

Abstract
Occupational exposure to 4,4′-methylene diphenyl diisocyanate (MDI), the most widely used monomeric diisocyanate, is one of the leading causes of occupational asthma (OA). Previously, we identified microRNA (miR)-206-3p/miR-381-3p-mediated PPP3CA/calcineurin signalling regulated iNOS transcription in macrophages and bronchoalveolar lavage cells (BALCs) after acute MDI exposure; however, whether PPP3CA/calcineurin signalling participates in regulation of other asthma-associated mediators secreted by macrophages/BALCs after MDI exposure is unknown.
Several asthma-associated, macrophage-secreted mediator mRNAs from MDI exposed murine BALCs and MDI-glutathione (GSH) conjugate treated differentiated THP-1 macrophages were analysed using RT-qPCR.
Endogenous IL1B, TNF, CCL2, CCL3, CCL5, and TGFB1 were upregulated in MDI or MDI-GSH conjugate exposed BALCs and macrophages, respectively. Calcineurin inhibitor tacrolimus (FK506) attenuated the MDI-GSH conjugate-mediated induction of CCL2, CCL3, CCL5, and CXCL8/IL8 but not others. Transfection of either miR-inhibitor-206-3p or miR-inhibitor-381-3p in macrophages induced chemokine CCL2, CCL3, CCL5, and CXCL8 transcription, whereas FK506 attenuated the miR-inhibitor-206-3p or miR-inhibitor-381-3p-mediated effects. Finally, MDI-GSH conjugate treated macrophages showed increased chemotactic ability to various immune cells, which may be attenuated by FK506.
In conclusion, these results indicate that MDI exposure to macrophages/BALCs may recruit immune cells into the airway via induction of chemokines by miR-206-3p and miR-381-3p-mediated calcineurin signalling activation.
Keywords: 4,4′-methylene diphenyl diisocyanate (MDI); calcineurin; macrophages; immune cell chemotaxis; microRNA (miR)
Introduction
Diisocyanates (dNCOs) are essential low molecular weight crosslinkers utilised in polyurethane production and are widely used in many applications. Currently, 4,4′-methylene diphenyl diisocyanate (MDI) is the most widely used dNCO globally (Allport et al. 2003), and it is utilised in diverse applications, such as spray foam insulation, truck bed liners, paints, adhesives, elastomers, coatings, and others (Munn et al. 2005). The demand for MDI is forecast to increase to ~8.6 million tons by 2022 (Statista 2021). In the occupational setting, MDI exposure has been identified as one of the leading causes for the development of occupational asthma (OA) (Bernstein et al. 1993; Redlich and Karol 2002; Lofgren et al. 2003; Jan et al. 2008; Engfeldt et al. 2013).
Alveolar macrophages are the most abundant immune cell type in the lung, accounting for 80–90% of all cells found in bronchoalveolar lavage (BAL) (Heron et al. 2012). These cells serve as one of the first immune responders against inhaled pathogens, particles (Hamilton et al. 2008; Hiraiwa and van Eeden 2013), and chemical allergens, such as dNCOs (Huffman et al. 1997; Wisnewski and Liu 2016). Upon encountering outside stimuli, alveolar macrophages react by phagocytosis as well as producing and secreting different mediators, such as cytokines, chemokines, bioactive lipids, nitric oxide (NO), reactive oxygen species (ROS), and others, into the alveoli microenvironment to orchestrate the initiation of inflammatory/immune responses (Balhara and Gounni 2012; Draijer and Peters-Golden 2017). Dysfunction of alveolar macrophages, including elevated production and secretion of pro-inflammatory cytokines and other immune mediators, has been shown to play an important role in asthma pathogenesis (Fricker and Gibson 2017). In the clinical setting, the levels of many immune mediators produced by macrophages were elevated in the asthmatic airway (Ackerman et al. 1994; Hoshi et al. 1995; Song et al. 2008). However, both the levels of these asthma-associated, macrophage-secreted inflammatory/immune mediators in MDI-OA patients’ airways and how the expression of these mediators change in response to MDI exposure in alveolar macrophages are largely undetermined.
Our previous report identified that MDI exposure upregulated iNOS transcription through miR-206-3p and miR-381-3p-mediated calcineurin signalling activation in macrophages (Lin et al. 2020). Other reports have indicated that the calcineurin/NFAT signalling pathway plays an important role in regulating the expression of cytokines, chemokines, and other immune system regulators in diverse immune cell types (Vaeth and Feske 2018; Park YJ et al. 2020). In addition, therapeutic strategies that target calcineurin signalling have been suggested to treat many immune diseases, including asthma (Kawano et al. 2004; Taniguchi et al. 2011; Vaeth and Feske 2018). Alveolar macrophages can secrete many immune mediators into the alveoli microenvironment upon exposure to outside stimuli; however, whether calcineurin signalling plays any role in the regulation of these mediators’ transcription in alveolar macrophages is currently unclear. Furthermore, whether these macrophage-secreted mediators can be regulated by calcineurin-mediated signalling activation upon exposure to MDI is unknown. Earlier studies from our laboratory showing that MDI exposure can upregulate iNOS transcription through upregulation of calcineurin-mediated signalling in BALCs and macrophages (Lin et al. 2020) led to the hypothesis that MDI exposure may upregulate several macrophage-secreted mediators through calcineurin signalling activation.
This report is focussed on first characterising possible MDI-mediated, macrophage-secreted mediator responses upon exposure to MDI, followed by investigating the involvement of calcineurin-mediated signalling in the regulation of MDI-mediated immune mediator expression and function. We determined candidate mediators’ transcriptional changes in response to MDI exposure using an in vivo murine MDI aerosol exposure model, then verified the role of calcineurin signalling in MDI-mediated immune mediator regulation using an in vitro THP-1 macrophage cell culture model. This report describes a putative miR-regulated calcineurin signalling mechanism by which MDI may recruit immune cells into alveoli to mediate early events of asthma pathogenesis.
Materials and methods
Chemicals and reagents
Butyric acid, high-performance liquid chromatography (HPLC) grade acetone, 3 Å molecular sieves (4–8 mesh), phosphate-buffered saline (PBS), dimethyl sulfoxide (DMSO), 98% 4,4′-methylene diphenyl diisocyanate (MDI), phorbol 12-myristate 13-acetate (PMA), ionomycin salt and reduced-glutathione (GSH) were acquired from MilliporeSigma (St. Louis, MO). Tacrolimus (FK506) was purchased from Selleckchem (Houston, TX, USA). Recombinant human (rh) proteins including interleukin (IL)-4, granulocyte-macrophage colony-stimulating factor (GMCSF), and tumour necrosis factor (TNF)-α were purchased from R&D Systems (Minneapolis, MN, USA). Roswell Park Memorial Institute (RPMI)-1640 culture medium and penicillin-streptomycin-glutamine (PSG; 100×) were purchased from ThermoFisher Scientific (Waltham, MA, USA). Hyclone™ foetal bovine serum (FBS) was obtained from Cytiva Life Sciences (Marlborough, MA, USA). Dry acetone was prepared by incubating 10 ml HPLC grade acetone on 3 Å molecular sieves for a minimum of 24 h to adsorb water.
Bronchoalveolar lavage cells (BALCs) from MDI aerosol exposed mice
Candidate macrophage-secreted mediator RNA expression studies were performed on the stored RNAs from prior studies, the BALCs RNAs were isolated from the BALB/c mice exposed to MDI aerosol using an in house developed nose-only aerosol inhalation exposure system as described previously (Hettick et al. 2018 ; Lin et al. 2019). Animals were exposed to 4580 ± 1497 μg/m3 MDI aerosol or pure house air control (Ctl) for 1 h followed by bronchoalveolar lavage at 24 h post-exposure.
Cell culture and cell differentiation
THP-1 (ATCC® TIB-202™), HL-60 (ATCC® CCL-240™), Clone 15 HL-60 (HL-60_C15; ATCC® CRL-1964™), and Jurkat Clone E6-1 (Jurkat_E6-1; ATCC® TIB-152™) cells were obtained from American Type Culture Collection (ATCC; Manassas, VA) and maintained at 0.5-1 × 106/ml in RPMI-1640 media supplement with 10% FBS, and 1 × PSG (Complete RPMI media) at 37 °C in a humidified atmosphere with 5% CO2. Enhanced differentiated THP-1 macrophages were prepared using PMA to induce differentiation as previously described (Lin et al. 2020). All macrophage in vitro experiments were performed using enhanced differentiated THP-1 macrophages. Other immune cell types used in chemotaxis experiments were differentiated as follows: For dendritic cell differentiation, a total of 3 × 107 naïve undifferentiated THP-1 cells were harvested by centrifugation at 300 × g for 5 min, washed twice with PBS, and resuspended in 30 ml of serum-free RPMI-1640 culture medium supplemented with 200 ng/ml rhIL-4, 100 ng/ml rhGM-CSF, 10 ng/ml rhTNF-α, and 200 ng/ml ionomycin and seeded into three 10 cm culture dishes for 3 days as described by Berges and colleagues (Berges et al. 2005). For neutrophil differentiation, HL-60 cells (5 × 105 cells/ml) were cultured in complete RPMI-1640 media containing 1.5% DMSO for 7 days as described by Millius and Weiner (2010). For eosinophil differentiation, HL-60_C15 cells (5 × 105 cells/ml) were cultured in complete RPMI-1640 media containing 0.5 mM butyric acid for 7 days as per previous reports (Fischkoff 1988; Tiffany et al. 1995; Badewa et al. 2002).
MDI-GSH conjugation and exposure
MDI-GSH conjugates were prepared as previously described (Lin et al. 2020). Briefly, a 10 mM GSH solution was prepared in sodium phosphate buffer (200 mM; pH = 7.4). Fifty microlitres of freshly prepared stock solution of 10% MDI (w/v) in dry acetone were added to 25 ml of GSH solution dropwise with stirring, to a final MDI concentration of 800 μM, after which the tube was subjected to end-over-end mixing for 1 h at 25 °C. Samples were centrifuged at 10 000 × g and filtered with a 0.2 μm syringe filter. MDI-GSH conjugates were prepared immediately before use and used to treat 1 × 106 enhanced differentiated THP-1 macrophages in a 6 well plate. After 24 h, the cell culture supernatant (conditioned media) was collected, centrifuged, and stored at −20 °C until ready to use. Cells were washed 2 times with warm PBS and cell lysates were prepared by adding 600 μl of lysis/binding solution from the mirVana™ miR Isolation Kit (ThermoFisher Scientific) and stored at −80 °C until ready to use.
Transient transfection and PPP3CA overexpression
To investigate chemokine expression in macrophages overexpressing PPP3CA, 1 × 106 enhanced differentiated THP-1 macrophages were transfected with 2.5 μg of either pcDNA3.1+/c-(k)dyk-PPP3CA expression plasmid (GenScript, Piscataway, NJ, USA) or pcDNA3.1+ empty vector (ThermoFisher Scientific) using Mirus TransIT-2020 transfection reagent (Mirus Corporation; Madison, WI, USA) in a 6-well plate for 24 h. After 24 h, total RNA was extracted and prepared for RT-qPCR expression analysis to determine the expression of candidate chemokines. For miR functional analyses, the following mirVana® miRNA inhibitors (MH) were obtained from ThermoFisher Scientific (Waltham, MA) and diluted to 20 μM in nuclease-free water: hsa-miR-206-3p (MH10409), hsa-miR-381-3p (MH10242), and MH-negative control #1 (4464076). A total of 2 × 105 enhanced differentiated THP-1 macrophages were subjected to two rounds of transfection in 6-well plates: an initial reverse transfection followed 24 h later by forward transfection as previously described (Lin et al. 2011). At 24 h after the start of the forward transfection, the cell culture media were collected, centrifuged, and stored at −20 °C until ready to use. Cells were washed twice with PBS and cell extracts were prepared for RNA isolation and RT-qPCR expression analysis.
