

Summary
It has been established that gut microbiota influences chicken growth performance and fat metabolism. However, whether gut microbiota affects chicken growth performance by regulating fat metabolism remains unclear. Therefore, seven‐week‐old chickens with high or low body weight were used in the present study. There were significant differences in body weight, breast and leg muscle indices, and cross‐sectional area of muscle cells, suggesting different growth performance. The relative abundance of gut microbiota in the caecal contents at the genus level was compared by 16S rRNA gene sequencing. The results of LEfSe indicated that high body weight chickens contained Microbacterium and Sphingomonas more abundantly (P < 0.05). In contrast, low body weight chickens contained Slackia more abundantly (P < 0.05). The results of H & E, qPCR, IHC, WB and blood analysis suggested significantly different fat metabolism level in serum, liver, abdominal adipose, breast and leg muscles between high and low body weight chickens. Spearman correlation analysis revealed that fat metabolism positively correlated with the relative abundance of Microbacterium and Sphingomonas while negatively correlated with the abundance of Slackia. Furthermore, faecal microbiota transplantation was performed, which verified that transferring faecal microbiota from adult chickens with high body weight into one‐day‐old chickens improved growth performance and fat metabolism in liver by remodelling the gut microbiota. Overall, these results suggested that gut microbiota could affect chicken growth performance by regulating fat metabolism.
16S rRNA gene sequencing indicated that high body weight chickens contained Microbacterium and Sphingomonas more abundantly (P < 0.05). In contrast, low body weight chickens contained Slackia more abundantly (P < 0.05). The fat metabolism level in liver between high and low body weight chickens was significantly different. Transferring faecal microbiota from adult chickens with high body weight into one‐day‐old chickens improved growth performance and fat metabolism in liver by remodeling the gut microbiota.

Introduction
In animal production, antibiotics have been used as feed additives to enhance growth performance. Poultry industry is one of the largest food industries worldwide, and chicken is the common species reared at farms for meat production (Agyare et al., 2018). Many countries are using a range of antibiotics for chicken production (Sahoo et al., 2010; Landers et al., 2012). However, long‐term use of antibiotics can produce antibiotic‐resistant zoonotic pathogens and cause antibiotic residues in food (Allen et al., 2014; Pamer, 2016). In addition, poorly absorbed antibiotics are excreted unchanged in faeces and urine, and dispersed in soil when manure is used as a fertilizer, resulting in an antibiotic pollution in the environment (Joy et al., 2014). Consequently, the use of antibiotics as a growth promoter has been banned in Sweden since 1986, in the European Union and other countries (Case well et al., 2003; Hoese et al., 2009; Maron et al., 2013). Several countries have withdrawn the use of some antibiotics, yet among all produced antibiotics, about 60% is estimated to be used in livestock production, especially in poultry and pigs (Van Boeckel et al., 2014; Van Boeckel et al., 2015). Therefore, the development of alternatives to antibiotic growth promoters (AGPs) is an important strategy in animal production and food safety.
The chicken gastrointestinal tract harbours a complex consortium of microbial communities, with the highest bacterial diversity in the caecum (Bjerrum et al., 2006; Wei et al., 2013; Awad et al., 2016; Saxena et al., 2016; Pandit et al., 2018). Multiple studies in chickens have established the importance of the gut microbiota, especially the caecal microbiota, in improving feed digestion, nutrient absorption and growth performance (Stanley et al., 2014; Stanley et al., 2016; Yan et al., 2017). Notably, alteration of the gut microbiota using faecal microbiota transplantation or probiotic supplementation as an alternative to antibiotics has also been reported to improve chicken growth performance, indicating that the gut microbiota is an essential resource for developing natural growth promoters (Angelakis, 2017; Siegerstetter et al., 2018; Videvall et al., 2019; Yadav and Jha, 2019).
Fat metabolism is an important and complex biochemical reaction, including the processes of digestion, absorption, synthesis and catabolism (D'Aquila et al., 2016). Balanced fat metabolism can improve host growth performance and meat quality, yet imbalanced fat metabolism results in obesity and disease. Accumulating investigations focussing on the relationship between gut microbiota and obesity suggested that the gut microbiota is an important environmental factor affecting energy harvest from the diet and energy storage in mammals (Bäckhed et al., 2004; Turnbaugh et al., 2006; Wang et al., 2017). It has been reported that 12 Lactobacillus strains reduced triglycerides, abdominal fat deposition and total serum cholesterol in broilers (Kalavathy et al., 2003; Wang et al., 2017). Independent to the host genetic factors, duodenal as well as caecal microbiota of chicken plays a critical role in fat deposition (Wen et al., 2019). Abundantly observed beneficial bacteria, that is Bacteroides and Lactobacillus in the chicken gut, were associated with high body weight gain, low abdominal fat, high breast muscle yield and increased growth performance (Zheng et al., 2019). Whether the gut microbiota affects chicken growth performance by regulating fat metabolism becomes an interesting question.
To tackle this question, chickens from the same group with different growth performance were used in the present study to compare fat metabolism levels and gut microbial communities. The correlation between fat metabolism and gut microbiota was analysed using Spearman correlation analysis. Besides, transferring faecal microbiota from adult chickens with high body weight into one‐day‐old chicks was performed to verify whether chicken gut microbiota affected growth performance by regulating fat metabolism.
Results
Difference in growth performance
Seven‐week‐old chickens from the same group with significantly different body weight (P = 0.001) (Fig. 1A) were selected. There were significant differences in breast muscle index (P = 0.014) (Fig. 1B) and leg muscle index (P = 0.0489) (Fig. 1C). Haematoxylin and eosin (H & E) staining results indicated that the average cross‐sectional areas of breast muscle cells (P = 0.0105) (Fig. 1D) and leg muscle cells (P = 0.0304) (Fig. 1E) were significantly larger in high than in low body weight chickens.
Fig. 1.
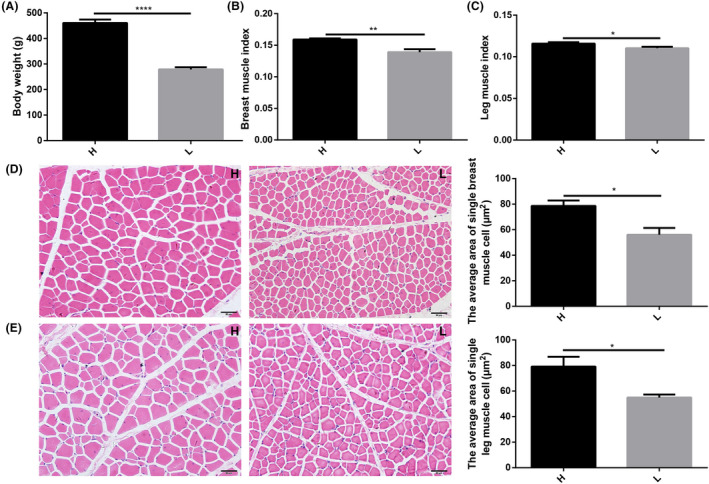
Different growth performance between high and low body weight chickens. Chickens (Turpan cockfighting × White Leghorn) at 7th week of age were selected to analyse the growth performance of high and low body weight chickens (n = 10). Each group contained five males and five females. (A) Body weight. (B) Breast muscle index. (C) Leg muscle index. (D) Section of breast muscle with H & E staining and the comparison of single cell’s cross‐sectional area. (E) Section of leg muscle with H & E staining and the comparison of single cell’s cross‐sectional area. H represented high body weight chickens and L represented low body weight chickens. Scale bars = 50 μm. All data were presented as the means ± SEM. P values were calculated using Student’s t‐test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Differences in caecal microbiota
The chickens’ caecal microbiota was analysed using 16S rRNA gene sequencing. A total of 1 341 412 high‐quality reads were obtained from 20 samples (an average of 67 070 reads per sample), which were clustered into 748 operational taxonomic units (OTUs) at 97% sequence similarity. The alpha diversity results exhibited no significant differences in microbial diversity in caecal contents between high and low body weight chickens (Fig. 2A‐B). However, beta diversity showed some separation between high and low body weight groups (Fig. 2C). At the phylum level, Firmicutes, Bacteroidetes and Proteobacteria were observed as the most abundant phyla. The relative abundance of Firmicutes was lower, while of Bacteroidetes was higher in high body weight chickens (Fig. 2D). The ratio of Firmicutes to Bacteroidetes was significantly lower in high body weight chickens (Fig. 2E). At the genus level, Bacteroides were observed as the dominant genus in caecal contents (Fig. 2F). Linear discriminant analysis effect size (LEfSe) analysis revealed that the relative abundances of Faecalibacterium, Microbacterium, Slackia, and Sphingomonas were significantly different in high and low body weight chickens (Fig. 2G).
Fig. 2.
