

Abstract
Inflammatory caspase sensing of cytosolic lipopolysaccharide (LPS) triggers pyroptosis and the concurrent release of damage-associated molecular patterns (DAMPs). Collectively, DAMPs are key determinants that shape the aftermath of inflammatory cell death. However, the identity and function of the individual DAMPs released are poorly defined. Our proteomics study revealed that cytosolic LPS sensing triggered the release of galectin-1, a β-galactoside-binding lectin. Galectin-1 release is a common feature of inflammatory cell death, including necroptosis. In vivo studies using galectin-1-deficient mice, recombinant galectin-1 and galectin-1-neutralizing antibody showed that galectin-1 promotes inflammation and plays a detrimental role in LPS-induced lethality. Mechanistically, galectin-1 inhibition of CD45 (Ptprc) underlies its unfavorable role in endotoxin shock. Finally, we found increased galectin-1 in sera from human patients with sepsis. Overall, we uncovered galectin-1 as a bona fide DAMP released as a consequence of cytosolic LPS sensing, identifying a new outcome of inflammatory cell death.
Introduction
Intracellular surveillance for pathogen-encoded products or activities by innate immune sensors licenses the activation of inflammasomes. The assembly of inflammasome complexes activate caspase-1, which subsequently cleaves and activates the inflammatory cytokines interleukin-1β (IL-1β) and IL-181. Caspase-1 also cleaves a pore-forming protein, gasdermin D (GSDMD), to release its N-terminal fragment, which permeabilizes the plasma membrane causing pyroptosis2,3. In addition, a noncanonical inflammasome monitors—by means of inflammatory caspases (caspase-11 in mice and caspase-4/5 in humans)4,5,6—for LPS that gains access to the cytosol upon bacterial invasion or via outer membrane vesicles7. Active caspases 11 and 4 also cleave GSDMD2,3; plasma membrane perforation by GSDMD is considered to trigger the activation of the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome and the maturation of caspase-18.
The cytosolic LPS sensing pathway can be host-protective during bacterial infections8. On the other hand, its excessive activation has the potential to cause tissue damage and compromise host survival as revealed in murine models of sepsis9,10,11,12. The effector mechanisms by which the noncanonical inflammasome exerts its deleterious effects are not clear. In addition to the well-characterized outcomes of IL-1 cytokine maturation and pyroptosis, caspase-11 detection of cytosolic LPS elicits the unconventional release of intracellular proteins that lack the leader sequence for secretion via the classical endoplasmic reticulum-Golgi apparatus route11. In the extracellular milieu, these proteins, including high mobility group protein B1 (HMGB1), act as alarmins or DAMPs and propagate inflammatory responses via a multitude of mechanisms13,14. The collective activities of DAMPs or alarmins orchestrate dysregulated inflammation, organ injury and poor outcomes in sepsis. Whereas the unconventional secretome associated with caspase-1 activation has been profiled15, the identity of DAMPs released as a result of caspase-11-dependent cytosolic LPS sensing and, more importantly, the functional roles of individual DAMPs, are poorly understood.
By utilizing a proteomic strategy involving a two-dimensional (2D) liquid phase fractionation system (PF2D) and mass spectrometry, we show that galectin-1 is released extracellularly in response to caspase-11 activation by cytosolic LPS. Galectin-1 is a prototype member of a family of β-galactoside-binding proteins with several immunoregulatory activities16. Although this lectin functions extracellularly by cross-linking N- and O-glycans on cell surface glycoconjugates, the mechanisms and molecular machinery involved in its secretion are uncertain since it lacks the signal sequence required for the classical endoplasmic reticulum-Golgi apparatus secretion17. We show that GSDMD pores are sufficient to mediate the cytosolic LPS-induced release of galectin-1 without requiring the NLRP3-caspase-1 inflammasome or cell lysis. Importantly, galectin-1 deficiency led to the attenuation of inflammatory responses and rendered mice less susceptible to LPS-induced lethality. Overall, these results identify galectin-1 as an inflammatory DAMP released as a consequence of intracellular LPS sensing, expanding the effector mechanisms underlying the functions of inflammatory caspases. Thus, this study represents a comprehensive characterization of the secretion as well as the function of a lectin DAMP in the context of intracellular LPS-induced inflammation.
