

Abstract
Background:
Chemokine (C- Cmotif) ligand 5 (CCL5) and its receptor C-C motif chemokine receptor 5 (CCR5), have been broadly studied in conjunction with infectious pathogens, however, their involvement in cardiovascular disease is not completely understood. NADPH oxidases (Noxs) are the major source of reactive oxygen species (ROS) in the vasculature. Whether the activation of Noxs is CCL5/CCR5 sensitive and whether such interaction initiates vascular injury is unknown. We investigated whether CCL5/CCR5 leads to vascular damage by activating Noxs.
Material and Methods:
We used rat aortic smooth muscle cells (RASMC) to investigate the molecular mechanisms by which CCL5 leads to vascular damage and carotid ligation (CL) to analyze the effects of blocking CCR5 on vascular injury.
Results:
CCL5 induced Nox1 expression in concentration and time-dependent manners, with no changes in Nox2 or Nox4. Maraviroc pre-treatment (CCR5 antagonist, 40uM) blunted CCL5-induced Nox1 expression. Furthermore, CCL5 incubation led to ROS production and activation of Erk1/2 and NFkB, followed by increased vascular cell migration, proliferation, and inflammatory markers. Notably, Nox1 inhibition (GKT771, 10uM) blocked CCL5-dependent effects. In vivo, CL induced pathological vascular remodeling and inflammatory genes and increased Nox1 and CCR5 expression. Maraviroc treatment (25mg/Kg/day) reduced pathological vascular growth and Nox1 expression.
Conclusions:
Our findings suggest that CCL5 activates Nox1 in the vasculature, leading to vascular injury likely via NFkB and Erk1/2. Herein, we place CCR5 antagonists and/or Nox1 inhibitors might be preeminent antiproliferative compounds to reduce the cardiovascular risk associated with medical procedures (e.g. angioplasty) and vascular diseases associated with vascular hyperproliferation.
Keywords: CCL5, CCR5, Nox1, Reactive Oxygen Species, Vascular
Graphical Abstract
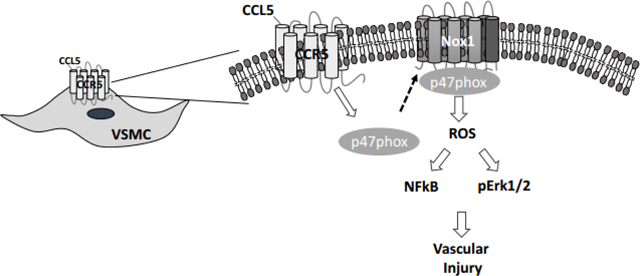
Introduction
Vascular smooth muscle cells (VSMC) are the most abundant component that constitute the arterial walls and are extremely important for vascular homeostasis. These cells contribute in regulating blood flow and vascular tone1,2. “Differentiated VSMC display a quiescent, contractile phenotype which is responsible for the majority of blood flow regulation within blood vessels. In response to injury VSMC can switch their differentiated phenotype into an undifferentiated migratory and proliferative phenotype. Such an event occurs in response to different agonists (cytokines, chemokines, and peptides) and environments (hypertension and post-angioplasty) which initiate proliferation and migration in VSMC3–7.
Chemokines are small chemotactic cytokines that assist leukocytes locate specific targets by binding to receptors positioned at the target cell surface8,9. Compelling evidence places chemokines and their receptors as key regulators of several cardiovascular diseases10–14. Among the chemokine family, chemokine (C-C motif) receptor 5 (CCR5) and its main ligand (C-C motif) ligand 5 (CCL5), also called regulated upon activation, normal T cell expressed and presumably secreted (RANTES), have been amply studied in infectious diseases due to their participation as co-receptors for pathogens in immune cells10,15–17.
However, CCR5 is expressed in many vascular cells including VSMC and endothelial cells (ECs) 18–20. CCR5 and CCL5 often participate in the genesis and progression of cardiovascular abnormalities in obesity, hypertension, and atherosclerosis11,19,21,22. We recently demonstrated that ritonavir, an antiretroviral drug, leads to vascular injury by regulating CCR5 expression in the vasculature19. However, the mechanisms downstream of the CCR5 and CCL5 interaction within the cardiovascular disease prognosis are still not fully deciphered.
NADPH oxidases (Noxs) are enzymes comprised of membrane and cytosolic subunits. Various stimuli cause the Noxs to become activated by forming an assembly among their subunits in order to generate reactive oxygen species (ROS)23,24. Noxs are divided into 7 different subunits, Nox1–5 and DUOX1 and 2. Nox1, Nox2, Nox4 and Nox5 (not expressed in mouse or rat) are the best characterized in cardiovascular cells and have shown to be involved in several cardiovascular events7,25,26. Whether CCL5/CCR5 activates Noxs in vascular cells and induces a vascular injury is still unknown.
Herein, we sought to test the hypothesis that CCL5 induces Nox1-derived ROS which leads to vascular migration, proliferation, and inflammation. Our study has a clinical direction by placing CCR5 and its ligand, as well as Nox1 as potential pharmacological targets in cardiovascular diseases or in medical interventions associated with high vascular proliferative status such as atherosclerosis and angioplasty.
