

Abstract
Gut microbiota plays important roles in host nutrition, metabolism and immunity, and is affected by multiple factors. However, the understandings of the gut microbiota in pigs within different breeds, growth periods and genders from a large cohort remain largely undefined. In the present study, the characteristics of the gut microbiota in 120 pigs of different breeds, growth periods and genders were investigated using the Illumina MiSeq PE300 combined with QIIME2 platform. A total of 7 388 636 raw reads and 16 411 features were obtained. Additionally, the microbial diversity, compositions and phenotypes were described. 66.53% microbiota belonged to the top 10 most abundant genera (pan gut bacteria), and 28 species were commonly identified (core gut bacteria, commonality ≥ 75%) among the pigs. Besides, the correlations within pan and core gut microbiota were firstly investigated. The metagenomic function was predicted by using PICRUSt2. Furthermore, the explanatory effects of the influencing factors suggested that growth period was the greatest contributor to the gut microbiota in pigs. These results expanded our knowledge of mammalian gut microbiota within different influencing factors and microbial‐related biological features in swine, which contributes to improving animal production and assisting animal model research.
The gut microbiota of 120 pigs of different breeds, growth periods and genders were characterized by newest platform. The pan and core microbiota of pigs were obtained. The effects of breeds, growth periods and genders on gut microbiota and their contributions were demonstrated.

Introduction
For recent decades, the microorganisms that colonize in human and animal intestines have earned extensive attentions, not only because of their value for microbiological research, but also because of their fundamental functions on host health, such as nutrient supplement, immune modulation, disease prevention and physical development (Li et al., 2019). Despite literatures have reported that the composition of the gut microbiota and functions were greatly influenced by various factors such as diet, breed, age, gender (Zhao et al., 2015; Frese et al., 2015; Kim et al., 2015; Yang et al., 2019; Wang et al., 2019a,b) and external environment (Patil et al., 2019), there is still lack of abundant understanding on these influencing factors. In addition, some microbes account for the majority of the total gut microbiota and exhibit critical functions, which are usually defined as pan microbes (Xue et al., 2018). Furthermore, some microbes are conserved and exhibit important properties during the mutual coevolution of the host and its intestinal microbes (Zhang et al., 2017). These shared core microbes represent a selected niche of health‐associated symbionts (Salonen et al., 2012). Both the gut pan and core microbes provide scientifically reasonable and economical strategies for assessing and regulating the gut microbes that are most relevant for host health.
Swine (Sus scrafa) is not only a major species for meat production but also an important animal research model that extensively used for a wide range of human physiological functions and diseases, due to its similar physiological functions and gut microbiota with human beings (Xiao et al., 2016). With the assistance of the intestinal microbiota, swine can remain healthy and supply meat to humans (Ramayo‐Caldas et al., 2016; Yan et al., 2016; Gresse et al., 2017; Knecht et al., 2020). The predominant gut microbes in pigs are bacteria (> 98% of the entire microbiota) (Xiao et al., 2016) that play crucial roles in nutrient digestion and metabolism, energy supply and immune homeostasis (Salonen et al., 2012; Lindberg, 2014; Huang et al., 2020). However, little specific attention has been paid to the pan and core microbes in gut microbiota of pigs, especially within different breeds, growth periods and genders from a large cohort, and how these factors affecting the swine gut microbiota lacks abundant understanding. Thus, expanding the available comprehensive references of gut microbiota in pigs contributes to global food security and human disease model research.
In the present study, a total of 120 pigs within different breeds (Chinese local Jinhua pigs, JH, and commercial Duroc × Landrace × Yorkshire crossbred pigs, CB), growth periods and genders were randomly selected to illustrate the following three key points: (ⅰ) the swine gut microbiota was characterized with Illumina MiSeq combined with QIIME2 platform; (ⅱ) the pan and core gut microbiota, correlations and predicted metagenomic functions were investigated; and (ⅲ) the effects of breeds, growth periods and genders on gut microbiota and their contributions were demonstrated. These results are of significances for putting insights into gut microbiota within different influencing factors, thus providing scientifically reasonable strategies for assessing the gut microbes that are most relevant for porcine health.
Results
Data assessment
Here, the sequence of V4 region of 16S RNA was performed by the Illumina MiSeq PE300 platform and processed by QIIME2 pipelines to collect gut microbiota data from 120 pigs of different breeds, growth periods and genders. A total of 7 388 636 raw reads were obtained, and each sample contained an average of 61 572 ± 11 340 sequences. After a series of data processing steps such as data filtering, denoising, merging and chimera removing, 47 612 ± 9350 filtered reads, 46 588 ± 9262 clean reads, 41 422 ± 9022 clean tags and 27 223 ± 14 154 effective tags per sample were generated (Fig. S1D). The frequency plots showed the frequency distribution of sequences and features. Most samples contained 15 000–25 000 effective sequences (Fig. S1A). The frequency of most features was below 1000 per sample (Fig. S1B). The quality scores of 93.85% sequence base reached 41 (Fig. S1C). The distributions of length and error rates were also analysed to assess the quality of sequencing data. Over 98% merged reads were at 255–256 nucleotides (nt), and the lengths of the effective tags were at an average level of 255.83 ± 0.11 nt (Fig. S1E). The error rates of the sequenced reads were completely at a low level (< 0.03%), although it was relatively high in the ending position (Fig. S1F).
