

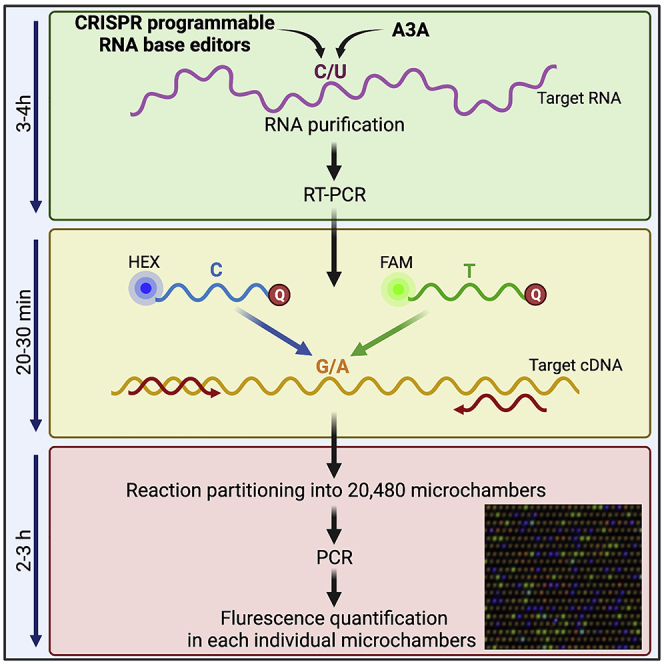
Summary
APOBEC3A, CRISPR programmable RNA base editors, or other enzymes can edit RNA transcripts at specific locations or hotspots. Precise quantification of these RNA-editing events is crucial to determine the activity and efficiency of these enzymes in cells. We have developed a quick method to quantify RNA-editing activity using digital PCR, a sensitive and quantitative technique to detect rare mutations by micro-partitioning bulk PCR reactions. This assay allows rapid absolute quantification of RNA editing events in cell lines or patient samples.
For complete details on the use and execution of this protocol, please refer to Jalili et al. (2020) and Oh et al. (2021).
Subject areas: Cancer, Cell Biology, CRISPR, Health Sciences, Molecular Biology
Graphical abstract
Highlights
-
•
Absolute quantification of RNA-editing events at hotspots
-
•
Sensitive assay to detect rare RNA-editing events in a sample
-
•
Adaptable to any type of cell lines or patient samples
-
•
Technique compatible with samples containing low amounts of RNA
APOBEC3A, CRISPR programmable RNA base editors, or other enzymes can edit RNA transcripts at specific locations or hotspots. Precise quantification of these RNA-editing events is crucial to determine the activity and efficiency of these enzymes in cells. We have developed a quick method to quantify RNA-editing activity using digital PCR, a sensitive and quantitative technique to detect rare mutations by micro-partitioning bulk PCR reactions. This assay allows rapid absolute quantification of RNA editing events in cell lines or patient samples.
Before you begin
This protocol aims to detect and quantify RNA editing events in culture cells or patient tumor samples using a digital PCR-based method. To illustrate this assay, we focus here on a specific RNA editing event caused by APOBEC3A activity in cells (Jalili et al., 2020; Isozaki et al., 2021; Oh et al., 2021). However, this protocol can be applied to monitor any RNA editing events from other types of enzymes or CRISPR programmable RNA base editors (Cox et al., 2017; Rees and Liu, 2018) that induce RNA mutations at a specific location.
Overview
APOBEC3A, a DNA and RNA cytidine deaminase that contributes to mutagenesis in tumors, is transiently expressed in cancer cells after viral infection or many other different genotoxic stresses (Stenglein et al., 2010; Sharma et al., 2015; Buisson et al., 2019; Oh et al., 2021). We have previously shown that APOBEC3A uniquely generates RNA mutations by promoting the deamination of C to U in specific RNA stem-loop structures (Jalili et al., 2020). Contrary to DNA mutations, RNA mutations are not inherited through genome duplications and are rapidly degraded. Thus, monitoring RNA mutations at hotspots provides a unique opportunity to measure the ongoing activities of APOBEC3A in cells or patient tumor samples (Jalili et al., 2020).
We previously found that the point mutation DDOST558C>U is the most frequent RNA hotspot mutation generated by APOBEC3A in patient tumors (Jalili et al., 2020). DDOST558C>U point mutation is located inside a loop of a stem-loop structure formed by DDOST mRNA and creates an optimal substrate for APOBEC3A activity (Figure 1A) (Jalili et al., 2020). For this reason, we selected DDOST558C>U as a target to monitor APOBEC3A-mediated RNA editing activity and developed a digital PCR (dPCR) assay to specifically and quantitatively measure the level of this RNA editing event in cancer cells or patient tumors (Jalili et al., 2020).
Figure 1.

