

Summary
Topoisomerase II (topo II) is essential for disentangling newly replicated chromosomes. DNA unlinking involves the physical passage of one duplex through another and depends on the transient formation of double-strand DNA breaks, a step exploited by frontline chemotherapeutics to kill cancer cells. Although anti-topo II drugs are efficacious, they also elicit cytotoxic side effects in normal cells; insights into how topo II is regulated in different cellular contexts is essential to improve their targeted use. Using chemical fractionation and mass spectrometry, we have discovered that topo II is subject to metabolic control through the TCA cycle. We show that TCA metabolites stimulate topo II activity in vitro and that levels of TCA flux modulate cellular sensitivity to anti-topo II drugs in vivo. Our work reveals an unanticipated connection between the control of DNA topology and cellular metabolism, a finding with ramifications for the clinical use of anti-topo II therapies.
eTOC Blurb
Lee et al. purify yeast metabolites that show activity against eukaryotic topoisomerase II (topo II). LC-MS/MS analysis identifies TCA cycle intermediates as stimulators of topo II activity. Modulating TCA cycle flux affects the cytotoxicity of topo II-targeting drugs, indicating that TCA cycle metabolism regulates topo II function in cells.
Introduction
The complement of chromosomes in a single human cell, laid out end-to-end, is ~10,000 times longer than the diameter of the nucleus. Efficient packaging, access, and duplication of this genetic material depends on the proper control of DNA superstructure. Some of the largest rearrangements of the genome occur during DNA replication and mitosis as sister chromatids are synthesized, condensed, and segregated (Barrington et al., 2017). The double-helical structure of DNA presents a challenge to these transformations, resulting in superhelical intertwinings and chromosomal entanglements (catenanes) (Peter et al., 1998; Postow et al., 2001; Sundin and Varshavsky, 1981). Enzymes known as topoisomerases are required to resolve topological stresses in DNA; of the two major classes, only type II topoisomerases are able to unlink tangled double-stranded segments to facilitate chromosome partitioning prior to cell division (Vos et al., 2011).
Beyond supporting supercoiling homeostasis and promoting DNA unlinking, mounting evidence suggests that topo II has additional roles in regulating chromatin structure throughout the eukaryotic cell cycle (Lee and Berger, 2019). Although most DNA regions are decatenated by the end of replication in S phase (Charbin et al., 2014; Lucas et al., 2001), the maintenance of some catenated regions, mostly in regions with repetitive sequences, appears to be important for proper chromosome condensation and sister chromatid cohesion (Bauer et al., 2012; Daniloski et al., 2019). Loss or gain of topo II function has been shown to disturb the precisely balanced catenation state of the genome and lead to detrimental effects on chromosome condensation (Cuvier and Hirano, 2003; Samejima et al., 2012; Uemura et al., 1987). In vertebrates, the cell cycle-dependent expression of topo IIα, one of two topo II isoforms found in such organisms, provides one means of calibrating levels of topoisomerase activity (Heck et al., 1988; Kimura et al., 1994; Woessner et al., 1991); however, invertebrates and lower-order eukaryotes (such as yeast and Drosophila) express only a single isoform of topo II for which cell cycle-dependent expression has not been observed (Eser et al., 2011; Goto and Wang, 1984; Spellman et al., 1998; Whalen et al., 1991). Post-translational modifications such as SUMOylation ubiquitylation, and phosphorylation have been reported to regulate enzyme stability, localization and activity, but how these marks exert such functions is poorly understood (Lee and Berger, 2019). Collectively, these and other observations demonstrate that there is a requirement for precise regulation of topo II activity. However, whether there exist fundamental regulatory mechanisms that are conserved throughout eukaryotes has not been established.
Because topo II is essential for cell proliferation, it is a target for many chemotherapeutics (Nitiss, 2009). A special class of topo II-targeting drugs, categorized as topoisomerase ‘poisons,’ triggers cell death by inducing topo II to generate cytotoxic DNA damage (Pommier et al., 2010; Wu et al., 2011). The major limitation of topoisomerase poisons is their potential to generate DNA damage in noncancerous cells that can lead to toxic side effects and therapy-related neoplasias (Felix, 1998; Turcotte et al., 2018). A greater understanding of the cellular mechanisms that diminish or enhance topo II activity levels is necessary to better match topoisomerase-targeted therapies against specific cancer types and to design combinatorial strategies that selectively enhance the potency of anti-topoisomerase drugs in cancer cells.
Previously, we found that a highly-conserved pocket in the ATPase domain of topo II that binds to ICRF-187, a clinically-approved drug, also associates with a natural, plant-derived antagonist of topo II known as resveratrol (Lee et al., 2017). This observation led us to wonder whether other small molecules found natively in cells might also modulate topo II function. To test this idea, we prepared crude metabolite extracts from S. cerevisiae and analyzed them for activity against topo II in vitro. We discovered that these extracts stimulated topo II activity, an effect that has not been observed previously for other agents that act on topo II. Using biochemical fractionation and LC-MS/MS analysis, we determined that di- and tri- carboxylate TCA cycle metabolites were responsible for the stimulatory effect, and that these compounds increase the efficiency of the topo II strand passage reaction. By monitoring the sensitivity of yeast to different classes of topo II inhibitors under distinct metabolic states, we further found that TCA cycle flux can directly influence topo II activity in vivo. Collectively, our results show that natural small molecules produced by metabolic processes of the cell can directly modulate topo II function and that manipulation of these processes affects the efficacy of clinically-approved topo II-targeting drugs. These findings reveal an unanticipated link between cellular metabolism and the regulation of a key central dogma process, a discovery that provides new directions for improving the safety and efficacy of anti-topo II chemotherapies.
Results
Small-molecule metabolites from S. cerevisiae stimulate topoisomerase II activity in vitro
To search for natural products that might regulate topo II, we first tested whether crude S. cerevisiae metabolite extracts could impact enzyme activity in vitro. Metabolic extracts were prepared from yeast grown in minimal media without ammonium sulfate, as sulfate ions were found to enhance topo II supercoil relaxation activity (Figure S1A) (all other media components were confirmed to have no significant effect on topo II activity (Figure S1B)). Extracts were taken from log-phase cultures to capture metabolic profiles representative of actively dividing cells in all cell cycle stages (Figure 1A). A sample of the spent media was also lyophilized as a control for media components and secreted compounds.
Figure 1. Crude metabolite extracts stimulate ScTop2 activity.
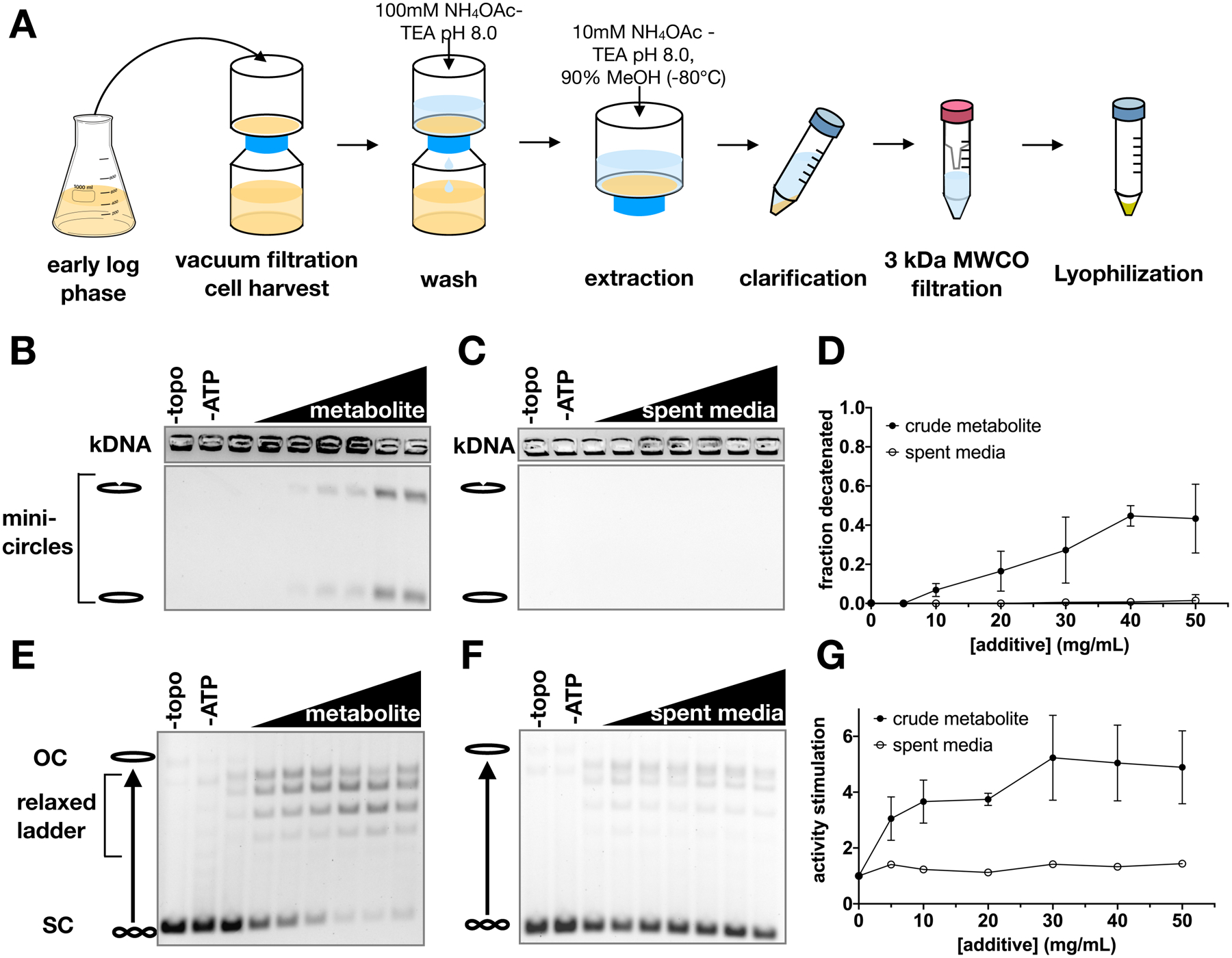
(A) Schematic of metabolite extraction procedure. (B-G) Representative gels and graphs of mean ± SD (n=3) for decatenation assays (B-D) and supercoil relaxation assays (E-G). Bands representing nicked minicircles and closed minicircles are indicated (B-C), as are bands representing unrelaxed substrate (SC), the relaxed topoisomer distribution, and nicked/open circle (OC) plasmids (E-F). No enzyme (‘-topo’) and no ATP (‘-ATP’) negative controls show the starting substrate. Metabolite extract and spent media were titrated from 0 to 50 mg/ml in 10 mg/ml increments.
We assessed the effects of the crude metabolite extract and spent media control on topo II using decatenation assays. Purified S. cerevisiae topo II (ScTop2) was incubated with kinetoplast DNA (kDNA) – a highly interlocked network of small circular DNAs – and ATP to catalyze strand passage. All experiments were conducted with excess ATP (5 mM) to eliminate any response that might have been elicited from the presence of nucleotide in the extracts. Although the spent media sample had little to no effect on DNA unlinking by ScTop2, the metabolite extract stimulated decatenation in a dose-dependent manner (Figures 1B–D). Similarly, the crude extract also stimulated DNA supercoil relaxation by ScTop2, as evidenced by the conversion of a negatively supercoiled plasmid substrate into a distribution of relaxed topoisomers (Figures 1E–G).
Biochemical characterization of active compounds from yeast metabolite extracts
Upon discovering a stimulatory activity in crude extracts, we set out to define the chemical properties of the active agent(s). Liquid-liquid extraction with butanol was first conducted under both acidic and basic conditions. In both cases, the stimulatory activity was recovered in the aqueous phase, indicating that the agent was highly polar (Figures S1C and S1D). Based on this finding and the nucleotide-binding capabilities of topo II, we surmised that the factor might contain phosphate groups or phosphodiester bonds. However, the stimulatory activity was resistant to treatment with Antarctic phosphatase or snake venom phosphodiesterase, suggesting that neither phosphates nor phosphodiester bonds were present in the stimulatory metabolite (Figures S1E and S1F).
To further characterize the active compound and prepare samples for mass spectrometry analysis, we developed a purification protocol for the agent (Figure 2A). Solid-phase extraction was used as a first-pass, bulk-fractionation method to remove contaminants and enrich the stimulatory activity roughly two-fold (Figure 2B). The active fraction was then further purified using reverse-phase HPLC, followed by normal phase HPLC, and the stimulatory activity was recovered in the fractions shown in Figures 2C–F (shaded yellow and green). For some reactions (see the right-most lane of the yellow-shaded fraction, Figures 2C and 2E), we observed the formation of precipitates, indicating that the resulting loss of activity was likely due to compromised protein solubility at high concentrations of metabolite. Interestingly, this procedure also revealed two separate fractions (shaded gray and pink in Figures 2D and 2F) that inhibited DNA supercoil relaxation by ScTop2 (Figure 2F) without causing precipitation; these latter activities appear to have been masked by the stimulatory activity in previous purification steps. The appearance of these distinct activities indicates that cells may contain more than one class of small molecules that can act on topo II.
Figure 2. Purification of stimulatory metabolites from crude metabolite extracts.
