

Abstract
This Review presents and discusses the current state of the art in “exchangeable liquid crystalline elastomers”, that is, LCE materials utilizing dynamically cross-linked networks capable of reprocessing, reprogramming, and recycling. The focus here is on the chemistry and the specific reaction mechanisms that enable the dynamic bond exchange, of which there is a variety. We compare and contrast these different chemical mechanisms and the key properties of their resulting elastomers. In the conclusion, we discuss the most promising applications that are enabled by dynamic cross-linking and present a summary table: a library of currently available materials and their main characteristics.
1. Introduction on LCEs and Their Alignment
The idea of a large-strain reversible mechanical actuator based on the intrinsic material properties of liquid crystalline elastomers (LCEs) has been understood for over 30 years.1−3 The key characteristics of LCE actuation are remarkable: fully reversible action,4 large amplitude, with a stroke of up to 500%,5,6 and the stress–strain–speed response matching or exceeding the human muscle.7 The origin of this effect lies in the direct coupling of the macroscopic shape of a cross-linked network and the underlying anisotropic order of its polymer strands, e.g., the length of a sample contracts along the director axis when the internal liquid crystal order is altered by heating into the isotropic phase (although other ways of altering the order parameter exist, extensively reviewed in the literature3,4). At the same time, separately from the natural order–shape connection leading to actuation, LCEs have a unique mechanical property of “soft elasticity” (when elastic deformation may occur at low or zero stress),8−10 which leads to a different strand of potential applications. These properties make LCEs highly attractive in biomedical engineering,11,12 robotics,11,13 smart textiles,14,15 damping,16,17 adhesive systems,11,18 surface modifications,14,19 and many other fields of modern engineering.20−23
After initially being envisioned as thermotropic mechanical actuators (artificial muscles) by de Gennes in 1975,24 the first LCEs were synthesized and investigated by Finkelmann in 1981.25 Finkelmann and co-workers were able to first make these new materials in the form of a nematic side-chain elastomer employing a hydrosilylation reaction to graft a vinyl mesogenic pendant groups onto a siloxane polymer backbone.25 A parallel approach used mesogenic monoacrylates to polymerize into a different kind of side-chain LCEs.26,27 In side-chain elastomers, the mesogens are attached to the flexible polymer backbone and their orientational order imposes anisotropy on the backbone chains linked into the rubbery networks.
To achieve actuation, the uniformly aligned LCE needs to be, e.g., heated above its isotropic transition temperature (although there are other ways of changing internal order). However, unlike in ordinary liquid crystals, the natural equilibrium configuration of LCE networks is a polydomain state with a characteristic domain size of ∼1 μm, unless special precautions are taken to cross-link it in an aligned configuration. A large body of knowledge exists on the origins of the polydomain state, which is due to the randomly quenched disorder in the liquid crystalline system,28,29 as well as on the polydomain–monodomain transition that occurs on uniaxial stretching of a polydomain LCE.30−32 Polydomain LCEs cannot produce actuation without external stress due to their lack of overall anisotropy. Therefore, LCEs with uniform equilibrium molecular alignment, i.e., the monodomain or the “single crystal” LCEs, were created to enable load-free reversible actuation.1 The average molecular alignment must be fixed permanently by network cross-linking, which is most commonly achieved through one of the following three methods: mechanical stretching with two-step cross-linking,1,33,34 surface alignment on a substrate,35,36 or cross-linking after shear extrusion5,37 (Figure 1).
Figure 1.

Schematic illustration of the three key methods of alignment in monodomain LCEs. Top line: alignment by two-step cross-linking.1,34 Middle line: surface alignment.36,48 Bottom line: alignment by shear on extrusion.5,37 Some graphics are adapted from ref (34), copyright 2015 Royal Society of Chemistry, from ref (5), copyright 2006 John Wiley and Sons, with permissions from ref (36) copyright 2015 AAAS, from ref (48) copyright 2017 Springer Nature.
Mechanical stretching via two-step cross-linking was the first technique to achieve the permanent uniform molecular alignment in LCEs, and it is this achievement that has “ignited” the whole field of LCE research and applications. Küpfer and Finkelmann originally utilized a two-step reaction to enable the stress-aligning of the weakly cross-linked LCE gel between the steps, based on different reaction rates. In the first step, a fast hydrosilylation reaction was used to attach side-chain vinyl mesogens to a siloxane backbone. The second step was to cross-link the network via a much slower reaction of siloxane with acrylate cross-links, the long window of time to full cross-linking allowing for the mechanical stretching and aligning of the partially cross-linked gel. The second reaction was allowed to complete in the sample under load to lock in the new aligned conformation (Figure 1). This technique has been extensively used by many research groups, in both side-chain and main-chain LCEs (in the latter, the mesogens are placed within the polymer backbone).1,2,33,38−40 The two-step hydrosilylation method produces lightly cross-linked elastomers with tunable thermomechanical properties and LC phase behavior and full actuation range.4,41
The hydrosilylation-based two-step cross-linking method had enormous success during the 1990s and early 2000s, and as a result, LCEs have become a relevant field in material science.4,42−46 However, this cross-linking method suffered from fundamental problems, which restricted the real-world applications for these materials. For example, the difficulty of making monodomain structures and producing scalable samples remained a major problem of the field of LCEs. The difficulties originated from the lack of control over the reaction kinetics during the alignment step: applying the load too soon to a still weak gel results in fracture, while applying the load too late in the continuously ongoing cross-linking process results in poor alignment and strong random disorder.
The concept of two-step cross-linking has been significantly improved in the past few years, after adapting thiol-based click chemistry to the production of LCEs (e.g., two-step thiol–acrylate reaction).34 Unlike for hydrosilylation, this chemistry relies on two independent reactions (nucleophilic Michael addition of thiol–acrylate and acrylate photopolymerization, which can be photoinitiated for additional control).34,47 Therefore, the process allows for much better separation of the reaction steps and the process parameters. Moreover, this chemistry uses well-established commercially available reacting monomers, which allows for producing scalable samples.
The second method to align LCEs is the surface alignment technique which has been used to align liquid crystalline (LC) molecules in LC displays since the 1970s.36,48,49 It was introduced to LC polymers in the 1980s by Broer and co-workers.50−52 This method of photopolymerization of diacrylate mesogenic monomers, aligned on a substrate, produces highly cross-linked and aligned networks with the glass transition (Tg) above 100 °C and an actuation strain of less than 5%. Polymerization of diacrylates was demonstrated as useful in many applications where induced surface bending could be utilized.53 Recently, White et al. added small difunctional isotropic monomers (e.g., primary amine or dithiol) to these diacrylate networks to form aligned elastomeric films with their Tg just below room temperature. The actuation of these new elastomers was about 40–100% strain.36,54 It is important to note that surface alignment techniques are only effective to produce planar systems (films). On the other hand, it could allow complex patterns of alignment compared to the conventional two-step alignment approach only giving a uniform uniaxial director. Nonetheless, the method remains limited to very few polymerization chemistries and to films of less than 100 μm in thickness.
The third method to align LCEs for actuation is by shear stress during extrusion. Shear has been traditionally employed to align fibers even in isotropic polymer composites. The method was introduced to the field of LCEs in 2006 when an extruder was used to extract well-aligned LCE fibers with high actuation strain (>400%) from physically cross-linked LC polymers.5 Recently, this technique was dramatically improved by the use of 3D printing, where printed LCE objects can be produced by extruding LC oligomers into filaments and then subsequently photo-cross-linking them to create complex shapes and structures.37,55−58
It is important to point out that all of these approaches to produce permanently aligned monodomain LCEs can be difficult to use in practice, especially when it comes to producing complex geometries and shapes of the elastomer, because the required molecular alignment must occur before the final cross-linking reaction is complete. This presents the unavoidable competition between the alignment (which needs low cross-linking to avoid quenched disorder) and the cross-linking (which is needed to give the material mechanical stability but prevents further alignment). Also, as with all thermosets, there is a problem of recycling or reprocessing.
This Review is organized as following. In the next two sections, we offer an overview of dynamically cross-linked LCE networks—which differ greatly from a “thermoplastic LCE” concept based on physical cross-linking (although thermoplastic elastomers are common in polymer science and technology, their application in the LCE field has not yet been successful, mainly because there is unavoidable creep at high temperatures). Section 2 gives a brief discussion of how the dynamically exchangeable covalent bonds can be used in forming the network, the idea that has led to the foundation of exchangeable liquid crystalline elastomers. Section 3 offers an overview of key physical properties that distinguish these materials from permanently cross-linked LCEs, which are common to all types of dynamic networks. Following that, the subsequent sections 4–12 list and discuss different chemistry advances (types of bond exchange and the resulting properties of the materials) achieved in the past few years—since this field has taken off.
2. Overiew of Dynamically Crosslinked xLCEs
The concept of LCE systems cross-linked using dynamic covalent chemistry (DCC) appears to solve these limitations by allowing the elastomers to be processed after full cross-linking. The aim of reprocessing cross-linked polymer networks with DCC is to gain access to well-developed processing methods used in the thermoplastic industry (extrusion, fiber drawing, and injection molding).59 Polymer networks that are cross-linked by DCC can be reprocessed through topological rearrangements at high temperature and under stress.60−66 The network topology can be changed on demand by activating the bond exchange. In the case of an exchange reaction with an associative mechanism, the total number of covalent bonds and the network integrity remain constant. For dissociative bond exchanges (and equivalently in physically cross-linked materials), the bond dissociation leads to a drop in viscosity during the process, though in the long term the mechanical properties are partially or fully recovered through bond reformation. This contrasts with standard thermoplastics, which melt on heating, retaining no structural integrity. Various stimuli can be used to activate the bond exchange: thermal, light, and chemical (including pH). For example, DCC reactions that can be thermally activated in polymer networks include the Diels–Alder reaction,67 the thiol–Michael adduct dynamic equilibrium,68 urea–amine exchange,69 transesterification,59 transcarbomylation,70 transcarbonation,71 thiourethane bond exchange,72 vinylogous urethane–amine exchange,73 siloxane exchange,74 olefin metathesis,75 triazolinedione click exchanges,76 transalkylation,77 and the nucleophilic exchange of quaternary anilinium salts.78 Examples of light-activated reactions include addition–fragmentation chain transfer (AFT, e.g., allyl sulfide–thiol exchange),79 cycloadditions [2 + 2],80−82 trithiocarbonates,83 and disulfides.84 There are several examples of multistimuli-activated bond exchanges (e.g., {light + temperature}), such as with disulfides, cycloadditions [4 + 4] (anthracene), or {temperature + pH} in boronic esters,85 aldehyde–NH2R condensations: hydrazone formation and exchange, imine formation and exchange, oxime formation and exchange, and disulfide.61,65
Choosing a right stimulus (temperature, light, or chemical) to trigger the DCC reaction is very important, as it dictates the processing conditions and the subsequent applications of the dynamically cross-linked LCE networks. For instance, elastomers cross-linked with thermally activated DCC can be processed (aligned, reshaped, welded, and recycled) at high temperature (as the exchange rate becomes faster, which helps facilitate plastic flow), which means in the isotropic state.86 On the other hand, LCEs cross-linked by light-triggered DCC can be processed at low temperature, in the nematic state.87 It is often beneficial to process LCEs (e.g., align or weld) in the nematic phase to enable better alignment of the nematic director, or preserve this alignment while reshaping or welding. Aligning LCEs at a high temperature (in the isotropic phase) is a more delicate process relying on embedding the seed anisotropy into the polymer network, which produces the “isotropic-genesis” LCEs,88 but may also lead to fracture of samples due to the internal stress generated by the subsequent phase change. In some cases, there could also be issues with the mesogen stability, e.g., when there is an ester group inside the mesogenic unit. Nevertheless, most of the thermally induced bond-exchangeable LCEs have been processed in the isotropic phase despite these issues.
Beyond thermal activation, the concept of chemically or solvent-activated/assisted DCC reactions is widely known in isotropic polymer networks. Solvent can be used to activate DCC either as a reactive chemical species (e.g., ethylene glycol dissolves transesterification-based vitrimers,89 2-mercaptoethanol dissolves disulfide networks, or primary amines dissolves siloxane networks)90 or as an inert chemical species serving as a swelling agent to stretch the existing bonds and thus reduce the activation energy (and thus activation temperature) in thermally induced DCC by local mechanical work in a mechanochemistry manner (e.g., dichloromethane or tetrahydrofuran reduces the activation temperature in transesterification-based vitrimers).91 Solvent has been used to trigger reprocessing of LCEs cross-linked by DCC reactions via swelling at low temperature. Even though the LCE processing was done in the isotropic state (since solvent swelling reduces the orientational order), this process avoids the use of high temperatures and possible degradation and also allows for spatial control of processing that is impossible to achieve in thermal activation by ambient conditions; see Figure 2.
Figure 2.
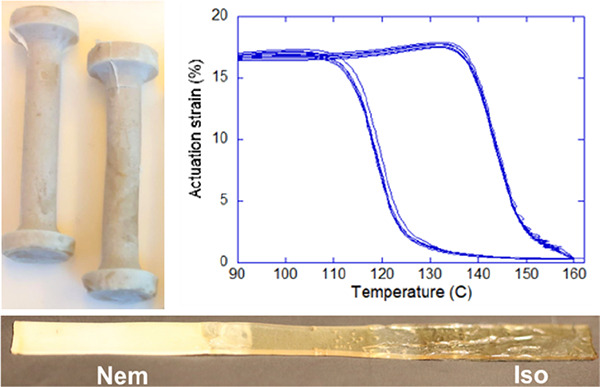
(a) The synthesis of the first xLCE, showing the difunctional epoxy mesogen and the spacer used at a 1:1 ratio; triazabicyclodecene (TBD) was used as catalyst at 180 °C. (b) The scheme of transesterification equilibrium, which keeps an equilibrium balance between reconnecting chains, 3-functional cross-links, and dangling ends. (c) Thermal actuation curves of aligned xLCEs at different prestress. The inset shows an example of complex shape molded from this material. Adapted with permission from ref (86). Copyright 2014 Springer Nature.
The aim of this article is to provide a broad “bird’s eye view” of the various DCC reactions that are utilized to enhance LCEs by imparting the covalent bond exchange. DCC offers a reliable means to process LCEs postpolymerization once the bond exchange is activated upon exposure to a stimulus.
The first examples of such “exchangeable LCEs” (named xLCEs)86 were based on transesterification in networks obtained through an epoxy–acid polymerization,86,92,93 shown in Figure 2a, mirroring the seminal work of Leibler et al.59 on the isotropic epoxy–acid vitrimer. This chemistry was utilized successfully to produce an xLCE capable of realignment and remolding. Since then, this field has rapidly expanded, and several other thermally induced DCC reaction strategies with different behavior and characteristics enabled by different stimuli have been introduced to achieve complex alignment or recycling of xLCEs. Examples include exchangeable urethane bonds,94 boronic transesterification,95 Diels–Alder dynamic bonds,96 and more recently the equilibrium siloxane bond exchange.97,98 Light-induced dynamic bond exchange, such as free-radical addition–fragmentation chain transfer (AFT),87 disulfide,99 cinnamate [2 + 2] cycloaddition, coumarin [2 + 2] cycloaddition,100 and anthracene [4 + 4] cycloaddition,101 has also been utilized to align the LCE networks by exposure to UV light at room temperature.
3. Distinct Physical Properties
At low temperatures, covalently cross-linked xLCEs behave no differently from the classical permanently cross-linked networks, showing the properties that have been well documented over the last 30 years. The differences emerge when on elevated temperature the bond exchange is initiated and the elastic–plastic transition occurs. Such a temperature is often called the “vitrification point” Tv, following the original terminology introduced by Lebler (which correctly draws a parallel between this rheological transformation and the vitrification of mineral glass).
