

Abstract
KNI-272 is a tripeptide protease inhibitor for treating human immunodeficiency virus type 1 (HIV-1). In in vitro stability studies using rat tissue homogenates, KNI-272 concentrations in the liver, kidney, and brain decreased significantly with time. Moreover, in tissue distribution studies, KNI-272 distributed highly to the liver, kidney, and small intestine in vivo. From these results and reported physiological parameters such as the tissue volume and tissue blood flow rate, we considered the liver to be the main organ which takes part in the metabolic elimination of KNI-272. Then the hepatic metabolism of KNI-272 was more thoroughly investigated by using rat liver microsomes. KNI-272 was metabolized in the rat liver microsomes, and five metabolites were found. The initial metabolic rate constant (kmetabolism) tended to decrease when the KNI-272 concentration in microsomal suspensions increased. The calculated Michaelis-Menten constant (Km) and the maximum velocity of KNI-272 metabolism (Vmax), after correction for the unbound drug concentration, were 1.12 ± 0.09 μg/ml (1.68 ± 0.13 μM) and 0.372 ± 0.008 μg/mg of protein/min (0.558 ± 0.012 nmol/mg of protein per min), respectively. The metabolic clearance (CLint,metabo), calculated as Vmax/Km, was 0.332 ml/mg of protein per min. Moreover, by using selective cytochrome P-450 inhibitors and recombinant human CYP3A4 fractions, KNI-272 was determined to be metabolized mainly by the CYP3A isoform. In addition, ketoconazole, a representative CYP3A inhibitor, inhibited KNI-272 metabolism competitively, and the inhibition constant (Ki) was 4.32 μM.
The human immunodeficiency virus type 1 (HIV-1) encodes a virus-specific aspartic protease that mediates crucial proteolytic processing of viral protein precursors at a late stage in the replication of the virus (14). Antiretroviral therapy directed at the reverse transcriptase of HIV-1 has had limited success because of drug toxicity and the emergence of viral resistance (22). With the recent appreciation of the continuous, high-level replication of HIV-1 which occurs in vivo (23, 53), the limitations of current therapies are better understood, as is the urgent need for new and effective antiretroviral agents. Therefore, the inhibition of HIV protease has attracted widespread interest because of its potential use in the therapeutic intervention of AIDS (14, 15, 43, 51). Several HIV-1 protease inhibitors (25, 36, 46) have been discovered based on the transition state analogue concept, which was known to be effective in studies of inhibitors of aspartic proteases such as renin and pepsin (20, 54). Recently, HIV-1 protease inhibitors, such as indinavir, ritonavir, and saquinavir, have generated worldwide interest as therapeutic agents for the control of HIV infection.
KNI-272, which was synthesized by Mimoto et al. (42), has one of the strongest HIV-1 protease-inhibitory activities in in vitro antiviral testing (27), and the highest bioavailability (BA) after intraduodenal (i.d.) administration to rats (29, 30), of the KNI series compounds. Moreover, we have already reported the pharmacokinetic characteristics of KNI-272. The BA values of KNI-272 after i.d. administration to rats were 24 to 45% (30, 49) when KNI-272 was dissolved in various solvents, and those after oral administration to beagles were 7 to 26% when KNI-272 was formulated in various capsules (32). The biliary and urinary excretions of KNI-272 were both around 1% as the intact drug in rats when KNI-272 was administered intravenously (i.v.) to rats at doses of 1.0 and 10.0 mg/kg of body weight (31), and excretion of KNI-272 was assumed not to be the main route of elimination from the circulatory system in rats. Furthermore, some HIV-1 protease inhibitors, HIV-1 reverse transcriptase inhibitors, and renin inhibitors were reported to be metabolized in rat, dog, and human livers (1–4, 16, 17, 35).
In this study, we found by an in vitro stability study using rat tissue homogenates and an in vivo tissue distribution study that the liver was the main eliminating organ for KNI-272 in the rat. Then, the metabolic characteristics of KNI-272, such as the metabolic kinetics, the P-450 isozymes responsible for KNI-272 metabolism, and their effects on the formation of metabolites of KNI-272, were investigated by using rat liver microsomes and various selective cytochrome P-450 inhibitors.
MATERIALS AND METHODS
Materials.
KNI-272 was kindly supplied by Japan Energy Co. (Tokyo, Japan). NADP, glucose-6-phosphate (G6P), and G6P dehydrogenase (G6PDH) were purchased from Sigma Chemical Company (St. Louis, Mo.). Ketoconazole (KCZ), chloramphenicol (CP), and quinidine (QND) were supplied by Janssen-Kyowa Co., Ltd. (Tokyo, Japan), Boehringer Mannheim (Tokyo, Japan), and Wako Pure Chemical Industries Ltd. (Osaka, Japan), respectively. Nalidixic acid (NA) and cimetidine (CIM) were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). Recombinant human CYP3A4 prepared from lymphoblastic cells was purchased from Daiichi Pure Chemicals Co., Ltd. (Osaka, Japan). The cytochrome P-450 content was about 68 pmol/mg of protein. Propylene glycol (PG) and acetonitrile (high-performance liquid chromatography [HPLC] grade) were obtained from Nacalai Tesque, Inc. and Kanto Chemical Co., Inc. (Tokyo, Japan), respectively. All other reagents used were of analytical grade and were commercially obtained.
Animal experiments.
Wistar male rats (SLC, Shizuoka, Japan), weighing 300 to 400 g, were used throughout the study. The rats were housed in pairs under controlled environmental conditions and fed commercial feed pellets. All rats had free access to food and water.
The biological samples obtained were frozen immediately after collection and stored in a freezer at −20°C until analysis.
In vitro stability in various rat tissue homogenates.