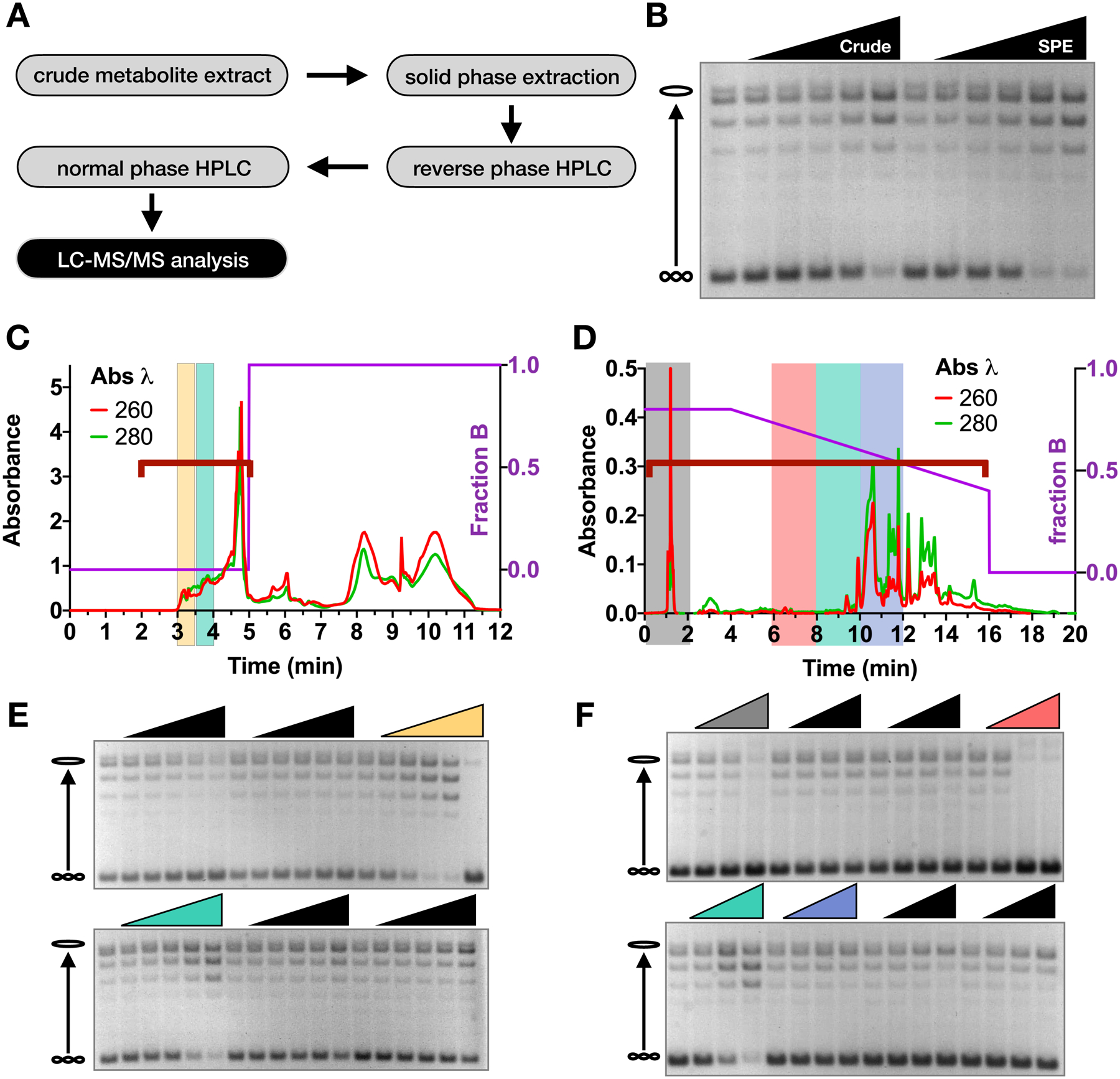
(A) Schematic of metabolite purification method. (B) Supercoil relaxation assay with metabolite samples before (‘Crude’) and after solid phase extraction (‘SPE’). Metabolite fractions were added from 0 to 40 mg/ml in two-fold increments. (C-D) Chromatograms of reverse phase (C) and normal phase (D) HPLC runs. The HPLC method is depicted by the purple line as indicated by the right y-axis. Red and green traces show absorbance values at 260 nm and 280 nm wavelengths respectively, as indicated on the left y-axis. (E-F) Supercoil relaxation assays of reverse-phase fractions (E) and normal-phase fractions (F), as indicated by the red brackets in (C) and (D). Fractions of interest are highlighted by corresponding colors in the chromatograms and relaxation assay gels. Lyophilized material from each fraction was titrated down from the maximum possible concentration in two-fold dilution steps.
Identification of topo II-stimulating metabolites
To identify the activating agent, the stimulatory fraction (highlighted ingreen in Figures 2D and 2F) and flanking fractions (shaded pink and blue in Figures 2D and 2F) from the last HPLC step were analyzed by LC-MS/MS. Spectra were first collected from a pooled sample to generate a peak-identification library representative of all metabolites present in the three fractions. Data were processed in Compound Discoverer™ by referencing mass spectral databases of natural products (Figure 3A). Next, the samples were analyzed individually and spectra from each sample were indexed based on the pooled library. By comparing peak intensities across samples, we generated a candidate list of compounds that were enriched in the stimulatory fraction as compared to its neighboring fractions (Figure 3B, Table S1).
Figure 3. Identification and structure activity relationship (SAR) analysis of stimulatory compounds.
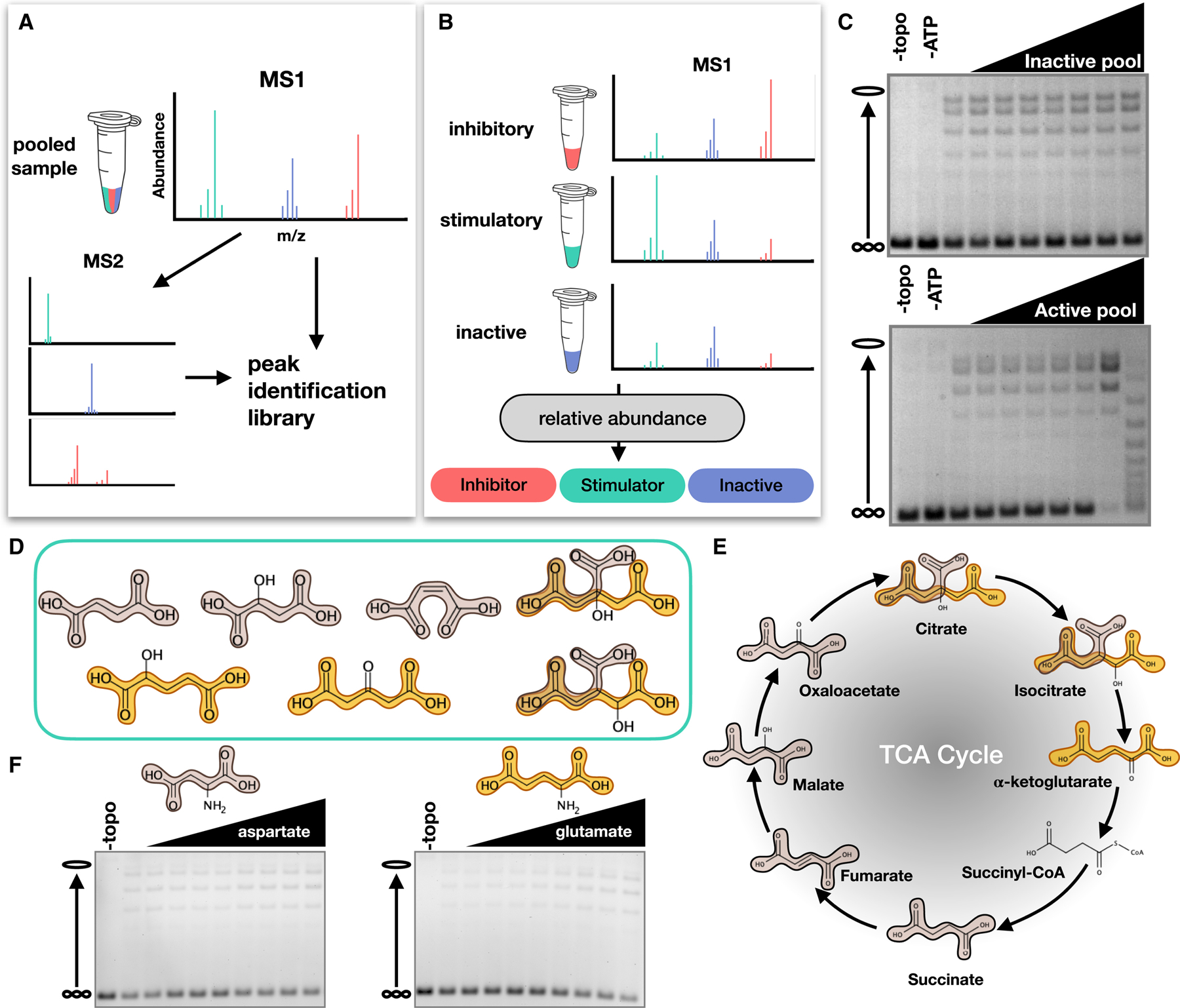
(A) Schematic depicting LC-MS/MS analysis of metabolite fractions to generate an ion peak-identification library. (B) Schematic depicting the individual analysis of metabolite fractions. (C) Example supercoil relaxation assays from testing an inactive and active pool of five candidate compounds. Each candidate compound was titrated from 0 to 20 mg/ml in two-fold steps. (D) Representative compounds with stimulatory activity. Succinic acid (tan) and glutaric acid (gold) motifs are highlighted. (E) TCA cycle metabolites. (F) Effects of dicarboxylic amino acids on topo II supercoil relaxation activity. Aspartate and glutamate were titration from 0 to 40 mM in 5 mM increments.
Thirty-two candidate compounds were initially tested in eight different pools for stimulatory activity in supercoil relaxation assays. Although most pools showed no effect on topo II, some pools did impact enzymatic activity (Figure 3C); compounds from active pools were then tested individually. Several compounds from the initial candidate list stimulated DNA supercoil relaxation by ScTop2. Notably, we identified two chemical motifs – succinic and glutaric acids – that were consistently present in the stimulatory compounds (Figure 3D). A substructure search of these motifs against the Human Metabolome Database revealed that they are most frequently represented in TCA cycle intermediates (Figure 3E and Table S2). Four TCA cycle intermediates (citrate, isocitrate, succinate, and malate) were identified as stimulatory compounds from our original candidate list (Figure 3D and Table S1). Aspartate, which was included in our candidate list (Table S1), and glutamate also contain succinic and glutaric acid substructures respectively. However, neither amino acid was able to stimulate topo II supercoil relaxation activity (Figure 3F), suggesting that the addition of an amine interferes with the stimulatory interaction.
TCA cycle intermediates stimulate topo II activity in vitro
Based on our observations, we predicted that other TCA cycle intermediates would have similar effects on topo II activity. All TCA cycle intermediates, except for succinyl-CoA, have succinic and/or glutaric acid substructures (Figure 3E). Having ascertained that such a moiety is present in compounds capable of stimulating topo II activity, we evaluated the effects of all TCA cycle intermediates on topo II. All reactions were conducted at a consistent pH and all metabolites were pre-pH’d to eliminate potential artifacts from variable protonation states. Consistent with our previous observations, these intermediates led to as much as a 4- to 12-fold stimulation of decatenation activity and to a 3- to 5-fold stimulation of supercoil relaxation activity at compound concentrations ranging from 25–40 mM (Figures 4A–J and S2). Glutamate, an inactive dicarboxylic acid (Figure 3F), was included as a non-TCA metabolite control for comparison.
Figure 4. TCA cycle intermediates stimulate strand passage by topo II without significant effects on ATP hydrolysis.
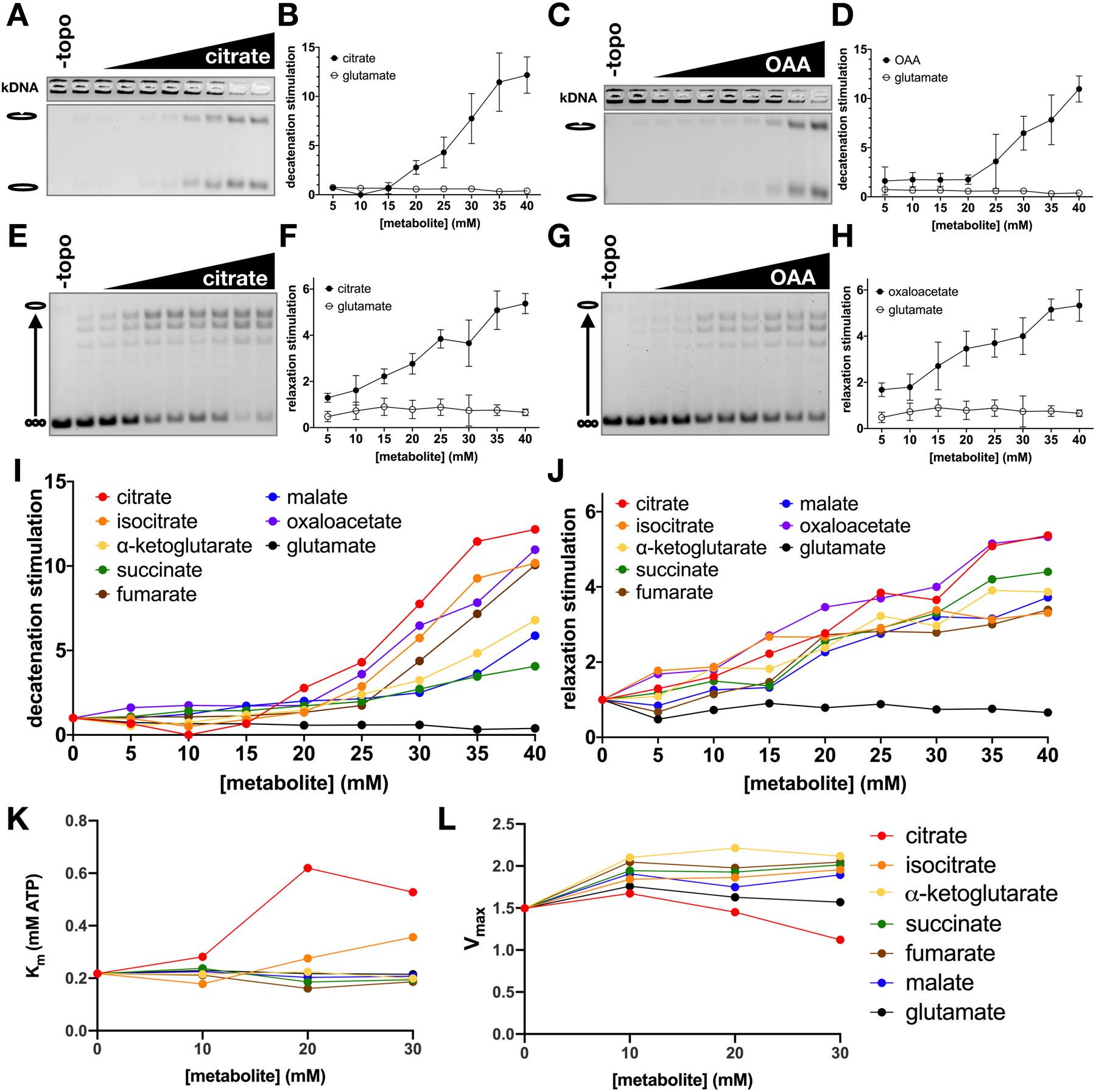
(A-H) Stimulation of topo II decatenation activity (A-D) and supercoil relaxation activity (E-H) by citrate and oxaloacetate (OAA). Metabolites were titrated from 0 to 40mM in 5mM increments. Graphs (B, D, F, and H) represent mean ± SD of n=3–5. (I-J) Stimulation of ScTop2 decatenation (I) and relaxation (J) activity by TCA metabolites and glutamate (negative control). Stimulation values represent mean of n=3–6 independent experiments. (See also Figure S3). (K-L) Effects of TCA metabolites on ATP hydrolysis activity. Km (K) and Vmax (μmol ATP • min−1 • nmol topo II−1) (L) values were derived from 3 replicates. (See also Figure S6).