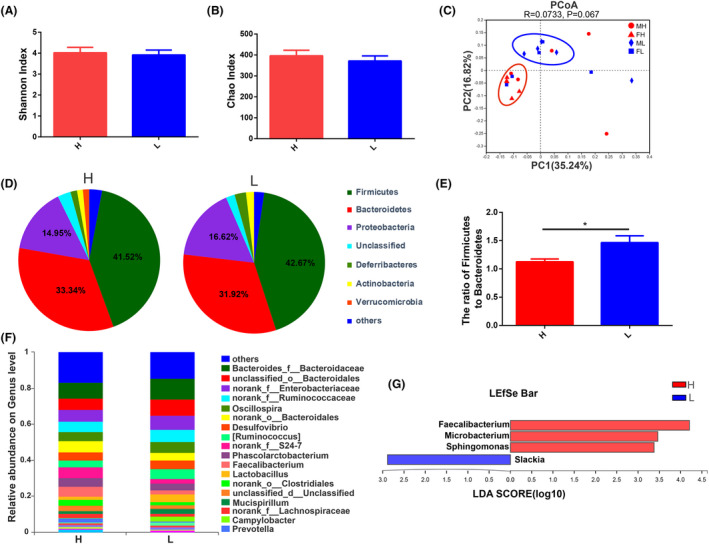
Differences in caecal microbiota between high and low body weight chickens.
A. Chickens (Turpan cockfighting × White Leghorn) at 7th week of age were sacrificed to perform 16S rRNA sequence of caecal microbiota (n = 10). Each group contained five male and five female chickens. Microbial community diversity (measured by the Shannon index).
B. Microbial community abundance (measured by the Chao index).
C. Beta diversity of high and low body weight chickens. MH: male with high body weight, FH: female with high body weight, ML: male with low body weight, FL: female with low body weight.
D. The composition of microbial communities at the phylum level.
E. The ratio of Firmicutes to Bacteroidetes.
F. The composition of microbial communities at the genus level.
G. LDA score for LEfSe analysis (LEfSe was done at LDA score of >2).
Differences in liver fat metabolism
H & E staining results indicated more vacuolar fat in the livers of high body weight chickens (Fig. 3A). The relative mRNA expression of fat synthesis‐related genes including cytochrome P450 2C45 (CYP2C45) (P = 0.0373), fatty acid desaturase 1 (FADS1) (P = 0.0256) and acyl‐CoA synthetase long chain family member 1 (ACSL1) (P = 0.0164) (Fig. 3B) and the fat catabolism‐related genes fasting‐induced adipose factor (fiaf) (P = 0.0384), peroxisome proliferator‐activated receptor alpha (PPARα) (P = 0.0234) and carnitine palmitoyl transferase I (CPT‐1) (P = 0.0319) (Fig. 3C) were significantly higher in the livers of high body weight chickens than in low body weight chickens. The relative mRNA expression of fat synthesis‐related genes acetyl CoA carboxylase (ACC) and fatty acid synthase (FAS) were higher in high body weight chickens as compared to low body weight chickens, but not significantly. The relative mRNA expression of fat transport‐related gene apolipoprotein A‐I (ApoAI) was significantly higher in the liver of high body weight chickens than in low body weight chickens (P = 0.0166) (Fig. 3D). Immunohistochemistry (IHC) staining indicated that phospho‐AMP‐activated protein kinase (P‐AMPK) was distributed in the cytoplasm and nucleus of hepatocytes in liver tissue (Fig. 3E), and the protein expression of P‐AMPK was significantly higher in high body weight chickens than that in low body weight chickens (P = 0.0095) (Fig. 3F). Western blot results also revealed a higher protein expression of P‐AMPK in high body weight chickens (P‐AMPK/GAPDH, P = 0.0045; P‐AMPK/AMPK, P = 0.0008) (Fig. 3G–I).
Fig. 3.
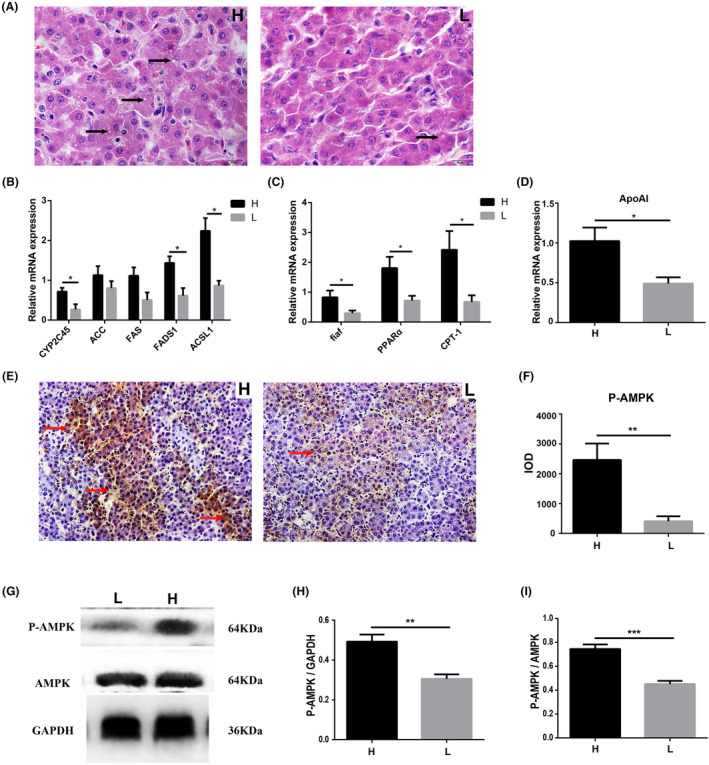
Differences of fat metabolism levels in liver between high and low body weight chickens. Chickens (Turpan cockfighting × White Leghorn) at 7th week of age were sacrificed to analyse fat metabolism level in liver (n = 10). Each group contained five males and five female chickens.
A. Fat content of hepatocytes (H & E staining). The white dots indicated by the arrows in the figure are fat droplets.
B. The relative mRNA expression of fat anabolism‐related genes (qPCR).
C. The relative mRNA expression of fat catabolism‐related genes (qPCR).
D. The relative mRNA expression of fat transport‐related genes (qPCR).
E. The distribution of P‐AMPK (IHC). The arrows in the figure indicated positive expression of P‐AMPK.
F. IOD comparison of IHC.
G–I. The protein expression of P‐AMPK (western blot). Scale bars = 50 μm. All data were presented as the means ± SEM. P values were calculated using Student’s t‐test, *P < 0.05, **P < 0.01.
Differences in fat metabolism in blood and abdominal adipose
In abdominal adipose, H & E staining results showed that adipocytes were vacuolar, and the number of adipocytes in abdominal adipose tissue was significantly higher in high body weight chickens rather than in low body weight chickens (P = 0.0056) (Fig. 4A). The average diameter of adipocytes was significantly smaller in high than in low body weight chickens (P = 0.0063) (Fig. 4B). In serum of high body weight chickens, the concentration of high‐density lipoprotein cholesterol (HDL‐C) was significantly (P = 0.0436) higher, while the concentration of low‐density lipoprotein cholesterol (P = 0.0273) (LDL‐C) was significantly lower compared with low body weight chickens (Fig. 4C). There were no significant differences in the concentrations of triglyceride (TG) or total cholesterol (TC) between high and low body weight chickens. qPCR results showed that the relative mRNA expression levels of ACC and FAS were lower in high body weight chickens than in low body weight chickens, but there were no significant differences (Fig. 4D). The relative mRNA expression levels of adipocyte differentiation‐related genes, including sterol regulatory element‐binding protein 1 (P = 0.0128) (SREBP1) and adiponectin (P = 0.0401), were significantly lower in high body weight chickens. The adipocyte protein 2 (AP2) and PPARG were also lower in high body weight chickens, but there were no significant differences (Fig. 4E).
Fig. 4.
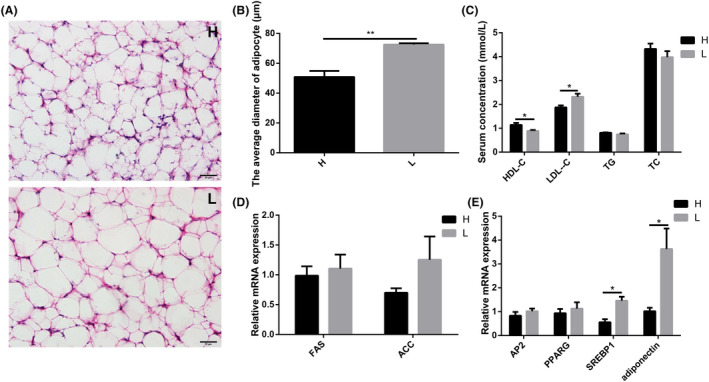
Different fat metabolism levels in blood and abdominal adipose tissue.
A. Chickens (Turpan cockfighting × White Leghorn) at 7th week of age were sacrificed to analyse fat metabolism level in blood and abdominal adipose tissue (n = 10). Each group contained five male and five female chickens. Different adipocyte structure by H & E staining.
B. The average diameter of adipocyte.
C. Different blood biochemical parameters related to lipid metabolism.
D. The relative mRNA expression of fat anabolism‐related genes in abdominal adipose tissue (qPCR).
E. The relative mRNA expression of adipocyte differentiation‐related genes in abdominal adipose tissue (qPCR). Scale bars = 50 μm. All data were presented as the means ± SEM. P values were calculated using Student’s t‐test, *P < 0.05, **P < 0.01.
Differences in fat metabolism in breast and leg muscles
There was no significant difference in the relative expression of ACC in breast muscle and leg muscle between high and low body weight chickens (Fig. 5A). The relative mRNA expression of the fat transport‐related gene (A‐FABP) was much lower (P = 0.0001) in the breast muscle of high body weight chickens (Fig. 5B), while the relative mRNA expression of the fat catabolism‐related gene CPT‐1 was significantly higher in the breast (P = 0.0205) and leg muscle (P = 0.0468) of high body weight chickens (Fig. 5C). IHC results showed that P‐AMPK was mainly distributed in the connective tissue of breast and leg muscle, and the protein expression of P‐AMPK was significantly higher in breast muscle (P = 0.0225) (Fig. 5D) and leg muscle (P = 0.0153) (Fig. 5E) of high body weight chickens. Western blot results also revealed higher protein expression of P‐AMPK in breast muscle (P = 0.0001) (Fig. 5F and G) and leg muscle (P = 0.0016) (Fig. 5H and I) of high body weight chickens.