After light ether anesthesia, rats were sacrificed by exsanguination and infused with 20 ml of Krebs-Henseleit buffer (pH 7.4) through the abdominal aorta in order to remove blood from all tissues. The liver, kidney, lung, spleen, brain, small intestine, and skeletal muscle were excised immediately, rinsed in ice-cold oxygenated (95% O2–5% CO2) Krebs-Henseleit buffer, weighed, and homogenized in 4 volumes of Krebs-Henseleit buffer. All procedures for the tissue preparation were carried out under ice-cold conditions. The assay mixture contained 1.0 μg of KNI-272/ml and 0.1 g of tissue homogenate/ml in Krebs-Henseleit buffer in a final volume of 1.0 ml. The reaction was started by addition of the tissue homogenate suspension to a preincubated drug solution at 37°C, and the reaction was carried out at 37°C with bubbling of 95% O2–5% CO2 gas to the incubation mixture for 2 h. The reaction was stopped by the addition of 2 volumes of methanol. The stability of KNI-272 in each tissue homogenate was estimated by comparison of the KNI-272 concentration after the incubation with its initial concentration. As a control experiment, samples without tissue homogenates and without bubbling gas were also run, and no decreases in KNI-272 concentration were observed in these samples.
In vivo tissue distribution studies.
After anesthesia by an intraperitoneal (i.p.) injection of urethane (1.0 g/kg), KNI-272 solution dissolved in 70% PG was administered i.v. to rats via the jugular vein, corresponding to a drug dose of 10.0 mg/kg of body weight. Four hours after drug administration, the rats were sacrificed by bleeding from the inferior vena cava and were infused with ice-cold saline to remove the blood from all tissues. This time was chosen based on our previous study with rats, which demonstrated that the plasma KNI-272 concentration reached the terminal elimination phase 4 h following i.v. administration at a 10.0-mg/kg dose (31). After whole blood was obtained from the inferior vena cava, the brain, lung, heart, liver, kidney, stomach, small intestine, spleen, adipose tissue, and skeletal muscle were excised immediately, rinsed in ice-cold saline, weighed, and homogenized in 4 volumes of saline, except for the liver and adipose tissue. The liver was homogenized in 3 volumes of saline, whereas the adipose tissue was cut into pieces, then extracted in 4 volumes of methanol. All procedures for tissue preparation were carried out under ice-cold conditions. KNI-272 concentrations in the blood, Cblood, and the tissues (organs), Ctissue, were determined by an HPLC method (see below).
The apparent tissue-to-blood distribution ratio, Kp,app, was determined from the relationship Kp,app = Ctissue/Cblood.
In vitro metabolism by rat liver microsomes. (i) Microsomal preparation.
Rat liver was freshly obtained and homogenized in ice-cold 1.15% KCl solution. The homogenate was centrifuged at 9,000 × g for 15 min, and the supernatant was centrifuged at 105,000 × g for 60 min to obtain a microsomal pellet. The microsomal pellet was resuspended in 0.1 M phosphate buffer (pH 7.4) to a concentration of 8.0 mg of protein per ml. The protein concentration was determined by the method of Lowry et al. (40).
(ii) Metabolic assay.
Typically, the incubation mixture contained an NADPH-generating system (0.5 mM NADP, 5 mM G6P, 2 U of G6PDH/ml, and 5 mM MgCl2), 0.5 to 20.0 μg of KNI-272/ml, and a microsomal suspension at 0.8 mg of protein per ml in 1.0 ml of 0.1 M phosphate buffer. After 5 min of preincubation at 37°C in a water bath, the reaction was initiated by the addition of 100 μl of a microsomal suspension at 8.0 mg of protein per ml. The reaction was stopped at the appropriate time by the addition of 200 μl of ice-cold 0.1 N NaOH solution. Moreover, a control incubation containing no NADPH was also run.
(iii) Identification of involved cytochrome P-450 isoforms.
To identify the cytochrome P-450 isoforms responsible for KNI-272’s hepatic metabolism, a number of cytochrome P-450 inhibitors thought to be selective for individual enzymes were used: NA for CYP1A (18), CP for CYP2B (11), QND for CYP2B6 (7, 12, 21, 26), KCZ for CYP3A (45) and CIM for CYP3A (8). Various concentrations of each inhibitor (final concentrations, 0.3 to 45.0 μM) were preincubated with the NADPH-generating system and KNI-272 (final concentrations, 0.5 to 3.0 μM) for 5 min at 37°C prior to the addition of 100 μl of a microsomal suspension at 8.0 mg of protein per ml. The reaction was stopped at each time (10 or 15 min) by the addition of 200 μl of ice-cold 0.1 N NaOH solution.
In addition, the metabolism of KNI-272 was investigated by using recombinant human CYP3A4. The incubation mixture contained the NADPH-generating system, 1.0 μg of KNI-272/ml, and recombinant human CYP3A4 at 0.5 mg of protein per ml in 0.5 ml of 0.1 M phosphate buffer. After 5 min of preincubation at 37°C, the reaction was initiated by the addition of 1 μl of 500 μg of KNI-272/ml and was carried out at 37°C with shaking in a water bath. After 30 min, the reaction was stopped by the addition of 200 μl of ice-cold 0.1 N NaOH solution.
(iv) Data analysis.
In vitro metabolic activities of KNI-272 were estimated from the loss of KNI-272 by using rat liver microsomes under linear conditions. The initial metabolic rate constants (kmetabolism) of KNI-272 were determined by linear regression using linear data points of the KNI-272 concentration-time plots, namely, those between 0 and 5 to 20 min. The apparent Michaelis-Menten constant (Km,app) and apparent maximum velocity of the metabolism (Vmax,app) of KNI-272 were estimated from the total KNI-272 concentration as a substrate concentration by fitting the obtained data to a Michaelis-Menten equation using a nonlinear least-squares regression analysis with a weighting factor of zero.
To obtain the unbound concentration of KNI-272 in microsomal suspensions, the results of binding experiments in rat liver microsomes were used. The total KNI-272 concentrations in rat liver microsomes were converted to unbound concentrations by substituting calculated binding parameters for the equations described under “Binding experiments in rat liver microsomes” below. Then the unbound concentration was obtained as a positive solution of an equation of the second degree and used as substrate concentrations to estimate the Michaelis-Menten constant (Km) and the maximum velocity of the metabolism (Vmax) of KNI-272 by the method described above.
The effects of various cytochrome P-450 inhibitors were estimated by the ratio of either KNI-272 decrease or metabolite formation in the presence and absence of inhibitors. The inhibition constant (Ki) by KCZ was estimated from the x-axis intercept in a plot of the slopes of Lineweaver-Burk analysis against the KCZ concentrations.