Material and Methods
Reagents
Rat Aortic Smooth Muscle cells (RASMC) (Catalog#: R-ASM-580, Lonza Walkersville, MD, USA); Dulbecco’s Modified Eagle Medium (DMEM) (Catalog#: 11965084 Gibco from Thermo Fisher Scientific, Waltham, MA, USA); Fetal bovine serum (FBS) (Catalog#: MT35010CV, Corning™, Glendale, AZ, USA); penicillin and streptomycin (Catalog#: 15140122, Gibco from Thermo Fisher Scientific, Waltham, MA, USA); recombinant mouse CCL5 (Catalog#: NB27623520U, R&D Systems, Minneapolis, MN, USA); (GKT771, Genkyotex, Switzerland); Lipopolysaccharide (LPS) (Catalog#: IAX100012M001, Adipogen Coorporation, San Diego, CA, U.S.A) Maraviroc (Catalog# NC1746377, Princeton, NJ, USA); 2-Mercaptoethanol (Catalog#: AC125472500, Thermo Fisher Scientific, Waltham, MA, USA) Laemmli Sample Buffer (Catalog#: 1610737, BioRad Hercules, CA – USA); Immobilon-P poly membranes (Catalog#: S2GVU11RE, Sigma Aldrich, St. Louis, MO, USA); Bovine serum albumin (BSA) (Catalog#: 9998 Cell Signaling, Danvers, MA, USA); Tween (Catalog#: AAJ20605AP, Fisher Scientific, Waltham, MA, USA); anti-Nox1 (Catalog#: SAB4200097) and anti-β-actin (Catalog#: A3854) from Sigma, St. Louis, MO, USA, anti-Nox4 (Catalog#: ab133303, Abcam Boston, MA, USA) anti-CyclinD1 (Catalog#: 55506), anti-PCNA (Catalog#: 13110), anti-phospho-Erk1/2 (Catalog#: 9101), and anti-Erk1/2 (Catalog#: 4695), anti-NFkB (Catalog#: 8242), from Cell Signaling, Danvers, MA, USA); anti-p47phox (Catalog#: sc-528334, Santa Cruz, Dallas, TX - USA), and Alexa Fluor 488 anti-rabbit secondary antibody (Catalog#: A11008), Alexa Fluor™ 488 Phalloidin (Catalog#: A12379); enhanced chemiluminescence luminol reagent (SuperSignal™ West Femto Maximum Sensitivity Substrate) (Catalog#: 34096), and NucBlue™ Fixed Cell ReadyProbes™ Reagent (DAPI), (Catalog#: R37606, Thermo Fisher Scientific, Waltham, MA, USA); RNeasy Mini Kit (Catalog #: 74106 Quiagen, Germantown, MD – USA); SuperScript III (Catalog#: 18080051, Thermo Fisher Waltham, MA USA); PowerTrack™ SYBR Green Master Mix (Catalog #: A46111 Thermo Fisher, Waltham, MA USA); [8-amino-5-chloro-7 phenylpyridol phenylpyridol [3,4-d]pyridazine1,4(2H,3H)dione] (L012) (Catalog #: 508510, Tocris Bioscience Minneapolis, MN, USA); Dihydroethidium (DHE) (Catalog#: D11347), 2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA) (Catalog#: D399); Hanks’ Balanced Salt Solution (Catalog#: 14025092), Mem-PER™ Plus Membrane Protein Extraction Kit (Catalog#: 89842), Click-iT™ EdU Cell Proliferation Kit for Imaging (Alexa Fluor™ 488 dye) (Catalog#: C10337), Dimethyl Sulfoxide (DMSO) (Catalog #: J66650.K2), corn oil (Catalog #: AC405435000), and triton X-100 (Catalog#: BP151500) from Thermo Fisher Scientific, Waltham, MA, USA.
Vascular Smooth Muscle Cells
RASMC were maintained in DMEM containing 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. The complete medium was replaced by DMEM containing 0.5% FBS before the treatments.
RASMC treatments
RASMC were stimulated with recombinant mouse CCL5 (50–300ng/mL) for 15–60 minutes for rapid events measurements and for 8–24h for other experiments. To study the impact of Nox1-derived ROS on CCL5-induced vascular damage, RASMC were pre-incubated with a specific Nox1 inhibitor25 (GKT771, 1μM) 30 min prior to CCL5 incubation. LPS (500 ng.ml−1 for 1h)27 was used as a positive control for NFkB activation measurement. Maraviroc (CCR5 antagonist, 40 uM) was used to analyze whether CCL5 mediates its effects via CCR5.
Western Blot analysis
RASMC samples were directly homogenized using 2x Laemmli Sample Buffer and supplemented with 2-Mercaptoethanol (β-mercaptoethanol) (BioRad Hercules, California – USA). Proteins were separated by electrophoresis on a polyacrylamide gel and transferred to Immobilon-P poly (vinylidene fluoride) membranes. Non-specific binding sites were blocked with 5% skim milk or 1% BSA in Tris-buffered saline solution with Tween for 1h at 24 °C. Membranes were then incubated with specific antibodies overnight at 4 °C. Antibodies were as follows: anti-Nox1 (1:1000), anti-Nox4 (1:1000), anti-CyclinD1 (1:1000), anti-PCNA (1:1000), anti-phospho-Erk1/2 (1:1000), anti-Erk1/2 (1:1000), and anti-β-actin (1:10000) which was used as a loading control to ensure that sample loading was consistent. After incubation with secondary antibodies, the enhanced chemiluminescence luminol reagent was used for antibody detection.
Real time RT (reverse transcription)-PCR
mRNA from carotid arteries and RASMC was extracted using RNeasy Mini Kit. Complementary DNA was generated by reverse transcription polymerase chain reaction (RT-PCR) with SuperScript III. Reverse transcription was performed at 58 °C for 50 min; the enzyme was heat inactivated at 85 °C for 5 min, and real-time quantitative RT-PCR was performed with the PowerTrack™ SYBR Green Master Mix. Sequences of genes as listed in the table. Experiments were performed in a QuantStudio™ 5 Real-Time PCR System, 384-well (Thermo Fisher, Waltham, MA USA). Data were quantified by 2ΔΔ Ct and are presented by fold changes indicative of either upregulation or downregulation.
ROS measurement in RASMC
Cells were seeded in specific well plates and incubated overnight in DMEM with 0.5% FBS. Cells were stimulated with recombinant mouse CCL5 (100ng/mL, 60 min) followed by ROS quantification through the following methods:
L012:
Cells were cultured on white clear-bottom 96-well culture plates and then were stimulated with CCL5. The media was replaced with PBS (pH 7.4) supplemented with CaCl2 (0.9 mM) and MgSO4 (0.49 mM) and chemiluminescence enhancer L012 (400 mM) was added and chemiluminescence measured in a LUMIstar OPTIMA Microplate Luminometer (BMG LABTECH, Cary, NC, USA). Superoxide production was quantified as relative light units and presented by % versus the average from the control group.
DHE:
After the stimulus in 24-wells plates, RASMC were washed twice with Phosphate Buffered Saline (PBS) and incubated with Hanks’ Balanced Salt Solution (HBSS) containing DHE (10 μM) for 30 min in a dark and humidified container at 37°C. The cells were then washed twice with PBS. The fluorescence signals were obtained with fluorescence microscopy (Revolve, Echo, San Diego, California USA)7.
DCFDA:
After the stimulus in 24-wells plates, RASMC were washed twice with PBS and incubated with HBSS containing DCFDA (20 μM) for 45 min in a dark and humidified container at 37°C. The cells were then washed twice with PBS. The fluorescence signals were obtained with fluorescence microscopy (Revolve, Echo, San Diego, California USA).