Fig. 5.
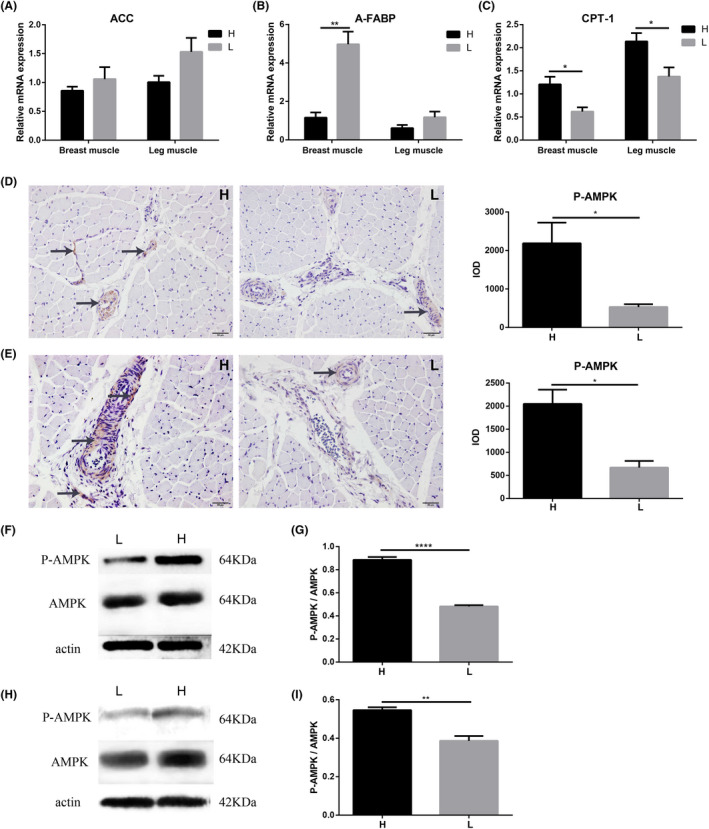
Different fat metabolism levels in muscle tissues. Chickens (Turpan cockfighting × White Leghorn) at 7th week of age were sacrificed to analyse fat metabolism level in muscles (n = 10). Each group contained five male and five female chickens.
A. The relative mRNA expression of fat anabolism‐related genes (qPCR).
B. The relative mRNA expression of fat transport‐related genes (qPCR).
C. The relative mRNA expression of fat catabolism‐related genes (qPCR). (D) The distribution and protein expression of P‐AMPK in breast muscle (IHC).
E. The distribution and protein expression of P‐AMPK in leg muscle (IHC). The arrows in the figures indicated positive signals.
F and G. The protein expression of P‐AMPK in breast muscle (western blot).
H and I. The protein expression of P‐AMPK in leg muscle (western blot). H represented high body weight chickens and L represented low body weight chickens. Scale bars = 50 μm. All data were presented as the means ± SEM. P values were calculated using Student’s t‐test, *P < 0.05, **P < 0.01.
Differential gut microbiota was related to fat metabolism in chickens
Spearman’s correlation analysis was used to analyse the correlation between fat metabolism and the differential gut microbiota. The results indicated that the abundance of Sphingomonas and Microbacterium was significantly and positively correlated with fat catabolism in serum, liver, breast muscle and leg muscle. The abundance of Slackia was significantly and negatively correlated with the fat catabolism in serum, liver, breast muscle and leg muscle (Fig. 6).
Fig. 6.
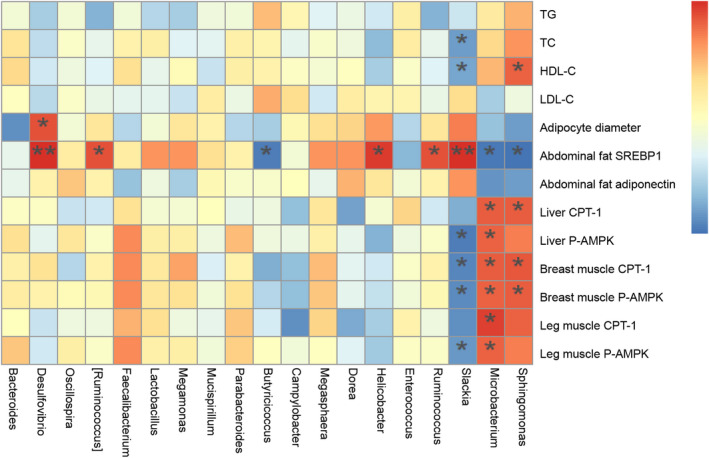
Heatmap of Spearman’s correlations between caecal microbe abundance and fat metabolism. The colours range from blue (negative correlation) to red (positive correlation). P values were calculated using Student’s t‐test, *P < 0.05 and **P < 0.01.
Faecal microbiota transplantation improved growth performance and hepatic fat metabolism in chicken
Faecal microbiota transplantation (FMT) was performed to investigate the effect of gut microbiota on growth performance and fat metabolism. The results showed that the body weight in FMT group was significantly (P < 0.05) higher than that in control group (Fig. 7A). The relative mRNA expression of fat synthesis‐related gene (Fig. 7B), fat catabolism‐related gene (Fig. 7C) and fat transport‐related gene (Fig. 7D) in liver was significantly (P < 0.05) up‐regulated in FMT group than the control group. 16S rRNA gene sequencing of FMT and control groups were also performed. For FMT and control groups, we obtained 995 072 high‐quality reads from 20 samples (an average of 49 753 reads per sample) that were clustered into 920 operational taxonomic units (OTUs) at 97% sequence similarity. These results revealed that alpha and beta diversities were significantly (P < 0.05) different in both groups (Fig. 8A–C). At the phylum level, Firmicutes, Bacteroidetes and Proteobacteria were observed as the most abundant phyla (Fig. 8D). The ratio of Firmicutes to Bacteroidetes was significantly different between FMT and control groups (Fig. 8E). LEfSe analysis revealed that the relative abundance of gut microbiota was also significantly (P < 0.05) different in FMT and control groups (Fig. 8G).
Fig. 7.
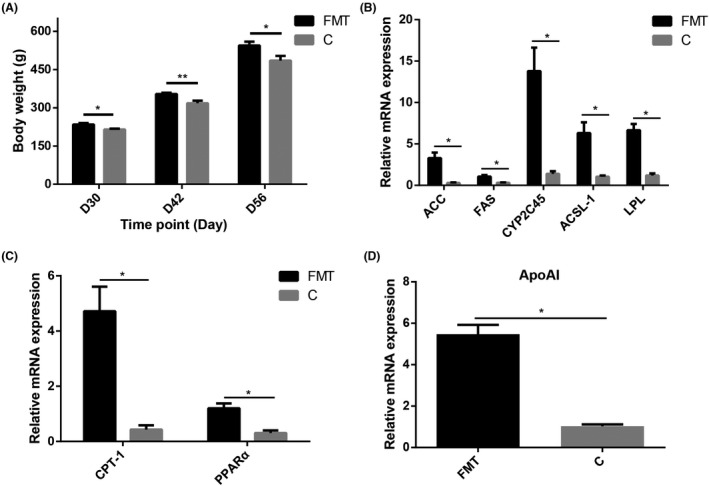
FMT influenced growth performance and fat metabolism level in liver.
Newly hatched chicks (Turpan cockfighting × White Leghorn) of FMT group and control group (C) were orally administrated with 1 ml faecal microbiota suspension and saline, respectively, for 30 days and sacrificed at the age of 31 days to analyse fat metabolism level in liver (n = 10).
A. Comparison of body weight between C and FMT group.
B. The relative mRNA expression of fat anabolism‐related genes (qPCR).
C. The relative mRNA expression of fat catabolism‐related genes (qPCR).
D. The relative mRNA expression of fat transport‐related genes (qPCR). All data were presented as the means ± SEM. P values were calculated using Student’s t‐test, *P < 0.05, **P < 0.01.
Fig. 8.
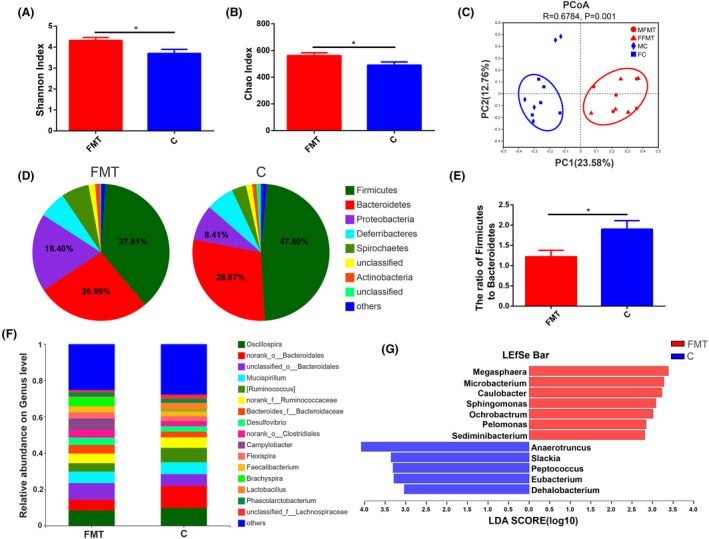
Differences in caecal microbiota between FMT and control groups.