Binding experiments in rat liver microsomes.
The binding characteristics of KNI-272 in rat liver microsomal suspensions were determined by an ultrafiltration method (37) using a disposable ultrafiltration cartridge (Centricon-10; 10,000-molecular-weight cutoff; Amicon, Inc., Beverly, Mass.). Various concentrations of KNI-272 (0.5 to 50.0 μg/ml) were added to the rat liver microsomes (0.8 mg of protein per ml), and then the microsomal suspensions were incubated at 37°C for 30 min. After sampling for the determination of total KNI-272 concentration (Ctotal), the aliquot was applied to the ultrafiltration cartridge, which was centrifuged at 1,500 × g for 30 min. The filtrate was used to determine the unbound KNI-272 concentration (Cunbound). The bound KNI-272 concentration in rat liver microsomal suspensions (Cbound) was determined by the equation Ctotal − Cunbound.
The association constant (Ka) and the total concentration of binding sites [n(P)] were estimated by fitting the obtained data to a model of specific and nonspecific binding sites (50) as described below by using a nonlinear least-squares regression analysis with a weighting factor of zero. The experimentally obtained Cbound and Cunbound values were substituted in the following equation, and binding parameters were obtained when the total square error between the experimentally obtained value and the value calculated by the equation became the minimum value.
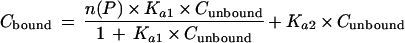 |
where subscripts 1 and 2 denote specific and nonspecific binding sites, respectively, and n and P are the number of binding sites per molecule of microsomal protein and the total protein concentration, respectively.
Analytical procedures. (i) Determination of KNI-272 concentrations.
The concentrations of KNI-272 in rat blood and plasma were measured by an HPLC assay method described in detail previously (30). The detection limit of this analytical method of KNI-272 was considered to be 2.0 ng (coefficient of variation = 8.5%). Briefly, 1 ml of 0.1 N NaOH, 200 μl of isoamyl alcohol, and 5 ml of dichloromethane were added to 100 μl of a biological sample. After extraction, the organic liquid was evaporated and the residue was dissolved in the mobile phase, of which an aliquot was injected into the HPLC system, which was equipped with a column-switching apparatus.
This HPLC system consisted of two pumps: P1, a Shimadzu (Kyoto, Japan) LC-6A pump, and P2, a Waters (Milford, Mass.) M45J pump. The flow rates of pumps P1 and P2 were both 1.0 ml/min. The UV detector was a Shimadzu SPD-10A, connected to a Shimadzu C-R4A Chromatopac. For column switching, a motor-actuated six-port column-switching valve (Kyoto Chromato Co., Ltd., Kyoto, Japan) was used. The precolumn (C2) was dry packed with Chemcosorb 5-octyldecyl silane (ODS)-H (4 mm [inner diameter] by 10 mm; Chemco Scientific Co., Ltd., Osaka, Japan). The analytical column (C1) was also a Chemcosorb 5-ODS-H (4.6 mm [inner diameter] by 250 mm) and was maintained at 65°C for all the separations. The compositions of the two mobile phases were as follows: mobile phase 1, acetonitrile–water (50:50), mobile phase 2, acetonitrile–water (25:75).
The pretreated sample was injected by using a Waters WISP 710B automatic sample injector on the precolumn, and KNI-272 was first adsorbed onto the precolumn with mobile phase 2 during 0.8 min. Thereafter, the line was switched to mobile phase 1, and KNI-272 was transferred to an analytical column with mobile phase 1. At 0.5 min after switching, mobile phase 1 was removed from the precolumn by mobile phase 2, and the HPLC system was ready for a new cycle. The KNI-272 that eluted from the analytical column was detected by UV absorption monitored at 208 nm. Levels were estimated by the chromatographic technique of comparing peaks obtained from plasma or blood to which were added known amounts of KNI-272. A set of six or seven calibration standards was run with each series of unknown samples.
To analyze the KNI-272 concentration in various tissue homogenates, KNI-272 was extracted by 2 volumes of methanol and the methanol extract was evaporated. Then a method similar to that described above was used.
(ii) Determination of KNI-272 metabolic activity.
The concentrations of KNI-272 and its metabolites in rat liver microsomal suspensions were also determined by a general HPLC method. After termination of the reaction, KNI-272 and its metabolites in the reaction mixture were extracted by 3.0 ml of ethyl acetate, except in the case of the inhibition study using NA, where diethyl ether was used, and the organic liquid was evaporated. The residue was then dissolved with the mobile phase, and an aliquot was injected into the HPLC system. The HPLC system consisted of a Shimadzu LC-10A pump, a Shimadzu SPD-10A variable-wavelength UV-VIS detector, and a Waters WISP 710B automatic sample injector. The analytical column was a Chemcosorb 5-ODS-H (4.6 mm [inner diameter] by 250 mm) and was maintained at 50°C for all separations. The composition of the mobile phase was water containing 0.01% trifluoroacetic acid–acetonitrile (70:30), and the flow rate was 0.8 ml/min. KNI-272 and metabolites eluted from the analytical column were detected by UV absorption monitored at 208 nm. In this study, the detectable metabolites in this experimental condition were evaluated, and the formation of each metabolite was estimated from the metabolite’s peak area, detected by the HPLC method.
Statistics.
Results were expressed as means or means ± standard errors (SE). The statistical difference was assumed to be significant when P < 0.05 (by the two-sided t test).
RESULTS
The in vitro stabilities of KNI-272 in rat tissue homogenates were determined, and the results are shown in Fig. 1. After incubation the KNI-272 concentrations in the liver, kidney, and brain homogenates decreased significantly to 62, 91, and 62% compared with the initial concentrations. The KNI-272 concentrations also decreased in the other tissue homogenates, although these differences were not significant.
FIG. 1.

In vitro stabilities of KNI-272 in various rat tissue homogenates. Each column represents the mean ± SE from three to seven experiments (one to three rats per experiment). ∗, significantly different from the initial concentration (1.0 μg/ml) (P < 0.05).