Porcine intestinal microbial communities and metabolic phenotypes
The α‐diversity and β‐diversity of gut microbiota from 120 samples of different breeds, growth periods and genders are shown in Fig. 1B and C. The rank–abundance curve revealed the species abundance and species uniformity of pigs among different factors (Fig. 1B). The faith‐pd, Shannon and Chao 1 scores of α‐diversity are provided in Fig. S2. The β‐diversity of gut microbiota was separated within the influencing factors (Fig. 1C). A total of 16 411 features were identified (according to 100% nucleotide sequence similarity), resulting in an average of 20 950 ± 6425 features per sample (Table S1). These features were clustered into 414 species, with an average of 76.13 ± 15.72 species per sample (data not shown). Sample‐drove box plot of species accumulation revealed the species amount climbing as the ascent number of samples (Fig. S1G). Firmicutes (68.65%), Bacteroidetes (20.94%), Proteobacteria (3.76%), Spirochaetes (2.59%), Tenericutes (2.32%), Actinobacteria (0.82%), Verrucomicrobia (0.20%), Cyanobacteria (0.17%), Planctomycetes (0.17%) and TM7 (0.16%) were the top 10 phyla (Table S2). A detailed overview of taxonomy bar plot at species level of 120 samples was presented (Fig. 1D). As to gut microbial metabolic phenotype, stress‐tolerant, Gram‐negative, forms biofilms, potentially pathogenic, aerobic, mobile elements, Gram‐positive, anaerobic and facultatively anaerobic bacteria were 75.59 ± 15.01%, 69.54 ± 20.24%, 64.74 ± 18.56%, 44.84 ± 19.68%, 40.30 ± 19.10%, 32.30 ± 20.34%, 30.46 ± 20.24%, 26.86 ± 17.18% and 12.83 ± 12.04% respectively (Fig. 1E).
Fig. 1.
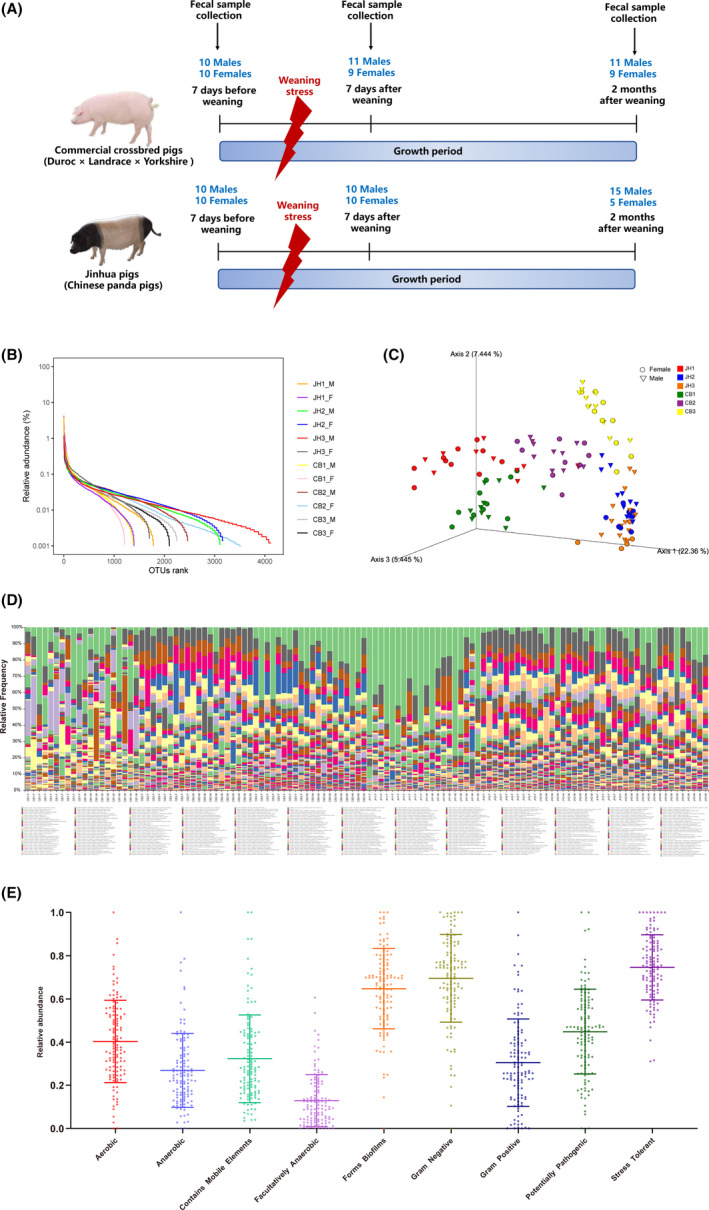
Experimental design and description of gut microbial diversity.
A. Experimental design.
B. Rank–abundance curve of α‐diversity.
C. PCA plot of β‐diversity based on unweighted UniFrac distance.
D. Composition of gut microbiota at species level from 120 samples.
E. The phenotypes of swine gut microbiota. JH1‐M: male Jinhua pigs at phase I; JH1‐F: female Jinhua pigs at phase I; JH2‐M: male Jinhua pigs at phase II; JH2‐F: female Jinhua pigs at phase II; JH3‐M: male Jinhua pigs at phase III; JH3‐F: female Jinhua pigs at phase III; CB1‐F: male crossbred pigs at phase I; CB1‐F: female crossbred pigs at phase I; CB2‐M: male crossbred pigs at phase II; CB2‐F: female crossbred pigs at phase II; CB3‐M: male crossbred pigs at phase III; CB3‐F: female crossbred pigs at phase III.
