

Summary
Despite recent advances in structural determination of individual proteins, elucidating the 3-dimensional architecture of large, multiprotein complexes remains challenging, partly because of issues related to structural integrity during purification. Here, we describe a protocol to determine the 3-dimensional architecture of the 11-constituent, multi-tRNA synthetase complex (MSC) using chemical cross-linking coupled with mass-spectrometry (XL-MS). The protocol does not require purification and is broadly applicable, facilitating determination of native structures in cell lysates and in non-disrupted cells as well as in purified complexes.
For complete details on the use and execution of this protocol, please refer to Khan et al. (2020).
Subject areas: Cell Biology, Molecular Biology, Protein Biochemistry, Structural Biology, Mass Spectrometry, Chemistry
Graphical abstract
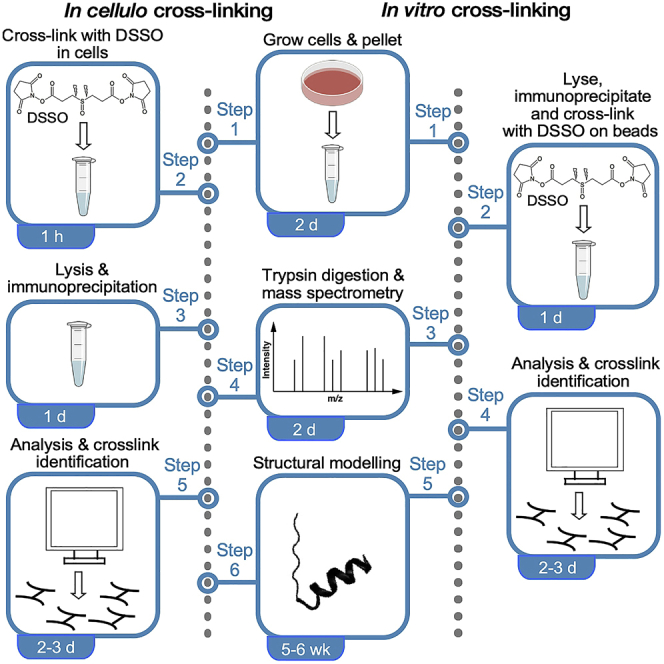
Highlights
-
•
Determines in vitro and in cellulo structures of multi-protein complexes
-
•
Facilitates analysis of multi-protein-complex architecture without purification
-
•
Reveals spatial relationships of disordered domains
-
•
Refines structures derived from X-ray crystallography which may be distorted by packing
Despite recent advances in structural determination of individual proteins, elucidating the 3-dimensional architecture of large, multiprotein complexes remains challenging, partly because of issues related to structural integrity during purification. Here, we describe a protocol to determine the 3-dimensional architecture of the 11-constituent, multi-tRNA synthetase complex (MSC) using chemical cross-linking coupled with mass-spectrometry (XL-MS). The protocol does not require purification and is broadly applicable, facilitating determination of native structures in cell lysates and in non-disrupted cells as well as in purified complexes.
Before you begin
Principal technologies used to determine protein structures include X-ray crystallography (XRD), cryo-electron microscopy (cryo-EM), and nuclear magnetic resonance (NMR). These approaches are challenged by the requirement for purification, instability of proteins and complexes during purification, low resolution of diffracting crystals, masses too small or too large for the specific technology, among other obstacles. Cross-linking mass-spectrometry (XL-MS) is a relatively recent addition to the armamentarium for protein structure elucidation. The technique combines chemical cross-linking of proteins and complexes followed by mass spectrometric analysis, revealing intra- and inter-protein cross-links indicative of proximate residues and domains. This “interaction” network can be used to model conformational states of protein and protein complexes (Mintseris and Gygi, 2020; Chavez et al., 2018). Disuccinimidyl sulfoxide (DSSO) is a MS-cleavable cross-linker, developed a decade ago, and cross-links Lys residues in peptides separated by about 10–27 Å (Sinz, 2017; Iacobucci et al., 2019). Cross-linkers with altered amino acid specificity, e.g., Cys or hydroxy-containing residues, or altered backbone length, can expand the coverage. The use of MS-cleavable cross-linkers facilitates the unambiguous determination of domain partners by improving specificity and reducing “false positives”, particularly in experiments where interacting partners are unknown. The advantages of XL-MS have permitted resolution of an array of refractory multi-protein complexes (Kao et al., 2011; Gutierrez et al., 2016, 2018). XL-MS is often used to resolve structures of purified proteins or multi-protein complexes; less common are applications of XL-MS for resolution of structures in complex mixtures such as cell lysates or intact cells and tissues (Chavez et al., 2013; Weisbrod et al., 2013). Here, we describe in detail an XL-MS-based protocol that we have developed to elucidate the 3-dimensional architecture of the multi-tRNA synthetase complex (MSC) using the Lys cross-linker DSSO in conjunction with molecular modeling. The MSC is a large, heteromeric complex consisting of nine tRNA synthetases and three auxiliary proteins (in eleven distinct polypeptides) with a combined mass of ∼1.2 MDa. We used the HEK293T cell-line in our experiments; however, the described method is general and can be adapted to analyze the structure of individual proteins and multi-protein complexes in diverse systems.