Expression analysis
Gene expression was determined by RT-qPCR analysis. Total RNA from BALCs or cultured THP-1 macrophages was extracted using mirVana™ miR Isolation Kit (ThermoFisher Scientific) as per manufacturer’s instructions. The mRNA and miR levels were determined as previously described (Lin et al. 2011). Candidate gene/miR expression was normalised to either human or mouse beta-2 microglobulin (B2M/B2m) for mRNA analysis, or to U6 snRNA for miR analysis. Gene expression assays and miR-specific assays used in this study were obtained from ThermoFisher Scientific (Waltham, MA) and include: human IL1B (Cat#:4331182/Assay ID:Hs01555410_m1), TNF (Hs00174128_m1), IL6 (Hs00174131_m1), CCL2/MCP-1 (Hs00234140_m1), CCL3/MIP-1α (Hs00234142_m1), CCL5/RANTES (Hs00982282_m1), CXCL8/IL8 (Hs00174103_m1), TGFB1 (Hs00998133_m1), B2M (Hs00187842_m1), mouse Il1b (Mm00434228_m1), Tnf (Mm00443258_m1), Il6 (Mm00446190_m1), Ccl2/Mcp-1 (Mm00441242_m1), Ccl3/Mip-1α (Mm00441259_g1), Ccl5/Rantes (Mm01302427_m1), Ccl11 (Mm00441238_m1), Ccl17/Tarc (Mm01244826_g1), Ccl22/Mdc (Mm00436439_m1), Tgfb1 (Mm01178820_m1), Gmcsf/Csf2 (Mm01290062_m1), Ptgs2/Cox2 (Mm03294838_g1), Alox5/5-Lox (Mm01182747_m1), Alox5ap/Flap (Mm00802100_m1) and B2m (Mm00437762_m1), hsa-miR-206-3p (Cat# 4427975; Assay ID#000510), hsa-miR-381-3p (#000571), and U6 snRNA (#001973). PCR reactions were performed on an ABI 7500 Real-Time PCR System from ThermoFisher Scientific (Waltham, MA). Expression of mRNAs and miRs were determined by using the ΔΔCT method as previous described (Lin et al. 2011).
Chemokine enzyme-linked immunosorbent assay
Conditioned media was collected from either MDI-GSH conjugate-treated THP-1 macrophages or miR-inhibitor transfected THP-1 macrophages as described above. The following enzyme-linked immunosorbent assay (ELISA) kits were obtained from ThermoFisher Scientific: Human CCL2/MCP-1 (Cat# 88739922), human CCL3/MIP-1α (#88703522), and human IL8/CXCL8 (#88808622). Human CCL5/RANTES ELISA kit (Cat# DY27805) was obtained from R&D systems (Minneapolis, MN). The assay sensitivity for each chemokine is as follows: CCL2 (7 pg/ml), CCL3 (16 pg/ml), CCL5 (15.6 pg/ml), and IL8/CXCL-8 (2 pg/ml). Human CCL2, CCL3, CCL5, and IL8 released into the conditioned media from THP-1 macrophages were measured by ELISA according to the manufacturer’s instructions.
In silico analysis of predicted interactions between CCL2, CCL3, CCL5, and CXCL8 transcripts and hsa-miR-206-3p/hsa-miR-381-3p
Potential interactions between the 3′ untranslated regions (3′UTRs) of human CCL2, CCL3, CCL5, and CXCL8/IL8 transcripts and hsa-miR-206-3p/hsa-miR-381-3p were first examined using the online in silico tool, TargetScanHuman 7.2 (http://www.targetscan.org/vert_72/) (Agarwal et al. 2015). Candidate miR-mRNA interactions were further examined with several in silico algorithms including miRanda (Enright et al. 2003), PicTar (Krek et al. 2005), PITA (Kertesz et al. 2007), and RNA22 (Miranda et al. 2006) using the web-based tool miRsystem (http://mirsystem.cgm.ntu.edu.tw/) (Lu et al. 2012). Furthermore, two online databases containing the most recent experimentally supported miR–gene interactions were queried to verify candidate miR binding to candidate mRNAs as followed: DIANA-TarBase v.8 (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8%2Findex/) (Karagkouni et al. 2018) and miRTarBase (https://mirtarbase.cuhk.edu.cn/~miRTarBase/miRTarBase_2022/php/search.php) (Huang et al. 2020).
Chemotaxis assays and quantification of migrated cells
Cell chemotaxis/migration assays were performed using 3 μm pore Transwell™ inserts in a 24-well plate format (Corning™ Transwell™ plates, ThermoFisher Scientific). Chemoattracted/migrating cells were quantified using the fluorescent cell counting dye CyQUANT® GR (ThermoFisher Scientific). The fluorescent cell counting method used with the Transwell™ assay counts all cells that migrate to the lower chamber—this method demonstrates a higher sensitivity than the traditional cell counting method using randomly selected high-power fields to count cells under a microscope (Gildea et al. 2000). Briefly, differentiated immune cells were collected, washed twice with PBS, and resuspended in serum-free RPMI 1640 media. A total of 1 × 106 cells in 100 μl serum-free RPMI 1640 media were added to each upper chamber and placed on the lower chamber containing chemoattractant (conditioned media). Five hundred microliters of cell-free conditioned media from either MDI-GSH conjugate treated or miR-inhibitor transfected THP-1 macrophages were placed in the lower chamber as a chemoattractant, and the cells were allowed to migrate for 6 h at 37 °C in a humidified atmosphere with 5% CO2. After 6 h, the lower chamber media containing migrated cells in suspension were collected in separate tubes and placed on ice. The media from the upper chamber was aspirated and discarded, and the upper chamber was transferred to a clean well containing PBS for washing. The upper chamber membrane and lower chamber surface were washed twice with PBS to collect migrated cells that remained attached to surfaces, 500 μl cell detaching media (0.25% Trypsin-EDTA, Cat#25200056, ThermoFisher Scientific) were added back to the lower chambers, and the upper chambers reinstalled. The whole plate was further incubated at 37 °C for 30 min to detach cells. After 30 min, the migrated cells in cell detaching media were combined with conditioned media/migrated cells collected previously, centrifuged at 300 × g for 5 min, washed with PBS twice, and stored at −80 °C before quantification using CyQUANT® Cell proliferation assay (ThermoFisher Scientific) as per manufacturer’s instructions.
Statistical analysis
Data were analysed using either the unpaired t-test (two-tailed; for Figure 1, and Figure 5(A)) or one-way analysis of variance followed by Tukey’s multiple comparison ad hoc post-test (All other figures). GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA) software was used for statistical analyses. Significant differences were considered when the analysis yielded p-value <0.05.
Results
In vivo MDI aerosol exposure upregulates expression of macrophage-secreted, asthma-associated mediators in BALCs
Dysfunction of macrophages, such as increased secretion of immune mediators (e.g. cytokines, chemokines, growth factors, bioactive lipids, etc.) in the lung plays an important role in asthma pathogenesis (Fricker and Gibson 2017; van der Veen et al. 2020). To investigate whether MDI aerosol exposure may affect macrophage-secreted mediators that are associated with asthma, we measured endogenous mRNA levels of cytokines (Il1b, Tnf, and Il6), chemokines (Ccl2, Ccl3, Ccl5, Ccl11, Ccl17, and Ccl22), growth factors (Tgfb1 and Gmcsf), and enzymes involves in de novo biosynthesis of bioactive lipids (Ptgs2, Alox5, and Alox5p) in the BALCs isolated from MDI aerosol exposed mice. Compared to mice exposed to house air, Il1b, Tnf, Il6, Ccl2, Ccl3, Ccl5, and Tgfb1 mRNAs were upregulated 2.41-, 1.39-, 30.06-, 45.01-, 4.27-, 24.33-, and 12.95-fold in BALCs collected at 24 h post-1 h MDI aerosol exposure, respectively (Figures 1(A–G)). Bioactive lipids, including prostaglandins (PGs), thromboxanes (TXs), leukotrienes (LTs), and others, can be de novo generated by macrophages via prostaglandin G/H synthase-2 (encoded by PTGS2), 5-lipoxygenases (5-Lox, encoded by Alox5), and arachidonate 5-lipoxygenase activating protein (FLAP, encoded by Alox5ap), and these bioactive lipids have been reported to play important roles in asthma pathogenesis (Diamant et al. 2019; Sokolowska et al. 2021). To examine whether MDI exposure affects de novo synthesis of bioactive lipids, we determined the expression of Ptgs2, Alox5, and Alox5ap expressions in BALCs from MDI aerosol exposed mice. The expression of Ptgs2, Alox5, and Alox5ap were unchanged in BALCs from MDI aerosol exposed mice compared to house air control exposed mice (Figures 1(H–J)), indicating that de novo synthesis of bioactive lipids in BALCs is not affected by MDI exposure. We did not measure the murine Il8 levels in these cells since a true human IL8 homolog has not been found in rodents. In addition, we were unable to detect any mRNA expression of Ccl11, Ccl17, Ccl22, or Gmcsf from BALCs isolated from either control or exposed mice (data not shown). We elected not to examine MDI regulation of PTGS2, ALOX5, ALOX5AP, CCL11, CCL17, CCL22, and GMCSF in in vitro THP-1 macrophage studies since these gene transcripts were unaffected/not detected in MDI aerosol exposures in vivo. These results indicate that MDI-aerosol exposure may increase the expression of several macrophage-secreted mediators in cells isolated from the alveoli of the lower airways.
Figure 1.
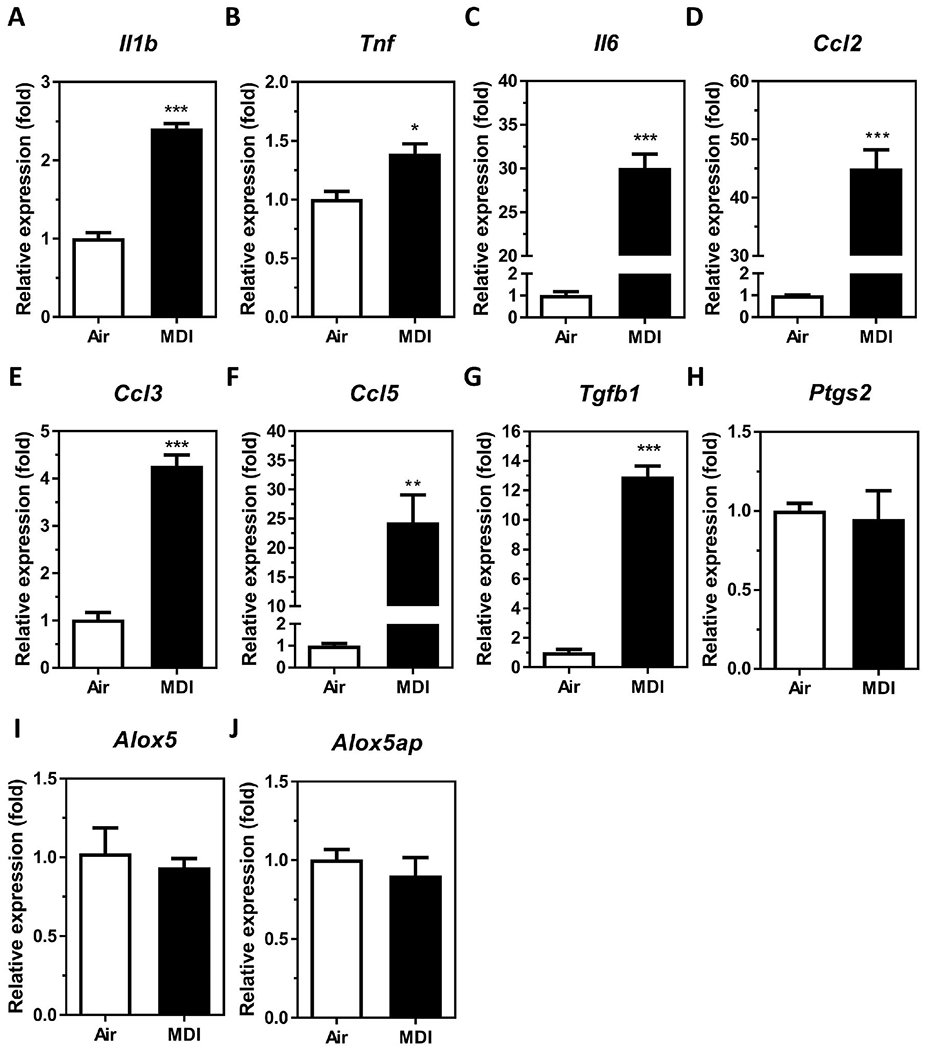
Candidate asthma-associated, macrophage-secreted mediator levels in BALCs isolated from MDI aerosol exposed mice. Total RNA was isolated from the cell fraction of BALF from MDI aerosol exposed mice by miRVana™ miR isolation kit, reverse transcribed, and subjected to gene specific TaqMan stem-loop RT-qPCR. Candidate mediator mRNA expression of (A) Il1b, (B) Tnf, (C) Il6, (D) Ccl2, (E) Ccl3, (F) Ccl5, (G) Tgfb1, (H) Ptgs2, (I) Alox5, and (J) Alox5ap were determined in BALCs isolated 24 h post-exposure [N = 3; bars, s.e.m). Air: house air; MDI: 4,4′-methylene diphenyl diisocyanate (*p < 0.05, **p < 0.01, ***p < 0.001).