Pan and core bacteria of swine gut, correlations and metagenome function prediction
Lactobacillus (20.95%, 252 features), Prevotella (6.41%, 861 features), Treponema (2.48%, 413 features), Oscillospira (1.96%, 610 features), Clostridium (1.69%, 167 features), Ruminococcus (1.36%, 427 features), Holdemania (1.28%, 97 features), Streptococcus (1.15%, 70 features), Bacteroides (1.06%, 211 features) and Coprococcus (0.87%, 301 features) were the top 10 highest relative abundance genera, which represented the pan gut bacteria of the pigs (Fig. S3, Table S3). These genera came from the three most abundant phyla: Firmicutes (68.56%), Bacteroidetes (20.94%) and Spirochaetes (2.59%) (Table S2). The phylogenetic relationships of these top 10 genera and their species were described (Fig. 2A). Scatter plot revealed the commonalities of gut microbiota among the 120 pigs (Fig. 2B). 28 bacterial taxa (commonality ≥ 75%) were commonly identified among the pig gut microbiota (Table S4). The correlations of top 10 abundance pan bacteria and 28 core species are shown in Fig 2C and D. Pan gut bacteria were clustered into 3 functional microbial groups. Bacteria in group II and group III were positively related to each other within the groups. Interestingly, Lactobacillus in group I had negative relationships with the bacteria in group II. Bacteroides in group III had negative correlation with Coprococcus and Ruminococcus in group I and group II respectively. As to core gut bacteria, 4 functional microbial groups were clustered. Strains in group I, group III and group IV were positively related to the others within the groups. Notably, Lactobacillus vaginalis ATCC49540 and Lactobacillus amylovorus ATCC33620 in group II showed significantly negative relationships with the most of strains in group I. Both Bamesiella intestinihominis YIT11860 and Intestinimonas butyriciproducens AP4 had positive correlations with the bacteria in group I and group III. The PCA plots of enzymes and pathways of pan bacteria are presented in Fig. 2E and F. The enzymes and pathways of the three clusters of pan bacteria were obviously separated. The distinguished enzyme and pathway information of the three groups is provided in Table S5.
Fig. 2.
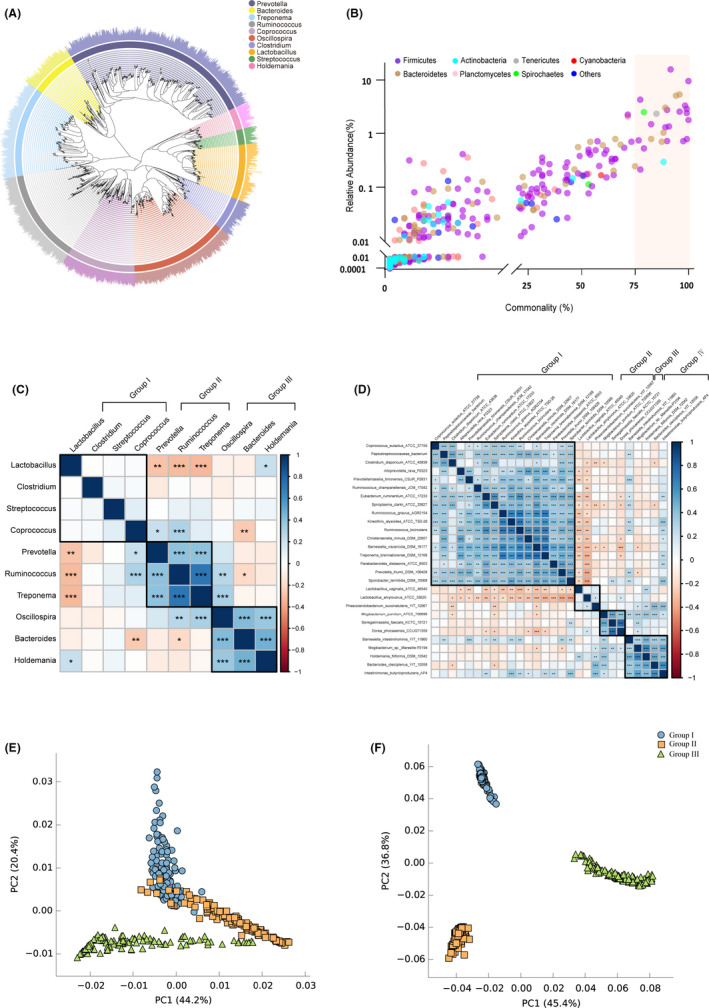
Pan and core gut microbiota of swine, correlations and metagenomic functions.
A. Phylogenetic tree of the top 10 most abundant genera.
B. Scatter plot of commonly identified microbes at species level. Commonality = number of observed samples/total samples. The colours of circles indicate the species from distinct bacterial phyla.
C. The correlation of top 10 abundance pan microbiota by Pearson’s rank.
D. The correlation of 28 core microbiota by Pearson’s rank. The black box in heat map plots represents the cluster of gut microbiota by ward method. Significant correlation is represented by ***: P < 0.001, **: 0.001 < P < 0.01 and *: 0.01 < P < 0.05 respectively.
E. PCA plot of predicted enzymes of pan microbiota based on Enzyme Commission numbers.
F. PCA plot of predicted pathways of pan microbiota based on KEGG.
Metagenomic function predictions of swine gut microbiota
The predicted metagenomic functions of pig gut microbes based on KEGG database are indicated in Fig 3A. The genes assigned to metabolism, genetic information processing, environmental information processing, unclassified functions, cellular processes and organismal systems were 46.24 ± 0.87%, 21.10 ± 1.06%, 14.22 ± 0.91%, 13.88 ± 0.45%, 2.99 ± 0.56% and 0.64 ± 0.07% respectively. The PCA plots of enzymes and pathways of 120 pigs within different influencing factors based on PICRUSt2 are presented in Fig. 3B and C. The distinguished functional information among three influencing factors is provided in Table S6.
Fig. 3.
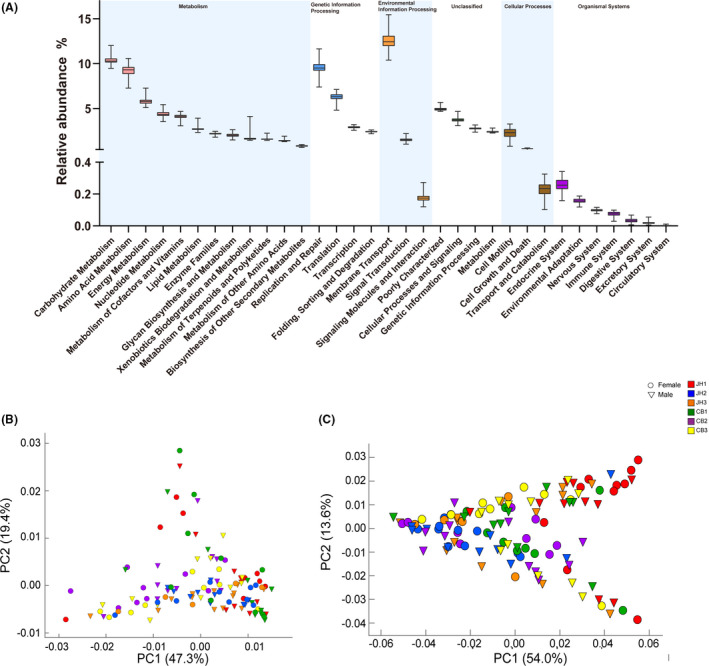
Metagenomic functional predications of gut microbiota of 120 pigs.
