967–982. doi: 10.1161/CIRCULATIONAHA.119.044484. [DOI] [PubMed] [Google Scholar]
- 533.Deshmukh V, Wang J, Martin JF. Leading progress in heart regeneration and repair. Curr. Opin. Cell Biol. 2019;61:79–85. doi: 10.1016/j.ceb.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Wang F, et al. LPA(3)-mediated lysophosphatidic acid signaling promotes postnatal heart regeneration in mice. Theranostics. 2020;10:10892–10907. doi: 10.7150/thno.47913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Aharonov A, et al. ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat. Cell Biol. 2020;22:1346–1356. doi: 10.1038/s41556-020-00588-4. [DOI] [PubMed] [Google Scholar]
- 536.Yu Y, et al. Yes-associated protein and transcriptional coactivator with PDZ-binding motif as new targets in cardiovascular diseases. Pharm. Res. 2020;159:105009. doi: 10.1016/j.phrs.2020.105009. [DOI] [PubMed] [Google Scholar]
- 537.Del Re DP. Beyond the cardiomyocyte: consideration of HIPPO pathway cell-type specificity. Circ. Res. 2018;123:30–32. doi: 10.1161/CIRCRESAHA.118.313383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Ramjee V, et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J. Clin. Investig. 2017;127:899–911. doi: 10.1172/JCI88759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Johansen AKZ, Molkentin JD. Hippo signaling does it again: arbitrating cardiac fibroblast identity and activation. Genes Dev. 2019;33:1457–1459. doi: 10.1101/gad.332791.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Francisco J, et al. Blockade of fibroblast YAP attenuates cardiac fibrosis and dysfunction through MRTF-A inhibition. JACC Basic Transl. Sci. 2020;5:931–945. doi: 10.1016/j.jacbts.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Landry NM, et al. SKI activates the Hippo pathway via LIMD1 to inhibit cardiac fibroblast activation. Basic Res. Cardiol. 2021;116:25. doi: 10.1007/s00395-021-00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Fan M, et al. Endothelial cell HSPA12B and yes-associated protein cooperatively regulate angiogenesis following myocardial infarction. JCI Insight. 2020;5:e139640. doi: 10.1172/jci.insight.139640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Hu J, et al. Luteolin alleviates post-infarction cardiac dysfunction by up-regulating autophagy through Mst1 inhibition. J. Cell. Mol. Med. 2016;20:147–156. doi: 10.1111/jcmm.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Hu, J. et al. Melatonin alleviates postinfarction cardiac remodeling and dysfunction by inhibiting Mst1. J. Pineal Res.62 (2017). [DOI] [PubMed]
- 545.Chen W, et al. TT-10-loaded nanoparticles promote cardiomyocyte proliferation and cardiac repair in a mouse model of myocardial infarction. JCI Insight. 2021;6:e151987. doi: 10.1172/jci.insight.151987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Feng Y, et al. rGO/Silk fibroin-modified nanofibrous patches prevent ventricular remodeling via Yap/Taz-TGFβ1/Smads signaling after myocardial infarction in rats. Front. Cardiovasc. Med. 2021;8:718055. doi: 10.3389/fcvm.2021.718055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Peng H, Shindo K, Donahue RR, Abdel-Latif A. Cardiac cell therapy: insights into the mechanisms of tissue repair. Int. J. Mol. Sci. 2021;22:1201. doi: 10.3390/ijms22031201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Balbi C, et al. An exosomal-carried short periostin isoform induces cardiomyocyte proliferation. Theranostics. 2021;11:5634–5649. doi: 10.7150/thno.57243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Ouyang Z, Wei K. miRNA in cardiac development and regeneration. Cell Regen. 