Cytosol and membrane fractions
To evaluate the Nox1 activation, RASMC were cultured in T25 flasks and stimulated with CCL5 (100ng/mL for 30 and 60 minutes). Cytosol and membrane fractions were isolated by using membrane protein extraction kit following the manufacturer instructions (please see reagents list). Western blot was utilized to detect the p47phox content in both fractions (anti-p47phox, 1:500, Santa Cruz, Dallas, TX - USA)26,28.
Immunofluorescence for NFkB activation
RASMC were cultured on glass coverslips in DMEM, then incubated with CCL5 for 1h either with or without GKKT771. LPS was used as a positive control. After washed, cells were fixed and permeabilized for 10 min at room temperature with PBS containing 2% paraformaldehyde and 0.3% Triton X-100, and then blocked with PBS/2% BSA. Anti-NFkB antibody (1:500) incubation was performed for 1 h at room temperature in PBS/2% BSA followed by incubation with Alexa Fluor 488 anti-rabbit secondary antibody plus phalloidin staining solution for 60 minutes at room temperature. PBS was used to wash residual solution between each different step. DAPI staining was used to determine cell nuclei. Images were acquired with a Revolve, Echo microscope (Echo, San Diego, California USA).
Phalloidin fluorescence
Cells were plated on coverslips overnight and incubated with CCL5 for 24h either with or without GKKT771. After this period, RASMC were fixed in 3.7% formaldehyde solution in PBS for 15 minutes at room temperature, permeabilized in 0.1% Triton™ X-100 in PBS for 15 minutes, blocked in solution containing 1% BSA for 45 minutes at room temperature and incubated with phalloidin staining solution for 60 minutes at room temperature. PBS was used to wash residual solution between each different step. DAPI staining was used to determine cell nuclei. Images were obtained using a fluorescence microscope (Revolve, Echo, San Diego, California USA).
Cell migration assay
Cells were incubated overnight in DMEM with 0.5% FBS. Cells were stimulated with recombinant mouse CCL5 (100ng/mL, 12h). The RASMC migration was evaluated by boyden chamber assay. Using Transwell 24-well cell culture inserts with 8 μm pores (Thermo Fisher Scientific, Waltham, MA, USA), RASMC were harvested and added to the insert (5 × 104 cells/well). Culture medium with or without CCL5 was added to the chamber. In some experiments, Nox1 inhibitor was added 30 min prior to CCL5 stimulation. After 12 h, non-migrating cells were removed from upper filter surfaces, and the filter was washed, fixed, and stained7,29. Six randomly selected filters were photographed and the number of cells that migrated in each was determined. 0.5% FBS was used during the experiment. A group was treated with 10% FBS was used as a positive control.
Cell proliferation assay
Click-iT™ EdU Cell Proliferation Kit for Imaging was used to measure the proliferative status of RASMC. Cells were plated on coverslips overnight. Afterwards, they were incubated with CCL5 for 24h with or without GKKT771. After 24h, EdU (at final concentration of 10 μM) was incubated for 2h in a humidified container at 37°C. The following steps of the protocol were performed according to manufacturer instruction. DAPI staining was used to determine cell nuclei (Thermo Fisher Scientific, Waltham, MA, USA). Images were obtained using a fluorescence microscope (Revolve, Echo, San Diego, California USA).
Animals
12-week-old male C57BL6/J mice were used. All mice were fed with standard mouse chow and tap water was provided ad libitum. Mice were housed in an American Association of Laboratory Animal Care–approved animal care facility in Rangos Research Building at Children’s Hospital of Pittsburgh of University of Pittsburgh. Institutional Animal Care and Use Committee approved all protocols (IACUC protocol # 19065333). All experiments were performed in accordance with The Care and Use of Laboratory Animals Guide.
Vascular Injury model
Vascular injury was performed using the carotid artery ligation model30. Under isoflurane anesthesia (3%), the left common carotid artery was ligated with 6–0 suture just proximal to the first carotid bifurcation. Carotid arteries were collected at 7 days after ligation. The unmanipulated right carotid artery served as a control. A group of mice were treated with a specific CCR5 antagonist compound (Maraviroc, 25mg/Kg/day i.p)17,31. Maraviroc was diluted in 10% DMSO and 90% corn oil. Approximately 50uL of this solution was injected in each mouse daily.
Morphometric analysis of the vascular wall
At 7 days after ligation, ligated arteries and their respective intact counterparts were collected, cleaned of connective tissue, immediately fixed in 4% phosphate-buffered paraformaldehyde at pH 7.4 and embedded in paraffin blocks. Four micrometer-thick slices were stained with hematoxylin and eosin. The lumen area (LA), lumen diameter external diameter (ED), and internal diameter (ID) of the arteries were calculated in millimeters using the ImageJ software. Media area/lumen area ratio was calculated as following: [(ED-ID)/LA]. Stained sections were examined with light microscopy (Revolve, Echo, San Diego, California USA).
Statistical Analysis
For comparisons between multiple groups, one-way or two-way analysis of variance (ANOVA) followed by the Tukey post-test was used. Analyses were performed using Prism 9.0 software (GraphPad). A difference was considered statistically significant when P ≤ 0.05.
Results
CCL5 increases Nox1 expression and activation in RASMC
To study whether CCL5 triggers Nox1 activation and expression, RASMC were stimulated with CCL5 and Nox1 expression/activity and ROS production were analyzed. First, we found that CCL5 incubation (50–300ng/ml for 8–24h) increased the protein and gene expression of Nox1, with not changes in Nox2 or Nox4 (gene and protein) (Fig. 1A–D). We also observed that CCL5 increased the p47phox protein content in the membrane fraction after 15–30 minutes of incubation (Fig. 1E) Antagonism of CCR5 with maraviroc inhibited CCL5-induced Nox1 expression (Fig. 1F). We measured the expression of CCR1, 3, and 5 (well-documented receptors that recognize CCL5) and CCL5 by RT-PCR to examine whether positive feedback regulation was present and no difference was observed in any CCRs or CCL5 (Fig. 1I).
Figure 1. CCL5 induces Nox1 expression and activation via CCR5 in vascular cells.