Results
Intracellular LPS sensing leads to the release of galectin-1
Cytosolic LPS sensing triggers the secretion of active IL-1β and pyroptosis (Fig. 1a) as well as the release of DAMPs11, with the latter remaining a poorly defined outcome. To identify the DAMPs that are released into the extracellular space as a consequence of intracellular LPS detection by caspase-11 during Gram-negative bacterial infections, we employed the ProteomeLab PF2D system. Here, proteins are separated according to their isoelectric point (pI) in the first dimension (isoelectric focusing covering a pH range of 4.0–8.0) followed by a second-dimension separation according to the hydrophobicity (reverse-phase chromatography)18. This 2D liquid phase fractionation followed by mass spectrometry enables accurate identification of even low-abundant proteins. We infected wild-type (WT) and Casp11−/− bone marrow-derived macrophages (BMDMs) with enterohemorrhagic Escherichia coli (EHEC), a known caspase-11 activator7; after 16 h, cell debris-free concentrated supernatants were fractionated by the PF2D platform into more than 500 fractions based on pI and hydrophobicity (Extended Data Fig. 1a). The 214-nm absorbance profiles of the fractions were compiled and integrated with the ProteoVue software to create a 2D map (pH versus hydrophobicity) of proteins in the supernatants of WT and Casp11−/− BMDMs (Fig. 1b). The comparative analysis of the whole proteomic maps of two samples with the DeltaVue program identified differential fingerprints and revealed peaks specific to the WT sample, indicating