2021;10:14. doi: 10.1186/s13619-021-00077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Braga L, Ali H, Secco I, Giacca M. Non-coding RNA therapeutics for cardiac regeneration. Cardiovasc. Res. 2021;117:674–693. doi: 10.1093/cvr/cvaa071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Diez-Cuñado M, et al. miRNAs that induce human cardiomyocyte proliferation converge on the Hippo pathway. Cell Rep. 2018;23:2168–2174. doi: 10.1016/j.celrep.2018.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Ma C, et al. MicroRNA-93 promotes angiogenesis and attenuates remodeling via inactivation of the Hippo/Yap pathway by targeting Lats2 after myocardial infarction. Mol. Med. Rep. 2020;22:483–493. doi: 10.3892/mmr.2020.11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Tian Y, et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 2015;7:279ra238. doi: 10.1126/scitranslmed.3010841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Han, C., Yang, J., Sun, J. & Qin, G. Extracellular vesicles in cardiovascular disease: biological functions and therapeutic implications. Pharmacol. Ther. 108025 (2021). [DOI] [PMC free article] [PubMed]
- 555.Grigorian-Shamagian L, et al. Insights into therapeutic products, preclinical research models, and clinical trials in cardiac regenerative and reparative medicine: where are we now and the way ahead. Current opinion paper of the ESC Working Group on Cardiovascular Regenerative and Reparative Medicine. Cardiovasc. Res. 2021;117:1428–1433. doi: 10.1093/cvr/cvaa337. [DOI] [PubMed] [Google Scholar]
- 556.Halladin NL, et al. Intracoronary and systemic melatonin to patients with acute myocardial infarction: protocol for the IMPACT trial. Dan. Med. J. 2014;61:A4773. [PubMed] [Google Scholar]
- 557.Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 558.Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 559.Wang D, et al. Aberrant activation of hedgehog signaling promotes cell proliferation via the transcriptional activation of forkhead Box M1 in colorectal cancer cells. J. Exp. Clin. Cancer Res. 2017;36:23. doi: 10.1186/s13046-017-0491-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Gerhardt C, Lier JM, Kuschel S, Rüther U. The ciliary protein Ftm is required for ventricular wall and septal development. PLoS ONE. 2013;8:e57545. doi: 10.1371/journal.pone.0057545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Xiao Q, et al. Impaired sonic hedgehog pathway contributes to cardiac dysfunction in type 1 diabetic mice with myocardial infarction. Cardiovasc. Res. 2012;95:507–516. doi: 10.1093/cvr/cvs216. [DOI] [PubMed] [Google Scholar]
- 562.Bangs F, Anderson KV. Primary cilia and mammalian hedgehog signaling. Cold Spring Harb. Perspect. Biol. 2017;9:a028175. doi: 10.1101/cshperspect.a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Ghaleh B, et al. Cardioprotective effect of sonic hedgehog ligand in pig models of ischemia reperfusion. Theranostics. 2020;10:4006–4016. doi: 10.7150/thno.40461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Lavine KJ, Kovacs A, Ornitz DM. Hedgehog signaling is critical for maintenance of the adult coronary vasculature in mice. J. Clin. Investig. 2008;118:2404–2414. doi: 10.1172/JCI34561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Kusano KF, et al. Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat. Med. 2005;11:1197–1204. doi: 10.1038/nm1313. [DOI] [PubMed] [Google Scholar]
- 566.Wang Y, Lu P, Zhao D, Sheng J. Targeting the hedgehog signaling pathway for cardiac repair and regeneration. Herz. 2017;42:662–668. doi: 10.1007/s00059-016-4500-y. [DOI] [PubMed] [Google Scholar]