Before initiating the experiment, prepare the solutions listed in the materials and equipment section. Cool the cell lysis buffer to 4°C and add protease inhibitor before cell lysis. Filter solutions with 0.22 μm filters. All steps are performed at 4°C unless otherwise described.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Normal rabbit IgG [Dilution (1:100)] | Cell Signaling Technology | 2729S |
| EPRS1, rabbit polyclonal [Dilution (1:100)] | BioSynthesis | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 1 M Tris-HCl (pH-7.5) | Boston BioProducts | BBT-75 |
| 1 M Tris-HCl (pH-7.8) | Boston BioProducts | BBT-78 |
| 5 M NaCl | Thermo Fisher Scientific | Cat#24740011 |
| Triton X-100 | Sigma-Aldrich | T8787 |
| Bovine serum albumin | Sigma-Aldrich | A8806 |
| Fetal Bovine Serum | Gemini | Cat#100-106 |
| 1 M HEPES (pH 7.8) | Boston BioProducts | BBH-78 |
| 1 M HEPES (pH 8.0) | Boston BioProducts | BBH-80 |
| 1 M MgCl2 | Alfa Aesar | J61014 |
| Dithiothreitol | Sigma-Aldrich | Cat#10708984001 |
| Disuccinimidyl sulfoxide (DSSO) | Thermo Fisher Scientific | A33545 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D8418 |
| Ammonium bicarbonate | Sigma-Aldrich | A6141 |
| Protease inhibitor | Thermo Fisher Scientific | 78429 |
| Acetic acid | Fisher Chemical | A113-50 |
| Formic acid | Fisher Chemical | A117-50 |
| Acetonitrile | Fisher Chemical | A955-4 |
| Sequencing Grade Modified Trypsin | Promega | V5111 |
| HBSS | Corning | T55-022-PB |
| Trypsin for cell culture | Caisson | T019 |
| EDTA | Sigma-Aldrich | E6511 |
| Sodium bicarbonate | Sigma-Aldrich | S6014 |
| Phenol red | Caisson | P024 |
| Penicillin | Sigma-Aldrich | P3032-100MU |
| Streptomycin | Gibco | 11860-038 |
| L-Glutamine | Caisson | G010 |
| DMEM base powder | Caisson | DMP08 |
| PBS base powder | Gibco | 21600-044 |
| 12N HCl | Fisher Scientific | A144-212 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | CRL-3216 |
| Critical commercial assays | ||
| Protein A/G magnetic beads | Thermo Fisher Scientific | Cat#88803 |
| C18 spin columns | Thermo Fisher Scientific | Cat#89873 |
| Mini Kleenpak | Pall | KA02DJLP2S |
| Supor DCF | Pall | CFS91SSPZK |
| Acropak 500 | Pall | Cat#12995 |
| MILLEX GP | Millipore | SLGP033RS |
| Software and algorithms | ||
| Proteome Discoverer | Thermo Fisher Scientific | https://planetorbitrap.com/proteome-discoverer |
| XlinkX | Thermo Fisher Scientific | https://www.hecklab.com/software/xlinkx/ |
| PyMol | Schrödinger | https://pymol.org/2/ |
| Swiss-Model | Protein Structure and Bioinformatics group | https://swissmodel.expasy.org/ |
| PatchDock | Wolfson Lab, Tel Aviv University | https://bioinfo3d.cs.tau.ac.il/PatchDock/ |
| Other | ||
| Microcentrifuge | Eppendorf | 5424R |
| Minicentrifuge | Benchmark | C1008 |
| Thermomixer | Eppendorf | Cat#4376600 |
| DynaMag-2 magnet | Thermo Fisher Scientific | 12321D |
| Plate reader | Molecular Devices | SpectraMax 190 |
| Mass spectrometer | Thermo Fisher Scientific | Fusion Lumos |
| NanoHPLC Pump: NCS-3500RS Nano ProFlow | Thermo Fisher Scientific | 5041.0010A |
| NanoHPLC Autosampler: WPS-3000TPL RS | Thermo Fisher Scientific | 5826.0020 |
| NanoHPLC Degasser: SRD-3400 | Thermo Fisher Scientific | 5035.9245 |
| Trapping column | Thermo Fisher Scientific | Cat#164564 |
| Analytical column | Thermo Fisher Scientific | Cat#164940 |
Materials and equipment
Composition of buffers:
| Cell lysis buffer (100 mL) | Final concentration | Amount |
|---|---|---|
| 1 M Tris-HCl (pH 7.5) | 100 mM | 10 mL |
| 5 M NaCl | 150 mM | 3 mL |
| Triton X-100 | 1% | 1 mL |
Add double-distilled (dd) H2O to 100 mL and store at 4°C for up to 3–6 months.
Add freshly made complete protease inhibitor cocktail to a 1× final concentration before use.
| Trypsin-EDTA (100 mL) | Final concentration | Amount |
|---|---|---|
| HBSS | 1× | 951 mg |
| Trypsin for cell culture | 0.05% | 50 mg |
| EDTA | 0.53 mM | 17.3 mg |
| NaHCO3 | 4.16 mM | 35 mg |
| Phenol red | 0.027 mM | 1.1 mg |
Add ddH2O to 100 mL. Bring the pH to 7.0 by bubbling the solution with CO2 gas. Filter the final solution using 0.1 micron Mini Kleenpak filter. Store at 4°C for up to 2–3 weeks.
| Phosphate-buffered saline (1 L) | Final concentration | Amount |
|---|---|---|
| PBS base powder | 1× | 9.55 g |
Add ddH2O to 1 L. Bring the pH to 7.0 with 12N HCl. Filter the final solution using 0.22 micron Acropak 500 filter. Store at 25°C for up to 2 months.