differences in the secretome of WT and Casp11−/− cells (Fig. 1c). To reveal the identity of protein(s) in one of the differential peaks in the WT sample, we selected the fraction containing that peak in the WT sample and the corresponding fraction in the Casp11−/− sample, which lacks the peak, as control and subjected them to mass spectrometry, which identified galectin-1 in the fraction from WT but not Casp11−/− supernatants (Fig. 1d). Thus, our proteomic approach revealed that caspase-11 activation by cytosolic LPS leads to galectin-1 release.
Fig. 1: Proteomic identification of intracellular LPS-elicited release of galectin-1.
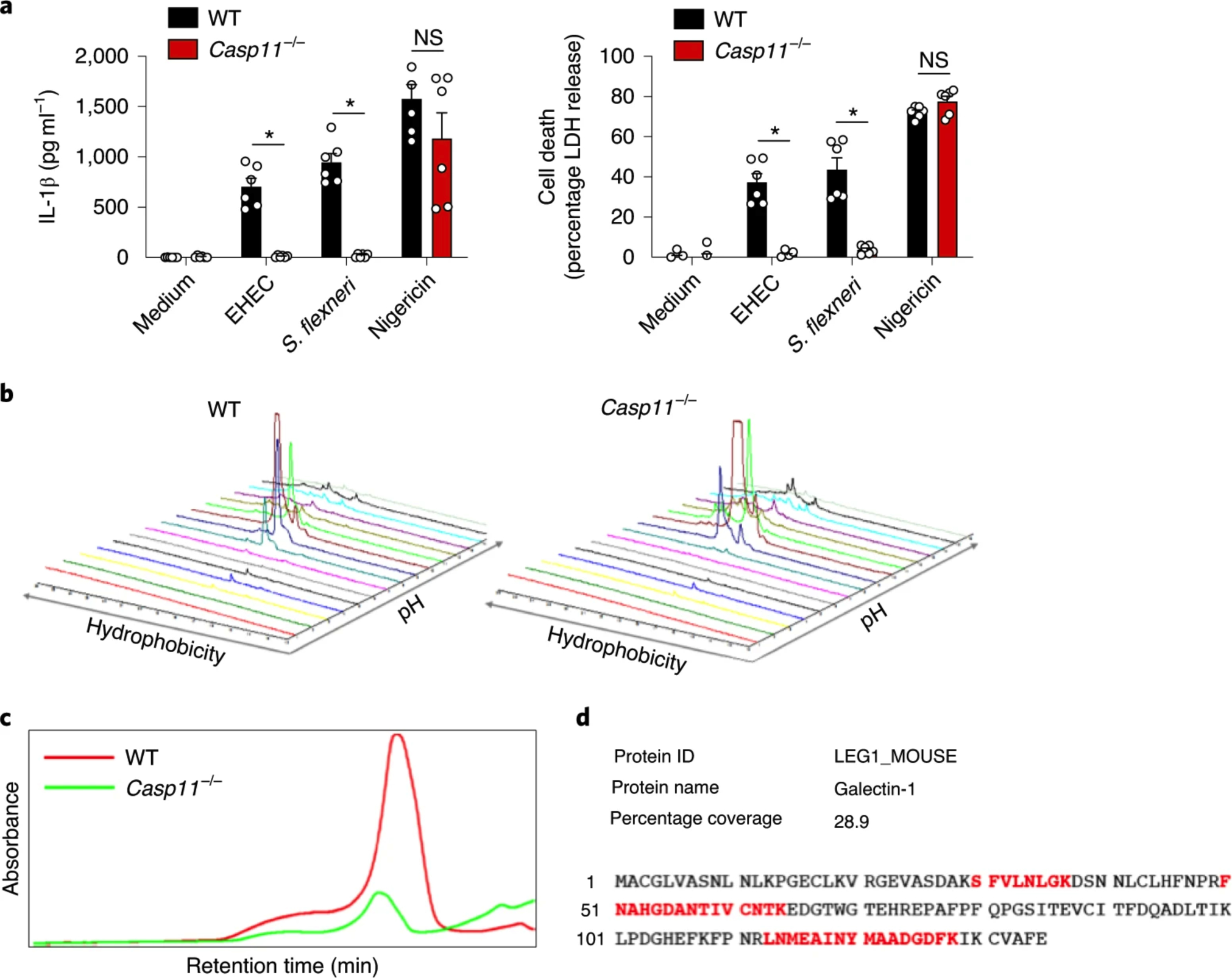
a, IL-1β and LDH release by Pam3CSK4 (0.5 μg ml−1)-primed WT and Casp11−/− BMDMs infected with EHEC or S. flexneri (MOI = 50) for 16 h or stimulated with 10 μM nigericin for 1 h. NS, not significant. b,c, 2D global proteomic maps of supernatants from EHEC-infected WT and Casp11−/− BMDMs generated by integrating the OD 214 nm absorbance of first- and second-dimension (hydrophobicity versus pH) fractions in the ProteoVue program (b). The chromatogram of a second-dimension fraction shows a peak in the WT but not in the Casp11−/− sample (c). d, LC– MS/MS analysis of the peaks shown in c identified galectin-1 only in the WT but not in the Casp11−/− sample. The amino acid sequence of galectin-1 and the matched peptides identified in LC–MS/MS (in bold red) are shown. a, The combined data from two independent experiments are shown as the mean ± s.e.m. *P < 0.05; two-way ANOVA followed by Šidák’s post-test.