In vitro exposure of differentiated THP-1 macrophages to MDI-GSH conjugate upregulates several macrophage-secreted mediators
Given that more than 80% of the cell population found in the BALCs are macrophages (Heron et al. 2012), and MDI has been predicted to react with the major antioxidant glutathione (GSH) that exists at a very high concentration (>100 μM) in the airway fluid (Cantin et al. 1987) to form MDI-GSH conjugates (Wisnewski et al. 2013; Wisnewski et al. 2015; Wisnewski et al. 2019), we used a THP-1 macrophage in vitro cell culture model to investigate the effects of MDI-GSH conjugate exposure on the expression of macrophage-derived, asthma-associated mediators. Exposure to MDI-GSH conjugates at 1 and 10 μM concentrations for 24 h upregulated the mRNA expression of IL1B, TNF, CCL2, CCL3, CCL5, CXCL8, and TGFB1 from 1.25- to 5.50-fold in differentiated THP-1 macrophages (Figure 2). However, the IL6 mRNA was not detected in either MDI-GSH conjugate- or control-exposed differentiated THP-1 macrophages (data not shown). These results indicate that MDI exposure in the form of MDI-GSH conjugates may increase IL1B, TNF, CCL2, CCL3, CCL5, CXCL8, and TGFB1 in macrophages.
Figure 2.
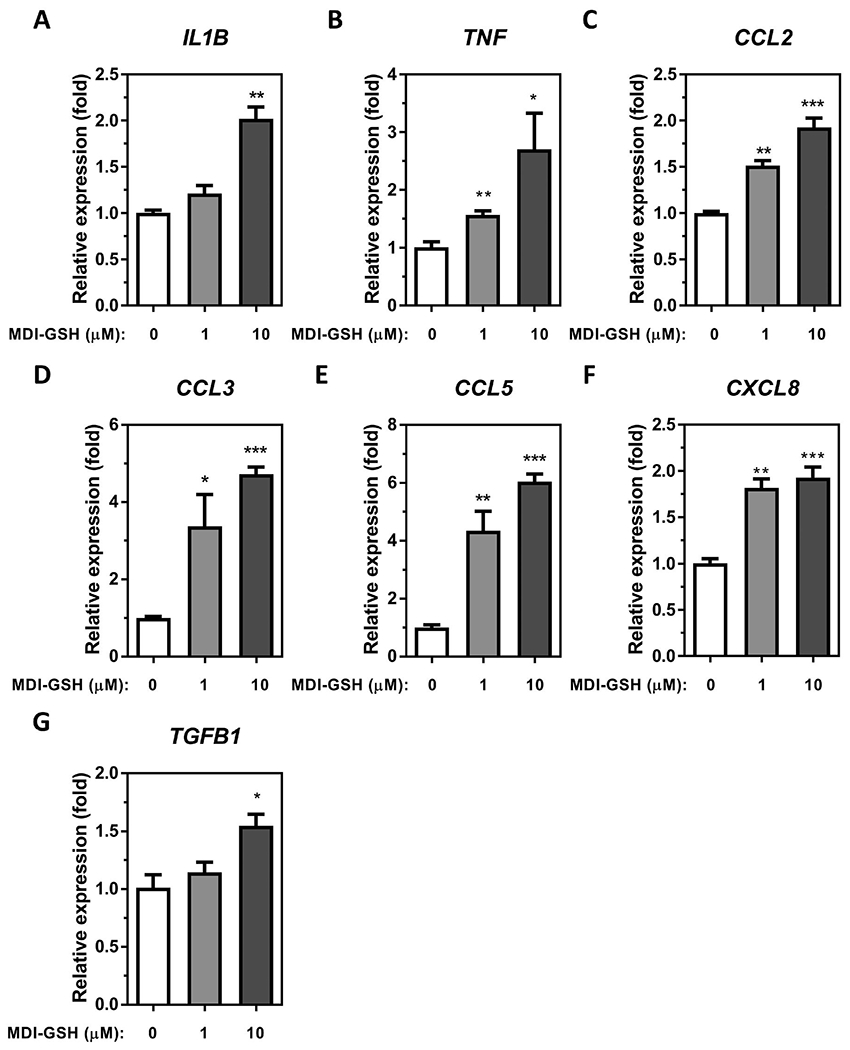
MDI-GSH conjugate induced macrophage secreted mediator expression in differentiated THP-1 macrophages. Total RNA was isolated from MDI-GSH conjugate exposed differentiated THP-1 macrophages at indicated concentration for 24 h by miRVana™ miR isolation kit, reverse transcribed, and subjected to TaqMan® stem-loop RT-qPCR. Candidate mediator mRNA expression of (A) IL1B, (B) TNF, (C) CCL2, (D) CCL3, (E) CCL5, (F) CXCL8, and (G) TGFB1 were determined at 24 h post-exposure (N = 3; bars, s.e.m). MDI: 4,4′-methylene diphenyl diisocyanate; GSH: glutathione (*p < 0.05, **p < 0.01, ***p < 0.001).
Calcineurin-mediated signalling is involved in the regulation of MDI-GSH conjugate-mediated chemokine upregulation in differentiated THP-1 macrophages
Activation of calcineurin-mediated NFAT signalling plays an important role in the upregulation of a variety of immune mediators in different immune cells (Fric et al. 2012). Our previous results demonstrated that exposure to MDI and/or MDI-GSH conjugates may activate this pathway through upregulation of endogenous PPP3CA/calcineurin A expression via decreased endogenous levels of miR-206-3p and miR-381-3p in macrophages (Lin et al. 2020). However, whether this PPP3CA/calcineurin-mediated signalling participates in the upregulation of macrophage-secreted mediators following MDI exposure is unknown. To determine whether calcineurin-mediated signalling participates in the regulation of macrophage-secreted mediators including IL1B, TNF, CCL2, CCL3, CCL5, CXCL8, and TGFB1, we used a loss-of-function strategy by using the specific calcineurin A inhibitor, tacrolimus (FK506), to suppress calcineurin-mediated signalling in MDI-GSH conjugate exposed THP-1 macrophages. Exposure to MDI-GSH conjugates upregulated IL1B, TNF, CCL2, CCL3, CCL5, CXCL8, and TGFB1 transcription (Figure 3(A–G)); however, only induction of chemokine CCL2, CCL3, CCL5, and CXCL8 transcripts by MDI-GSH conjugate exposure was observed to be suppressed by treatment with FK506 (Figure 3(C–F)). To determine whether calcineurin-mediated signalling is involved in the regulation of MDI-GSH conjugate-induced chemokine secretion in macrophages, we utilised ELISA to measure the secreted protein levels of CCL2, CCL3, CCL5, and IL8 in media collected from MDI-GSH conjugate treated macrophages. Consistent with the observation that MDI-GSH conjugate-induced chemokine mRNA levels were attenuated by FK506, the MDI-GSH conjugate-induced secreted chemokine protein levels in the media were decreased by the treatment of FK506 (Figure 3(H–K)). These results indicate that calcineurin-mediated signalling plays a role in the regulation of the MDI-mediated upregulation of chemokines CCL2, CCL3, CCL5, and IL8 in macrophages.
Figure 3.
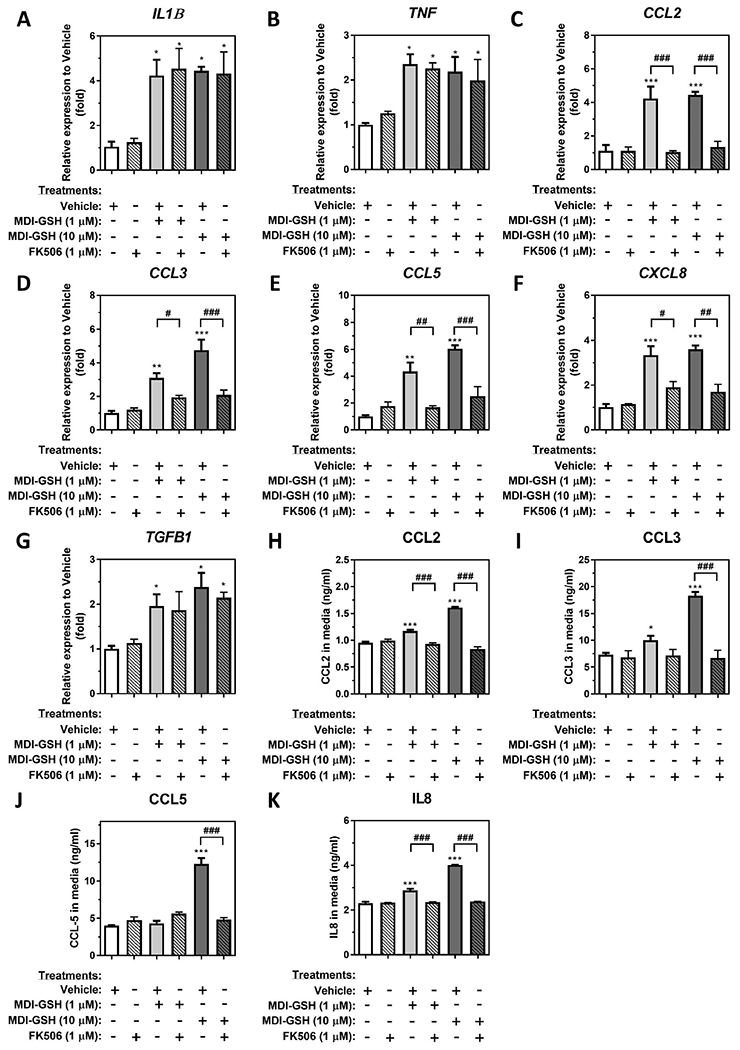
MDI-GSH conjugate induced macrophage-secreted chemokine expression through Calcineurin-mediated signalling activation in differentiated TMP-1 macrophages. Total RNA was isolated from MDI-GSH conjugate exposed differentiated THP-1 macrophages with or without 1 μM FK506 at indicated concentration for 24 h by miRVana™ miR isolation kit, reverse transcribed, and subjected to TaqMan® stem-loop RT-qPCR. Candidate mediator mRNA expression of (A) IL1B, (B) TNF, (C) CCL2, (D) CCL3, (E) CCL5, (F) CXCL8, and (G) TGFB1 were determined at 24 h post-exposure (N = 3; bars, s.e.m). To examine secreted protein levels, the conditioned media were collected from MDI-GSH conjugate exposed differentiated THP-1 macrophages with or without 1 μM FK506 at indicated concentration for 24 h. The protein levels of (H) CCL2, (I) CCL3, (J) CCL5, and (K) IL8 were assayed by ELISA (N = 3; bars, s.e.m). Vehicle: complete media containing 0.01% DMSO; MDI: 4,4′-methylene diphenyl diisocyanate; GSH: glutathione (*p < 0.05, **p < 0.01, ***p < 0.001, when compared to vehicle control; #p < 0.05, ##p < 0.01, ###p < 0.001, when compared to MDI-GSH conjugate exposed macrophages without FK506 inhibition).