| Penicillin-streptomycin solution, 10,000 U/mL (100 mL) | Final concentration | Amount |
|---|---|---|
| Penicillin | 10,000 U/mL | Depends on lot potency |
| Streptomycin | 10,000 μg/mL | Depends on lot potency |
| NaCl | 0.85% | 0.85g |
Add ddH2O to 100 mL. The pH is ∼5.5. Filter the final solution using 0.1 micron Mini Kleenpak filter. Store at −20°C for up to 1 year. Thaw at 37°C water bath just before supplementing DMEM media.
| L-glutamine solution (100 mL) | Final concentration | Amount |
|---|---|---|
| L-glutamine | 200 mM | 2.923g |
| NaCl | 0.85% | 0.85g |
Add ddH2O to 100 mL. The pH is ∼5.5. Filter the final solution using 0.1 micron Mini Kleenpak filter. Store at −20°C for up to 1 year. Thaw at 37°C water bath just before supplementing DMEM media.
| Dulbecco’s modified Eagle’s Medium/DMEM (1 L) | Final concentration | Amount |
|---|---|---|
| Base Media | 1× | 11.49 g |
| Sodium bicarbonate | 44 mM | 3.7 g |
| Penicillin-streptomycin solution | 100 U/mL | 10 mL |
| L-glutamine | 2 mM | 10 mL |
| FBS | 10% | 100 mL |
Add ddH2O to 1 L. Bring the pH to 7.0 by bubbling the solution with CO2 gas. Filter the final solution using 0.1 micron Supor DCF filter. Add FBS, penicillin-streptomycin and L-glutamine solution once you open a new bottle of DMEM. Store at 4°C for up to 4 weeks.
| Pre-blocking buffer (10 mL) | Final concentration | Amount |
|---|---|---|
| 1 M Tris-HCl (pH 7.5) | 100 mM | 1 mL |
| 5 M NaCl | 150 mM | 0.3 mL |
| Triton X-100 | 1% | 0.11 mL |
| Bovine serum albumin (BSA) | 0.02% | 2 mg |
Add ddH2O to 10 mL. Prepare just before use as BSA is highly susceptible to microbial contamination. Add complete protease inhibitor cocktail to a 1× final concentration prior to use.
| Hypotonic buffer (100 mL) | Final concentration | Amount |
|---|---|---|
| 1 M HEPES (pH 8.0) | 10 mM | 1 mL |
| 5 M NaCl | 10 mM | 2 mL |
| 1 M MgCl2 | 1 mM | 0.1 mL |
| Dithiothreitol (DTT) | 0.5 mM | 0.05 mL |
Add ddH2O to 100 mL. Store at 4°C for up to 3–6 months.
Add DTT and complete protease inhibitor cocktail to a 1× final concentration just before use.
| Wash buffer (100 mL) | Final concentration | Amount |
|---|---|---|
| 1 M Tris-HCl (pH 7.5) | 50 mM | 5 mL |
| 5 M NaCl | 150 mM | 3 mL |
| Triton X-100 | 0.5% | 0.5 mL |
Add ddH2O to 100 mL. Store at 4°C for up to 3–6 months.
| Cross-linking buffer (100 mL) | Final concentration | Amount |
|---|---|---|
| 1 M HEPES (pH 7.8) | 20 mM | 2 mL |
| 5 M NaCl | 150 mM | 3 mL |
Add ddH2O to 100 mL. Store at 4°C for up to 3–6 months.
| Quenching buffer | Final concentration | Amount |
|---|---|---|
| 1 M Tris-HCl (pH 7.8) | 20 mM | As needed based on bead resuspension volume |
Can store at 4°C for 3–6 months.
Crosslinker:
Prepare working stock (50 mM) of DSSO by dissolving in DMSO.
Step-by-step method details
Cell culture and preparation of pellet
Timing: 2 days before start of experiment
-
1.
Seed two 150-mm dishes with 5 × 106 HEK293T cells (between passages 2–10). Culture until about 90% confluent (∼48 h) in DMEM containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, L-glutamine (2 mM) and sodium bicarbonate (44 mM) in incubator with 5% CO2 at 37°C.
-
2.
Aspirate medium and wash adherent cells twice with phosphate-buffered saline (PBS). Detach cells with 3 mL of trypsin-EDTA, 5 min at 37°C.
-
3.
Combine cells from both dishes, and resuspend in 10 mL of DMEM containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, L-glutamine (2 mM), and sodium bicarbonate (44 mM) in a 15 mL centrifuge tube, and pellet by centrifuging at 1,000×g for 5 min.
-
4.
Remove medium and wash cells twice with ice-cold PBS, and re-pellet cells at 1,000×g for 5 min.
Note: For in situ cross-linking of live cells (i.e., not lysates) proceed to step 24 (i.e., skip steps 5–23).
CRITICAL: The cells can be snap-frozen at this stage and stored at −80°C. Altered interaction of MSC constituents was not observed by freezing. However, the stability of other protein-complexes may be affected by the freeze-thaw process, and should be determined by immunoprecipitation experiments before proceeding to cross-linking.
Lysis of cells and preparation of lysate
Note: Protein A/G magnetic beads (Pierce) should be blocked in pre-blocking buffer for 1 h during cell lysis procedure in readiness for pre-clearing step when cell lysates are being prepared.
-
5.
Add protease inhibitor to 1× final concentration in lysis buffer.
-
6.
Add a 1.4 mL volume of lysis buffer to the cell pellet and mix gently by trituration until particulates are solubilized to homogeneity.
-
7.
Transfer lysate to a 2-mL eppendorf tube and mix it in a rotator end-to-end for 30 min at 4°C.
-
8.
Centrifuge at 21,000 g for 20 min at 4°C.
-
9.
Transfer by pipette the clarified cell lysate into a 2 mL eppendorf tube.
-
10.
Estimate lysate protein concentration by BCA or Bradford assay.