APOBEC3A promotes RNA editing at a specific stem-loop structure in the gene DDOST
(A) Schematic representation of the stem-loop structure in the mRNA of DDOST, the most highly APOBEC-edited RNA site in patient tumor samples.
(B) Design of the PCR primers and probes used to monitor DDOST558C>U RNA editing event. The primer and probe sequences are designed from the genomic sequence of DDOST.
dPCR is a sensitive and quantitative method to detect SNPs, rare mutations, copy number variations, and gene rearrangements. dPCR is based on the PCR reaction being partitioned into thousands of individual microchambers, making it equivalent to running thousands of PCR reactions simultaneously. After PCR amplification, the microchambers are individually assessed for fluorescence, and following the Poisson distribution that describes incidence rate, an absolute quantification of the template sequence is determined. Importantly, two or more fluorescent probes can be used simultaneously to determine the absolute proportions of two sequence variants in a population of molecules (Dobnik et al., 2018; Quan et al., 2018).
To quantify RNA editing induced by APOBEC3A or other types of RNA editing systems, we have developed a dPCR-based method in which we explain how to (1) purify RNAs and reverse transcribe to DNA; (2) combine the sample with two fluorescence probes that recognize either the wild-type or the edited sequence of the region of interest; (3) run the digital PCR to partition the reactions into thousands of individual micro-reactions and individually assess the fluorescence in each of these reactions; (4) analyze the fluorescent signal to quantify the level of edited RNA in the samples using a Poisson correction.
Primer and probe design for digital PCR
Timing: 1–3 weeks before the experiment
The dPCR assay to quantify RNA editing activity consists of a set of primers and a set of fluorescent probes. The primers amplify the full region surrounding the edited event. For example, the primer sequences used to amplify the surrounding region of the DDOST edited site by APOBEC3A (DDOST558C>U) are highlighted in yellow (Figure 1B). Meanwhile, the fluorescent probes (highlighted in blue) bind directly to the sequence including the edited site (distinguished in red). These probes are designed to specifically recognize either the wild-type (WT) or mutated sequence and have a reporter dye (HEX or FAM) attached at the 5′end and a quencher dye (Q: Iowa Black) attached at the 3′ end of it. (Figure 1B). During the PCR reaction, the DNA polymerase will degrade the probes that are hybridized to the target sequence through its 5′-exonuclease activity. When the FAM or HEX dye molecules are released from the probes and thus no longer in close proximity to the quencher, they can be detected and quantified.
CRITICAL: Both probes need to be very specific to their respective target sequences that differ in only one nucleotide. We recommend testing several sets of probes on a synthetic substrate (e.g. 200 nucleotides synthetic oligonucleotide) (see “primer and probe optimization for digital PCR” section for an example of how to test the specificity of the probes and to determine false positive signal).
Note: We recommend that the probes have less than 30 nucleotides between the fluorophore and the quencher to avoid affecting baseline signal intensity. The probe must not have a G at its 5′ end because this may quench the fluorescent signal even after hydrolysis. When possible, it is recommended that the fluorescent probes targeting the point mutation have a higher melting temperature (Tm) than the primers used to amplify the region of interest. The sequence length of the probes can be slightly different to increase the specificity of each probe [e.g. underlined C at the 5′ end of the probe targeting DDOST T558 (Figure 1B)]
Culture cell lines and cell treatment
To mimic the induction of APOBEC3A expression by exogenous or endogenous stresses, we transfect cells with 3p-hpRNA oligonucleotide, a well-characterized agonist of RIG-I. RIG-I activation triggers APOBEC3A expression through an interferon response by recruiting STAT2 to APOBEC3A promoter (Oh et al., 2021). Cells overexpressing APOBEC3A can be used as a positive control when monitoring its activity in samples with unknown APOBEC3A levels.
-
1.
Plate cell line of interest in a 20 mm culture dish (180,000 cells).
Note: The number of cells needs to be adjusted in the function of the cell line used to obtain between 60% and 70% confluency the following day.
-
2.The next day, transfect the cells with 3p-hpRNA (e.g., 400 ng/mL for BICR6 cells)Note: We adjust the concentration of 3p-hpRNA according to each cell line. We have found that MCF10A is more sensitive to 3p-hpRNA, so we have lowered the concentration of 3p-hpRNA to 40 ng/mL.
-
a.Change the media of the cells about 1 h before transfection with 2 mL of fresh culture media without penicillin/streptomycin.Note: We established this protocol with the BICR6 or the MCF10A cell lines. BICR6 are cultured in DMEM/F12 GlutaMAX-I supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. MCF10A are cultured in DMEM/F12 GlutaMAX-I supplemented with 5% horse serum, 20 ng/mL epidermal growth factor, 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, 10 μg/mL insulin, and 1% penicillin/streptomycin.Note: We have found that culture media with penicillin/streptomycin affects the transfection efficiency of 3p-hpRNA.Alternatives: Other types of oligonucleotides such as poly(I:C) can be used to trigger APOBEC3A expression. However, we have noticed that the transfection of poly(I:C) causes higher level of cell death.