Of the TCA intermediates, citrate and isocitrate are distinguished by the presence of a third carboxylate. Citrate chelates Mg2+ ions (Yamagami et al., 2018), which are required for topo II catalytic activity (Osheroff, 1987). To evaluate the potential role of Mg2+-ion chelation in the stimulation of topo II, we assessed the ability of the tricarboxylate compounds to stimulate decatenation activity at varying concentrations of Mg(OAc)2 (Figure S3). Increasing the concentration of Mg2+ alone led to a small but detectable increase in decatenation. However, the addition of 30 mM citrate or isocitrate significantly stimulated decatenation beyond the effect of adding Mg2+ alone (Figure S3). These data indicate that the chelation of free Mg2+ ions by citrate or isocitrate does not appreciably contribute to the stimulation of topo II strand passage activity.
To further eliminate the possibility of indirect solute effects, we tested the two most active compounds, citrate and oxaloacetate, in a reaction buffer containing potassium chloride (KCl) (Figures S4A–D & I–L) or potassium glutamate (KGlu) (Figures S4E–H) to compare to our original experiments, which were performed with potassium acetate (KOAc) as a salt (Figures 4A–J & S2). Interestingly, citrate maintained stimulatory activity in both conditions, but oxaloacetate failed to stimulate topo II when KCl was present as a salt (Figure S4A–H). Isocitrate also maintained stimulatory activity in the absence of acetate or glutamate (Figure S4I–L). These results suggest that the relevant metabolite-binding site on topo II associates most effectively with short chain tricarboxylic acids, but is responsive to dicarboxylic acids providing that a small, high-abundance carboxylic acid is also available to occupy the third site (Figure S4M). In support of such a model, and to survey the stereospecificity of the topo II-tricarboxylate interaction, we compared the effects of racemic isocitrate to D-threo-isocitrate, the naturally occurring isomer. D-threo-isocitrate indeed proved significantly more effective in stimulating topo II supercoil relaxation as compared to the racemic mixture, indicating the topo II preferentially binds the natural isomer (Figure S4I–L). This finding further supports the idea that there exists one or more specific binding sites for activating molecules whose chemical features match those of TCA cycle intermediates.
We next biochemically characterized the ability of TCA metabolites to modulate the ATPase activity of ScTop2 (Figures 4K, 4L and S5). Oxaloacetate was excluded from the ATPase activity experiment because as a substrate for lactate dehydrogenase, it interferes with the coupled reaction. We observed that, except for citrate, all evaluated metabolites led to only a very slight increase in the maximum rate of ATP hydrolysis (< 40%) as compared to the glutamate control. The dicarboxylate intermediates had no significant effect on the Km of enzyme affinity, whereas an increase in Km (~2–3 fold) was seen with tricarboxylate metabolites (a response that may relate to the chelating activity of these compounds, as Mg2+ is a co-factor for the topo II ATPase reaction). Overall, our data indicate that TCA metabolites have relatively little effect on ATP turnover by topo II as compared to the degree of stimulation they exert on the enzyme’s strand passage activity, demonstrating that the topo II-metabolite interaction increases enzyme efficiency.
The interaction between topo II and TCA cycle metabolites is specific to eukaryotic type II topoisomerases
Having established a stimulatory interaction between TCA intermediates and yeast topo II, we assessed whether this effect was maintained in human homologs. We tested citrate as a representative tricarboxylic acid and succinate as representative dicarboxylic acid against human topo IIα (HsTop2A) and topo IIβ (HsTop2B). Both compounds stimulated DNA decatenation by the human topo IIs (Figures 5A–H). HsTop2B decatenation activity was increased by ~10- to 15-fold by both compounds (Figures 5A–D). Succinate had a similar effect on HsTop2A, whereas stimulation of HsTop2A by citrate was biphasic (Figures 5E–H), reaching a peak level of decatenation activity at 30mM citrate that was enhanced ~20-fold compared to the lowest amount of metabolite tested (5 mM) (Figure 5F). Because the TCA cycle is present in prokaryotes as well as eukaryotes, we also tested the activity of citrate and succinate against E. coli topo IV (Figures 5I–L). Interestingly, both compounds behaved like glutamate and had little to no effect on kDNA decatenation by topo IV. Finally, the effects of citrate and succinate were tested on DNA supercoil relaxation activity by yeast topo I, a eukaryotic type IB topoisomerase (Figures 5M–P). Both citrate and succinate were seen to give rise to a very slight stimulatory effect on the enzyme at the highest concentrations tested, but to a much lesser extent than seen for topo II (< 2-fold compared >5-fold); however, unlike with topo II, a similar increase in activity was seen for topo I with the glutamate negative control. This pattern indicates that the mild enhancement of relaxation activity seen for the metabolites with topo I is likely due to a salt effect, as opposed to a specific interaction. These findings demonstrate that the action of TCA metabolites is particular to eukaryotic type II topoisomerases and is evolutionarily conserved from yeast to humans.
Figure 5. Stimulatory effect of TCA metabolites is eukaryotic specific.
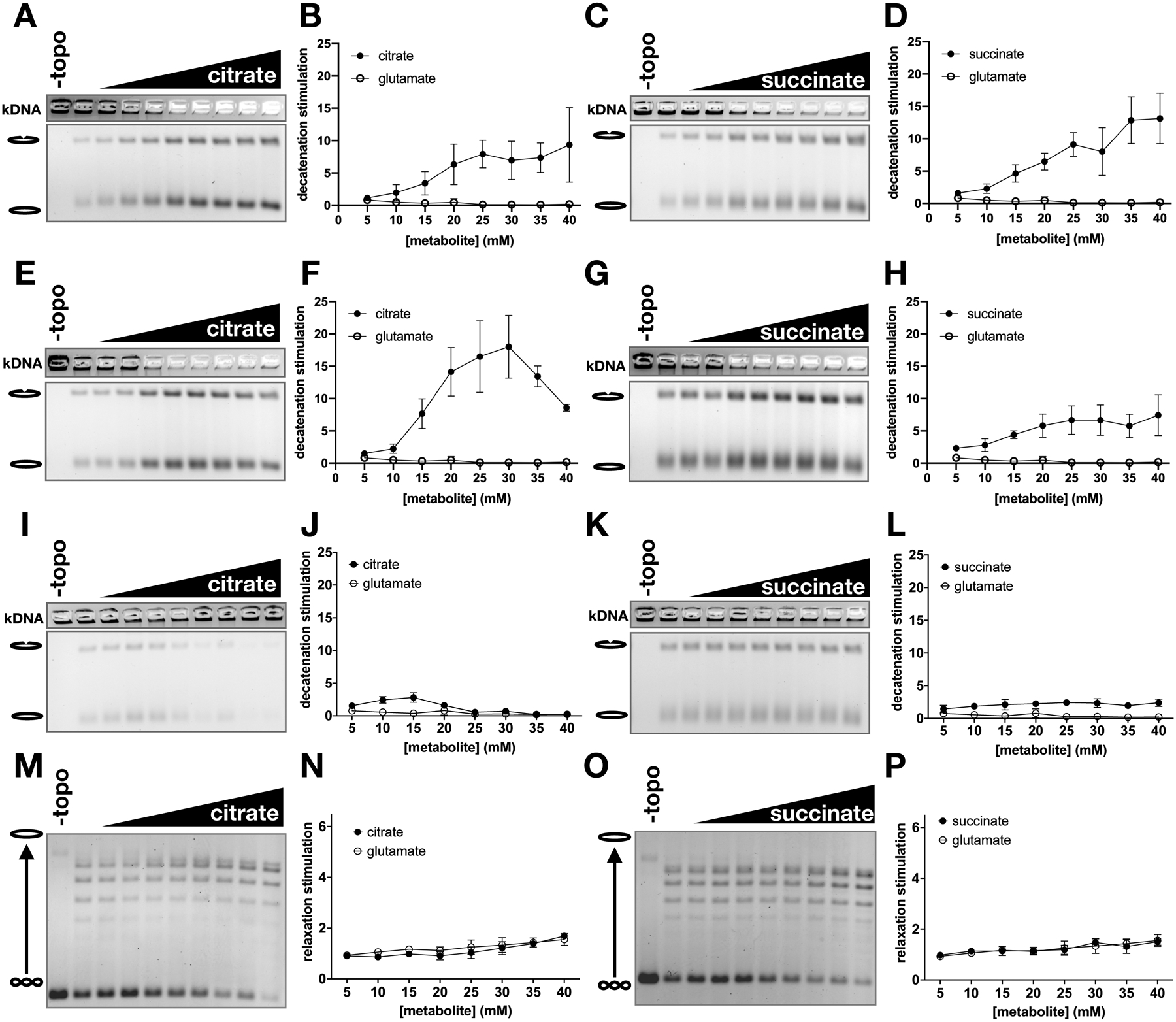
Citrate and succinate were titrated from 0 to 40 mM in 5 mM increments and added to decatenation assays with HsTop2B (A-D), HsTop2A (E-H), and Ec topo IV (I-L), and to supercoil relaxation assays with Sc topo I. Graphs represent mean ± SD of n=3–5.
TCA cycle flux affects levels of topo II activity in vivo
Having established that TCA cycle metabolites exert a stimulatory biochemical effect on topo II, we proceeded to explore the physiological relevance of this activity. Our in vitro data predicted that an increase or decrease in the abundance of TCA cycle intermediates should lead to commensurate changes in topo II activity in the cell. To test this hypothesis, we devised two approaches to modulate TCA cycle flux and assess topo II function in vivo using drug sensitivity. Experiments were conducted with a drug-efflux deficient (ED) strain of S. cerevisiae to improve the dynamic range of the growth assays (Stepanov et al., 2008).
We first assessed the effects of lowered TCA metabolite abundance by deleting mpc1, a subunit of the mitochondrial pyruvate carrier (MPC), in the ED background. Transport of pyruvate into the mitochondrial matrix replenishes TCA cycle intermediates; loss of mpc1 leads to a significant decrease in the concentration of these molecules (Herzig et al., 2012; Morita et al., 2019). Given our biochemical data, we hypothesized that topo II activity levels should be lower in the mpc1Δ strain as compared to the wildtype MPC1 strain on account of the diminished metabolite levels (Figure 6A). To test this idea, we evaluated the sensitivity of the two strains to chemical agents that antagonize topo II. Type II topoisomerase antagonists are broadly classified as either poisons or general catalytic inhibitors (Nitiss, 2009). Poisons stabilize the cleavage complex of topo II with DNA, leading the enzyme to form persistent protein-DNA adducts and DNA breaks. As a result, cells that overexpress topo II are hypersensitive to poisoning agents (Nitiss et al., 1992). In contrast, general catalytic inhibitors reduce enzyme activity without directly stimulating DNA breakage. As a consequence of these different modes of action, changes in topo II activity will have opposing effects on the sensitivity of cells to poisons or catalytic inhibitors: increasing topo II activity is expected to lead to hypersensitivity to poisons (due to the elevated formation of toxic cleavage complexes) and resistance to catalytic inhibitors (because stimulation of enzyme activity overcomes general inhibition), whereas decreasing topo II activity should display the converse (Figure 6A). Thus, changes in drug cytotoxicity report on changes in topo II activity.
Figure 6. Changes in TCA cycle flux affect sensitivity of yeast to topo II inhibitors.