-
11.
Divide lysate equally (generally between 2-3 mg protein) into two 1.5 mL eppendorf tubes.
-
12.
Add a 30 μL volume of pre-blocked Protein A/G magnetic beads to each tube and mix in end-to-end rotator for 1 h at 4°C.
-
13.
Separate lysate from magnetic beads using a DynaMag-2 magnet and transfer to fresh 1.5 mL eppendorf tube. The lysate is ready for immunoaffinity pulldown.
Immunoaffinity pulldown using antibody targeting bait protein, and chemical cross-linking of bait and associated proteins
-
14.
Add target-specific rabbit polyclonal antibody in one tube and IgG control antibody in the other, and mix in an end-to-end rotator for 4 h at 4°C. In our experiments we used an antibody against human glutamyl-prolyl tRNA synthetase (EPRS1) custom generated using the human EPRS1 linker domain (Leu753 to Thr956).
-
15.
Add a 60 μL volume of pre-blocked Protein A/G magnetic beads suspended in lysis buffer to each tube, and mix for 12–16 h in an end-to-end rotator at 4°C.
Note: There are multiple ways to do the pull-down. The antibody can be first immobilized on Protein A/G magnetic beads to be added directly to the cell lysate followed by overnight incubation. Alternatively, the antibody can be incubated overnight with the cell lysate, and Protein A/G magnetic beads added the next morning for 2–4 h incubation in an end-to-end rotator.
-
16.
Next day, the beads are collected using a DynaMag-2 magnet and the lysate carefully withdrawn by pipet without disturbing the beads.
-
17.
The beads are washed with a 1 mL volume of ice-cold wash buffer for 15 min on an end-to-end rotator at 4°C.
-
18.
Collect beads again using DynaMag-2 magnet and repeat the wash step three more times.
-
19.
After removing the final wash buffer, wash the beads again with 1 mL of cross-linking buffer two times to completely remove residual Tris buffer.
Note: During the final washing step with cross-linking buffer, prepare a 50 mM DSSO solution by dissolving 0.5 mg of DSSO in 26 μL of DMSO, followed by a brief vortex to dissolve. For in cellulo cross-linking skip steps (20) to (23).
-
20.
After the final wash, resuspend beads in a 400 μL volume of cross-linking buffer and add an 8 μL volume of the 50 mM DSSO solution to yield a final concentration of 1 mM. Discard leftover DSSO solution.
-
21.
Vortex beads gently to resuspend and mix for 45 min at 25°C in a thermomixer.
-
22.
Stop the reaction by addition of a 8 μL volume of quenching buffer (20 mM final concentration) for 30 min at 25°C in thermomixer.
-
23.
Collect cross-linked beads using a DynaMag-2 magnet and proceed to on-bead tryptic digestion for mass spectrometry (step 28).
Alternate procedure for cross-linking in live cells
-
24.
Resuspend pelleted HEK293T cells in a 1.88 mL volume of hypotonic buffer containing protease inhibitor cocktail.
-
25.
Add a 120 μL volume of DSSO (50 mM in DMSO; prepared by dissolving 2.5 mg DSSO in 130 μL volume of DMSO) to yield a final concentration of 3 mM, and mix for 1 h in an end-to-end rotator at 4°C. Discard leftover DSSO solution.
-
26.
The cross-linking reaction is stopped by addition of a 40 μL volume of quenching buffer (1 mM Tris-HCl, pH 7.8 to final concentration of 20 mM) for 30 min on an end-to-end rotator.
-
27.
Return to steps (5) through (19). After completion of step (19) proceed to step (28).
Trypsin digestion and sample preparation for mass spectrometry
-
28.
Resuspend beads in a 10 μL volume of 100 mM ammonium bicarbonate containing 100 ng trypsin (10 ng/μL).
-
29.
Vortex samples for 15 s every 2–3 min for 15 min at room temperature (∼25°C).
-
30.
Continue digestion for 12–16 h at 37°C in thermomixer.
-
31.
Next morning, add a second aliquot of 100 ng trypsin in 100 mM ammonium bicarbonate, and incubate in thermomixer at 37°C for 4 h.
-
32.
Collect supernatant using a DynaMag-2 magnet, and dilute with formic acid (5% v/v, final concentration).
-
33.
Remove salts from supernatants with PepClean C-18 spin columns, and dry sample completely with a vacuum concentrator.
-
34.
Reconstitute dried samples in a 30 μL volume of 1% acetic acid.
Mass spectrometry and identification of proximate cross-linked peptides
-
35.
Perform LC-MS experiments using an Orbitrap Fusion Lumos Tribrid MS instrument coupled to an Ultimate 3000 UHPLC system equipped with a 2-cm reverse-phase C18 trapping column and a 15-cm reverse-phase C18 analytical column.
-
36.
Load a 5 μL volume of digested peptides (1–2 μg protein) onto the trapping column with solvent A (0.1% formic acid), at a flow rate of 10 μL/min for 5 min. After desalting, elute the peptide onto the analytical column and fractionate for 140 min using a gradient of solvent A/solvent B (80% acetonitrile/20% water/0.1% formic acid) at a flow rate of 0.3 μL/min.
Gradient:
| Time (min) | % Solvent A | % Solvent B |
|---|---|---|
| 0 | 98 | 2 |
| 5 | 98 | 2 |
| 110 | 65 | 35 |
| 120 | 10 | 90 |
| 127 | 10 | 90 |
| 129 | 98 | 2 |
| 140 | 98 | 2 |
-
37.