Conceptually, Tv corresponds to a temperature range, above which the covalently bonded network starts flowing plastically under stress due to the acceleration of the bond exchange within the material. Below this temperature, the exchange reaction rate is negligible within the time scale of most experiments, resulting in a “fixed” network structure. The concept of Tv is vague, both experimentally and theoretically, as this is not a “transition” of any kind but an entirely kinetic phenomenon based on the sharp exponential variation of the bond-exchange rate; it is a property that is hard to quantify, as this elastic–plastic transition is affected by the rates of heating, the stress applied, and also the total time allowed for experiment.
The most common and illustrative tests of this elastic–plastic transition are carried out at constant stress applied to the material, while temperature is increased (iso-stress transition), or at constant high temperature and stress, while monitoring the gradual plastic flow (creep test). Although there are a few theoretical studies of transient networks under stress, they are mostly applicable to thermoplastic systems, and there is still no clear understanding of many aspects of this elastic–plastic transition driven by dynamic exchange of covalent bonds. For instance, the classical iso-stress transition in a network with transesterification bond exchange shows that the onset of fast flow (the divergence in the plots) depends on the applied stress (Figure 3a), while in a network with equilibrium siloxane bond exchange the same test shows the same temperature of rapid flow, regardless of stress (Figure 3b). Note that both of these illustrations only show the elastic–plastic transition, always above the liquid crystal–isotropic transition point Ti. Naturally, if the bond-exchange reaction requires a catalyst (as most of them do, see further in this Review), the concentration of catalyst plays a role not dissimilar to temperature (Figure 3c): the catalyst lowers the activation energy of the reaction, while the temperature overcomes this barrier in a thermally activated fashion, rate ∼ exp[−ΔE/kBT].
Figure 3.

An illustration of the elastic–plastic transition. (a) The iso-stress heating curves for a dynamic network controlled by transesterification. (b) The iso-stress heating curves for a dynamic network controlled by equilibrium siloxane exchange. (c) The role of the catalyst (in this illustration: TBD) in the iso-stress heating elastic–plastic transition.
The other aspect of the stress-induced plastic deformation of dynamic covalent networks is the creep at constant stress and temperature. Strictly, a small degree of bond exchange, and the resulting creep, would occur at all temperatures, even quite low, as long as the network remains rubbery (above its glass transition). However, if the activation barrier for the bond exchange is sufficiently high, such creep is effectively not seen at reasonable times and low temperatures. In the sections below, we will often quote the activation energy determined for various types of covalent bond exchange, but the possible range is very wide: as far as we are aware, the activation energy ΔE could be as low as 5–10 kJ/mol (compare this with a thermal energy at, e.g., 100 °C equal to 3.1 kJ/mol) and as high as 160–180 kJ/mol. Depending on the activation energy of the bond exchange (in turn, determined by the type of exchange and the catalyst), temperature, and the applied stress—the creep flow could take different forms: it is misleading to represent it as a regular viscous flow, in which case the flow would proceed at a constant rate. There are examples when a constant rate of creep is observed but is accidental. The examples illustrated in Figure 4 show two very different systems, both with highly nonlinear creep: one the concave curve and the other the accelerating convex curve.
Figure 4.
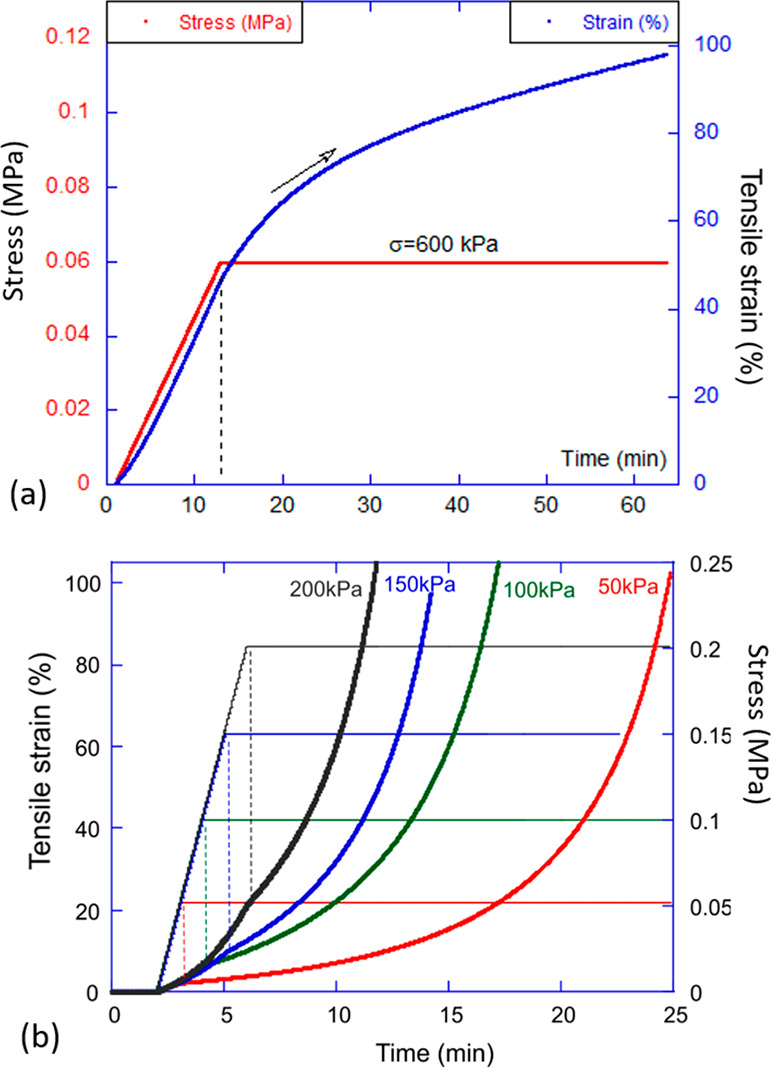
An illustration of plastic creep. (a) The plastic flow (creep) test: at a constant (high) temperature, the tensile stress is ramped and then kept constant. The strain continues to increase at a variable rate depending on the system. (b) An illustration of the rate of nonlinear plastic creep in the xLCE network depending on the level of constant stress kept. Such a test informs about the conditions and the time required to “program” the uniaxial alignment in xLCEs.
The creep experiment is particularly useful in xLCE systems, because it offers a reliable way to align the materials into amonodomain state, or “program” the elastomer. Figure 4b shows an example of such programming, which may take a short or long time, depending on the chosen conditions (the temperature and the stress). During such plastic creep, the network acquires a weak intrinsic anisotropy, and when the temperature is lowered—and the bond exchange stops—this anisotropy remains and acts as a “seed” for the formation of the aligned monodomain liquid crystalline order.
Many if not most of the physical properties of xLCEs are not yet fully understood. Despite a few attempts, there is no adequate theory that would combine the physics of liquid crystalline order and that of bond-exchange “vitrimers”. Equally, although many experimental findings have been reported in the past few years, there is no consistent picture yet: some facts do not agree with some others. There is a lot of basic research that is still to be done in this new field. In the remainder of this Review, we present and discuss the different chemistry approaches that have been developed in the past few years and have led to a variety of new and diverse xLCE systems.
4. Hydroxyl Transesterification
The xLCE breakthrough achieved by Ji et al. in 201486 has proven the possibility to design a LCEs with dynamic covalent bonds within its structure and obtain a monodomain alignment postpolymerization, leading to actuation. To achieve this, they make use of the separation between the characteristic temperatures of the material: the liquid crystalline transition temperature Ti (smectic-A to isotropic, in their case) and the temperature beyond which the bond-exchange reaction (illustrated in Figure 2b) is activated.
For a stable actuation to occur in xLCEs, the bond exchange needs to be absent in the temperature range around Ti, so that the material behaves as a thermoset. If this were not the case, the stress induced by actuation could produce creep: a further network rewiring that would erase or distort the properties programmed into it. Hence, a sufficient gap is necessary between the operating temperature of the material (dictated by the actuation and so Ti) and the temperature of onset of the plastic flow at which the material becomes malleable (Tv), for the two phenomena to not interfere with each other. Transesterification as a source of DCC in covalent polymer networks has been the center of many studies, due to its importance as a type of vitrimer chemistry, which showed that the Tv of such systems could be tuned through factors such as the nature and concentration of the catalyst used,102 or by changing the activation energy of the exchangeable bonds. This gives a degree of control over the gap between Ti and Tv. When Ti is much lower than Tv, one could expect all of the LCE effects, including actuation, to proceed as in a normal thermoset—yet the possibility of remolding (and thus resetting the reference state of the thermoset) at a much higher temperature opens many new opportunities. Apart from the ability to form complex shapes, the key effect was the programming of monodomain alignment achieved during the uniaxial plastic flow above Tv. It has turned out that, when the elongation plastic flow (creep) was induced by external stress at a temperature not much higher than Tv, the plastic flow is quite slow and the induced chain anisotropy high (even though the material is in the isotropic state, well above its Ti). When rapidly cooling below Tv, the network retains this anisotropy, and the liquid crystal order emerges in the well-aligned monodomain fashion. In this way, one could make the “usual” thermal actuators (see Figure 2c) but then completely remold and reprogram the material into a different shape or alignment. Working with a biphenyl epoxy-functionalized mesogen and sebacic acid as the spacer under TBD catalysis, Ji et al. were able to program LCE samples at 180 °C in under a minute. Different sections of a complex shape could be selectively and differentially aligned, leading to structures capable of folding themselves from a flat sheet into complex shapes reversibly.
The same group investigated ways to make the actuator network more stable thermally and presented a catalyst-free version of the material.103 It was possible to trigger sample reprogramming through the sole effect of high temperatures over long periods of time. This increases the stability of the material with regard to actuation but on the other hand decreases the practicality of the programming (as very high temperatures and long times are required).
Another important discovery made by the same group investigating these first epoxy–acid xLCE networks was the ability to achieve the plastic flow and thus reprogramming of xLCEs at a much lower (even ambient) temperature by the use of a solvent swelling the network.91 In that case, the nominal vitrification point was Tv = 105 °C, since the energy barrier for transesterification was measured to be ΔG ≈ 80 kJ/mol with 5 mol % of TBD. The solvent-induced shape programming was found not only to avoid higher temperatures but also to afford simple implementation and versatility: effects are achieved just by selectively coating solvent onto the vitrimer surface. A path to accelerating a thermally activated reaction without changing the temperature is by effectively reducing the energy barrier. If the material is swollen (Figure 5), the additional stretching of network chains provides an extra tension on the cross-linking. In the classical fashion of mechanochemistry, the associated mechanical work is calculated from the chain tension and chain extension (both easily estimated in the physics of swelling gels), giving ΔW ≈ 17 kJ/mol for the equilibrium swelling ratio of 1.75. This shift in the activation energy (ΔG–ΔW) is enough to bring the vitrification temperature Tv from 105 °C down to 25 °C, and thus explains the observed elastic–plastic transition, which allows reprogramming in the swollen network at ambient temperatures.
Figure 5.
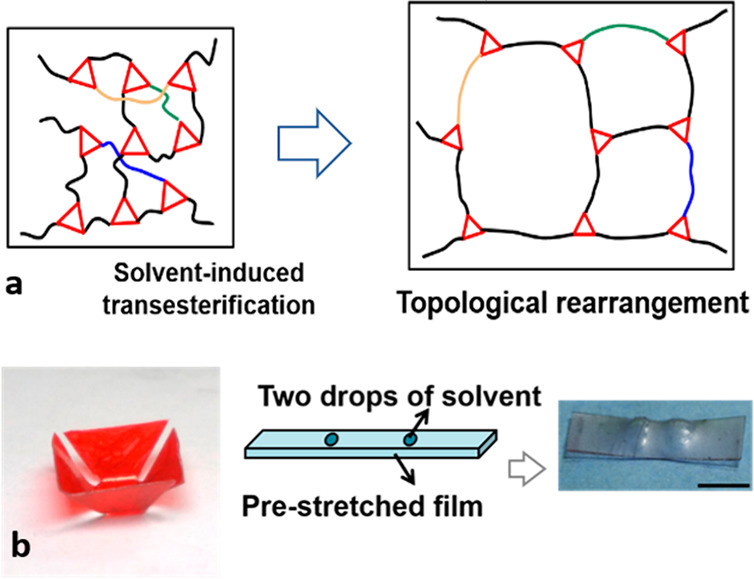
(a) Schematic of topological rearrangement induced by solvent-activated transesterification, which enables shape programming, reconfiguration, welding, and healing of xLCEs. (b) Illustrations of a kirigami structure achieved by depositing a thin strip of solvent at the base of each petal and of a two-dot pattern by depositing two drops of THF onto a prestrained film. Scale bar: 5 mm. Adapted with permission from ref (91) under CCBY license, Springer Nature.
Once the concept of epoxy–acid xLCEs was established and the practical benefits of postpolymerization processing demonstrated, a number of subsequent studies presented additional important findings. In particular, one needs to mention the methods of rendering the LCE actuation sensitive to light, which is traditionally done by introducing chromophores into the network: either as a dopant (with dyes or as composites with nanoparticles) or inserting a light-sensitive unit into the LCE structure directly. The latter approach, following from a 2001 work using azobenzene,44,104 inserted an azobenzene mesogenic unit into the polymer chain, replacing the biphenyl core of the original xLCE93 (see Figure 6). This produced UV-responsive xLCEs with a particularly strong response coefficient, since the isomerizing units in the main chain have the greatest effect on the chain anisotropy. The attractive idea of linear locomotion (Figure 6b) was enabled by using a helical actuator band converting local photobending into the perpendicular motion.
Figure 6.
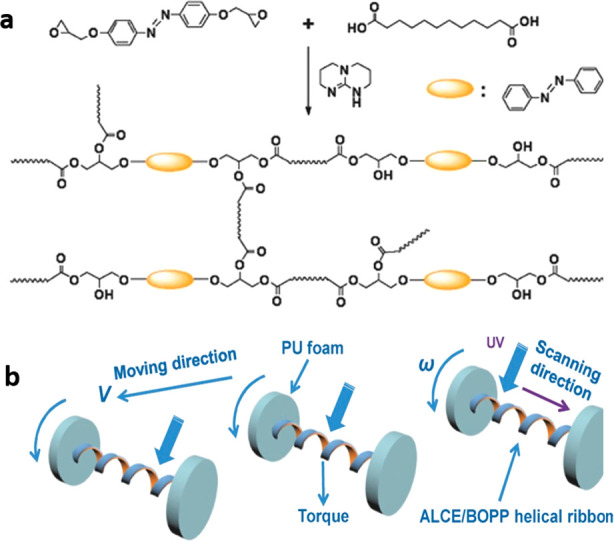
(a) Synthesis of photoisomerizing xLCE by the epoxy–acid polymerization method, in the presence of TBD, at 180 °C. (b) Illustration of a rolling engine propelled by illuminating the helical strip with UV light. Adapted with permission from ref (93). Copyright 2017 John Wiley and Sons.
One problem with the “classical” epoxy–acid polymerization is that the reaction is quite slow, even with a potent catalyst such as TBD, and takes hours even at an elevated temperature. One also can never be sure of the network topology, since the cross-linking occurs as a byproduct of transesterification (illustrated in Figure 2b) and, therefore, even the degree of cross-linking becomes a dynamic variable. Finally, there is a glass transition factor. We already described the two key temperatures “competing” in xLCEs: the vitrification point Tv and the phase transition to the isotropic phase Ti. However, if the glass transition (Tg) is in the temperature range of interest, then all actuation effects will cease below Tg. In most −C–O– based polymers, the glass transiton is relatively high, up to Tg ∼ 50 °C.86 That restricts the use of the material as an elastomer at room temperature.