Endogenous hsa-miR-206-3p and hsa-miR-381-3p regulate CCL2, CCL3, CCL5, and IL8 expression
Our previous report demonstrated that endogenous hsa-miR-206-3p and hsa-miR-381-3p were downregulated by MDI-GSH conjugate exposure, resulting in the upregulation of PPP3CA/calcineurin A expression and calcineurin signalling activation (Lin et al. 2020). Because the chemokine CCL2, CCL3, CCL5, and IL8 were upregulated by MDI-GSH conjugate treatment in macrophages, we hypothesised that either hsa-miR-206-3p or hsa-miR-381-3p may regulate the expression of chemokine CCL2, CCL3, CCL5, and IL8. We first examined the endogenous hsa-miR-206-3p, hsa-miR-381-3p, and PPP3CA in MDI-GSH conjugate treated macrophages by RT-qPCR (Figure 4(A)). Consistent with previous findings (Lin et al. 2020), treatment of MDI-GSH conjugate in macrophages significantly downregulated endogenous hsa-miR-206-3p from 1.75- to 2.99-fold (Figure 4(A), left panel), and hsa-miR-381-3p from 6.15- to 10.88-fold (Figure 4(A), middle panel), whereas MDI-GSH conjugates upregulated endogenous PPP3CA from 1.64- to 2.13-fold (Figure 4(A), right panel). To investigate whether endogenous hsa-miR-206-3p or hsa-miR-381-3p may serve as downstream effectors of MDI-GSH conjugates to regulate chemokines CCL2, CCL3, CCL5, and IL8 expression in macrophages, we transfected differentiated THP-1 macrophages with either miR-inhibitor-206-3p, miR-inhibitor-381-3p or non-targeting miR-inhibitor-control (Ctl) to mimic the down-regulation of either hsa-miR-206-3p or hsa-miR-381-3p caused by MDI-GSH conjugate exposure in macrophages. Transfection of either miR-inhibitor-206-3p or miR-inhibitor-381-3p into differentiated macrophages significantly upregulated endogenous PPP3CA transcript levels by 1.69- or 2.53-fold, respectively, when compared to macrophages transfected with miR-inhibitor non-targeting control (Figure 4(B)). Furthermore, transfection of either miR-inhibitor-206-3p or miR-inhibitor-381-3p upregulated endogenous CCL2, CCL3, CCL5, and CXCL8 mRNAs in macrophages (Figure 4(C–F), left panels), as well as the secreted protein levels of CCL2, CCL3, CCL5, and IL8 in the conditioned media collected from either miR-inhibitor-206-3p or miR-inhibitor-381-3p transfected macrophages (Figure 4(C–F), right panels). These results indicate that either endogenous hsa-miR-206-3p or hsa-miR-381-3p may regulate chemokines CCL2, CCL3, CCL5, and IL8 in macrophages.
Figure 4.
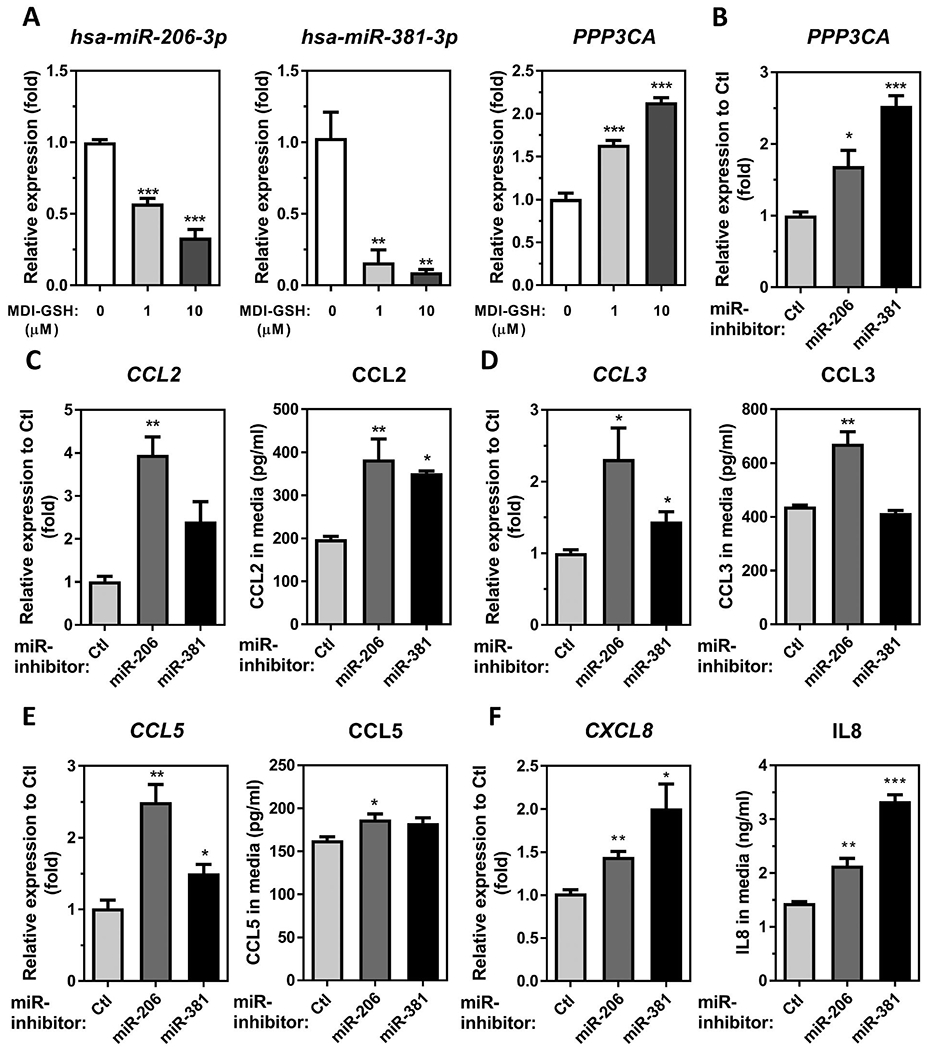
MDI mediated downregulation of either hsa-miR-206-3p or hsa-miR-381-3p is involved in chemokine upregulation in macrophages. (A) Total RNA was isolated from MDI-GSH conjugate exposed differentiated THP-1 macrophages at indicated concentration for 24 h by miRVana™ miR isolation kit, and reverse transcribed. The endogenous levels of hsa-miR-206-3p, hsa-miR-381-3p, and PPP3CA levels were determined by TaqMan® miR stem-loop RT-qPCR or TaqMan stem-loop RT-qPCR (N = 3; bars, s.e.m). (B–F) Total RNA was isolated from differentiated THP-1 macrophages transfected with either miR-inhibitor-206-3p, miR-inhibitor-381-3p or miR-inhibitor non-targeting control (Ctl) by miRVana™ miR isolation kit, reverse transcribed, and subjected to TaqMan® stem-loop RT-qPCR. The media were collected from miR inhibitor-transfected macrophages for measuring protein concentrations by ELISA. (B) Endogenous PPP3CA mRNA levels were determined by TaqMan® stem-loop RT-qPCR [N = 3; bars, s.e.m). Candidate chemokine mRNA expression and secreted protein concentrations of (C) CCL2, (D) CCL3, (E) CCL5, and (F) CXCL8/IL8, were determined at 24 h after macrophages subjected to forward miR-inhibitors transfection (N = 3; bars, s.e.m). MDI: 4,4′-methylene diphenyl diisocyanate; GSH: glutathione (*p < 0.05, **p < 0.01, ***p < 0.001).
Either hsa-miR-206-3p or hsa-miR-381-3p indirectly regulates chemokine CCL2, CCL3, CCL5, and IL8 expression partially through calcineurin-mediated signalling activation in THP-1 macrophages
The ability of hsa-miR-206-3p or hsa-miR-381-3p to regulate chemokines CCL2, CCL3, CCL5, and IL8 expression may either be through direct binding to the 3′UTR of these chemokine transcripts or through other indirect regulatory mechanisms. To determine possible interactions between hsa-miR-206-3p/hsa-miR-381-3p and candidate chemokine transcripts, we first used the in-silico algorithm TargetScanHuman 7.2 to retrieve candidate miRs that are predicted to target human CCL2, CCL3, CCL5, and CXCL8 transcripts (Supplemental Tables 1–4). TargetScanHuman 7.2 precited that hsa-miR-206-3p but not hsa-miR-381-3p can target CCL2 3′UTR (Supplemental Table 1), whereas neither hsa-miR-206-3p nor hsa-miR-381-3p were predicted to bind the 3′UTRs of CCL3, CCL5, and CXCL8 transcripts (Supplemental Tables 2–4). To further support the predicted interaction of the CCL2 3′UTR as one of the hsa-miR-206-3p targets, we used several in silico algorithms including miRanda, PicTar, PITA, and RNA22. However, the miRanda, PicTar, PITA, and RNA22 algorithms failed to predict any miR-mRNA interaction between the CCL2 3′UTR and hsa-miR-206-3p (data not shown). Similarly, neither hsa-miR-206-3p nor hsa-miR-381-3p were predicted to exhibit binding between any 3′UTRs of CCL3, CCL5, and CXCL8 transcripts when analysed by miRanda, PicTar, PITA, and RNA22 (data not shown). To confirm the suggested interaction between hsa-miR-206-3p and CCL2 transcripts as predicted by TargetScanHuman 7.2 in THP-1 macrophages, we performed an RNA-induced silencing complex (RISC)-immunoprecipitation (IP) experiment by using an anti-panArgonaute(AGO) antibody. The CCL2 mRNA was not enriched in RISC-IP of THP-1 macrophages transfected with miR-mimic-206-3p, suggesting that there is no interaction between the CCL2 transcript and hsa-miR-206-3p in macrophages (Supplemental Figure 1). Furthermore, we cannot find any experimentally validated record that demonstrates interactions between CCL2 3′UTR and hsa-miR-206-3p using two miR-mRNA interaction databases, DIANA-TarBase v8.0, and miRTarBase. Based on the combination of in silico miR-mRNA interaction analysis and RISC-IP experiments, we conclude that chemokines CCL2, CCL3, CCL5, and CXCL8 are not directly targeted by either hsa-miR-206-3p or hsa-miR-381-3p in THP-1 macrophages. Therefore, the demonstrated ability of hsa-miR-206-3p/hsa-miR-381-3p to upregulate CCL2, CCL3, CCL5, and IL8 mRNA expression likely utilises indirect regulatory mechanisms, such as targeting other important signalling molecules which leads to transcriptional activation of these genes.
Our previous report demonstrated that endogenous hsa-miR-206-3p and hsa-miR-381-3p regulate iNOS transcription through PPP3CA/calcineurin/NFAT signalling activation in macrophages (Lin et al. 2020), calcineurin/NFAT-mediated signalling pathways have been reported to regulate chemokine CCL2, CCL3, CCL5, CXCL8 expression in many different cell types (Staruch et al. 1998; Maldonado-Perez et al. 2009; Sales et al. 2009; Lawrence et al. 2011; Busch et al. 2016). We, therefore, hypothesise that endogenous hsa-miR-206-3p/hsa-miR-381-3p may be capable of regulating CCL2, CCL3, CCL5, CXCL8 transcription through indirect regulatory mechanism(s) by targeting PPP3CA/calcineurin-mediated signalling in macrophages. To test this hypothesis, we performed both a gain-of-function study by overexpression of PPP3CA and a loss-of-function study by using FK506 to suppress PPP3CA/calcineurin signalling in either miR-inhibitor-206-3p or miR-inhibitor-381-3p transfected THP-1 macrophages. Compared to vector-transfected differentiated THP-1 macrophages, PPP3CA overexpression induced endogenous CCL2, CCL3, CCL5, and CXCL8 mRNA by 2.28-, 7.04-, 7.02-, and 3.85-fold, respectively (Figure 5(A)). These results indicate that calcineurin-mediated pathways are involved in the regulation of CCL2, CCL3, CCL5, and CXCL8 transcription. Furthermore, we utilised FK506 to suppress calcineurin signalling in miR-inhibitor-206-3p and miR-inhibitor-381-3p transfected THP-1 macrophages. Consistent with the finding that transfection of miR-inhibitor-206-3p and miR-inhibitor-381-3p upregulated endogenous CCL2, CCL3, CCL5, and CXCL8 mRNA in THP-1 macrophages as well as secreted proteins in conditioned media (Figure 4), independent transfection of miR-inhibitor-206-3p upregulated CCL2, CCL3, CCL5, and CXCL8 mRNAs by 4.85-, 2.17-, 2.99-, and 1.80-fold, respectively, when compared to the miR-inhibitor-Ctl transfected THP-1 macrophages (Figure 5(B–E)). Similarly, transfection of miR-inhibitor-381-3p in macrophages upregulated CCL2, CCL3, CCL5, and CXCL8 mRNAs by 1.95-, 1.53-, 2.04-, and 2.27-fold, respectively (Figure 5(B–E)). Furthermore, treatment of 1 μM FK506 attenuated the induction of CCL2, CCL3, CCL5, and CXCL8 mRNA upregulation by transfection of either miR-inhibitor-206 or miR-inhibitor-381-3p in THP-1 macrophages (Figure 5(B–E)). These results indicate that PPP3CA/calcineurin-mediated signalling is important for CCL2, CCL3, CCL5, and CXCL8 transcriptional activation by inhibition of either hsa-miR-206-3p or hsa-miR-381-3p.