A. Chickens (Turpan cockfighting × White Leghorn) at 31st day of age were sacrificed to perform 16S rRNA sequence of caecal microbiota (n = 10). Each group contained five males and five female chickens. Microbial community diversity (Shannon index).
B. Microbial community abundance (Chao index).
C. Beta diversity of FMT and control groups. MFMT: male in FMT group, FFMT: female in FMT group, MC: male in control group, FC: female in control group.
D. The composition of microbial communities at the phylum level.
E. The ratio of Firmicutes to Bacteroidetes.
F. The composition of microbial communities at the genus level.
G. LDA score for LEfSe analysis (LEfSe was done at LDA score of >2).
Discussion
The fat metabolic profile associated with gut microbiota is a strong determinant to regulate host physiology. The current study aimed to investigate the critical role of gut microbiota in the growth performance of chicken via regulating fat metabolism.
Fat metabolism has considerable significance in biological processes. It is an essential and complex biochemical reaction, including digestion, absorption, synthesis and catabolism. Digested fat in the form of glycerol and fatty acid is absorbed into the bloodstream and transported to the liver, adipose tissue and other organs (Hermier, 1997; Margetak et al., 2012). Fat synthesized in the liver is bound to apolipoprotein or cholesterol to form very low‐density lipoprotein (VLDL), which is transported through blood to other tissues for storage or usage (Alves‐Bezerra and Cohen, 2017). However, fat synthesized in adipose tissue is stored there. In chickens, adipose tissue plays a vital role to regulate homoeostasis and metabolic energy (Di Somma et al., 2019). Blood biochemical indicators are also closely related to fat metabolism and have been used as criteria to select lean chicken lines (Zhang et al., 2018). In the present study, significantly higher concentrations of HDL‐C were observed in high body weight chickens, suggesting that it might effectively carry cholesterol through blood and transport it to the liver (Oldoni et al., 2014; Manthei et al., 2018).
The liver is the largest solid organ and plays a critical role in lipid metabolism, providing significant energy resources for host growth (Grijalva and Vakili, 2013; Parry and Hodson, 2017). The chicken liver is reported as the leading site for de novo synthesis of fatty acid (Mellouk et al., 2018). Hepatic lipid metabolism effectively regulates the fat synthesis and catabolic related genes (Na et al., 2018). In our study, high body weight chickens exhibited higher expression of fat synthesis‐related genes (P450 2C45 (CYP2C45), FADS1 and ACSL1) in the liver, as well as greater numbers of fat vacuoles in hepatocytes, suggesting a potent fat synthesis in the livers of high body weight chickens. At the same time, the expression of fat catabolism‐related genes (fiaf, PPARα and CPT‐1) increased, ultimately promoting the oxidative catabolism of fatty acids, thereby providing energy for the growth and development of chickens (Witczak et al., 2008; Niu et al., 2010; Kim et al., 2017). Significantly higher expression of fat transport‐related hepatic ApoAI gene in high body weight chickens also indicated an appropriate regulation in fat transport (Liu et al., 2016; Zheng et al., 2016). Additionally, a remarkably prominent expression of hepatic phosphorylated‐AMPK in hepatocytes of higher body weight chickens maintained the energy balance through fatty acid synthesis and oxidation (Yang et al., 2018; Hu et al., 2019a; Li et al., 2020a). Abdominal adipose tissue is the main organ of fat synthesis and deposition. The up‐regulated expression of adipocyte differentiation‐related genes can mediate excessive proliferation and differentiation of adipocytes, causing excessive deposition of fat in animal bodies (Xiao et al., 2018). Abdominal fat deposition impacts negatively on carcass quality and feed efficiency (Demeure et al., 2013). In the present study, the expression of fat synthesis‐ and adipocyte differentiation‐related genes in low body weight chickens was significantly higher than in high body weight chickens, suggesting increased fat synthesis in abdominal adipose tissue. In addition, the average diameter of adipocytes in low body weight chickens was much larger, indicating that more synthesized fat was stored in abdominal adipose tissue in low body weight chickens. As a result, excessive fat deposition affected growth performance (Xiao et al., 2018). These results indicated that the differential fat metabolism level is a key factor leading to differences in growth performance.
It has been reported that the composition of the gut microbiota is closely related to fat metabolism in mammals and chickens (Ridaura et al., 2013; Yang et al., 2016; Pascale et al., 2019; Wen et al., 2019). In human and mice, obese individuals had a lower relative abundance of the Bacteroidetes and higher relative abundance of the Firmicutes than lean controls (Ley et al., 2006; Turnbaugh et al., 2008). The ratio of Firmicutes/Bacteroidetes is vital for optimum nutritional physiology (De Filippo et al., 2010; Bervoets et al., 2013). Further findings indicated that the higher ratio of Firmicutes and Bacteroidetes can induce fat deposition (Ley et al., 2006; Compare et al., 2016; Koliada et al., 2017; Indiani et al., 2018). In addition, recent studies described Sphingomonas as an abundant bacterium in the gut microbiota of chicken (Chen et al., 2018a; Li et al., 2020b). Another study reported that sphingolipids from Sphingomonas could control different cellular processes, that is migration, apoptosis and proliferation (Bryan et al., 2016). For instance, Sphingomonas synthesized sphingosine, which later with fatty acids derivatives formed sphingomyelin, thus reduced fat deposition in the chicken liver. These findings suggested that the abundance of Sphingomonas genus was significantly correlated with hepatic fat metabolism (Li et al., 2020b). Microbacterium and Sphingomonas related to lipid metabolism were significantly observed across all chicken embryonic stages, which suggested a significant contribution in the development of gut microbiota for chicken growth (Akinyemi et al., 2020). In another study, the clusters of orthologous groups (COG) analysis revealed 20 lipid metabolism genes associated with Microbacterium (Chen et al., 2018b). A few other studies also reported that Microbacterium was important in lipid metabolism (Hadjadj et al., 2016; Akinyemi et al., 2020). Slackia could also be involved in an unbalanced compositional signature of gut microbiota. Therefore, its role in imbalancing gut microbiota and disease susceptibility is critical. Contradictory findings have been reported in different studies about Slackia. For instance, a recent study reported that Slackia isolated from chicken intestine was the best detoxifying agent against mycotoxin and helps improve chicken intestinal functions (Gao et al., 2020). However, several other studies reported that Slackia was found to be significantly enriched in the microbiota of gastric carcinogenesis and not only caused disease progression but also gut dysbiosis (Coker et al., 2018; Kharrat et al., 2019). Another recent study also reported that Slackia‐related lipid metabolism abnormality may abruptly change the levels of HDL, LDL and Apo A (Han et al., 2019). In the current study, higher Firmicutes/Bacteroidetes ratio in low body weight group revealed that chickens in this group stored more fat in reservoir instead of using it as energy for their growth. Sphingomonas, Microbacterium and Slackia were significantly different in high and low body weight chickens. Spearman’s analysis demonstrated positive correlations between HDL‐C, CPT‐1, and P‐AMPK and the relative abundances of Sphingomonas and Microbacterium and negative correlations between HDL‐C, CPT‐1, and P‐AMPK and the relative abundance of Slackia. These results indicated that Sphingomonas and Microbacterium might promote the oxidative decomposition of fatty acids to stimulate growth and development of animals, yet Slackia promoted the deposition of fat in animals.
FMT is an effective way to reshape gut microbiota (Metzler‐Zebeli et al., 2019; Guo et al., 2020). Transferring faecal microbiota from adult chickens with high body weight into one‐day‐old chicks was performed to verify whether chicken gut microbiota affected growth performance by regulating fat metabolism. The body weight in FMT group was significantly higher than that in control group. The relative mRNA expressions of hepatic anabolic (ACC, FAS, CYP2C45, ACSL1 and LPL), catabolic (PPARα and CPT‐1) and transportation (ApoA1) fat genes were significantly up‐regulated. The ratio of Firmicutes to Bacteroidetes was significantly different between FMT and control groups. The relative abundance of Sphingomonas and Microbacterium was significantly higher in FMT group, yet Slackia was significantly higher in control group. The results indicated that FMT increased chicken growth performance by remodelling gut microbiota which could regulate fat metabolism.
Conclusion
In summary, the levels of fat metabolism in liver, abdominal adipose tissue, breast muscle and leg muscle differ between high and low body weight chickens. Sphingomonas, Microbacterium and Slackia were significantly different in high and low body weight chickens. FMT improved chicken growth performance and changed fat metabolism level in liver. These findings provided novel insights into the gut microbiota increased chicken growth performance by regulating fat metabolism and contributed to the development of alternatives to AGPs for improving chicken productive efficiency.
Experimental procedures
Animals
The Institutional Animal Care and Use Committee of Huazhong Agricultural University approved all the animal procedures, and all methods were performed in accordance with the relevant guidelines and regulations.