For all of these reasons, the next round of material development has explored the “click” chemistry of thiols.95 Thiol chemistry has proven to be very attractive for polymer synthesis due to its ease of implementation, high conversion rates, and mild reaction conditions. Additionally, replacing some −O– units by −S– in the polymer chain increases its flexibility and significantly lowers the glass transition.105,106
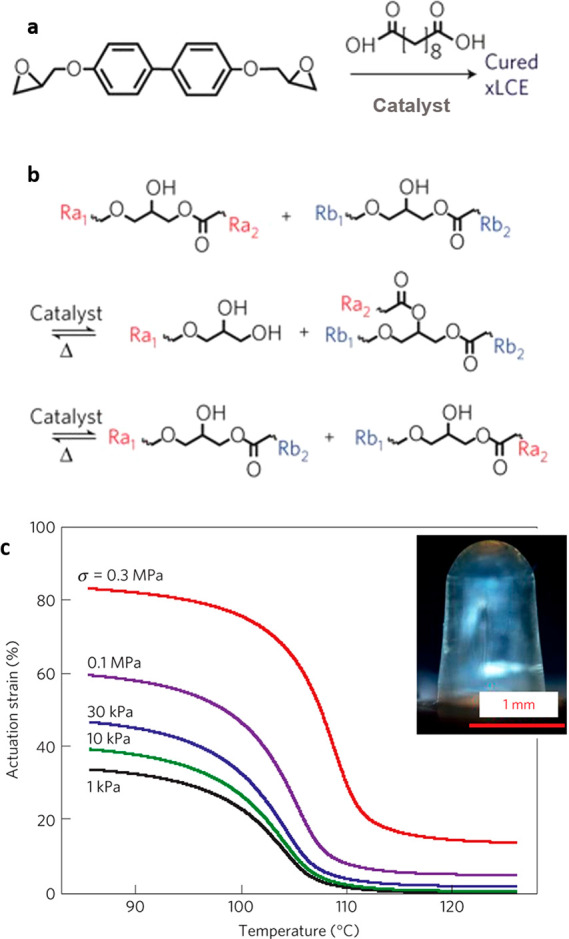
In contrast to the epoxy–acid polymerization, the cross-linking in the epoxy–thiol system is produced during polymerization by the fixed amount of 4-functional thiol monomer (see Figure 7a), as there is no competing side reaction. Therefore, the network has an overall more controlled and homogeneous structure. However, once the network is brought above the DCC activation temperature Tv, new 3-functional cross-links form through the chain branching from the hydroxyl–ester transesterification, until a final equilibrium network topology is attained after a sufficient amount of time for the reaction (with a mixture of 4-functional thiol cross-linkers, 3-functional branching points, and the matching number of dangling ends acting as network plasticizers).
Figure 7.
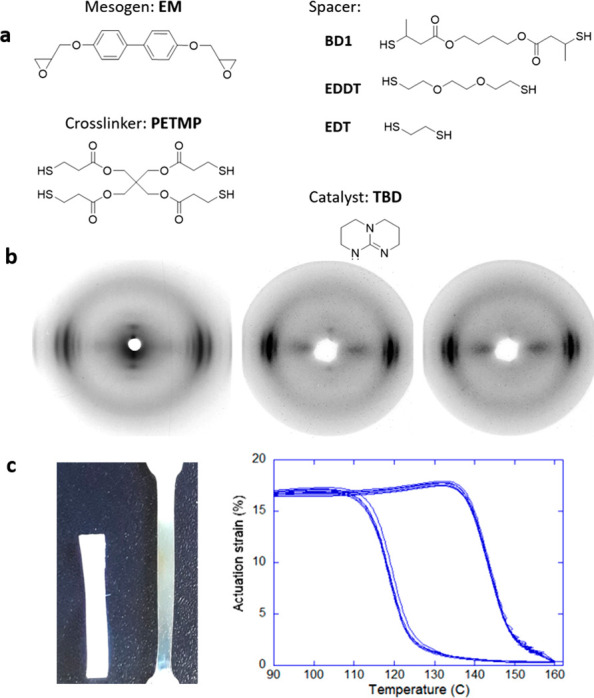
(a) Monomers used for the synthesis of the epoxy–thiol xLCE network. An epoxy mesogen was reacted in equimolar proportion with 15% of 4-functional cross-linker PETMP and 85% of 2-functional spacer, from a selection of options. (b) X-ray images showing the complex smectic phase down to the nematic phase depending on the spacer selection. (c) An illustration of an xLCE aligned by uniaxial plastic flow and its highly stable actuation cycles. Adapted from ref (142). Copyright 2020 American Chemical Society.
Using this approach, the −OH groups required for transesterification are generated in situ through the epoxy ring opening during the polymerization, like in the epoxy–acid approach. The ester groups, on the other hand, are incorporated into the system only through the structure of selected thiol monomers (spacers and/or cross-linker, Figure 7a). This greater control over the network structure allowed the understanding of factors influencing Tv in transesterification-based dynamic networks to be deepened. It was found that, on top of the factors listed previously, the concentration of reactive groups for the exchange in the network (esters and hydroxyls), as well as the overall elastic modulus of the material, had a strong influence on the final flow properties of the network once the bond exchange was activated. The value of Tv could be modulated by acting upon any of these factors. In this respect, the nature of the spacer used in conjunction with the mesogen was found to enable a great liberty over the material properties. Study of stress relaxation obtained the activation energy of bond exchange for EDDT as ΔG = 134 kJ/mol, while for the BD1 the activation energy was 92 kJ/mol. Indeed, using the ester-rich spacer BD1, Tv ∼ 165 °C was obtained, while for ester-poor EDDT/EDT spacers the plastic flow started at 195 and 205 °C, respectively. Mixing of spacers was also found to affect the nature of the liquid crystalline phase, with pure spacers invariably giving the smectic order, same as in other biphenyl epoxy-based main-chain polymers, but the nematic phase has emerged for the mixture of EDT/EDDT spacers in the network (see Figure 7b for an illustration of different LC phases by X-ray and Figure 7c for an illustration of reversible actuation).
5. Boronic Transestrification
The concept of boronic cross-coupling (well-studied in small molecules107−109) has been recently applied to polymers.110,111 Unlike most other dynamic bond systems, boronic-based exchanges do not require a catalyst. The reversible bond exchange can occur in these systems by boronic transesterification85,112 or via the exchange of boroxine bonds.113,114 Boronic transesterification is much more interesting for preparing vitrimers due to the strength and kinetic tunability of the switchable B–O bonds. The bond exchange requires chain mobility and low energy barriers, so it could only occur above the melt or glass transitions.112,115,116
Figure 8a,b illustrates a versatile synthetic route using a Michael addition thiol–acrylate reaction to yield a class of xLCEs with a fraction of exchangeable boronic ester bonds.95 The use of acrylate mesogens is especially attractive, as these are widely commercially available. A fraction of permanent cross-links is used to ensure the structural stability of the material. When the fraction of exchangeable spacers is sufficiently high (which is verified by testing that stress relaxation can progress to zero), the vitrimer networks can be molded into complex shapes, with their plasticity and actuation carefully controlled. Note that, in xLCEs with a moderate amount of boronic bond-exchange spacers, for instance, 15%, the stress relaxation rate is similar to that of the fully plastic sample with 100% exchangeable spacers, but the equilibrium stress never reaches zero; see Figure 8c. Surprisingly, the studies of stress relaxation at different temperatures have shown that the activation energy of this boronic transesterification is very low (certainly below 5 kJ/mol, that is, readily activated at room temperature), which means that the rate of this bond exchange is diffusion, rather than reaction controlled. Overall, there is a trade-off dilemma between the processability and actuation stability in boronic ester xLCE actuators (i.e., readily processable actuators can be unstable, whereas thermally stable actuators can be very difficult to process).
Figure 8.

(a) Monomers used for the boronic-exchange xLCE: diacrylate, dithiols, and the 4-functional thiol cross-linker. Isotropic and LCE networks are formulated with various boronic ester concentrations through Michael addition. (b) Boronic ester exchange reaction through transesterification. (c) Iso-strain stress relaxation (a signature of vitrimer exchange) depends on the fraction of bond-exchange spacers in the network (labeled on the plot): at lower concentrations of BDB, there is an increasing fraction of permanent covalent network in the material. Adapted with permission from ref (95). Copyright 2020 John Wiley and Sons.
As with all xLCE materials, the preferred route to their programming (e.g., creating a permanently monodomain uniaxially aligned elastomer) is via the stress-induced plastic flow just above the vitrification point. Selecting the temperature and the stress such that the resulting creep is sufficiently slow to easily monitor and control, we need to allow the creep to reach the steady-flow regime and then cool the network well below Tv before removing the stress. In this way, the induced network anisotropy is preserved and results in the liquid crystalline ordering emerging below Ti being a monodomain aligned state.
This study also demonstrated the manufacturing and assembly capability of xLCEs with fast bond-exchange reaction through remolding and welding of partial vitrimer material. By welding differently aligned parts (or welding LCEs with an isotropic vitrimer), a variety of complex shape-morphing actuators were obtained in that study.
6. Polyurethane Transcarbamoylation
Polyurethanes are an important class of polymeric materials, widely used in applications ranging from damping materials to coatings.117 Typically, polyurethanes are produced by cross-linking isocyanates and alcohols into thermoset networks. Naturally, thermoset polyurethanes are hard to process postpolymerization. Recently, several studies have shown that cross-linked polyurethanes can be converted into DCC networks through the transcarbamoylation reaction.118 Bowman and co-workers have developed an LCE with exchangeable carbamate (urethane) bonds, starting from mesogenic oligomers obtained using a thiol–Michael addition reaction (Figure 9a).94 The final polyurethane LCE network was obtained using three reaction steps, where the exchangeable urethane bonds were locally placed within the network. The bond exchange (see Figure 9b) was induced by the use of 1 wt % catalyst (DBTDL) and occurred rapidly at 160 °C. The overall programming procedure to prepare monodomain samples was done via stretching at 160 °C for 60 min. An unusual feature of this network is the presence of two separate LC phase transitions (isotropic–nematic and nematic–smectic), which produces a two-stage freestanding triple shape memory effect in a uniformly aligned sample (Figure 9c).
Figure 9.
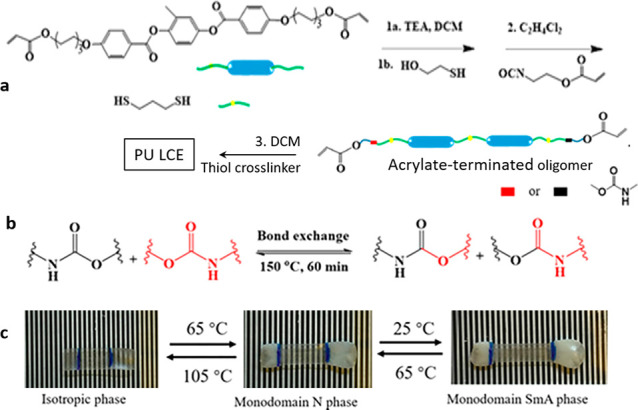
(a) Chemical structures of mesogenic and isotropic monomers. (b) Mechanism and illustration of the urethane bond-exchange reaction via transcarbamoylation. (c) Multireversible shape change, where a load-free strain was actuated by isotropic–nematic (shape 1 to shape 2) and nematic–smectic phase transitions (shape 2 to shape 3) upon thermal cycling. Adapted with permission from ref (94). Copyright 2018 American Chemical Society.
7. Siloxane Exchange
Silicone-based elastomers (partially replacing carbon with silicone, which results in a significant lowering of the glass transition) are another important class of polymers, widely used in low-temperature environments, often utilized as sealants, and in microfluidic fabrication due to their extreme hydrophobic nature and ideal mechanical properties.119 The original work of Finkelmann used siloxane-based elastomers as a key material strategy due to their incredible properties (high failure strain, low glass transitions, and low modulus), attributed to the highly flexible Si–O–Si linkages within the polymer backbone. In principle, all of the methods used to add network plasticity to hydrocarbon-based elastomers can be incorporated in silicone-based elastomers as well. For example, vinylogous urethane exchange,120 transesterification,121 boroxine bonds,114 or Meldrum’s acid-derived bonds122 were used with cross-linked polydimethylsiloxane (PDMS) systems. However, there are certain types of dynamic bond exchanges unique to silicone-based systems, such as the bond exchange in the siloxane adaptable networks74,123,124 and the silyl ether metathesis.125
Recently, a new class of xLCEs using robust click chemistry (“double-click” of thiol–acrylate and thiol–ene) was introduced (Figure 10a).97,126 This material design has several important advantages over the earlier generations of LCEs, which also use commercial off-the-shelf starting monomers. First, the presence of thiols and siloxanes makes the glass transition naturally low. Second, it allows for a good control of the nematic transition, including bringing the Ti down to the “human range” of 30–40 °C that allows control of actuation by body heat. Third, the siloxane bond-exchange reaction (Figure 10b) imparts the bond exchange into the network, in a manner similar to vitrimers: the plastic flow under stress at a high temperature allows both the programming of monodomain textures in the xLCEs and the (re)molding of plastic samples into desired structures.
Figure 10.

(a) The summary of “double-click” chemistry: the mesogenic diacrylate (RM82) first reacts with a dithiol (EDDT), which is in excess. Following that, the thiol-terminated oligomer chains are photopolymerized with the vinyl bonds of the ring-siloxane, leading to the permanent network with 4-functional cross-links. (b) The general scheme of siloxane exchange enabled by an acid or base catalysis. (c) The programmed shape of a helix reversibly unwinds into a flat strip in the isotropic phase. (d) A thermally molded continuous strip combining three different xLCE materials with increasing Ti. Adapted with permission from ref (97) under CCBY license, Springer Nature.
Figure 10d illustrates the result of remolding using this chemistry, in this case the welding of three xLCE strips with different nematic–isotropic transitions into one, where one cannot distinguish the initial overlap regions. In this photo, at room temperature, all three sections are in the polydomain nematic state, and thus white (strongly scattering light). Then, on heating this strip, we see the sequential phase transitions into the isotropic phase that take place in different sections of the otherwise continuous polymer strip: first one section becomes isotropic (transparent, no longer scattering light), then the middle section, until finally the whole strip becomes isotropic.
It is important to note that, like for the transesterification reaction, the rate of the siloxane bond exchange can easily be tuned by the catalysts. The study has shown that the nature and the amount of catalyst have a profound effect on the elastic–plastic transition temperature, the stress relaxation, and the monodomain programming temperature of the LCE networks.126 For example, the activation energy of the exchange varied from quite a low value of ΔG ∼ 83 kJ/mol for 1 wt % TBD, all the way to the high 164 kJ/mol for 1% of nucleophilic catalyst triphenylphosphine (PPh3).
The same bond-exchange reaction strategy in xLCEs has been presented by Ji and co-workers.98 These authors incorporated an anionic catalyst (e.g., bis(tetramethylammonium) oligodimethylsiloxanediolate, TMA–DMSiO) in a “classical” main-chain LCE, where both the cross-links and the flexible spacers between mesogens were siloxane; see Figure 11a. The catalyst enabled this xLCE network to change its topology at elevated temperature by siloxane exchange, as illustrated in Figure 10b, which allowed the xLCE to be processed post-cross-linking. The obtained network had a smectic LC phase due to the microphase separation of siloxane spacers and mesogens but with the same capability to be remolded and reprogrammed (Figure 11b,c). The paper demonstrated the possibility to manufacture 3D actuators with highly complex shapes, such as cones and flowers, despite the material’s “classical” chemistry.
Figure 11.