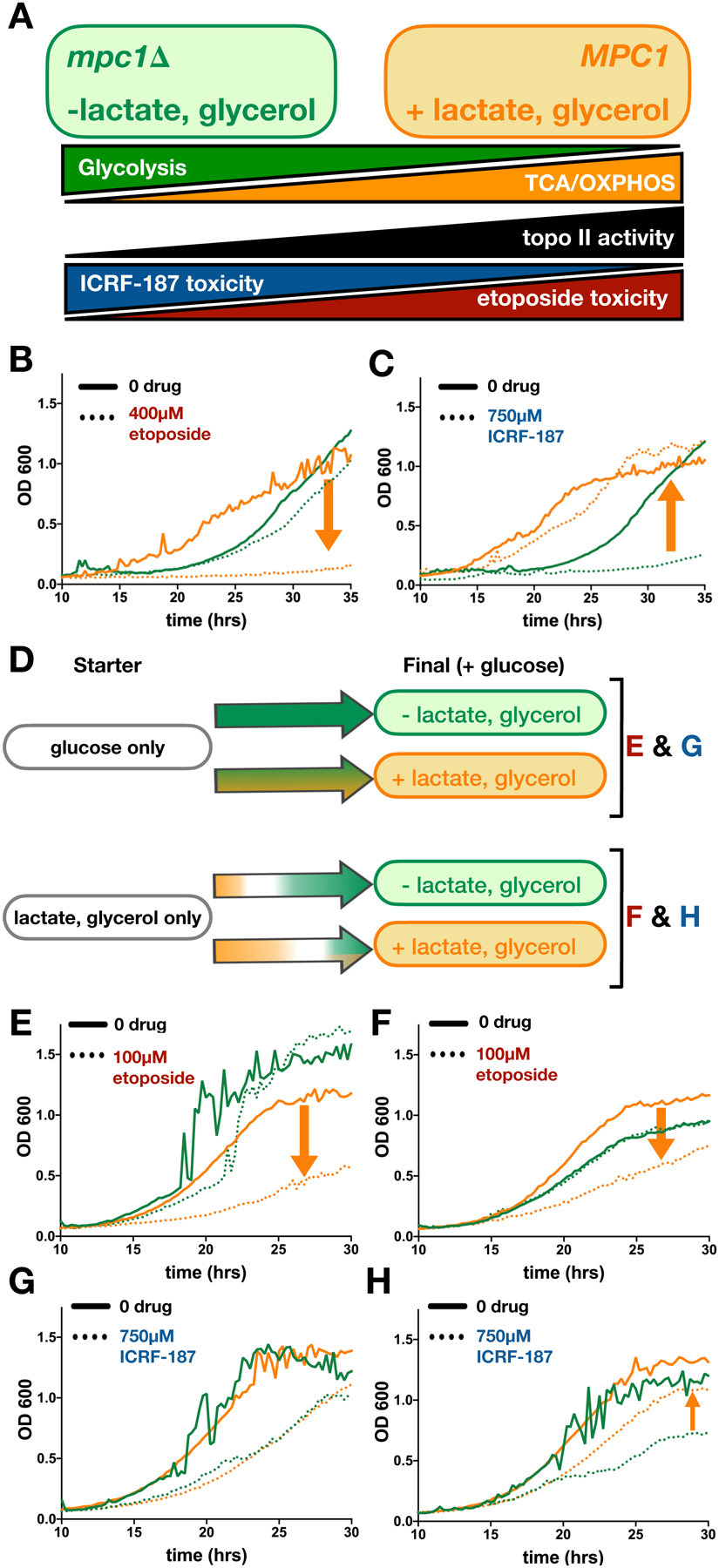
Green indicates conditions in which TCA flux is decreased. Orange indicates conditions in which TCA flux is increased. (A) Predicted correlation between ATP-generating metabolic pathways and topo II activity. Cells generate ATP by glycolysis (green) and oxidative phosphorylation (TCA/OXPHOS, orange). Our model predicts that topo II activity (black) is directly correlated to TCA metabolism; thus, etoposide toxicity (red) should directly correlate with TCA flux and ICRF-187 toxicity (blue) should inversely correlate with TCA flux. (B-C) Growth curves of mpc1Δ (green) compared to MPC1 (orange) in the presence of etoposide (B) and ICRF-187 (C). Orange arrows show the effect of increasing TCA flux (down indicates sensitization and up indicates rescue, see also Figures S7A–B). (D) Schematic of the effects of nutrient changes on flux through glycolysis and TCA cycle. Colored arrows show shifts in metabolism over time. Letters to the right of (D) indicate the nutrient conditions of the growth curves shown in (E-H). (E-H) Green lines show growth in glucose-only media and orange lines show growth in media with glucose and LG. Cultures were inoculated from starters grown in glucose-only media (E and G) or LG-only media (F and H). As before, orange arrows show the effect of increasing TCA flux on cytotoxicity of etoposide (E-F) and ICRF-187 (G-H). See also Figures S7F–I.
For our experiments, we chose etoposide as a topo II poison and ICRF-187 as a catalytic inhibitor (Ross et al., 1984; Tanabe et al., 1991). The effects of these drugs on the growth of the mpc1Δ strain and the MPC1 strain were observed by measuring optical density over the course of 35 hours. The mpc1Δ strain proved resistant to etoposide and sensitized to ICRF-187 as compared to the control, consistent with the idea that decreasing TCA intermediate abundance leads to a decrease in topo II activity (Figures 6B, 6C, S6A, and S6B).
To test the effect of increasing TCA metabolite abundance, we developed a strategy to enhance TCA cycle flux by adding non-fermentable carbon nutrients. As described by the Crabtree effect (Crabtree, 1928), yeast will opt to generate ATP through glycolysis rather than oxidative phosphorylation under high glucose conditions, regardless of oxygen availability. However, when fermentable carbon sources are limited, yeast will generate ATP by oxidative phosphorylation (OXPHOS), which is coupled to the TCA cycle. To induce different metabolic states, we cultured yeast in minimal media either with glucose only or glucose supplemented with lactate and glycerol (LG). Lactate and glycerol are non-fermentable and must be metabolized through the TCA cycle to generate ATP. By LC-MS analysis, we were able to qualitatively compare the levels of five TCA metabolites in yeast grown in glucose media with and without LG. We detected significant increases in succinate and malate in the presence of LG and no significant changes in the other metabolites (Figure S6C). To further check that the addition of LG increases TCA flux, we compared the effect of UK-5099, a chemical inhibitor of MPC that inhibits pyruvate entry into the TCA cycle, on colony growth in the two different nutrient environments. Yeast cells proved more resistant to UK-5099 in media containing LG, confirming that the addition of non-fermentable carbon sources increases TCA flux to counteract the effects of UK-5099 (Figure S6D).
We initially examined the effects of adding LG on the sensitivity of yeast cells to anti-topo II agents using a spot-growth assay. The addition of LG sensitized yeast to etoposide as expected (Figure S6E) but we were unable to reach cytotoxic concentrations of ICRF-187 necessary to see rescue. We therefore designed a liquid media growth assay incorporating a diauxic shift to assess the impact of TCA metabolism on cellular sensitivity to topo II antagonists (Figure 6D). Starter cultures were grown to saturation in media containing either glucose alone, or in LG alone, to allow the yeast to adjust to either a glycolysis-dominant or an OXPHOS-dominant expression profile. These cells were then shifted into glucose-containing media with and without LG to monitor growth in the presence of anti-topo II drugs. Because glucose is the preferred carbon source, yeast from the glucose-only starter condition will primarily rely on glycolysis and show only a slight increase in TCA metabolism in the presence of LG (Figure 6D). By contrast, yeast grown in LG-only media will undergo a diauxic shift as glucose is introduced. If LG are present, this diauxic shift will be delayed, and the concentration of TCA intermediates will be elevated as the cells attune to metabolizing non-glycolytic carbon substrates (Figure 6D). In agreement with the spot test, the addition of LG sensitized cells to etoposide (Figures 6E–F and S6F–G). Solution-based DNA cleavage assays with etoposide also show that both citrate and D-threo-isocitrate increase plasmid linearization in vitro (Figure S7), demonstrating that increasing topo II activity by metabolite action directly increases the toxicity of topo II poisons.
In assaying the metabolic-coupled response in vivo to a catalytic inhibitor, the addition of LG did not rescue cells that were acclimated to glucose-only nutrient conditions from ICRF-187 (Figures 6G and S6H). However, growth with LG did partially rescue yeast from ICRF-187 toxicity when the starter culture was grown without glucose (Figures 6H and S6I). Having observed the effect of acclimatizing cells to metabolize to either glucose or LG prior to drug treatment, we repeated this experiment on solid media with ICRF-193, a more potent form of ICRF-187 (Ishida et al., 1995; Tanabe et al., 1991). In this assay, regardless of whether the cells were exposed to drug on plates with glucose-only or glucose supplemented with LG, cells that had been grown to saturation in a starter culture containing only LG were almost completely rescued from ICRF-193 toxicity (Figure S6J). Collectively, these results show that changes in TCA status modulate cellular responses to topo II inhibitors and poisons, consistent with their ability to directly impact enzyme function in vitro.
To confirm that the changes in sensitivity to topo II-targeted drugs were due to catalytic stimulation and not a result of differences in protein expression levels, we generated two yeast strains with a 3xHA-tag at either the N-terminal or C-terminal end of endogenous topo II. These strains were then cultured in glucose-only media or glucose media supplemented with LG to compare topo II expression levels (Figure S7). Cells were harvested in mid-log phase to capture topo II expression in actively dividing cells and assessed by Western blot. These data show that the change in relative topo II protein abundance across the different growth conditions was insignificant, indicating that the differences in topo II activity observed in the drug toxicity experiments derive from biochemical stimulation of the enzyme and not due to alterations in protein concentration.
Discussion
Regulatory interactions between endogenous metabolites and DNA machinery
TCA cycle intermediates are some of the most abundant metabolites in the cell, with compounds such as citrate, succinate, and malate each reaching concentrations in the 1–5 mM range (Park et al., 2016; Wittmann et al., 2005). Here, we show that topo II is subject to regulatory control by TCA cycle metabolites. The metabolite binding site on topo II appears to preferentially engage tricarboxylic acids, but because small molecules containing carboxylates are also abundant in the cell (e.g., acetate and glutamate), fluctuations in the concentrations of the dicarboxylic TCA cycle intermediates may also influence topo II activity.
Given that seven TCA cycle intermediates stimulate topo II activity, a shift in the collective concentration of this metabolite pool could exert a functional effect. In vitro, supercoil relaxation was stimulated ~5-fold and DNA decatenation stimulated ~12-fold at metabolite concentrations ≥ 20 mM (Figure 4). However, up to 3-fold increases in activity were evident at lower, more physiological levels (between 5–15 mM). Many biological systems maintain homeostatic setpoints where modest (sub-twofold) biochemical changes can have profound physiological consequences. For example, a < 2-fold increase in Km and < 20% decrease in kcat of the tetracycline resistance protein, TetX2, imparts robust resistance to tetracycline in bacteria (Walkiewicz et al., 2012). Similarly, a ~15% decrease in the expression of the MSH2 repair protein is reported to confer chemoresistance to temozolomide, in glioblastoma cells (McFaline-Figueroa et al., 2015). Regulatory processes can also be highly sensitive to minor changes in protein activity. A < 10% difference in the concentration of Bicoid, a morphogen, determines cell fate during D. melanogaster embryonic development through transcriptional regulation (Driever and Nüsslein-Volhard, 1988; Fradin, 2017). Based on these examples, it is not unreasonable that even a two-fold change in topo II activity resulting from changes in TCA cycle flux would have physiologic effects. Consistent with this idea, we show that TCA cycle status directly impacts topo II function in vivo, with changes in TCA flux altering the cytotoxicity of topo II-targeted drugs in yeast (Figure 6).
Why link topo II activity to metabolic status? One possibility is that metabolic sensing by topo II may provide a means for coordinating enzyme function with the cell cycle, as metabolism is highly responsive to environmental changes (Brauer et al., 2008; Lee and Finkel, 2013; Wellen and Thompson, 2010; Zhu and Thompson, 2019). Progression through the cell cycle requires the activation of anabolic pathways to accommodate increased transcription, translation, and DNA replication. As such, metabolism has been observed to fluctuate in accordance with the cell cycle (Almeida et al., 2010; Buchakjian and Kornbluth, 2010; Colombo et al., 2010; Liu et al., 2020; Salazar-Roa and Malumbres, 2017; Tudzarova et al., 2011; Wang et al., 2014). Proliferating cells up-regulate glycolysis in G1 to accumulate ATP and carbon substrates for anabolism (Buchakjian and Kornbluth, 2010; Diaz-Moralli et al., 2013; Pavlova and Thompson, 2016), but as cells enter S phase, metabolism shifts towards the pentose phosphate pathway to support DNA replication (Diaz-Moralli et al., 2013; Da Veiga Moreira et al., 2015). As transcription ramps down upon chromosome condensation (Johnson and Holland, 1965; Littau et al., 1964; Prescott and Bender, 1962; Taylor, 1960), cells depend on the TCA cycle and OXPHOS as a more efficient source of ATP to fuel mitotic progression (Wang et al., 2014). The transition in energy metabolism towards TCA cycle/OXPHOS at mitotic entry mirrors the upregulated expression pattern of vertebrate topo IIα. We hypothesize that TCA cycle fluctuations coordinate an increase of topo II activity at the G2-to-M transition to ensure full decatenation of chromosomes. This mechanism may be particularly important in lower-order eukaryotes that do not display cell cycle-dependent expression of topo II and could have been preserved in vertebrates capable of upregulating topo IIα expression to contend with their larger chromosomal content.
Cancer-specific metabolic profiles may impact efficacy of topo II-targeted chemotherapeutics
Cancer cells have distinguishing metabolic features, one of the first of which was observed by Otto Warburg (Warburg, 1925). The Warburg Effect, characterized by increased aerobic glycolysis in cancer cells, was initially thought to indicate a shift away from TCA cycle/OXPHOS to favor the rapid production of ATP through glycolysis; however, growing evidence shows that elevated glycolysis serves to provide anabolic intermediates for biosynthesis, and that TCA cycle/OXPHOS remain active in cancer cells (Locasale et al., 2011; Pavlova and Thompson, 2016). TCA cycle metabolites are key anabolic precursors and citrate, in particular, is critical to support elevated lipid biosynthesis in cancer cells (Li and Cheng, 2014; Menendez and Lupu, 2007). Recent studies have also found that the impairment of TCA cycle/OXPHOS can decrease tumorigenic and metastatic potential (Cai et al., 2020; Cavalli et al., 1997; Lebleu et al., 2014; Morais et al., 1994; Tan et al., 2015). These findings demonstrate that cancer cells not only maintain active TCA cycle metabolism but are dependent on it.