GSDMD mediates cytosolic LPS-induced galectin-1 release
Galectin-1 is a leaderless protein belonging to a family of β-galactoside-binding lectins and its mechanism of secretion is unknown16,17. To validate the PF2D–mass spectrometry identification of galectin-1, we stimulated WT and Casp11−/− BMDMs with LPS or EHEC for 16 h and assessed galectin-1 levels in supernatants. Unstimulated BMDMs release low amounts of galectin-1, and Toll-like receptor 4 (TLR4) activation by LPS did not enhance it further (Fig. 2a,b). In contrast, EHEC infection, which activates cytosolic LPS sensing via outer membrane vesicles7, stimulated the release of very high amounts of galectin-1 in WT but not in Casp11−/− BMDMs (Fig. 2a,b). Similarly, Francisella novicida, a cytosolic bacterium that activates the absent in melanoma 2 (AIM2) inflammasome19, also induced galectin-1 release. The specificity of galectin-1 ELISA and immunoblotting was verified by using cells from galectin-1-deficient mice (Lgals1−/−) (Extended Data Fig. 1b,c). To gain insights into the mechanisms and molecular machinery involved in galectin-1 release, we determined the contribution of each of the noncanonical inflammasome components, namely caspase-11, GSDMD, NLRP3 and caspase-1 to this event. Gram-negative bacterial pathogens such as EHEC and Shigella flexneri stimulated galectin-1 release from WT BMDMs but not from Casp11−/− BMDMs (Fig. 2c,d). WT and Casp11−/− BMDMs stimulated with nigericin to activate the canonical NLRP3 inflammasome released similar amounts of galectin-1 (Fig. 2c,d). Unlike in WT BMDMs, EHEC- and S. flexneri-induced galectin-1 release from Gsdmd−/− BMDMs was significantly reduced (Fig. 2c,d). Surprisingly, galectin-1 release by Nlrp3−/− and Casp1−/− BMDMs after EHEC and S. flexneri infection was comparable to that of WT BMDMs (Fig. 2c–e). Similar to galectin-1 release, the release of IL-1α, a well-characterized DAMP, was dependent on caspase-11 rather than caspase-1 during infection with EHEC and S. flexneri (Fig. 2e and Extended Data Fig. 1d). In this regard, the molecular requirement for cytosolic LPS-induced galectin-1 release resembles that of pyroptosis and IL-1α and HMGB1 release (Fig. 2c–e)11. Therefore, cytosolic LPS-driven galectin-1 release depends on the caspase-11–GSDMD axis but not the secondary NLRP3-caspase-1 signaling.