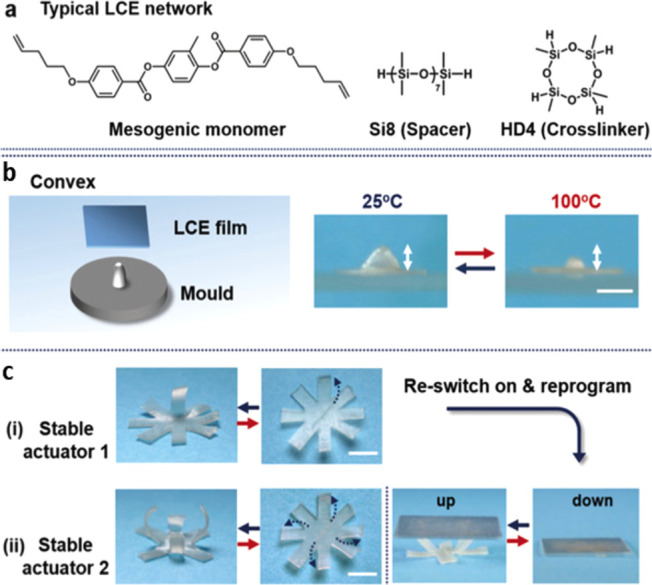
(a) Chemical structure of the monomers used to synthesize siloxane xLCEs by Ji et al. (b) Remolding a flat xLCE sheet into a 3D convex actuator. Scale bar: 2 mm. (c) Demonstration of reprogramming of the one-arm bending motion (i) into four-arm bending motion (ii) for lifting objects. Adapted with permission from ref (98). Copyright 2020 John Wiley and Sons.
It should be noted that, in the presented studies of siloxane-based xLCEs, the authors used old and well-known chemistry and avoided sophisticated synthesis of new materials. They just added a catalyst and achieved a dynamically exchangeable network. This approach invites a much broader application base and can be directly applicable to ordinary silicone elastomers (like PDMS), which have been broadly explored over 30 years and have many advantageous characteristics. Making the siloxane bonds dynamic offers an alternative way to process these materials compared to the current one-use thermosets. It also opens a whole library of siloxane-based LCE materials, that have been studied for decades, to DCC. This promises exciting prospects and new applications from these well-known LCEs due to a new level of processability.
In addition to showing that standard siloxane LCEs could become dynamic exchangeable networks merely with the addition of the right catalyst, the studies demonstrated a possibility to selectively render the networks dynamic only when and where desired. This was done through the choice of a specific catalyst: TMA–DMSiO decomposes into volatile gases at high temperatures. When added into the network through solvent swelling (and removal), the network becomes homogeneously dynamic and can be reprogrammed at 100 °C. However, if the temperature is raised to 150 °C, the catalyst can be removed from the sample through its thermal decomposition, and the network reverted back to a standard thermoset. If possible to have only the local heating, then such a transformation could be achieved selectively in certain regions of the sample. Hence, no catalyst is present in the LCE during its use, removing any risk of creep or triggering inadvertently the bond exchange during actuation, which is a non-negligible concern when the Ti and Tv are so close. To reactivate the dynamic bond exchange, it is enough to reload the catalyst into the network through the same procedure. This method is innovative and guarantees network stability in the materials at all times, which is a concern that cannot be ignored for thermally or photoactivated DCC networks; however, reprogramming requires a number of additional steps and chemicals through the need to load in the catalyst, which could be limiting for large scale applications.
8. Diels–Alder Dynamic Networks
Zhao and co-workers have reported a new material system for xLCE actuators, using Diels–Alder dynamic bond exchange for actuator programming and reprocessing (Figure 12).96 The thermally reversible nature of the Diels–Alder (DA) reaction has long been utilized for designing recyclable thermosets.67 DA reactions have been extensively studied for various dynamic material applications because of the wide window of processing temperatures it requires. Several DA additions form stable adducts from room temperature to 60 °C, whereas the retro-DA reaction becomes significant at temperatures above 110 °C. During the retro-DA reaction, the polymer can be processed.127,128 The authors have shown that the xLCE systems based on DA exchange can be reprogrammed at 125 °C, which is more than 30 °C higher than the isotropic transition temperature in their case. This enables stable actuation without the network starting to creep due to it becoming dynamic.
Figure 12.
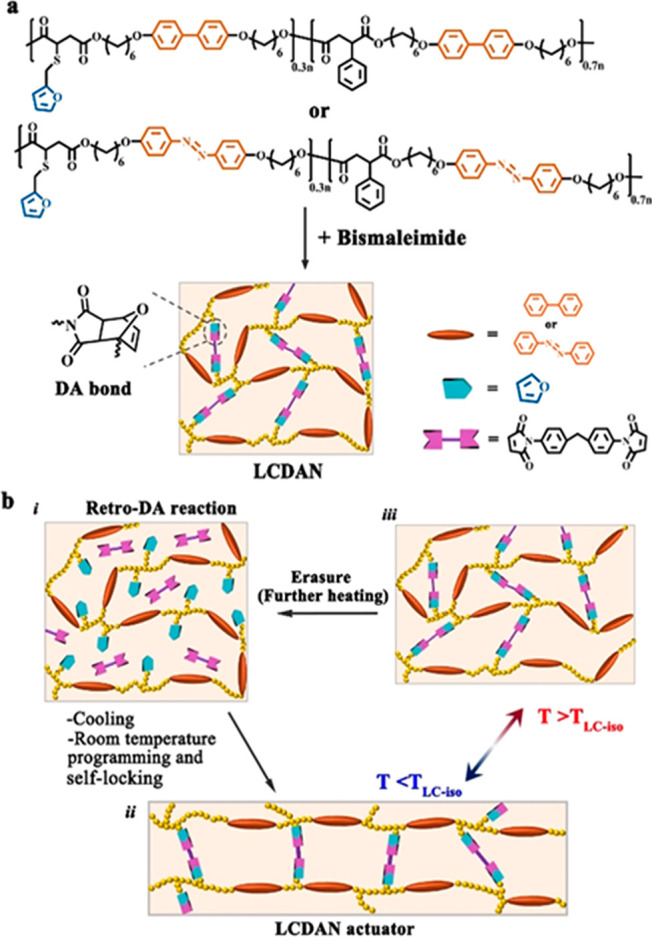
(a) Chemical structures of two main-chain liquid crystalline polymers bearing furan side groups and containing either biphenyl or azobenzene as mesogenic moieties, as well as the preparation of their cross-linked networks through DA-bonded cross-links. (b) A schematic of programming and self-locking of xLCE actuators utilizing the DA exchange reaction. Adapted with permission from ref (96). Copyright 2020 John Wiley and Sons.
The retro-DA reaction is a dissociative exchange process. Using the intrinsic dissymmetry in the rates of DA association/dissociation depending on the temperature, a very interesting possibility for sample processing was presented by Zhao and co-workers. The retro-DA is triggered once the sample is brought to a high temperature (125 °C) to activate bond dynamicity; the sample can then be quickly cooled down to room temperature, to yield a network with partially dissociated cross-links. At room temperature, DA bond reforming between the furan and maleimide groups is a slow process, giving plenty of time for sample processing and molding. After multiple hours, all bonds have formed again, locking in the order and shape imparted to the sample. This locked-in network structure can be erased by heating the sample to 125 °C again. Room-temperature sample manipulation is ideal, as it removes a number of experimental restrictions associated with having a high-temperature environment. Additionally, using a thermal trigger rather than photoactivation of bond exchange guarantees a homogeneous activation throughout the sample, which can be a limiting factor when light is used on thicker samples. In that respect, this new dynamic chemistry applied to xLCEs is exciting, as this makes it unique compared to previous chemistries discussed and opens a novel approach to material processing. A similar process was used to process xLCEs containing disulfide groups by Cai and co-workers (see below).
As with most xLCEs we are presenting here, the actuation can be induced via either heat or light (if a photochromic dye is added or an azobenzene mesogen is included).
9. Disulfide Exchange
Disulfide metathesis is a unique type of DCC where the dynamic bond exchange can occur either under UV light irradiation or upon heating without any catalyst or initiators.84,129,130 In 2017, Kessler and co-workers have demonstrated xLCEs with exchangeable disulfide bonds, synthesized by polymerizing a biphenyl-based epoxy mesogen with an aliphatic dicarboxylic acid that contains disulfide bonds (Figure 13a).99,131 The authors have shown that the reprocessability and recyclability can be controlled through adjusting the molar ration of disulfide-containing monomer with respect to a regular flexible spacer (sebacic acid, the same spacer as in the first xLCE system).85 The biphenyl epoxy mesogen inherently produces a smectic phase, and these samples were programmed for actuation at 160 °C and recycled at 200 °C (Figure 13b,c).
Figure 13.
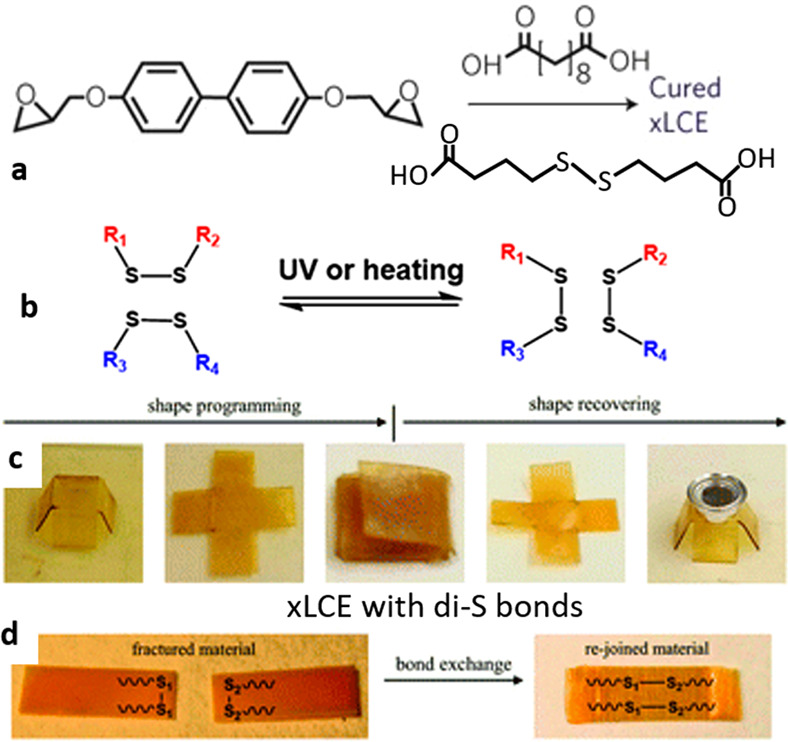
(a) The epoxy monomer and the cebacic acid are the same as those in the original work introducing xLCEs by Ji et al.,86 but a new function is added by incorporating disulfide bonds in a fraction of spacers. (b) Illustration of the disulfide exchange. (c) Demonstration of triple shape memory in the xLCE. (d) Demonstration of welding of two disulfide-containing xLCEs. Adapted with permission from ref (99) under CCBY license, Royal Society of Chemistry.
Almost concurrently to this report, a study by Cai and co-workers has shown similar results, using a thiol–acylate network containing disulfide bonds.132 Instead of following the earlier epoxy–acid route, they used thiol–acrylate click chemistry. In the first stage, using an excess of thiol spacers (as in Figure 8a), thiol-terminated mesogenic oligomers are obtained, which are then reacted with the tetra-functional thiol cross-linker in the presence of peroxide and sodium iodine. This generates disulfide bonds in a different way from the work of Kessler, but the resulting xLCE has the same properties. In the paper, the authors presented an xLCE with a lower glass transition (−5 °C) and a lower liquid crystalline transition temperature compared to epoxy-based xLCEs (56 °C). The xLCE samples could be reprogrammed to the monodomain aligned state either under UV illumination or at high temperature (180 °C) in the absence of any catalyst. Apart from producing xLCEs that are reprocessable and self-healing at high temperature, the use of disulfide bonds has several additional important advantages. First, the disulfide xLCE can be reprogrammed locally, with good spatial resolution, at ambient temperature by localized UV irradiation, which selectively activates the disulfide exchange.84 Second, complex microstructures can be easily fabricated on the surface of the disulfide LCEs through imprint lithography to impart some novel functions. Third, this reaction strategy produces xLCEs with a more desirable nematic phase, which is difficult to produce using epoxy-based disulfide LCEs due to the commonly used biphenyl core that favors smectic ordering. Fourth, unlike most other dynamic covalent bonds, the disulfide metathesis can proceed without a catalyst, which gives these materials an excellent long-term functionality, without degradation of material properties often associated with catalysts. This is not the only example: other xLCEs with exchangeable bonds may not need a catalyst to facilitate the reaction, e.g., with boronic transesterification or DA exchange. However, they often produce xLCEs that are not thermally stable and prone to unwanted plastic creep, whereas the disulfide xLCE has been shown to have highly reproducible results.
10. Cycloaddition [4 + 4] of Anthracene
The reversible photoinitiated and thermally initiated [4 + 4] cycloadditions of anthracene groups have been known since 1867.133,134 Recently, this dynamic covalent bond approach has been applied to reversibly cross-link or de-cross-link polymers using light or heat.134 The reversible photodimerization (for cross-linking) and photocleavage (for de-cross-linking) of anthracene groups occur under UV light at two wavelengths (365 nm for cross-linking and 254 nm for de-cross-linking) (Figure 12). Other studies have reported that an elevated temperature between 110 and 180 °C could also be used to achieve cross-linking or de-cross-linking.
A study by Zhao et al. has utilized this reaction strategy to produce 3D LCE actuator structures, where the cross-linking and de-cross-linking was achieved via UV light.101 The use of optical cross-linking allowed for fine spatial control of the regions where the cross-linking density was changed in the anthracene-containing LCE, which gives rise to unusual actuation patterns. The light can be used to reconfigure some parts of the LCE network while preserving the alignment in the rest of the material. For example, a flat LCE film can be aligned and spatially programmed at room temperature to achieve multiple actuation modes, such as simple contraction and expansion (Figure 14a), flat to “roll up–roll down” shape (Figure 14b), flat to “roll up–bend down” (Figure 14c), flat to “bend up–bend down” (“S”-like) shape (Figure 14d), “bend up–wrinkle” (Figure 14e), and flat to all-wrinkle (Figure 14f). The spatial control was achieved using a photomask and irradiating with 254 nm UV light for 2 h.
Figure 14.