-
b.Transfect each well with 3p-hpRNA oligonucleotide by using Lipofectamine 2000 (Thermo Fisher Scientific) per manufacturer’s instruction. Per reaction:
-
i.Mix 250 μL of Opti-MEM medium and 5 μL of Lipofectamine 2000 in Tube 1
-
ii.Mix 250 μL of Opti-MEM medium and 3p-hpRNA in Tube 2
-
iii.Add Tube 1 to Tube 2 and mix well by pipetting or slowly vortexing for 5s
-
iv.Incubate for 20 min at room temperature (18°C–22°C)
-
v.Add drop by drop the transfection complex to the cells
-
i.
-
c.Exchange the cell media with fresh media containing penicillin/streptomycin 8 h–16 h after transfection.Note: We have found that APOBEC3A expression is optimal between 24 h and 48 h following 3p-hpRNA transfection.Alternatives: To overexpress APOBEC3A, cells can be also transiently transfected with a plasmid or through the use of an integrated doxycycline (DOX)-inducible system. We have previously generated doxycycline (DOX)-inducible U2OS-derived cell lines that express wild-type A3A (A3AWT) or catalytically inactive APOBEC3A mutant (A3AE72A) to demonstrate the specificity of APOBEC3A RNA editing activity at the DDOST558 hotspot (Buisson et al., 2017; Jalili et al., 2020).Note: Similar cell culture and transfection methods can be used for other CRISPR programmable RNA base editors or other RNA editing enzymes but have not yet been tested.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 3p-hpRNA | InvivoGen | Cat#tlrl-hprna |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#11668-019 |
| RNAse free ddH2O | Thermo Fisher Scientific | Cat#AM9937 |
| Fetal bovine serum | Thermo Fisher Scientific | Cat#10437-036 |
| Penicillin/streptomycin | Corning | Cat#30-002-Cl |
| Opti-MEM | Thermo Fisher Scientific | Cat# 31985070 |
| DMEM/F12 GlutaMAX-I | Thermo Fisher Scientific | Cat#10565-018 |
| Horse serum | Thermo Fisher Scientific | Cat#16050-122 |
| Epidermal growth factor | PeproTech | Cat#af-100-15 |
| Hydrocortisone | Millipore Sigma | Cat#H0888 |
| Cholera toxin from Vibrio cholerae | Millipore Sigma | Cat#C8052 |
| Insulin from bovine pancreas | Millipore Sigma | Cat#I-1882 |
| Critical commercial assays | ||
| Quick-RNA MiniPrep Kit | Zymo Research | Cat# R1055 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat#4368813 |
| QuantStudio Absolute Q Isolation Buffer | Thermo Fisher Scientific | Cat#A52730 |
| Absolute Q DNA Digital PCR Master Mix | Thermo Fisher Scientific | Cat#A52490 |
| Experimental models: Cell lines | ||
| BICR6 | Millipore Sigma | Cat#05070501 |
| MCF10A | Daniel Haber laboratory | n/a |
| U2OS complemented with A3A wild-type | Buisson et al. (2017) | n/a |
| U2OS complemented with A3A E72A | Buisson et al. (2017) | n/a |
| Oligonucleotides | ||
| DDOST Forward | Bio-Rad | n/a |
| DDOST Reverse | Bio-Rad | n/a |
| DDOSTC558 Probe | Bio-Rad | n/a |
| DDOSTT558 Probe | Bio-Rad | n/a |
| Software and algorithms | ||
| QuantStudio Absolute Q Analysis Software | Thermo Fisher Scientific | Cat# A52864 |
| Other | ||
| QuantStudio Absolute Q Digital PCR System | Thermo Fisher Scientific | Cat# A52864 |
| QuantStudio MAP16 Digital PCR Plate and Gasket Strips | Thermo Fisher Scientific | Cat# A52865 |
Materials and equipment
DDOST primer and probe sequences
DDOST Forward Sequence: ACTGAGAACCTGCTGAAG
DDOST Reverse Sequence: AAGAGGATGGGATTTAGAGA
DDOSTC558 Probe Sequence: 5’-(HEX)-CAACCATCGTTGGGAAATC-(Q)-3′
DDOSTT558 Probe Sequence: 5’-(FAM)-CCAACCATTGTTGGGAAATC-(Q)-3′
Note: Primers and probes with fluorophores and quenchers can be obtained directly from different manufacturers such as IDT, Bio-Rad, or Thermo Fisher Scientific.