Different cancer cell types have distinct metabolic dependencies that may influence their response to various treatments (Diaz-Ruiz et al., 2011; Guppy et al., 2002; Martin et al., 1998; Pasdois et al., 2003). With respect to the TCA cycle, mutations in several TCA cycle enzymes are linked to specific cancers. Isocitrate dehydrogenase (IDH1/2) mutations are frequently found in gliomas, cholangiocarcinomas, and acute myeloid leukemias (Balss et al., 2008; Borger et al., 2012; Yan et al., 2009; Yang et al., 2012). Inactivating mutations in succinate dehydrogenase and fumarate hydratase have also been shown to cause an accumulation of succinate and fumarate in paragangliomas and pheochromocytomas, as well as in some gastrointestinal and renal cell cancers (Astuti et al., 2001; Baysal et al., 2000; Janeway et al., 2011; King et al., 2006; Letouzé et al., 2013; Selak et al., 2005; Tomlinson et al., 2002). The accumulation of oncometabolites (e.g., succinate and fumarate) are thought to affect cell fate determination pathways by disrupting chromatin modification status and DNA repair processes (Baksh and Finley, 2020; Intlekofer and Finley, 2019; Sulkowski et al., 2020; Xiao et al., 2012). Our demonstration that elevated levels of TCA intermediates sensitize yeast cells to topo II poisons (Figure 7) suggests that topo II-targeted therapies may be more effective against cancers that accumulate oncometabolites. Future efforts will be needed to test this supposition.
Figure 7. Schematic depicting how sensitivity to topo II-targeting drugs is influenced by metabolic state.
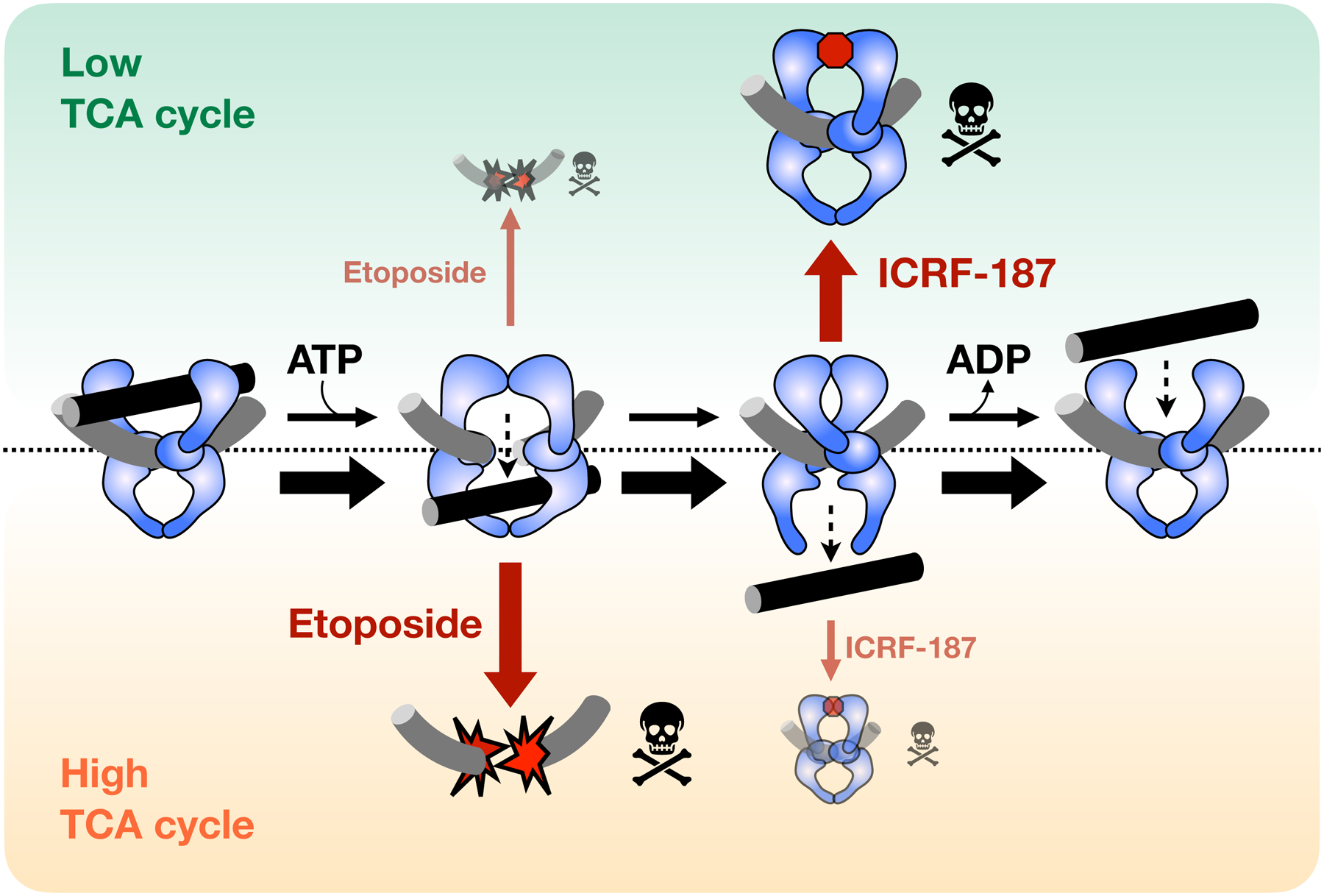
When TCA cycle flux is low (green) topo II strand passage activity is not stimulated. General catalytic inhibitors (e.g., ICRF-187) that decrease topo II activity are more effective in this state, as compared to a high TCA state (orange). By contrast, topo II poisons (e.g., etoposide) are more toxic in the high TCA state because elevated topo II activity leads to increased formation of DNA cleavage complexes.
Concluding remarks
The present work describes the ability of native small-molecule metabolites to directly activate eukaryotic topo II in vitro and in vivo. Many topo II inhibitors have been studied, but small molecules capable of stimulating topo II are far rarer. Polyamines, such as spermine and spermidine, can enhance topo II supercoil relaxation activity by condensing DNA but these compounds also affect the activity of type I and bacterial type II topoisomerases (Pommier et al., 1989; Srivenugopal et al., 1987). By contrast, the specificity of TCA metabolites for stimulating eukaryotic topo II indicates that the effect is due to a direct enzyme-metabolite interaction (Figure 5). These findings also represent a remarkable example of an allosteric interaction between a non-nucleotide metabolite and an enzyme involved in controlling a ‘central dogma’ process. Small molecule ‘alarmones,’ such as (p)ppGpp and cGAMP, can regulate translation, transcription, and DNA replication (Srivatsan and Wang, 2008; Sun et al., 2013); however, these agents are nucleotide analogs and are produced in response to stress. Metabolites such as α-ketoglutarate, succinate, and fumarate can act as cofactors or competitive inhibitors for chromatin-modifying enzymes (Baksh and Finley, 2020; Intlekofer and Finley, 2019), but prior to this study, none were known to allosterically regulate an enzyme involved in nucleic acid transactions. The discovery of a regulatory topo II-TCA cycle connection opens the possibility that other such interactions between nuclear enzymes and endogenous small molecules may modulate events such as transcription and/or DNA replication. Our approach for isolating metabolite-enzyme interactions is transferable to any system with an enzymatic activity assay. Future applications of the approach may reveal regulatory connections between cellular metabolism and other DNA-dependent machineries.
With regard to anti-topo II agents as cancer therapeutics, the off-target poisoning of topo IIβ appears to be a major source of negative side effects, including cardiotoxicity and secondary malignancies (Azarova et al., 2007; Felix, 1998; Turcotte et al., 2018; Yi et al., 2007). Because topo IIα and topo IIβ are highly similar, increasing the specificity of topo II inhibitors is challenging. Our data show a difference in the biochemical response of topo IIα and topo IIβ to di- and tricarboxylic acids (Figure 5); defining the biochemical underpinnings of these differences may provide new insights toward increasing the specificity of topo II drugs. To this end, we note that some fractions of our metabolic extracts showed inhibitory activity against topo II, indicating other biologically relevant metabolite interactions may exist that have yet to be uncovered. The identification of these agents could serve as a starting point for such investigations.
STAR Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James M. Berger (jmberger@jhmi.edu).
Materials Availability
All reagents generated for this study are available on request through the Lead Contact.
Data and Code Availability
The data obtained in this study is accessible at the NIH Common Fund’s NMDR (supported by NIH grant, U01-DK097430) website, the Metabolomics Workbench (Sud et al., 2016), http://dx.doi.org/10.21228/M89695. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Yeast strains and growth conditions
Crude metabolite samples were extracted from cultures of BY4741 yeast (MATα his3Δ1 leu2Δ met15Δ0 ura3Δ0). Growth experiments to assess the sensitivity of yeast to topo II-targeted drugs were performed with a drug-efflux deficient (ED) strain (pdr1Δ::pdr1:Cyc8 LEU2) described in (Stepanov et al., 2008). To generate the ED-mpc1Δ strain, the KANMX resistance marker was PCR amplified from the pUG6 plasmid (Güldener et al., 1996) with the following primers containing homology regions outside of the MPC1 gene: (5’)CAGCAAACGTCAATACATCTACATATATACGTATAGATTTTATTGCACTGTGATCGACATGGAGGCCCAGAATACC and (5’)GTTTCCATCTAGTCACCTACTTCAGGTTCTTAGACTGCTCGTTTTACCAGTATAGCGACCAGCATTC. The deletion construct was then transformed into ED yeast. After recovery on YPD plates followed by selection on G418 containing replica plates, mpc1Δ mutants were verified by PCR.
CRISPR/Cas9 technology was used to incorporate a 3xHA-tag at the N-terminus and C-terminus of endogenous topo II in the BY4741 background. The following gRNA sequences were integrated in the CRISPR/Cas9 vector, pJH2972 (Anand et al., 2017): (5’)GCAGTGAAAGATAAATGATCGTTGACATGGTTAGCCGTGCGTTTTAGAGCTAGAAATAGC (N-terminal gRNA) and (5’)GCAGTGAAAGATAAATGATCAAAAAGAATGGCGCTTTCTCGTTTTAGAGCTAGAAATAGC (C-terminal gRNA). The CRISPR/Cas9 constructs were then transformed into BY4741 yeast with their corresponding repair templates: (5’)TTTCAGTTAAAGGAGTTTATAACGACGAGCACGCCTAACCATGTACCCATACGATGTTCCTGACTATGCGGGCTATCCCTATGACGTCCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTACGCTGGTACGATGTCAACTGAACCGGTAAGCGCCTCTGATAAATATCAGA (N-tag) and (5’)GGAAAACCAAGGATCAGATGTTTCGTTCAATGAAGAAGATGGTACGTACCCATACGATGTTCCTGACTATGCGGGCTATCCCTATGACGTCCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTACGCTTGAATAATATTTATCGAGAGAAAGCGCCATTCTTTTTATA (C-tag). Integration of the repair constructs was verified by colony PCR followed by BamHI digest of the product (only the PCR product from a strain with correct integration of the 3xHA-tag will have a BamHI cut site).
Cultures for metabolite extraction were grown in synthetic media in which ammonium sulfate was replaced with monosodium glutamate as the nitrogen source (SC-sulfate). Per liter, SC-sulfate media contains 75 mg of each amino acid, 20 mg of adenine, 75 mg of uracil, 25 mg of inositol, 1.71 g of YNB-sulfate (Sunrise Science), 1 g monosodium glutamate, and 20 g dextrose. Growth assays were conducted in SC media (Sunrise Science). Glucose (20 g/L), lactate (2%) and glycerol (1.5%) were added to the media after autoclaving as required for each experiment.
Method Details
S. cerevisiae metabolite extraction
BY4741 cultures grown in SC-sulfate media were harvested at early log phase, OD600 = 0.4–0.8, by vacuum filtration through a surfactant-free cellulose acetate membrane with a 0.2 μm pore size (ThermoFisher Scientific). When ~5 mL of media was left in the filter, 30 mL of 100 mM NH4OAc – TEA (pH 8.0) was added to wash the cells and remove any remaining extracellular material. Immediately after the wash solution was passed through the filter, the filter apparatus was released from vacuum and 10 mL of 90% MeOH, 10 mM NH4OAc – TEA (pH 8.0) (chilled to −80 °C) was added to the filter for rapid quenching and extraction (Boer et al., 2010; Crutchfield et al., 2010). The extract was then centrifuged at 4000g for 10–15 min to remove cell debris. The extract supernatant was passed through a 3 kDa MWCO filter (Cytiva) to remove large macromolecules. The filtrate was diluted 2-fold with water and lyophilized to remove the extraction solvent. The remaining extracts were stored at −80 °C for up to one month.
Supercoil relaxation, decatenation, DNA cleavage and ATPase activity assays
Recombinant eukaryotic topo II proteins were prepared as described previously (Lee et al., 2017). Briefly, ScTop2, HsTop2A, and HsTop2B were expressed in the S. cerevisiae strain BCY123 (Wasserman and Wang, 1994), and cell pellets were lysed by cryogenic grinding. Proteins were first purified by Ni-affinity purification (HisTrap FF, GE) followed by cation exchange (HiTrap SP, GE). Affinity tags were removed by His6-tagged TEV protease (QB3 MacroLab). The digested proteins were passed over a second Ni-affinity column to remove the cleaved His6-tag and the His6-TEV protease. For the final purification step, proteins were run on a gel-filtration column (S-400, GE). Recombinant Ec topoIV was prepared as described previously (Vos et al., 2013). His6-tagged subunits of Ec topoIV (ParE and ParC) were expressed in BL21 codon-plus (DE3) RIL cells and cells were lysed by sonication. Proteins were first purified by Ni-affinity purification (HiTrap Ni2+, GE). After removing the affinity tags by TEV cleavage and a second Ni-affinity step as described above, the proteins were purified by gel filtration (S-300, GE). Recombinant His6-MBP-tagged S. cerevisiae topo I was cloned into and expressed from the 12UraC vector (QB3 Macrolab) in BCY123 cells as described for eukaryotic topo II proteins. Cells were lysed by cryogenic grinding and resuspended in 50 mM Tris-HCl (pH 7.4), 200 mM KCl, 20 mM imidazole (pH 8.0), 10% glycerol, 0.5 mM TCEP, 1 mM PMSF, 2.34 μM leupeptin, and 1.45 μM pepstatin. After clarification, the lysate was loaded onto a HisTrap FF (GE) column, washed in the resuspension buffer, and eluted with 200 mM imidazole (other buffer components were equal to the resuspension buffer). The elution was treated with His6-TEV (QB3 Macrolab) while dialyzing back into the resupension buffer and then repassed over the HisTrap FF column to isolate the cleaved protein. The protein was run over a gel filtration column (S-400, GE) in 50 mM Tris-HCl (pH 7.4), 500 mM KCl, 10% glycerol, and 0.5 mM TCEP. Purified proteins were concentrated (10 to 20 μM for eukaryotic proteins and 0.5 to 1.0 mM for topo IV subunits) and flash frozen in gel filtration buffer supplemented with 30% glycerol. Frozen aliquots were stored at −80 °C.