Newly hatched chickens (Turpan cockfighting × White Leghorn chickens for both meat and egg) were reared under the same husbandry conditions and were fed a corn‐soybean diet in pellet form in a poultry farm with no medication or vaccination. The birds had ad libitum access of water and feed. When they were 7 weeks old, a total of 20 chickens with the highest (n = 10, five males and five females) and the lowest (n = 10, five males and five females) body weights were selected for the next study. In faecal microbiota transplantation experiment, two adult female chickens (ranked 5th and 6th), which harboured the top 10 body weight in the same batch, with no gastrointestinal diseases or drug treatment, were selected as faecal donors. Fresh faecal samples were collected every day for 30 days during morning time in sterile 50‐mL tube and 0.75% saline were mixed in 1:6 ratios (6 ml of 0.75% saline for each gram of faeces). Then collect the supernatant and filter it with sterile gauze to get faecal suspension. Then, a total of 60 one‐day‐old chickens with the same genetic background were selected as recipients. The recipients were randomly divided into two groups: saline control group (C group) and faecal microbiota transplantation group (FMT group). Each bird in FMT group was orally administrated with 1 ml faecal microbiota suspension for 30 days while the birds in control group were orally administrated with 1 ml 0.75% saline.
Samples collection
Samples were collected from the high and low body weight chickens (five males and five females in each group) at 7 weeks of age, and samples from the chickens in the FMT and control groups were collected at the age of 31st day. After fasting for 12 h, the chickens were sacrificed and blood, liver and abdominal adipose were harvested. Breast muscle and leg muscle were also collected and weighed. To ensure the comparability of research results, the same part of each organ was chosen for further analysis. For gut microbiota analysis, the contents of the caeca from the selected twenty chickens were snap‐frozen in liquid nitrogen and stored at −80 °C. For histo‐morphological analysis, freshly harvested breast muscle, leg muscle and liver tissues were fixed in 4% paraformaldehyde solution. Similarly, abdominal adipose tissues from each chicken were fixed in optimal cutting temperature (OCT) compound. For protein and gene expression analysis, the parts of freshly harvested muscle, adipose and liver tissues were snap‐frozen in liquid nitrogen and then stored at −80 °C. For blood biochemical parameters analysis, blood samples from birds were centrifuged at 1500 g for 15 min and serum samples were snap‐frozen in liquid nitrogen and stored at −80 °C for subsequent analysis.
Muscle index calculation
The muscle index was calculated using the following formula: muscle index = muscle weight (g)/ body weight (g).
Microbial genomic DNA extraction and 16S rRNA gene sequencing
16S rRNA gene sequencing was used to compare the microbial composition between high and low body weight chickens. Total bacterial genomic DNA was extracted from samples using Fast DNA SPIN extraction kits (MP Biomedicals, Santa Ana, CA, USA), following the manufacturer’s instructions and as described by (Hu et al., 2019b). The sample (500 mg) was mixed in Lysing Matrix E tube with 978 μl sodium phosphate buffer and MT buffer (122 μl). Then, the mixture was homogenized at 6.0 m s−1 speed for 40 s and centrifuged for 10 min at 14 000 g. The obtained supernatant was mixed with protein precipitation solution (250 μl) in a clean 2.0‐ml micro‐centrifuge tube. This mixture was mixed thoroughly in the tube, 10 times by shaking with hands. To remove the protein, the mixture was again centrifuged for 5 min at 14 000 g. Subsequently, the supernatant was transferred to 15‐ml tube, 1 ml Binding Matrix Solution was added, suspended it for 2 min and placed the tube on a rack for 3 min. After discarding 500 μl of supernatant, 600 μl of DNA Solution was transferred to a SPIN™ Filter Tube followed by centrifuging for 1 min at 14 000 g. Then, 500 μl SEWS‐M wash Solution was added and centrifuged for 1 min at 14 000 g. To remove the residual SEWS‐M wash solution, it was again centrifuged for 2 min at 14 000 g followed by drying for 5 min at room temperature. Finally, 50 μl of DES was added for the resuspension of binding matrix and centrifuged for 1 min at 14 000 g to obtain DNA. The extracted DNA was stored at −20 °C for further analysis. The quantity and quality of extracted DNA fragments were measured using a NanoDrop ND‐1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and agarose gel electrophoresis respectively.
PCR amplification of the bacterial 16S rRNA gene V3‐V4 region was performed using the forward primer 338F (5’‐ACTCCTACGGGAGGCAGCA‐3’) and the reverse primer 806R (5’‐GGACTACHVGGGTWTCTAAT‐3’). Sample‐specific 7‐bp barcodes were incorporated into the primers for multiplex sequencing. The PCR components contained 5 μl of Q5 reaction buffer (5×), 5 μl of Q5 High‐Fidelity GC buffer (5×), 0.25 μl of Q5 High‐Fidelity DNA Polymerase (5 U μl−1), 2 μl (2.5 mM) of dNTPs, 1 μl (10 μM) of each forward and reverse primer, 2 μl of DNA Template and 8.75 μl of dd H2O. Thermal cycling consisted of an initial denaturation at 98 °C for 2 min, followed by 25 cycles of denaturation at 98 °C for 15 s, annealing at 55 °C for 30 s and extension at 72 °C for 30 s, with a final extension of 5 min at 72 °C. PCR amplicons were purified with Agencourt AMPure Beads (Beckman Coulter, Indianapolis, IN) and quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). After the individual quantification step, amplicons were pooled in equal amounts, and paired‐end 2 × 300 bp sequencing was performed using the Illumina MiSeq platform with the MiSeq Reagent Kit v3 at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China).
Sequencing data analysis
The Quantitative Insights Into Microbial Ecology (QIIME, v1.8.0) pipeline was employed to process the sequencing data, as previously described by (Caporaso et al., 2010). Briefly, raw sequencing reads with exact matches to the barcodes were assigned to respective samples and identified as valid sequences. The low‐quality sequences were filtered through the following criteria (Gill et al., 2006; Chen and Jiang, 2014): sequences that had a length of < 150 bp, sequences that had average Phred scores of < 20, sequences that contained ambiguous bases and sequences that contained mononucleotide repeats of > 8 bp. Paired‐end reads were assembled using Fast Length Adjustment of SHort reads (FLASH), a fast computational tool (Magoč and Salzberg, 2011). After chimera detection, the remaining high‐quality sequences were clustered into Operational Taxonomic Units (OTUs) at 97% sequence identity by UCLUST (Edgar, 2010). A representative sequence was selected from each OTU using default parameters. OTU taxonomic classification was performed with BLAST, searching the representative sequences set against the Greengenes Database (DeSantis et al., 2006) using the best hit method (Altschul et al., 1997). Further, to record the abundance and taxonomy of these OTUs in every sample, an OTU table was generated. OTUs containing less than 0.001% of total sequences across all samples were discarded. To minimize the difference in sequencing depth across samples, an average, rounded rarefied OTU table was made. As this table was based on taking an average of 100 evenly resampled OTU subsets under 90% of the minimum sequencing depth, so, it was used for further analysis.
Blood biochemical parameters analysis
To test the fat metabolism level in blood, the concentrations of serum TG, TC, HDL‐C and LDL‐C were determined using a Rayto Chemistry Analyzer Chemray 240 (Chemray 240, China) according to the manufacturer’s instructions with the commercial diagnostic kits (Changchun Huili Biotec. Co., Ltd). The serum sample was mixed thoroughly with reaction solution in recommend ratio and kept at 37 °C for 10 min. After that the absorbance was measured and total concentration was calculated following the formula: Total concentration = Absorbance of sample / Absorbance of calibration solution × Calibration concentration (mmol l−1).
Haematoxylin and eosin (H & E) staining
To compare the morphological changes, liver, breast muscle and leg muscle tissue samples, embedded in paraffin, were cut into 3‐μm‐thick sections with a rotary slicer (LEICARM2245, Leica, Germany). Abdominal adipose tissue samples fixed in OCT were cut into 10‐μm‐thick frozen slice with a microtome cryostat (Shandon Cryotome FSE, Thermo Fisher Scientific, Waltham, MA, USA). H & E staining was performed using a routine protocol, and the examination of stained tissue sections was accomplished by light microscopy (Olympus BX51, Tokyo, Japan) with a digital camera (DP72; Olympus). The average diameter of adipocytes and the average areas of breast muscle cells and leg muscle cells were quantitated using Image‐Pro Plus 6.0.
Immunohistochemical (IHC) staining
To test the distribution and protein expression of P‐AMPK, immunohistochemical staining was performed following the same steps as described in earlier studies (Ansari et al., 2016; Rahman Ansari et al., 2016). Briefly, the tissue sections were deparaffinized twice in xylene and rehydrated in a graded series of ethanol. A microwave oven (MYA‐2270M, Haier, Qingdao, China) was used for heat antigen retrieval in citrate acid buffer solution (pH 6.0) for 20 min (5 min at high level, i.e. 700 W and 15 min at low level, i.e. 116 W). After cooling at room temperature for 2–3 h, 3% H2O2 was used to block endogenous peroxidase. For blocking of non‐specific antibody binding, the tissue sections were incubated with 5% bovine serum albumin (BSA) at 37 °C for 30 min. Sections were then incubated with primary antibodies using rabbit anti‐P‐AMPK antibody (1:100) (Cell Signaling Technology, Inc., USA). Subsequently, tissue sections were incubated at 37 °C with suitable horseradish peroxidase (HRP)‐conjugated secondary antibodies (Boster, Wuhan, China) for 30 min. Immunostaining for all the tissue sections was accomplished using the chromogenic marker Diaminobenzidine (DAB) (Boster, Wuhan, China), and counterstaining was performed using haematoxylin. Then, the sections were washed, dried, dehydrated, cleared and finally mounted with a coverslip.