- 567.Bueno-Betí C, et al. Microparticles harbouring Sonic hedgehog morphogen improve the vasculogenesis capacity of endothelial progenitor cells derived from myocardial infarction patients. Cardiovasc. Res. 2019;115:409–418. doi: 10.1093/cvr/cvy189. [DOI] [PubMed] [Google Scholar]
- 568.Yao Q, et al. Sonic hedgehog mediates a novel pathway of PDGF-BB-dependent vessel maturation. Blood. 2014;123:2429–2437. doi: 10.1182/blood-2013-06-508689. [DOI] [PubMed] [Google Scholar]
- 569.Mackie AR, et al. Sonic hedgehog-modified human CD34+ cells preserve cardiac function after acute myocardial infarction. Circ. Res. 2012;111:312–321. doi: 10.1161/CIRCRESAHA.112.266015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Kanaya K, et al. Sonic Hedgehog signaling regulates vascular differentiation and function in human CD34 positive cells: vasculogenic CD34(+) cells with Sonic Hedgehog. Stem Cell Res. 2015;14:165–176. doi: 10.1016/j.scr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 571.Tang T, Wu M, Yang J. Transplantation of MSCs transfected with SHH gene ameliorates cardiac dysfunction after chronic myocardial infarction. Int. J. Cardiol. 2013;168:4997–4999. doi: 10.1016/j.ijcard.2013.07.126. [DOI] [PubMed] [Google Scholar]
- 572.Ahmed RP, Haider KH, Shujia J, Afzal MR, Ashraf M. Sonic Hedgehog gene delivery to the rodent heart promotes angiogenesis via iNOS/netrin-1/PKC pathway. PLoS ONE. 2010;5:e8576. doi: 10.1371/journal.pone.0008576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Ueda K, et al. Sonic hedgehog is a critical mediator of erythropoietin-induced cardiac protection in mice. J. Clin. Investig. 2010;120:2016–2029. doi: 10.1172/JCI39896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Lavine KJ, et al. Fibroblast growth factor signals regulate a wave of Hedgehog activation that is essential for coronary vascular development. Genes Dev. 2006;20:1651–1666. doi: 10.1101/gad.1411406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Roncalli J, et al. Sonic hedgehog-induced functional recovery after myocardial infarction is enhanced by AMD3100-mediated progenitor-cell mobilization. J. Am. Coll. Cardiol. 2011;57:2444–2452. doi: 10.1016/j.jacc.2010.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Li SH, Zhang YY, Sun YL, Zhao HJ, Wang Y. Inhibition of microRNA-802-5p inhibits myocardial apoptosis after myocardial infarction via Sonic Hedgehog signaling pathway by targeting PTCH1. Eur. Rev. Med. Pharm. Sci. 2021;25:326–334. doi: 10.26355/eurrev_202101_24398. [DOI] [PubMed] [Google Scholar]
- 577.Sharma S, Kaur A, Sharma S. Preconditioning potential of purmorphamine: a hedgehog activator against ischaemic reperfusion injury in ovariectomised rat heart. Perfusion. 2018;33:209–218. doi: 10.1177/0267659117732401. [DOI] [PubMed] [Google Scholar]
- 578.Xiao Q, et al. Increased expression of Sonic hedgehog restores diabetic endothelial progenitor cells and improves cardiac repair after acute myocardial infarction in diabetic mice. Int. J. Mol. Med. 2019;44:1091–1105. doi: 10.3892/ijmm.2019.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Agouni A, et al. Sonic hedgehog carried by microparticles corrects endothelial injury through nitric oxide release. FASEB J. 2007;21:2735–2741. doi: 10.1096/fj.07-8079com. [DOI] [PubMed] [Google Scholar]