Figure 5.
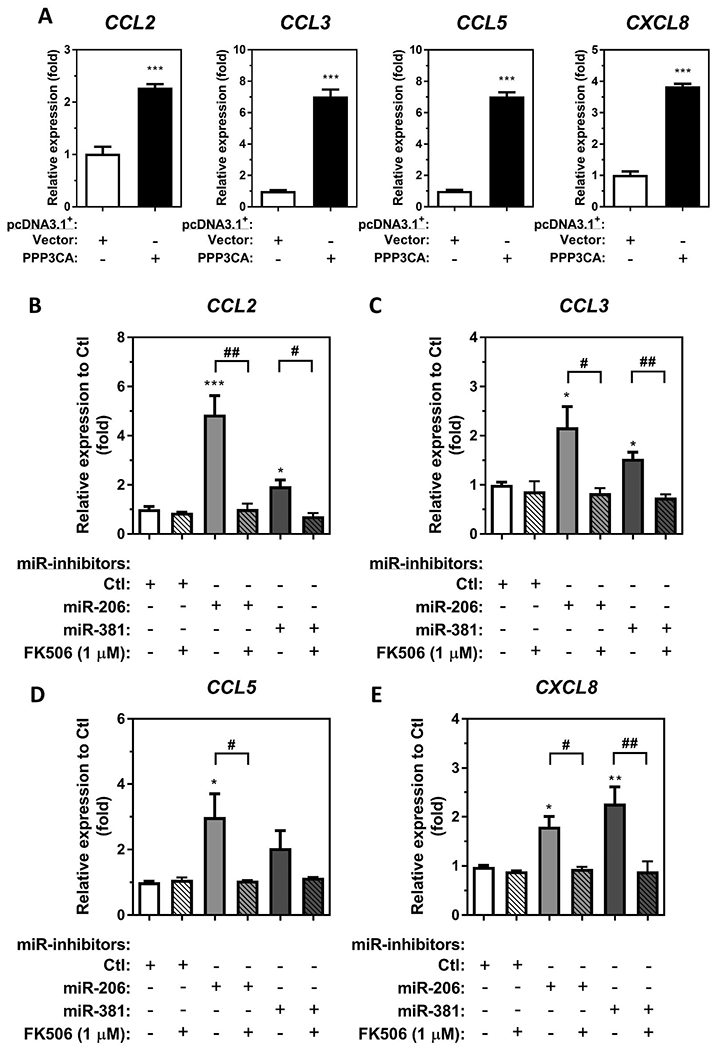
Transfection of miR-inhibitors of either hsa-miR-206-3p or hsa-miR-381-3p upregulate chemokine expression through Calcineurin-mediated signalling activation in differentiated THP-1 macrophages. (A), Differentiated THP-1 macrophages were transfected with 2.5 μg of either a pcDNA3.1+/c-(k)dyk-PPP3CA expression plasmid (PPP3CA) or a pcDNA3.1+ empty vector control (Vector). Total RNA was isolated, and the enrichment of CCL2, CCL3, CCL5, and CXCL8 transcripts was measured by RT-qPCR (N = 3; bars, s.e.m). Total RNA was isolated from differentiated THP-1 macrophages transfected with either miR-inhibitor-206-3p, miR-inhibitor-381-3p or miR-inhibitor non-targeting control (Ctl) with or without 1 μM FK506 for 24 h by miRVana™ miR isolation kit, reverse transcribed, and subjected to TaqMan® stem-loop RT-qPCR. Candidate chemokine mRNA expression of (B) CCL2, (C) CCL3, (D) CCL5, and (E) CXCL8 were determined at 24 h after miR-inhibitor transfection (N = 3; bars, s.e.m) (*p < 0.05, **p < 0.01, ***p < 0.001, when compared to macrophages transfected with miR-inhibitor non-targeting control; #p < 0.05, ##p < 0.01, when compared to miR-inhibitor transfected macrophages without FK506 treatment).
Chemotaxis/cell migration of macrophages, eosinophils, neutrophils, and T-cells is induced by macrophages exposed to MDI-GSH conjugates
Increased levels of chemokines CCL2, CCL3, CCL5, and IL8 from macrophages treated with MDI-GSH conjugates suggests that these chemokines may recruit immune cells including T-cells, macrophages, dendritic cells, eosinophils, and neutrophils that are associated with asthma pathogenesis. To determine whether MDI-GSH conjugate exposed macrophages can attract immune cells, we used in vitro transwell/cell migration assays to investigate chemotactic abilities of undifferentiated monocytes (untreated THP-1 cells), differentiated dendritic cells (dendritic cell differentiation cocktail differentiated THP-1 cells), differentiated macrophages (PMA treated, and enhanced THP-1 cells), eosinophils (butyric acid differentiated HL-60_C15 cells), neutrophils (DMSO differentiated HL-60 cells), and T-cells (Jurkat_E6-1 cells) in response to conditioned media obtained from MDI-GSH conjugate treated THP-1 macrophages. Cell-free conditioned media obtained from THP-1 macrophages treated with 10 μM MDI-GSH conjugates for 24 h significantly increased the cell migration of macrophages, eosinophils, neutrophils, and T-cells when compared to control (Figure 6(A–D)) but failed to attract monocytes and dendritic cells (Figure 6(E,F)). To investigate whether calcineurin-mediated signalling pathways may participate in MDI-GSH conjugate induced immune cell chemotaxis/migration, the cell-free conditioned media collected from macrophages treated with FK506/MDI-GSH conjugates were used as chemoattractant. Treatment with 1 μM FK506 significantly suppressed MDI-GSH conjugate-induced cell chemotaxis/migration of immune cells including macrophages, neutrophils, and T-cells (Figure 6(A,C,D)) but not eosinophils (Figure 6(B)) when compared to the cells exposed to the conditioned media from only MDI-GSH conjugate treated macrophages. Given that either hsa-miR-206-3p or hsa-miR-381-3p are involved in the regulation of chemokines CCL2, CCL3, CCL5, and IL8 expression as shown in Figure 4; down-regulation of either endogenous hsa-miR-206-3p or hsa-miR-381-3p may contribute to the chemoattraction of immune cells including macrophages, eosinophils, neutrophils, and T-cells. To verify that either endogenous hsa-miR-206-3p or hsa-miR-381-3p inhibition mediated signalling may play a role in chemoattraction of immune cells, the cell-free conditioned media collected from macrophages transfected with either miR-inhibitor-206-3p or miR-inhibitor-381-3p were used as a chemoattractant for macrophages, eosinophils, neutrophils, and T-cells chemotaxis/migration assays. Conditioned media collected from macrophages transfected with miR-inhibitor-206-3p significantly attracted neutrophils and T-cells (Figure 6(I,J)), but not macrophages or eosinophils (Figure 6(G,H)). Furthermore, conditioned media collected from macrophages transfected with miR-inhibitor-381-3p significantly attracted macrophages, neutrophils, and T-cells (Figure 6(G,I,J)) but not eosinophils (Figure 6(H)). These results indicated that MDI exposure in the form of MDI-GSH conjugate may increase chemotactic abilities of differentiated immune cells including macrophages, neutrophils, and undifferentiated T-cells partially through hsa-miR-206-3p or hsa-miR-381-3p regulated calcineurin signalling pathway activation in macrophages.
Figure 6.
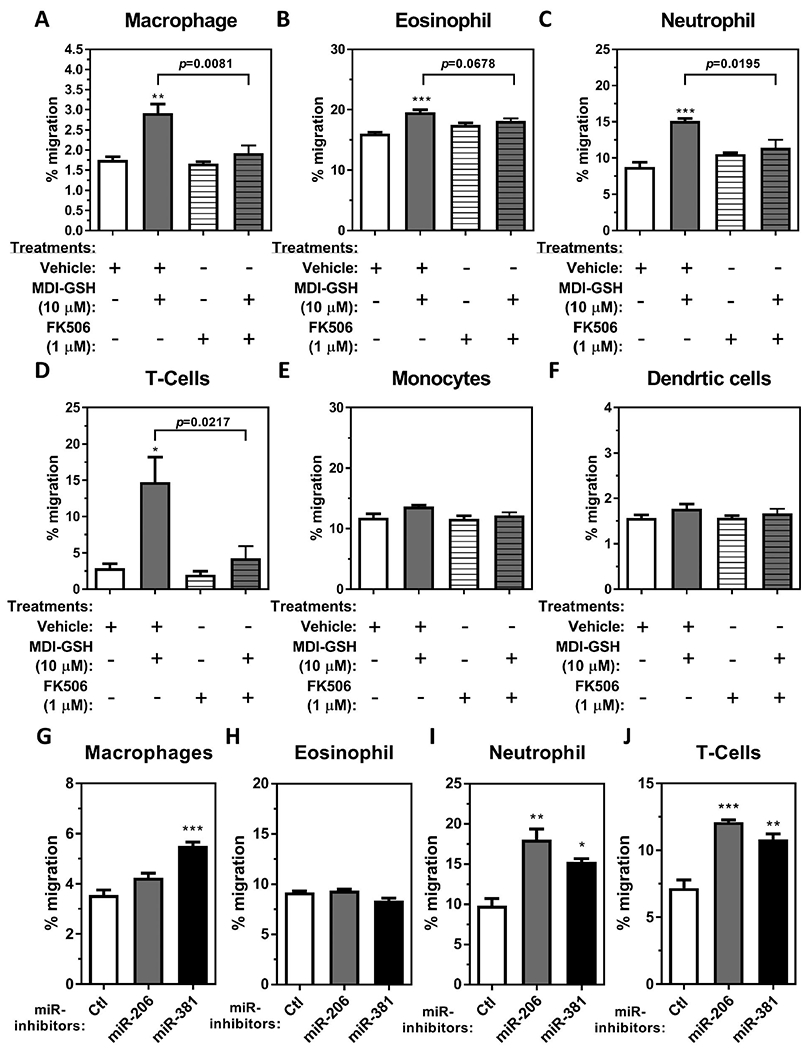
THP-1 macrophages treated with MDI-GSH conjugate induced macrophage, neutrophil, eosinophil, and T-cell migration. Cell-free conditioned media were obtained from THP-1 macrophages treated with 10 μM MDI-GSH conjugates with or without 1 μM FK506 for 24 h. Cell-free conditioned media were also collected 24 h after differentiated THP-1 macrophages subjected two rounds of miR-inhibitors transfection where the macrophages were transfected with either miR-inhibitor-206-3p or miR-inhibitor-381-3p or miR-inhibitor non-targeting control (Ctl). These isolated conditioned media were used as chemoattractant to attract immune cells as indicated. Immune cell migration responding to the conditioned media from MDI conjugate exposed macrophages with or without FK506 was measured after 6 h. Percent of cells migrated towards the bottom chamber are shown for (A) differentiated macrophages, (B) eosinophils, (C) neutrophils, (D), T-cells, (E) undifferentiated monocytes, and (F) dendritic cells (N = 3; bars, s.e.m). Immune cell migration in response to conditioned media isolated from macrophages transfected with miR-inhibitors are shown for (G) differentiated macrophages, (H) eosinophils, (I) neutrophils, and (J) T-cells (N = 3; bars, s.e.m). Vehicle: complete media containing 0.01% DMSO; MDI: 4,4′-methylene diphenyl diisocyanate; GSH: glutathione (*p < 0.05, **p < 0.01, ***p < 0.001).