Nox1, 2, and 4 gene expression from Rat Aortic Vascular Smooth Muscle Cells (RASMC) treated with CCL5 (100ng/mL, 8h and 24h) (A); Nox1 (B) and Nox4 (C) protein expression from RASMC treated with CCL5 (100ng/mL, 8h and 24h); Nox1 protein expression from RASMC treated with CCL5 (30–300ng/mL for 24 (D); p47phox protein content in membrane and cytosol fractions isolated from RASMC treated with CCL5 (100ng/mL, 30min and 60min) (E); Nox1 protein expression from RASMC treated with CCL5 (100ng/mL) in presence of maraviroc (40uM) (F); CCR1, 3, 5, and CCL5 gene expression from RASMC treated with CCL5 (100ng/mL, 8h and 24h). N=4–12. Data are presented as mean ± standard error of the mean (SEM). *P<0.05 vs vehicle; #P<0.05 vs CCL5+GKT771.
Furthermore, CCL5increased ROS production, which was analyzed by DHE and DCFDA staining and GKT771 pre-treatment abrogated CCL5-induced ROS production (Fig. 2A–C), suggesting that CCL5 activates Nox1 and leads to ROS production in RASMC.
Figure 2. CCL5 induces Nox1-derived ROS in vascular cells.

Reactive Oxygen Species analyzed by Dihydroethidium (DHE) (A), 2’,7’ – dichlorofluorescin diacetate (DCFDA) (B), and L012 (C) from RASMC treated CCL5 (100ng/mL, 60min) in presence of GKT771 (10uM); N=4–12. Data are presented as mean ± standard error of the mean (SEM). *P<0.05 vs vehicle; #P<0.05 vs CCL5+GKT771.
CCL5 induces vascular migration, proliferation, and cytoskeleton rearrangement via Nox-1-dependent mechanisms
Pathological vascular remodeling is characterized by high migratory rate30,32. To identify whether CCL5 induces vascular migration via Nox1, we stimulated RASMC with CCL5 and studied the migratory profile via scratch and Boyden chamber assays. We observed that CCL5 induced vascular migration (Fig. 3A), cytoskeletal reorganization, and Erk1/2 phosphorylation, which were blunted by inhibiting Nox1 (Fig. 2B–C). Another aspect involved in vascular remodeling is the elevated cell proliferation levels in the injury site. Herein, we found that CCL5 increased the Edu+ cells, as well as PCNA, Cyclin D1, and Ki67 (all markers for proliferation and cell cycle progression), whereas GKT771 pre-treatment blocked CCL5-induced vascular proliferation (Fig. 4A–E).
Figure 3. CCL5 induces vascular migration dependent on Nox1.

Effects of CCL5 (100ng/mL, 12h) on Rat Aortic Vascular Smooth Muscle Cells (RASMC) migration analyzed by boyden chamber assay (A). Phalloidin staining (red) in RASMC treated with CCL5 (100ng/mL for 24h) (B); Erk1/2 phosphorylation levels in RASMC treated with CCL5 (100ng/mL for 30min and 60min). Nox1 was inhibited by GKT771 (10uM). N=4–8. White arrows mean changes in F-actin organization. Data are presented as mean ± standard error of the mean (SEM). *P<0.05 vs vehicle; #P<0.05 vs vehicle. #P<0.05 vs CCL5+GKT771.
Figure 4. CCL5 induces vascular proliferation by activating Nox1.

Rat Aortic Vascular Smooth Muscle Cells (RASMC) staining for Edu (red) and nuclei (blue) after 24h of CCL5 treatment (100ng/mL) (A, B); PCNA (C) and Cyclin D1 (D) protein expression from RASMC treated with CCL5 (100ng/mL, 8h and 24h); Ki67 gene expression from RASMC treated with CCL5 (100ng/mL, 8h and 24h). Nox1 was inhibited by GKT771 (10uM). N=4–6. Data are presented as mean ± standard error of the mean (SEM). *P<0.05 vs vehicle; #P<0.05 vs vehicle. #P<0.05 vs CCL5+GKT771.
CCL5 activates NFkB and increases inflammatory genes dependent on Nox1 activation
NFkB participates in inflammatory responses in multiples cardiovascular risks 33–35, we investigated whether CCL5 induces NFkB activation dependent on Nox1 and whether such interaction might be leading to an inflammatory response in RASMC. We first used LPS (NFkB inducer) to validate our method of fluorescence, LPS treatment (60 min) induced more NFkB (green) migration to the nucleus (blue). Next, we found that CCL5, in a manner similar to LPS, also induced NFkB translocation into the nucleus (Fig. 5A), whereas GKT771 inhibited CCL5-induced NFkB activation. We also observed that CCL5 treatment increased the IL1β, TNFα, VCAM, and ICAM gene expression and Nox1 inhibition blunted the inflammatory genes expression caused by CCL5 (Fig. 5B).
Figure 5. CCL5 induces vascular inflammation and NFkB activation via Nox1.

NFkB activation analyzed by immunostaining NFkB (green), phalloidin (red), nuclei (blue) in Rat Aortic Vascular Smooth Muscle Cells treated with CCL5 (100ng/mL, 60 min). Lipopolysaccharides (500ng/mL, 60 min) was used as a positive control (A); IL1β, TNFα, VCAM, and ICAM gene expression from RASMC treated with CCL5 (100ng/mL, 60 min). Nox1 was inhibited by GKT771 (10uM). N=3–4. Data are presented as mean ± standard error of the mean (SEM). *P<0.05 vs vehicle.
CCR5 blockage attenuates Nox1 and CL-induced vascular remodeling and inflammation
To investigate our in vitro findings in vivo, we analyzed whether CCR5 may be involved in pathological vascular remodeling and vascular inflammation in a well-characterized model of vascular injury (carotid artery ligation, CL). 7 days post-injury, we first observed that CL significantly elevated CCR3 and CCR5 with a pronounced increase in CCR5. No changes were observed for CCR1 (Fig. 6). Next, we blocked CCR5, by treating mice with maraviroc (CCR5 antagonist), and observed a significant protection from CL-induced vascular injury, characterized by reduction in the lumenal area and medial/lumenal areas ratio, as well as increase in the lumen diameter (Fig. 6A–D). Furthermore, we observed that 2 mice from ligated group developed neointima formation, whereas none in ligated + maraviroc group did. Multiple studies have shown that 7 days of injury is not enough to induce a consistent neointima formation36–38. CL induced vascular inflammatory markers followed by an increase in Nox1 gene expression with no changes in Nox2 and Nox4 (Fig. 7). Interestingly, CCR5 antagonist treatment minimized the vascular inflammation and blocked elevation of injury-induced Nox1 expression.