- 580.Xiao Q, et al. Oxidative stress contributes to the impaired sonic hedgehog pathway in type 1 diabetic mice with myocardial infarction. Exp. Ther. Med. 2015;10:1750–1758. doi: 10.3892/etm.2015.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Przyklenk K, et al. Acute induction of autophagy as a novel strategy for cardioprotection: getting to the heart of the matter. Autophagy. 2011;7:432–433. doi: 10.4161/auto.7.4.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Paulis L, et al. Activation of Sonic hedgehog signaling in ventricular cardiomyocytes exerts cardioprotection against ischemia reperfusion injuries. Sci. Rep. 2015;5:7983. doi: 10.1038/srep07983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Weyers JJ, et al. Sonic Hedgehog upregulation does not enhance the survival and engraftment of stem cell-derived cardiomyocytes in infarcted hearts. PLoS ONE. 2020;15:e0227780. doi: 10.1371/journal.pone.0227780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Feng L, et al. Simvastatin relieves myocardial ischemia/reperfusion injury in rats through hedgehog signaling pathway. Eur. Rev. Med. Pharm. Sci. 2020;24:6400–6408. doi: 10.26355/eurrev_202006_21538. [DOI] [PubMed] [Google Scholar]
- 585.Dunaeva M, Waltenberger J. Hh signaling in regeneration of the ischemic heart. Cell. Mol. Life Sci. 2017;74:3481–3490. doi: 10.1007/s00018-017-2534-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Johnson NR, et al. Coacervate delivery of growth factors combined with a degradable hydrogel preserves heart function after myocardial infarction. ACS Biomater. Sci. Eng. 2015;1:753–759. doi: 10.1021/acsbiomaterials.5b00077. [DOI] [PubMed] [Google Scholar]
- 587.Wang C, et al. Tetramethylpyrazine and Astragaloside IV synergistically ameliorate left ventricular remodeling and preserve cardiac function in a rat myocardial infarction model. J. Cardiovasc. Pharm. 2017;69:34–40. doi: 10.1097/FJC.0000000000000437. [DOI] [PubMed] [Google Scholar]
- 588.Povsic TJ, et al. The RENEW Trial: efficacy and safety of intramyocardial autologous CD34(+) cell administration in patients with refractory angina. JACC Cardiovasc. Int. 2016;9:1576–1585. doi: 10.1016/j.jcin.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 589.Lehtinen M, et al. Prospective, randomized, double-blinded trial of bone marrow cell transplantation combined with coronary surgery—perioperative safety study. Interact. Cardiovasc. Thorac. Surg. 2014;19:990–996. doi: 10.1093/icvts/ivu265. [DOI] [PubMed] [Google Scholar]
- 590.Seo WW, et al. Efficacy of IntraCoronary Erythropoietin Delivery BEfore Reperfusion-Gauging infarct size in patients with acute ST-segment elevation myocardial infarction (ICEBERG) Int Heart J. 2019;60:255–263. doi: 10.1536/ihj.18-035. [DOI] [PubMed] [Google Scholar]
- 591.Minamino T, et al. Low-dose erythropoietin in patients with ST-segment elevation myocardial infarction (EPO-AMI-II)—a Randomized Controlled Clinical Trial. Circ. J. 2018;82:1083–1091. doi: 10.1253/circj.CJ-17-0889. [DOI] [PubMed] [Google Scholar]
- 592.Steppich B, et al. Effect of Erythropoietin in patients with acute myocardial infarction: five-year results of the REVIVAL-3 trial. BMC Cardiovasc. Disord. 2017;17:38. doi: 10.1186/s12872-016-0464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Chitimus DM, et al. Melatonin’s impact on antioxidative and anti-inflammatory reprogramming in homeostasis and disease. Biomolecules. 2020;10:1211. doi: 10.3390/biom10091211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Luo R, Sun X, Shen F, Hong B, Wang Z. Effects of high-dose rosuvastatin on ventricular remodelling and cardiac function in ST-segment elevation myocardial infarction. Drug Des. Dev. Ther. 2020;14:3891–3898. doi: 10.2147/DDDT.S254948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Bao JW, et al. Rosuvastatin inhibits inflammatory response and resists fibrosis after myocardial infarction. Eur. Rev. Med. Pharm. Sci. 2018;22:238–245. doi: 10.26355/eurrev_201801_14123. [DOI] [PubMed] [Google Scholar]