A. Level 2 of KEGG pathways predicted by PICRUSt1.
B. PCA plot of predicted enzymes based on PICRUSt2.
C. PCA plot of predicted pathways based on PICRUSt2.
Influencing factor investigation and their contributions to gut microbiota
The most differentially abundant taxa from the linear discriminant analysis (LDA) effect size (LEfSe) results showed the details of the microbiota varied between breeds, growth periods and genders (Fig. 4). Jinhua pigs had more abundance of Ruminococcaceae, Lanchnospiraceae and Bacteroidales compared with commercial pigs. On the contrary, commercial pigs had more abundance of Clostridium, Erysipelotrichaceae and Flavobateriia (Fig. 4A). Pigs before weaning had relatively high richness of Lactobacillus, Enterobacteriaceae and Dorea, while weaned pig contained higher abundant level of Prevotella, Burkholderiales and Ruminococcaceae. As pig grown up, high amount of Erysipelotrichaceae, Clostridials and Clostridium was found in their gut microbiota (Fig. 4B). Additionally, male pigs had more abundance of Fusobacteria compared with the females (Fig. 4C). The FDR corrections were used to verify the results of LEfSe for statistical analysis of metagenomic profiles (STAMP) (Fig. S4). Venn plots demonstrated the contribution of breed, gender and growth period to the pig gut microbiota (Fig. 4D). The individual contribution of breed, gender and growth period was 2.02%, 0.81% and 16.38% respectively. The combined explanation of breed and gender, gender and growth period and growth period and breed was 0.80%, 8.30% and 5.52%. The explanation of the above three factors reached 53.08%. 13.09% of gut microbial contribution remained unexplained.
Fig. 4.
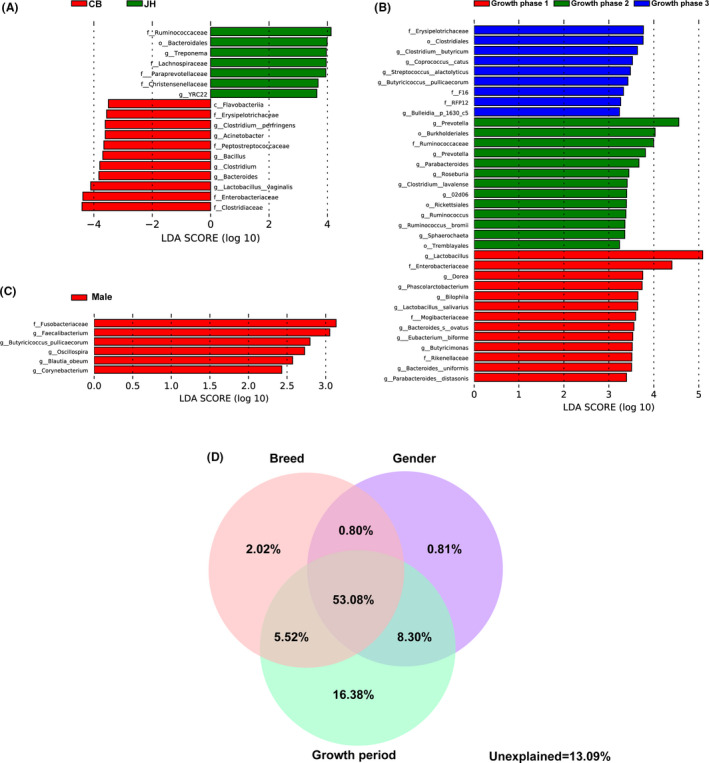
The effects of breeds, growth periods and genders on swine gut microbiota.
A‐C. Histograms of the LDA scores reveal the most differentially abundant taxa among different breeds (A), growth periods (B) and genders (C).
D. The contributions of individual influencing factors and their interaction effects.
Discussion
As one of the Chinese indigenous species, Jinhua pig is famous for high intramuscular fat deposition, stress resistance and tolerance of crude feed (Wu et al., 2013). Duroc × Landrace × Yorkshire crossbred pig is one of the most used commercial pigs around the world due to the characteristics of high growth rate and feed conversion rate. The gut microbiota of Jinhua and Duroc × Landrace × Yorkshire with different growth periods and genders was successfully analysed in this study. The data were systemically filtered by quality scores, length of reads and error rates to ensure the quality. A gradually stable platform was observed in the curve as the species amount reach saturation and a growing smaller number of novel species were identified in the samples (Fig. S1G), showing that sufficient sequencing depth to reveal gut microbiota exactly (Colwell et al., 2004). Therefore, this data set contained superior quality and quantity to reflect the gut microbiome of pigs of different breeds, growth periods and genders.