Figure 6. Vascular injury elevates CCR5 and CCL5 gene expression.

CCR1, CCR3, CCR5, and CCL5 gene expression in non-ligated carotid arteries and ligated carotid arteries (7 days of ligation) from the same animal. N=4. Data are presented as mean ± standard error of the mean (SEM). *P<0.05 vs non-ligated.
Figure 7. CCR5 antagonist treatment alleviates vascular injury.

Representative images from non-ligated and ligated carotid arteries from animals treated or not with maraviroc for 7 days (20mg/kg/day, i.p) (A); Quantification is presented by lumen area (B), Lumen diameter (C), and media area/lumen area. N=5. Data are presented as mean ± standard error of the mean (SEM). *P<0.05 vs non-ligated; #P<0.05 vs ligated + maraviroc.
Discussion
Vascular remodeling is a complex process that involves physical, biochemical and genetic components39,40. VSMC migration and proliferation are key phenomena in the processes of vascular remodeling associated with vascular disease, medical procedures, and therapeutic treatments. Changes in vascular structure are typically triggered by alterations to blood flow, growth and apoptotic factors, inflammation, or fibrotic stimulus4,40–43. Although it is well known that these events and mediators lead to vascular structural changes, understanding the mechanisms by which such abnormality occurs is still to be elucidated.
CCR5 is a chemokine receptor broadly studied in infection diseases research44–46. However, its participation in cardiovascular disease is not fully understood. Studies in obesity, hypertension, and antiretroviral treatment have shown that CCR5 plays important roles in each process, making it a pleiotropic receptor with the ability to govern cardiovascular structure and function11,19,22,47. Recently we found that ritonavir, an antiretroviral drug used in HIV infection34,48,49 which was recently shown to reduce the viral load in COVID-1950, increases CCR5 expression in the vasculature and induces endothelial dysfunction, seemingly mediated by Nox1-derived ROS19. Herein, we pursued experiments to better understand whether CCR5 has any role in vascular remodeling and whether CCR5 leads to Nox1 activation.
Generally, chemokines and their receptors are potent oxidant agents that stimulate different sources of ROS and regulate antioxidant machinery. 51–56. The Nox family has been implicated in several cardiovascular alterations, including endothelial dysfunction, vascular inflammation and vascular growth7,19,25,26,28,57. We found that CCL5 also has the capability of activating the Nox family, particularly Nox1, by elevating its expression and increasing p47phox translocation from the cytosol to the membrane (index to measure oxidase activity)7,26,28.
Although numerous reports have shown that diverse chemokines and their receptors induce changes in vascular profile, there is a lack of knowledge regarding their downstream signaling2,4,6,47. Nox1 propagates cellular responses in cardiovascular disease inducing a high migratory and proliferative profile in VSMC32,58. Besides CCR5, other chemokines receptors recognize CCL5 including CCR1 and CCR347,59. In this study, CCL5 regulated Nox1 expression via CCR5, since maraviroc (CCR5 antagonist) blunted CCL5-induced Nox1 expression. Interestingly, CCL5 seems to orchestrate the vascular profile, increasing the migration and proliferation rates, changing the cytoskeleton organization, as well as increasing Erk1/2 phosphorylation levels – (key protein on vascular cells profile) by activating CCR5 and Nox1 enzyme. Multiple studies including our own 60,61,7, have demonstrated the key participation of Erk1/2 in the establishment of VSMC profile. These data suggest that CCL5 triggers a vascular injury by inducing vascular migration, proliferation, and cytoskeleton arrangement likely via Nox1-derived ROS.
In obesity, hypertension, and atherosclerosis, chronic low-grade inflammation is a condition frequently observed in blood vessels22,40,62,63. CCR5 seems to be an attractive pharmacological target to minimize vascular inflammation11,19,22, however it is not clear by which downstream mechanisms CCL5/CCR5 induces such change. Herein, we found that CCL5 promotes vascular inflammation via activating Nox1, this oxidase plays a major role in adjusting vascular inflammation in obesity, acquired or congenital lipodystrophies, and hypertension19,25,26,57,64. In our study we also demonstrate that CCL5 activates NFkB, a complex that regulates transcription of inflammatory genes and cell survival65–67, via Nox1. Some studies have shown that NFkB is a ROS-and-Nox-sensitive complex65,66, whereas others have presented that NFkB promotes Nox expression/regulation68, indicating that a positive feedback loop between Noxs and NFkB may exist in inflammatory diseases. In the present study, we did not study whether NFkB inhibition confers protection from CCL5-induced vascular inflammatory and/or injury, but others have reported that blocking NFkB blunts the inflammatory effects under diverse environments and treatments65,67. In our future studies, we will seek to fill this gap.
Interestingly, we observed that CL-induced vascular injury is partially dependent on CCR5, as maraviroc treatmentpartly inhibited pathological vascular remodeling and inflammation induced by CL. The role of CCR5 on vascular remodeling has been previously reported in abdominal aortas of mice exposed to high-fat diet which was attributed to the diet-induced hyperlipidemia47. Here, our study focused local blood flow disturb caused by CL. Furthermore, we observed that vascular remodeling was associated with upregulation of Nox1 gene expression and that maraviroc treatment blunted the Nox1 expression. Using balloon injury, Xu et al. 2012 observed an increase in Nox1 expression and activity in VSMC70. In another study, Lee et al 2009, showed that inhibition or loss of Nox1 was protective for injury-induced neointimal formation and stretch-induced phenotypic modulation of VSMC58. In our study it appears that CCR5 antagonism confers vascular protection by attenuating Nox1 expression and activity. Further investigations are necessary to confirm such speculation.
Rodríguez et al32. suggest that oxidant enzymes, other than Nox1, might also be controlling the vascular status under stretch-induced phenotypic modulation of VSMC. Thus, we cannot exclude that Nox2 and/or Nox4 are involved in CCL5-induced vascular changes. Based on our ex vivo model, we found a trending increase in Nox2 and a decrease in Nox4 in carotid arteries after injury. In our study, we did not confirm the direct effects of CCL5 on Nox2 expression/activity in RASMC due to its lack in this cell line32. Furthermore, Nox4 has shown vascular protective effects on vascular phenotype control32,71, which suggests that Nox4 downregulation might also participate in CL-induced vascular injury. Further studies are necessary to determine whether reduced Nox4 has any involvement on CL-induced vascular remodeling.