Perform three LC-MS/MS experiments on the cross-linked samples i.e., a data-dependent acquisition (DDA), MS2-MS3, and electron-transfer and higher-energy collision dissociation (EThcD).
Note: See Table 1 for parameters used in LC-MS/MS experiments.
-
38.
For the DDA method, switch between MS1 scans for molecular weight measurement and MS/MS experiments for peptide sequencing.
-
39.
For the MS2-MS3 experiments, perform low energy MS2 experiments on ions with charges between 4 and 8. Perform MS3 experiments on ions with a mass difference of 31.9721 Da, i.e., the mass difference between the alkene and thiol forms of DSSO cross-linked peptides.
-
40.
For EthcD experiments, perform two MS2 experiments, EThcD and low-energy collision-induced dissociation (CID) experiments on ions with charges between 4 and 8.
Table 1.
Parameters for LC-MS/MS analysis
| DDA | MS2-MS3 | EthcD | |
|---|---|---|---|
| LC parameters | |||
| Mobile phase A | 0.1% Formic acid | 0.1% Formic acid | 0.1% Formic acid |
| Mobile phase B | 80% ACN, 0.1% formic acid | 80% ACN, 0.1% formic acid | 80% ACN, 0.1% formic acid |
| Trapping column | Dionex (2 cm × 100 μm id) Acclaim Pepmap C18, 5 μm, 100 Å | Dionex (2 cm × 100 μm id) Acclaim Pepmap C18, 5 μm, 100 Å | Dionex (2 cm × 100 μm id) Acclaim Pepmap C18, 5 μm, 100 Å |
| Analytical column | Dionex (25 cm × 75 μm id) Acclaim Pepmap C18, 2 μm, 100 Å | Dionex (25 cm × 75 μm id) Acclaim Pepmap C18, 2 μm, 100 Å | Dionex (25 cm × 75 μm id) Acclaim Pepmap C18, 2 μm, 100 Å |
| Flow rate: trapping, loading time | 5 μL/min, 5 min | 5 μL/min, 5 min | 5 μL/min, 5 min |
| Flow rate: analytical | 0.3 μL/min | 0.3 μL/min | 0.3 μL/min |
| Column oven temperature | 36°C | 36°C | 36°C |
| Gradient | 0 min, 2% B 5 min, 2% B 110 min, 35% B 120 min, 90% B 127 min, 90% B 129 min, 2% B 140 min, 2% B |
0 min, 2% B 5 min, 2% B 110 min, 35% B 120 min, 90% B 127 min, 90% B 129 min, 2% B 140 min, 2% B |
0 min, 2% B 5 min, 2%B 110 min, 35% B 120 min, 90% B 127 min, 90% B 129 min, 2% B 140 min, 2% B |
| MS1 parameters | |||
| Spray voltage | 2.3 kV | 2.3 kV | 2.3 kV |
| MS1: detection | Orbitrap | Orbitrap | Orbitrap |
| MS1: resolution | 120,000 | 60,000 | 60,000 |
| MS1: scan range | 350–1,500 Da | 350–1,500 Da | 350–1,500 Da |
| MS1: mode | Profile | Profile | Profile |
| MS1: AGC target | 4.0 e5 | 4.0 e5 | 4.0 e5 |
| Maximum injection time | 50 msec | 50 msec | 50 msec |
| MS2: CID parameters | |||
| MS2: charge states | 2–7 | 4–8 | 4–8 |
| MS2: cycle time | 3 s | 5 s | 5 s |
| MS2: isolation window | 0.7 | 1.6 Da | 1.6 Da |
| MS2 fragmentation method | CID | CID | CID |
| MS2: collision energy | 35% | 25% | 25% |
| MS2: ETD activation | False | False | False |
| MS2: detection | Ion trap | Orbitrap | Orbitrap |
| MS2: resolution | Unit | 30,000 | 30,000 |
| MS2: AGC target | 3.0 e3 | 5.0 e4 | 5.0 e4 |
| MS2: mode | Centroid | Centroid | Profile |
| Maximum injection time | 300 msec | 100 msec | 100 msec |
| Dynamic exclusion | Enabled | Enabled | Enabled |
| Exclude after n times | 1 | 1 | 1 |
| Exclusion duration | 60 s | 60 s | 60 s |
| MS2: Ethcd parameters | |||
| MS2: charge States | – | – | 4–10 |
| MS2: cycle time | – | – | 5 s |
| MS2: isolation window | – | – | 1.6 Da |
| MS2 fragmentation method | – | – | ETD |
| MS2: ETD activation | – | – | True |
| ETD: collision energy | – | – | 15% |
| MS2: detection | – | – | Orbitrap |
| MS2: resolution | – | – | 30,000 |
| MS2: AGC Target | – | – | 1.0 e5 |
| MS2: mode | – | – | Centroid |
| Maximum injection time | – | – | 120 msec |
| Dynamic exclusion | – | – | Enabled |
| Exclude after n times | – | – | 1 |
| Exclusion duration | – | – | 60 s |
| MS3: parameters | DDA | MS2-MS3 | EthcD |
| Target mass difference | – | 31.9721 Da | – |
| Ions must be same charge | – | Yes | – |
| MS2: charge states | – | 2–6 | – |
| MS2: scans | – | 4 | – |
| MS2: isolation window | – | 2.0 Da | – |
| MS2 fragmentation method | – | HCD | – |
| MS2: collision energy | – | 30% | – |
| MS2: ETD activation | – | False | – |
| MS2: detection | – | Ion trap | – |
| MS2: resolution | – | Nominal | – |
| MS2: AGC target | – | 2.0 e4 | – |
| MS2: mode | – | Centroid | – |
| Maximum injection time | – | 150 s | – |
Data and error analysis for mass spectrometry
-
41.