Synthetic DDOST oligonucleotide sequences
DDOSTC558: 5-AAACGGCTGTCATTGACCATCACAACTATGACATCTCAGACCTTGGCCAGCATACGCTCATCGTGGCTGACACTGAGAACCTGCTGAAGGCCCCAACCATCGTTGGGAAATCATCTCTAAATCCCATCCTCTTTCGAGGTGTTGGGATGGTGGCCGATCCTGATAACCCTTTGGTGCTGGACATCCTGACGGGCTCTTCCA-3′
DDOSTT558: 5′-AAACGGCTGTCATTGACCATCACAACTATGACATCTCAGACCTTGGCCAGCATACGCTCATCGTGGCTGACACTGAGAACCTGCTGAAGGCCCCAACCATTGTTGGGAAATCATCTCTAAATCCCATCCTCTTTCGAGGTGTTGGGATGGTGGCCGATCCTGATAACCCTTTGGTGCTGGACATCCTGACGGGCTCTTCCA-3′
Reverse transcription (RT) reaction mix
| Reagent | Final concentration | Amount |
|---|---|---|
| RT buffer (10×) | 2× | 2 μL |
| RT Random Primers (10×) | 2× | 2 μL |
| dNTP Mix (25×) | 2× | 0.8 μL |
| ddH2O | n/a | 4.2 μL |
| MultiScribe Reverse Transcriptase (20×; 50 U/μL) | 2× | 1 μL |
| Total | n/a | 10 μL |
Note: The given reaction mix is for one sample. We recommend preparing a master mix for the number of samples to analyze plus one. 10 μL of RNA will be added to this 2× reaction mixture as described in step 2 under “RNA purification and generation of complementary DNA” section.
Alternatives: In this protocol, we used High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). However, other types of RT-PCR master mix can be used but have not been tested.
Digital PCR (dPCR) reaction mix
| Reagent | Final concentration | Amount |
|---|---|---|
| Absolute Q DNA Digital PCR Master Mix (5×) | 1× | 1.8 μL |
| DDOST WT target probe (HEX) (10×) | 1× | 0.9 μL |
| DDOST mutant target probe (FAM) (10×) | 1× | 0.9 μL |
| ddH2O | n/a | 4.4 μL |
| cDNA | n/a | 1 μL |
| Total | n/a | 9 μL |
Note: We recommend preparing a master mix (8 μL per sample without cDNA) for all the samples to analyze plus one. cDNA (1 μL) will be added later in step 4 under “digital PCR reaction mix” section.
Note: The final concentration (1×) of the primers is 900 nM each and 250 nM for each probe.
Alternatives: In this protocol, we used Absolute Q DNA Digital PCR Master Mix (Thermo Fisher Scientific). However, other types of digital PCR master mix can be used but have not been tested.
Step-by-step method details
Note: The method described below has been specifically optimized for the QuantStudio Absolute Q Digital PCR platform from Thermo Fisher Scientific.
Alternatives: The method described below can be successfully adapted to other commercially available digital PCR systems such as the QIAcuity dPCR platform from Qiagen or the droplet digital PCR platform from Bio-Rad. The QIAcuity dPCR platform uses similar technology as the one described here with the QuantStudio Absolute Q Digital PCR platform but has not been tested. Additional information on how to adapt this method to the QX200 Droplet Digital PCR (ddPCR) System from Bio-Rad can be found in Jalili et al. (2020).
RNA purification and generation of complementary DNA
This step describes how to purify RNAs from cells and perform reverse transcription to generate the cDNA required for the dPCR reaction step.
-
1.Collect the cells for RNA purification.
-
a.Wash the cells twice with 2 mL of PBS.
-
b.Add 300 μL of RNA lysis buffer (R1060-1-100; Zymo) directly onto the cells, mix, and collect lysed cells in a Non-Stick RNAse-free 1.5 mL microfuge tube.
 Pause point: Lysed cells can be stored at −80°C for at least a month without compromising the results.
Pause point: Lysed cells can be stored at −80°C for at least a month without compromising the results. -
c.Perform RNA purification according to the manufacturer’s protocol. This protocol uses a Zymo Quick RNA mini-prep kit.Alternatives: RNA purification using a TRIzol-based method as well as RNA purification kits from other manufacturers can also be used but have not been tested.
-
d.Quantify purified RNA using a Nanodrop spectrometer (Thermo Fisher Scientific) or equivalent.Note: We recommend using a Qubit fluorometer (Thermo Fisher Scientific) or equivalent to determine the RNA concentration for samples with low RNA concentrations (below 10 ng/μL).CRITICAL: When working with RNA, it is important to avoid contamination with RNases. We recommend adopting laboratory practices that minimize contamination with RNases such as wearing gloves and using RNase-free tubes, filter tips, water, and other reagents. We also recommend using RNase decontamination solution to clean the workspace.Pause point: Purified RNA can be stored at −80°C for at least a month without compromising the results.
-
a.
-
2.Perform a reverse transcription reaction on the purified RNA samples.