Discussion
Although elevated levels of macrophage-secreted mediators in asthmatic airways may contribute to asthma pathogenesis (Balhara and Gounni 2012; Draijer and Peters-Golden 2017); the level of these mediators in the MDI-OA airways and the downstream molecular mechanism(s) that regulate these mediators’ expression after MDI exposure in alveolar macrophages is currently unclear. In the current study, we identified a potential miR-206-3p and miR-381-3p regulated calcineurin-mediated signalling activation that may be involved in the upregulation of chemokines CCL2, CCL3, CCL5, and CXCL8 transcription after MDI exposure. We found that the endogenous chemokine Ccl2/CCL2, Ccl3/CCL3, Ccl5/CCL5, and CXCL8 transcripts were upregulated in the BALCs from an in vivo MDI-aerosol exposure murine model and an in vitro human THP-1 macrophage model. The upregulation of chemokine CCL2, CCL3, CCL5, and CXCL8 transcription by MDI exposure was partially through calcineurin-mediated signalling activation in macrophages. In addition, conditioned media isolated from MDI-GSH conjugate treated macrophages induced the chemotactic activity in immune cells including T-cells, neutrophils, macrophages, and eosinophils (Figure 7). The induction of immune cell chemotaxis/migration abilities by macrophages exposed to MDI may account for one of the multiple pathophysiological mechanisms leading to MDI-OA development by recruiting immune cells into the airway microenvironment.
Figure 7.
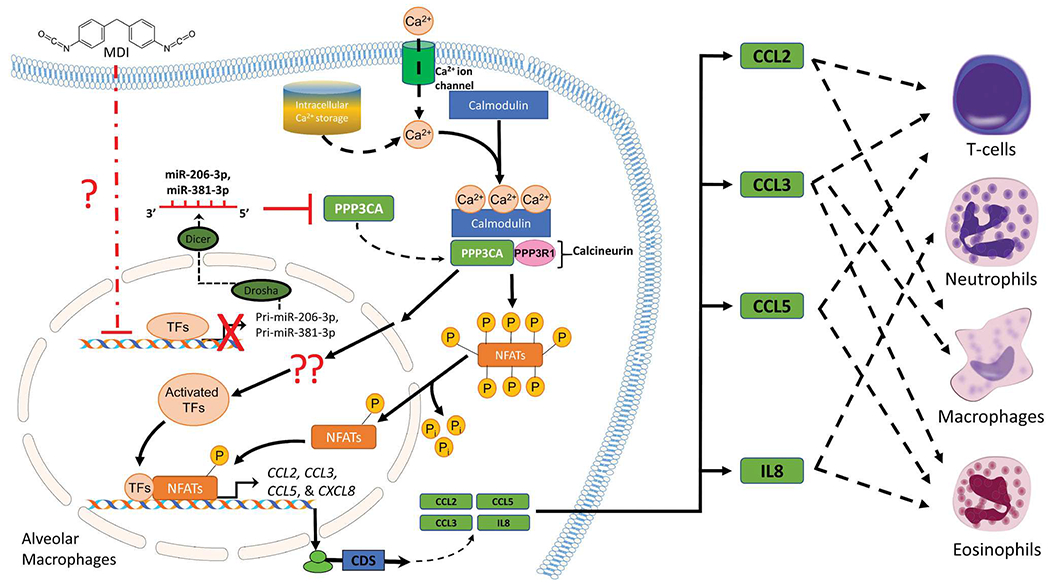
Proposed mechanisms by which MDI exposure induces chemokine CCL2, CCL3, CCL5, and IL8 expression and chemotactic activity in immune cells via miR-206-3p and/or miR-381-3p mediated PPP3CA/Calcineurin signalling pathway activation in macrophages. MDI: 4,4′-methylene diphenyl diisocyanate; TFs: transcription factors; CDS: coding sequences; NFATs: nuclear factor of activated T-cells. Note. Some illustrated schematics were obtained from motifolio templates (www.motifollo.com, Accessed 1 September 2021) or openclipart website (www.openclipart.org, Accessed 1 September 2021).
Macrophages can participate in altering asthmatic inflammation in several ways, including changes in inflammatory cytokine/chemokine production, polarisation status, cellular processes, such as phagocytosis and efferocytosis, as well as induction of the inflammasome in the asthmatic airways (van der Veen et al. 2020). These cells contribute to chronic airway inflammation in asthma through increased production/secretion of proinflammatory cytokines, such as IL-1β, TNF-α, and IL6 (Gosset et al. 1991; Goleva et al. 2008; Kim et al. 2017). MDI aerosol exposure induced proinflammatory cytokines Il1b, Tnf, and Il6 mRNAs in an in vivo MDI inhalation mouse model whereas the MDI-GSH conjugate treatments only induced IL1B and TNF transcription in an in vitro THP-1 macrophage culture model (Figures 1 and 2). We were not able to detect any IL6 mRNA expression in differentiated THP-1 macrophages. Although the ability of MDI to induce IL6 in human macrophages remains unclear from the current study, our results indicate that MDI exposure may induce the production of proinflammatory cytokines including IL-1β and TNF-α, which may contribute to MDI-OA pathogenesis. This current report investigates the role of calcineurin-mediated signalling in MDI-regulated macrophage-secreted mediator transcription regulation by using a specific calcineurin A inhibitor, FK506. FK506 treatment failed to attenuate the induction of IL1B, and TNF transcription by MDI-GSH conjugate treatments in macrophages (Figure 3(A,B)), indicating that MDI upregulation of IL1B, and TNF does not proceed via calcineurin-mediated NFAT signalling but rather other signalling pathways. Although the molecular mechanism(s) by which MDI induced IL1B and TNF is currently unknown, studies from other closely related diisocyanate-induced occupational asthma models may give some insight into how MDI contributes to the induction of these cytokines. Liang et al. reported that TDI exposure induced airway IL-1β levels through activation of a PI3K-signalling mediated lung caspase-1 and HMGB1 upregulation (Liang et al. 2015). Could MDI exposure result in similar PI3K-mediated signalling to upregulate IL-1β expression in the airways? Further studies are required to understand how MDI modulates IL-1β transcription in the macrophages.
In addition, TNF-α has been shown to play important roles in the regulation of both non-specific processes and specific immune events (such as Th2 responses following TDI exposure) that may contribute to TDI-OA pathogenesis (Matheson et al. 2002); however, the detailed mechanism(s) by which dNCOs upregulate TNF-α expression after exposure is still unclear. Activation of TNF transcription requires binding of nudeoprotein complexes (Falvo et al. 2000; Tsai et al. 2000; Tsytsykova and Goldfeld 2002), which contain sets of transcription factors and coactivators including NFATs, Ets/Elk, AP-1, NFκB, CEBP/β, and others (Barthel et al. 2003; Falvo et al. 2010) to the enhancer or promoter region of corresponding DNA elements located on the promoter region of the TNF gene. There are at least six putative NFAT binding elements (Goldfeld et al. 1993; McCaffrey et al. 1994; Tsai et al. 1996; Tsytsykova and Goldfeld 2002), along with four Ets/Elk binding sites (Kramer et al. 1995; Tsai et al. 2000; Tsytsykova and Goldfeld 2002), and two SP-1 binding sites (Goldfeld et al. 1991; Kramer et al. 1994; Tsytsykova and Goldfeld 2002) in the human TNF promoter region, indicating potential NFAT involvement in transcriptional regulation of TNF. However, three of the NFAT binding sites overlap with Ets/Elk sites while one NFAT binding site overlaps with one of the SP-1 binding sites (Tsai et al. 2000; Tsytsykova and Goldfeld 2002) located in the promoter region, suggesting that intracellular active NFATs may compete with Ets/Elk or SP-1 transcription factors on the TNF promoter region to regulate TNF transcription. The binding of NFATs on the putative NFAT binding sites may be cell type and stimuli dependent. For example, when monocytes are stimulated with LPS, the putative NFAT binding sites are occupied with SP1 and Ets/Elk proteins, whereas the same NFAT binding sites of the TNF promoter were occupied by NFAT proteins in T-cells with the same LPS stimulus (Tsai et al. 2000; Tsytsykova and Goldfeld 2002). In the current report, FK506 treatment failed to attenuate the induction of TNF transcription by MDI-GSH conjugates (Figure 3(B)) which may indicate that NFAT proteins are not involved in MDI-GSH conjugate-mediated TNF upregulation but rather other transcription factors, such as Ets/Elk or SP-1 transcription factors in macrophages. Future studies will investigate the possible role of MDI-mediated signalling leading to Ets/Elk and/or SP1 activation. Furthermore, the levels of these proinflammatory cytokines in the airways from MDI-OA patients have not been reported in the literature. Determination of these cytokines in MDI-OA patient airways may be beneficial to understand the pathogenesis of this disease.
A recent microarray-based study examining gene expression changes from lung tissues isolated from an MDI-OA murine model identified that chemokines, including Ccl6, Ccl8, Ccl9, Ccl11, Ccl12, Ccl17, and Ccl22, were among the most changed genes induced by MDI exposure (Wisnewski et al. 2020). Interestingly, our previous study identified that the chemokine signalling pathways were among the most enriched biological pathways that were targeted by both miR-206-3p and miR-381-3p (Lin et al. 2019), indicating that either miR-206-3p or miR-381-3p may participate in the regulation of chemokine expression and/or chemokine-induced signalling pathways. Our current results showed that CCL2, CCL3, CCL5, and CXCL8 transcription are induced by MDI-GSH conjugates through miR-206-3p and miR-381-3p mediated signalling pathways—potentially through calcineurin-mediated signalling activation in macrophages; it will be interesting to examine whether the same mechanism could apply to MDI-induced chemokines, such as CCL6, CCL8, CCL9, CCL11, CCL12, CCL17, and CCL22.
Immune cells, including T-cells, macrophages, dendritic cells, eosinophils, neutrophils, and other cell types have been shown to be involved in asthma pathogenesis (Boonpiyathad et al. 2019; Zhu et al. 2020). Infiltration of these immune cells into the lung following irritation or allergen exposure in the airways and the interactions between these immune cells and airway cells within lung microenvironments play critical roles during the development of asthma (Barnes 2008; Holgate 2008). Among the immune cells that infiltrate into the lung during the early development of asthma, T-cells have been identified as an important cell type that can become activated and differentiated into different classes of T helper (Th) cells (e.g. Th1, Th2, Th17, etc.), Treg cells, memory T cells, natural killer T cells, and others; these differentiated and activated T-cells can orchestrate different aspects of immune processes leading to asthma (Medoff et al. 2008). Early reports have shown that increased numbers of activated T-cell were displayed in the airways from workers with dNCO-OA (Bentley et al. 1992; Redlich et al. 1996), indicating that lung T-cell infiltration may play an important role in dNCO-OA pathogenesis. In the occupational clinic, more activated T-cells were found infiltrated into airway mucosa after MDI-specific challenge of the patients with MDI-OA than those from asymptomatic normal control subjects with a history of exposure to MDI (Hur et al. 2008), indicating that T-cells can be attracted and activated in the airways after MDI exposure. In addition, neutrophils are among the first responsive innate immune cell types recruited to the lung associated with asthma-specific events, such as asthma exacerbations (Norzila et al. 2000). After neutrophils are recruited into the lung, they can produce many neutrophilic mediators that contribute to early and late asthma responses and have been associated with severe asthma development (Monteseirín 2009; Radermecker et al. 2018). Several reports have shown predominant lung neutrophilia inflammation in TDI-OA patients and many TDI-OA animal models (Fabbri et al. 1987; Park H et al. 1999; Vanoirbeek et al. 2004; Matheson et al. 2005; Tarkowski et al. 2007). Furthermore, neutrophil infiltration into the lung is also identified in MDI-OA animal models (Pauluhn et al. 2005; Pollaris et al. 2016), indicating that neutrophil recruitment may play an important role in MDI-OA pathogenesis. The current report demonstrates that undifferentiated naïve Jurkat T-cells (induced by 5.06-fold; Figure 6(D)), as well as neutrophils (induced by 1.72-fold; Figure 6(C)) were the two most responsive cell types that can be attracted by MDI-GSH conjugate exposed macrophages or miR-inhibitor transfected macrophages (Figure 6(I,J)). Interestingly, macrophages treated with a calcineurin inhibitor, FK506, inhibited the chemotaxis/migration of these immune cells (Figure 6). Previous reports have suggested the use of calcineurin/NFAT signalling pathway inhibitors, such as cyclosporine and tacrolimus (FK506), to treat asthma (Lock et al. 1996; Kawano et al. 2004; Taniguchi et al. 2011). The current report suggests a potential therapeutic strategy by using calcineurin-mediated signalling inhibitors, such as FK506, or using hsa-miR-206-3p/hsa-miR-381-3p agonist/supplements as therapeutic drugs to treat MDI-OA; further studies will be needed to investigate this hypothesis.