The α‐diversity, β‐diversity, species‐level taxonomy and gut microbial phenotypes of the pigs were presented, revealing the gut microbiota variations among individuals and overall status of gut microbiota of the pigs. Pan and core gut microbiota drive researchers to investigate due to their potentials for regulating health. In the present study, Prevotella, Ruminococcus, Lactobacillus, Clostridium, Bacteroides, Treponema, Coprococcus, Oscillospira, Streptococcus and Holdemania were detected in the top 10 genera as pan gut microbiota of pigs. Xiao et al. (2016) reported that Prevotella, Ruminococcus, Lactobacillus, Eubacterium and Helicobacter were predominant bacteria in swine gut. Lactobacillus, Streptococcus, Clostridium, SMB53 and Bifidobacterium were found as the most abundant genera in the gut microbiota of Jinhua pigs (Yang et al., 2018). These results suggest some pan bacteria such as Prevotella, Ruminococcus, Lactobacillus and Clostridium are relatively constant in swine gut microbiota. Notably, Bacteroides, Treponema, Coprococcus, Oscillospira, Streptococcus and Holdemania were newly identified as pan gut microbiota in JH and CB pigs in the present study. Additionally, the phylogenetic tree of top 10 pan bacteria of pig gut was firstly investigated (Fig. 3A). Few studies have defined the core microbiota of porcine gut. Our study identified 28 gut core genera (commonality ≥ 75%), which commonly present in swine gut, including Senegalimassilia, Barnesiella, Bacteroides, Xylanivirga, Intestinimonas, Holdemania, Mogibacterium, Phascolarctobacterium, Coprococcus, Dorea, Treponema and Spiroplasma. To date, only a meta‐analysis of core bacteria in swine gut has demonstrated that Clostridium, Blautia, Lactobacillus, Prevotella, Ruminococcus, Roseburia, RC9 and Subdoligranulum accounted for 99% of swine faeces (Holman et al., 2017). The differences in identified core gut microbiota in the present and previous studies may be the various commonality ranges and samples used. Therefore, the present study has provided a more comprehensive reference of the swine gut microbiota using a large cohort of pigs within different breeds, growths periods and genders. Remarkably, Prevotella, Bacteroides, Ruminococcus, Lactobacillus and Helicobacter were identified in both pan and core bacteria in swine gut, suggesting their fundamental functions on immunity, metabolism and gut homeostasis (Wang et al., 2019a,b). The correlations and clusters of pan and core pig gut microbiota and the predicted metagenomic functions of pan bacteria were firstly reported. The results demonstrated both pan and core bacteria can inhibit or facilitate the growth of other bacteria to maintain the micro‐environment homeostasis of the porcine gut microbiota by their alone or clustering into functional groups. These evidences provide vital information of porcine gut microbiota and potentials to manipulate pig gut bacteria by external intervention.
Metabolism accounted for the most proportion in metabolic functions of gut microbiota, which suggested the main task of gut microbiota of pigs was metabolizing feed and providing nutrients for host. Genetic information processing and environmental information process accounted for the second and third high percentage of metagenomic functions, revealing that porcine gut microbiota plays important roles in genome processing and response to environmental changes. Additionally, the properties of microbial physiological activities and interaction with other organisms, which were known as the microbial functions of cellular processes and organismal systems, were observed.
Several studies have investigated the variations of pig gut microbiota within different influencing factors (Benson et a l., 2010; Round and Mazmanian, 2009; Leamy et al., 2014). However, few demonstrated the contributions of these factors to host gut microbiota. Our results demonstrated that breeds, growth periods and genders affected swine gut microbiota at various degrees. Notably, we found that growth period is the greatest contribution among these three factors due to the various host physiological status (such as energy demand, feed intake, hormone and metabolism). Also, diets, environment and farm management (weaning) contributed to the prominent effects of growth period on gut microbiota. Piglets suffered huge stress during weaning, which is critical window phase for gut microbiota manipulations (Li et al., 2018). The alpha diversity of gut microbiota climbed as pig growing up (Fig. 1B). Similarly, Frese et al (2015) found the alpha diversity of microbiota in faecal sample of pigs increased from nursing to weaned period. Therefore, the richness and maturity of gut microbiota increased when pig grown up. The high relative abundance of Bacteroidaceae, Clostridiaceae, Lachnospiraceae, Lactobacillaceae and Enterobacteriaceae existed in nursing piglets, while the amount of Bacteroides and Enterobacteriaceae reduced and the richness of Lactobacillaceae, Ruminococcaceae, Veillonellaceae and Prevotellaceae improved after weaning. The great population of Bacteroides, Lactobacillaceae and Enterobacteriaceae before weaning and high level of Ruminococcaceae and Prevotellaceae after weaning were found in piglets. Kim et al (2015) reported Bacteroidetes reduced with swine growing up. Additionally, Clostridium, Coprococcus, Streptococcus and Butyricicoccus were found to composite major microbial community in swine gut 2 months after weaning. The evidences confirmed the level of Bacteroides and Enterobacteriaceae decreased and population of Ruminococcaceae and Prevotellaceae increased when piglets go through weaning period due to physical, psychological and dietary changes. To date, information of the effects of breed on the intestinal microbiota in pig is limited, especially in local and commercial pigs. In the present study, great population of Ruminococcaceae, Bacteroidaceae, Spirochaetaceae, Lachnospiraceae, Paraprevotellaceae and Christensenellaceae in Jinhua pigs and high levels of Clostridiaceae, Enterobacteriaceae, Lactobacillaceae and Planococcaceae in crossbreed pigs were observed. Xiao et al (2018) have compared the gut microbiota between Jinhua and Landrace pigs. They found Jinhua pigs contained abundant amount of Lactobacillaceae, Clostridiaceae, Peptostreptococcaceae and Erysipelotrichaceae, while Landrace pigs had high levels of Enterobacteriaceae, Pasteurellaceae, Campylobacteraceae and Bacteroidaceae. However, they did not investigate the contribution of breed on gut microbiota. The variations in gut microbiota found in Jinhua pigs may be caused by different growth periods and locations of intestinal tracts used. Gender had the minimum impact on pig gut microbiota in our study. Male pigs gut microbiota contained high level of Fusobacteriaceae, Facalibacterium, Butyricicoccus and Oscillospira compared with females, suggesting significantly different gut microbiota and microbial functions between genders.
In summary, we described the characteristics of gut microbiota from different breeds, growth stages and genders of pigs, revealed the relationships within pan and core bacteria and explained the contributors of gut microbiota. This investigation provides a critical resource for microbiological research and expands our knowledge of mammalian gut microbiota among different influencing factors. It can be used to study other topics in the gut microbiota of swine to help increasing farm animal productivity and assisting animal model research.