Negatively supercoiled pSG483, a derivative of pBluescript SK, was prepared from XL1-Blue E. coli cultures by maxi prep with NucleoBond Xtra Maxi columns (Macherey-Nagel) (Lee et al., 2017). The purified plasmid substrate was diluted to 250 ng/μL in water, flash frozen in single-use aliquots, and stored at −20 °C. Metabolite extracts were solubilized in 12.5% DMSO and metabolite stocks prepared from commercial compounds were solubilized in water and adjusted to pH 6.5–7.5 with HOAc or KOH.
For strand passage activity assays and DNA cleavage assays, enzyme stocks were first diluted in two-fold steps in 30 mM Tris-HCl (pH 7.9), 500 mM KOAc, 10% glycerol and 0.5 mM TCEP. Ec topo IV subunits were incubated together on ice at high concentrations (200 – 300 μM of each subunit) for 10 min prior to dilution to allow formation of the holoenzyme. The enzyme was then added to the appropriate DNA substrate, negatively supercoiled pSG483 plasmid for supercoil relaxation and DNA cleavage assays or kDNA (Inspiralis) for decatenation assays, followed by metabolites. Reactions were initiated by the addition of ATP (5 mM for assays with extracts and 1 mM for assays with commercial metabolites) and shifted to 30 °C for ScTop2 and Sc topo I or 37 °C for HsTop2A/B and topo IV. All final reaction conditions contained 32 mM Tris-HCl (pH 7.9), 0.05 mg/ml BSA, 0.6 mM TCEP, and 10% glycerol. Assays conducted in acetate buffers contained 100 mM KOAc, 20 mM Mg(OAc)2; assays conducted in glutamate buffers contained 100 mM monopotassium glutamate, 20 mM magnesium diglutamate; and assays conducted in chloride buffers contained 100 mM KCl and 20 mM MgCl2. For assays with metabolite extracts, solid material left after solvent removal from each fraction was solubilized in equal volumes of 12.5% DMSO and titrated into relaxation assays in 2-fold dilution steps. The final concentration DMSO in reactions with metabolite extracts was 2.5%. Decatenation reactions were incubated for 3 min and supercoil relaxation and DNA cleavage assays were incubated for 5 min. Relaxation and decatenation reactions were quenched with 20 mM EDTA and 1% SDS and DNA cleavage reactions were quenched with only 1% SDS. All reactions were treated with 0.2mg/mL Proteinase K to remove any remaining protein bound to DNA. Final products were resolved and visualized by gel electrophoresis (1.4% agarose, 1X TAE) run at 2–2.5 V/cm for 15–20h. Supercoil relaxation products were run on native gels whereas decatenation and cleavage products were run on gels containing 0.4 μg/mL ethidium bromide.
ATP hydrolysis by topo II was measured by an NADH-coupled ATPase assay. Reactions were prepared with 50 nM ScTop2, 200 ng/μL sheared salmon sperm DNA (Fisher Scientific), 0.5 mM NADH, 0.2% rabbit muscle pyruvate kinase/lactate dehydrogenase (Sigma-Aldrich), 2 mM PEP, 36 mM Tris-HCl (pH 7.9), 100 mM KOAc, 10 mM Mg(OAc)2, 0.05 mg/mL BSA, 0.6 mM TCEP, and 2% glycerol in a final reaction volume of 75 μL. ATP concentrations were titrated from 0–4 mM. Depletion of NADH was observed over the course of one hour at 30°C by monitoring absorbance at 340 nm in a CLARIOStar microplate reader (BMB LAB TECH). ATP hydrolysis rates were calculated based on an NADH standard curve and fit for Km and Vmax values in PRISM (GraphPad).
Fractionation and purification of active compounds from crude metabolite extracts
Lyophilized samples of metabolite extracts were resuspended with water and adjusted to pH 3.5 with formic acid or pH 10 with TEA. The metabolite solution was transferred to a separatory funnel and mixed with an equal volume of butanol. The mixture was then left until the separation of aqueous and organic layers was complete. The aqueous solvent was removed by lyophilization and the organic solvent was removed in a rotary evaporator. The activities of the aqueous and organic fractions from acid- or base- extraction were assessed in a supercoil relaxation assay to determine the hydrophilic nature of the active compound.
Enrichment and purification of the stimulatory metabolite from crude extracts for LC-MS/MS analysis began with solid phase extraction (SPE). Crude extracts were solubilized in water and adjusted to pH 10 with TEA. A mixed-mode anion exchange cartridge was equilibrated as per the manufacturer’s instructions (Oasis MAX, Waters). The flow-through from the passage of the crude extract through the SPE column (i.e., unbound material) was lyophilized for further purification.
The extract was then further purified by reverse-phase HPLC on a C18 column (Atlantis T3, Waters). One column volume of water with 0.1% formic acid followed by eight column volumes of MeOH were passed through the column over the course of the HPLC run. Absorbance at 260 nm and 280 nm wavelengths was observed by a Waters 2996 Photodiode array. Fractions from the aqueous phase were lyophilized and assessed for stimulation of supercoil relaxation. Active fractions were then further purified through normal-phase HPLC on an amide column (XBridge BEH Amide, Waters). One column volume of 80% acetonitrile with 0.1% formic acid followed by a three-column volume gradient down to 40% acetonitrile with 0.1% formic acid were passed through the column. Afterwards, one column volume of water with 0.1% formic acid was passed over the column as the last step. The separated fractions were lyophilized and assessed similarly to all prior purification steps.
Antarctic phosphatase and phosphodiesterase treatment of metabolites
Snake venom phosphodiesterase I (SVPD, Sigma-Aldrich) was reconstituted at 1 mg/mL in 20 mM Tris-HCl (pH 7.9), 100 mM NH4OAc, 20 mM Mg(OAc)2, 50% glycerol (Konokhova et al., 2016). Crude extracts were first fractionated by SPE as described above. The lyophilized sample was then resuspended in water and split into four samples of 44 μL each. Next, 5μl of 10X reaction buffer was added to each sample: 10x Antarctic phosphatase (AP) reaction buffer (New England BioLabs) was added to two samples and 200 mM Tris-HCl (pH 7.9), 1 M NaCl, 200 mM MgCl2 was added to the remaining two samples for SVPD treatment. For each pair of samples, 1 μl of water was added to the no enzyme control and 1 μl of enzyme (AP or SVPD) was added to the second sample. All reactions were incubated at 37 °C for two hours. Samples were then diluted to a 1 mL final volume and passed through a 3 kDa MWCO filter. The filtrate was lyophilized and assessed for activity against topo II in a supercoil relaxation assay.
Untargeted LC-MS/MS analysis of metabolite samples
Lyophilized samples were reconstituted in water plus 0.1% (v/v) formic acid such that samples reached a final concentration of 20 mg/ml. A pooled sample was generated by combining equal volume aliquots from each sample. Untargeted metabolomics uHPLC-MS/MS data acquisition was performed in both positive and negative ion modes and using both reversed-phase and normal-phase separations.
Liquid chromatographic separation was performed using a Dionex Ultimate 3000 uHPLC system (Thermo) for reversed-phase and normal-phase methods. For both methods, mobile phase A was 1mM ammonium acetate + 0.1% (v/v) formic acid in water and mobile phase B was 1mM ammonium acetate + 0.1% (v/v) formic acid in 20% water / 80% acetonitrile (v/v). Reversed-phase experiments were performed with a Hypersil GOLD aQ polar endcapped C18 column (150 mm × 2.1 mm, 1.9 μm particle size) (Thermo) using the following gradient of mobile phases: 0% B from 0–2 min, 0–60% B from 2–13 min, 60–90% B from 13–15 min, 90–0% B from 15–16 min, and 0% B from 16–20 minutes. Normal-phase experiments were performed with a Polyhydroxyethyl A hydrophilic interaction chromatography column (200 mm × 2.1 mm, 5 μm particle size) (PolyLC) using the following gradient of mobile phases: 80% B from 0–2 min, 80–40% B from 2–13 min, 40–10% B from 13–15 min, 10–80% B from 15–16 min, and 80% B from 16–20 min. For both uHPLC methods, the flow rate was set to 0.2 mL/min. Samples were each injected 3 times under each unique set of conditions. A blank injection of 0.1% formic acid in water (v/v) was run in between each sample injection to minimize carry over.
After separation by uHPLC, metabolites were ionized and detected by a Q-Exactive quadrupole-orbitrap mass spectrometer (Thermo) using a heated electrospray ionization source. Samples were run separately in negative and positive ion modes. In positive mode, ionization was performed with a spray voltage of 4000 V, capillary temperature of 275°C, sheath gas flow rate of 35 Arb, and auxiliary gas flow rate of 8 Arb. Negative mode ionization was performed with a spray voltage of 4000 V, capillary temperature of 350°C, sheath gas flow rate of 25 Arb, and auxiliary gas flow rate of 2 Arb.
For each set of separation and ion modes, mass spectra were acquired by scans in data-dependent MS/MS mode for the pooled samples and full-MS mode for individual fraction replicates. Full-MS scans were performed at a resolution of 140,000 and a mass range of m/z 65–850. Data-dependent MS/MS scans were carried out for the top-5 abundant ions at a resolution of 140,000 FWHM with a dynamic exclusion of 6.0 seconds and stepped normalized collision energy of 20, 40, and 100. MS/MS fragments were observed at a resolution of 17,500 FWHM.
The LC-MS and LC-MS/MS data were processed using Compound Discoverer 3.1 software (ThermoFisher Scientific). Spectral features retention times were aligned with a permitted mass deviation of 5 ppm. Background peaks were subtracted based on features in a blank sample. The data-dependent MS/MS files were analyzed for compound identification using mzCloud and the ChemSpider yeast metabolome database. The full-MS scan data was used for comparison of metabolite abundance by average peak area. Analysis of metabolite enrichment was performed by comparing replicates from fractions with an effect on topo II activity to those with no effect on topo II activity.
Targeted Metabolomics Analysis of TCA Metabolites
Lyophilized samples of crude extracts from yeast grown in different nutrient conditions were reconstituted in water + 0.1% formic acid, such that the ratio of milligrams of cell mass to microliters of acidified water was 0.5. Samples were analyzed on a Dionex Ultimate 3000 uHPLC (Thermo) coupled to a TSQ Vantage triple quadrupole mass spectrometer (Thermo). Analytes were separated on a Hypersil GOLD aQ polar endcapped C18 column (150 mm × 2.1 mm, 1.9 μm particle size) using an isocratic flow of 100% mobile phase A (water +0.1% formic acid) at a flow rate of 0.2 mL/min over five minutes. Analytes were ionized by heated electrospray ionization with a spray voltage of 3000 V, capillary temperature of 204°C, sheath gas flow rate of 50 Arb, and auxiliary gas flow rate of 55 Arb. Metabolites were detected in negative ion mode using distinct single reaction monitoring scans with the following transitions and collision energies - citric acid: m/z 191 → 111 (CE 12), alpha-ketoglutaric acid: m/z 145 → 101 (CE 10), succinic acid: 117 → 73 (CE 12), fumaric acid: 115 → 71 (CE 10), malic acid: 133 → 71 (CE 15). Chromatographic peak areas were used for comparisons of metabolite abundance. Data were normalized to the average of the control condition and unpaired, two-tailed t-tests were performed in PRISM (GraphPad) to determine significant differences.
S. cerevisiae growth assays
Stock solutions of etoposide (Sigma-Aldrich), ICRF-187 (TCI), and ICRF-193 (Sigma) were prepared at 100 mM concentration in 100% DMSO then aliquoted and stored at −20 °C. DMSO was added to all no drug experimental controls to match the DMSO content of the drug containing conditions. For spot growth assays, ED yeast were first grown to saturation at 30 °C overnight in SC media. The starter culture was then diluted to OD600 = 0.1 in sterile water. In a sterile 96-well plate, the culture was serially diluted down from OD600 = 0.1 in 5-fold dilution steps. A multichannel pipette was then used to spot 5 μL of each dilution on an SC agar plate with the appropriate drug condition for each experiment. Plates were incubated for 2–3 days at 30 °C and imaged by normal photography. For liquid media growth assays, ED or ED-mpc1Δ yeast were first grown to saturation at 30 °C overnight in SC media with the appropriate carbon source for each experiment. The cultures were then diluted to OD600 0.01 in the growth assay media condition and placed in a sterile 24-well plate with 1.5ml of culture in each well. The plates were incubated at 30 °C with constant shaking inside a Bio-Tek Synergy HT plate reader for 30 – 35 h. OD600 measurements were taken every 15 min to generate a growth curve for each well.