Perform data analysis in Proteome Discoverer 2.4.
Note: See Figure 1 for processing and workflow for analysis of XL-MS datasets using Proteome Discoverer 2.4.
-
42.
For analysis of the DDA dataset, search against a species-specific sequence database such as human SwissProtKB (see parameters in Table 1).
-
43.
For DDA searches, export protein identifications for all proteins identified by at least 2 peptides at a peptide and protein false discovery rate (FDR) of 1%.
Note: Peptide FDR rates are estimated using the Percolator algorithm, a semi-supervised machine learning approach that compares peptide scores in searches performed against the forward and reverse (decoy) databases. Only high-confidence peptides are considered.
-
44.
For analysis of XL-MS datasets, use the XlinkX node bundled into Proteome Discoverer 2.4 (see parameters in Table 2) to search against the database generated in the DDA analysis.
Note: FDR is estimated using XlinkX validator node. Only consider high-confidence peptides.
-
45.
Cross-links identified in the MS2-MS3 analysis are interrogated to ensure that 3 or more of the 4 expected peptides are fragmented, and the spectra for these peptides contain sequence-specific ions consistent with the identified peptides.
-
46.
Cross-links identified from the EthcD analysis are interrogated to ensure that sequence-specific ions consistent with both cross-linked peptides are identified in the spectrum.
Figure 1.

Workflow for processing and for analysis of XL-MS datasets using Proteome Discoverer 2.4
Table 2.
Parameters for proteome discoverer searches
| LC parameters | DDA | MS2-MS3 | EthcD |
|---|---|---|---|
| Database | Sequence-specific SwissProt | Generated from DDA experiment | Generated from DDA experiment |
| Precursor selection | Use MS1 precursor | Use MS (n-1) with parent precursor | Use MS (n-1) parent precursor |
| Acquisition strategy | Not applicable | MS2_MS3 | MS2_MS2 |
| Mass tolerance MS1 | 10 ppm | 10 ppm | 10 ppm |
| Mass tolerance MS1 | 0.5 Da (ITMS) | 20 ppm (FTMS) 0.5 Da (ITMS) |
20 ppm (FTMS) |
| Dynamic modifications | Oxidation (M) DSSO modification (K, 158.004 Da) Acetylation (Protein N-term) |
Oxidation (M) DSSO modification (K, 158.004 Da) Acetylation (Protein N-term) |
Oxidation (M) DSSO modification (K, 158.004 Da) Acetylation (Protein N-term) |
| Peptide FDR (Percolator) | 0.01 | – | – |
| Peptide FDR (Xlinkx Validator) | – | 0.01 | 0.01 |
| Additional criteria for identification | Two peptides per protein | High quality MS/MS spectra identified for 3 out of the 4 DSSO (alkene and thiol) peptides | Able to identify sequence specific ions consistent with both cross-linked peptides in the spectrum. |
Cross-link validation and modeling individual complex constituents
-
47.
Our XL-MS-based analysis of the MSC revealed a total of 137 cross-links, including both intra- and inter-protein cross-links (Figure 2). In vitro and in cellulo analyses yielded 131 and 57 cross-links, respectively, consistent with a substantially greater efficiency of the former (Tables S1 and S2). However, the overlap between the groups was high as 51 of 57 cross-links detected in cellulo were also present in the in vitro analysis. Importantly, no inconsistencies, e.g., steric conflicts, were introduced by cross-links detected by either approach.
-
48.
For cross-links in constituents or sub-complexes of known structure, use reported crystal or NMR structure from Protein Data Bank (PDB) to validate newly obtained intra- and inter-protein cross-links (e.g., in our experiment, EPRS1 and KARS1-AIMP2, respectively (Figure 3).
-
49.
To build structures for proteins where homologous structures are available for non-human species only, perform homology modeling using SWISS-MODEL program.
-
50.
For individual constituents of unknown or incomplete structure, perform de novo modeling with SWISS-MODEL to build full-length structure. SWISS-MODEL uses a library of previously published structures to search for proteins with significant sequence similarity compared to the sequence of the modelled protein. Construct model from structurally conserved regions extracted from selected template structures; missing residues can be modeled de novo. Accommodate the distance restraints provided by intra-protein crosslinks during modeling.
Note: Detailed information regarding the modeling of individual constituents of the MSC has been reported (Khan et al., 2020).
Figure 2.

XL-MS-derived crosslinks for human MSC
(A) Intra-protein cross-links within constituents of the MSC (black curved lines).
(B) Inter-protein cross-links between constituents of the MSC (dashed lines).
Figure 3.

Validation of XL-MS-derived crosslinks for human MSC
(A) Intra-protein cross-link distances within structure of human EARS1 domain of EPRS1. EARS1 was modeled based on homology to the archaebacterium Methanothermobacter thermautotrophicus EARS1 (PDB ID: 3AII). Cross-linked Lys residues are shown as atom-level structures (yellow), and cross-links are indicated (line of yellow spheres).
(B) Inter-protein cross-link distances between AIMP2 and KARS1 in X-ray crystal structure (PDB ID: 6ILD). XL-MS-derived cross-linked Lys residues are highlighted (yellow spheres) and Inter-protein cross-links indicated (line of orange spheres). Figure reprinted with permission from Khan et al. (2020).