Experimental procedures
Animal treatment and sample collection
All the procedures were carried out in accordance with the university’s relevant regulations for animal experiments, and the guidelines of the Institutional Animal Care and Use Committee of Zhejiang University (Hangzhou, China) were strictly enforced during the entire experimental period.
A total of 120 JH pigs (Chinese domestic pigs, well known for their meat quality) and commercial Duroc × Landrace × Yorkshire CB pigs were selected for the experiment from three growth periods: approximate 7 days before weaning (phase I, n = 40), approximate 7 days after weaning (phase II, n = 40) and approximate 2 months after weaning (phase III, n = 40) (Fig. 1A). All pigs were housed in pens under consistent management conditions and were allowed to free access to feed and water. The pigs in each growth period were fed the same diet (Table S7). The phenotypes including the body weight and gender were recorded (Table S8).
Sterile swabs (Biosigma Inc., Cona, Italy) were used to collect fresh internal faecal samples to avoid contamination and obtain the most typical samples, and then were quickly transferred to 2.0‐ml cryogenic vials (Sigma‐Aldrich®, Los Angeles, CA, USA) and immediately frozen at −80°C in liquid nitrogen.
DNA extraction and amplicon, library construction and 16S sequencing
Briefly, the sample was transferred to a 5‐mL tube after thawing and 4.5 ml of TN150 buffer and sterile zirconium beads (0.3 g) was added. The mixture was vigorously vortexed for 30 s and centrifuged at 200 g for 5 min at 4°C. Then, the upper phase of the sample mixture (1 ml) was transferred. The pellet was obtained and resuspended in 1 ml TN150 buffer, followed by centrifugation at 500 rpm for 5 min. After purification via phenol–chloroform (25:1) extraction, DNA was precipitated at −20°C for 4 h and dissolved in 60 µl nuclease‐free TE buffer. The concentration was detected in a NanoDrop One/Oneᶜ system (Thermo Fisher Scientific Inc., Wilmington, NC, USA), and 2% agarose gel electrophoresis (Biowest Inc., Madrid, Spain) was performed to assess DNA purity. The DNA sample was then diluted to the proper concentration.
PCR amplifications were conducted targeting the V4 region of the 16S rRNA gene with the primers (F: 5’‐GTGCCAGCMGCCGCGG‐3’ and R: 5’‐GGACTACHVGGGTWTCTAAT‐3’) according to the following parameters: initial denaturation at 95 °C for 3 min, followed by 27 cycles of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec, elongation at 72°C for 45 sec and a final extension at 72°C for 10 min. The final products were extracted from 2% agarose gels, purified using a MinElute Gel Extraction Kit (Qiagen Inc., Dusseldorf, Germany) and assessed on an Invitrogen Qubit 4 System (Thermo Fisher).
Before the preparation of the library, the PCR products were mixed and purified by using the GenElute™ PCR Clean‐Up Kit (Sigma). The construction of sequencing libraries was conducted by using a DNA Seq Library Preparation Kit (Illumina Inc., Madison, WI, USA). Digital PCR Library Quantification Kit (Bio‐Rad, Los Angeles, CA, USA) and Qsep Series Bio‐Fragment Analyzers (BiOptic Inc., Taiwan, China) were used to assess the quality of library. The Illumina MiSeq PE 300 platform was applied for sequencing to generate paired‐end reads (2 × 300 bp) from the sequence library based on a standard protocol (Caporaso et al., 2012).
Bioinformatic analyses
The sequence data have been submitted to the NCBI database under Accession Number SRR11485509‐11485628. The analysis of sequencing data was performed based on the QIIME2 (Quantitative Insight Into Microbial Ecology 2) analysis (version 2020.2, http://QIIME2.org/index.html). DADA2 pipelines including read filtering, sequence denoising, data assembly and chimaera removal were applied (Callahan et al., 2016).
To improve the accuracy, extensiveness and reproducibility of the data analysis (Martino et al., 2019), we classified these features by using QIIME2’s q2‐feature‐classifier plugin (Bokulich et al., 2018). A feature table was generated from the effective tags that did not contain non‐biological nucleotides, which presented the number of every precise feature observed in each sample. We regarded the most abundant sequence in each feature as the most representative sequence and used it for taxonomic annotation at the phylum to species levels against the greenGene 13.8 database (DeSantis et al., 2006) with 100% sequence identification on the basis of the Ribosomal Database Project (RDP) Multiclassifier tool (http://sourceforge.net/projects/rdp‐classifier/) (Wang et al., 2007). Unclassified features were further identified with NCBI BLAST method (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
α‐ and β‐diversities of bacterial community were calculated by QIIME2 diversity plugin base on faith‐pd, Chao1 and Shannon scores and unweighted UniFrac distance. The phylogenetic relationships among distinct features were analysed. Sequence alignment was performed by applying Molecular Evolutionary Genetics Analysis software (MEGA) (Tamura et al., 2011) with the MUSCLE method (neighbour‐joining tree, bootstrap method 1000 and Poisson’s model) (Edgar, 2004), and the results were visualized by iTOL (version 5, https://itol.embl.de/) (Letunic and Bork, 2019). Correlations plots were generated by R studio corrplot package. Furthermore, the predicted microbial metabolic functions were conducted by PICRUSt1 (https://huttenhower.sph.harvard.edu/galaxy/) and PICRUST2 (Douglas et al., 2020).
Statistical analysis
SAS software (version 9.2; SAS Inc., Chicago, IL, USA) was applied to analyse the data. Data were expressed as means with standard deviation (SD). The analysis of the non‐parametric Pearson correlation coefficient and significance between pan and core bacteria of swine gut were performed using the ‘corrplot’ package in R (R Core Team, 2014). Welch’s test and the Benjamini–Hochberg FDR‐adjusted method for STAMP (version 2.1.3) were used to perform the multiple testing corrections of the most differentially abundant taxa within breeds, growth periods and genders. The explanations of the influencing factors were calculated by variance partitioning analyses by ‘varpart’ package of R.