In summary, these findings show a new signaling pathway originated by CCL5 and CCR5 which stimulates Nox1-derived ROS, NFkB, and Erk1/2 phosphorylation to cause a migratory and proliferative VSMC phenotype. We provide new insight into vascular proliferative mechanisms and place CCR5 and Nox1 as possible therapeutic targets for vascular diseases associated with uncontrolled vascular growth. In addition, CCR5 antagonists or Nox1 inhibitors might be useful as anti-proliferative agents in drug-eluting stents to ameliorate the vascular damage following angioplasty procedures.
Figure 8. CCR5 antagonist treatment reduces vascular inflammatory genes and Nox1 expression.

IL1β, TNFα, VCAM, and ICAM gene expression (A); Nox1, Nox2, and Nox4 gene expression (B) in non-ligated and ligated carotid arteries from animals treated or not with maraviroc for 7 days (20mg/kg/day, i.p) N=4. Data are presented as mean ± standard error of the mean (SEM). *P<0.05 vs non-ligated; #P<0.05 vs ligated + maraviroc.
Table 1.
List and sequence of primers
| Target gene | Primers | ||
|---|---|---|---|
| Mouse | Rat | ||
| Nox1 | FW | GGGTCAAACAGAGGAGAGCT | TTCCCTGGAACAAGAGATGG |
| RV | ATGCCTGCAACTCCCCTTAT | GACGTCAGTGGCTCTGTCAA | |
| Nox2 | FW | CAAGATGGAGGTGGGACAGT | CAAAGCCTGTGGCTGTGATA |
| RV | GCTTATCACAGCCACAAGCA | TCCCAGCTCCCACTAACATC | |
| Nox4 | FW | TGTTGCATGTTTCAGGTGGT | CGGGTGGCTTGTTGAAGTAT |
| RV | AAAACCCTCGAGGCAAAGAT | AAAACCCTCCAGGCAAAGAT | |
| CCR1 | FW | GCCAAAAGACTGCTGTAAGAGCC | AAAGGCATGTGGCTATACCG |
| RV | GCTTTGAAGCCTCCTATGCTGC | ATCAGAAGCCCCCAGAAAGT | |
| CCR3 | FW | CCACTGTACTCCCTGGTGTTCA | CAGCAGAGCATACACCTGGA |
| RV | GGACAGTGAAGAGAAAGAGCAGG | CTGTGGAAAAAGAGCCGAAG | |
| CCR5 | FW | GGTTCCTGAAAGCGGCTGTAAATA | CTGCCTCAACCCTGTCATCT |
| RV | CTGTTGGCAGTCAGGCACATC | GTGTTTGCAGAAGCGTTTGA | |
| CCL5 | FW | AGATCTCTGCAGCTGCCCTCA | ATATGGCTCGGACACCACTC |
| RV | GGAGCACTTGCTGCTGGTGTAG | TGACAAAGACGACTGCAAGG | |
| IL1 | FW | TGACGGACCCCAAAAGATGA | CACCTTCTTTTCCTTCATCTTTG |
| RV | GCTCTTGTTGATGTGCTGCT | GTCGTTGCTTGTCTCTCCTTGTA | |
| TNF | FW | AATGGCCTCCCTCTCATCAG | GCTGTCGCTACATCACTGAACCT |
| RV | CCTAACTGCCCTTCCTCCAT | TGACCCGTAGGGCAATTACA | |
| VCAM | FW | TGACAAGTCCCCATCGTTGA | CGGTCATGGTCAAGTGTTTG |
| RV | ACCTCGCGACGGCATAATT | GACGGTCACCCTTGAACAGT | |
| ICAM | FW | ATCACATGGGTCGAGGGTTT | GGTATCCATCCATCCCACAG |
| RV | AACCACTGCCAGTCCACATA | CTCGCTCTGGGAACGAATAC | |
| Ki67 | FW | CTTTGCGCCATGCTGAAACT | |
| RV | ATGACGACCTGGAACATCGG` | ||
| GAPDH | FW | GAGAGGCCCTATCCCAACTC | GAGAGGCCCTATCCCAACTC |
| RV | TCAAGAGAGTAGGGAGGGCT | TCAAGAGAGTAGGGAGGGCT | |
Acknowledgments
We thank Genkyotex for providing us the GKT771 compound (Nox1 inhibitor).
Funding information
This work was supported by a NHLBI-R00 (R00HL14013903), AHA-CDA (CDA857268), and startup funds from University of Pittsburgh to TBN.