Adjustment of reported structures to conform to cross-link data
Note: Cross-links of Lys residues at distances between about 10 and 27 Å in crystal or NMR structures validate the approach and methodology. Occasionally, distances between cross-linked amino acids in published X-ray structures can substantially exceed the ∼27 Å distance limit. The discrepancy between the X-ray and XL-MS data can result from low-confidence data in the crystal structure due to mobile domains, distortion of the structure due to high-density packing in the crystal, or altered conformation of the target when present under intracellular conditions, e.g., in a complex.
-
51.
When sufficient justification is provided, e.g., by multiple discrepant cross-links, modify the conformation to constrain the lengths of these cross-links to less than ∼27 Å (Figure 4). Biochemical activity data can provide support for the altered conformation, as was the case with the XL-MS-based structural modification of QARS1 (Ognjenovic et al., 2016; Khan et al., 2020).
Figure 4.

Adjustment of QARS1 structure by XL-MS-derived intra-links
(Top) XL-MS-derived intra-links in QARS1; cross-links between N-terminal domain (NTD) and catalytic domain (CAT) are highlighted (red). (Left) Crystal structure (PDB ID: 4YE6) of the NTD (red) and CAT (pink) of QARS1 showing 3 cross-links much greater than the ∼27 Å limit (left). (Right) Amended model of QARS1 with XL-MS-derived cross-links within distance constraints of the cross-linker. Figure reprinted with permission from Khan et al. (2020).
Figure 5.

XL-MS-derived cross-links can provide evidence for homo-dimers
(Left) Cross-link (red dashed line) in monomeric structure exceeds distance constraint of the cross-linker. (Right) Cross-link in dimeric structure is within distance constraint of the cross-linker.
Modeling of the holo-MSC
-
52.
Using the protein-protein docking program PatchDock, assemble the MSC in a step-wise manner by one-at-a-time docking constituents or sub-complexes to the partially assembled complex retaining the constraints provided by inter-molecular cross-links. For example, the modified QARS1 model was docked with the pentameric AARS core (comprising EPRS1, MARS1, AIMP2, AIMP3, and DARS1) using the distant constraints obtained from QARS1-EPRS1 and QARS1-AIMP2 intermolecular cross-links.
-
53.
Visualize model using the molecular visualization program PyMOL (see Figure 6 for example).
Figure 6.

XL-MS-derived 3-D architecture of the human MSC
(Left) XL-MS-derived ribbon model of the human MSC. (Right) Space-fill model of the human MSC. Figure reprinted with permission from Khan et al. (2020).
Expected outcomes
The current protocol is derived from scoring cross-links between MSC components pulled down from HEK293T cells using an antibody against one constituent, EPRS1. It identifies proximate Lys residues both within and between the protein constituents of the human MSC. Intra-protein cross-link data can append domains or refine individual protein structures, and can be combined with inter-protein cross-link data for modeling the entire complex.
Limitations
This protocol takes advantage of a well-characterized, highly specific antibody used extensively in our laboratory, and efficiently pulls down endogenous EPRS1. We have not tested this protocol using over-expressed epitope-tagged proteins. However, we do not anticipate substantial technical issues with this approach beyond the possibility that the tag interferes with protein-protein interactions. Because the MSC is a relatively stable complex, we could use stringent conditions for lysis and washing steps. Possibly, such stringent condition will disrupt weak or transient interactions in less stable complexes. We recommend initially using less stringent conditions for lysis and washing, and testing the presence of the bait and target proteins following immuno-pull down by western blot analysis before proceeding with XL-MS.
Troubleshooting
Problem 1
Low expression of target protein or complex (Check before starting the experiment).
Potential solution
Increase the amount of starting material
Increase bait protein expression by induction or transfection. If the complex contains two proteins, then co-express both components of the complex.
Problem 2
Absence of interacting proteins following pull-down (steps 5–18).
Potential solution
The absence of interacting proteins can be due to transient nature of the complex, or instability during lysis and pull-down. In this case, lysis conditions can be explored by varying the buffer, salt, and detergent composition to determine the optimal lysis buffer for isolation of the complex. Also, use of a milder wash buffer under these conditions is recommended.
Problem 3
No cross-links are detected (steps 35–46).
Potential solution
This might be due to absence of accessible or proximate target residues, e.g., Lys in case of DSSO, in the bait or target proteins. In this case, MS-cleavable cross-linkers with alternative amino acid specificity, e.g., BMSO for Cys cross-links or DHSO for acidic amino acid cross-links can be considered (Gutierrez et al., 2016, 2018). In the case of poor accessibility of a residue, for example if the reactive amine group is not near the surface, increasing the concentration of the cross-linker or increasing the incubation time can improve reaction efficiency. If the distance between residues is greater than the cross-linker distance limit, then cross-linkers with longer backbones can be considered. Unpaired, covalent interaction of DSSO with a single Lys residue on a bait or target peptide indicates formation of a mono-link. Analysis of mono-links can provide information on surface accessibility of Lys residues.
DSSO in powder form is theoretically stable for years; however, if cross-links are not detected a new lot of DSSO can be used for cross-linking reactions.
Problem 4
No or low abundance of cross-links obtained by in cellulo cross-linking (steps 24–27).
Potential solution
If few or no cross-links are detected by in cellulo cross-linking compared to in vitro cross-linking, a different permeabilization technique, higher cross-linker concentration, or increased incubation time can be adopted to increase cross-linking efficiency. Also, certify that the cross-linker is membrane permeable. To enrich low-abundance cross-linked peptides and separate them from unmodified peptides – a common problem associated with in cellulo cross-linking - other approaches can be used, e.g., biotinylated cross-linkers trapped on avidin beads (Kang et al., 2009; Tang and Bruce, 2010).
Problem 5
Cross-links are not detected during mass spectrometry analysis (steps 35–46).