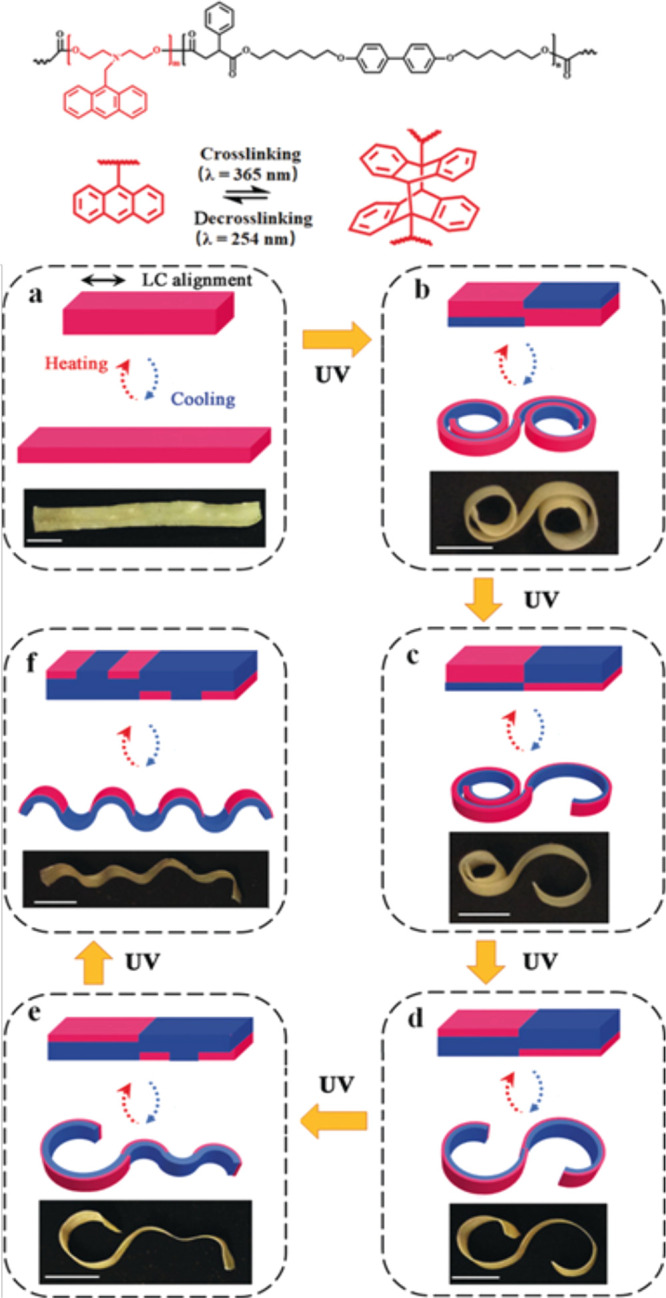
Chemical structure of the anthracene-containing xLCE and the reversible thermal or photodimerization. Demonstration of the optical reconfiguration process on a single xLCE film. The reversible switch between flat (in isotropic phase) and various shapes (in LC phase) upon photo-de-cross-linking in selected areas (blue) in succession: from (a) elongation, to (b) “roll up–roll down”, to (c) “roll up–bend down”, to (d) “bend up–bend down” (S shape), to (e) “bend up–wrinkle”, and to (f) all-wrinkle. The magenta regions represent cross-linked actuating domains, and the blue regions represent de-cross-linked non-actuating domains. Adapted with permission from ref (101). Copyright 2019 John Wiley and Sons.
11. Cycloadditions [2 + 2]
Cinnamyl groups are also photosensitive chemical species and have also been utilized to synthesize photoresponsive shape memory polymers.135,136 The double bond in the cinnamyl group, when subject to UV light irradiation of wavelength above 280 nm (e.g., 365 nm), can form a four-membered ring, whereas the newly formed bonds of the four-membered ring can be cleaved back to its original double bond state by the irradiation with a harder UV light with a wavelength below 280 nm (e.g., 254 nm).82,136
This reaction strategy was applied successfully to reversibly photo-cross-link LC networks (Figure 15a).100,137 Zhao and co-workers have demonstrated simple planar alignment for uniaxial actuation via mechanical stretching of the liquid crystal polymer and subsequent photo-cross-linking using a 350 nm UV light. They achieved a 33% strain actuation by thermally cycling their xLCE between the Tg (22 °C) and Ti (60 °C). They have also reported complex shape programming that enabled 3D shapes to reversibly switch on actuation.
Figure 15.
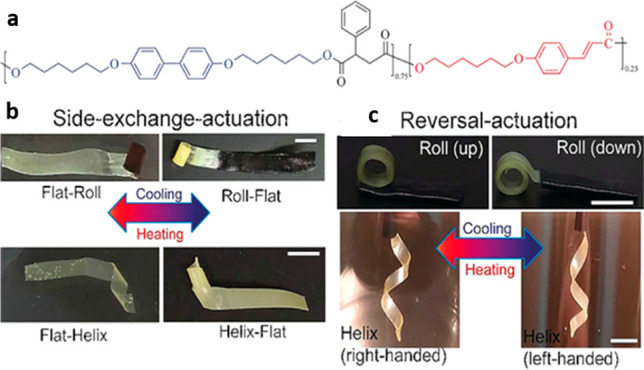
(a) Chemical structure of the liquid crystal polymer before photo-cross-linking. (b and c) Photographs showing 3D shape change starting from flat LCE films: side-exchange actuation (the two sides exchange their respective shape) (b) and reversal actuation (the actuator reversibly switches between two opposite configurations) (c). Scale bars: 3 mm. Adapted with permission from ref (100). Copyright 2017 John Wiley and Sons.
xLCEs cross-linked by cinnamyl groups have similar thermomechanical characteristics to the dynamic LCE network cross-linked by the anthracene group (that is, comparable glass and isotropic transition temperatures), due to their identical polymer backbone and the similar nature of cross-linking. Therefore, a similar programming technique can be applied to achieve complex 3D actuation shapes, such as special spatial patterning of the cross-linking. Examples of this are side-exchange actuation, reversal actuation, and hyperbolic paraboloid shapes (Figure 15b,c). Remarkably, all of these shape changes can be obtained from a single piece of the polymer. It is important to note that many other light-induced DCC reactions exist, and similar strategies can be used to effect spatial pattering of alignment and cross-linking density. This concept leads to a nonuniform inscription of actuation domains in xLCEs, which simplifies the fabrication of actuators capable of complex and precise shape change, which is an important step toward applications.
12. Addition–Fragmentation Chain Transfer
In 2005, Bowman and co-workers introduced the radical-mediated addition–fragmentation chain transfer reaction (AFT) to effect plasticity in a thiol–ene polymer network using light.79 The AFT utilized relies on allyl sulfides placed in the elastomeric backbone to induce rapid and controlled stress relaxation in the polymer network. The reaction occurs when an allyl sulfide is reacted with a thiol fragment, stimulated by light in the presence of a photoinitiator. The reaction can be repeated multiple times until total consumption of the photoinitiator in the polymer network is achieved.
The same reaction principle was introduced in an allyl-sulfide-containing LCE network.87,138 Light initiation allowed for altering the local alignment, simultaneously enabling spatial patterning. Light-facilitated bond exchange is achieved here by incorporating AFT-capable functionalities into the backbone of acrylate-terminated LC oligomers (Figure 16a). The allyl sulfide AFT exchange mechanism is illustrated in Figure 16b. Extending this concept beyond simple mechanical stretching, complex folding shapes, such as the Miura fold pattern, were programmed, as shown in Figure 16c. In comparison to other photoinduced DCC, allyl sulfide AFT produces LCEs with desirable physical properties, that is, well-controlled and uniform network topology formed by click chemistry, i.e., robust and reliable. Also, this material gives the nematic phase and a low glass transition, which are both preferable in designing xLCE networks. However, photoresponsive DCC reactions are difficult to recycle compared to more traditional thermally activated bond-exchange elastomers. Indeed, reactions relying on a radical mechanism, such as this one, require the presence of an initiator (photo or thermal). Once the initiator present in the network has been fully consumed, the exchange reaction can no longer be triggered, which in turn limits the number of reprogramming attempts possible for a given material. The method seen for the siloxane network in which the catalyst is reloaded into the network through swelling could circumvent this limitation, but this once again requires time and can be limiting in terms of applications accessible.
Figure 16.
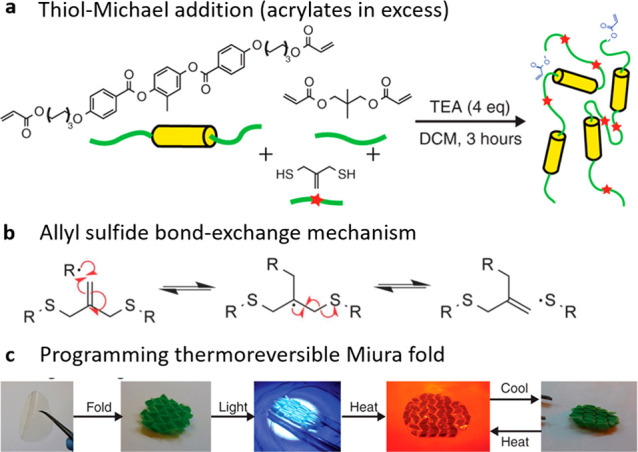
(a) Thiol–Michael addition reaction scheme to install RAFT functional groups into photopolymerizable acrylic oligomers. (b) Schematic of radical-mediated allyl sulfide bond-exchange mechanism through light-facilitated cleavage of a photoinitiator. (c) Example of the programming process starting from a polydomain 250 μm-thick film. The polymer was folded by hand and programmed with 320–500 nm (100 mW/cm2) coupled with gentle heating (30–40 °C). Subsequent thermal cycling resulted in the network unfolding at high temperatures and refolding on cooling. Adapted with permission from ref (87). Copyright 2018 AAAS.
Remarkably, Davidson et al. turned this limitation to their advantage. In recent work, they utilized allyl sulfide AFT to create 3D printable and reconfigurable LCEs that reversibly shape-morph when heated above and below their isotropic temperature, whose actuated shape can be locked-in on demand at any desirable temperature upon UV exposure.139 For the actuation, the molecular alignment was obtained through the shear extrusion force associated with the 3D printing process. Through the UV exposure, the allyl sulfide exchange reaction was triggered, resulting in a remodeling of the network to reach a new equilibrium state based on the particular shape the material is in at the given temperature. The use of light-controlled network reconfiguration allows postprocessing reprogramming of the actuating material without an imposed mechanical field. Using this integrated method, they were able to construct 3D LCEs in both monolithic and heterogeneous layouts that exhibited complex shape changes and whose transformed shapes could be locked-in on demand (Figure 17). The locked-in network structure is stable, as the exchange can no longer be triggered.
Figure 17.
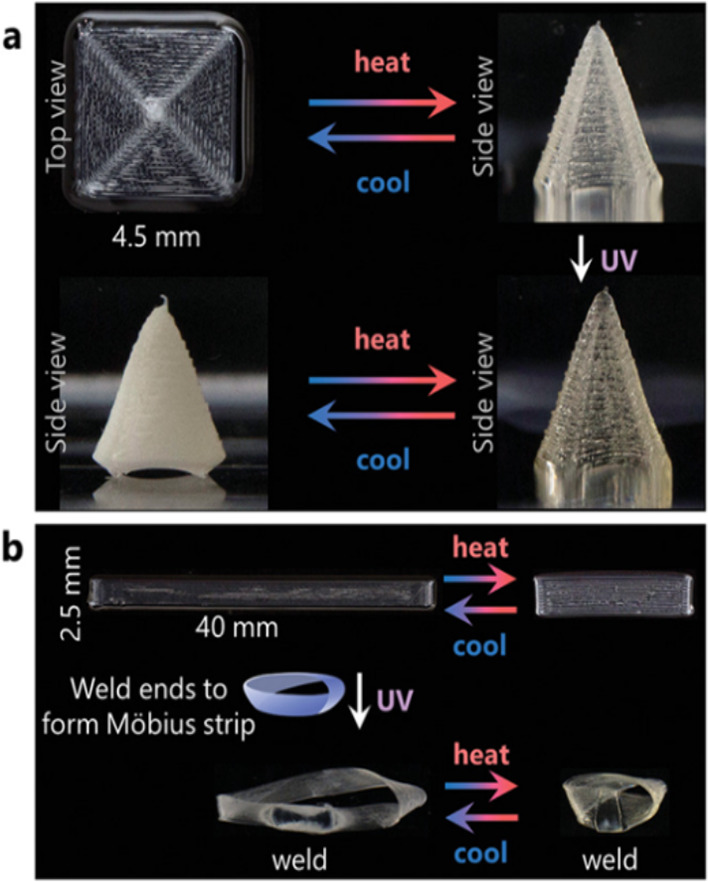
Reconfigurable xLCEs exhibit complex shapes. (a) Printed concentric squares reversibly actuate into a square cone until exposed to UV light, resulting in a locked-in, opaque cone under ambient conditions. (b) A printed strip exhibits reversible linear actuation until the ends are welded together at 60 °C via dynamic bond exchange to form an actuating Möbius strip. Adapted with permission from ref (139). Copyright 2020 John Wiley and Sons.
13. Conclusions and Outlook
The field of liquid crystal elastomers and their applications has been exploding in the past few years, driven by the robust and accessible chemistries leading to a set of standard “benchmark” materials, and inviting real application development. The latter development is happening in several directions but very noticeably in the 3D printing using the direct writing of LC “ink”. After permanent cross-linking (often induced by UV, after extrusion), the LCE structures become fully reversible shape-morphing 3D objects, where the actuation is controlled by the local alignment.
On this background, dynamic xLCE networks represent the next turn of this development—a radical departure from the idea of permanent networks, leading to a different approach to precision shaping and local programming of alignment, and as a result offering a different spectrum of applications. The inherent ability to be remolded and recycled is the key appeal of xLCEs. This Review attempted to present the main directions of materials exploration, where a variety of exchangeable chemistries have been introduced with the bond exchange induced/triggered by a range of stimuli (temperature, light, solvent). Table 1 below gives a summary of these materials and methods. However, we feel the standard “benchmark” in the xLCE field is not yet set. The field of xLCEs continues to grow, and new chemistries are being introduced all the time.
Table 1. Examples of DCC Reactions That Have Been Used to Produce xLCEsa.
| DCC reactions (reference) | activation stimulus | activation temperature (°C) | LC phase temperature (°C) | Tg (°C) | actuation (%) |
|---|---|---|---|---|---|
| transesterification from epoxy–acid86,93,140 | thermal or solvent | 150–180 | SmC-100-I | 55 | 30:80 |
| transesterification from thiol–acrylate92 | thermal or solvent | 80 | N-50-I | 18 | 10:30 |
| transesterification from hydrosilylation121 | thermal | 120 | N-150-I | 4 | |
| transesterification of epoxy–thiol141,142 | thermal | 170–210 | SmC/N-40:140-I | 4–14 | 7:25 |
| boronic transesterification95 | thermal | 40 | N-90-I | –5 | 90 |
| transcarbamoylation of urethane94 | thermal | 150 | SmA-42-N-80-I | –8 | 100 |
| siloxane exchange from thiol–ene97 | thermal | 250 | N-75:32-I | –35 | 60 |
| siloxane exchange hydrosilylation98 | thermal | 125 | SmC-72-I | –5 | 55 |
| Diels–Alder dynamic networks96 | thermal | 125 | SmA-88-I | 25 | 48 |
| disulfide exchange from epoxy acid99 | thermal, photo, or solvent | 160 or 22 | SmC-100:135-I | 10–48 | 40 |
| disulfide exchange from thiol acrylate132 | thermal or photo | 180 or 22 | N-56-I | –5 | 38 |
| reversible [4 + 4] cycloaddition of anthracene101 | thermal or photo | 200 or 22 | N-63-I | 22 | |
| reversible [2 + 2] cycloaddition of cinnamon100,137 | photo | 22 | SmC-50-N-60-I | 22 | 50 |
| radical-mediated addition–fragmentation chain transfer87 | photo | 22 | N-80-I | 0 | 60 |
The bond-exchange types, activation conditions (e.g., temperature, light), thermal transition temperature, liquid crystalline phase type, and actuation performance. The actuation is recalculated using the following formula: actuation strain (%) = (L/Liso – 1) × 100.
In the range of different chemistries explored, each have demonstrated their own unique appeal, advantages, and also limitations. When considering practical applications, these have to be factored in for the choice of dynamic chemistry for the xLCE network. The constraints of the application envisaged will determine the specifications the xLCE has to meet, from the type of stimulus for the actuation and for the activation of the bond exchange (which can be identical or orthogonal), the presence or not of a catalyst and its nature, the reprogramming conditions and methods, the value of Ti, etc. The mechanism of the exchange reaction is important when considering the overall properties desired for a material. In the case of reactions such as transesterification, an associative bond exchange occurs: the new bond forms before the old one breaks, guaranteeing a conservation of the structural integrity of the network at all times and resulting in a material that is insoluble even when the bond exchange is activated. This is excellent to guarantee a robust material under all conditions. In the case of reactions such as the disulfide exchange, the DA reaction, and other cycloadditions, the exchange occurs through a dissociative mechanism, when the bond rupture and the bond formation are two independent phenomena. Despite a bond dissociation resulting in a decrease in network connectivity (often undesirable in general polymer networks), this mechanism has proven quite appealing for xLCEs, as it enabled sample reprogramming at room temperature and can also enable solvent use to facilitate reprocessing if desired. Finally, in other reactions, such as transcarbamoylation, either mechanism is possible, and the predominant one depends on a range of factors, such as the structure of the chemical groups involved and the nature of the catalyst used in the material if any is used. These factors influence the material behavior and properties during and after network exchange.