Funding Information
This work was funded by the National Natural Science Foundation of China (Grant Nos. 32022079, 31702123 and 31630075), Zhejiang Provincial Natural Science Foundation of China (Grant No. LZ20C170005), Fundamental Research Funds for the Central Universities (Grant No. 2020‐KYY‐517102‐0001), Earmarked Fund for Modern Agro‐industry Technology Research System (Grant No. CARS‐35) and Seed Foundation of Innovation and Creation for Graduate Students in Northwestern Polytechnical University (Grant No. CX2020269).
Conflict of interest
None declared.
Supporting information
Fig. S1. Statistic information of the sequencing data through QIIME2 pipelines. (A) The details of quality‐controlled reads generation through DADA2 method. (B) The frequency plots of the sequence number per sample. (C) The frequency plots of the feature number per feature. (D) The quality score of sequenced data. (E) The length variations of merged sequences. (F) The error rate of at positions along reads. (G) The species accumulation curve.
Fig. S2. α diversity of gut microbiota based on faith‐pd score (A), Shannon (B) and Chao1 (C).
Fig. S3. The phylogenetic tree of porcine gut microbiota. The top 10 most abundant genera and each taxa’s proportions were listed. Each colour in pie charts represents one sample of 120 pigs.
Fig. S4. The STAMP test results of significantly different swine gut microbiota within breeds, ages and genders. (A) the FDR correction of the most differentially abundant taxa within breeds. (B) the FDR correction of the most differentially abundant taxa within genders. (B, C, D) the FDR correction of the most differentially abundant taxa within genders, phase I vs phase II, phase I vs phase III and phase II vs phase III respectively.
Table S1. Feature table of swine gut microbiota.
Table S2. Phylum table of swine gut microbiota.
Table S3. Genera table of swine gut microbiota.
Table S4. A list of the commonly identified strains (commonality ≥ 75%) in 120 pigs.
Table S5. Significantly different metagenome functions prediction of pan microbiota.
Table S6. Significantly different metagenome functions prediction of pig gut microbiota.
Table S7. Diet information in experiment.
Table S8. Phenotypic information of 120 pigs.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant Nos. 32022079, 31702123 and 31630075), Zhejiang Provincial Natural Science Foundation of China (Grant No. LZ20C170005), Fundamental Research Funds for the Central Universities (Grant No. 2020‐KYY‐517102‐0001), Earmarked Fund for Modern Agro‐industry Technology Research System (Grant No. CARS‐35) and Seed Foundation of Innovation and Creation for Graduate Students in Northwestern Polytechnical University (Grant No. CX2020269).
Microb. Biotechnol. (2022) 15(00), 793–804
References
- Benson, A.K. , Kelly, S.A. , Legge, R. , Ma, F. , Low, S.J. , Kim, J. , et al. (2010) Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA 107: 18933–18938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , Kaehler, B.D. , Rideout, J.R. , Dillon, M. , Bolyen, E. , Knight, R. , et al. (2018) Optimizing taxonomic classification of marker‐gene amplicon sequences with QIIME 2 ' s q2‐feature‐classifier plugin. Microbiome 6: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B.J. , McMurdie, P.J. , Rosen, M.J. , Han, A.W. , Johnson, A.J. , and Holmes, S.P. (2016) DADA2: High‐resolution sample inference from Illumina amplicon data. Nat Methods 13: 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G., Lauber C.L., Walters W.A., Berg‐Lyons D., Huntley J., Fierer N., et al. (2012) Ultra‐high‐throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J, 6(8), 1621–1624. 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell, R.K. , Mao, C.X. , and Chang, J. (2004) Interpolating, extrapolating, and comparing incidence‐based species accumulation curves. Ecology 85: 2717–2727. [Google Scholar]
- DeSantis, T.Z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E.L. , Keller, K. , et al. (2006) Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas, G.M. , Maffei, V.J. , Zaneveld, J.R. , Yurgel, S.N. , Brown, J.R. , Taylor, C.M. , et al. (2020) PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 38: 685–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese, S.A. , Parker, K. , Calvert, C.C. , and Mills, D.A. (2015) Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 3: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresse, R. , Chaucheyras‐Durand, F. , Fleury, M.A. , Van de Wiele, T. , Forano, E. , and Blanquet‐Diot, S. (2017) Gut microbiota dysbiosis in postweaning piglets: understanding the keys to health. Trends Microbiol 25: 851–873. [DOI] [PubMed] [Google Scholar]
- Holman, D.B. , Brunelle, B.W. , Trachsel, J. , and Allen, H.K. (2017) Meta‐analysis to define a core microbiota in the swine gut. mSystems 2: e00004‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S.M. , Wu, Z.H. , Li, T.T. , Liu, C. , Han, D.D. , Tao, S.Y. , et al. (2020) Perturbation of the lipid metabolism and intestinal inflammation in growing pigs with low birth weight is associated with the alterations of gut microbiota. Sci Total Environ 719: 137382. [DOI] [PubMed] [Google Scholar]
- Kim, J. , Nguyen, S.G. , Guevarra, R.B. , Lee, I. , and Unnoet, T. (2015) Analysis of swine fecal microbiota at various growth stages. Arch Microbiol 197: 753–759. [DOI] [PubMed] [Google Scholar]