Potential solution
Presence of strong chromatogram peaks derived from protein complex of interest but no cross-links: Look for mono-links in bait or target protein as described in Problem 3. Absence of mono-links will indicate either no surface exposed Lys residues or failure of cross-linking reaction and will need further optimization.
Presence of mono-links will indicate absence of proximate intra- or inter-molecular Lys residues.
Absence of strong chromatogram peaks from protein complex of interest and no cross-links: This indicates insufficient enrichment of protein-complex components. More starting material or efficient pull-down conditions needs to be established under these scenarios.
Problem 6
Accommodation of all cross-links results in prohibited steric interference of domains during modeling (step 51).
Potential solution
The cross-link can be due to an alternative conformation of the holo-complex or to the presence of the target protein in a distinct, but related complex. If the former, an alternative conformation of one or more constituents, or a major alteration to the holo-model, can relieve the interference. If the latter, isolation, capture and analysis of the alternative complex might be necessary. Lastly, there remains the possibility of a cross-link artifact; in this case a low-reliability cross-link, e.g., weakly detected by mass spectrometry, might need to be discarded.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact, Paul Fox (foxp@ccf.org).
Materials availability
This study did not generate specialized materials.
Acknowledgments
This project was supported by NIH grants R01 DK123236, R01 DK118085, R01 AG067146, and R01 NS124547; a VeloSano Pilot Research Award; and a Lerner Research Institute Research Accelerator Program Award (to P.L.F.).
Author contributions
Conceptualization, K.K. and P.F.; Methodology, K.K.; Investigation, K.K.; Analysis, K.K. and B.W.; Writing – O.D., K.K.; Writing - Review & Editing, K.K. and P.F.; Bioinformatic Analysis and Structural Modeling, C.B.-G. and V.G.; Supervision, Project Administration, and Funding Acquisition, P.F.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101201.
Contributor Information
Krishnendu Khan, Email: khank3@ccf.org.
Paul L. Fox, Email: foxp@ccf.org.
Supplemental information
Data and code availability
This study did not generate datasets or code for any algorithms.
References
- Chavez J.D., Lee C.F., Caudal A., Keller A., Tian R., Bruce J.E. Chemical crosslinking mass spectrometry analysis of protein conformations and supercomplexes in heart tissue. Cell Syst. 2018;6:136–141.e5. doi: 10.1016/j.cels.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez J.D., Weisbrod C.R., Zheng C., Eng J.K., Bruce J.E. Protein interactions, post-translational modifications and topologies in human cells. Mol. Cell Proteomics. 2013;12:1451–1467. doi: 10.1074/mcp.M112.024497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez C.B., Block S.A., Yu C., Soohoo S.M., Huszagh A.S., Rychnovsky S.D., Huang L. Development of a novel sulfoxide-containing ms-cleavable homobifunctional cysteine-reactive cross-linker for studying protein-protein interactions. Anal. Chem. 2018;90:7600–7607. doi: 10.1021/acs.analchem.8b01287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez C.B., Yu C., Novitsky E.J., Huszagh A.S., Rychnovsky S.D., Huang L. Developing an acidic residue reactive and sulfoxide-containing MS-cleavable homobifunctional cross-linker for probing protein-protein interactions. Anal. Chem. 2016;88:8315–8322. doi: 10.1021/acs.analchem.6b02240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobucci C., Piotrowski C., Aebersold R., Amaral B.C., Andrews P., Bernfur K., Borchers C., Brodie N.I., Bruce J.E., Cao Y., et al. First community-wide, comparative cross-linking mass spectrometry study. Anal. Chem. 2019;91:6953–6961. doi: 10.1021/acs.analchem.9b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S., Mou L., Lanman J., Velu S., Brouillette W.J., Prevelige P.E., Jr. Synthesis of biotin-tagged chemical cross-linkers and their applications for mass spectrometry. Rapid Commun. Mass Spectrom. 2009;23:1719–1726. doi: 10.1002/rcm.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao A., Chiu C.L., Vellucci D., Yang Y., Patel V.R., Guan S., Randall A., Baldi P., Rychnovsky S.D., Huang L. Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.002212. M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K., Beleanu Gogonea C., Willard B., Gogonea V., Fox P.L. 3-Dimensional architecture of the human multi-tRNA synthetase complex. Nucleic Acids Res. 2020;48:8740–8754. doi: 10.1093/nar/gkaa569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintseris J., Gygi S.P. High-density chemical cross-linking for modeling protein interactions. Proc. Natl. Acad. Sci. U S A. 2020;117:93–102. doi: 10.1073/pnas.1902931116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ognjenovic J., Wu J., Matthies D., Baxa U., Subramaniam S., Ling J., Simonovic M. The crystal structure of human GlnRS provides basis for the development of neurological disorders. Nucleic Acids Res. 2016;44:3420–3431. doi: 10.1093/nar/gkw082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A. Divide and conquer: cleavable cross-linkers to study protein conformation and protein-protein interactions. Anal. Bioanal. Chem. 2017;409:33–44. doi: 10.1007/s00216-016-9941-x. [DOI] [PubMed] [Google Scholar]
- Tang X., Bruce J.E. A new cross-linking strategy: protein interaction reporter (PIR) technology for protein-protein interaction studies. Mol. Biosyst. 2010;6:939–947. doi: 10.1039/b920876c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod C.R., Chavez J.D., Eng J.K., Yang L., Zheng C., Bruce J.E. In vivo protein interaction network identified with a novel real-time cross-linked peptide identification strategy. J. Proteome Res. 2013;12:1569–1579. doi: 10.1021/pr3011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or code for any algorithms.