-
a.Add 500 ng of purified RNA diluted in 10 μL of RNAse free ddH2O to 10 μL RT-PCR reaction mix (total volume 20 μL). This protocol uses a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific).Note: While we recommend using 500 ng of purified RNA in the RT reaction, it is possible to use less RNA from a few ng to 500 ng without compromising the results.
-
b.Start the RT-PCR reaction (step 1: 10 min at 20°C, step 2: 2h at 37°C, step 3: 5 min at 85°C, step 4: hold at 4°C).Note: The generation of cDNA may be verified by performing PCR or qPCR with primers targeting the gene of interest or a housekeeping gene.Pause point: cDNA can be stored at −20°C or −80°C for at least a month without compromising the results.
-
a.
Digital PCR reaction mix
This step describes how to prepare a digital PCR reaction and properly load the samples into a digital PCR plate.
-
3.
Dilute the cDNA 4 to 32-fold in RNAse free ddH2O.
Note: This dilution factor has been optimized for cDNA obtained from BICR6 cells (see step 2). Because the expression level of DDOST mRNA may vary between cell lines, we recommend adjusting the dilution when using other cell lines.
-
4.
Add 1 μL of diluted cDNA to 8 μL of dPCR reaction mix.
-
5.
Load 9 μL of the PCR mix to the bottom of the well of the dPCR plate following the steps described in Figures 2A and 2B.
-
6.
Load 15 μL of the isolation buffer overlay for each well (Figure 2B).
-
7.
Apply 5 gaskets to close all the wells from the plates (Figure 2C)
Figure 2.

Samples loading on a digital PCR plate
(A) Right; Picture of a digital PCR plate and its indicated compartments. Left; Zoom in on some of the 20,480 microchambers present in the partition array.
(B) Schematic explanation of how to load a sample in a well of the digital PCR plate.
(C) Picture of a digital PCR plate with the gaskets.
Digital PCR set up
This step describes how to load a digital PCR plate and use the QuantStudio Absolute Q Digital PCR System to analyze the samples by digital PCR.
-
8.
Insert the plates into the QuantStudio Absolute Q Digital PCR System.
-
9.
Open the QuantStudio Absolute Q Digital PCR Software and select the column(s) to be run and assign each partition array on the software to a specific sample (Figure 3).
-
10.
Select Preheat Two-Step and set up the reaction as below (Table 1):
Note: The PCR steps for this digital PCR-based method differ from a regular PCR reaction with both the annealing and extension steps merged into a single step. Moreover, the time for the annealing/extension step is very short due to the close proximity of the 2 primers (separated by only 24 base pairs) (Figure 1B).
Note: The PCR condition has been optimized for DDOST probes on a QuantStudio Absolute Q Digital PCR platform (Thermo Fisher Scientific). Optimization of the PCR reaction is required if different dPCR platforms are used. For example, the PCR reaction for the DDOST probes needs to be modified to: 5 min at 95°C, 40 cycles of 94°C for 30 s, 53°C for 1 min and then 98°C for 10 min (ramp rate: 2°C s−1) when used in a droplet-based digital PCR equipment from Bio-Rad.
-
11.
Assign a fluorescence (e.g., FAM or HEX) channel to each probe (Figure 3).
Alternatives: Other channels such as VIC and Cy5 can be selected instead of FAM and HEX depending on the fluorophores present on the probes of interest.
-
12.
Assign a name of the run and start the reaction.
Figure 3.

Digital PCR set up
Example of a setup page obtained from QuantStudio Absolute Q Digital PCR Software to assign the samples to be analyzed on the digital plate, the fluorescence dyes associated with the probes, and the PCR conditions.
Table 1.
Digital PCR steps
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Preheat | 96°C | 10 min | 1 |
| Denaturation | 95°C | 5 s | 45 cycles |
| Annealing/extension | 56°C | 10 s |
Data analysis
-
13.
Once the PCR reaction is completed, open QuantStudio Absolute Q Digital PCR Software.
-
14.
Select the run to be analyzed.
-
15.
Assign the samples into groups if running different sets of samples that need to be analyzed independently.
-
16.
Adjust the thresholds on the x and y-axis to assign positive FAM and HEX signals as shown in Figure 4.
-
17.
The data can be exported as an Excel spreadsheet or generated as a report (Figure 5).
Note: During a digital PCR, the target DNAs in the samples are randomly partitioned into the 20,480 microchambers (Figure 2A). As the result of this random distribution, microchambers will contain zero, one, or several copies of the target DNA depending on the overall concentration. The microchambers are assigned as positive or negative based on their fluorescence amplitude (partitions with two identical templates of DNAs cannot be differentiated from those with one template DNA). To account for the fraction of partitions with one or more copies (where both cases are assigned as positive microchambers), the concentration of samples is corrected using Poisson modeling (Whale et al., 2013; Dobnik et al., 2018; Quan et al., 2018). The formula used for Poisson modeling is:
-
18.