Overall, the conditional network malleability, leading to the reprogramming, reprocessing, and recycling the materials, opens new doors for xLCE applications; a range of innovative processing methods have been demonstrated to enable this. xLCEs could be used to fabricate aligned composites that are responsive to temperature, light, electricity, or magnetic field to generate mechanical actuation. In the near future, we hope to see xLCEs utilized in 3D printing applications based on the fused filament fabrication (FFF) technique. xLCEs with thermally induced bond exchange can plastically flow under stress at high temperature, which allows them to be extruded into aligned filaments and printed into active 3D objects. FFF is a better and cheaper 3D printing technique compared to direct ink writing methods, because it does not rely on UV cross-linking (as the current used methods do).
In addition to the actuation response, the damping and adhesion applications promise better results with xLCEs, since the material has an additional energy dissipation mechanism due to the dynamic cross-linking (separate from the independently mobile nematic director, which is already making the damping of LCEs anomalous compared to standard elastomers). It is possible that the impact and vibration damping efficiency, and the strength of the dynamic adhesion associated with it, could increase in xLCEs compared to permanent thermosets. However, this depends on how significant the bond-exchange rate is at an operating temperature—and in some dynamic networks with a higher activation energy of exchange, this increase could be small. Much additional research is needed into these dynamic properties of xLCEs. Similarly, the modulation of the adhesive powers of xLCE materials through the modulation of the number of loose dangling chains generated through reversible dissociative bond cleavage is an additional direction of interest for these materials, compared to standard LCE thermosets. It would be interesting to see whether the damping efficiency is enhanced by the presence of potentially dynamic bonds within the network. Indeed, dynamicity is expected to confer to the material an additional dissipation mechanism due to the stress relief through the bond exchange. In surface modification applications, xLCEs offer reliable ways to imprint surface topography or composite structures that would reversibly alter the surface behavior.
Interestingly, the theoretical understanding, modeling, and predictions of xLCE behavior are somewhat behind, which is in contrast to the classical field of LCEs where the theory went hand in hand, or even ahead of experiments and the material development. Active fully recyclable polymers, which are doing mechanical work by themselves, is certainly a future of plastics in the 21st century.
Acknowledgments
This work was supported by the European Research Council AdG No. 786659.
Biographies
Mohand O. Saed is a senior postdoctoral researcher in the Cavendish Laboratory at Cambridge University. He earned his Ph.D. in Mechanical Engineering from University of Colorado Denver, USA, in 2017 and B.S. degree in Chemical Engineering from University of Gezira, Sudan, in 2008. His current research interests range from materials design to additive manufacturing which includes vitrimer chemistry and physics, responsive polymers, plastic compounding, adhesive materials, liquid crystalline elastomers, actuators, 3D printing, and soft robotics.
Alexandra Gablier is a Ph.D. student in the Biological and Soft Systems group in the Cavendish Laboratory. She graduated from ESPCI Paris in 2017 with a Master in Engineering, majoring in Chemistry, after which she earned an M.Phil. from the University of Cambridge working in inorganic polymers. Her research interests include liquid crystalline elastomers, vitrimers, and polymers more generally.
Eugene M. Terentjev is the Professor of Polymer Physics at the University of Cambridge, working in the Cavendish Laboratory since 1992. He obtained his M.Sc. in Physics from Moscow State University in 1981 and Ph.D. in 1985, before taking a postdoctoral position at Case Western Reserve University in 1990. He actively works on a broad range of topics, ranging from LCEs and dynamic polymer networks to molecular and cell biophysics, using both theoretical and experimental approaches.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
References
- Küpfer J.; Finkelmann H. Nematic Liquid Single Crystal Elastomers. Makromol. Chem., Rapid Commun. 1991, 12 (12), 717–726. 10.1002/marc.1991.030121211. [DOI] [Google Scholar]
- Tajbakhsh A. R.; Terentjev E. M. Spontaneous Thermal Expansion of Nematic Elastomers. Eur. Phys. J. E: Soft Matter Biol. Phys. 2001, 6 (2), 181–188. 10.1007/s101890170020. [DOI] [Google Scholar]
- White T. J.; Broer D. J. Programmable and Adaptive Mechanics with Liquid Crystal Polymer Networks and Elastomers. Nat. Mater. 2015, 14 (11), 1087–1098. 10.1038/nmat4433. [DOI] [PubMed] [Google Scholar]
- Warner M.; Terentjev E. M.. Liquid Crystal Elastomers; Oxford University Press: 2007; Vol. 120. [Google Scholar]
- Ahir S. V.; Tajbakhsh A. R.; Terentjev E. M. Self-assembled Shape-memory Fibers of Triblock Liquid-crystal Polymers. Adv. Funct. Mater. 2006, 16 (4), 556–560. 10.1002/adfm.200500692. [DOI] [Google Scholar]
- Saed M. O.; Volpe R. H.; Traugutt N. A.; Visvanathan R.; Clark N. A.; Yakacki C. M. High Strain Actuation Liquid Crystal Elastomers via Modulation of Mesophase Structure. Soft Matter 2017, 13, 7537–7547. 10.1039/C7SM01380A. [DOI] [PubMed] [Google Scholar]
- Huber J. E.; Fleck N. A.; Ashby M. F. The Selection of Mechanical Actuators Based on Performance Indices. Proc. R. Soc. London, Ser. A 1997, 453 (1965), 2185–2205. 10.1098/rspa.1997.0117. [DOI] [Google Scholar]
- Stenull O.; Lubensky T. C. Phase Transitions and Soft Elasticity of Smectic Elastomers. Phys. Rev. Lett. 2005, 94 (1), 18304. 10.1103/PhysRevLett.94.018304. [DOI] [PubMed] [Google Scholar]
- Clarke S. M.; Tajbakhsh A. R.; Terentjev E. M.; Remillat C.; Tomlinson G. R.; House J. R. Soft Elasticity and Mechanical Damping in Liquid Crystalline Elastomers. J. Appl. Phys. 2001, 89 (11), 6530–6535. 10.1063/1.1368177. [DOI] [Google Scholar]
- Forrest J. A. A Decade of Dynamics in Thin Films of Polystyrene: Where Are We Now?. Eur. Phys. J. E 2002, 8 (2), 261–266. [DOI] [PubMed] [Google Scholar]
- Wani O. M.; Zeng H.; Priimagi A. A Light-Driven Artificial Flytrap. Nat. Commun. 2017, 8 (1), 15546. 10.1038/ncomms15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe R. H.; Mistry D.; Patel V. V.; Patel R. R.; Yakacki C. M. Dynamically Crystalizing Liquid-Crystal Elastomers for an Expandable Endplate-Conforming Interbody Fusion Cage. Adv. Healthcare Mater. 2020, 9 (1), 1901136. 10.1002/adhm.201901136. [DOI] [PubMed] [Google Scholar]
- Kotikian A.; McMahan C.; Davidson E. C.; Muhammad J. M.; Weeks R. D.; Daraio C.; Lewis J. A. Untethered Soft Robotic Matter with Passive Control of Shape Morphing and Propulsion. Sci. Robot. 2019, 4 (33), eaax7044. 10.1126/scirobotics.aax7044. [DOI] [PubMed] [Google Scholar]
- Ohzono T.; Saed M. O.; Yue Y.; Norikane Y.; Terentjev E. M. Dynamic Manipulation of Friction in Smart Textile Composites of Liquid-Crystal Elastomers. Adv. Mater. Interfaces 2020, 7 (7), 1901996. 10.1002/admi.201901996. [DOI] [Google Scholar]
- Roach D. J.; Yuan C.; Kuang X.; Li V. C.-F.; Blake P.; Romero M. L.; Hammel I.; Yu K.; Qi H. J. Long Liquid Crystal Elastomer Fibers with Large Reversible Actuation Strains for Smart Textiles and Artificial Muscles. ACS Appl. Mater. Interfaces 2019, 11 (21), 19514–19521. 10.1021/acsami.9b04401. [DOI] [PubMed] [Google Scholar]
- Merkel D. R.; Shaha R. K.; Yakacki C. M.; Frick C. P. Mechanical Energy Dissipation in Polydomain Nematic Liquid Crystal Elastomers in Response to Oscillating Loading. Polymer 2019, 166, 148–154. 10.1016/j.polymer.2019.01.042. [DOI] [Google Scholar]
- Traugutt N. A.; Mistry D.; Luo C.; Yu K.; Ge Q.; Yakacki C. M. Liquid-Crystal-Elastomer-Based Dissipative Structures by Digital Light Processing 3D Printing. Adv. Mater. 2020, 32 (28), 2000797. 10.1002/adma.202000797. [DOI] [PubMed] [Google Scholar]
- Ohzono T.; Saed M. O.; Terentjev E. M. Enhanced Dynamic Adhesion in Nematic Liquid Crystal Elastomers. Adv. Mater. 2019, 31, 1902642. 10.1002/adma.201902642. [DOI] [PubMed] [Google Scholar]
- Feng W.; Broer D. J.; Liu D. Oscillating Chiral-Nematic Fingerprints Wipe Away Dust. Adv. Mater. 2018, 30 (11), 1704970. 10.1002/adma.201704970. [DOI] [PubMed] [Google Scholar]
- Mistry D.; Connell S. D.; Mickthwaite S. L.; Morgan P. B.; Clamp J. H.; Gleeson H. F. Coincident Molecular Auxeticity and Negative Order Parameter in a Liquid Crystal Elastomer. Nat. Commun. 2018, 9 (1), 5095. 10.1038/s41467-018-07587-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jampani V. S. R.; Volpe R. H.; de Sousa K. R.; Machado J. F.; Yakacki C. M.; Lagerwall J. P. F. Liquid Crystal Elastomer Shell Actuators with Negative Order Parameter. Sci. Adv. 2019, 5 (4), eaaw2476 10.1126/sciadv.aaw2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry D.; Nikkhou M.; Raistrick T.; Hussain M.; Jull E. I. L.; Baker D. L.; Gleeson H. F. Isotropic Liquid Crystal Elastomers as Exceptional Photoelastic Strain Sensors. Macromolecules 2020, 53 (10), 3709–3718. 10.1021/acs.macromol.9b02456. [DOI] [Google Scholar]
- Donovan B. R.; Fowler H. E.; Matavulj V. M.; White T. J. Mechanotropic Elastomers. Angew. Chem. 2019, 131 (39), 13882–13886. 10.1002/ange.201905176. [DOI] [PubMed] [Google Scholar]
- de Gennes P. G. Réflexions Sur Un Type de Polymères Nématiques. C. R. Acad. Sci., Ser. B 1975, 281, 101–103. [Google Scholar]
- Finkelmann H.; Kock H.; Rehage G. Investigations on Liquid Crystalline Polysiloxanes 3. Liquid Crystalline Elastomers—a New Type of Liquid Crystalline Material. Makromol. Chem., Rapid Commun. 1981, 2 (4), 317–322. 10.1002/marc.1981.030020413. [DOI] [Google Scholar]
- Talroze R. V.; Gubina T. I.; Shibaev V. P.; Platé N. A.; Dakin V. I.; Shmakova N. A.; Sukhov F. F. Peculiarities of the Thermoelastic Behaviour of Liquidcrystalline Elastomers. Makromol. Chem., Rapid Commun. 1990, 11 (2), 67–71. 10.1002/marc.1990.030110204. [DOI] [Google Scholar]
- Mitchell G. R.; Davis F. J.; Guo W.; Cywinski R. Coupling between Mesogenic Units and Polymer Backbone in Side-Chain Liquid Crystal Polymers and Elastomers. Polymer 1991, 32 (8), 1347–1353. 10.1016/0032-3861(91)90412-C. [DOI] [Google Scholar]
- Petridis L.; Terentjev E. M. Nematic-Isotropic Transition with Quenched Disorder. Phys. Rev. E 2006, 74 (5), 51707. 10.1103/PhysRevE.74.051707. [DOI] [PubMed] [Google Scholar]
- Clarke S. M.; Nishikawa E.; Finkelmann H.; Terentjev E. M. Light-scattering Study of Random Disorder in Liquid Crystalline Elastomers. Macromol. Chem. Phys. 1997, 198 (11), 3485–3498. 10.1002/macp.1997.021981113. [DOI] [Google Scholar]
- Hikmet R. A. M.; Broer D. J. Dynamic Mechanical Properties of Anisotropic Networks Formed by Liquid Crystalline Acrylates. Polymer 1991, 32 (9), 1627–1632. 10.1016/0032-3861(91)90398-3. [DOI] [Google Scholar]
- Legge C. H.; Davis F. J.; Mitchell G. R. Memory Effects in Liquid Crystal Elastomers. J. Phys. II 1991, 1 (10), 1253–1261. 10.1051/jp2:1991131. [DOI] [Google Scholar]
- Fridrikh S. V.; Terentjev E. M. Polydomain-Monodomain Transition in Nematic Elastomers. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 1999, 60 (2), 1847. 10.1103/PhysRevE.60.1847. [DOI] [PubMed] [Google Scholar]
- Küupfer J.; Finkelmann H. Liquid Crystal Elastomers: Influence of the Orientational Distribution of the Crosslinks on the Phase Behaviour and Reorientation Processes. Macromol. Chem. Phys. 1994, 195 (4), 1353–1367. 10.1002/macp.1994.021950419. [DOI] [Google Scholar]
- Yakacki C. M.; Saed M.; Nair D. P.; Gong T.; Reed S. M.; Bowman C. N. Tailorable and Programmable Liquid-Crystalline Elastomers Using a Two-Stage Thiol-Acrylate Reaction. RSC Adv. 2015, 5 (25), 18997–19001. 10.1039/C5RA01039J. [DOI] [Google Scholar]
- Broer D. J.; Boven J.; Mol G. N.; Challa G. In-situ Photopolymerization of Oriented Liquid-crystalline Acrylates, 3. Oriented Polymer Networks from a Mesogenic Diacrylate. Makromol. Chem. 1989, 190 (9), 2255–2268. 10.1002/macp.1989.021900926. [DOI] [Google Scholar]
- Ware T. H.; McConney M. E.; Wie J. J.; Tondiglia V. P.; White T. J. Voxelated Liquid Crystal Elastomers. Science (Washington, DC, U. S.) 2015, 347 (6225), 982–984. 10.1126/science.1261019. [DOI] [PubMed] [Google Scholar]