Fig. 2: Cytosolic LPS-induced galectin-1 release in vitro is dependent on caspase-11/4 and GSDMD.
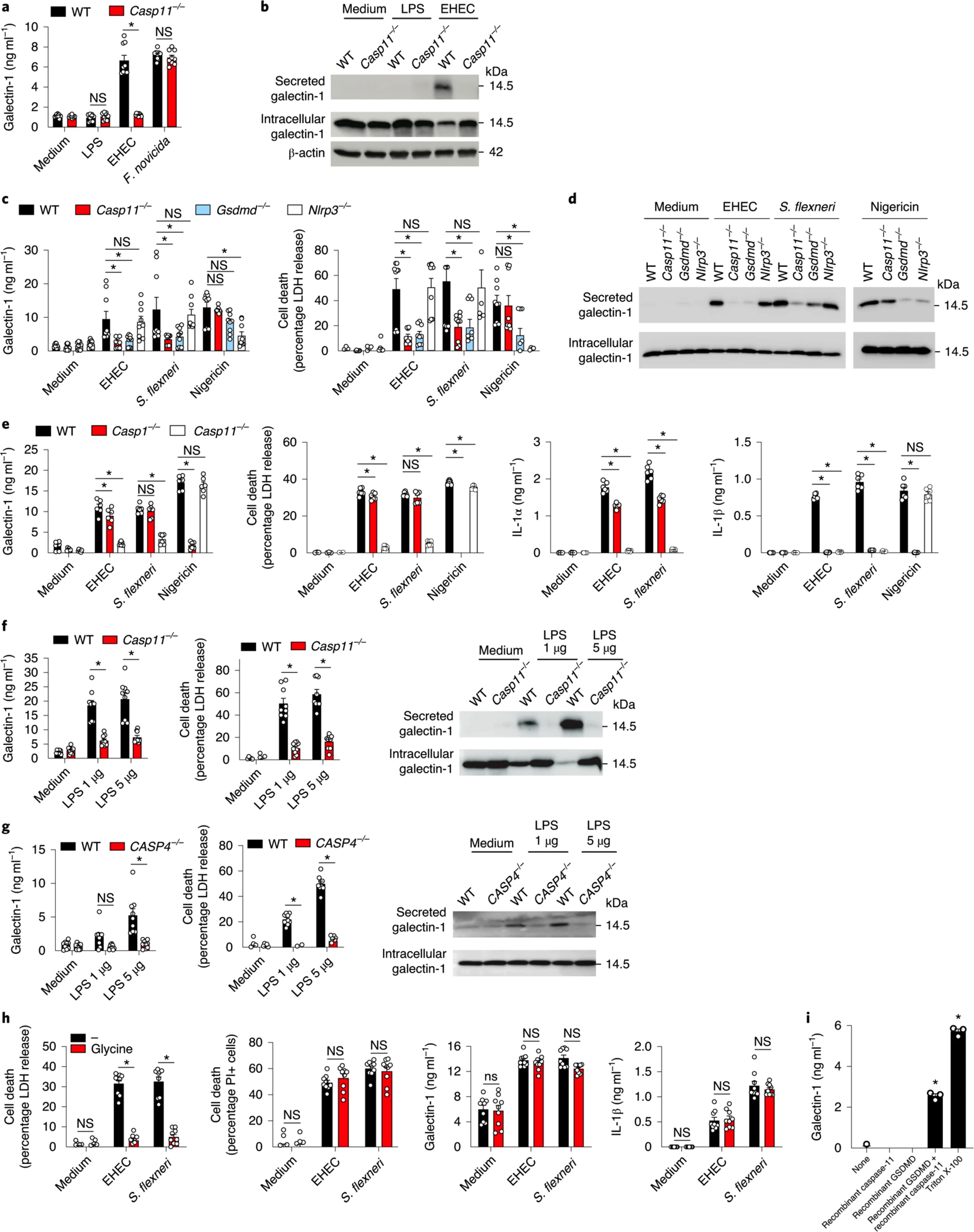
a, Galectin-1 release by Pam3CSK4-primed WT and Casp11−/− BMDMs stimulated with LPS (1 μg ml−1) to activate TLR4 or infected with EHEC (MOI = 50) to activate caspase-11 or infected with F. novicida (MOI = 50) to activate AIM2 for 16 h as measured by ELISA. b, Immunoblot of galectin-1 (14.5 kDa) in the supernatant and lysates of BMDMs stimulated with LPS (1 μg ml−1) or EHEC (MOI = 50) for 16 h. c, Galectin-1 release and cell death (LDH release) in Pam3CSK4-primed WT, Casp11−/−, Gsdmd−/− and Nlrp3−/− BMDMs infected with EHEC or S. flexneri (MOI = 50) for 16 h or stimulated with 10 μM nigericin for 1 h as measured by ELISA. d, Immunoblot of galectin-1 (14.5 kDa) in the supernatants and lysates of BMDMs stimulated as in c. e, Galectin-1, IL-1α and IL-1β release and LDH release by Pam3CSK4-primed WT, Casp11−/− and Casp1−/− BMDMs infected with EHEC or S. flexneri (MOI = 50) for 16 h or stimulated with 10 μM nigericin for 1 h. f, Galectin-1 and LDH release by LPS-primed WT and Casp11-deficient MS1 endothelial cells electroporated with LPS (1 or 5 μg) for 2 h. Immunoblot of galectin-1 (14.5 kDa) in the supernatants and lysates of WT and Casp11-deficient MS1 endothelial cells stimulated as above. g, Galectin-1 and LDH release by LPS-primed WT and CASP4-deficient HeLa cells electroporated with LPS (1 or 5 μg) for 2 h. Immunoblot of galectin-1 (14.5 kDa) in the supernatants and lysates of WT and CASP4-deficient HeLa cells stimulated as above. h, LDH release, PI uptake, galectin-1 release and IL-1β release by Pam3CSK4-primed WT BMDMs infected with EHEC or S. flexneri (MOI = 50) for 16 h in the presence or absence of 50 mM glycine. i, Galectin-1 release from liposomes packaged with galectin-1 and incubated with either 1 μg of recombinant active caspase-11, 2 μg of recombinant GSDMD, both caspase-11 and GSDMD or 0.1% Triton X-100. Combined data from three (a,c,f–h) or two (e) independent experiments are shown as the mean ± s.e.m. i, Data are presented as mean ± s.e.m. of one experiment representative of two (data from the replicate experiment are shown in Extended Data Fig. 1h). b,d,f,g, Immunoblots are representative of two independent experiments. *P < 0.05 for WT versus indicated knockouts; one-way (i) or two-way ANOVA (a,c,e,f–h) followed by Šidák’s post-test.