Calculate the percentage of RNA editing events present in each sample (Figure 6). The formula used for calculating the fraction of editing events is:
Note: The dynamic range of the dPCR extends from around 0.25 copies/μL to several thousands of copies/μL without affecting the absolute quantification of the fraction of the target DNA in the samples as illustrated in Figure 6C for DDOST. The strong advantage of having an extended dynamic range is the direct comparison of samples with different levels of concentration of the target DNA.
Alternatives: Similar analysis and calculation can be performed when using other dPCR platforms such as the QX200 Droplet Digital PCR System from Bio-Rad (Jalili et al., 2020; Oh et al., 2021).
Figure 4.

Quantification of the positive and negative micro-chambers
(A–C) Example of scatter plots displaying HEX (WT) signals shown in orange and FAM (Mutant) signals shown in purple in MCF10A (A) and BICR6 (B) cell lines transfected with 3p-hpRNA for 48 h and 36 h respectively, or U2OS-A3A/A3AE72A DOX-inducible cell lines (C) treated with doxycycline (DOX) (200 ng/mL) for 48 h. Partitions with double-positive signals are shown in green and partitions without any amplification are shown in black. The thresholds to determine positive HEX and FAM signals are applied uniformly among samples from the same group.
Figure 5.

Example of a table generated after the digital PCR reaction is completed
The positive FAM and HEX signals in the microchambers are assigned based on their fluorescence amplitude.
Figure 6.

Quantification of RNA editing at DDOST558C>U
(A) RNA editing events at DDOST558C>U were quantified in MCF10A or BICR6 cells after 3p-hpRNA transfection for 48 h and 36 h, respectively. Error bar: S.D. (n = 3). N.D.: not detected (signal below LoD).
(B) U2OS-derived cell lines inducibly expressing A3A or A3A-E72A were induced with doxycycline (DOX) for 48h or left uninduced. The level of edited DDOST558C>U in U2OS cells was quantified by dPCR assay. Error bar: S.D. (n = 3).
(C) Indicated serial dilution of cDNA obtained from BICR6 cell line transfected with 3p-hpRNA for 36 h. The Poisson corrected concentration of DDOST T558 (left y-axis) and the fraction of RNA-editing event DDOST558C>U (right y-axis) were monitored. Error bar: S.D. (n = 3).
Primer and probe optimization for digital PCR
This protocol can be readily adapted to monitor additional RNA editing events induced by A3A or by other RNA editing systems. To successfully detect RNA editing events, it is important to first test the primers and probes to verify their specificity to their respective target sequences as they differ in only one nucleotide. This is crucial when determining false-positive signals.
To test the specificity of the primers and probes, we advise synthesizing two oligonucleotides of 200 nucleotides containing the wild-type or mutated sequence of the region of interest. Then, use these two oligonucleotides as templates instead of adding cDNA as described in step 4.
As shown in Figure 7, we recommend testing the probes in 4 different dPCR reactions with (A) no templates, (B) wild-type oligonucleotide template only (DDOST C558), (C) mutated oligonucleotide template only (DDOST T558), and (D) both wild-type and mutated oligonucleotide template. Specific probes should only amplify their respective target (Figures 7B and 7C) and equal fluorescence signal (50%) should be obtained when both oligonucleotides are equally mixed together (Figures 7D and 8). In these reactions, using 0.1–1 pg/mL of oligonucleotides is recommended.
Figure 7.

Example of dPCR results used to determine the specificity of the DDOST primers and probes
(A–D) No templates (A), DDOST C558 oligonucleotide template (B), DDOST T558 oligonucleotide template (C), or both DDOST C558 and DDOST T558 oligonucleotide templates (D) were added to the dPCR mix containing the DDOST primers and probes as indicated in step 4. The dPCR reactions were then run as described in steps 5 to 18.
Figure 8.

Quantification of the DDOST C558 and DDOST T558 oligonucleotides ratio by dPCR
DDOST C558 and DDOST T558 oligonucleotides were mixed together (1:1 ratio) at different concentrations and quantified by dPCR. The concentrations and the ratio highlighted in blue were obtained from the panel shown in Figure 6D. S.D.: Standard deviation.
Additionally, the annealing/extension temperature and time must be optimized for every set of primers/probes to limit non-specific amplification of the target. We suggest increasing or decreasing the annealing/extension temperature to improve the specificity of the primers and probes and limit false positive if such results are not obtained. Troubleshooting 3, 4, and 5.
Other than testing for the specificity of the probes, this optimization step is also crucial for determining the Limit of Detection (LoD) or the threshold for significance to establish the level of false-positive signals generated by each probe, which allows to separate positive signals from background noises (see quantification and statistical analysis section for more details).