Footnotes
Declarations of interest
None
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Brozovich FV et al. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol Rev 68, 476–532, doi: 10.1124/pr.115.010652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petsophonsakul P et al. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arterioscler Thromb Vasc Biol 39, 1351–1368, doi: 10.1161/ATVBAHA.119.312787 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Osman I et al. TEAD1 (TEA Domain Transcription Factor 1) Promotes Smooth Muscle Cell Proliferation Through Upregulating SLC1A5 (Solute Carrier Family 1 Member 5)-Mediated Glutamine Uptake. Circ Res 124, 1309–1322, doi: 10.1161/CIRCRESAHA.118.314187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rensen SS, Doevendans PA & van Eys GJ Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J 15, 100–108, doi: 10.1007/BF03085963 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu M & Gomez D Smooth Muscle Cell Phenotypic Diversity. Arterioscler Thromb Vasc Biol 39, 1715–1723, doi: 10.1161/ATVBAHA.119.312131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J et al. Smooth muscle cell phenotypic diversity between dissected and unaffected thoracic aortic media. J Cardiovasc Surg (Torino) 54, 511–521 (2013). [PubMed] [Google Scholar]
- 7.Bruder-Nascimento T et al. Angiotensin II induces Fat1 expression/activation and vascular smooth muscle cell migration via Nox1-dependent reactive oxygen species generation. J Mol Cell Cardiol 66, 18–26, doi: 10.1016/j.yjmcc.2013.10.013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vinader V & Afarinkia K A beginner’s guide to chemokines. Future Med Chem 4, 845–852, doi: 10.4155/fmc.12.49 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Hughes CE & Nibbs RJB A guide to chemokines and their receptors. FEBS J 285, 2944–2971, doi: 10.1111/febs.14466 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones KL, Maguire JJ & Davenport AP Chemokine receptor CCR5: from AIDS to atherosclerosis. Br J Pharmacol 162, 1453–1469, doi: 10.1111/j.1476-5381.2010.01147.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cipriani S et al. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation 127, 2114–2124, doi: 10.1161/CIRCULATIONAHA.113.001278 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Dusi V, Ghidoni A, Ravera A, De Ferrari GM & Calvillo L Chemokines and Heart Disease: A Network Connecting Cardiovascular Biology to Immune and Autonomic Nervous Systems. Mediators Inflamm 2016, 5902947, doi: 10.1155/2016/5902947 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aukrust P et al. Chemokines in cardiovascular risk prediction. Thromb Haemost 97, 748–754 (2007). [PubMed] [Google Scholar]
- 14.Rothenbacher D, Muller-Scholze S, Herder C, Koenig W & Kolb H Differential expression of chemokines, risk of stable coronary heart disease, and correlation with established cardiovascular risk markers. Arterioscler Thromb Vasc Biol 26, 194–199, doi: 10.1161/01.ATV.0000191633.52585.14 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Meanwell NA & Kadow JF Maraviroc, a chemokine CCR5 receptor antagonist for the treatment of HIV infection and AIDS. Curr Opin Investig Drugs 8, 669–681 (2007). [PubMed] [Google Scholar]
- 16.Larena M, Regner M & Lobigs M The chemokine receptor CCR5, a therapeutic target for HIV/AIDS antagonists, is critical for recovery in a mouse model of Japanese encephalitis. PLoS One 7, e44834, doi: 10.1371/journal.pone.0044834 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fatkenheuer G et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med 11, 1170–1172, doi: 10.1038/nm1319 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Li G et al. Neferine inhibits the upregulation of CCL5 and CCR5 in vascular endothelial cells during chronic high glucose treatment. Inflammation 36, 300–308, doi: 10.1007/s10753-012-9547-1 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Bruder-Nascimento T, Kress TC, Kennard S & Belin de Chantemele EJ HIV Protease Inhibitor Ritonavir Impairs Endothelial Function Via Reduction in Adipose Mass and Endothelial Leptin Receptor-Dependent Increases in NADPH Oxidase 1 (Nox1), C-C Chemokine Receptor Type 5 (CCR5), and Inflammation. J Am Heart Assoc 9, e018074, doi: 10.1161/JAHA.120.018074 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z et al. Chemokine Receptor 5, a Double-Edged Sword in Metabolic Syndrome and Cardiovascular Disease. Front Pharmacol 11, 146, doi: 10.3389/fphar.2020.00146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Podolec J et al. Chemokine RANTES is increased at early stages of coronary artery disease. J Physiol Pharmacol 67, 321–328 (2016). [PMC free article] [PubMed] [Google Scholar]
- 22.Mikolajczyk TP et al. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J 30, 1987–1999, doi: 10.1096/fj.201500088R (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panday A, Sahoo MK, Osorio D & Batra S NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol 12, 5–23, doi: 10.1038/cmi.2014.89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drummond GR, Selemidis S, Griendling KK & Sobey CG Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 10, 453–471, doi: 10.1038/nrd3403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruder-Nascimento T et al. Leptin Restores Endothelial Function via Endothelial PPARgamma-Nox1-Mediated Mechanisms in a Mouse Model of Congenital Generalized Lipodystrophy. Hypertension 74, 1399–1408, doi: 10.1161/HYPERTENSIONAHA.119.13398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruder-Nascimento T et al. Vascular injury in diabetic db/db mice is ameliorated by atorvastatin: role of Rac1/2-sensitive Nox-dependent pathways. Clin Sci (Lond) 128, 411–423, doi: 10.1042/CS20140456 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Cau SB et al. Angiotensin-II activates vascular inflammasome and induces vascular damage. Vascul Pharmacol 139, 106881, doi: 10.1016/j.vph.2021.106881 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruder-Nascimento T et al. Atorvastatin inhibits pro-inflammatory actions of aldosterone in vascular smooth muscle cells by reducing oxidative stress. Life Sci 221, 29–34, doi: 10.1016/j.lfs.2019.01.043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou R, Liu L, Anees S, Hiroyasu S & Sibinga NE The Fat1 cadherin integrates vascular smooth muscle cell growth and migration signals. J Cell Biol 173, 417–429, doi: 10.1083/jcb.200508121 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao LL et al. Control of mitochondrial function and cell growth by the atypical cadherin Fat1. Nature 539, 575–578, doi: 10.1038/nature20170 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mencarelli A et al. Highly specific blockade of CCR5 inhibits leukocyte trafficking and reduces mucosal inflammation in murine colitis. Sci Rep 6, 30802, doi: 10.1038/srep30802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez AI et al. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 35, 430–438, doi: 10.1161/ATVBAHA.114.304936 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang CH, Fang IM, Lin CP, Yang CM & Chen MS Effects of the NF-kappaB inhibitor pyrrolidine dithiocarbamate on experimentally induced autoimmune anterior uveitis. Invest Ophthalmol Vis Sci 46, 1339–1347, doi: 10.1167/iovs.04-0640 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Westendorp MO et al. HIV-1 Tat potentiates TNF-induced NF-kappa B activation and cytotoxicity by altering the cellular redox state. EMBO J 14, 546–554 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan MJ & Liu ZG Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 21, 103–115, doi: 10.1038/cr.2010.178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moura R, Tjwa M, Vandervoort P, Cludts K & Hoylaerts MF Thrombospondin-1 activates medial smooth muscle cells and triggers neointima formation upon mouse carotid artery ligation. Arterioscler Thromb Vasc Biol 27, 2163–2169, doi: 10.1161/ATVBAHA.107.151282 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Zhang X et al. Fasudil, a Rhokinase inhibitor, prevents intimamedia thickening in a partially ligated carotid artery mouse model: Effects of fasudil in flowinduced vascular remodeling. Mol Med Rep 12, 7317–7325, doi: 10.3892/mmr.2015.4409 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Godin D, Ivan E, Johnson C, Magid R & Galis ZS Remodeling of carotid artery is associated with increased expression of matrix metalloproteinases in mouse blood flow cessation model. Circulation 102, 2861–2866, doi: 10.1161/01.cir.102.23.2861 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Wang X & Khalil RA Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv Pharmacol 81, 241–330, doi: 10.1016/bs.apha.2017.08.