Serial sections were examined under a light microscope (BH‐2; Olympus, Japan) with a digital camera (DP72; Olympus), and the fields of vision were chosen according to different regions of the liver and muscle tissue in each section. The distributions and expression levels of different proteins were measured in high‐power fields selected at random. All of the images were taken using the same microscope and camera set. Image‐Pro Plus (IPP) 6.0 software (Media Cybernetics, USA) was used to calculate the mean density for positive staining.
Western blotting (WB)
To test the protein expression of P‐AMPK, Western blotting was performed following previously described methods (Hnasko and Hnasko, 2015). Briefly, the frozen specimens were powdered in liquid nitrogen and homogenized in lysis buffer with a protease inhibitor. The supernatants were vortexed, incubated on ice and centrifuged at 12 000 g for 5 min. Protein concentrations were measured using the BCA protein quantification kit (Beyotime, Jiangsu, China). Equal amounts of total proteins (40 μg) were subjected to 10% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) (30 min at 80 volts, following 80 min at 120 volts). Then, the separated proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Merck Millipore, USA). The membranes were incubated with rabbit anti‐P‐AMPK (1:1000) (Cell Signaling Technology, Inc., USA), rabbit anti‐AMPK (1:1000) (ABclonal, China), mouse anti‐GAPDH (1:10 000) (Proteintech Group, Inc., USA) and rabbit anti‐β‐actin (1:5000) (ABclonal, China) antibodies for 12 h. After washing in 1X TBST buffer three times, samples were incubated with peroxidase‐conjugated secondary antibody (1:5000) for 120 min (Boster, China). The blots were developed with Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA) and visualized using ChemiDoc‐It™ Imaging System. Western blot results were analysed using IPP 6.0 software.
Real‐time quantitative polymerase chain reaction (qPCR)
To measure the expression of fat metabolism‐related genes at the mRNA level, total RNA was isolated from the liver, abdominal adipose tissue, breast muscle and leg muscle with TRIzol reagent (Takara, Japan) according to the manufacturer’s instructions. The cDNA was synthesized using the RevertAid First‐Strand cDNA Synthesis Kit (Takara, Japan). The reaction mixture (10 μl) for qPCR contained 5 μl of SYBR Select Master Mix for CFX (Takara, Japan), 0.4 μl of each forward and reverse primer, 3.2 μl of ddH2O and 1 μl of template cDNA. The qPCR reactions were performed on a Bio‐Rad CFX Connect real‐time qPCR detection system (Bio‐Rad, Hercules, CA, USA).
The qPCR conditions were as follows: pre‐denaturation at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s and elongation at 72 °C for 20 s. The primer sequences are listed in Table 1. β‐Actin was chosen as a reference for qPCR. All samples were run in triplicate, and gene expression levels were quantified using the ΔΔCt method.
Table 1.
Primers used for real‐time qPCR
| Gene | Primer sequences (5’ to 3’) | Accession no. |
|---|---|---|
| β‐actin | f‐TTGTTGACAATGGCTCCGGT | NM_205518.1 |
| r‐TCTGGGCTTCATCACCAACG | ||
| ApoAI | f‐GTGACCCTCGCTGTGCTCTT | NM_205525.4 |
| r‐CACTCAGCGTGTCCAGGTTGT | ||
| LPL | f‐TGGACATTGGTGACCTGCTTATGC | NM_205282.1 |
| r‐TCGCCTGACTTCACTCTGACTCTC | ||
| ACC | f‐TCCAGCAGAACCGCATTGACAC | NM_205505.1 |
| r‐GTATGAGCAGGCAGGACTTGGC | ||
| FAS | f‐GCTCTGCGTCTGCTTCAGTCTAC | NM_001199487.1 |
| r‐GGTACAGGACTCTGCCATCAATGC | ||
| FADS1 | f‐CCGTGCCACTGTGGAGAAGATG | LC061145.1 |
| r‐GCCTAGAAGCAACGCAGAGAAGAG | ||
| CYP2C45 | f‐AACAAGCACCACCACACGATACG | AJ430583.1 |
| r‐GGTCAGCCACGCAAGGTCTTC | ||
| ACSL1 | f‐GACTAATGGTCACAGGAGCAGCAC | NM_001012578.1 |
| r‐CCAGGCATTGACAGTGAGCATCC | ||
| PPARα | f‐TGCTGTGGAGATCGTCCTGGTC | AF163809.1 |
| r‐CTGTGACAAGTTGCCGGAGGTC | ||
| CPT‐1 | f‐GCCAAGTCGCTCGCTGATGAC | DQ314726.1 |
| r‐ACGCCTCGTAGGTCAGACAGAAC | ||
| fiaf | f‐AGATCAAGCAGCAGCAGTACAAGC | XM_001232283.5 |
| r‐ACGCTCACATTATGGCTCTGGTTG | ||
| A‐FABP | f‐ACAATGGCACACTGAAGCAGG | FJ493543.1 |
| r‐AGCAGGTTCCCATCCACCAC | ||
| SREBP1 | f‐GGTCCGGGCCATGTTGA | AJ310768.1 |
| r‐CAGGTTGGTGCGGGTGA | ||
| PPARG | f‐GAATGCCACAAGCGGAGAAGGAG | NM_001001460.1 |
| r‐GCTCGCAGATCAGCAGATTCAGG | ||
| AP2 | f‐ACTGAAGCAGGTGCAGAAGTGG | NM_204290.1 |
| r‐TGCATTCCACCAGCAGGTTCC | ||
| Adiponectin | f‐TACGTGTACCGCTCCGCCTTC | KP729052.1 |
| r‐GTGCTGCTGTCGTAGTGGTTCTG |
Statistical analysis
Sequence data analyses were mainly performed using QIIME and R packages (v3.2.0). OTU‐level alpha diversity indices, such as the Chao richness index and Shannon diversity index, were calculated using the OTU table in QIIME. The taxonomic compositions and abundances were visualized using Excel. LEfSe was performed to detect differentially abundant taxa across groups using the default parameters (LDA > 2) (Segata et al., 2011). Spearman’s correlations between the gut microbiota and fat metabolism were determined using the R software package. All data are presented as the means ± standard error of mean (SEM). All analyses and graphic representations were performed with Prism software 5.01 (GraphPad Software, Inc., San Diego, CA, USA). The statistical significance of the mean values in two‐group comparisons was determined using Student’s t‐test. A P value < 0.05 was considered statistically significant.
Conflict of interest
The authors declare that they have no conflicts of interest.
Author contributions
XL Zhang, YF Hu and HZ Liu designed the research. XL Zhang, YF Hu, Y Chen, RR Cheng, L Cui and AA Nafady performed the research. XL Zhang, YF Hu, AA Elokil and HZ Liu analysed the data. XL Zhang, YF Hu, AR Ansari, M Akhtar, El‐SM Abdel‐Kafy and HZ Liu wrote the paper with the help of all authors. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The current scientific investigation was conducted in accordance with the rules and regulations of the ethics committee for use of animals, Huazhong Agricultural University (HZAUCH‐2018‐008), Wuhan, China.
Consent for publication
Not applicable.
Acknowledgements
We sincerely thank Xinyun Li and Yunlong Ma for their suggestions and revisions on this manuscript and Shijun Li for his assistance in animal management and sample collections.
Microb. Biotechnol. (2022) 15(00), 844–861
Funding information
The National Key Research and Development Program of China (2017YFE0113700), the Hubei Provincial Natural Science Foundation of China (2017CFB514) and the National Natural Science Foundation of China (30800808) supported this work.
Data availability statement
The raw 16S rRNA gene sequencing data are available at the NCBI Sequence Read Archive (SRA), under BioProject PRJNA637407.