Conclusion
In conclusion, this report demonstrates that many macrophage mediators including proinflammatory cytokines IL1B and TNF as well as chemokines CCL2, CCL3, CCL5, and CXCL8 were upregulated after MDI aerosol or MDI-GSH conjugate exposure in BALCs/macrophages. The miR-206-3p and miR-381-3p-mediated calcineurin signalling pathway was identified as an important pathway to upregulate chemokine CCL2, CCL3, CCL5, and CXCL8 transcription in airway macrophages after MDI exposure. The chemotaxis/cell migration of macrophages, eosinophils, neutrophils, and T-cells is attributed to increased expression of chemokines CCL2, CCL3, CCL5, and IL8. This miR-206-3p/miR-381-3p regulated mechanism may be ultimately responsible for the pathogenesis of MDI-OA.
Supplementary Material
Funding
This work was supported by intramural funds (CAN#19390BN8) from the National Institute for Occupational Safety and Health, Centres for Disease Control and Prevention.
Footnotes
This work was authored as part of the Contributor’s official duties as an Employee of the United States Government and is therefore a work of the United States Government. In accordance with 17 USC. 105, no copyright protection is available for such works under US Law.
Supplemental data for this article can be accessed here.
Disclosure statement
The authors declare that they have no conflicting financial interests. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the National Institute for Occupational Safety and Health, Centres for Disease Control and Prevention.
References
- Ackerman V, Marini M, Vittori E, Bellini A, Vassali G, Mattoli S. 1994. Detection of cytokines and their cell sources in bronchial biopsy specimens from asthmatic patients. Relationship to atopic status, symptoms, and level of airway hyperresponsiveness. Chest. 105(3):687–696. [DOI] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam J-W, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allport DC, Gilbert DS, Outterside SM. 2003. MDI and TDI: a safety, health and the environment: a source book and practical guide. New York (NY): J. Wiley. [Google Scholar]
- Badewa AP, Hudson CE, Heiman AS. 2002. Regulatory effects of eotaxin, eotaxin-2, and eotaxin-3 on eosinophil degranulation and superoxide anion generation. Exp Biol Med. 227(8):645–651. [DOI] [PubMed] [Google Scholar]
- Balhara J, Gounni AS. 2012. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol. 5(6):605–609. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. 2008. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 8(3):183–192. [DOI] [PubMed] [Google Scholar]
- Barthel R, Tsytsykova AV, Barczak AK, Tsai EY, Dascher CC, Brenner MB, Goldfeld AE. 2003. Regulation of tumor necrosis factor alpha gene expression by mycobacteria involves the assembly of a unique enhanceosome dependent on the coactivator proteins CBP/p300. Mol Cell Biol. 23(2):526–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley AM, Maestrelli P, Saetta M, Fabbri LM, Robinson DS, Bradley BL, Jeffery PK, Durham SR, Kay AB. 1992. Activated T-lymphocytes and eosinophils in the bronchial mucosa in isocyanate-induced asthma. J Allergy Clin Immunol. 89(4):821–829. [DOI] [PubMed] [Google Scholar]
- Berges C, Naujokat C, Tinapp S, Wieczorek H, Hoh A, Sadeghi M, Opelz G, Daniel V. 2005. A cell line model for the differentiation of human dendritic cells. Biochem Biophys Res Commun. 333(3):896–907. [DOI] [PubMed] [Google Scholar]
- Bernstein DI, Korbee L, Stauder T, Bernstein JA, Scinto J, Herd ZL, Bernstein IL. 1993. The low prevalence of occupational asthma and antibody-dependent sensitization to diphenylmethane diisocyanate in a plant engineered for minimal exposure to diisocyanates. J Allergy Clin Immunol. 92(3):387–396. [DOI] [PubMed] [Google Scholar]
- Boonpiyathad T, Sözener ZC, Satitsuksanoa P, Akdis CA. 2019. Immunologic mechanisms in asthma. Semin Immunol. 46:101333. [DOI] [PubMed] [Google Scholar]
- Busch R, Murti K, Liu J, Patra AK, Muhammad K, Knobeloch KP, Lichtinger M, Bonifer C, Wortge S, Waisman A, et al. 2016. NFATc1 releases BCL6-dependent repression of CCR2 agonist expression in peritoneal macrophages from Saccharomyces cerevisiae infected mice. Eur J Immunol. 46(3):534–646. [DOI] [PubMed] [Google Scholar]
- Cantin AM, North SL, Hubbard RC, Crystal RG. 1987. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol. 63(1):152–157. [DOI] [PubMed] [Google Scholar]
- Diamant Z, Aalders W, Parulekar A, Bjermer L, Hanania NA. 2019. Targeting lipid mediators in asthma: time for reappraisal. Curr Opin Pulm Med. 25(1):121–127. [DOI] [PubMed] [Google Scholar]
- Draijer C, Peters-Golden M. 2017. Alveolar macrophages in allergic asthma: the forgotten cell awakes. Curr Allergy Asthma Rep. 17(2):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engfeldt M, Isaksson M, Zimerson E, Bruze M. 2013. Several cases of work-related allergic contact dermatitis caused by isocyanates at a company manufacturing heat exchangers. Contact Dermatitis. 68(3): 175–180. [DOI] [PubMed] [Google Scholar]
- Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. 2003. MicroRNA targets in Drosophila. Genome Biol. 5(1):R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri LM, Boschetto P, Zocca E, Milani G, Pivirotto F, Plebani M, Burlina A, Licata B, Mapp CE. 1987. Bronchoalveolar neutrophilia during late asthmatic reactions induced by toluene diisocyanate. Am Rev Respir Dis. 136(1):56–42. [DOI] [PubMed] [Google Scholar]
- Falvo JV, Tsytsykova AV, Goldfeld AE. 2010. Transcriptional control of the TNF gene. Curr Dir Autoimmun. 11:27–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falvo JV, Uglialoro AM, Brinkman BM, Merika M, Parekh BS, Tsai EY, King HC, Morielli AD, Peralta EG, Maniatis T, et al. 2000. Stimulus-specific assembly of enhancer complexes on the tumor necrosis factor alpha gene promoter. Mol Cell Biol. 20(6):2239–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischkoff SA. 1988. Graded increase in probability of eosinophilic differentiation of HL-60 promyelocytic leukemia cells induced by culture under alkaline conditions. Leuk Res. 12(8):579–686. [DOI] [PubMed] [Google Scholar]
- Fric J, Zelante T, Wong AY, Mertes A, Yu HB, Ricciardi-Castagnoli P. 2012. NFAT control of innate immunity. Blood. 120(7):1380–1389. [DOI] [PubMed] [Google Scholar]
- Fricker M, Gibson PG. 2017. Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J. 50(3):1700196. [DOI] [PubMed] [Google Scholar]
- Gildea JJ, Harding MA, Guiding KM, Theodorescu D. 2000. Transmembrane motility assay of transiently transfected cells by fluorescent cell counting and luciferase measurement. Biotechniques. 29(1):81–86. [DOI] [PubMed] [Google Scholar]
- Goldfeld AE, McCaffrey PG, Strominger JL, Rao A. 1993. Identification of a novel cyclosporin-sensitive element in the human tumor necrosis factor alpha gene promoter. J Exp Med. 178(4):1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfeld AE, Strominger JL, Doyle C. 1991. Human tumor necrosis factor alpha gene regulation in phorbol ester stimulated T and B cell lines. J Exp Med. 174(1 ):73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goleva E, Hauk PJ, Hall CF, Liu AH, Riches DW, Martin RJ, Leung DY. 2008. Corticosteroid-resistant asthma is associated with classical anti-microbial activation of airway macrophages. J Allergy Clin Immunol. 122(3):550–559.e553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset P, Tsicopoulos A, Wallaert B, Vannimenus C, Joseph M, Tonnel AB, Capron A. 1991. Increased secretion of tumor necrosis factor alpha and interleukin-6 by alveolar macrophages consecutive to the development of the late asthmatic reaction. J Allergy Clin Immunol. 88(4):561–571. [DOI] [PubMed] [Google Scholar]
- Hamilton RF Jr., Thakur SA, Holian A. 2008. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med. 44(7):1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron M, Grutters JC, ten Dam-Molenkamp KM, Hijdra D, van Heugten-Roeling A, Claessen AM, Ruven HJ, van den Bosch JM, van Velzen-Blad H. 2012. Bronchoalveolar lavage cell pattern from healthy human lung. Clin Exp Immunol. 167(3):523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettick JM, Law BF, Lin C-C, Wisnewski AV, Siegel PD. 2018. Mass spectrometry-based analysis of murine bronchoalveolar lavage fluid following respiratory exposure to 4,4′-methylene diphenyl diisocyanate aerosol. Xenobiotica. 48(6):626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraiwa K, van Eeden SF. 2013. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediators Inflamm. 2013:619523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgate ST. 2008. Pathogenesis of asthma. Clin Exp Allergy. 38(6): 872–897. [DOI] [PubMed] [Google Scholar]
- Hoshi H, Ohno I, Honma M, Tanno Y, Yamauchi K, Tamura G, Shirato K. 1995. IL-5, IL-8 and GM-CSF immunostaining of sputum cells in bronchial asthma and chronic bronchitis. Clin Exp Allergy. 25(8):720–728. [DOI] [PubMed] [Google Scholar]
- Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC, Tang Y, Chen YG, Jin CN, Yu Y, et al. 2020. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 48(D1):D148–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman LJ, Judy DJ, Frazer D, Shapiro RE, Castranova V, Billie M, Dedhia HV. 1997. Inhalation of toluene diisocyanate is associated with increased production of nitric oxide by rat bronchoalveolar lavage cells. Toxicol Appl Pharmacol. 145(1):61–67. [DOI] [PubMed] [Google Scholar]
- Hur GY, Sheen SS, Kang YM, Koh DH, Park HJ, Ye YM, Yim HE, Kim KS, Park HS. 2008. Histamine release and inflammatory cell infiltration in airway Mucosa in methylene diphenyl diisocyanate (MDI)-induced occupational asthma. J Clin Immunol. 28(5):571–580. [DOI] [PubMed] [Google Scholar]
- Jan RL, Chen SH, Chang HY, Yeh HJ, Shieh CC, Wang JY. 2008. Asthma-like syndrome in school children after accidental exposure to xylene and methylene diphenyl diisocyanate. J Microbiol Immunol Infect. 41(4):537–341. [PubMed] [Google Scholar]
- Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, Papadimitrlou D, Kavakiotis I, Manlou S, Skoufos G, et al. 2018. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 46(D1):D239–D245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Matsuse H, Kondo Y, Machida I, Saeki S, Tomari S, Mitsuta K, Fukushima C, Obase Y, Shimoda T, et al. 2004. Tacrolimus reduces urinary excretion of leukotriene E(4) and inhibits aspirin-induced asthma to threshold dose of aspirin. J Allergy Clin Immunol. 114(6): 1278–1281. [DOI] [PubMed] [Google Scholar]
- Kertesz M, lovino N, Unnerstall U, Gaul U, Segal E. 2007. The role of site accessibility in microRNA target recognition. Nat Genet. 39(10): 1278–1284. [DOI] [PubMed] [Google Scholar]
- Kim RY, Pinkerton JW, Essilfie AT, Robertson AAB, Baines KJ, Brown AC, Mayall JR, Ali MX, Starkey MR, Hansbro NG, et al. 2017. Role for NLRP3 inflammasome-mediated, IL-1 β-dependent responses in severe, steroid-resistant asthma. Am J Respir Crit Care Med. 196(3):283–297. [DOI] [PubMed] [Google Scholar]