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Intengan HD & Schiffrin EL Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension 38, 581–587, doi: 10.1161/hy09t1.096249 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Raffetto JD & Khalil RA Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol 75, 346–359, doi: 10.1016/j.bcp.2007.07.004 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vorpahl M, Virmani R, Ladich E & Finn AV Vascular remodeling after coronary stent implantation. Minerva Cardioangiol 57, 621–628 (2009). [PubMed] [Google Scholar]
- 43.Serruys PW et al. Arterial Remodeling After Bioresorbable Scaffolds and Metallic Stents. J Am Coll Cardiol 70, 60–74, doi: 10.1016/j.jacc.2017.05.028 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Lopalco L CCR5: From Natural Resistance to a New Anti-HIV Strategy. Viruses 2, 574–600, doi: 10.3390/v2020574 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gulick RM et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med 359, 1429–1441, doi: 10.1056/NEJMoa0803152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hutter G et al. CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape. Viruses 7, 4186–4203, doi: 10.3390/v7082816 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin CS et al. The CCL5/CCR5 Axis Promotes Vascular Smooth Muscle Cell Proliferation and Atherogenic Phenotype Switching. Cell Physiol Biochem 47, 707–720, doi: 10.1159/000490024 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Reyskens KM & Essop MF HIV protease inhibitors and onset of cardiovascular diseases: a central role for oxidative stress and dysregulation of the ubiquitin-proteasome system. Biochim Biophys Acta 1842, 256–268, doi: 10.1016/j.bbadis.2013.11.019 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Pasi KJ et al. The effects of the 32-bp CCR-5 deletion on HIV transmission and HIV disease progression in individuals with haemophilia. Br J Haematol 111, 136–142, doi: 10.1046/j.1365-2141.2000.02325.x (2000). [DOI] [PubMed] [Google Scholar]
- 50.Cao B et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N Engl J Med, doi: 10.1056/NEJMoa2001282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Deken X, Corvilain B, Dumont JE & Miot F Roles of DUOX-mediated hydrogen peroxide in metabolism, host defense, and signaling. Antioxid Redox Signal 20, 2776–2793, doi: 10.1089/ars.2013.5602 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Furie MB & Randolph GJ Chemokines and tissue injury. Am J Pathol 146, 1287–1301 (1995). [PMC free article] [PubMed] [Google Scholar]
- 53.Tan JH et al. Tyrosine sulfation of chemokine receptor CCR2 enhances interactions with both monomeric and dimeric forms of the chemokine monocyte chemoattractant protein-1 (MCP-1). J Biol Chem 288, 10024–10034, doi: 10.1074/jbc.M112.447359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee YS et al. Loss of extracellular superoxide dismutase induces severe IL-23-mediated skin inflammation in mice. J Invest Dermatol 133, 732–741, doi: 10.1038/jid.2012.406 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Ansar M, Ivanciuc T, Garofalo RP & Casola A Increased Lung Catalase Activity Confers Protection Against Experimental RSV Infection. Sci Rep 10, 3653, doi: 10.1038/s41598-020-60443-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishi T, Maier CM, Hayashi T, Saito A & Chan PH Superoxide dismutase 1 overexpression reduces MCP-1 and MIP-1 alpha expression after transient focal cerebral ischemia. J Cereb Blood Flow Metab 25, 1312–1324, doi: 10.1038/sj.jcbfm.9600124 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Silva MA et al. Mineralocorticoid receptor blockade prevents vascular remodelling in a rodent model of type 2 diabetes mellitus. Clin Sci (Lond) 129, 533–545, doi: 10.1042/CS20140758 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Lee MY et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol 29, 480–487, doi: 10.1161/ATVBAHA.108.181925 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi SW et al. CCR1/CCL5 (RANTES) receptor-ligand interactions modulate allogeneic T-cell responses and graft-versus-host disease following stem-cell transplantation. Blood 110, 3447–3455, doi: 10.1182/blood-2007-05-087403 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu M et al. Small interfering RNA against ERK1/2 attenuates cigarette smoke-induced pulmonary vascular remodeling. Exp Ther Med 14, 4671–4680, doi: 10.3892/etm.2017.5160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshizumi M, Kyotani Y, Zhao J & Nakahira K Targeting the mitogen-activated protein kinase-mediated vascular smooth muscle cell remodeling by angiotensin II. Ann Transl Med 8, 157, doi: 10.21037/atm.2019.12.145 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faulkner JL & Belin de Chantemele EJ Sex Differences in Mechanisms of Hypertension Associated With Obesity. Hypertension 71, 15–21, doi: 10.1161/HYPERTENSIONAHA.117.09980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zimnol A, Spicker N, Balhorn R, Schroder K & Schupp N The NADPH Oxidase Isoform 1 Contributes to Angiotensin II-Mediated DNA Damage in the Kidney. Antioxidants (Basel) 9, doi: 10.3390/antiox9070586 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dikalova A et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112, 2668–2676, doi: 10.1161/CIRCULATIONAHA.105.538934 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Kabe Y, Ando K, Hirao S, Yoshida M & Handa H Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal 7, 395–403, doi: 10.1089/ars.2005.7.395 (2005). [DOI] [PubMed] [Google Scholar]
- 66.O’Leary DP et al. TLR-4 signalling accelerates colon cancer cell adhesion via NF-kappaB mediated transcriptional up-regulation of Nox-1. PLoS One 7, e44176, doi: 10.1371/journal.pone.0044176 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cau SB et al. Pyrrolidine dithiocarbamate down-regulates vascular matrix metalloproteinases and ameliorates vascular dysfunction and remodelling in renovascular hypertension. Br J Pharmacol 164, 372–381, doi: 10.1111/j.1476-5381.2011.01360.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Manea A, Tanase LI, Raicu M & Simionescu M Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem Biophys Res Commun 396, 901–907, doi: 10.1016/j.bbrc.2010.05.019 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Nam D et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol 297, H1535–1543, doi: 10.1152/ajpheart.00510.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu S et al. Increased expression of Nox1 in neointimal smooth muscle cells promotes activation of matrix metalloproteinase-9. J Vasc Res 49, 242–248, doi: 10.1159/000332958 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang M et al. Both cardiomyocyte and endothelial cell Nox4 mediate protection against hemodynamic overload-induced remodelling. Cardiovasc Res 114, 401–408, doi: 10.1093/cvr/cvx204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]