Table 1 shows the obtained Kp,app of KNI-272 and the physiological parameters (6), such as the tissue volume (V) and tissue blood flow rate (Q), in various rat tissues. KNI-272 easily distributed into the liver, kidney, and small intestine but was not detected in the muscle. Although the liver and kidney are the main organs from which KNI-272 was eliminated, KNI-272 highly distributed to these organs. In addition, the liver has a 4.5-times-large V and a 1.3-times-larger Q than the kidney. Consequently, we considered the liver to be the main organ which takes part in the metabolic elimination of KNI-272, and we investigated the hepatic metabolism of KNI-272 more fully, using rat liver microsomes.
TABLE 1.
Physiological parameters and Kp,app of KNI-272 in various rat tissues after i.v. administration to rats at 10.0 mg/kg
| Tissue | Q (ml/min)a | V (ml)a | Kp,appb |
|---|---|---|---|
| Lung | 43.00 | 1.0 | 0.92 |
| Brain | 1.33 | 1.7 | 0.44 |
| Heart | 3.92 | 0.8 | 0.24 |
| Liver | 11.80c | 10.3 | 5.72 |
| Kidney | 9.23 | 2.3 | 4.12 |
| Stomach | 1.14 | 1.1 | 2.36 |
| Spleen | 0.63 | 0.6 | 0.53 |
| Small intestine | 7.52 | 10.0 | 7.53 |
| Adipose tissue | 0.40 | 10.0 | 2.29 |
| Skeletal muscle | 7.50 | 121.9 | ND |
Values for Q and V are those observed by Bernareggi et al. for a 250-g rat (6).
Each value is the mean from three experiments. ND, not detected.
Sum of hepatic artery plus portal vein flow.
In Fig. 2, chromatographic peaks of three major metabolites (M1, M2, and M4) and two minor metabolites (M3 and M5) were detected after 10 min of incubation of KNI-272 with rat liver microsomes. The presence of these peaks was NADPH dependent and their sizes increased with continued incubation, whereas the chromatographic peak of KNI-272 decreased with time. The retention times on the HPLC of M1, M2, M3, M4, M5, and KNI-272 were 8.0, 9.5, 11.8, 16.0, 31.0, and 20.3 min, respectively.
FIG. 2.

Reversed-phase HPLC chromatograms of KNI-272 and its metabolites after incubation in rat liver microsomes. Two micrograms per milliliter of KNI-272 was incubated with rat liver microsomes (0.8 mg of protein per ml) in the absence (a) or presence (b) of an NADPH-generating system at 37°C for 10 min.
In Fig. 3, the metabolic profile of KNI-272 in the reconstituted rat liver microsome system is shown. The kmetabolism of KNI-272 were 0.195, 0.130, 0.037, 0.025, 0.016, and 0.007 min−1 at the initial concentrations of 0.5, 1.0, 2.0, 5.0, 10.0, and 20.0 μg/ml, respectively. Thus, as the KNI-272 concentration increased, the metabolic rate of KNI-272 tended to decrease in the examined concentration range.
FIG. 3.

Metabolic profile of KNI-272 in rat liver microsomes. Each point represents the mean ± SE from three experiments (two to three rats per experiment). Initial concentrations of KNI-272 in rat liver microsomal reaction mixture are 0.5 (■), 1.0 (●), 2.0 (▴), 5.0 (□), 10.0 (○), and 20.0 (▵) μg/ml.
Fig. 4 shows the rate of KNI-272 metabolism normalized to the protein content as a function of KNI-272 concentration and its double-reciprocal plot (inset) in the rat liver microsomes after 5 min of incubation. The obtained Km,app and Vmax,app, which were calculated from total KNI-272 concentrations in rat liver microsomes, were 3.03 ± 0.24 μg/ml (4.54 ± 0.36 μM) and 0.404 ± 0.010 μg/mg of protein per min (0.606 ± 0.015 nmol/mg of protein per min), respectively, and the apparent metabolic intrinsic clearance (CLint,metab), CLint,metab,app, calculated as Vmax,app/Km,app, was 0.133 ml/min/mg of protein. Moreover, we determined the binding characteristics of KNI-272 in the rat liver microsomes to calculate the Km and Vmax. In Fig. 5, the binding profile of KNI-272 in the rat liver microsomal suspensions is shown. The calculated Ka1, Ka2, and n(P) were 0.935 ± 0.207 ml/μg, 0.396 ± 0.011 ml/μg, and 1.801 ± 0.184 μg/ml, respectively. After the KNI-272 concentration in the rat liver microsomes was corrected for its unbound concentration, the Km and Vmax were calculated to be 1.12 ± 0.09 μg/ml (1.68 ± 0.13 μM) and 0.372 ± 0.008 μg/mg of protein/min (0.558 ± 0.012 nmol/mg of protein per min), respectively. Then the CLint,metab was calculated as 0.332 ml/mg of protein/min.
FIG. 4.

Rate of KNI-272 metabolism as a function of KNI-272 concentration and its Lineweaver-Burk plot (inset) in rat liver microsomes. Each point represents the mean ± SE from three experiments (two to three rats per experiment). The incubation time was 5 min at 37°C with shaking.
FIG. 5.

Binding profile of KNI-272 in rat liver microsomes. The protein concentration of rat liver microsomes was 0.8 mg/ml in 0.1 M phosphate buffer (pH 7.4).