- Ambulo C. P.; Burroughs J. J.; Boothby J. M.; Kim H.; Shankar M. R.; Ware T. H. Four-Dimensional Printing of Liquid Crystal Elastomers. ACS Appl. Mater. Interfaces 2017, 9 (42), 37332–37339. 10.1021/acsami.7b11851. [DOI] [PubMed] [Google Scholar]
- Thomsen D. L.; Keller P.; Naciri J.; Pink R.; Jeon H.; Shenoy D.; Ratna B. R. Liquid Crystal Elastomers with Mechanical Properties of a Muscle. Macromolecules 2001, 34 (17), 5868–5875. 10.1021/ma001639q. [DOI] [Google Scholar]
- Donnio B.; Wermter H.; Finkelmann H. A Simple and Versatile Synthetic Route for the Preparation of Main-Chain, Liquid-Crystalline Elastomers. Macromolecules 2000, 33 (21), 7724–7729. 10.1021/ma0002850. [DOI] [Google Scholar]
- Sánchez-Ferrer A.; Finkelmann H. Uniaxial and Shear Deformations in Smectic-C Main-Chain Liquid-Crystalline Elastomers. Macromolecules 2008, 41 (3), 970–980. 10.1021/ma7025644. [DOI] [Google Scholar]
- Ohm C.; Brehmer M.; Zentel R. Liquid Crystalline Elastomers as Actuators and Sensors. Adv. Mater. 2010, 22 (31), 3366–3387. 10.1002/adma.200904059. [DOI] [PubMed] [Google Scholar]
- Wermter H.; Finkelmann H.. Liquid Crystalline Elastomers as Artificial Muscles. e-Polym. 2001, 1 ( (1), ). 10.1515/epoly.2001.1.1.111 [DOI] [Google Scholar]
- Warner M.; Terentjev E. M. Nematic Elastomers—a New State of Matter?. Prog. Polym. Sci. 1996, 21 (5), 853–891. 10.1016/S0079-6700(96)00013-5. [DOI] [Google Scholar]
- Finkelmann H.; Nishikawa E.; Pereira G. G.; Warner M. A New Opto-Mechanical Effect in Solids. Phys. Rev. Lett. 2001, 87 (1), 15501. 10.1103/PhysRevLett.87.015501. [DOI] [PubMed] [Google Scholar]
- Warner M.; Bladon P.; Terentjev E. M. Soft Elasticity”—Deformation without Resistance in Liquid Crystal Elastomers. J. Phys. II 1994, 4 (1), 93–102. 10.1051/jp2:1994116. [DOI] [Google Scholar]
- Finkelmann H.; Greve A.; Warner M. The Elastic Anisotropy of Nematic Elastomers. Eur. Phys. J. E: Soft Matter Biol. Phys. 2001, 5 (3), 281–293. 10.1007/s101890170060. [DOI] [Google Scholar]
- Saed M. O.; Torbati A. H.; Nair D. P.; Yakacki C. M. Synthesis of Programmable Main-Chain Liquid-Crystalline Elastomers Using a Two-Stage Thiol-Acrylate Reaction. J. Visualized Exp. 2016, (107), e53546. 10.3791/53546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelebart A. H.; Jan Mulder D.; Varga M.; Konya A.; Vantomme G.; Meijer E. W.; Selinger R. L. B.; Broer D. J. Making Waves in a Photoactive Polymer Film. Nature 2017, 546 (7660), 632. 10.1038/nature22987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson W. J. Jr.; Kuhfuss H. F. Liquid Crystal Polymers. I. Preparation and Properties of p-Hydroxybenzoic Acid Copolyesters. J. Polym. Sci., Polym. Chem. Ed. 1976, 14 (8), 2043–2058. 10.1002/pol.1976.170140820. [DOI] [Google Scholar]
- Zentel R. Liquid Crystal Elastomers. Angew. Chem. 1989, 101, 1437. 10.1002/ange.19891011041. [DOI] [Google Scholar]
- Hikmet R. A. M.; Lub J.; Broer D. J. Anisotropic Networks Formed by Photopolymerization of Liquid-crystalline Molecules. Adv. Mater. 1991, 3 (7–8), 392–394. 10.1002/adma.19910030713. [DOI] [Google Scholar]
- Broer D. J. On the History of Reactive Mesogens: Interview with Dirk J. Broer. Adv. Mater. 2020, 32 (20), 1905144. 10.1002/adma.201905144. [DOI] [PubMed] [Google Scholar]
- Tabiryan N.; Serak S.; Dai X.-M.; Bunning T. Polymer Film with Optically Controlled Form and Actuation. Opt. Express 2005, 13 (19), 7442–7448. 10.1364/OPEX.13.007442. [DOI] [PubMed] [Google Scholar]
- Ware T. H.; Perry Z. P.; Middleton C. M.; Iacono S. T.; White T. J. Programmable Liquid Crystal Elastomers Prepared by Thiol-Ene Photopolymerization. ACS Macro Lett. 2015, 4 (9), 942–946. 10.1021/acsmacrolett.5b00511. [DOI] [PubMed] [Google Scholar]
- López-Valdeolivas M.; Liu D.; Broer D. J.; Sánchez-Somolinos C. 4D Printed Actuators with Soft-Robotic Functions. Macromol. Rapid Commun. 2018, 39 (5), 1700710. 10.1002/marc.201700710. [DOI] [PubMed] [Google Scholar]
- Kotikian A.; Truby R. L.; Boley J. W.; White T. J.; Lewis J. A. 3D Printing of Liquid Crystal Elastomeric Actuators with Spatially Programed Nematic Order. Adv. Mater. 2018, 30 (10), 1706164. 10.1002/adma.201706164. [DOI] [PubMed] [Google Scholar]
- Roach D. J.; Kuang X.; Yuan C.; Chen K.; Qi H. J. Novel Ink for Ambient Condition Printing of Liquid Crystal Elastomers for 4D Printing. Smart Mater. Struct. 2018, 27 (12), 125011. 10.1088/1361-665X/aae96f. [DOI] [Google Scholar]
- Saed M. O.; Ambulo C. P.; Kim H.; De R.; Raval V.; Searles K.; Siddiqui D. A.; Cue J. M. O.; Stefan M. C.; Shankar M. R. Molecularly-Engineered, 4D-Printed Liquid Crystal Elastomer Actuators. Adv. Funct. Mater. 2019, 29 (3), 1806412. 10.1002/adfm.201806412. [DOI] [Google Scholar]
- Montarnal D.; Capelot M.; Tournilhac F.; Leibler L. Silica-like Malleable Materials from Permanent Organic Networks. Science 2011, 334 (6058), 965–968. 10.1126/science.1212648. [DOI] [PubMed] [Google Scholar]
- McBride M. K.; Worrell B. T.; Brown T.; Cox L. M.; Sowan N.; Wang C.; Podgorski M.; Martinez A. M.; Bowman C. N. Enabling Applications of Covalent Adaptable Networks. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 175–198. 10.1146/annurev-chembioeng-060718-030217. [DOI] [PubMed] [Google Scholar]
- Podgórski M.; Fairbanks B. D.; Kirkpatrick B. E.; McBride M.; Martinez A.; Dobson A.; Bongiardina N. J.; Bowman C. N. Toward Stimuli-Responsive Dynamic Thermosets through Continuous Development and Improvements in Covalent Adaptable Networks (CANs). Adv. Mater. 2020, 32 (20), 1906876. 10.1002/adma.201906876. [DOI] [PubMed] [Google Scholar]
- Kloxin C. J.; Bowman C. N. Covalent Adaptable Networks: Smart, Reconfigurable and Responsive Network Systems. Chem. Soc. Rev. 2013, 42, 7161–7173. 10.1039/C3CS60046G. [DOI] [PubMed] [Google Scholar]
- Winne J. M.; Leibler L.; Du Prez F. E. Dynamic Covalent Chemistry in Polymer Networks: A Mechanistic Perspective. Polym. Chem. 2019, 10 (45), 6091–6108. 10.1039/C9PY01260E. [DOI] [Google Scholar]
- Denissen W.; Winne J. M.; Du Prez F. E. Vitrimers: Permanent Organic Networks with Glass-like Fluidity. Chem. Sci. 2016, 7 (1), 30–38. 10.1039/C5SC02223A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zee N. J.; Nicolaÿ R. Vitrimers: Permanently Crosslinked Polymers with Dynamic Network Topology. Prog. Polym. Sci. 2020, 104, 101233. 10.1016/j.progpolymsci.2020.101233. [DOI] [Google Scholar]
- Zou W.; Dong J.; Luo Y.; Zhao Q.; Xie T. Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations. Adv. Mater. 2017, 29 (14), 1606100. 10.1002/adma.201606100. [DOI] [PubMed] [Google Scholar]
- Chen X.; Dam M. A.; Ono K.; Mal A.; Shen H.; Nutt S. R.; Sheran K.; Wudl F. A Thermally Re-Mendable Cross-Linked Polymeric Material. Science 2002, 295 (5560), 1698–1702. 10.1126/science.1065879. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Digby Z. A.; Flum J. A.; Chakma P.; Saul J. M.; Sparks J. L.; Konkolewicz D. Dynamic Thiol-Michael Chemistry for Thermoresponsive Rehealable and Malleable Networks. Macromolecules 2016, 49 (18), 6871–6878. 10.1021/acs.macromol.6b01061. [DOI] [Google Scholar]
- Erice A.; de Luzuriaga A. R.; Matxain J. M.; Ruipérez F.; Asua J. M.; Grande H.-J.; Rekondo A. Reprocessable and Recyclable Crosslinked Poly (Urea-Urethane) s Based on Dynamic Amine/Urea Exchange. Polymer 2018, 145, 127–136. 10.1016/j.polymer.2018.04.076. [DOI] [Google Scholar]
- Fortman D. J.; Brutman J. P.; Cramer C. J.; Hillmyer M. A.; Dichtel W. R. Mechanically Activated, Catalyst-Free Polyhydroxyurethane Vitrimers. J. Am. Chem. Soc. 2015, 137 (44), 14019–14022. 10.1021/jacs.5b08084. [DOI] [PubMed] [Google Scholar]
- Snyder R. L.; Fortman D. J.; De Hoe G. X.; Hillmyer M. A.; Dichtel W. R. Reprocessable Acid-Degradable Polycarbonate Vitrimers. Macromolecules 2018, 51 (2), 389–397. 10.1021/acs.macromol.7b02299. [DOI] [Google Scholar]
- Li L.; Chen X.; Torkelson J. M. Reprocessable Polymer Networks via Thiourethane Dynamic Chemistry: Recovery of Cross-Link Density after Recycling and Proof-of-Principle Solvolysis Leading to Monomer Recovery. Macromolecules 2019, 52 (21), 8207–8216. 10.1021/acs.macromol.9b01359. [DOI] [Google Scholar]
- Denissen W.; Rivero G.; Nicolaÿ R.; Leibler L.; Winne J. M.; Du Prez F. E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25 (16), 2451–2457. 10.1002/adfm.201404553. [DOI] [Google Scholar]
- Zheng P.; McCarthy T. J. A Surprise from 1954: Siloxane Equilibration Is a Simple, Robust, and Obvious Polymer Self-Healing Mechanism. J. Am. Chem. Soc. 2012, 134 (4), 2024–2027. 10.1021/ja2113257. [DOI] [PubMed] [Google Scholar]
- Lu Y.-X.; Guan Z. Olefin Metathesis for Effective Polymer Healing via Dynamic Exchange of Strong Carbon-Carbon Double Bonds. J. Am. Chem. Soc. 2012, 134 (34), 14226–14231. 10.1021/ja306287s. [DOI] [PubMed] [Google Scholar]
- Billiet S.; De Bruycker K.; Driessen F.; Goossens H.; Van Speybroeck V.; Winne J. M.; Du Prez F. E. Triazolinediones Enable Ultrafast and Reversible Click Chemistry for the Design of Dynamic Polymer Systems. Nat. Chem. 2014, 6 (9), 815–821. 10.1038/nchem.2023. [DOI] [PubMed] [Google Scholar]
- Obadia M. M.; Mudraboyina B. P.; Serghei A.; Montarnal D.; Drockenmuller E. Reprocessing and Recycling of Highly Cross-Linked Ion-Conducting Networks through Transalkylation Exchanges of C-N Bonds. J. Am. Chem. Soc. 2015, 137 (18), 6078–6083. 10.1021/jacs.5b02653. [DOI] [PubMed] [Google Scholar]
- Chakma P.; Digby Z. A.; Shulman M. P.; Kuhn L. R.; Morley C. N.; Sparks J. L.; Konkolewicz D. Anilinium Salts in Polymer Networks for Materials with Mechanical Stability and Mild Thermally Induced Dynamic Properties. ACS Macro Lett. 2019, 8 (2), 95–100. 10.1021/acsmacrolett.8b00819. [DOI] [PubMed] [Google Scholar]
- Scott T. F.; Schneider A. D.; Cook W. D.; Bowman C. N. Photoinduced Plasticity in Cross-Linked Polymers. Science (Washington, DC, U. S.) 2005, 308 (5728), 1615–1617. 10.1126/science.1110505. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Ren B.; Liu S.; Liu X.; Tong Z. A Novel Photoreversible Poly (Ferrocenylsilane) with Coumarin Side Group: Synthesis, Characterization, and Electrochemical Activities. Chem. Commun. 2006, (7), 779–781. 10.1039/b515413h. [DOI] [PubMed] [Google Scholar]
- Trenor S. R.; Shultz A. R.; Love B. J.; Long T. E. Coumarins in Polymers: From Light Harvesting to Photo-Cross-Linkable Tissue Scaffolds. Chem. Rev. 2004, 104 (6), 3059–3078. 10.1021/cr030037c. [DOI] [PubMed] [Google Scholar]
- Frisch H.; Marschner D. E.; Goldmann A. S.; Barner-Kowollik C. Wavelength-Gated Dynamic Covalent Chemistry. Angew. Chem., Int. Ed. 2018, 57 (8), 2036–2045. 10.1002/anie.201709991. [DOI] [PubMed] [Google Scholar]
- Amamoto Y.; Kamada J.; Otsuka H.; Takahara A.; Matyjaszewski K. Repeatable Photoinduced Self-healing of Covalently Cross-linked Polymers through Reshuffling of Trithiocarbonate Units. Angew. Chem. 2011, 123 (7), 1698–1701. 10.1002/ange.201003888. [DOI] [PubMed] [Google Scholar]
- Fairbanks B. D.; Singh S. P.; Bowman C. N.; Anseth K. S. Photodegradable, Photoadaptable Hydrogels via Radical-Mediated Disulfide Fragmentation Reaction. Macromolecules 2011, 44 (8), 2444–2450. 10.1021/ma200202w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash J. J.; Kubo T.; Bapat A. P.; Sumerlin B. S. Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters. Macromolecules 2015, 48 (7), 2098–2106. 10.1021/acs.macromol.5b00210. [DOI] [Google Scholar]
- Pei Z.; Yang Y.; Chen Q.; Terentjev E. M.; Wei Y.; Ji Y. Mouldable Liquid-Crystalline Elastomer Actuators with Exchangeable Covalent Bonds. Nat. Mater. 2014, 13 (1), 36. 10.1038/nmat3812. [DOI] [PubMed] [Google Scholar]
- McBride M. K.; Martinez A. M.; Cox L.; Alim M.; Childress K.; Beiswinger M.; Podgorski M.; Worrell B. T.; Killgore J.; Bowman C. N. A Readily Programmable, Fully Reversible Shape-Switching Material. Sci. Adv. 2018, 4 (8), eaat4634 10.1126/sciadv.aat4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins J. S.; Warner M.; Bhattacharya K. Supersoft Elasticity in Polydomain Nematic Elastomers. Phys. Rev. Lett. 2009, 103 (3), 37802. 10.1103/PhysRevLett.103.037802. [DOI] [PubMed] [Google Scholar]
- Shi Q.; Yu K.; Kuang X.; Mu X.; Dunn C. K.; Dunn M. L.; Wang T.; Qi H. J. Recyclable 3D Printing of Vitrimer Epoxy. Mater. Horiz. 2017, 4 (4), 598–607. 10.1039/C7MH00043J. [DOI] [Google Scholar]
- Hsiao Y. -C; Hill L. W.; Pappas S. P. Reversible Amine Solubilization of Cured Siloxane Polymers. J. Appl. Polym. Sci. 1975, 19 (10), 2817–2820. 10.1002/app.1975.070191017. [DOI] [Google Scholar]