Expected outcomes
Successful detection of RNA editing events should result in a clear separation of FAM fluorescence from the HEX fluorescence as well as from the partition without amplification. The percentage of RNA editing events detected using this dPCR method will fluctuate depending on the enzyme activity or the efficiency of the CRISPR programmable RNA base editor used. In the example of monitoring the endogenous expression of APOBEC3A in cancer cells, we expect to detect between 2% and 6% of DDOST558C>U events (Figure 6A). When APOBEC3A is expressed using a DOX-inducible system (Buisson et al., 2017), higher levels of RNA edited events are expected (>30% of DDOST558C>U events) (Figure 6B).
Quantification and statistical analysis
To distinguish positive signals from background noise, it is important to establish a threshold for significance or limit of detection (LoD). The LoD for a given target is the highest number of partitions that can be detected in a sample with no target DNA. The LoD needs to be calculated for every set of probes and adjusted in the function of the number of partitions analyzed.
Run three or more dPCR reactions on a control sample (e.g., a synthetic oligonucleotide containing the wild type sequence only or mutant sequence only as described in Figures 7B and 7C) and calculate the LoD using the following formula:
Any concentration with positive signals less than or equal to this calculated mean should be considered as negative or false positive. For example, the LoD for DDOST T558 is 0.060% (Table 2). It means that APOBEC3A-induced RNA editing activity below 0.060% for the fraction of DDOST 558 C>U (or below 0.060% of DDOST T558) will be considered as negative for RNA editing events.
Table 2.
Example of limit of detection calculation for DDOST probes
| Samples | LoD for DDOST T558 (using DDOST C558 oligonucleotide template) | LoD for DDOST C558 (using DDOST T558 oligonucleotide template) |
|---|---|---|
| Replicate 1 | 0.049% | 0.020% |
| Replicate 2 | 0.000% | 0.078% |
| Replicate 3 | 0.130% | 0.058% |
| Mean | 0.060% | 0.052% |
Three independent dPCR were run using the DDOST C558 oligonucleotide template only, or the DDOST T558 oligonucleotide template only. dPCR results for replicate 1 were shown as an example in Figures 6B and 6C and Table 2. For each dPCR, the LoD is calculated from the fraction of DDOST C558 or DDOST T558 cp/μL divided by the total Poisson corrected cp/ μL (e.g., DDOST T558 LoD obtained from the replicate 1 shown in Figure 6B: Poisson corrected 0.35 cp/μL (from 3 DDOST C558 positive partitions) divided by total Poisson corrected 712.02 cp/μL (from 5232 total positive partitions) = 0.049%.
In addition to calculating the LoD of each probe and determining the level of false positives, it is also essential to obtain enough partitions with positive fluorescence signals to accurately calculate the fraction of RNA editing events in a sample. Because we observed a loss of linearity when a sample had a low level of positive partitions, we recommend having at least 1000 positive partitions in each sample and excluding samples that do to reach this abundance.
Limitations
The specific location of the RNA editing events can be a limitation for the design of the PCR primers and probes because the sequence around the RNA editing events may not be optimal for the PCR reaction (e.g., high GC content or sequences forming secondary structures) and will directly affect the efficiency the amplification of the target sequence or the specificity of the set of probes for the point mutation. Troubleshooting 4 and 5
Troubleshooting
Problem 1
Absence of partition without any fluorescence signal on the scatter plots.
Potential solution
The sample was not sufficiently diluted in step 3 and all the microchambers contain one or several DNA targets. The Poisson correction cannot be applied if all the partitions contain a fluorescence signal. Increase the dilution factor of the cDNA before starting the dPCR reaction.
Problem 2
A cluster of signals across the scatter plot (e.g., Figure 9A) without clear separation of FAM and HEX fluorescence signals in step 16.
Figure 9.

Examples of bad partitioning during the loading of the samples
(A) Example of a sample without a clear separation of the FAM and HEX fluorescence signal in the scatter plot graph.
(B) Examples of a proper (left) or improper (right) sample partitioning across a partition array. The quality of the partitioning is determined by monitoring the even distribution of the ROX fluorescence.
Potential solution
A cluster of negative signals without any separation of FAM and HEX signals may appear when the partitioning in the 20,480 microchambers of the partition array is not performed properly. Check the ROX-QC array on the analysis software to determine whether the sample was improperly loaded in the partition array (Figure 9B). Improper filling of the microchambers may be caused by any micro debris in the sample. This may be solved by centrifuging the samples at 10,000g for 1–2 min to pellet any micro debris that may be present in the samples before their loading.
Problem 3
Positive partition on the scatter plots in the no template control sample in step 16.
Potential solution
Digital PCR is a very sensitive method and can detect very low levels of target DNA. Positive fluorescence signal in the no template control sample is often caused by cross-contamination between samples or the reagents used for the dPCR reaction. It is advised to use fresh buffers and dedicated pipettes and filter tips, and prepare the samples under a PCR workstation or PCR hood to prevent any undesired contamination.