To investigate the involvement of cytochrome P-450 isozymes, the effects of various specific cytochrome P-450 inhibitors on KNI-272 metabolism by rat liver microsomes were examined, and the alterations of KNI-272 metabolic activity after 10 min of incubation are shown as percentages of the control activity (without the inhibitor). The initial KNI-272 concentration in the reaction mixture was 3.0 μM. The metabolic activity of KNI-272 varied to 94.5 ± 18.0, 108.6 ± 9.1, 100.5 ± 13.8, and 74.0 ± 11.2% by the addition of NA, CP, QND, and CIM at 3.0 μM, respectively, and significant decreases were not observed. Only with the addition of KCZ did the metabolic activity decrease significantly, to 29.8 ± 1.7, 22.6 ± 3.5, and 23.7 ± 2.4% at 0.3, 3.0, and 30.0 μM KCZ, respectively (n = 3 to 5 experiments). Although CIM decreased the metabolic activity, the results were not significant. Both KCZ and CIM are known to be the inhibitors of the CYP3A subfamily. Furthermore, the Lineweaver-Burk plot and the Dixon plot of KNI-272 metabolism, shown in Fig. 6a and b, respectively, were determined in order to kinetically investigate the effect of KCZ. As the reaction velocity was insufficient to reach the Vmax,app at the highest KCZ concentration in this inhibition study, the correct metabolic parameters were not calculated. Therefore, we determined Vmax,app (in micrograms per milligram of protein per minute) and Km,app (in micrograms per milliliter) as follows: 0.351 ± 0.028 and 7.93 ± 0.92, 0.291 ± 0.033 and 8.78 ± 1.42, 0.302 ± 0.038 and 11.8 ± 1.9, and 0.109 ± 0.005 and 3.49 ± 0.30 at the KCZ concentrations of 4.5, 15.0, 30.0, and 45.0 μM, respectively. Except for the parameter value at 45.0 μM KCZ, Km,app increased significantly with increasing KCZ concentration. In contrast, Vmax,app was independent of KCZ concentration. Therefore, KCZ is considered to be a competitive inhibitor, with a Ki of 4.32 μM.
FIG. 6.

Inhibition kinetics of KNI-272 metabolism by KCZ in rat liver microsomes. Each point represents the mean from three experiments (two to three rats per experiment). (a) Lineweaver-Burk plot. KCZ concentrations in the rat liver microsomal reaction mixture are 4.5 (●), 15.0 (▴), 30.0 (■), and 45.0 (⧫) μM. ○, control (no KCZ). (Inset) Plot of the slopes from the Lineweaver-Burk plot against the KCZ concentration. The Ki was estimated from the x-axis intercept. (b) Dixon plot. KNI-272 concentrations in the rat liver microsomal reaction mixture are 0.5 (●), 1.0 (▴), 2.0 (■), and 3.0 (⧫) μM.
We then investigated the change in KNI-272 metabolite formation in the presence of KCZ (Fig. 7). The formation of M1, M2, and M4 decreased as the KCZ concentration increased. The changes in M5 formation were not determined at 3.0 and 30.0 μM of KCZ, because M5 was the minor metabolite and the peaks of M5 and KCZ on our HPLC chromatogram were too close to each other for adequate resolution. As the KCZ concentration increased, the two peaks overlapped. M3 was the smallest among the five metabolites formed (as shown in Fig. 2). Therefore, the effect of KCZ on M3 formation could not be investigated.
FIG. 7.

Inhibitory effect of ketoconazole on KNI-272 metabolite formations in rat liver microsomes. (a) M1; (b) M2; (c) M4; (d) M5. N.D., not determined. Each column represents the mean ± SE from three experiments (two to three rats per experiment). ∗, significantly different from the control (P < 0.05). The concentration of KNI-272 was 3.0 μM. The incubation time was 15 min. The formation of each metabolite was determined from the metabolite’s peak area, detected by the HPLC method.
The recombinant human CYP3A4 was used instead of rat liver microsomes to confirm that CYP3A4 is responsible for KNI-272 metabolism. After 30 min of incubation, the KNI-272 concentration decreased significantly, to 37.6%, and the metabolic activity was 0.0416 ± 0.0024 μg/mg of protein per min.
DISCUSSION
By now, many HIV-1 protease inhibitors have been developed (5, 13, 19, 25, 28, 36, 41, 46, 52). Some HIV-1 protease inhibitors and renin inhibitors, which are aspartic protease inhibitors similar to HIV-1 protease inhibitors, were reported to be metabolized in the liver, and several metabolites were identified (3, 4, 16, 17, 35). The most important drug-metabolizing enzymes are the cytochrome P-450s, a group of monooxygenases. They are located in almost all tissues, with the highest concentration by far found in the liver. These enzymes catalyze the biotransformation of lipophilic drugs to more-polar compounds, which are readily excreted by the kidney into the urine. Therefore, we tried to determine which tissue metabolically eliminates KNI-272 by using various rat tissue homogenates. KNI-272 concentrations decreased mainly in the liver, kidney, and brain homogenates. In general, the liver is known to be the main metabolic tissue in which various drugs are metabolized. Although there is a bulk blood flow through the lungs, and although the small intestine has a large tissue volume, a large surface area, and a long residence time of drugs, decreases in KNI-272 concentration were not observed in these tissues. If KNI-272 concentrations in these tissue homogenates had decreased, then they could not be ignored as eliminating tissues for KNI-272. Although KNI-272 concentrations decreased significantly in brain homogenates, it is known that a blood-brain barrier exists in vivo. Therefore, peptidic drugs, such as KNI-272, presumably are not able to enter the brain through blood circulation. In fact, KNI-272 distributed highly to the liver, kidney, and small intestine, but little or hardly at all to the brain, heart, spleen, and skeletal muscle. Accordingly, we concluded that KNI-272 is mainly eliminated from the liver in rats, based on the above results and the physiological parameters (the tissue volume and tissue blood flow), and we more thoroughly investigated KNI-272 metabolism, using rat liver microsomes.
In our previous study (33), KNI-272 bound about 90% in rat and human plasma. As KNI-272 is also considered to be highly bound in the microsomal suspensions, the KNI-272 concentration in microsomal suspensions was corrected back to its unbound concentration, and the Km and Vmax were calculated. The Vmax obtained from in vitro experiments, expressed in micrograms per milligram of protein per minute, was multiplied by the term 464 (45 mg of protein per g of liver × a 10.3-g liver/250-g rat) as a scaling factor (24) to yield estimates for the whole liver. Consequently, the Vmax was estimated to be 172 μg/min in a 250-g rat, and the CLint,metab obtained in an in vitro study was 154 ml/min in a 250-g rat. We previously investigated the hepatic extraction ratio (E) using rats by comparing areas under the concentration-time curve (AUC) obtained after i.v. infusion with that obtained after portal venous infusion at 10.0 mg/kg/h, and E was estimated to be 0.368. Then CLint,metab was calculated to be 166 ml/min in a 250-g rat, assuming the well-stirred model (44); thus, the two CLint,metab values were very similar.