References
- Agyare, C. , Boamah, V. , Zumbi, C. , and Osei, F. (2018) Antibiotic use in poultry production and its effects on bacterial resistance. In Antimicrobial Resistance‐A Global Threat. Kumar, Y. (ed.). London, UK: IntechOpen, pp. 33–51. [Google Scholar]
- Akinyemi, F.T. , Ding, J. , Zhou, H. , Xu, K.e. , He, C. , Han, C. , et al. (2020) Dynamic distribution of gut microbiota during embryonic development in chicken. Poult Sci 99: 5079–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, H.K. , Trachsel, J. , Looft, T. , and Casey, T.A. (2014) Finding alternatives to antibiotics. Ann N Y Acad Sci 1323: 91–100. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F. , Madden, T.L. , Schäffer, A.A. , Zhang, J. , Zhang, Z. , Miller, W. , and Lipman, D.J. (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves‐Bezerra, M. , and Cohen, D.E. (2017) Triglyceride metabolism in the liver. Compr Physiol 8: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelakis, E. (2017) Weight gain by gut microbiota manipulation in productive animals. Microb Pathog 106: 162–170. [DOI] [PubMed] [Google Scholar]
- Ansari, A.R. , Ge, X.‐H. , Huang, H.‐B. , Huang, X.‐Y. , Zhao, X. , Peng, K.‐M. , et al. (2016) Effects of lipopolysaccharide on the histomorphology and expression of toll‐like receptor 4 in the chicken trachea and lung. Avian Pathol 45: 530–537. [DOI] [PubMed] [Google Scholar]
- Awad, W.A. , Mann, E. , Dzieciol, M. , Hess, C. , Schmitz‐Esser, S. , Wagner, M. , and Hess, M. (2016) Age‐related differences in the luminal and mucosa‐associated gut microbiome of broiler chickens and shifts associated with campylobacter jejuni infection. Front Cell Infect Microbiol 6: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed, F. , Ding, H. , Wang, T. , Hooper, L.v. , Koh, G.y. , Nagy, A. , et al. (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101: 15718–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bervoets, L. , Van Hoorenbeeck, K. , Kortleven, I. , Van Noten, C. , Hens, N. , Vael, C. , et al. (2013) Differences in gut microbiota composition between obese and lean children: a cross‐sectional study. Gut Pathog 5: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerrum, L. , Engberg, R.M. , Leser, T.D. , Jensen, B.B. , Finster, K. , and Pedersen, K. (2006) Microbial community composition of the ileum and cecum of broiler chickens as revealed by molecular and culture‐based techniques. Poult Sci 85: 1151–1164. [DOI] [PubMed] [Google Scholar]
- Bryan, P.‐F. , Karla, C. , Edgar Alejandro, M.‐T. , Sara Elva, E.‐P. , Gemma, F. , and Luz, C. (2016) Sphingolipids as mediators in the crosstalk between microbiota and intestinal cells: implications for inflammatory bowel disease. Mediators Inflamm 2016: 9890141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J.G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F.D. , Costello, E.K. , et al. (2010) QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casewell, M. , Friis, C. , Marco, E. , McMullin, P. , and Phillips, I. (2003) The European ban on growth‐promoting antibiotics and emerging consequences for human and animal health. J Antimicrob Chemother 52: 159–161. [DOI] [PubMed] [Google Scholar]
- Chen, H. , and Jiang, W. (2014) Application of high‐throughput sequencing in understanding human oral microbiome related with health and disease. Front Microbiol 5: 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M. , Qin, N. , Pei, W. , Li, Q. , Yang, Q. , Chen, Y. , et al. (2018b) Draft whole‐genome sequences of Zhihengliuella halotolerans La12 and Microbacterium kitamiense Sa12, strains with cellulase activity, isolated from the Qinghai‐Tibetan Plateau. Genome Announc 6: e01531‐01517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z.M. , Chang, W. , Zheng, A.J. , Zhang, S. , Cai, H. , and Liu, G. (2018a) Comparison of gut microbial diversity in Beijing oil and Arbor acres chickens. Rev Brasil Ciência Avícola 20: 37–44. [Google Scholar]
- Coker, O.O. , Dai, Z. , Nie, Y. , Zhao, G. , Cao, L. , Nakatsu, G. , et al. (2018) Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 67: 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compare, D. , Rocco, A. , Sanduzzi Zamparelli, M. , and Nardone, G. (2016) The gut bacteria‐driven obesity development. Dig Dis 34: 221–229. [DOI] [PubMed] [Google Scholar]
- D'Aquila, T. , Hung, Y.H. , Carreiro, A. , and Buhman, K.K. (2016) Recent discoveries on absorption of dietary fat: presence, synthesis, and metabolism of cytoplasmic lipid droplets within enterocytes. Biochim Biophys Acta 1861: 730–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo, C. , Cavalieri, D. , Di Paola, M. , Ramazzotti, M. , Poullet, J.b. , Massart, S. , et al. (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA 107: 14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeure, O. , Duclos, M.J. , Bacciu, N. , Le Mignon, G. , Filangi, O. , Pitel, F. , et al. (2013) Genome‐wide interval mapping using SNPs identifies new QTL for growth, body composition and several physiological variables in an F2 intercross between fat and lean chicken lines. Genet Sel Evol 45: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis, T.z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E.l. , Keller, K. , et al. (2006) Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Somma, M. , Schaafsma, W. , Grillo, E. , Vliora, M. , Dakou, E. , Corsini, M. , et al. (2019) Natural histogel‐based bio‐scaffolds for sustaining angiogenesis in beige adipose tissue. Cells 8: 1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- Gao, X. , Mu, P. , Zhu, X. , Chen, X. , Tang, S. , Wu, Y. , et al. (2020) Dual function of a novel bacterium, Slackia sp. D‐G6: detoxifying deoxynivalenol and producing the natural estrogen analogue, equol. Toxins 12: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, S.R. , Pop, M. , DeBoy, R.T. , Eckburg, P.B. , Turnbaugh, P.J. , Samuel, B.S. , et al. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312: 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grijalva, J. , and Vakili, K. (2013) Neonatal liver physiology. Semin Pediatr Surg 22: 185–189. [DOI] [PubMed] [Google Scholar]
- Guo, W. , Ren, K.e. , Ning, R. , Li, C. , Zhang, H. , Li, D. , et al. (2020) Fecal microbiota transplantation provides new insight into wildlife conservation. Glob Ecol Conserv 24: e01234. [Google Scholar]
- Hadjadj, L. , Rathored, J. , Keita, M.B. , Michelle, C. , Levasseur, A. , Raoult, D. , et al. (2016) Non contiguous‐finished genome sequence and description of Microbacterium gorillae sp. nov. Stand Genomic Sci 11: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, S. , Pan, Y. , Yang, X.i. , Da, M. , Wei, Q. , Gao, Y. , et al. (2019) Intestinal microorganisms involved in colorectal cancer complicated with dyslipidosis. Cancer Biol Ther 20: 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermier, D. (1997) Lipoprotein metabolism and fattening in poultry. J Nutr 127: 805s–808s. [DOI] [PubMed] [Google Scholar]
- Hnasko, T.S. , and Hnasko, R.M. (2015) The western blot. Methods Mol Biol 1318: 87–96. [DOI] [PubMed] [Google Scholar]
- Hoese, A. , Clay, S.A. , Clay, D.E. , Oswald, J. , Trooien, T. , Thaler, R. , and Carlson, C.G. (2009) Chlortetracycline and tylosin runoff from soils treated with antimicrobial containing manure. J Environ Sci Health B 44: 371–378. [DOI] [PubMed] [Google Scholar]
- Hu, T.G. , Wen, P. , Fu, H.Z. , Lin, G.Y. , Liao, S.T. , and Zou, Y.X. (2019a) Protective effect of mulberry (Morus atropurpurea) fruit against diphenoxylate‐induced constipation in mice through the modulation of gut microbiota. Food Funct 10: 1513–1528. [DOI] [PubMed] [Google Scholar]
- Hu, X. , Wang, Y. , Sheikhahmadi, A. , Li, X. , Buyse, J. , Lin, H. , and Song, Z. (2019b) Effects of glucocorticoids on lipid metabolism and AMPK in broiler chickens' liver. Comp Biochem Physiol B: Biochem Mol Biol 232: 23–30. [DOI] [PubMed] [Google Scholar]
- Indiani, C. , Rizzardi, K.F. , Castelo, P.M. , Ferraz, L.F.C. , Darrieux, M. , and Parisotto, T.M. (2018) Childhood obesity and firmicutes/bacteroidetes ratio in the gut microbiota: a systematic review. Child Obes 14: 501–509. [DOI] [PubMed] [Google Scholar]
- Joy, S.R. , Li, X. , Snow, D.D. , Gilley, J.E. , Woodbury, B. , and Bartelt‐Hunt, S.L. (2014) Fate of antimicrobials and antimicrobial resistance genes in simulated swine manure storage. Sci Total Environ 481: 69–74. [DOI] [PubMed] [Google Scholar]
- Kalavathy, R. , Abdullah, N. , Jalaludin, S. , and Ho, Y.W. (2003) Effects of Lactobacillus cultures on growth performance, abdominal fat deposition, serum lipids and weight of organs of broiler chickens. Br Poult Sci 44: 139–144. [DOI] [PubMed] [Google Scholar]
- Kharrat, N. , Assidi, M. , Abu‐Elmagd, M. , Pushparaj, P.N. , Alkhaldy, A. , Arfaoui, L. , et al. (2019) Data mining analysis of human gut microbiota links Fusobacterium spp. with colorectal cancer onset. Bioinformation 15: 372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.H. , Jeong, D. , Kang, I.B. , Kim, H. , Song, K.Y. , and Seo, K.H. (2017) Dual function of Lactobacillus kefiri DH5 in preventing high‐fat‐diet‐induced obesity: direct reduction of cholesterol and upregulation of PPAR‐α in adipose tissue. Mol Nutr Food Res 61: 1700252. [DOI] [PubMed] [Google Scholar]
- Koliada, A. , Syzenko, G. , Moseiko, V. , Budovska, L. , Puchkov, K. , Perederiy, V. , et al. (2017) Association between body mass index and Firmicutes/Bacteroidetes ratio in an adult Ukrainian population. BMC Microbiol 17: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landers, T.F. , Cohen, B. , Wittum, T.E. , and Larson, E.L. (2012) A review of antibiotic use in food animals: perspective, policy, and potential. Public Health Rep 127: 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R.E. , Turnbaugh, P.J. , Klein, S. , and Gordon, J.I. (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444: 1022–1023. [DOI] [PubMed] [Google Scholar]