- 596.Dominguez-Rodriguez, A. et al. Effect of intravenous and intracoronary melatonin as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: results of the Melatonin Adjunct in the acute myocaRdial Infarction treated with Angioplasty trial. J. Pineal Res. 62 (2017). [DOI] [PubMed]
- 597.Losordo DW, et al. Gene therapy for myocardial angiogenesis: initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation. 1998;98:2800–2804. doi: 10.1161/01.cir.98.25.2800. [DOI] [PubMed] [Google Scholar]
- 598.Kukula K, et al. Long-term follow-up and safety assessment of angiogenic gene therapy trial VIF-CAD: transcatheter intramyocardial administration of a bicistronic plasmid expressing VEGF-A165/bFGF cDNA for the treatment of refractory coronary artery disease. Am. Heart J. 2019;215:78–82. doi: 10.1016/j.ahj.2019.06.009. [DOI] [PubMed] [Google Scholar]
- 599.Grajek S, et al. Influence of bone marrow stem cells on left ventricle perfusion and ejection fraction in patients with acute myocardial infarction of anterior wall: randomized clinical trial: impact of bone marrow stem cell intracoronary infusion on improvement of microcirculation. Eur. Heart J. 2010;31:691–702. doi: 10.1093/eurheartj/ehp536. [DOI] [PubMed] [Google Scholar]
- 600.Kim SH, et al. Improvement in left ventricular function with intracoronary mesenchymal stem cell therapy in a patient with anterior wall ST-segment elevation myocardial infarction. Cardiovasc. Drugs Ther. 2018;32:329–338. doi: 10.1007/s10557-018-6804-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Fernández-Avilés F, et al. Safety and efficacy of intracoronary infusion of allogeneic human cardiac stem cells in patients with ST-segment elevation myocardial infarction and left ventricular dysfunction. Circ. Res. 2018;123:579–589. doi: 10.1161/CIRCRESAHA.118.312823. [DOI] [PubMed] [Google Scholar]
- 602.Traverse JH, et al. TIME Trial: effect of timing of stem cell delivery following ST-elevation myocardial infarction on the recovery of global and regional left ventricular function: final 2-year analysis. Circ. Res. 2018;122:479–488. doi: 10.1161/CIRCRESAHA.117.311466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Blau HM, Daley GQ. Stem cells in the treatment of disease. N. Engl. J. Med. 2019;380:1748–1760. doi: 10.1056/NEJMra1716145. [DOI] [PubMed] [Google Scholar]
- 604.Miao C, Lei M, Hu W, Han S, Wang Q. A brief review: the therapeutic potential of bone marrow mesenchymal stem cells in myocardial infarction. Stem Cell Res. Ther. 2017;8:242. doi: 10.1186/s13287-017-0697-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Fu J, et al. Gastrin exerts a protective effect against myocardial infarction via promoting angiogenesis. Mol. Med. 2021;27:90. doi: 10.1186/s10020-021-00352-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Lee JM, et al. Physiological and clinical assessment of resting physiological indexes. Circulation. 2019;139:889–900. doi: 10.1161/CIRCULATIONAHA.118.037021. [DOI] [PubMed] [Google Scholar]
- 607.Xu F, et al. MicroRNA-302d promotes the proliferation of human pluripotent stem cell-derived cardiomyocytes by inhibiting LATS2 in the Hippo pathway. Clin. Sci. 2019;133:1387–1399. doi: 10.1042/CS20190099. [DOI] [PubMed] [Google Scholar]
- 608.Munarin F, Kant RJ, Rupert CE, Khoo A, Coulombe KLK. Engineered human myocardium with local release of angiogenic proteins improves vascularization and cardiac function in injured rat hearts. Biomaterials. 2020;251:120033. doi: 10.1016/j.biomaterials.2020.120033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Riaud M, et al. Pharmacology active microcarriers delivering HGF associated with extracellular vesicles for myocardial repair. Eur. J. Pharm. Biopharm. 2021;169:268–279. doi: 10.1016/j.ejpb.2021.10.018. [DOI] [PubMed] [Google Scholar]