The present study showed that KNI-272 was metabolized in the rat liver microsomes, and the kmetabolism decreased with the increase in initial concentration. On the other hand, we investigated the effects of the KNI-272 dose, between 1.0 and 50.0 mg/kg, on the plasma kinetics of KNI-272 (34) As a result, the total clearance (CLtotal) decreased and the AUC per dose increased after i.v. administration of KNI-272 at a 50.0-mg/kg dose. In addition, the biliary and urinary excretions of KNI-272 after i.v. administration were about 1%. If these dose-dependent pharmacokinetics were caused by nonlinear plasma protein binding (33), the CLtotal would increase inversely with the dose. Consequently, we concluded that these results were partly caused by the saturation of hepatic metabolism.
The metabolism of KNI-272 was thought to involve the CYP3A isozyme, at least in the formation of M1, M2, M4, and M5 in this study. Some HIV-1 protease inhibitors and renin inhibitors have been reported to be metabolized in rats, dogs, and humans, and some metabolites were identified (3, 4, 16, 17, 35). Furthermore, several compounds, containing indinavir and ritonavir, which are potent and selective HIV-1 protease inhibitors (13, 28, 41, 52), were found to be mostly or partly metabolized by CYP3A isoforms (9, 10, 16, 35, 38, 39). The chemical structures of HIV-1 protease inhibitors and renin inhibitors show similarities. Therefore, these drugs are theoretically metabolized through the same cytochrome P-450 isozymes. Moreover, it is reported that CYP3A is partly involved in the metabolism of L-696,229 and L-738,372, both HIV-1 reverse transcriptase inhibitors (47, 48). At present, it is general practice that more than two drugs are administered to a patient in clinical situations. Therefore, it is possible that such drugs and KNI-272 may interact with each other through hepatic metabolism. In this study, we detected five KNI-272 metabolites on an HPLC chromatogram generated in rat liver microsomes, although their chemical structures have not been determined. After the metabolites of KNI-272 are identified, we will be able to consider the metabolic kinetics, drug interactions, and species differences in metabolism, etc., more precisely.
REFERENCES
- 1.Balani S K, Pitzenberger S M, Kauffman L R, Arison B H, Ramjit H G, Goldman M E, O’Brien J A, King J D, Hoffman J M, Rooney C S, Theoharides A D. Metabolism of a new HIV-1 reverse transcriptase inhibitor, 3-[2-(benzoxazol-2-yl)ethyl]-5-ethyl-6-methylpridine-2(1H)-one (L-696,229), in rat and liver slices. Drug Metab Dispos. 1992;20:869–876. [PubMed] [Google Scholar]
- 2.Balani S K, Kauffman L R, Arison B H, Olah T V, Goldman M E, Varga S L, O’Brien J A, Ramjit H G, Rooney C S, Hoffman J M, Pitzenberger S M. Metabolism of 3-[2-(benzoxazol-2-yl)ethyl]-5-ethyl-6-methylpridine-2(1H)-one (L-696,229), an HIV-1 reverse transcriptase inhibitor, by rat liver slices and in humans. Drug Metab Dispos. 1994;22:200–205. [PubMed] [Google Scholar]
- 3.Balani S K, Pitzenberger S M, Schwartz M S, Ramjit H G, Thompson W J. Metabolism of L-689,502 by rat liver slices to potent HIV-1 protease inhibitors. Drug Metab Dispos. 1995;23:185–189. [PubMed] [Google Scholar]
- 4.Balani S K, Arison B H, Mathai L, Kauffman L R, Miller R R, Stearns R A, Chen I-W, Lin J H. Metabolites of L-735,524, a potent HIV-1 protease inhibitor, in human urine. Drug Metab Dispos. 1995;23:266–270. [PubMed] [Google Scholar]
- 5.Barry M, Gibbons S, Bach D, Mulcahy F. Protease inhibitors in patients with HIV disease: clinically important pharmacokinetic considerations. Clin Pharmacokinet. 1997;32:194–209. doi: 10.2165/00003088-199732030-00003. [DOI] [PubMed] [Google Scholar]
- 6.Bernareggi A, Rowland M. Physiologic modeling of cyclosporin kinetics in rat and man. J Pharmacokinet Biopharm. 1991;19:21–50. doi: 10.1007/BF01062191. [DOI] [PubMed] [Google Scholar]
- 7.Brøsen K, Zeugin T, Meyer U A. Role of P450 II D6, the target of the spartein-debrisoquin oxidation polymorphism, in the metabolism of imipramine. Clin Pharmacol Ther. 1991;49:609–617. doi: 10.1038/clpt.1991.77. [DOI] [PubMed] [Google Scholar]
- 8.Chang T, Levine M, Bellward G D. Selective inhibition of rat hepatic microsomal cytochrome P-450. II. Effect of the in vitro administration of cimetidine. J Pharmacol Exp Ther. 1992;260:1450–1455. [PubMed] [Google Scholar]
- 9.Chiba M, Hensleigh M, Nishime J A, Balani S K, Lin J H. Role of cytochrome P450 3A4 in human metabolism of MK-639, a potent human immunodeficiency virus protease inhibitor. Drug Metab Dispos. 1996;24:307–314. [PubMed] [Google Scholar]
- 10.Chiba M, Hensleigh M, Lin J H. Hepatic and intestinal metabolism of indinavir, an HIV protease inhibitor, in rat and human microsomes. Biochem Pharmacol. 1997;53:1187–1195. doi: 10.1016/s0006-2952(97)00100-7. [DOI] [PubMed] [Google Scholar]
- 11.Ciaccio P J, Duignan D B, Halpert J R. Selective inactivation by chloramphenicol of the major phenobarbital-inducible isozyme of dog liver cytochrome P-450. Drug Metab Dispos. 1987;15:852–856. [PubMed] [Google Scholar]
- 12.Crewe H K, Lennard M S, Tucker G T, Woods F R, Haddock R E. The effect of paroxetine and other specific 5-HT re-uptake inhibitors on cytochrome P450 II D6 activity in human liver microsomes. Br J Clin Pharmacol. 1991;32:658–659. [Google Scholar]
- 13.Danner S A, Carr A, Leonard J M, Lehman L M, Gudiol F, Gonzales J, Raventos A, Rubio R, Bouza E, Pintado V, Aguado A G, de Lomas J G, Delgado R, Borleffs J C C, Hsu A, Valdes J M, Boucher C A B, Cooper D A. A short-term study of the safety, pharmacokinetics, and efficacy of ritonavir, an inhibitor of HIV-1 protease. N Engl J Med. 1995;333:1528–1533. doi: 10.1056/NEJM199512073332303. [DOI] [PubMed] [Google Scholar]
- 14.Debouck C, Metcalf B W. Human immunodeficiency virus protease: a target for AIDS therapy. Drug Dev Res. 1990;21:1–17. [Google Scholar]
- 15.Debouck C. The HIV-1 protease as a therapeutic target for AIDS. AIDS Res Hum Retroviruses. 1992;8:153–164. doi: 10.1089/aid.1992.8.153. [DOI] [PubMed] [Google Scholar]
- 16.Denissen J F, Grabowski B A, Johnson M K, Boyd S A, Uchic J T, Stein H, Cepa S, Hill P. The orally active renin inhibitor A-74273: in vivo and in vitro morpholine ring metabolism in rats, dogs, and humans. Drug Metab Dispos. 1994;22:880–888. [PubMed] [Google Scholar]
- 17.Denissen J F, Grabowski B A, Johnson M K, Buko A M, Kempf D J, Thomas S B, Surber B W. Metabolism and disposition of the HIV-1 protease inhibitor ritonavir (ABT-538) in rats, dogs, and humans. Drug Metab Dispos. 1997;25:489–501. [PubMed] [Google Scholar]
- 18.Fuhr U, Anders E-M, Mahr G, Sörgel F, Staib A H. Inhibitory potency of quinolone antibacterial agents against cytochrome P450 I A2 activity in vivo and in vitro. Antimicrob Agents Chemother. 1992;36:942–948. doi: 10.1128/aac.36.5.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galpin S, Roberts N A, O’Connor T, Jeffries D J, Kinchington D. Antiviral properties of the HIV-1 proteinase inhibitor Ro 31-8959. Antivir Chem Chemother. 1994;5:43–45. [Google Scholar]
- 20.Grenlee W J. Renin inhibitors. Pharm Res. 1987;4:364–374. doi: 10.1023/a:1016422009662. [DOI] [PubMed] [Google Scholar]
- 21.Guengerich F P, Müller-Enoch D, Blair I A. Oxidation of quinidine by human liver cytochrome P-450. Mol Pharmacol. 1986;30:287–295. [PubMed] [Google Scholar]
- 22.Hirsch M S, D’Aquila R T. Therapy for human immunodeficiency virus infection. N Engl J Med. 1993;328:1686–1695. doi: 10.1056/NEJM199306103282307. [DOI] [PubMed] [Google Scholar]
- 23.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J, Markowits M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 24.Houston J B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47:1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 25.Huff J R. HIV protease: a novel chemotherapeutic target for AIDS. J Med Chem. 1991;34:2305–2314. doi: 10.1021/jm00112a001. [DOI] [PubMed] [Google Scholar]
- 26.Inaba T, Jurima M, Mahon W A, Kalow W. In vitro inhibition studies of two isozymes of human liver cytochrome P-450: mephenitoin p-hydroxylase and sparteine monooxygenase. Drug Metab Dispos. 1985;13:443–448. [PubMed] [Google Scholar]
- 27.Kageyama S, Mimoto T, Murakawa Y, Nomizu M, Ford H, Jr, Shirasaka T, Gulnik S, Erickson J, Takada K, Hayashi H, Broder S, Kiso Y, Mitsuya H. In vitro anti-human immunodeficiency virus (HIV) activities of transition state mimetic HIV protease inhibitors containing allophenylnorstatine. Antimicrob Agents Chemother. 1993;37:810–817. doi: 10.1128/aac.37.4.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kempf D J, Marsh K C, Denissen J F, McDonald E, Vasavanonda S, Flentge C A, Green B E, Fino L, Park C H, Kong X-P, Wideburg N E, Saldivar A, Ruiz L, Kati W M, Sham H L, Robins T, Stewart K D, Hsu A, Plattner J J, Leonard J M, Norbeck D W. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci USA. 1995;92:2484–2488. doi: 10.1073/pnas.92.7.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kiriyama A, Mimoto T, Kiso Y, Takada K. Pharmacokinetic study of a tripeptide HIV-1 protease inhibitor, KNI-174, in rats after intravenous and intraduodenal administrations. Biopharm Drug Dispos. 1993;14:199–207. doi: 10.1002/bdd.2510140303. [DOI] [PubMed] [Google Scholar]
- 30.Kiriyama A, Mimoto T, Kisanuki S, Kiso Y, Takada K. Comparison of a new orally potent tripeptide HIV-1 protease inhibitor (anti-AIDS drug) based on pharmacokinetic characteristics in rats after intravenous and intraduodenal administrations. Biopharm Drug Dispos. 1993;14:697–707. doi: 10.1002/bdd.2510140806. [DOI] [PubMed] [Google Scholar]
- 31.Kiriyama A, Fujita K, Takemura S, Kuramoto H, Kiso Y, Takada K. Plasma pharmacokinetics and urinary and biliary excretion of a new potent tripeptide HIV-1 protease inhibitor, KNI-272, in rats after intravenous administration. Biopharm Drug Dispos. 1994;15:617–626. doi: 10.1002/bdd.2510150709. [DOI] [PubMed] [Google Scholar]
- 32.Kiriyama A, Sugahara M, Yoshikawa Y, Kiso Y, Takada K. The bioavailability of oral dosage forms of a new HIV-1 protease inhibitor, KNI-272, in beagle dogs. Biopharm Drug Dispos. 1996;17:125–134. doi: 10.1002/(SICI)1099-081X(199603)17:2<125::AID-BDD940>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 33.Kiriyama A, Nishiura T, Ishino M, Yamamoto Y, Ogita I, Kiso Y, Takada K. Binding characteristics of KNI-272 to plasma proteins, a new potent tripeptide HIV protease inhibitor. Biopharm Drug Dispos. 1996;17:739–751. doi: 10.1002/(SICI)1099-081X(199612)17:9<739::AID-BDD987>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Kiriyama, A., T. Nishiura, H. Yamaji, and K. Takada. Physiologically based pharmacokinetics of KNI-272, a tripeptide HIV-1 protease inhibitor. Submitted for publication. [DOI] [PubMed]
- 35.Kumar G N, Rodrigues A D, Buko A M, Denissen J F. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J Pharmacol Exp Ther. 1996;277:423–431. [PubMed] [Google Scholar]
- 36.Lang M, Roesel J. HIV-1 protease inhibitors: development, status, and potential role in the treatment of AIDS. Arch Pharm (Weinheim) 1993;326:921–924. doi: 10.1002/ardp.19933261202. [DOI] [PubMed] [Google Scholar]
- 37.Lin J H, Hayashi M, Awazu S, Hanano M. Correlation between in vitro and in vivo drug metabolism rate: oxidation of ethoxybenzamide in rat. J Pharmacokinet Biopharm. 1978;6:327–337. doi: 10.1007/BF01060096. [DOI] [PubMed] [Google Scholar]
- 38.Lin J H, Chiba M, Chen I-W, Vastag K J, Nishime J A, Dorsey B D, Michelson S R, McDaniel S L. Time- and dose-dependent pharmacokinetics of L-754,394, an HIV protease inhibitor, in rats, dogs and monkeys. J Pharmacol Exp Ther. 1995;274:264–269. [PubMed] [Google Scholar]
- 39.Lin J H, Chiba M, Balani S K, Chen I-W, Kwei G Y-S, Vastag K J, Nishime J A. Species differences in the pharmacokinetics and metabolism of indinavir, a potent human immunodeficiency virus protease inhibitor. Drug Metab Dispos. 1996;24:1111–1120. [PubMed] [Google Scholar]
- 40.Lowry O H, Rosebrough N J, Farr A L, Randall R J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 41.Markowitz M, Saag M, Powderly W G, Hurley A M, Hsu A, Valdes J M, Henry D, Sattler F, Marca A L, Leonard J M, Ho D D. A preliminary study of ritonavir, an inhibitor of HIV-1 protease, to treat HIV-1 infection. N Engl J Med. 1995;333:1534–1539. doi: 10.1056/NEJM199512073332204. [DOI] [PubMed] [Google Scholar]
- 42.Mimoto T, Imai J, Kisanuki S, Enomoto H, Hattori N, Akaji K, Kiso Y. Kynostatine (KNI)-227 and -272, highly potent anti-HIV agents: conformationally constrained tripeptide inhibitors of HIV protease containing allophenylnorstatine. Chem Pharm Bull. 1992;40:2251–2253. doi: 10.1248/cpb.40.2251. [DOI] [PubMed] [Google Scholar]
- 43.Norbeck D W, Kempf D J. HIV protease inhibitors. Annu Rep Med Chem. 1991;26:141–150. [Google Scholar]
- 44.Pang K S, Rowland M. Hepatic clearance of drugs. I. Theoretical consideration of a “well-stirred” and a “parallel tube” model. Influence of hepatic blood, plasma and blood binding and hepatocellular enzyme activity on hepatic drug clearance. J Pharmacokinet Biopharm. 1977;5:625–653. doi: 10.1007/BF01059688. [DOI] [PubMed] [Google Scholar]
- 45.Pichard L, Fabre I, Fabre G, Domergue J, Aubert B S, Mourad G, Maurel P. Cyclosporin A drug interactions: screening for inducers of cytochrome P-450 (cyclosporin A oxidase) in primary cultures of human hepatocytes and in liver microsomes. Drug Metab Dispos. 1990;18:595–606. [PubMed] [Google Scholar]
- 46.Pillay D, Bryant M, Getman D, Richman D D. HIV-1 protease inhibitors: their development, mechanism of action and clinical potential. Med Virol. 1995;5:23–33. [Google Scholar]
- 47.Prueksaritanont T, Dwyer L M, Balani S K, Theoharides A D. In vitro metabolism of L-696,229, an HIV-1 reverse transcriptase inhibitor in rats and humans. Hepatic and extrahepatic metabolism and identification of enzymes involved in the hepatic metabolism. Drug Metab Dispos. 1994;22:281–288. [PubMed] [Google Scholar]
- 48.Prueksaritanont T, Balani S K, Dwyer L M, Ellis J D, Kauffman L R, Varga S L, Pitzenberger S M, Theoharides A D. Species differences in the metabolism of a potent HIV-1 reverse transcriptase inhibitor, L-738,372. In vivo and in vitro studies in rats, dogs, monkeys, and humans. Drug Metab Dispos. 1995;23:688–695. [PubMed] [Google Scholar]
- 49.Sugahara M, Kiriyama A, Hamada Y, Kiso Y, Takada K. Absorption of new HIV-1 protease inhibitor, KNI-272, after intraduodenal and intragastric administrations to rats: effect of solvent. Biopharm Drug Dispos. 1995;16:269–277. doi: 10.1002/bdd.2510160403. [DOI] [PubMed] [Google Scholar]
- 50.Taira Z, Terada H. Specific and non-specific ligand binding to serum albumin. Biochem Pharmacol. 1985;34:1999–2005. doi: 10.1016/0006-2952(85)90322-3. [DOI] [PubMed] [Google Scholar]
- 51.Tomasselli, A. G., W. J. Howe, T. K. Sawyer, A. Wlodawer, and R. L. Heinrickson. 1991. The complexities of AIDS: an assessment of the HIV protease as a therapeutic target. Chim. Oggi:6–27.
- 52.Vacca J P, Dorsey B D, Schleif W A, Levin R B, McDaniel S L, Darke P L, Zugay J, Quintero J C, Blahy O M, Roth E, Sardana V V, Schlabach A J, Graham P I, Condra J H, Gotlib L, Holloway M K, Lin J, Chen I-W, Vastag K, Ostovic D, Anderson P S, Emini E A, Huff J R. L-735,524: an orally bioavailable human immunodeficiency virus type 1 protease inhibitor. Proc Natl Acad Sci USA. 1994;91:4096–4100. doi: 10.1073/pnas.91.9.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, Saag M S, Shaw G M. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 54.Wolfenden R. Analog approaches to the structure of the transition state in enzyme reactions. Acc Chem Res. 1972;5:10–18. [Google Scholar]