- Knecht, D. , Cholewinska, P. , Jankowska‐Makosa, A. , and Czyz, K. (2020) Development of swine's digestive tract microbiota and its relation to production indices‐a review. Animals 10: 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leamy, L.J. , Kelly, S.A. , Nietfeldt, J. , Legge, R.M. , Ma, F. , Hua, K. , et al. (2014) Host genetics and diet, but not immunoglobulin A expression, converge to shape compositional features of the gut microbiome in an advanced intercross population of mice. Genome Biol 15: 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , and Bork, P. (2019) Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47: W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Zhou, R. , Zhu, J.X. , Huang, X.D. , and Qu, J.P. (2019) Environmental filtering increases with elevation for the assembly of gut microbiota in wild pikas. Microb Biotechnol 12: 976–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Guo, Y. , Wen, Z. , Jiang, X. , Ma, X. , and Han, X. (2018) Weaning stress perturbs gut microbiome and its metabolic profile in piglets. Sci Rep 8: 18068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg, J.E. (2014) Fiber effects in nutrition and gut health in pigs. J Anim Sci Biotechnol 5: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino, C. , Morton, J. T. , Marotz, C. A. , Thompson, L. R. , Tripathi, A. , Knight, R. et al. (2019) A novel sparse compositional technique reveals microbial perturbations. mSystems 4: e00016‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, Y. , Gooneratne, R. , and Ju, X.H. (2019) Interactions between host and gut microbiota in domestic pigs: a review. Gut Microbes 3: 310–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2014). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. http://www.R‐project.org/ [Google Scholar]
- Ramayo‐Caldas, Y. , Mach, N. , Lepage, P. , Levenez, F. , Denis, C. , Lemonnier, G. , et al. (2016) Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J 10: 2973–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round, J.L. , and Mazmanian, S.K. (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9: 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen, A. , Salojarvi, J. , Lahti, L. , and de Vos, W.M. (2012) The adult intestinal core microbiota is determined by analysis depth and health status. Clin Microbiol Infec 18: 16–20. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Peterson, D. , Peterson, N. , Stecher, G. , Nei, M. , and Kumar, S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G.M. , Tiedje, J.M. , and Cole, J.R. (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb 73: 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W.L. , Hu, H.F. , Zijlstra, R.T. , Zheng, J.S. , and Gänzle, M.G. (2019a) Metagenomic reconstructions of gut microbial metabolism in weanling pigs. Microbiome 7: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X.F. , Tsai, T. , Deng, F.L. , Wei, X.Y. , Chai, J.M. , Knapp, J. , et al. (2019b) Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 7: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, T. , Zhang, Z.H. , Yuan, Z.Q. , Lo, L.J. , Chen, J. , Wang, Y.Z. , et al. (2013) Distinctive genes determine different intramuscular fat and muscle fiber ratios of the longissimus dorsi muscles in Jinhua and landrace pigs. PLoS One 8: e53181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, L. , Estellé, J. , Kiilerich, P. , Ramayo‐Caldas, Y. , Xia, Z.K. , Feng, Q. , et al. (2016) A reference gene catalogue of the pig gut microbiome. Nat Microbiol 1: 16161. [DOI] [PubMed] [Google Scholar]
- Xiao, Y.P. , Kong, F.L. , Xiang, Y. , Zhou, W.D. , Wang, J.J. , Yang, H. , et al. (2018) Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci Rep 8: 5985‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, M.Y. , Sun, H.Z. , Wu, X.H. , Guan, L.L. , and Liu, J.X. (2018) Assessment of rumen microbiota from a large dairy cattle cohort reveals the pan and core bacteriomes contributing to varied phenotypes. Appl Environ Microbiol 84: e00970–e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, H.L. , Diao, H. , Xiao, Y. , Li, W.X. , Yu, B. , He, J. , et al. (2016) Gut microbiota can transfer fiber characteristics and lipid metabolic profiles of skeletal muscle from pigs to germ‐free mice. Sci Rep 6: 31786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Xiao, Y.P. , Wang, J.J. , Xiang, Y. , Gong, Y.J. , Wen, X.T. , et al. (2018) Core gut microbiota in Jinhua pigs and its correlation with strain, farm and weaning age. J Microbiol 56: 346–355. [DOI] [PubMed] [Google Scholar]
- Yang, Q.L. , Huang, X.Y. , Wang, P.F. , Yan, Z.Q. , Sun, W.Y. , Zhao, S.G. , et al. (2019) Longitudinal development of the gut microbiota in healthy and diarrheic piglets induced by age‐related dietary changes. Microbiologyopen 8: e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Berg, M. , Dierking, K. , Félix, M.A. , Shapira, M. , Samuel, B.S. , et al. (2017) Caenorhabditis elegans as a model for microbiome research. Front Microbiol 8: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, W.J. , Wang, Y.P. , Liu, S.Y. , Huang, J.J. , Zhai, Z.X. , He, C. , et al. (2015) The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS One 10: e0117441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Statistic information of the sequencing data through QIIME2 pipelines. (A) The details of quality‐controlled reads generation through DADA2 method. (B) The frequency plots of the sequence number per sample. (C) The frequency plots of the feature number per feature. (D) The quality score of sequenced data. (E) The length variations of merged sequences. (F) The error rate of at positions along reads. (G) The species accumulation curve.
Fig. S2. α diversity of gut microbiota based on faith‐pd score (A), Shannon (B) and Chao1 (C).
Fig. S3. The phylogenetic tree of porcine gut microbiota. The top 10 most abundant genera and each taxa’s proportions were listed. Each colour in pie charts represents one sample of 120 pigs.
Fig. S4. The STAMP test results of significantly different swine gut microbiota within breeds, ages and genders. (A) the FDR correction of the most differentially abundant taxa within breeds. (B) the FDR correction of the most differentially abundant taxa within genders. (B, C, D) the FDR correction of the most differentially abundant taxa within genders, phase I vs phase II, phase I vs phase III and phase II vs phase III respectively.
Table S1. Feature table of swine gut microbiota.
Table S2. Phylum table of swine gut microbiota.
Table S3. Genera table of swine gut microbiota.
Table S4. A list of the commonly identified strains (commonality ≥ 75%) in 120 pigs.
Table S5. Significantly different metagenome functions prediction of pan microbiota.
Table S6. Significantly different metagenome functions prediction of pig gut microbiota.
Table S7. Diet information in experiment.
Table S8. Phenotypic information of 120 pigs.