Western Blot
Approximately 1.5 × 108 cells or 15 OD600 units of log phase yeast (OD600 = 0.8–1.2) were harvested by centrifugation. Pellets were then resuspended in 200 μL of 10% trichloroacetic acid (TCA) and incubated at room temperature for 30 min. The TCA solution was then removed by centrifugation and the pellets were washed with 1 mL of 1 M HEPES•KOH (pH 7.5). After removal of the wash solution by centrifugation, the pellets were then resuspended in 50 μL 2x SDS-PAGE loading buffer with ~50 μL of 0.5 mM glass beads and vortexed for 3 min. An additional 50 μL of 2x SDS-PAGE loading buffer was added before the samples were boiled for 5 min and then vortexed for 15 sec. After centrifugation to pellet the beads, 20 μL of each supernatant sample were run on an SDS-PAGE gradient gel and transferred onto a PVDF membrane. The membranes were blotted with rat anti-HA (1:5000, Roche 11867423001) and rabbit anti-tubulin (1:5000, abcam ab184970) followed by IRDye 800CW goat anti-Rat (1:15,000, Li-Cor 926–32219) and IRDye 680CW goat anti-rabbit (1:15,000, Li-Cor 926–68071). Blots were imaged on a Li-Cor Odyssey system and band intensities were analyzed in ImageJ.
Quantification and Statistical Analysis
Data are presented as the mean ± SD of a minimum of three independent experiments, indicated by the n value described in the figure legends.
Supplementary Material
Table S2. Results of substructure search of succinic and glutaric acid motifs on the Human Metabolome Database, related to Figure 3.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rat anti-HA | Roche | CAT# 11867423001 (RRID:AB_390918) |
| rabbit anti-tubulin | abcam | CAT# ab184970 |
| IRDye 800CW goat anti-Rat | Li-Cor | CAT# 926–32219 (RRID:AB_1850025) |
| IRDye 680CW goat anti-rabbit | Li-Cor | CAT# 926–68071 (RRID:AB_10956166) |
| Bacterial and Virus Strains | ||
| XL1Blue (cloning strain) | QB3 MacroLab | N/A |
| BL21- CodonPlus (DE3) RIL | Agilent | CAT# 230245 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| YNB-sulfate Powder | Sunrise Science | CAT# 1538 |
| SC complete -Glucose powder | Sunrise Science | CAT# 1486 |
| Monosodium glutamate | Sigma-Aldrich | CAT# G1626 |
| Geneticin Selective Antibiotic (G418 sulfate) | ThermoFisher Scientific | CAT# 10131035 |
| Acetonitrile, Optima LC/MS grade | ThermoFisher Scientific | CAT# A955–4 |
| Methanol, Optima LC/MS Grade | ThermoFisher Scientific | CAT# A4561 |
| Ammonium acetate | Sigma-Aldrich | CAT# 238074 |
| Triethylamine (TEA) | Sigma-Aldrich | CAT# 471283 |
| Formic acid 98–100% | Fisher Scientific | CAT# 33015 |
| Trichloroacetic acid, ACS, ≥99% | Sigma-Aldrich | CAT# T6399 |
| kDNA | Inspiralis | N/A |
| Adenosine 5’-triphosphate disodium | Sigma-Aldrich | CAT# A2383 |
| Salmon Sperm DNA, sheared | ThermoFisher Scientific | CAT# AM9680 |
| NADH | Sigma-Aldrich | CAT# 10107735001 |
| Pyruvate kinase/lactate dehydrogenase | Sigma-Aldrich | CAT# P0294 |
| Phosphodiesterase I from Crotalus adamanteus venom | Sigma-Aldrich | CAT# P3243 |
| Antarctic phosphatase | NEB | CAT# M0289 |
| BamHI | NEB | CAT# R0136S |
| TEV protease | QB3 MacroLab | N/A |
| ScTop2 | Berger Lab | N/A |
| HsTop2A | Berger Lab | N/A |
| HsTop2B | Berger Lab | N/A |
| Ec topo IV | Berger Lab | N/A |
| Etoposide, synthetic, ≥98%, powder | Sigma-Aldrich | CAT# E1383 |
| Dexrazoxane (ICRF-187) | TCI Chemicals | CAT# D4227 |
| ICRF-193 | Sigma | CAT# I4659 |
| D-threo-isocitrate | ChiroBlock | CAT# 40056 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| ST001658 (mass spectrometry data set) | Metabolomics Workbench | http://dx.doi.org/10.21228/M89695 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Yeast strain: BY4741 MATα his3Δ1 leu2Δ met15Δ0 ura3Δ0 | ATCC | CAT# 201388 |
| Drug Efflux Deficient (ED) yeast strain (pdr1Δ::pdr1:Cyc8 LEU2) | Bjornsti Lab (Stepanov et al., 2008) | N/A |
| Yeast strain: ED- mpc1Δ (mpc1Δ pdr1Δ::pdr1:Cyc8 LEU2) | Berger lab | N/A |
| Yeast strain: BCY123 (eukaryotic topo II expression strain) | Berger lab | N/A |
| Oligonucleotides | ||
| Mpc1 deletion construct forward primer: CAGCAAACGTCAATACATCTACATATATACGTATAGATTTTATTGCACTGTGATCGACATGGAGGCCCAGAATACC | IDT DNA | N/A |
| Mpc1 deletion construct reverse primer: GTTTCCATCTAGTCACCTACTTCAGGTTCTTAGACTGCTCGTTTTACCAGTATAGCGACCAGCATTC | IDT DNA | N/A |
| TOP2 N-terminal CRISPR/Cas9 gRNA oligo: GCAGTGAAAGATAAATGATCGTTGACATGGTTAGCCGTGCGTTTTAGAGCTAGAAATAGC | IDT DNA | N/A |
| TOP2 C-terminal CRISPR/Cas9 gRNA oligo: GCAGTGAAAGATAAATGATCAAAAAGAATGGCGCTTTCTCGTTTTAGAGCTAGAAATAGC | IDT DNA | N/A |
| N-terminal 3xHA tag CRISPR/Cas9 repair template: TTTCAGTTAAAGGAGTTTATAACGACGAGCACGCCTAACCATGTACCCATACGATGTTCCTGACTATGCGGGCTATCCCTATGACGTCCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTACGCTGGTACGATGTCAACTGAACCGGTAAGCGCCTCTGATAAATATCAGA | IDT DNA | N/A |
| C-terminal 3xHA tag CRISPRI/Cas9 repair template: GGAAAACCAAGGATCAGATGTTTCGTTCAATGAAGAAGATGGTACGTACCCATACGATGTTCCTGACTATGCGGGCTATCCCTATGACGTCCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTACGCTTGAATAATATTTATCGAGAGAAAGCGCCATTCTTTTTATA | IDT DNA | N/A |
| Recombinant DNA | ||
| pUG6 | Berger lab (Güldener et al., 1996) | N/A |
| pJH2972 | Addgene (Anand et al., 2017) | CAT# 100956 (RRID:Addgene_100956) |
| pSG483 | Berger lab (Corbett et al. 2005) | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | https://graphpad.com/scientific-software/prism/ (RRID:SCR_002798) |
| ImageJ | NIH | https://imagej.nih.gov/ij/ (RRID:SCR_003070) |
| Compound Discoverer 3.0 | ThermoScientific | OPTON-30925 |
| Other | ||
| NucleoBond Xtra Maxi Columns | Macherey-Nagel | CAT # 740414 |
| Oasis MAX Plus Short Cartridge | Waters | CAT# 186003517 |
| Atlantis T3 OBD Prep Column (100Å, 5μm, 10mm × 250mm) | Waters | CAT# 186008205 |
| XBridge BEH Amide OBD Pre Column (130Å., 5μm 10mm × 250mm) | Waters | CAT# 186008263 |
| Hypersil GOLD aQ polar endcapped C18 column | ThermoFisher Scientific | CAT# 25305 |
| Polyhydroxyethyl A hydrophilic interaction chromatography column (200 mm × 2.1 mm, 5 μm particle size) | PolyLC | CAT# 3.54HY05-- |
Significance.
As with most chemotherapeutics, anti-topoisomerase agenst walk a fine line between acting as a medicine versus a poison. To more effectively target topo II in cancer cells, it is necessary to understand how changing cellular environments impact topo II function. The relationship between TCA cycle metabolism and topo II activity described here raises new questions regarding how cells regulate topo II activity and how anti-topo II drugs interact with cancerous and noncancerous cells. A deeper understanding of the metabolic regulation of topo II may be useful for improving for patient selection strategies and topo II-targeted chemotherapies. Future studies expanding into mammalian systems will clarify the conserved link between cellular metabolism and topo II regulation with the ultimate goal of improving the safety and efficacy of patient-specific cancer treatments.
Highlights.
TCA cycle intermediates stimulate the strand passage activity of S. cerevisiae topo II
Stimulation of enzyme activity by TCA metabolites is specific to eukaryotic topo IIs
Topo II activity and drug response is impacted by changes in TCA cycle flux in cells
Acknowledgments
We thank B. Cormack and members of C. Wolberger’s lab for access to and guidance on the usage of equipment employed for this study. We thank M. Bjornsti for sharing the ED yeast strain, G. Hauk for providing purified E. coli topo IV, and J. Jeong for help in preparing S. cerevisiae topo I. We are grateful to J. Stivers, B. Cormack, J. Nitiss and T. Lee for advice, as well as M. Wolfgang and S. Yegnasubramanian for critical reading of the manuscript. These studies were supported by the NIH (T32-GM007445 and F31-CA224896 to JL, R01-CA168653 to YSL, R01-GM103853 to NNB, and R01-CA077373 to JMB).
Footnotes
Declaration of interests
The authors declare no competing interests
References
- Almeida A, Bolaños JP, and Moncada S (2010). E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc. Natl. Acad. Sci. U. S. A 107, 738–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Memisoglu G, and Haber J (2017). Cas9-mediated gene editing in Saccharomyces cerevisiae. Protoc. Exch 1–6. [Google Scholar]
- Astuti D, Latif F, Dallol A, Dahia PLM, Douglas F, George E, Sko F, Husebye ES, Eng C, and Maher ER (2001). Gene Mutations in Succinate Dehydrogenase Subunit SDHB Cause Susceptibility to Familial Pheochromocytoma and to Familial Paranganglioma. Am. J. Hum. Genet 69, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau JYN, Wang JC, and Liu LF (2007). Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. U. S. A 104, 11014–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baksh SC, and Finley LWS (2020). Metabolic Coordination of Cell Fate by α-Ketoglutarate-Dependent Dioxygenases. Trends Cell Biol. 31, 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, and von Deimling A (2008). Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 116, 597–602. [DOI] [PubMed] [Google Scholar]
- Barrington C, Pezic D, and Hadjur S (2017). Chromosome structure dynamics during the cell cycle: a structure to fit every phase. EMBO J. 36, 2661–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DLV, Marie R, Rasmussen KH, Kristensen A, and Mir KU (2012). DNA catenation maintains structure of human metaphase chromosomes. Nucleic Acids Res. 40, 11428–11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, Van Der Mey A, Taschner PEM, Rubinstein WS, Myers EN, et al. (2000). Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science (80-. ). 287, 848–851. [DOI] [PubMed] [Google Scholar]
- Boer VM, Crutchfield CA, Bradley PH, Botstein D, and Rabinowitz JD (2010). Growth-limiting Intracellular Metabolites in Yeast Growing under Diverse Nutrient Limitations. Mol. Biol. Cell 21, 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, et al. (2012). Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad‐Based Tumor Genotyping. Oncologist 17, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, and Bots (2008). Coordination of Growth Rate, Cell Cycle, Stress Response, and Metabolic Activity in Yeast. Mol. Biol. Cell 19, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchakjian MR, and Kornbluth S (2010). The engine driving the ship: Metabolic steering of cell proliferation and death. Nat. Rev. Mol. Cell Biol 11, 715–727. [DOI] [PubMed] [Google Scholar]
- Cai Z, Li CF, Han F, Liu C, Zhang A, Hsu CC, Peng D, Zhang X, Jin G, Rezaeian AH, et al. (2020). Phosphorylation of PDHA by AMPK Drives TCA Cycle to Promote Cancer Metastasis. Mol. Cell 80, 263–278.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli LR, Varella-Garcia M, and Liang BC (1997). Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 8, 1189–1198. [PubMed] [Google Scholar]
- Charbin A, Bouchoux C, and Uhlmann F (2014). Condensin aids sister chromatid decatenation by topoisomerase II. Nucleic Acids Res. 42, 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo SL, Palacios-Callender M, Frakich N, De Leon J, Schmitt CA, Boorn L, Davis N, and Moncada S (2010). Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc. Natl. Acad. Sci. U. S. A 107, 18868–18873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree HG (1928). The carbohydrate metabolism of certain pathological overgrowths. Biochem. J 22, 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutchfield CA, Lu W, Melamud E, and Rabinowitz JD (2010). Mass spectrometry-based metabolomics of yeast (Elsevier Inc.). [DOI] [PubMed] [Google Scholar]
- Cuvier O, and Hirano T (2003). A role of topoisomerase II in linking DNA replication to chromosome condensation. J. Cell Biol 160, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniloski Z, Bisht KK, McStay B, and Smith S (2019). Resolution of human ribosomal dna occurs in anaphase, dependent on tankyrase 1, condensin II, and topoisomerase IIα. Genes Dev. 33, 276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, and Cascante M (2013). Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther 138, 255–271. [DOI] [PubMed] [Google Scholar]
- Diaz-Ruiz R, Rigoulet M, and Devin A (2011). The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta - Bioenerg 1807, 568–576. [DOI] [PubMed] [Google Scholar]
- Driever W, and Nüsslein-Volhard C (1988). A gradient of bicoid protein in Drosophila embryos. Cell 54, 83–93. [DOI] [PubMed] [Google Scholar]
- Eser U, Falleur-Fettig M, Johnson A, and Skotheim JM (2011). Commitment to a Cellular Transition Precedes Genome-wide Transcriptional Change. Mol. Cell 43, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix CA (1998). Secondary leukemias induced by topoisomerase-targeted drugs. Biochim. Biophys. Acta -Gene Struct. Expr 1400, 233–255. [DOI] [PubMed] [Google Scholar]
- Fradin C (2017). On the importance of protein diffusion in biological systems: The example of the Bicoid morphogen gradient. Biochim. Biophys. Acta -Proteins Proteomics 1865, 1676–1686. [DOI] [PubMed] [Google Scholar]
- Goto T, and Wang JC (1984). Yeast DNA topoisomerase II is encoded by a single-copy, essential gene. Cell 36, 1073–1080. [DOI] [PubMed] [Google Scholar]
- Güldener U, Heck S, Fiedler T, Beinhauer J, and Hegemann JH (1996). A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24, 2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guppy M, Leedman P, Zu XL, and Russell V (2002). Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J 364, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck MMS, Hittelman WN, and Earnshaw WC (1988). Differential expression of DNA topoisomerases I and II during the eukaryotic cell cycle. Proc. Natl. Acad. Sci. U. S. A 85, 1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ERS, and Martinou JC (2012). Identification and functional expression of the mitochondrial pyruvate carrier. Science (80-. ). 336, 93–96. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, and Finley LWS (2019). Metabolic signatures of cancer cells and stem cells. Nat. Metab 1, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida R, Hamatake M, Wasserman RA, Nitiss JL, Wang JC, and Andoh T (1995). DNA Topoisomerase II Is the Molecular Target of Bisdioxopiperazine Derivatives ICRF-159 and ICRF-193 in Saccharomyces cerevisiae. Cancer Res. 55, 2299–2303. [PubMed] [Google Scholar]
- Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P, Gaal J, Dahia PLM, Liegl B, Ball ER, Raygada M, et al. (2011). Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. U. S. A 108, 314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TC, and Holland JJ (1965). Ribonucleic acid and protein synthesis in mitotic HeLa cells. J. Cell Biol 27, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Saijo M, Ui M, and Enomoto T (1994). Growth state- and cell cycle-dependent fluctuation in the expression of two forms of DNA topoisomerase II and possible specific modification of the higher molecular weight form in the M phase. J. Biol. Chem 269, 1173–1176. [PubMed] [Google Scholar]
- King A, Selak MA, and Gottlieb E (2006). Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682. [DOI] [PubMed] [Google Scholar]
- Konokhova Y, Spendiff S, Jagoe RT, Aare S, Kapchinsky S, MacMillan NJ, Rozakis P, Picard M, Aubertin-Leheudre M, Pion CH, et al. (2016). Failed upregulation of TFAM protein and mitochondrial DNA in oxidatively deficient fibers of chronic obstructive pulmonary disease locomotor muscle. Skelet. Muscle 6, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, De Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. (2014). PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol 16, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, and Finkel T (2013). Metabolic regulation of the cell cycle. Curr. Opin. Cell Biol 25, 724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, and Berger JM (2019). Cell cycle-dependent control and roles of DNA topoisomerase II. Genes (Basel). 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Wendorff TJ, and Berger JM (2017). Resveratrol: A novel type of topoisomerase II inhibitor. J. Biol. Chem 292, 21011–21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, et al. (2013). SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 23, 739–752. [DOI] [PubMed] [Google Scholar]
- Li J, and Cheng JX (2014). Direct visualization of de novo lipogenesis in single living cells. Sci. Rep 4, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littau VC, Allfrey VG, Frenster JH, and Mirsky AE (1964). Active and Inactive Regions of Nuclear Chromatin As Revealed By Electron Microscope Autoradiography. Proc. Natl. Acad. Sci 52, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Peng Y, Shi L, Wan L, Inuzuka H, Long J, Guo J, Zhang J, Yuan M, Zhang S, et al. (2021). Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle. Cell Res. 31, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet 43, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas I, Germe T, Chevrier-Miller M, and Hyrien O (2001). Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J. 20, 6509–6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Beauvoit B, Voisin PJ, Canioni P, Guérin B, and Rigoulet M (1998). Energetic and morphological plasticity of C6 glioma cells grown on 3-D support; Effect of transient glutamine deprivation. J. Bioenerg. Biomembr 30, 565–578. [DOI] [PubMed] [Google Scholar]
- McFaline-Figueroa JL, Braun CJ, Stanciu M, Nagel ZD, Mazzucato P, Sangaraju D, Cerniauskas E, Barford K, Vargas A, Chen Y, et al. (2015). Minor changes in expression of the mismatch repair protein MSH2 exert a major impact on glioblastoma response to temozolomide. Cancer Res. 75, 3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, and Lupu R (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777. [DOI] [PubMed] [Google Scholar]
- Morais R, Zinkewich-Péotti K, Parent M, Wang H, Zollinger M, and Babai F (1994). Tumor-forming Ability in Athymic Nude Mice of Human Cell Lines Devoid of Mitochondrial DNA. Cancer Res. 54, 3889–3896. [PubMed] [Google Scholar]
- Morita K, Matsuda F, Okamoto K, Ishii J, Kondo A, and Shimizu H (2019). Repression of mitochondrial metabolism for cytosolic pyruvate-derived chemical production in Saccharomyces cerevisiae. Microb. Cell Fact 18, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL (2009). Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL, Liu Y-X, Harbury P, Jannatipour M, Wasserman R, and Wang JC (1992). Amsacrine and Etoposide Hypersensitivity of Yeast Cells Overexpressing DNA Topoisomerase II. Cancer Res. 52, 4467–4472. [PubMed] [Google Scholar]
- Osheroff N (1987). Role of the Divalent Cation in Topoisomerase II Mediated Reactions. Biochemistry 26, 6402–6406. [DOI] [PubMed] [Google Scholar]
- Park JO, Rubin SA, Xu YF, Amador-Noguez D, Fan J, Shlomi T, and Rabinowitz JD (2016). Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat. Chem. Biol 12, 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdois P, Deveaud C, Voisin P, Bouchaud V, Rigoulet M, and Beauvoit B (2003). Contribution of the Phosphorylable Complex I in the Growth Phase-Dependent Respiration of C6 Glioma Cells in Vitro. J. Bioenerg. Biomembr 35, 439–450. [DOI] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BJ, Ullsperger C, Hiasa H, Marians KJ, and Cozzarelli NR (1998). The structure of supercoiled intermediates in DNA replication. Cell 94, 819–827. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Kerrigan D, and Kohn K (1989). Topological Complexes between DNA and Topoisomerase II and Effects of Polyamines. Biochemistry 28, 995–1002. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Leo E, Zhang H, and Marchand C (2010). DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol 17, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow L, Crisona NJ, Peter BJ, Hardy CD, and Cozzarelli NR (2001). Topological challenges to DNA replication: Conformations at the fork. Proc. Natl. Acad. Sci. U. S. A 98, 8219–8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott DM, and Bender MA (1962). Synthesis of RNA and protein during mitosis in mammalian tissue culture cells. Exp. Cell Res 26, 260–268. [DOI] [PubMed] [Google Scholar]
- Ross W, Rowe T, Glisson B, Yalowich J, and Liu L (1984). Role of TopoisomeraseII in MediatingEpipodophyllotoxin-induced DMA Cleavage1. Cancer Res. 44, 5857–5860. [PubMed] [Google Scholar]
- Salazar-Roa M, and Malumbres M (2017). Fueling the Cell Division Cycle. Trends Cell Biol. 27, 69–81. [DOI] [PubMed] [Google Scholar]
- Samejima K, Samejima I, Vagnarelli P, Ogawa H, Vargiu G, Kelly DA, Alves F. de L., Kerr A, Green LC, Hudson DF, et al. (2012). Mitotic chromosomes are compacted laterally by KIF4 and condensin and axially by topoisomerase IIα. J. Cell Biol 199, 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, and Gottlieb E (2005). Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7, 77–85. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Eisen MB, Brown PO, Botstein D, and Futcher B (1998). Comprehensive Identification of Cell Cycle-regulated Genes of the Yeast Saccharomyces cerevisiae by Microarray Hybridization. Mol. Biol. Cell 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivatsan A, and Wang JD (2008). Control of bacterial transcription, translation and replication by (p)ppGpp. Curr. Opin. Microbiol 11, 100–105. [DOI] [PubMed] [Google Scholar]
- Srivenugopal KS, Wemmer DE, and Morris DR (1987). Aggregation of DNA by analogs of spermidine, enzymatic and structural studies. Nucleic Acids Res. 15, 2563–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanov A, Nitiss KC, Neale G, and Nitiss JL (2008). Enhancing drug accumulation in Saccharomyces cerevisiae by repression of pleiotropic drug resistance genes with chimeric transcription repressors. Mol. Pharmacol 74, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Nair KS, et al. (2016). Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 44, D463–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski PL, Oeck S, Dow J, Economos NG, Mirfakhraie L, Liu Y, Noronha K, Bao X, Li J, Shuch BM, et al. (2020). Oncometabolites suppress DNA repair by disrupting local chromatin signalling (Springer; US: ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor that Activates the Type I Interferon Pathway. Science (80-. ). 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundin O, and Varshavsky A (1981). Arrest of segregation leads to accumulation of highly intertwined catenated dimers: Dissection of the final stages of SV40 DNA replication. Cell 25, 659–669. [DOI] [PubMed] [Google Scholar]
- Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, et al. (2015). Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 21, 81–94. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Ikegami Y, Ishida R, and Ando (1991). Inhibition of Topoisomerase II by Antitumor Agents Bis(2,6-dioxopiperazine) Derivatives. Cancer Res. 51, 4903–4908. [PubMed] [Google Scholar]
- Taylor JH (1960). Nucleic Acid Synthesis in Relation to the Cell Division Cycle. Ann. N. Y. Acad. Sci 90, 409–421. [DOI] [PubMed] [Google Scholar]
- Tomlinson IPM, Alam NA, Rowan AJ, Barclay E, Jaeger EEM, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, et al. (2002). Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer the multiple leiomyoma consortium. Nat. Genet 30, 406–410. [DOI] [PubMed] [Google Scholar]
- Tudzarova S, Colombo SL, Stoeber K, Carcamo S, Williams GH, and Moncada S (2011). Two ubiquitin ligases, APC/C-Cdh1 and SKP1-CUL1-F (SCF)-β-TrCP, sequentially regulate glycolysis during the cell cycle. Proc. Natl. Acad. Sci. U. S. A 108, 5278–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte LM, Neglia JP, Reulen RC, Ronckers CM, Van Leeuwen FE, Morton LM, Hodgson DC, Yasui Y, Oeffinger KC, and Henderson TO (2018). Risk, risk factors, and surveillance of subsequent malignant neoplasms in survivors of childhood cancer: A review. J. Clin. Oncol 36, 2145–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Ohkura H, Adachi Y, Morino K, Shiozaki K, and Yanagida M (1987). DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell 50, 917–925. [DOI] [PubMed] [Google Scholar]
- Da Veiga Moreira J, Peres S, Steyaert JM, Bigan E, Paulevé L, Nogueira ML, and Schwartz L (2015). Cell cycle progression is regulated by intertwined redox oscillators. Theor. Biol. Med. Model 12, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SM, Tretter EM, Schmidt BH, and Berger JM (2011). All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol 12, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SM, Lee I, and Berger JM (2013). Distinct regions of the escherichia coli ParC C-terminal domain are required for substrate discrimination by topoisomerase IV. J. Mol. Biol 425, 3029–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkiewicz K, Benitez Cardenas AS, Sun C, Bacorn C, Saxer G, and Shamoo Y (2012). Small changes in enzyme function can lead to surprisingly large fitness effects during adaptive evolution of antibiotic resistance. Proc. Natl. Acad. Sci. U. S. A 109, 21408–21413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, et al. (2014). Cyclin B1/Cdk1 coordinates mitochondrial respiration for Cell-Cycle G2/M progression. Dev. Cell 29, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1925). The metabolism of carcinoma cells. J. Cancer Res 9, 148–163. [Google Scholar]
- Wasserman RA, and Wang JC (1994). Mechanistic studies of amsacrine-resistant derivatives of DNA topoisomerase II. Implications in resistance to multiple antitumor drugs targeting the enzyme. J. Biol. Chem 269, 20943–20951. [PubMed] [Google Scholar]
- Wellen KE, and Thompson CB (2010). Cellular Metabolic Stress: Considering How Cells Respond to Nutrient Excess. Mol. Cell 40, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen AM, McConnell M, and Fisher PA (1991). Developmental regulation of Drosophila DNA topoisomerase II. J. Cell Biol 112, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann C, Hans M, Van Winden WA, Ras C, and Heijnen JJ (2005). Dynamics of intracellular metabolites of glycolysis and TCA cycle during cell-cycle-related oscillation in Saccharomyces cerevisiae. Biotechnol. Bioeng 89, 839–847. [DOI] [PubMed] [Google Scholar]
- Woessner RD, Mattern MR, Mirabelli CK, Johnson RK, and Drake FH (1991). Proliferation- and Cell Cycle-dependent Differences in Expression of the 170 Kilodalton and 180 Kilodalton Forms of Topoisomerse II in NIH-3T3 Cells. Cell Growth Differ. 2, 209–214. [PubMed] [Google Scholar]
- Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW, and Chan NL (2011). Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science (80-. ). 333, 459–462. [DOI] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, et al. (2012). Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami R, Bingaman JL, Frankel EA, and Bevilacqua PC (2018). Cellular conditions of weakly chelated magnesium ions strongly promote RNA stability and catalysis. Nat. Commun 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. (2009). IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ye D, Guan KL, and Xiong Y (2012). IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res 18, 5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi LL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, and Liu LF (2007). Topoisomerase IIβ-mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 67, 8839–8846. [DOI] [PubMed] [Google Scholar]
- Zhu J, and Thompson CB (2019). Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol 20, 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Results of substructure search of succinic and glutaric acid motifs on the Human Metabolome Database, related to Figure 3.
Data Availability Statement
The data obtained in this study is accessible at the NIH Common Fund’s NMDR (supported by NIH grant, U01-DK097430) website, the Metabolomics Workbench (Sud et al., 2016), http://dx.doi.org/10.21228/M89695. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.