- Li, L. , Jiang, Z. , Yao, Y. , Yang, Z. , and Ma, H. (2020a) (−)‐Hydroxycitric acid regulates energy metabolism by activation of AMPK ‐ PGC1α ‐ NRF1 signal pathway in primary chicken hepatocytes. Life Sci 254: 117785. [DOI] [PubMed] [Google Scholar]
- Li, S. , Yan, C. , Liu, T. , Xu, C. , Wen, K. , Liu, L. , et al. (2020b) Research Note: Increase of bad bacteria and decrease of good bacteria in the gut of layers with vs. without hepatic steatosis. Poult Sci 99: 5074–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Fu, R. , Liu, R. , Zhao, G. , Zheng, M. , Cui, H. , et al. (2016) Protein profiles for muscle development and intramuscular fat accumulation at different post‐hatching ages in chickens. PLoS One 11: e0159722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč, T. , and Salzberg, S.L. (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27: 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthei, K.A. , Yang, S.‐M. , Baljinnyam, B. , Chang, L. , Glukhova, A. , Yuan, W. , et al. (2018) Molecular basis for activation of lecithin:cholesterol acyltransferase by a compound that increases HDL cholesterol. elife 7: e41604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margetak, C. , Travis, G. , Entz, T. , Mir, P.S. , Wei, S. , and Dodson, M.V. (2012) Fatty acid composition of phospholipids and in the central and external positions of triacylglycerol in muscle and subcutaneous fat of beef steers fed diets supplemented with oil containing n6 and n3 fatty acids while undergoing one of three 48 h feed withdrawal treatments. J Lipids 2012: 543784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron, D.F. , Smith, T.J. , and Nachman, K.E. (2013) Restrictions on antimicrobial use in food animal production: an international regulatory and economic survey. Global Health 9: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellouk, N. , Ramé, C. , Barbe, A. , Grandhaye, J. , Froment, P. , and Dupont, J. (2018) Chicken is a useful model to investigate the role of adipokines in metabolic and reproductive diseases. Int J Endocrinol 2018: 4579734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler‐Zebeli, B.U. , Siegerstetter, S.‐C. , Magowan, E. , Lawlor, P.G. , O′Connell, N.E. , and Zebeli, Q. (2019) Fecal microbiota transplant from highly feed efficient donors affects cecal physiology and microbiota in low‐ and high‐feed efficient chickens. Front Microbiol 10: 1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na, W. , Wu, Y.‐Y. , Gong, P.‐F. , Wu, C.‐Y. , Cheng, B.‐H. , Wang, Y.‐X. , et al. (2018) Embryonic transcriptome and proteome analyses on hepatic lipid metabolism in chickens divergently selected for abdominal fat content. BMC Genom 19: 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, Y. , Yuan, H. , and Fu, L. (2010) Aerobic exercise's reversal of insulin resistance by activating AMPKα‐ACC‐CPT1 signaling in the skeletal muscle of C57BL/6 mice. Int J Sport Nutr Exerc Metab 20: 370–380. [DOI] [PubMed] [Google Scholar]
- Oldoni, F. , Sinke, R.J. , and Kuivenhoven, J.A. (2014) Mendelian disorders of high‐density lipoprotein metabolism. Circ Res 114: 124–142. [DOI] [PubMed] [Google Scholar]
- Pamer, E.G. (2016) Resurrecting the intestinal microbiota to combat antibiotic‐resistant pathogens. Science 352: 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, R.J. , Hinsu, A.T. , Patel, N.V. , Koringa, P.G. , Jakhesara, S.J. , Thakkar, J.R. , et al. (2018) Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing. Microbiome 6: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry, S.A. , and Hodson, L. (2017) Influence of dietary macronutrients on liver fat accumulation and metabolism. J Investig Med 65: 1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascale, A. , Marchesi, N. , Govoni, S. , Coppola, A. , and Gazzaruso, C. (2019) The role of gut microbiota in obesity, diabetes mellitus, and effect of metformin: new insights into old diseases. Curr Opin Pharmacol 49: 1–5. [DOI] [PubMed] [Google Scholar]
- Rahman Ansari, A. , Ge, X.‐H. , Huang, H. , Wang, J.‐X. , Peng, K.‐M. , Yu, M. , and Liu, H.‐Z. (2016) Expression patterns of toll‐like receptor 4 in pig uterus during pregnancy. Pak Vet J 35: 466–469. [Google Scholar]
- Ridaura, V.K. , Faith, J.J. , Rey, F.E. , Cheng, J. , Duncan, A.E. , Kau, A.L. , et al. (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo, K.C. , Tamhankar, A.J. , Johansson, E. , and Lundborg, C.S. (2010) Antibiotic use, resistance development and environmental factors: a qualitative study among healthcare professionals in Orissa, India. BMC Public Health 10: 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, S. , Saxena, V.K. , Tomar, S. , Sapcota, D. , and Gonmei, G. (2016) Characterisation of caecum and crop microbiota of Indian indigenous chicken targeting multiple hypervariable regions within 16S rRNA gene. Br Poult Sci 57: 381–389. [DOI] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W.S. , and Huttenhower, C. (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegerstetter, S.‐C. , Petri, R.M. , Magowan, E. , Lawlor, P.G. , Zebeli, Q. , O'Connell, N.E. , and Metzler‐Zebeli, B.U. (2018) Fecal microbiota transplant from highly feed‐efficient donors shows little effect on age‐related changes in feed‐efficiency‐associated fecal microbiota from chickens. Appl Environ Microbiol 84: e02330‐02317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley, D. , Hughes, R.J. , and Moore, R.J. (2014) Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Appl Microbiol Biotechnol 98: 4301–4310. [DOI] [PubMed] [Google Scholar]
- Stanley, D. , Hughes, R.J. , Geier, M.S. , and Moore, R.J. (2016) Bacteria within the gastrointestinal tract microbiota correlated with improved growth and feed conversion: challenges presented for the identification of performance enhancing probiotic bacteria. Front Microbiol 7: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh, P.J. , Bäckhed, F. , Fulton, L. , and Gordon, J.I. (2008) Diet‐induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh, P.J. , Ley, R.E. , Mahowald, M.A. , Magrini, V. , Mardis, E.R. , and Gordon, J.I. (2006) An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031. [DOI] [PubMed] [Google Scholar]
- Van Boeckel, T.P. , Brower, C. , Gilbert, M. , Grenfell, B.T. , Levin, S.A. , Robinson, T.P. , et al. (2015) Global trends in antimicrobial use in food animals. Proc Natl Acad Sci USA 112: 5649–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Boeckel, T.P. , Gandra, S. , Ashok, A. , Caudron, Q. , Grenfell, B.T. , Levin, S.A. , and Laxminarayan, R. (2014) Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect Dis 14: 742–750. [DOI] [PubMed] [Google Scholar]
- Videvall, E. , Song, S.J. , Bensch, H.M. , Strandh, M. , Engelbrecht, A. , Serfontein, N. , et al. (2019) Major shifts in gut microbiota during development and its relationship to growth in ostriches. Mol Ecol 28: 2653–2667. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Ni, X. , Qing, X. , Zeng, D. , Luo, M. , Liu, L. , et al. (2017) Live probiotic Lactobacillus johnsonii BS15 promotes growth performance and lowers fat deposition by improving lipid metabolism, intestinal development, and gut microflora in broilers. Front Microbiol 8: 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, S. , Morrison, M. , and Yu, Z. (2013) Bacterial census of poultry intestinal microbiome. Poult Sci 92: 671–683. [DOI] [PubMed] [Google Scholar]
- Wen, C. , Yan, W. , Sun, C. , Ji, C. , Zhou, Q. , Zhang, D. , et al. (2019) The gut microbiota is largely independent of host genetics in regulating fat deposition in chickens. Isme j 13: 1422–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witczak, C.A. , Sharoff, C.G. , and Goodyear, L.J. (2008) AMP‐activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci 65: 3737–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Y. , Kong, F. , Xiang, Y. , Zhou, W. , Wang, J. , Yang, H. , et al. (2018) Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci Rep 8: 5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav, S. , and Jha, R. (2019) Strategies to modulate the intestinal microbiota and their effects on nutrient utilization, performance, and health of poultry. J Anim Sci Biotechnol 10: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, W. , Sun, C. , Yuan, J. , and Yang, N. (2017) Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Sci Rep 7: 45308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Qian, K. , Zhang, W. , Xu, Y. , and Wu, Y. (2016) Effects of chromium‐enriched bacillus subtilis KT260179 supplementation on chicken growth performance, plasma lipid parameters, tissue chromium levels, cecal bacterial composition and breast meat quality. Lipids Health Dis 15: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Liu, Y. , Li, M. , Wu, H. , Wang, Y. , You, Y.u. , et al. (2018) Predictive and preventive significance of AMPK activation on hepatocarcinogenesis in patients with liver cirrhosis. Cell Death Dis 9: 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H.l. , Xu, Z.q. , Yang, L.l. , Wang, Y.x. , Li, Y.m. , Dong, J.q. , et al. (2018) Genetic parameters for the prediction of abdominal fat traits using blood biochemical indicators in broilers. Br Poult Sci 59: 28–33. [DOI] [PubMed] [Google Scholar]
- Zheng, A. , Luo, J. , Meng, K. , Li, J. , Bryden, W.L. , Chang, W. , et al. (2016) Probiotic (Enterococcus faecium) induced responses of the hepatic proteome improves metabolic efficiency of broiler chickens (Gallus gallus). BMC Genom 17: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, M. , Mao, P. , Tian, X. , Guo, Q. , and Meng, L. (2019) Effects of dietary supplementation of alfalfa meal on growth performance, carcass characteristics, meat and egg quality, and intestinal microbiota in Beijing‐you chicken. Poult Sci 98: 2250–2259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw 16S rRNA gene sequencing data are available at the NCBI Sequence Read Archive (SRA), under BioProject PRJNA637407.