- Yang Y.; Terentjev E. M.; Wei Y.; Ji Y. Solvent-Assisted Programming of Flat Polymer Sheets into Reconfigurable and Self-Healing 3D Structures. Nat. Commun. 2018, 9 (1), 1906. 10.1038/s41467-018-04257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzon D. W.; Traugutt N. A.; McBride M. K.; Bowman C. N.; Yakacki C. M.; Yu K. Adaptable Liquid Crystal Elastomers with Transesterification-Based Bond Exchange Reactions. Soft Matter 2018, 14 (6), 951–960. 10.1039/C7SM02110K. [DOI] [PubMed] [Google Scholar]
- Lu X.; Guo S.; Tong X.; Xia H.; Zhao Y. Tunable Photocontrolled Motions Using Stored Strain Energy in Malleable Azobenzene Liquid Crystalline Polymer Actuators. Adv. Mater. 2017, 29 (28), 1606467. 10.1002/adma.201606467. [DOI] [PubMed] [Google Scholar]
- Wen Z.; McBride M. K.; Zhang X.; Han X.; Martinez A. M.; Shao R.; Zhu C.; Visvanathan R.; Clark N. A.; Wang Y. Reconfigurable LC Elastomers: Using a Thermally Programmable Monodomain to Access Two-Way Free-Standing Multiple Shape Memory Polymers. Macromolecules 2018, 51 (15), 5812–5819. 10.1021/acs.macromol.8b01315. [DOI] [Google Scholar]
- Saed M. O.; Gablier A.; Terentejv E. M. Liquid Crystalline Vitrimers with Full or Partial Boronic-Ester Bond Exchange. Adv. Funct. Mater. 2020, 30 (3), 1906458. 10.1002/adfm.201906458. [DOI] [Google Scholar]
- Jiang Z.; Xiao Y.; Yin L.; Han L.; Zhao Y. Self-Lockable” Liquid-Crystalline Diels-Alder Dynamic Network Actuators with Room Temperature Programmability and Solution-Reprocessability. Angew. Chem. 2020, 132 (12), 4955–4961. 10.1002/ange.202000181. [DOI] [PubMed] [Google Scholar]
- Saed M. O.; Terentjev E. M. Siloxane Crosslinks with Dynamic Bond Exchange Enable Shape Programming in Liquid-Crystalline Elastomers. Sci. Rep. 2020, 10 (1), 6609. 10.1038/s41598-020-63508-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Yang Y.; Qian X.; Chen Q.; Wei Y.; Ji Y. Liquid-Crystalline Soft Actuators with Switchable Thermal Reprogrammability. Angew. Chem., Int. Ed. 2020, 59 (12), 4778–4784. 10.1002/anie.201915694. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zhang Y.; Rios O.; Keum J. K.; Kessler M. R. Photo-Responsive Liquid Crystalline Epoxy Networks with Exchangeable Disulfide Bonds. RSC Adv. 2017, 7 (59), 37248–37254. 10.1039/C7RA06343A. [DOI] [PubMed] [Google Scholar]
- Yang R.; Zhao Y. Non-uniform Optical Inscription of Actuation Domains in a Liquid Crystal Polymer of Uniaxial Orientation: An Approach to Complex and Programmable Shape Changes. Angew. Chem. 2017, 129 (45), 14390–14394. 10.1002/ange.201709528. [DOI] [PubMed] [Google Scholar]
- Jiang Z.; Xiao Y.; Tong X.; Zhao Y. Selective Decrosslinking in Liquid Crystal Polymer Actuators for Optical Reconfiguration of Origami and Light-Fueled Locomotion. Angew. Chem., Int. Ed. 2019, 58 (16), 5332–5337. 10.1002/anie.201900470. [DOI] [PubMed] [Google Scholar]
- Capelot M.; Unterlass M. M.; Tournilhac F.; Leibler L. Catalytic Control of the Vitrimer Glass Transition. ACS Macro Lett. 2012, 1 (7), 789–792. 10.1021/mz300239f. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Terentjev E. M.; Zhang Y.; Chen Q.; Zhao Y.; Wei Y.; Ji Y. Reprocessable Thermoset Soft Actuators. Angew. Chem. 2019, 131 (48), 17635–17640. 10.1002/ange.201911612. [DOI] [PubMed] [Google Scholar]
- Hogan P. M.; Tajbakhsh A. R.; Terentjev E. M. UV Manipulation of Order and Macroscopic Shape in Nematic Elastomers. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2002, 65 (4), 41720. 10.1103/PhysRevE.65.041720. [DOI] [PubMed] [Google Scholar]
- Fernández-Francos X.; Konuray A.-O.; Belmonte A.; De la Flor S.; Serra À.; Ramis X. Sequential Curing of Off-Stoichiometric Thiol-Epoxy Thermosets with a Custom-Tailored Structure. Polym. Chem. 2016, 7 (12), 2280–2290. 10.1039/C6PY00099A. [DOI] [Google Scholar]
- Lotti N.; Siracusa V.; Finelli L.; Marchese P.; Munari A. Sulphur-Containing Polymers: Synthesis and Thermal Properties of Novel Polyesters Based on Dithiotriethylene Glycol. Eur. Polym. J. 2006, 42 (12), 3374–3382. 10.1016/j.eurpolymj.2006.08.022. [DOI] [Google Scholar]
- Suzuki A. Recent Advances in the Cross-Coupling Reactions of Organoboron Derivatives with Organic Electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576 (1–2), 147–168. 10.1016/S0022-328X(98)01055-9. [DOI] [Google Scholar]
- Roy C. D.; Brown H. C. Stability of Boronic Esters-Structural Effects on the Relative Rates of Transesterification of 2-(Phenyl)-1, 3, 2-Dioxaborolane. J. Organomet. Chem. 2007, 692 (4), 784–790. 10.1016/j.jorganchem.2006.10.013. [DOI] [Google Scholar]
- Cote A. P.; Benin A. I.; Ockwig N. W.; O’keeffe M.; Matzger A. J.; Yaghi O. M. Porous, Crystalline, Covalent Organic Frameworks. Science 2005, 310 (5751), 1166–1170. 10.1126/science.1120411. [DOI] [PubMed] [Google Scholar]
- Niu W.; O’Sullivan C.; Rambo B. M.; Smith M. D.; Lavigne J. J. Self-Repairing Polymers: Poly (Dioxaborolane) s Containing Trigonal Planar Boron. Chem. Commun. 2005, (34), 4342–4344. 10.1039/b504634c. [DOI] [PubMed] [Google Scholar]
- Niu W.; Smith M. D.; Lavigne J. J. Self-Assembling Poly (Dioxaborole) s as Blue-Emissive Materials. J. Am. Chem. Soc. 2006, 128 (51), 16466–16467. 10.1021/ja065986c. [DOI] [PubMed] [Google Scholar]
- Cromwell O. R.; Chung J.; Guan Z. Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc. 2015, 137 (20), 6492–6495. 10.1021/jacs.5b03551. [DOI] [PubMed] [Google Scholar]
- Ogden W. A.; Guan Z. Recyclable, Strong, and Highly Malleable Thermosets Based on Boroxine Networks. J. Am. Chem. Soc. 2018, 140 (20), 6217–6220. 10.1021/jacs.8b03257. [DOI] [PubMed] [Google Scholar]
- Lai J.; Mei J.; Jia X.; Li C.; You X.; Bao Z. A Stiff and Healable Polymer Based on Dynamic-covalent Boroxine Bonds. Adv. Mater. 2016, 28 (37), 8277–8282. 10.1002/adma.201602332. [DOI] [PubMed] [Google Scholar]
- Cash J. J.; Kubo T.; Dobbins D. J.; Sumerlin B. S. Maximizing the Symbiosis of Static and Dynamic Bonds in Self-Healing Boronic Ester Networks. Polym. Chem. 2018, 9 (15), 2011–2020. 10.1039/C8PY00123E. [DOI] [Google Scholar]
- Röttger M.; Domenech T.; van der Weegen R.; Breuillac A.; Nicolaÿ R.; Leibler L. High-Performance Vitrimers from Commodity Thermoplastics through Dioxaborolane Metathesis. Science 2017, 356 (6333), 62–65. 10.1126/science.aah5281. [DOI] [PubMed] [Google Scholar]
- Krol P. Synthesis Methods, Chemical Structures and Phase Structures of Linear Polyurethanes. Properties and Applications of Linear Polyurethanes in Polyurethane Elastomers, Copolymers and Ionomers. Prog. Mater. Sci. 2007, 52 (6), 915–1015. 10.1016/j.pmatsci.2006.11.001. [DOI] [Google Scholar]
- Zheng N.; Fang Z.; Zou W.; Zhao Q.; Xie T. Thermoset Shape-memory Polyurethane with Intrinsic Plasticity Enabled by Transcarbamoylation. Angew. Chem. 2016, 128 (38), 11593–11597. 10.1002/ange.201602847. [DOI] [PubMed] [Google Scholar]
- Jo B.-H.; Van Lerberghe L. M.; Motsegood K. M.; Beebe D. J. Three-Dimensional Micro-Channel Fabrication in Polydimethylsiloxane (PDMS) Elastomer. J. Microelectromech. Syst. 2000, 9 (1), 76–81. 10.1109/84.825780. [DOI] [Google Scholar]
- Stukenbroeker T.; Wang W.; Winne J. M.; Du Prez F. E.; Nicolaÿ R.; Leibler L. Polydimethylsiloxane Quenchable Vitrimers. Polym. Chem. 2017, 8 (43), 6590–6593. 10.1039/C7PY01488K. [DOI] [Google Scholar]
- Ube T.; Kawasaki K.; Ikeda T. Photomobile Liquid-crystalline Elastomers with Rearrangeable Networks. Adv. Mater. 2016, 28 (37), 8212–8217. 10.1002/adma.201602745. [DOI] [PubMed] [Google Scholar]
- Ishibashi J. S. A.; Kalow J. A. Vitrimeric Silicone Elastomers Enabled by Dynamic Meldrum’s Acid-Derived Cross-Links. ACS Macro Lett. 2018, 7 (4), 482–486. 10.1021/acsmacrolett.8b00166. [DOI] [PubMed] [Google Scholar]
- Osthoff R. C.; Bueche A. M.; Grubb W. T. Chemical Stress-Relaxation of Polydimethylsiloxane Elastomers1. J. Am. Chem. Soc. 1954, 76 (18), 4659–4663. 10.1021/ja01647a052. [DOI] [Google Scholar]
- Kantor S. W.; Grubb W. T.; Osthoff R. C. The Mechanism of the Acid-and Base-Catalyzed Equilibration of Siloxanes. J. Am. Chem. Soc. 1954, 76 (20), 5190–5197. 10.1021/ja01649a076. [DOI] [Google Scholar]
- Nishimura Y.; Chung J.; Muradyan H.; Guan Z. Silyl Ether as a Robust and Thermally Stable Dynamic Covalent Motif for Malleable Polymer Design. J. Am. Chem. Soc. 2017, 139 (42), 14881–14884. 10.1021/jacs.7b08826. [DOI] [PubMed] [Google Scholar]
- Saed M. O.; Terentjev E. M. Catalytic Control of Plastic Flow in Siloxane-Based Liquid Crystalline Elastomer Networks. ACS Macro Lett. 2020, 9, 749–755. 10.1021/acsmacrolett.0c00265. [DOI] [PubMed] [Google Scholar]
- Polgar L. M.; Van Duin M.; Broekhuis A. A.; Picchioni F. Use of Diels-Alder Chemistry for Thermoreversible Cross-Linking of Rubbers: The next Step toward Recycling of Rubber Products?. Macromolecules 2015, 48 (19), 7096–7105. 10.1021/acs.macromol.5b01422. [DOI] [Google Scholar]
- Berto P.; Pointet A.; Le Coz C.; Grelier S.; Peruch F. Recyclable Telechelic Cross-Linked Polybutadiene Based on Reversible Diels-Alder Chemistry. Macromolecules 2018, 51 (3), 651–659. 10.1021/acs.macromol.7b02220. [DOI] [Google Scholar]
- Canadell J.; Goossens H.; Klumperman B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44 (8), 2536–2541. 10.1021/ma2001492. [DOI] [Google Scholar]
- Michal B. T.; Jaye C. A.; Spencer E. J.; Rowan S. J. Inherently Photohealable and Thermal Shape-Memory Polydisulfide Networks. ACS Macro Lett. 2013, 2 (8), 694–699. 10.1021/mz400318m. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zhang Y.; Rios O.; Keum J. K.; Kessler M. R. Liquid Crystalline Epoxy Networks with Exchangeable Disulfide Bonds. Soft Matter 2017, 13, 5021–5027. 10.1039/C7SM00934H. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Tian H.; He Q.; Cai S. Reprogrammable, Reprocessible, and Self-Healable Liquid Crystal Elastomer with Exchangeable Disulfide Bonds. ACS Appl. Mater. Interfaces 2017, 9 (38), 33119–33128. 10.1021/acsami.7b09246. [DOI] [PubMed] [Google Scholar]
- Van Damme J.; van den Berg O.; Brancart J.; Vlaminck L.; Huyck C.; Van Assche G.; Van Mele B.; Du Prez F. Anthracene-Based Thiol-Ene Networks with Thermo-Degradable and Photo-Reversible Properties. Macromolecules 2017, 50 (5), 1930–1938. 10.1021/acs.macromol.6b02400. [DOI] [Google Scholar]
- Xu J.-F.; Chen Y.-Z.; Wu L.-Z.; Tung C.-H.; Yang Q.-Z. Dynamic Covalent Bond Based on Reversible Photo [4+ 4] Cycloaddition of Anthracene for Construction of Double-Dynamic Polymers. Org. Lett. 2013, 15 (24), 6148–6151. 10.1021/ol403015s. [DOI] [PubMed] [Google Scholar]
- Wu L.; Jin C.; Sun X. Synthesis, Properties, and Light-Induced Shape Memory Effect of Multiblock Polyesterurethanes Containing Biodegradable Segments and Pendant Cinnamamide Groups. Biomacromolecules 2011, 12 (1), 235–241. 10.1021/bm1012162. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yang X.; Chen H.; Gong T.; Li W.; Yang G.; Zhou S. Design of Triple-Shape Memory Polyurethane with Photo-Cross-Linking of Cinnamon Groups. ACS Appl. Mater. Interfaces 2013, 5 (21), 10520–10528. 10.1021/am402091m. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Lu X.; Fei G.; Wang Z.; Xia H.; Zhao Y. 4D Printing of a Liquid Crystal Elastomer with a Controllable Orientation Gradient. ACS Appl. Mater. Interfaces 2019, 11 (47), 44774–44782. 10.1021/acsami.9b18037. [DOI] [PubMed] [Google Scholar]
- McBride M. K.; Hendrikx M.; Liu D.; Worrell B. T.; Broer D. J.; Bowman C. N. Photoinduced Plasticity in Cross-Linked Liquid Crystalline Networks. Adv. Mater. 2017, 29 (17), 1606509. 10.1002/adma.201606509. [DOI] [PubMed] [Google Scholar]
- Davidson E. C.; Kotikian A.; Li S.; Aizenberg J.; Lewis J. A. 3D Printable and Reconfigurable Liquid Crystal Elastomers with Light-Induced Shape Memory via Dynamic Bond Exchange. Adv. Mater. 2020, 32, 1905682. 10.1002/adma.201905682. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Li Y.; Yang Y.; Xu Y.; Qian X.; Wei Y.; Ji Y. Durable Liquid-Crystalline Vitrimer Actuators. Chem. Sci. 2019, 10 (10), 3025–3030. 10.1039/C8SC05358H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gablier A.; Saed M. O.; Terentjev E. Rates of Transesterification in Epoxy-Thiol Vitrimers. Soft Matter 2020, 16, 5195–5202. 10.1039/D0SM00742K. [DOI] [PubMed] [Google Scholar]
- Gablier A.; Saed M. O.; Terentjev E. M. Transesterification in Epoxy-Thiol Exchangeable Liquid Crystalline Elastomers. Macromolecules 2020, 53 (19), 8642. 10.1021/acs.macromol.0c01757. [DOI] [Google Scholar]