- Kramer B, Meichle A, Hensel G, Charnay P, Kronke M. 1994. Characterization of an Krox-24/Egr-1-responsive element in the human tumor necrosis factor promoter. Biochim Biophys Acta. 1219(2):413–421. [DOI] [PubMed] [Google Scholar]
- Kramer B, Wiegmann K, Kronke M. 1995. Regulation of the human TNF promoter by the transcription factor ETS. J Biol Chem. 270(12): 6577–6583. [DOI] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. 2005. Combinatorial microRNA target predictions. Nat Genet. 37(5):495–500. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Naziruddin B, Levy MF, Jackson A, McGlynn K. 2011. Calcineurin/nudear factor of activated T cells and MAPK signaling induce TNF-{alpha} gene expression in pancreatic islet endocrine cells. J Biol Chem. 286(2):1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zhao H, Yao L, Tang H, Dong H, Wu Y, Liu L, Zou F, Cai S. 2015. Phosphatidylinositol 3-kinases pathway mediates lung caspase-1 activation and high mobility group box 1 production in a toluene-diisocyanate induced murine asthma model. Toxicol Lett. 236(1):25–33. [DOI] [PubMed] [Google Scholar]
- Lin CC, Law BF, Hettick JM. 2020. Acute 4,4′-methylene diphenyl diisocyanate exposure-mediated downregulation of miR-206-3p and miR-381-3p activates inducible nitric oxide synthase transcription by targeting calcineurin/NFAT signaling in macrophages. Toxicol Sci. 173(1): 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Law BF, Siegel PD, Hettick JM. 2019. Circulating miRs-183–5p, −206–3p and −381–3p may serve as novel biomarkers for 4,4′-methy-lene diphenyl diisocyanate exposure. Biomarkers. 24(1):76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Liu LZ, Addison JB, Wonderlin WF, Ivanov AV, Ruppert JM. 2011. A KLF4-miRNA-206 autoregulatory feedback loop can promote or inhibit protein translation depending upon cell context. Mol Cell Biol. 31 (12):2513–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock SH, Kay AB, Barnes NC. 1996. Double-blind, placebo-controlled study of cyclosporin A as a corticosteroid-sparing agent in corticosteroid-dependent asthma. Am J Respir Crit Care Med. 153(2):509–514. [DOI] [PubMed] [Google Scholar]
- Lofgren DJ, Walley TL, Peters PM, Weis ML. 2003. MDI exposure for spray-on truck bed lining. Appl Occup Environ Hyg. 18(10):772–779. [DOI] [PubMed] [Google Scholar]
- Lu TP, Lee CY, Tsai MH, Chiu YC, Hsiao CK, Lai LC, Chuang EY. 2012. miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets. PLOS One. 7(8):e42390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Perez D, Brown P, Morgan K, Millar RP, Thompson EA, Jabbour HN. 2009. Prokineticin 1 modulates IL-8 expression via the calcineurin/NFAT signaling pathway. Biochim Biophys Acta. 1793(7): 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson JM, Johnson VJ, Vallyathan V, Luster MI. 2005. Exposure and immunological determinants in a murine model for toluene diisocyanate (TDI) asthma. Toxicol Sci. 84(1):88–98. [DOI] [PubMed] [Google Scholar]
- Matheson JM, Lemus R, Lange RW, Karol MH, Luster MI. 2002. Role of tumor necrosis factor in toluene diisocyanate asthma. Am J Respir Cell Mol Biol. 27(4):396–405. [DOI] [PubMed] [Google Scholar]
- McCaffrey PG, Goldfeld AE, Rao A. 1994. The role of NFATp in cyclosporin A-sensitive tumor necrosis factor-alpha gene transcription. J Biol Chem. 269(48):30445–30450. [PubMed] [Google Scholar]
- Medoff BD, Thomas SY, Luster AD. 2008. T cell trafficking in allergic asthma: the ins and outs. Annu Rev Immunol. 26:205–232. [DOI] [PubMed] [Google Scholar]
- Millius A, Weiner OD. 2010. Manipulation of neutrophil-like HL-60 cells for the study of directed cell migration. In: Papkovsky DB , editor. Live cell imaging: methods and protocols. Totowa (NJ): Humana Press; p. 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. 2006. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 126(6):1203–1217. [DOI] [PubMed] [Google Scholar]
- Monteseirin J 2009. Neutrophils and asthma. J Investig Allergol Clin Immunol. 19(5):340–354. [PubMed] [Google Scholar]
- Munn SA, Aschberger K, Cosgrove O, Pakalin S, PayaPerez A, Pellegrini G, Schwarz-Schulz B, Vegro S. 2005. European Union Risk Assessment Report: methylenediphenyl diisocyanate (MDI). Luxembourg: European Commission, Office for Official Publications of the European Communities. [Google Scholar]
- Norzila MZ, Fakes K, Henry RL, Simpson J, Gibson PG. 2000. Interleukin-8 secretion and neutrophil recruitment accompanies induced sputum eosinophil activation in children with acute asthma. Am J Respir Crit Care Med. 161(3):769–774. [DOI] [PubMed] [Google Scholar]
- Park H, Jung K, Kim H, Nahm D, Kang K. 1999. Neutrophil activation following TDI bronchial challenges to the airway secretion from subjects with TDI-induced asthma. Clin Exp Allergy. 29(10):1395–1401. [DOI] [PubMed] [Google Scholar]
- Park YJ, Yoo SA, Kim M, Kim WU. 2020. The role of calcium-calcineurin-NFAT signaling pathway in health and autoimmune diseases. Front Immunol. 11:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauluhn J, Woolhiser MR, Bloemen L. 2005. Repeated inhalation challenge with diphenyl methane-4,4′-diisocyanate in brown Norway rats leads to a time-related increase of neutrophils in bronchoalveolar lavage after topical induction. Inhal Toxicol. 17(2):67–78. [DOI] [PubMed] [Google Scholar]
- Pollaris L, Devos F, De Vooght V, Seys S, Nemery B, Hoet PH, Vanoirbeek JA. 2016. Toluene diisocyanate and methylene diphenyl diisocyanate: asthmatic response and cross-reactivity in a mouse model. Arch Toxicol. 90(7):1709–1717. [DOI] [PubMed] [Google Scholar]
- Radermecker C, Louis R, Bureau F, Marichal T. 2018. Role of neutrophils in allergic asthma. Curr Opin Immunol. 54:28–34. [DOI] [PubMed] [Google Scholar]
- Redlich CA, Homer RJ, Smith BR, Wirth JA, Cullen MR. 1996. Immunologic responses to isocyanates in sensitized asthmatic subjects. Chest. 109(3 Suppl):6S–8S. [DOI] [PubMed] [Google Scholar]
- Redlich CA, Karol MH. 2002. Diisocyanate asthma: clinical aspects and immunopathogenesis. Int Immunopharmacol. 2(2—3):213–224. [DOI] [PubMed] [Google Scholar]
- Sales KJ, Maldonado-Perez D, Grant V, Catalano RD, Wilson MR, Brown P, Williams AR, Anderson RA, Thompson EA, Jabbour HN. 2009. Prostaglandin F(2alpha)-F-prostanoid receptor regulates CXCL8 expression in endometrial adenocarcinoma cells via the calcium-calcineurin-NFAT pathway. Biochim Biophys Acta. 1793(12):1917–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowska M, Rovati GE, Diamant Z, Untersmayr E, Schwarze J, Lukasik Z, Sava F, Angelina A, Palomares O, Akdis CA, et al. 2021. Current perspective on eicosanoids in asthma and allergic diseases: EAACI Task Force consensus report, part I. Allergy. 76(1):114–130. [DOI] [PubMed] [Google Scholar]
- Song C, Luo L, Lei Z, Li B, Liang Z, Liu G, Li D, Zhang G, Huang B, Feng ZH. 2008. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. J Immunol. 181 (9):6117–6124. [DOI] [PubMed] [Google Scholar]
- Statista. 2021. Methylene diphenyl diisocyanate demand worldwide from 2011 to 2020 with a forecast for 2022; [accessed 2021 Nov 16]. https://www.statista.com/statistics/750809/mdi-demand-worldwide/.
- Staruch MJ, Camacho R, Dumont FJ. 1998. Distinctive calcineurin-dependent (FK506-sensitive) mechanisms regulate the production of the CC chemokines macrophage inflammatory protein (MIP)-1alpha, MIP-1beta, and RANTES vs IL-2 and TNF-alpha by activated human T cells. Cell Immunol. 190(2):121–131. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, Tokui K, Iwata Y, Abo H, Izumi S. 2011. A case of severe bronchial asthma controlled with tacrolimus. J Allergy. 2011:479129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski M, Vanoirbeek JA, Vanhooren HM, De Vooght V, Mercier CM, Ceuppens J, Nemery B, Hoet PH. 2007. Immunological determinants of ventilatory changes induced in mice by dermal sensitization and respiratory challenge with toluene diisocyanate. Am J Physiol Lung Cell Mol Physiol. 292(1):L207–L214. [DOI] [PubMed] [Google Scholar]
- Tiffany HL, Li F, Rosenberg HF. 1995. Hyperglycosylation of eosinophil ribonudeases in a promyelocytic leukemia cell line and in differentiated peripheral blood progenitor cells. J Leukoc Biol. 58(1):49–54. [DOI] [PubMed] [Google Scholar]
- Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, Fenton MJ, Gordon DC, Dunn IF, Goldfeld AE. 2000. A lipopolysaccharide-specific enhancer complex involving Ets, Elk-1, Sp1, and CREB binding protein and p300 is recruited to the tumor necrosis factor alpha promoter in vivo . Mol Cell Biol. 20(16):6084–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai EY, Yie J, Thanos D, Goldfeld AE. 1996. Cell-type-specific regulation of the human tumor necrosis factor alpha gene in B cells and T cells by NFATp and ATF-2/JUN. Mol Cell Biol. 16(10):3232–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsytsykova AV, Goldfeld AE. 2002. Inducer-specific enhanceosome formation controls tumor necrosis factor alpha gene expression in T lymphocytes. Mol Cell Biol. 22(8):2620–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Feske S. 2018. NFAT control of immune function: new frontiers for an abiding trooper. F1000Res. 7:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen TA, de Groot LES, Melgert BN. 2020. The different faces of the macrophage in asthma. Curr Opin Pulm Med. 26(1):62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoirbeek JA, Tarkowski M, Ceuppens JL, Verbeken EK, Nemery B, Hoet PH. 2004. Respiratory response to toluene diisocyanate depends on prior frequency and concentration of dermal sensitization in mice. Toxicol Sci. 80(2):310–321. [DOI] [PubMed] [Google Scholar]
- Wisnewski AV, Liu J, Colangelo CM. 2015. Glutathione reaction products with a chemical allergen, methylene-diphenyl diisocyanate, stimulate alternative macrophage activation and eosinophilic airway inflammation. Chem Res Toxicol. 28(4):729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisnewski AV, Liu J, Nassar AF. 2019. LC-UV-MS and MS/MS characterize glutathione reactivity with different isomers (2,2′ and 2,4′ vs. 4,4′) of methylene diphenyl-diisocyanate. EC Pharmacol Toxicol. 7(3):205–219. [PMC free article] [PubMed] [Google Scholar]
- Wisnewski AV, Liu J, Redlich CA. 2013. Connecting glutathione with immune responses to occupational methylene diphenyl diisocyanate exposure. Chem Biol Interact. 205(1):38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisnewski AV, Liu J, Redlich CA. 2020. Analysis of lung gene expression reveals a role for Cl− channels in diisocyanate-induced airway eosinophilia in a mouse model of asthma pathology. Am J Respir Cell Mol Biol. 63(1):25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisnewski AV, Liu J. 2016. Immunochemical detection of the occupational allergen, methylene diphenyl diisocyanate (MDI), in situ. J Immunol Methods. 429:60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Cui J, Yi L, Qin J, Tulake W, Teng F, Tang W, Wei Y, Dong J. 2020. The role of T cells and macrophages in asthma pathogenesis: a new perspective on mutual crosstalk. Mediators Inflamm. 2020:7835284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.