Problem 4
The HEX or FAM Fluorescence is not detected in step 16.
Potential solution
The amplification primers are not working properly. Optimize the sequence of the primers to increase the amplification efficiency. We recommend testing first the different sets of primers on cDNA to identify the ones that amplify the region of interest most efficiently.
RNA or cDNA samples were too diluted (see steps 2 and 3). Increase the amount of RNA used in the reverse transcription reaction or dilute less the cDNA before starting the dPCR reaction.
Problem 5
The HEX or FAM Fluorescence signals are curved on the scatter plots in step 16.
Potential solution
HEX or FAM Fluorescence signals are curved when the two probes (that recognize the WT sequence or the mutated sequence) are not specific enough and start hybridizing to the wrong sequence. Increasing the annealing/extension temperature will limit the unspecific hybridization of the probes. Alternatively, modify the sequence of the probes to increase sequence specificity (e.g., sequence with a lower Melting temperature, longer probe, or higher GC content).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rémi Buisson (rbuisson@uci.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We thank Andrew Babbin, Elodie Bournique, and Pedro Ortega for their helpful discussions. We thank Christina Bouwens and Adam Langston (Thermo Fisher Scientific) for technical advice. R.B. is supported by an NIH MERIT Award (R37-CA252081), an NIH Pathway to Independence Award (R00-CA212154), the California Breast Cancer Research Program, the Concern Foundation, and a University of California Cancer Research Coordinating Committee (CRCC) award grant. This work was also made possible, in part, through access to the Genomics High Throughput Facility Shared Resource of the Chao Family Comprehensive Cancer Center Support Grant (P30-CA062203). Cartoons in the Graphical abstract were created with BioRender.com. Image captions included in the Graphical Abstract (bottom right), Figure 2A (right), and Figure 2B were used by permission of Thermo Fisher Scientific, the copyright owner.
Author contributions
S.O. performed all the experiments. S.O. and R.B. developed the method and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate/analyze datasets
References
- Buisson R., Lawrence M.S., Benes C.H., Zou L. APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res. 2017;77:4567–4578. doi: 10.1158/0008-5472.CAN-16-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R., Langenbucher A., Bowen D., Kwan E.E., Benes C.H., Zou L., Lawrence M.S. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019;364:eaaw2872. doi: 10.1126/science.aaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D.B., Gootenberg J.S., Abudayyeh O.O., Franklin B., Kellner M.J., Joung J., Zhang F. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–1027. doi: 10.1126/SCIENCE.AAQ0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobnik D., Spilsberg B., Štebih D., Morisset D., Holst-Jensen A., Žel J. In: Methods in Molecular Biology. Karlin-Neumann George, Bizouarn Francisco., editors. Springer; 2018. Multiplex droplet digital PCR protocols for quantification of GM maize events; pp. 69–98. [DOI] [PubMed] [Google Scholar]
- Isozaki H., Abbasi A., Nikpour N., Langenbucher A., Su W., Stanzione M., Frisco-Cabanos H., Siddiqui F.M., Phan N., Jalili P., Oh S. APOBEC3A Drives Acquired Resistance to Targeted Therapies in Non-small Cell Lung Cancer. bioRxiv. 2021 doi: 10.1101/2021.01.20.426852. [DOI] [Google Scholar]
- Jalili P., Bowen D., Langenbucher A., Park S., Aguirre K., Corcoran R.B., Fleischman A.G., Lawrence M.S., Zou L., Buisson R. Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat. Commun. 2020;11:2971. doi: 10.1038/s41467-020-16802-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S., Bournique E., Bowen D., Jalili P., Sanchez A., Ward I., Dananberg A., Manjunath L., Tran G.P., Semler B.L., Maciejowski J. Genotoxic stress and viral infection induce transient expression of APOBEC3A and pro-inflammatory genes through two distinct pathways. Nat. Commun. 2021;12:1–17. doi: 10.1038/S41467-021-25203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan P.-L., Sauzade M., Brouzes E. dPCR: a technology review. Sensors. 2018;18:1271. doi: 10.3390/S18041271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770. doi: 10.1038/S41576-018-0059-1. [DOI] [PubMed] [Google Scholar]
- Sharma S., Patnaik S.K., Taggart R.T., Kannisto E.D., Enriquez S.M., Gollnick P., Baysal B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015;6:6881. doi: 10.1038/ncomms7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein M.D., Burns M.B., Li M., Lengyel J., Harris R.S., Stenglein M.D., Burns M.B., Li M., Lengyel J., Harris R.S. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010;17:222–229. doi: 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whale A.S., Cowen S., Foy C.A., Huggett J.F. Methods for applying accurate digital PCR analysis on low copy DNA samples. PLoS ONE. 2013;8:e58177. doi: 10.1371/journal.pone.0058177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets