

Abstract
The practice of serial X-ray crystallography (SX) depends on efficient, continuous delivery of hydrated protein crystals while minimizing background scattering. Of the two major types of sample delivery devices, fixed-target devices offer several advantages over widely adopted jet injectors, including: lower sample consumption, clog-free delivery, and the ability to control on-chip crystal density to improve hit rates. Here we present our development of versatile, inexpensive, and robust polymer microfluidic chips for routine and reliable room-temperature serial measurements at both synchrotrons and X-ray free electron lasers (XFELs). Our design includes highly X-ray-transparent enclosing thin film layers tuned to minimize scatter background, adaptable sample flow layers tuned to match crystal size, and a large sample area compatible with both raster scanning and rotation based serial data collection. The optically transparent chips can be used both for in situ protein crystallization (to eliminate crystal handling) or crystal slurry loading, with prepared samples stable for weeks in a humidified environment and for several hours in ambient conditions. Serial oscillation crystallography, using a multi-crystal rotational data collection approach, at a microfocus synchrotron beamline (SSRL, beamline 12–1) was used to benchmark the performance of the chips. High-resolution structures (1.3–2.7 Å) were collected from five different proteins- hen egg white lysozyme, thaumatin, bovine liver catalase, concanavalin-A (type VI), and SARS-CoV-2 nonstructural protein NSP5. Overall, our modular fabrication approach enables precise control over the cross-section of materials in the X-ray beam path and facilitates chip adaption to different sample and beamline requirements for user-friendly, straightforward diffraction measurements at room temperature.
Keywords: X-ray crystallography, synchrotrons, sample delivery, fixed-targets, cyclic olefin copolymer (COC), microfluidics, serial crystallography (SX), room temperature
Graphical Abstract
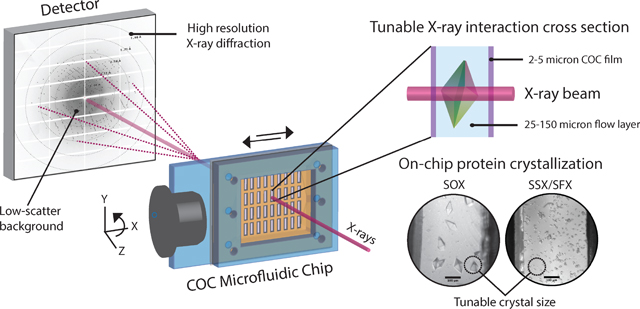
This work presents our development of versatile, inexpensive, and robust polymer microfluidic chips for routine and reliable room-temperature serial X-ray crystallography measurements.
Introduction
As a result of continued developments in the field of X-ray crystallography, the number of X-ray structures deposited annually in the Protein Data Bank (rcsb.org) has continued to increase rapidly from 135 in 1990 to 11234 in 2020. Alongside the rise of cryo-crystallography1, these include improvements in crystallization methods2, approaches to high throughput screening3, and the emergence of highly brilliant, microfocus synchrotron beamlines enabling the collection of data from ever smaller crystals.4,5 Meanwhile, the development of femtosecond XFELs has ushered in a new era of structural biology, with radiation damage-free data collection made possible at room temperature (RT) using serial femtosecond crystallography (SFX), due to the diffraction before destruction principle.6–8 While single crystal cryogenic measurements continue to be the workhorse of macromolecular crystallography, technological development to support XFEL experiments has elicited a resurgence of interest in RT data collection at synchrotron facilities in recent years, with the parallel appearance of serial synchrotron crystallography (SSX) and serial oscillation crystallography (SOX) methods. In contrast with cryo measurements, RT synchrotron studies are advantageous because they eliminate the need to screen cryoprotectants and freeze samples.9 Importantly, measurements at RT have opened avenues to study not just static structures but also protein dynamics over a broad time-range, from picosecond to seconds, using time-resolved measurements. This new frontier in capabilities is paving the path for making molecular movies involving ligand binding, photoactivation, and catalysis to better understand dynamic structure-function relationships but requires facile and functional sample delivery.10–14
The high intensity of 3rd and 4th generation microfocus X-ray beamlines, fast onset of radiation damage at RT, and the use of microcrystals limit the number of high-resolution diffraction frames that can be collected from a single crystal. To address this limitation, serial crystallography (SX) can be used, where data from multiple crystals is combined.6,15,16 For SOX, in which small rotation wedges (1–20°) are collected from small crystals (~10s of μm in size), data from tens to hundreds of crystals is typically sufficient.17 For SSX or SFX, each diffraction volume is exposed only once and diffraction data from thousands of crystals is often required to obtain a complete dataset with high redundancy and good signal-to-noise at high resolution.15,18 In most cases, these techniques require a much larger amount of crystalline material than conventional crystallography, using crystals that may be too small to successfully mount using conventional cryo-loops. This necessitates the development of specialized crystal delivery methods which are continuous, robust, and keep the crystals hydrated over the course of the measurement. Therefore, an optimal sample delivery platform should: (1) maintain crystal quality and hydration by minimizing sample stresses during sample preparation, characterization, and delivery; (2) minimize scatter contribution from the delivery device and excess buffer surrounding crystals; (3) optimize crystal density and crystal size to the beam and data collection requirements; (4) allow efficient use of beamtime by minimizing down time (e.g. from clogging or sample alignment for rastering) with fast/automated sample changes (e.g. sample preparation in advance); and (5) allow experiments to probe structural dynamics by allowing different triggering methods.19,20 Various sample delivery approaches have been proposed and demonstrated including liquid-jets21–24, droplet-on-demand tape drives25,26, and fixed-target devices27–32. The work presented here focuses on a new fixed-target platform for SOX or SSX/SFX. Fixed-target devices are advantageous compared to other approaches because they require very low sample volumes. Our novel geometries are compatible with in situ crystal growth which eliminates crystal handling and enable tuning of crystal densities and sizes to achieve high hit rates and high-quality diffraction images. Furthermore, they allow for the use of non-jetable crystal morphologies, like needles or plates, or crystals of heterogeneous size. They can also be used to facilitate dynamic experiments using stimuli like electric fields gradients, temperature jumps, pH gradients, ligand exchange and photo-activation.
For fixed targets, the choice of construction materials and fabrication strategy are important considerations as they significantly impact the cost, fragility, and stability of the devices. Several materials such as low-Z polymers (COC, PMMA, PDMS, Kapton, Mylar, epoxies), silicon, silicon nitride, glass and quartz have been used to make devices that balance these attributes with the measurement requirements. The two most common design formats are (1) microgrids and (2) microfluidic chips. Microgrids are chips with a 2D array of micropatterned holes for crystal entrapment upon deposition, sometimes used with thin films supports (0.1–10 μm) to improve sample retention and/or protect against dehydration.17,27–30,33–46 These chips can offer advantages of high hit-rates and ultra-low background if wicking or vacuum application is used to localize crystals into pores and remove excess crystallization solution, but the exposed sample (even with sandwiching thin films) is sensitive to dehydration, requiring sample preparation and assembly shortly before measurements (< 1–2 hours). Microfluidic chips, on the other hand, are enclosed devices offering precise control over sample volume and thickness, long-term stability against evaporation, and in situ crystal growth to eliminate crystal handling. But, the thick flow channels (25–300 μm) and capping layers (25–600 μm) often used to construct these devices contribute significantly to background scatter.18,47–57 While several such devices have demonstrated high resolution data collection from large, well-ordered protein crystals, for many proteins obtaining large crystals often proves intractable. In the case of these small or weakly-diffracting microcrystals (desired for SX), the X-ray attenuation and scatter background from typical microfluidic chips can limit the resolution attained. Therefore, there is a need for the development of novel fixed targets with the stability and ease-of-use of microfluidic approaches, that are inexpensively fabricated and easily modified to match different sample and beamline requirements, while maintaining the advantages of high hit-rates and low background of microgrid approaches.
In this paper we describe the design of polymer fixed-target chips that address this need, for routine and reliable RT SOX and SSX/SFX experiments. The polymer materials used were selected for low water permeability, high X-ray transparency and high optical transparency (for on-chip imaging and light triggering of structural changes for future time-resolved structural studies). A modular, layered fabrication process enabled control over the cross section of materials in the beam path and easy design modification or adaption to different sample and beamline requirements. The chip is compatible with in situ crystallization using micro-batch and vapor diffusion methods. Pilot X-ray data collection with the chips was performed using SOX. The robustness and versatility of the chips was demonstrated, showing that they allow long-term storage, stability, easy transportation, and straightforward on-chip crystallization to diffraction measurements. Preliminary results also indicate that the chip is amenable for SFX measurements without further alteration.
Materials and methods
A layered assembly process was used to construct the polymer microfluidic chip from hot-embossed COC supports, spin-coated COC thin films, laser-cut PMMA frames, and an adhesive sample flow layer. Five proteins (hen egg white lysozyme, thaumatin, bovine liver catalase, concanavalin-A, and SARS-CoV-2 nonstructural protein NSP5) were crystallized in situ and diffraction data directly collected on-chip at RT at beamline 12–1 at SSRL. The minimum beam spot size was 55 μm × 5 μm (X-Y, FWHM) and a SOX data collection approach was used to benchmark the performance of the chip.
Microfluidic chip fabrication and assembly
A schematic of the chip construction layers, and the final assembled chip are shown in Figure 1. The X-ray imaging regions of the chip were made of cyclic olefin copolymer (TOPAS® COC, Grade 8007, Tg = 75 °C). The sample flow layer (layer 1) consisted of a CO2 laser cut, tunable acrylic or silicone pressure sensitive adhesive (25 μm AR92734 or 48 μm AR 92712, Adhesives Research Inc., Glen Rock, PA, USA) used to bond the two microfabricated sides (top and bottom) together. 2–5 μm COC thin films (layer 2) provided low-background sample enclosing layers. These were prepared by spin-coating solutions of 10–20 wt.% COC dissolved in sec-butylbenzene on UV-ozone treated silicon wafers. Films ranging from 500 nm to 10 μm in thickness could be easily produced by varying the COC concentration and spin speed (Figure S1). The 200 μm thick COC supports with windows (layer 3) were hot embossed using molds made of elastic polydimethylsiloxane (PDMS, details of the mold fabrication and hot embossing below). And finally, 0.5 or 1 mm polymethyl methacrylate (PMMA) frames (layer 4) with an adhesive layer (3M™ F9460PC) were produced by CO2 laser cutting.
Figure 1:

A schematic of (A) the different construction layers used to assemble the polymer microfluidic chip; (B) the cross-section view showing the different layer thicknesses; and (C) the final assembled chip
The use of hard mold materials like steel, silicon or high-temperature epoxy used to emboss COC in previous works49,58,50 proved difficult due to feature entanglement and warping issues while demolding the COC sheets (with 200 μm deep through-holes) from the rigid molds. PDMS molds proved successful due to their elasticity and low adhesion to COC. These molds were fabricated by casting a replica of a silicon master mold with an array of 300 μm deep X-ray window features using a 5:1 monomer:curing agent mixture of Dow Sylgard™ 184 (fabrication of Si master and additional process details provided in SI Section 1). The PDMS replica was demolded and bonded to a silicon wafer using oxygen plasma treatment (50 W, 25 sccm O2, 0.79 Torr, 1 minute). Hot embossing was performed using a semi-automated EVG 501 wafer bonder. The PDMS mold was brought into contact with a 240 μm COC sheet (Europlex 0F304, Roehm America LLC, Sanford, ME, USA) attached to a polyvinyl alcohol (9 wt. % PVA, 1500 rpm, 60 s) coated silicon wafer. The assembly was heated to 120 °C and a force of 12 kN was applied for 15 minutes to emboss window in the supports. The embossed sheet was demolded after cooling below the glass transition temperature of COC. Obtaining perfect through holes with hot embossing was difficult due to the flexibility of the PDMS mold. Instead, the ~20–30 μm residual thin film in the windows features was removed by an oxygen plasma reactive-ion etching process (500 W, 25 sccm O2, 330 mTorr, 30 minutes) to yield the COC window support with through-holes (layer 3). A schematics of the fabrication steps is shown in Figure 2A,B.
Figure 2:

A schematic of the fabrication scheme used to produce the top and bottom sides of the microfluidic chip (as shown in Figure 1). (A) Hot embossing was performed using an elastic PDMS mold to imprint an array of “X-ray window” features in COC sheets (240 μm). (B) The residual thin film (20–30 μm) from the embossing step was removed using oxygen plasma reactive ion etching (RIE) to create COC supports (~200 μm) with through-hole windows. The supports were detached from the silicon wafer by dissolving the PVA sacrificial layer in water. (C) To assemble each side (top and bottom), a COC support was solvent treated to facilitate bonding to COC thin films of desired thicknesses (2–5 μm), followed by attachment of a 0.5–1 mm thick PMMA frame using an adhesive layer to provide rigidity and flatness.
After fabricating the various layers as described above, Figure 2C shows a schematic of the processing steps involved in assembling the two symmetric sides of the chip (top and bottom, Figure 1). First, a COC window support (layer 3) was solvent treated with 35:65 acetone: cyclohexane solution for 1 minute to render the surface tacky59, dried with a nitrogen stream, and brought into contact with a spin-coated COC thin film (layer 2) on a silicon wafer. This process created a strong room-temperature bond between layers while maintaining the integrity of the thin film (layer 2). The assembly was further reinforced by attaching 0.5 or 1 mm thick PMMA frames (layer 4) on the other side to improve planarity, prevent bowing of the thinner layers, and make the chip easy to handle. At this stage, the assembled layers (layers 2–4) were robust and could be stored until needed. This was advantageous as the flow-layer (layer 1) could be varied in thickness to tailor the chip for a particular protein crystal size/beamline spot size on demand. Both thin (25–50 μm) or thick (80–150 μm) flow layers could be used depending on sample requirements, e.g., ultra-small protein crystals benefit from thinner flow layers to decrease background from excess crystallization solution, while efficient slurry loading of larger crystals necessitates thicker flow layers. The hydrophobic COC films needed to be rendered hydrophilic prior to final assembly to facilitate fast and complete solution loading into the chip. To do this, assembled top and bottom sides (layers 2–4) were exposed to atmospheric plasma treatment for 3 minutes in a Harrick PDC-32G Basic Plasma Cleaner. Advancing water contact angles using a Ramé-Hart goniometer were 83 ± 6° for the native COC surface, 22 ± 5° after plasma treatment, and recovered to ~60° upon storage in ambient conditions for 2–4 weeks (Figure S2). To complete the chip fabrication process, the two chip sides were bonded using a laser-cut, double-sided adhesive sample flow layer of the desired thickness. The holes or “window” features (0.5 × 1.75 mm or 1 mm × 1 mm) in the two sides of the chip were aligned by maximizing light transmission through the features using a backlight. The aligned sides were manually pressed together to create an enclosed microfluidic chip. A contact dwell time of 24–72 hours was required to ensure strong adhesion between all the layers before using the chip. A more detailed protocol on the individual microfabrication steps is provided in the SI Section S1.
Water permeability measurements
Water vapor transmission rate as a function of COC film thickness was measured using a slightly modified version wet-cup tests described elsewhere60. In brief, free standing COC thin films of different thickness were solvent bonded to a 240 μm COC sheet with a 1 cm diameter hole in the center and affixed to the opening of a 3 mL clear glass vial containing Millipore water using Dow Corning® high vacuum grease. Sample weight loss was monitored over a period of one week from five replicates. Steady-state water vapor transmission rates (WVTR) were calculated from the weight loss measurements using , where Δm was the weight loss, A is the area of the membrane, and t is the time. The water vapor permeability (WVP) of COC, which is a function of the solubility and diffusivity of water in the material, was calculated by fitting the experimental data to where ΔP was the differential pressure of water vapor across the membrane of thickness L. The measurements were carried out in a climate-controlled room with a relative humidity of 20 ± 2% at 23 ± 2 °C. The relative humidity gradient (ΔRH) was approximately 80% assuming the relative humidity inside the sealed vial enclosure was close to 100%, resulting in ΔP = P × ΔRH ≅ 2.2 kPa, using a water vapor saturation pressure of 2.8 kPa at 23 °C.
Protein expression, purification, and crystallization
SARS-CoV-2 main protease nonstructural protein NSP5 was expressed and purified using a modified protocol as previously described61. In short, NSP5 was expressed from PGEX-6p-1-NSP5 plasmid (kindly provided by R. Hilgenfeld, University of Lübeck, Germany), from E. coli BL21 DE3 Gold cells in 2YT media overnight at 18 °C. Freshly streaked plates from transformed glycerol stocks were used for inoculation. The harvested cell pellets were stored at −80 °C until purification. The pellet was thawed in Buffer A (20 mM Tris pH 7.8, 150 mM NaCl, 5 mM imidazole) and disrupted by sonication at 4 °C (2 min total) and two passes through a cell disruptor (12–14 kPa). The suspension was clarified by centrifugation (30 kg, 45 min), filtered (0.8 um) and loaded onto a 5 mL HisTrap FF column (Cytiva). The column was washed with 5 CV of Buffer A and the protein eluted in 5mL fractions of Buffer B (20 mM Tris pH 7.8, 150 mM NaCl, 300 mM imidazole). The protein was dialyzed against Buffer C (20 mM Tris pH 7.8, 150 mM NaCl, 0.5 mM TCEP) concurrently with GST tag removal by overnight digestion with 10% w/w HRV3C protease. The protein was loaded into a 5 mL HisTrap FF column and the flow through collected in 12.5 mL fractions. The protein was further purified by size exclusion chromatography (Superdex 200 pg, Cytiva) in Buffer D (20 mM Tris pH 7.8, 150 mM NaCl, 0.5 mM TCEP, 1 mM EDTA). The protein was concentrated to 10 mg/mL and crystallized via vapor diffusion with 100 mM Bis-Tris pH 6.5, 17.5% w/v PEG 3350, 175 mM Li2SO4. Seed stock was generated from the resulting large plate clusters using Hampton Research’s PTFE seed beads and diluted by a factor of one hundred with crystallization buffer. The vapor diffusion conditions were spiked with 10% 1:100 seed stock and this process was repeated to generate second generation seeds. To crystallize NSP5 on-chip, the same condition used to generate second generation seeds was loaded into a microfluidic chip with 10% second generation seeds (Table 1) and crystallized via vapor diffusion as described below.
Table 1:
A summary of crystallization conditions used in this work and a comparison of crystal size, morphology, density, and time to first crystals, obtained from on-chip crystallization and comparable crystallization in 24-well crystallization plates. For on-chip crystallization, ~8 μL of solution was loaded into the chip and the chip was stored either sealed (micro-batch) or unsealed (vapor diffusion) in an enclosed environment with the precipitant solution in the reservoir. Similar droplet volumes were used for crystallization in a 24-well plate format using micro-batch under oil (paraffin) or hanging drop vapor diffusion methods for comparison, as appropriate. Crystal density (n= # of crystals/mm2) was measured using optical microscopy and qualitatively described as low for n≤ 10, medium for 10 < n < 100, and high for n ≥ 100.
| Protein | Protein solution | Precipitant solution | Incubation Temperature | Crystallization method | Crystal size, morphology, density, crystallization time | |
|---|---|---|---|---|---|---|
| On-chip | 24-well Crystallization plate | |||||
|
| ||||||
| Lysozyme | 30 mg/mL in 20 mM Sodium Acetate buffer, pH 4.6 | 1M Sodium Chloride, 0.1M Sodium Acetate buffer, pH 4.6 | 4°C | Micro-batch | 25–35 μm, cuboid, high, ~2–3 hours. | 25–35 μm, cuboid, high, ~2–3 hours. |
| 50 mg/mL in DI water | 2M Sodium Chloride, 0.1M Sodium Acetate buffer, pH 4.6 | 21°C | Micro-batch | 60–100 μm, cuboid, low, 12–24 hours. | 30–70 μm, cuboid, medium, 2–4 hours. | |
|
| ||||||
| Thaumatin | 25 mg/ml in DI water | 1M L-Sodium Potassium Tartrate, 0.1M ADA buffer, pH 6.5 | 4°C | Micro-batch | ~25 μm, tetragonal bipyramidal, low, ~12 hours. Crystals grow to 70–100 μm in size over 2–3 days. | ~20 μm, tetragonal bipyramidal, high, ~12 hours. |
| 21°C | Micro-batch | 50–100 μm, tetragonal bipyramidal, low, 3–4 days. | Did not crystallize | |||
| 21°C | Vapor diffusion | 50–100 μm, tetragonal bipyramidal, low, ~24 hours. | 50–150 μm, tetragonal bipyramidal, low, ~24 hours. | |||
| 21°C | Micro-batch (with seeding using 1° seed stock 1:5:5 seed: protein: precipitant) | 15–20 μm, tetragonal bipyramidal, high, ~24 hours. | 15–20 μm, tetragonal bipyramidal, high, ~24 hours | |||
|
| ||||||
| Concanavalin-A (Type VI) | 70 mg/mL in 20 mM Tris buffer, pH 8.0 | 2.8M Ammonium Sulfate in 0.1M Tris buffer, pH 8.5 | 21°C | Micro-batch | 150–300 μm, round base rhombic tetrahedron, low, ~12 hours. | 30–60 μm, cubic, high, 0–2 hours initially; equilibrates to 150–300 μm, round base rhombic tetrahedron, low, over the next 3–7 days. |
|
| ||||||
| Bovine Liver Catalase | 40 mg/mL in 50 mM Sodium Phosphate buffer, pH 6.8 | 22.5% PEG 4000 in 0.1M Tris buffer, pH 8.5 | 21°C | Micro-batch | 70–120 μm, prism shaped, low, <12 hours | 70–120 μm, prism shaped, low, 2–4 hours |
| 21°C | Vapor diffusion | 30–100 μm, prism shaped, medium, 0–4 hours | 30–100 μm, prism shaped, medium, 0–2 hours | |||
|
| ||||||
| NSP5 | 5mg/ml in 20 mM Tris pH 7.8, 150 mM NaCl, 0.5 mM TCEP, 1 mM EDTA | 100 mM Bis-Tris pH 6.5, 17.5% w/v PEG 3350, 175 mM; Li2SO4; 10% 1:100 2’ seeds | 21°C | Vapor diffusion, with seeding | 300 um, large thin plate clusters, low, ~12 hours | 150–200 μm large single plates or plate clusters, low, ~12 hours |
Chicken egg-white lysozyme (#L6876), thaumatin from Thaumatococcus daniellii (#T7638), concanavalin A from Canavalia ensiformis (Jack bean) Type VI (#L7647) and catalase from bovine liver (#C40) were purchased from Millipore Sigma (St. Louis, MO, USA) and dissolved in MilliQ water or low ionic strength buffers as listed in Table 1. The protein solutions were gently vortexed for a few seconds until the solution was well-mixed, centrifuged at 10,000 rpm for 5 minutes to remove any insoluble materials, and the supernatant was removed and stored at 4 °C. All buffers and precipitant solutions were filtered through a 0.22 μm syringe filter prior to use. The crystallization conditions used in this work were adapted from previous literature62–64 and are also reported in Table 1.
Direct crystal slurry loading requires filtration to ensure removal of large crystals which can otherwise clog the inlet channel. While this was tested, this work mainly focusses on demonstration of on-chip crystallization. The microfluidic chips were loaded with a well-mixed 1:1 solution of protein and precipitant solution by pipetting ~8–10 μL into one of the inlet holes in the PMMA frame. Corner vents in the spacer flow layer ensured that the solution filled the wide fluid chamber (~10 mm × 10 mm × 25–50 μm) uniformly while minimizing bubble entrapment. After filling, the inlets and outlets were sealed using Hampton crystal clear sealing tape for micro-batch crystallization or stored unsealed for vapor diffusion crystallization to allow for equilibration with the reservoir chamber. The filled chips were then placed in a Falcon 6-well tissue culture plate either on a microbridge-like pedestal or affixed to a small magnet on the wall of the well plate using magnetic chip holder pin bases (Crystal Positioning Systems, Jamestown, NY, USA). The well was filled with 1.5–2 mL of precipitant solution and the plate was sealed using crystal clear sealing tape to ensure that the chamber remained humidified to prevent sample desiccation during storage over several days to weeks. The optically transparent microfluidic chip and well sealing tape enabled on-chip sample monitoring without disturbing the equilibrated enclosure.
On-chip X-ray diffraction
X-ray diffraction data were collected at RT on the 12–1 beamline at SSRL, equipped with an Eiger X 16M detector (Dectris AG). A magnetic chip holder pin base with a slot and set screw was used to securely hold the microfluidic chip before magnetically mounting it on the goniometer. Inline high-resolution cameras at ~0° and 90° orientation to the beam were used to position the chip in the beam path, scan through sample regions/windows, and to center the protein crystals along the rotational axis. The beamline allowed for a translation range of ± 2.5 mm along the Y-direction (vertical) and greater than ± 7.5 mm along the X-direction (horizontal). The close proximity of an upstream microcollimator limited the range of rotation about the axis (Z) to ± 35°. The smallest beam size available was 55 μm × 5 μm (X-Y, FWHM), with Y varied between 5 to 50 μm based on crystal size and morphology to maximize the sample-beam interaction cross-section.
Data was collected remotely using the Blu-Ice package65. Individual single crystals were manually centered and 30° rotation wedges with 1° oscillation at 0.1s exposure per frame were collected at 10–20 % transmission (full photon flux 4 to 5.6 × 1012 photons per second at 12.5 keV) with a 200 mm detector distance. A drop off in diffraction resolution was observed by the 20–25th frame due to cumulative radiation damage. Data from 15–30 crystals was collected from each chip with the final number of crystals merged for each protein reported in Table 2. Diffraction data from multiple crystals was processed in xia266 (multiplexing mode) running the CCP467 and DIALS68 packages to perform indexing, merging, and scaling. Structures were solved with Phaser69, part of the PHENIX package70, using PDB entries 1VED, 1RQW, 8CAT, 1SCR, and 6XR3 as templates for phasing via molecular replacement for lysozyme, thaumatin, concanavalin-A, catalase and NSP5 respectively. Iterative refinement was performed with phenix.refine71 alternating with molecular modeling performed with Coot72. Final data processing and structure refinement statistics are provided in Table 2.
Table 2:
Crystallographic statistics obtained from Lysozyme, Thaumatin and Concanavalin-A, Catalase and NSP5 crystals grown in the microfluidic chip. Values in parentheses correspond to the highest resolution shell.
| Protein | Lysozyme (fresh) | Lysozyme (aged) | Thaumatin | Concanavalin-A | Catalase | NSP5 |
|---|---|---|---|---|---|---|
|
| ||||||
| Number of crystals used | 7 | 6 | 16 | 12 | 7 | 11 |
| Average crystal size | ~30–35 μm | ~70 μm | ~ 100 μm | ~ 200 μm | ~ 90–100 μm | ~ 200 μm |
| (Largest dimension) | ||||||
| Resolution range | 56.05 – 1.87 | 35.25 – 1.5 | 58.61 – 1.45 | 51.18 – 1.30 | 52.8 – 2.274 | 31.64 – 2.7 |
| (1.94 – 1.87) | (1.55 – 1.5) | (1.48 – 1.45) | (1.35 – 1.30) | (2.355 – 2.274) | (2.797 – 2.7) | |
| Unit cell dimensions | a = b = 79.26 , | a = b = 78.82 , | a = b = 58.60 , | a = 63.20 , b = 87.22 , | a = b = 141.77 , | a = 114.29 , b = 54.81 , |
| c = 37.94 , | c = 38.207 , | c = 151.44 , | c = 89.12 , | c =103.47 , | c =45.33 , | |
| α = β =γ = 90° | α = β =γ = 90° | α = β =γ = 90° | α = β =γ = 90° | α = β = 90°, γ = 120° | α = γ = 90°, β = 101.45° | |
| Space group | P43212 | P43212 | P41212 | I222 | P3221 | C121 |
| Data processing statistics | ||||||
| Total reflections | 120310 (12080) | 331623 (33168) | 1392515 (65410) | 786259 (24955) | 1040538 (97971) | 49766 (5027) |
| Unique reflections | 10449 (1003) | 19849 (1949) | 47884 (2332) | 60428 (5732) | 55166 (5256) | 7530 (747) |
| Multiplicity | 11.5 (12.0) | 16.7 (17.0) | 29.1 (28.0) | 13.0 (4.4) | 18.9 (18.6) | 6.6 (6.7) |
| Completeness (%) | 99.82 (98.91) | 99.97 (99.95) | 100.0 (100.0) | 98.8 (89.5) | 99.56 (95.89) | 96.30 (93.18) |
| Mean I/σ(I) | 11.47 (2.02) | 16.80 (1.59) | 15.4 (0.9) | 17.6 (0.6) | 10.64 (1.05) | 8.24 (1.78) |
| Rmerge | 0.159 (1.63) | 0.09752 (1.953) | 0.146 (3.606) | 0.072 (2.107) | 0.218 (2.587) | 0.248 (2.01) |
| Rmeas | 0.167 (1.703) | 0.1007 (2.013) | 0.149 (3.671) | 0.075 (2.384) | 0.224 (2.658) | 0.2691 (2.183) |
| Rpim | 0.048 (0.481) | 0.02467 (0.4847) | 0.027 (0.677) | 0.019 (1.055) | 0.051 (0.606) | 0.097 (0.795) |
| CC1/2 | 0.994 (0.602) | 0.999 (0.773) | 0.999 (0.647) | 0.993 (0.159) | 0.997 (0.576) | 0.968 (0.267) |
| Wilson B-factor | 28.48 | 22.45 | 19.9 | 17.8 | 48.65 | 55.59 |
| Refinement statistics | ||||||
| Rwork (%) | 15.61 | 15.02 | 15.2 | 14.6 | 16.2 | 18.0 |
| Rfree (%) | 17.9 | 17.13 | 16.0 | 16.5 | 20.1 | 21.8 |
| RMS (bonds, Å) | 0.005 | 0.008 | 0.022 | 0.004 | 0.003 | 0.003 |
| RMS (angles, °) | 0.59 | 0.84 | 1.59 | 0.83 | 0.58 | 0.53 |
| Ramachandran | ||||||
| favored (%) | 99.21 | 99.21 | 98.0 | 97.8 | 95.85 | 96.04 |
| allowed (%) | 0.79 | 0.79 | 2.0 | 2.2 | 3.94 | 3.96 |
| outliers (%) | 0 | 0 | 0 | 0 | 0.20 | 0 |
| Average B-factor | ||||||
| macromolecule | 33.30 | 28.13 | 21.9 | 22.7 | 56.46 | 59.64 |
| ligands | - | - | - | 14.6 | 55.17 | - |
| solvent | 36.39 | 36.13 | 38.6 | 34.6 | 46.48 | 49.38 |
Results and discussion
Microfluidic chip fabrication and device performance
The goal of this work was to develop robust, user-friendly, low-background microfluidic chips to support room-temperature serial crystallography experiments at synchrotrons or XFEL facilities. A modular fabrication process was designed and used to construct large-area polymer chips that could deliver hundreds to thousands of hydrated protein microcrystals to the X-ray beam. An exemplary chip design (overall dimensions 25 mm × 15 mm) with a 4 × 9 array of rectangular X-ray windows (0.5 mm × 1.75 mm) is shown in Figure 3A. The X-ray imaging regions of the chip were made of COC, a thermoplastic material with excellent water barrier properties, optical transmissivity, chemical compatibility with acids, bases and alcohols73, and low X-ray attenuation58. Microfluidic chips based on this material have been reported previously, with designs using either injection molding to make mm-thick devices, or hot embossing to produce few-hundred μm-thick devices.48,49,51,52,58 The characterization of small or weakly-diffracting microcrystals in these devices can prove challenging due to significant background from thick enclosing and flow layers, limiting the diffraction resolution attained. Therefore, a key focus of our design was to minimize the X-ray cross section thickness to maximize signal to noise from crystals. This was achieved by using a layered assembly process to produce physically robust and easy-to-handle chips with a total thickness of ~1.5 mm but an effective cross section thickness of only 30–60 μm in the X-ray window regions.
Figure 3:

(A) A macroscopic view of the assembled polymer microfluidic chip with a 4 × 9 array of rectangular X-ray windows in the COC supports, to which COC thin films are attached. Features for sample introduction are highlighted in the outermost layer of the chip, the PMMA frame. (B) Optical microscopy images of tetragonal thaumatin crystals grown directly on-chip using micro-batch crystallization as observed through the optically transparent X-ray windows in the chip. The top and bottom figures correspond to crystallization without and with seeding to control crystal size and density. The scale bar is 100 μm. (C) The chip magnetically mounted on the SMB goniometer at SSRL using a chip holder pin base. Experiments were performed at room temperature without the need for any external humidification.
The chip consisted of two sides (Figure 1, layers 2–4) mirror symmetric in construction, bonded using a tunable pressure-sensitive adhesive spacer layer that defined the flow layer thickness (layer 1, 25–48 μm in this work). Windows in the 200 μm hot-embossed COC support had ultra-thin 2–5 μm COC films attached that served as the low-background X-ray interaction regions. The outermost 0.5–1 mm thick PMMA frame attached to the COC support imparted rigidity and included inlet, outlet, and vent holes for introduction of crystallization cocktails or crystal slurries into the chip by micro-pipetting. A major advantage of this approach was the straightforward incorporation of tunable-background, wrinkle-free thin films in an enclosed flow-chip design. Solvent bonding eliminated the need to handle fragile films, as is typically required for alternatives like the single/few-layer graphene (<1–3 nm, expensive and laborious to produce)42,45,74, freestanding commercially-available Mylar (2.5–3.5 μm)38,40 or Kapton (3–8 μm)34 films used as enclosing layers in open-format fixed-target chips. The modular construction of the chip allows rapid modifications to the chip design, e.g., the sample area available for rastering, sample volume, and thickness of enclosing and films, depending on sample and experimental requirements. Sample fabrication and assembly of 12 chips took approximately 1 hour, but parallel processing could further reduce the fabrication time. For applications that do not require ultra-thin supports (<5 μm), the reactive-ion etch step could be omitted after hot-embossing COC sheets to retain ~20–30 μm residual thin films over X-ray windows. This decreases the number of fabrication steps and manufacturing time, but at the cost of proportionally higher background scatter. Hydrophilic oxygen plasma surface treatment provided facile loading of aqueous solutions into the chip as shown in Figure S3. The advancing contact angle of atmospheric plasma treated COC films was ~ 22° for freshly treated films and increased to ~ 60° over a period of 4 weeks as shown in Figure S2. Thus, fully assembled chips could be stored for at least one month before use without significantly impacting solution loading.
To identify the timescales over which diffraction measurements could be carried out on our chips without external humidification, measurements of water evaporative loss through thin COC films were performed using a modified wet-cup method. Figure 4 shows the steady-state water vapor transmission rate (WVTR) through COC films as a function of film thickness. The WVTR measured was inversely proportional to the film thickness, as expected for diffusive transport. Film thicknesses ranging from 2.5 to 140 μm were included to serve as a guideline for users to choose barrier properties as required for their application, keeping in mind that scatter background would scale linearly with film thickness. For the 2–5 μm films used in this work, a WVTR of ~ translated to a low evaporative loss of 0.5–1 wt.% per hour, ensuring that crystals would remain hydrated without external humidity control for several hours during diffraction measurements. To extend sample stability to several weeks, we stored the chips in individually sealed wells with saturated salt slurries of K2SO4 (RH set point ~97.3% at 25 °C75) or with the precipitant solution, to reduce the relative humidity gradient against which the chips were equilibrating. We found that under these conditions, chips with crystals could be stored over a period of 2–4 weeks without visible or measurable loss of crystallinity despite the small sample volume in the chip (<10 μL). In comparison, previous microfluidic chips used thick (>50–200 μm) sealing layers of COC, Kapton or Mylar and were stable for weeks in ambient conditions. Thin support films (~2.5–3.5 μm Mylar or 3–8 μm Kapton) have been used previously as enclosing layers with microgrids, but these approaches are seldom designed or characterized for long term stability, requiring sample preparation at the beamline34,38,40.
Figure 4:

Steady-state water vapor transmission rates (WVTR) through COC films measured using a modified wet-cup method at 23°C, against a ΔRH gradient of approximately 80 percent. COC films in the thickness range of 2–5 microns that were used in this work as sample enclosing layers (layer 2) have a low WVTR of ~ or ~0.5–1% sample wt. loss per hour of storage in ambient room temperature conditions without external humidity control. The fitted line corresponds to a water vapor permeability (WVP) of .
On-chip protein crystallization
To test the utility of the chips for in situ crystal growth and long-term storage, vapor diffusion and micro-batch crystallization trials were conducted for five different proteins by adapting existing conditions from the literature61–64. Table 1 summarizes the crystallization conditions used and compares observations of crystal size, morphology, densities, and time-to-crystallization between on-chip and on-crystallization plate methods (hanging drop vapor diffusion or micro-batch under oil methods were used for comparison as appropriate). For most of the conditions tested, similar crystal sizes and morphologies were obtained on and off chip without the need to modify established crystallization conditions. On average, slightly lower nucleation rates and slower crystallization kinetics were observed on-chip leading to larger crystals with sparser densities. Compared to the large droplet solution geometry in plate-based methods, solution in a microfluidic chip is confined to the micro-scale in one or more dimensions. In the near-2D geometry of our chip, a large interfacial contact area between the solution and the polymer surface is expected to affect crystal nucleation. The slower crystallization kinetics are due to the smaller evaporation rate in the chips and that mass transport is primarily driven by molecular diffusion as buoyancy driven convection is suppressed in this geometry.76,77
The 30–200 μm crystal sizes obtained were ideal for proof-of-principle SOX measurements due to the comparable minimum beam size (5 × 55 μm) at the 12–1 microfocus beamline at SSRL. Seeding can be used to control the nucleation rates to produce a high density of smaller 10–20 μm microcrystals. Figure 3B shows fully hydrated, randomly oriented tetragonal thaumatin crystals grown on chip using micro-batch crystallization (without and with seeding, top and bottom respectively) and stored for three days. Figure S4 shows exemplary images of the other protein crystals measured in this work. It is worth noting that the thin spacer film (25–48 μm) not only reduced the sample volume requirements (4–8 μL) but also restricted crystal growth in the Z-direction to span the “set” spacer thickness. This minimized scatter contribution from the crystallization solution surrounding the microcrystals. Preferential alignment of 200–300 μm long plate-like crystals of concanavalin-A and NSP5 was observed microscopically. The concanavalin-A crystals appeared to grow to span the entire spacer thickness ~50 μm whereas individual NSP5 plates in the crystal clusters were ~10–15 μm thick. Lysozyme and catalase crystals, which were cuboid in appearance, did not have any visible preferential alignment.
X-ray scatter background measurements
To quantify and compare the X-ray scatter background contributions from different materials in the beam interaction cross section, background measurements of ambient air scattering (5 mm path length), chips with different COC films thicknesses, and buffer-filled chips (0.1 M Na Acetate buffer, 1M NaCl) with two different spacer thicknesses were carried out. Radial averages of the scattered intensity for air and buffer filled chips (Figure 5) are dominated at low angles by air-scatter background. Contributions to background scattering from the enclosing amorphous COC films represent an increase of approximately 20 percent over ambient air scattering at the peak of the COC-associated scattering at approximately 1.1 Å−1, corresponding to 5.8 Å in real space (seen as a broad “halo”). This is in agreement with previous observations of increased scattering ~ 5–6 Å for different COC grades58,74 and indicates there is some degree of systematic packing between adjacent polymer backbone strands78. COC also contributes a broad, featureless scattering signal at low resolution (>6 Å). At higher angles, the enclosed buffer layer was the most significant background contribution with buffer filled chips exhibiting a “water ring” at 1.8 Å−1 corresponding to 3.4 Å in real space. This contribution can be decreased by using a thinner spacer layer as demonstrated by the yellow curve in Figure 5, corresponding to a chip with a 25 μm spacer layer. Because of the inherent flexibility of the enclosing thin COC films, some variability (10–20 percent) in the buffer or flow layer thickness is expected. This explains why the scattering signal (around 1.8 Å−1) for the 25- and 48-μm spacer samples does not scale linearly. Minor changes in peak intensities at ~ 0.3 and ~ 1.1 Å−1 were also observed between the 25 and 48 μm spacer samples, that cannot be explained sufficiently by COC film thickness variation between different batches (< 0.2 μm). Since the two samples were characterized during different beamtimes using different batches of COC from Polysciences, Inc., we suspect minor batch to batch compositional variation in the supplied COC material and subsequent spin-coating solution preparation and processing steps (filtration, postbake) could influence the degree of short-range chain packing, resulting in slightly different amorphous scatter signal at low q. Regardless, both the enclosing film thickness and the flow spacer thickness are parameters that can be changed to control background scatter to match sample requirements. While a direct background comparison with other microfluidic chips from literature is made challenging due to differences in material attenuation properties, film and flow layer thicknesses, and beam characteristics, the inset in Figure 5 shows the drastic reduction in background that results from using thin films over X-ray windows (~3.7 μm per side) vs. shooting through thick supports (~200 μm per side) that are on the order of film thicknesses sometimes used as capping layers in microfluidic chips.
Figure 5:

Radial averages of scattering associated with crystal-free chips (or instrument-associated air scattering) to quantify background scattering contributions. Air (cyan, blue) and buffer-filled (red, yellow) chips with nominal 2.7 μm (cyan) or 3.7 μm (yellow, blue, red) COC enclosing films and 50 μm (red) or 25 μm (yellow) spacer layers are shown. Note: The data represented by the yellow curve, collected during a separate beamtime using a different batch of COC material from Polysciences Inc., shows a slightly different scatter profile at low q. Inset: Comparison of scattering intensity from the 200 μm COC support frame material and a 3.7 μm enclosing film.
In situ X-ray diffraction at room temperature
Figure 3C shows the COC microfluidic chip mounted on a slotted magnetic pin base and attached to the goniometer for room-temperature data collection at the 12–1 beamline at SSRL. The device’s large measurable area and facile mounting allowed rapid collection of data in a semi-serial mode where 30° rotation wedges were collected on a series of manually centered crystals, eliminating the need for mounting of individual crystals. Individual wedges were collected in 15–20 seconds. The average data collection time for each chip was between 30 min and one hour, where the limiting step was manual centering of the crystals. Minimal settling or crystal movement was observed for most in situ grown crystals where the crystal size was well matched to the spacer layer thickness due to contact with the COC films. Some movement was observed when the crystal size was well below the spacer thickness (~50 μm spacer vs. 10–20 μm lysozyme crystals).
Data from 5 to 16 individual crystals were merged to constitute complete data sets for the measured proteins. Data collection and refinement statistics for each protein are shown in Table 2. The lysozyme, thaumatin, and concanavalin-A crystals generated high resolution datasets, diffracting to 1.5 Å, 1.45 Å, and 1.30 Å respectively, with high multiplicity and excellent merging statistics including overall CC1/2 of 0.999, 0.999, and 0.993 respectively. The catalase dataset diffracted to a slightly lower resolution of 2.27 Å (CC1/2 0.997) but is comparable in quality to other room temperature structures reported for the protein79. Random crystal orientation for lysozyme, thaumatin and catalase was confirmed by a high degree of completeness, both in the total dataset and in the highest resolution shell. Concanavalin-A had only slightly less favorable completeness statistics because of its lower symmetry space group and apparent preferentially orientated crystal growth to span the spacer thickness (Figure S4F).
In cases where in situ growth limited crystal size to the full spacer thickness, background scattering around the water ring (1.8 Å−1) was lower due to displacement of excess crystallization solution from the X-ray cross section (Figure S5). Moreover, when contact between crystals and the enclosing COC films was high, mother liquor background could also be reduced by physically removing excess buffer from the chip, leaving film-supported crystals surrounded by a minimal amount of mother liquor as observed in the case of catalase (Figure S6). The differences in the unit cell dimensions between the fully hydrated and excess solvent removed or partially “dehydrated” crystals were within the measurement error with ~0.1 Å larger a- and b- unit cell dimensions for the fully hydrated crystals. The merging statistics for both these datasets is reported in Table S1.
Plate-like or plate-cluster morphologies for the larger, thinner NSP5 crystals, however, proved more challenging as the chip geometry limited possible crystal orientations. (Figure S4C). This made collection of an adequately redundant data set with sufficient completeness in all resolution shells difficult. Some of the crystals in the NSP5 dataset diffracted up to 2.3 Å which is comparable to other room temperature data sets for the protein that use similar crystallization conditions, PDB entries 6WQF (single crystal, 2.3 Å) and 7JVZ (SFX, 2.5 Å). But ultimately, a lower resolution cut-off of 2.7 Å was used for the final structure refinement due to constraints imposed by the crystal morphology and the low symmetry space group (C2), although only one chip with this sample was measured. We expect that additional samples and tuning of on chip crystal density and crystal size would improve these results. Overall, each of the solved structures were in excellent agreement with the PDB references used for molecular replacement except for a previously unreported carboxymethylation on the Cys376 residue observed in our catalase structure. The RMSD values ranged from 0.121–0.416 Å, which includes minor movements of sidechains and loops and are shown in Table S2. Representative 2mFo-DFc electron density maps of the concanavalin-A and catalase actives sites are shown in Figure 6.
Figure 6:

Active sites of two structures determined from on-chip crystallization and synchrotron data collection. The 2mFo-DFc density maps at 1.5σ are show in purple mesh. (A) The saccharide binding site and calcium coordination site of concanavalin A. (B) The heme active site of catalase bound to a proximal tyrosine residue.
To study the effects of long-term storage on chip, data was also collected on lysozyme crystallized under similar conditions but stored in a humidified environment at 21 °C for 17 days (“aged”). These crystals showed minimal signs of dehydration based on unit cell shrinkage, and diffracted to similar resolutions, as much as 1.5 Å for the aged samples compared to 1.87 Å for freshly prepared crystals (stored for 3 days on chip). This difference in resolution can be attributed to slight differences in crystal dimensions and normal variation in crystal quality, especially given that each of these datasets was obtained from multiple crystals which diffracted to differing resolutions. Thus, long term storage effects such as evaporative dehydration did not degrade crystal quality. The RMSD between the two structures was 0.094 Å, demonstrating that the two structures were identical. A detailed comparison of crystallographic statistics for the aged and fresh lysozyme sample is included in Table 2.
To assess the effect of both the polymer thin film and the mother liquor surrounding the crystals on the structures, we made a comparison between the I/σ(I) vs resolution and the measured chip background (purely buffer-filled) vs resolution for both fresh and aged lysozyme, as shown in Figure 7. Both samples appear to have no features in I/σ(I) that correlate with background scattering and no deleterious effects of the COC scatter peak at 5.8 Å are apparent. While there is a small drop in I/σ(I) around the peak of COC scattering, in both cases it is within the level of noise in the data.
Figure 7:

A comparison between the I/σ(I) for the two lysozyme structures solved in this work and the total scattering background from the chip. The fresh sample shown in light blue (~30 μm crystals) diffracted to 1.87 Å and the aged sample shown in dark blue (~70 μm crystals) diffracted to 1.5 Å confirming that long term storage of crystals on the chip does not affect crystal integrity. A small dip in signal to noise is observed around scattering peak for COC around 5.8 Å (dashed line). The larger dip around 5 Å corresponds to the location of a gap in the detector panel (masked out during analysis) and is confirmed by a decrease in the number of reflections in this resolution bin.
Conclusion
In summary, this work demonstrates the development of robust, easy-to-use polymer microfluidic chips that can be used for routine and reliable room temperature diffraction measurements on fully hydrated protein microcrystals. Our large area, optically transparent chips are compatible with in situ crystallization, sample monitoring, crystal storage and transport, and diffraction measurements directly on the chip, eliminating the need to handle small or fragile crystals. The chips are stable over several weeks in a humidified environment, and for several hours in ambient conditions removing the stress of on-the-fly sample preparation. The modular device construction allows design flexibility to tune sample or flow layer thickness, enclosing film thickness, chip volume, or rastering area to match sample and experimental requirements. In this work, the chips were designed to be directly compatible with the standard goniometer setup at SSRL. High resolution structures (1.3–2.7 Å) for five different proteins (including one “non-model” protein, NSP5) were collected on chip using SOX. Looking forward, we envision these low-cost chips could be made available to users, to allow crystallization and sample screening well ahead of beam-time, followed by streamlined plug-and-play experiments with minimal sample handling or mounting requirements. The platform can be expanded to add functionalities to study not just static structures but dynamics by leveraging microfluidic capabilities to allow ligand introduction, electric field application, temperature-jumps, or pump-probe time-resolved experiments.
Outlook: XFEL measurements
We are currently expanding this work and are applying the design and fabrication principles to produce and demonstrate chips that are tailored for XFEL SFX applications. The diffraction before destruction principle of operation at XFEL sources demands fresh sample at every beam shot. Based on the crystal symmetry and the data analysis techniques used80,81, hundreds to tens of thousands of crystals must be delivered to the beam in random orientations. Since XFEL beamtime is limited and precious, our goal is to develop a “shelf-stable”, plug-and-play microfluidic chip for XFEL sample introduction that maintains crystal hydration for up to 24 hours without the need for external humidity control, while still contributing minimally to background. This would address some of the limitations of existing approaches like the need for on-the-fly sample preparation (open-format chips) and high background (microfluidic chips) to enable routine, high-resolution SFX with minimal downtime. Validation of the first XFEL chip prototypes in preliminary experiments is promising. The current design allows collection of ~104 useful, low background shots per chip when continuously rastering through the thick and thin regions of the chip support with minimal alignment requirements due to the diffuse scattering of the amorphous target material. Our ongoing efforts focus on (1) maximizing the X-ray window regions on the chip to increase the fraction of area useful for sample characterization (beyond current limit of 50 % due to thick supports), (2) controlling crystal nucleation and crystal densities on the chip leveraging surface chemistry modifications, and (3) demonstrating the use of this platform for more non-model proteins of interest.
Supplementary Material
Acknowledgements
This work was performed, in part, under the auspices of the U.S. DOE by LLNL under Contract DE-AC52-07NA27344. This work was also supported by National Science Foundation (NSF) BioXFEL STC Grant 1231306, NIH grants R01GM117342 (NIGMS) and U19 AI144184 (NIAID). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. Use of the LCLS, SLAC National Accelerator Lab, is supported by the U.S. DOE, Office of Science, under contract no. DE-AC02-76SF00515.
The PGEX-6p-1-NSP5 plasmid was kindly provided by R. Hilgenfeld (University of Lübeck, Germany).
Footnotes
Conflict of Interest
There are no conflicts to declare.
References
- 1.Hope H, Acta Crystallogr. B, 1988, 44, 22–26. [DOI] [PubMed] [Google Scholar]
- 2.Moreno A, in Protein Crystallography, eds. Wlodawer A, Dauter Z and Jaskolski M, Springer New York, New York, NY, 2017, vol. 1607, pp. 51–76. [Google Scholar]
- 3.Skarina T, Xu X, Evdokimova E and Savchenko A, in Structural Genomics and Drug Discovery, ed. Anderson WF, Springer New York, New York, NY, 2014, vol. 1140, pp. 159–168. [Google Scholar]
- 4.Cusack S, Belrhali H, Bram A, Burghammer M, Perrakis A and Riekel C, Nat. Struct. Biol, 1998, 5, 634–637. [DOI] [PubMed] [Google Scholar]
- 5.Smith JL, Fischetti RF and Yamamoto M, Curr. Opin. Struct. Biol, 2012, 22, 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, Doak RB, Maia FRNC, Martin AV, Schlichting I, Lomb L, Coppola N, Shoeman RL, Epp SW, Hartmann R, Rolles D, Rudenko A, Foucar L, Kimmel N, Weidenspointner G, Holl P, Liang M, Barthelmess M, Caleman C, Boutet S, Bogan MJ, Krzywinski J, Bostedt C, Bajt S, Gumprecht L, Rudek B, Erk B, Schmidt C, Hömke A, Reich C, Pietschner D, Strüder L, Hauser G, Gorke H, Ullrich J, Herrmann S, Schaller G, Schopper F, Soltau H, Kühnel K-U, Messerschmidt M, Bozek JD, Hau-Riege SP, Frank M, Hampton CY, Sierra RG, Starodub D, Williams GJ, Hajdu J, Timneanu N, Seibert MM, Andreasson J, Rocker A, Jönsson O, Svenda M, Stern S, Nass K, Andritschke R, Schröter C-D, Krasniqi F, Bott M, Schmidt KE, Wang X, Grotjohann I, Holton JM, Barends TRM, Neutze R, Marchesini S, Fromme R, Schorb S, Rupp D, Adolph M, Gorkhover T, Andersson I, Hirsemann H, Potdevin G, Graafsma H, Nilsson B and Spence JCH, Nature, 2011, 470, 73–77.21293373 [Google Scholar]
- 7.Schlichting I and Miao J, Curr. Opin. Struct. Biol, 2012, 22, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirata K, Shinzawa-Itoh K, Yano N, Takemura S, Kato K, Hatanaka M, Muramoto K, Kawahara T, Tsukihara T, Yamashita E, Tono K, Ueno G, Hikima T, Murakami H, Inubushi Y, Yabashi M, Ishikawa T, Yamamoto M, Ogura T, Sugimoto H, Shen J-R, Yoshikawa S and Ago H, Nat. Methods, 2014, 11, 734–736. [DOI] [PubMed] [Google Scholar]
- 9.Fischer M, Q. Rev. Biophys, 2021, 54, e1. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt M, Adv. Condens. Matter Phys, 2013, 2013, 1–10. [Google Scholar]
- 11.Tenboer J, Basu S, Zatsepin N, Pande K, Milathianaki D, Frank M, Hunter M, Boutet S, Williams GJ, Koglin JE, Oberthuer D, Heymann M, Kupitz C, Conrad C, Coe J, Roy-Chowdhury S, Weierstall U, James D, Wang D, Grant T, Barty A, Yefanov O, Scales J, Gati C, Seuring C, Srajer V, Henning R, Schwander P, Fromme R, Ourmazd A, Moffat K, Van Thor JJ, Spence JCH, Fromme P, Chapman HN and Schmidt M, Science, 2014, 346, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawate AS, Šrajer V, Schieferstein J, Guha S, Henning R, Kosheleva I, Schmidt M, Ren Z, Kenis PJA and Perry SL, Acta Crystallogr. Sect. F Struct. Biol. Commun, 2015, 71, 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orville AM, Curr. Opin. Struct. Biol, 2020, 65, 193–208. [DOI] [PubMed] [Google Scholar]
- 14.Pearson AR and Mehrabi P, Curr. Opin. Struct. Biol, 2020, 65, 168–174. [DOI] [PubMed] [Google Scholar]
- 15.Boutet S, Lomb L, Williams GJ, Barends TRM, Aquila A, Doak RB, Weierstall U, DePonte DP, Steinbrener J, Shoeman RL, Messerschmidt M, Barty A, White TA, Kassemeyer S, Kirian RA, Seibert MM, Montanez PA, Kenney C, Herbst R, Hart P, Pines J, Haller G, Gruner SM, Philipp HT, Tate MW, Hromalik M, Koerner LJ, van Bakel N, Morse J, Ghonsalves W, Arnlund D, Bogan MJ, Caleman C, Fromme R, Hampton CY, Hunter MS, Johansson LC, Katona G, Kupitz C, Liang M, Martin AV, Nass K, Redecke L, Stellato F, Timneanu N, Wang D, Zatsepin NA, Schafer D, Defever J, Neutze R, Fromme P, Spence JCH, Chapman HN and Schlichting I, Science, 2012, 337, 362–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W, Ishchenko A and Cherezov V, Nat. Protoc, 2014, 9, 2123–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wierman JL, Paré-Labrosse O, Sarracini A, Besaw JE, Cook MJ, Oghbaey S, Daoud H, Mehrabi P, Kriksunov I, Kuo A, Schuller DJ, Smith S, Ernst OP, Szebenyi DME, Gruner SM, Miller RJD and Finke AD, IUCrJ, 2019, 6, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyubimov AY, Murray TD, Koehl A, Araci IE, Uervirojnangkoorn M, Zeldin OB, Cohen AE, Soltis SM, Baxter EL, Brewster AS, Sauter NK, Brunger AT and Berger JM, Acta Crystallogr. D Biol. Crystallogr., 2015, 71, 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martiel I, Müller-Werkmeister HM and Cohen AE, Acta Crystallogr. Sect. Struct. Biol, 2019, 75, 160–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng R, Crystals, 2020, 10, 215. [Google Scholar]
- 21.DePonte DP, Weierstall U, Schmidt K, Warner J, Starodub D, Spence JCH and Doak RB, J. Phys. Appl. Phys, 2008, 41, 195505. [Google Scholar]
- 22.Weierstall U, James D, Wang C, White TA, Wang D, Liu W, Spence JCH, Bruce Doak R, Nelson G, Fromme P, Fromme R, Grotjohann I, Kupitz C, Zatsepin NA, Liu H, Basu S, Wacker D, Won Han G, Katritch V, Boutet S, Messerschmidt M, Williams GJ, Koglin JE, Marvin Seibert M, Klinker M, Gati C, Shoeman RL, Barty A, Chapman HN, Kirian RA, Beyerlein KR, Stevens RC, Li D, Shah STA, Howe N, Caffrey M and Cherezov V, Nat. Commun, 2014, 5, 3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sierra RG, Gati C, Laksmono H, Dao EH, Gul S, Fuller F, Kern J, Chatterjee R, Ibrahim M, Brewster AS, Young ID, Michels-Clark T, Aquila A, Liang M, Hunter MS, Koglin JE, Boutet S, Junco EA, Hayes B, Bogan MJ, Hampton CY, Puglisi EV, Sauter NK, Stan CA, Zouni A, Yano J, Yachandra VK, Soltis SM, Puglisi JD and DeMirci H, Nat. Methods, 2016, 13, 59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oberthuer D, Knoška J, Wiedorn MO, Beyerlein KR, Bushnell DA, Kovaleva EG, Heymann M, Gumprecht L, Kirian RA, Barty A, Mariani V, Tolstikova A, Adriano L, Awel S, Barthelmess M, Dörner K, Xavier PL, Yefanov O, James DR, Nelson G, Wang D, Calvey G, Chen Y, Schmidt A, Szczepek M, Frielingsdorf S, Lenz O, Snell E, Robinson PJ, Šarler B, Belšak G, Maček M, Wilde F, Aquila A, Boutet S, Liang M, Hunter MS, Scheerer P, Lipscomb JD, Weierstall U, Kornberg RD, Spence JCH, Pollack L, Chapman HN and Bajt S, Sci. Rep, 2017, 7, 44628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuller FD, Gul S, Chatterjee R, Burgie ES, Young ID, Lebrette H, Srinivas V, Brewster AS, Michels-Clark T, Clinger JA, Andi B, Ibrahim M, Pastor E, de Lichtenberg C, Hussein R, Pollock CJ, Zhang M, Stan CA, Kroll T, Fransson T, Weninger C, Kubin M, Aller P, Lassalle L, Bräuer P, Miller MD, Amin M, Koroidov S, Roessler CG, Allaire M, Sierra RG, Docker PT, Glownia JM, Nelson S, Koglin JE, Zhu D, Chollet M, Song S, Lemke H, Liang M, Sokaras D, Alonso-Mori R, Zouni A, Messinger J, Bergmann U, Boal AK, Bollinger JM, Krebs C, Högbom M, Phillips GN, Vierstra RD, Sauter NK, Orville AM, Kern J, Yachandra VK and Yano J, Nat. Methods, 2017, 14, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beyerlein KR, Dierksmeyer D, Mariani V, Kuhn M, Sarrou I, Ottaviano A, Awel S, Knoska J, Fuglerud S, Jönsson O, Stern S, Wiedorn MO, Yefanov O, Adriano L, Bean R, Burkhardt A, Fischer P, Heymann M, Horke DA, Jungnickel KEJ, Kovaleva E, Lorbeer O, Metz M, Meyer J, Morgan A, Pande K, Panneerselvam S, Seuring C, Tolstikova A, Lieske J, Aplin S, Roessle M, White TA, Chapman HN, Meents A and Oberthuer D, IUCrJ, 2017, 4, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zarrine-Afsar A, Barends TRM, Müller C, Fuchs MR, Lomb L, Schlichting I and Miller RJD, Acta Crystallogr. D Biol. Crystallogr., 2012, 68, 321–323. [DOI] [PubMed] [Google Scholar]
- 28.Frank M, Carlson DB, Hunter MS, Williams GJ, Messerschmidt M, Zatsepin NA, Barty A, Benner WH, Chu K, Graf AT, Hau-Riege SP, Kirian RA, Padeste C, Pardini T, Pedrini B, Segelke B, Seibert MM, Spence JCH, Tsai C-J, Lane SM, Li X-D, Schertler G, Boutet S, Coleman M and Evans JE, IUCrJ, 2014, 1, 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunter MS, Segelke B, Messerschmidt M, Williams GJ, Zatsepin NA, Barty A, Benner WH, Carlson DB, Coleman M, Graf A, Hau-Riege SP, Pardini T, Seibert MM, Evans J, Boutet S and Frank M, Sci. Rep, 2015, 4, 6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feld GK, Heymann M, Benner WH, Pardini T, Tsai C-J, Boutet S, Coleman MA, Hunter MS, Li X, Messerschmidt M, Opathalage A, Pedrini B, Williams GJ, Krantz BA, Fraden S, Hau-Riege S, Evans JE, Segelke BW and Frank M, J. Appl. Crystallogr, 2015, 48, 1072–1079. [Google Scholar]
- 31.Cohen AE, Soltis SM, González A, Aguila L, Alonso-Mori R, Barnes CO, Baxter EL, Brehmer W, Brewster AS, Brunger AT, Calero G, Chang JF, Chollet M, Ehrensberger P, Eriksson TL, Feng Y, Hattne J, Hedman B, Hollenbeck M, Holton JM, Keable S, Kobilka BK, Kovaleva EG, Kruse AC, Lemke HT, Lin G, Lyubimov AY, Manglik A, Mathews II, McPhillips SE, Nelson S, Peters JW, Sauter NK, Smith CA, Song J, Stevenson HP, Tsai Y, Uervirojnangkoorn M, Vinetsky V, Wakatsuki S, Weis WI, Zadvornyy OA, Zeldin OB, Zhu D and Hodgson KO, Proc. Natl. Acad. Sci, 2014, 111, 17122–17127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halsted TP, Yamashita K, Hirata K, Ago H, Ueno G, Tosha T, Eady RR, Antonyuk SV, Yamamoto M and Hasnain SS, IUCrJ, 2018, 5, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baxter EL, Aguila L, Alonso-Mori R, Barnes CO, Bonagura CA, Brehmer W, Brunger AT, Calero G, Caradoc-Davies TT, Chatterjee R, Degrado WF, Fraser JS, Ibrahim M, Kern J, Kobilka BK, Kruse AC, Larsson KM, Lemke HT, Lyubimov AY, Manglik A, McPhillips SE, Norgren E, Pang SS, Soltis SM, Song J, Thomaston J, Tsai Y, Weis WI, Woldeyes RA, Yachandra V, Yano J, Zouni A and Cohen AE, Acta Crystallogr. Sect. Struct. Biol, 2016, 72, 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray TD, Lyubimov AY, Ogata CM, Vo H, Uervirojnangkoorn M, Brunger AT and Berger JM, Acta Crystallogr. D Biol. Crystallogr., 2015, 71, 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roedig P, Vartiainen I, Duman R, Panneerselvam S, Stübe N, Lorbeer O, Warmer M, Sutton G, Stuart DI, Weckert E, David C, Wagner A and Meents A, Sci. Rep, 2015, 5, 10451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coquelle N, Brewster AS, Kapp U, Shilova A, Weinhausen B, Burghammer M and Colletier J-P, Acta Crystallogr. D Biol. Crystallogr., 2015, 71, 1184–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Axford D, Aller P, Sanchez-Weatherby J and Sandy J, Acta Crystallogr. Sect. F Struct. Biol. Commun, 2016, 72, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oghbaey S, Sarracini A, Ginn HM, Pare-Labrosse O, Kuo A, Marx A, Epp SW, Sherrell DA, Eger BT, Zhong Y, Loch R, Mariani V, Alonso-Mori R, Nelson S, Lemke HT, Owen RL, Pearson AR, Stuart DI, Ernst OP, Mueller-Werkmeister HM and Miller RJD, Acta Crystallogr. Sect. Struct. Biol, 2016, 72, 944–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roedig P, Ginn HM, Pakendorf T, Sutton G, Harlos K, Walter TS, Meyer J, Fischer P, Duman R, Vartiainen I, Reime B, Warmer M, Brewster AS, Young ID, Michels-Clark T, Sauter NK, Kotecha A, Kelly J, Rowlands DJ, Sikorsky M, Nelson S, Damiani DS, Alonso-Mori R, Ren J, Fry EE, David C, Stuart DI, Wagner A and Meents A, Nat. Methods, 2017, 14, 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doak RB, Nass Kovacs G, Gorel A, Foucar L, Barends TRM, Grünbein ML, Hilpert M, Kloos M, Roome CM, Shoeman RL, Stricker M, Tono K, You D, Ueda K, Sherrell DA, Owen RL and Schlichting I, Acta Crystallogr. Sect. Struct. Biol, 2018, 74, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren Z, Ayhan M, Bandara S, Bowatte n, Kumarapperuma I, Gunawardana S, Shin H, Wang C, Zeng X and Yang X, Lab. Chip, 2018, 18, 2246–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seuring C, Ayyer K, Filippaki E, Barthelmess M, Longchamp J-N, Ringler P, Pardini T, Wojtas DH, Coleman MA, Dörner K, Fuglerud S, Hammarin G, Habenstein B, Langkilde AE, Loquet A, Meents A, Riek R, Stahlberg H, Boutet S, Hunter MS, Koglin J, Liang M, Ginn HM, Millane RP, Frank M, Barty A and Chapman HN, Nat. Commun, 2018, 9, 1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee D, Baek S, Park J, Lee K, Kim J, Lee SJ, Chung WK, Lee J-L, Cho Y and Nam KH, Sci. Rep, 2019, 9, 6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karpik A, Martiel I, Kristiansen PM and Padeste C, Micro Nano Eng, 2020, 7, 100053. [Google Scholar]
- 45.Shelby ML, Gilbile D, Grant TD, Seuring C, Segelke BW, He W, Evans AC, Pakendorf T, Fischer P, Hunter MS, Batyuk A, Barthelmess M, Meents A, Coleman MA, Kuhl TL and Frank M, IUCrJ, 2020, 7, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shelby M, Gilbile D, Grant T, Bauer W, Segelke B, He W, Evans A, Crespo N, Fischer P, Pakendorf T, Hennicke V, Hunter M, Batyuk A, Barthelmess M, Meents A, Kuhl T, Frank M and Coleman M, Crystals, 2020, 10, 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansen CL, Classen S, Berger JM and Quake SR, J. Am. Chem. Soc, 2006, 128, 3142–3143. [DOI] [PubMed] [Google Scholar]
- 48.Ng JD, Clark PJ, Stevens RC and Kuhn P, Acta Crystallogr. D Biol. Crystallogr., 2008, 64, 189–197. [DOI] [PubMed] [Google Scholar]
- 49.Dhouib K, Khan Malek C, Pfleging W, Gauthier-Manuel B, Duffait R, Thuillier G, Ferrigno R, Jacquamet L, Ohana J, Ferrer J-L, Théobald-Dietrich A, Giegé R, Lorber B and Sauter C, Lab. Chip, 2009, 9, 1412. [DOI] [PubMed] [Google Scholar]
- 50.Jena RK, Yue CY, Lam YC, Tang PS and Gupta A, Sens. Actuators B Chem, 2012, 163, 233–241. [Google Scholar]
- 51.Pinker F, Brun M, Morin P, Deman A-L, Chateaux J-F, Oliéric V, Stirnimann C, Lorber B, Terrier N, Ferrigno R and Sauter C, Cryst. Growth Des, 2013, 13, 3333–3340. [Google Scholar]
- 52.de Wijn R, Hennig O, Roche J, Engilberge S, Rollet K, Fernandez-Millan P, Brillet K, Betat H, Mörl M, Roussel A, Girard E, Mueller-Dieckmann C, Fox GC, Olieric V, Gavira JA, Lorber B and Sauter C, IUCrJ, 2019, 6, 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Popov AM, Dorovatovskii PV, Mamichev DA, Marchenkova MA and Yu A. Nikolaeva, Crystallogr. Rep, 2019, 64, 282–286. [Google Scholar]
- 54.Zhao F-Z, Sun B, Yu L, Xiao Q-J, Wang Z-J, Chen L-L, Liang H, Wang Q-S, He J-H and Yin D-C, Lab. Chip, 2020, 20, 3888–3898. [DOI] [PubMed] [Google Scholar]
- 55.Maeki M, Ito S, Takeda R, Ueno G, Ishida A, Tani H, Yamamoto M and Tokeshi M, Chem. Sci, 2020, 11, 9072–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Junius N, Jaho S, Sallaz-Damaz Y, Borel F, Salmon J-B and Budayova-Spano M, Lab. Chip, 2020, 20, 296–310. [DOI] [PubMed] [Google Scholar]
- 57.Gavira JA, Rodriguez-Ruiz I, Martinez-Rodriguez S, Basu S, Teychené S, McCarthy AA and Mueller-Dieckman C, Acta Crystallogr. Sect. Struct. Biol, 2020, 76, 751–758. [DOI] [PubMed] [Google Scholar]
- 58.Guha S, Perry SL, Pawate AS and Kenis PJA, Sens. Actuators B Chem, 2012, 174, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keller N, Nargang TM, Runck M, Kotz F, Striegel A, Sachsenheimer K, Klemm D, Länge K, Worgull M, Richter C, Helmer D and Rapp BE, Lab. Chip, 2016, 16, 1561–1564. [DOI] [PubMed] [Google Scholar]
- 60.Su Y, Kravets VG, Wong SL, Waters J, Geim AK and Nair RR, Nat. Commun, 2014, 5, 4843. [DOI] [PubMed] [Google Scholar]
- 61.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K and Hilgenfeld R, Science, 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Asherie N, Ginsberg C, Greenbaum A, Blass S and Knafo S, Cryst. Growth Des, 2008, 8, 4200–4207. [Google Scholar]
- 63.Pham T, Lai D, Ji D, Tuntiwechapikul W, Friedman JM and Lee TR, Colloids Surf. B Biointerfaces, 2004, 34, 191–196. [DOI] [PubMed] [Google Scholar]
- 64.Foroughi LM, Kang Y-N and Matzger AJ, Cryst. Growth Des, 2011, 11, 1294–1298. [Google Scholar]
- 65.McPhillips TM, McPhillips SE, Chiu H-J, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM and Kuhn P, J. Synchrotron Radiat, 2002, 9, 401–406. [DOI] [PubMed] [Google Scholar]
- 66.Winter G, J. Appl. Crystallogr, 2010, 43, 186–190. [Google Scholar]
- 67.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A and Wilson KS, Acta Crystallogr. D Biol. Crystallogr., 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winter G, Waterman DG, Parkhurst JM, Brewster AS, Gildea RJ, Gerstel M, Fuentes-Montero L, Vollmar M, Michels-Clark T, Young ID, Sauter NK and Evans G, Acta Crystallogr. Sect. Struct. Biol, 2018, 74, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC and Read RJ, J. Appl. Crystallogr, 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC and Zwart PH, Acta Crystallogr. D Biol. Crystallogr., 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH and Adams PD, Acta Crystallogr. D Biol. Crystallogr., 2012, 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Emsley P and Cowtan K, Acta Crystallogr. D Biol. Crystallogr., 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 73.PACKAGING WITH TOPAS® COC, TOPAS Advanced Polymers/Polyplastics. [Google Scholar]
- 74.Sui S, Wang Y, Kolewe KW, Srajer V, Henning R, Schiffman JD, Dimitrakopoulos C and Perry SL, Lab. Chip, 2016, 16, 3082–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Greenspan L, J. Res. Natl. Bur. Stand. Sect. Phys. Chem, 1977, 81A, 89. [Google Scholar]
- 76.Squires TM and Quake SR, Rev. Mod. Phys, 2005, 77, 977–1026. [Google Scholar]
- 77.Carruthers CW Jr, Gerdts C, Johnson MD and Webb P, PLoS ONE, 2013, 8, e82298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller RL and Boyer RF, J. Polym. Sci. Polym. Phys. Ed, 1984, 22, 2043–2050. [Google Scholar]
- 79.Ko T-P, Day J, Malkin AJ and McPherson A, Acta Crystallogr. D Biol. Crystallogr., 1999, 55, 1383–1394. [DOI] [PubMed] [Google Scholar]
- 80.White TA, Kirian RA, Martin AV, Aquila A, Nass K, Barty A and Chapman HN, J. Appl. Crystallogr, 2012, 45, 335–341. [Google Scholar]
- 81.Uervirojnangkoorn M, Zeldin OB, Lyubimov AY, Hattne J, Brewster AS, Sauter NK, Brunger AT and Weis WI, eLife, 2015, 4, e05421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.