Cytosolic LPS sensing exists in several cell types, such as myeloid, epithelial and endothelial cells2,4,9. To determine if cytosolic recognition of LPS also leads to galectin-1 release in nonimmune cells, we electroporated purified LPS into WT and Casp11-deficient murine endothelial cells (MS1) and into WT and CASP4-deficient HeLa cells and measured galectin-1 release. Casp11-deficient MS1 cells and CASP4-deficient HeLa cells were generated using CRISPR–Cas9 and verified by immunoblotting (Extended Data Fig. 1e,f). Upon the delivery of LPS into the cytosol of WT endothelial and HeLa cells, there was an enhanced release of galectin-1 into the supernatants, which was reduced tremendously by the deficiency of caspase-11 or caspase-4 (Fig. 2f,g). Like in BMDMs, galectin-1 release upon LPS electroporation correlated with pyroptosis in both endothelial and HeLa cells (Fig. 2f,g). These data indicate that inflammatory caspase-driven galectin-1 release upon intracellular LPS sensing is common to both immune and nonimmune cells.
Several mechanisms mediate the release of the leaderless proteins during inflammasome activation. These include release via GSDMD pores and microvesicles and passive release upon cell lysis20,21,22,23. To determine if the formation of GSDMD pores without cell lysis is sufficient for galectin-1 release after caspase-11 activation, we utilized glycine, which inhibits terminal cell lysis but not the initial plasma membrane perforation19,22. As expected, without affecting GSDMD cleavage (Extended Data Fig. 1g) and propidium iodide (PI) uptake (Fig. 2h), glycine treatment significantly reduced cell lysis and the release of large lactate dehydrogenase (LDH) complexes from BMDMs after infection with EHEC and S. flexneri (Fig. 2h). In contrast, glycine did not affect galectin-1 release from EHEC- and S. flexneri-infected BMDMs (Fig. 2h). Furthermore, glycine-treated cells also released normal levels of IL-1β after infection with Gram-negative bacteria (Fig. 2h). These data suggest that the formation of GSDMD pores is sufficient to permit galectin-1 release after noncanonical inflammasome activation. To test if galectin-1 is released via GSDMD pores directly, we performed a liposome leakage assay. In this assay, galectin-1 was encapsulated into liposomes and these liposomes were incubated with recombinant GSDMD and active caspase-11 proteins. GSDMD N-terminal fragments released upon cleavage by caspase-11 bound and formed pores on the liposome bilayer leading to the leakage of liposome cargo24. The incubation of galectin-1-containing liposomes with GSDMD and active caspase-11 together, but not with caspase-11 or GSDMD alone, resulted in the release of galectin-1 (Fig. 2i and Extended Data Fig. 1h). These results clearly demonstrate that plasma membrane perforation by GSDMD directly permits galectin-1 release.
Cytosolic LPS sensing by caspase-11 triggers the systemic release of galectin-1 in vivo
To determine if in vitro galectin-1 release driven by cytosolic LPS also occurs in vivo, we injected WT mice with LPS and assessed galectin-1 release into the circulation and peritoneal cavity. Consistent with our observations in cultured cells, LPS induced galectin-1 release into the plasma and peritoneal cavity in a dose-dependent manner (Fig. 3a,b). The amount of galectin-1 released into the plasma and peritoneal cavity upon LPS administration also increased over time (Fig. 3c,d). The cytosolic LPS sensing pathway was responsible for the release of galectin-1 in LPS-injected mice since galectin-1 amounts in the plasma and peritoneal cavity were reduced in LPS-injected Casp11−/− mice compared to LPS-injected WT mice (Fig. 3e,f). Similarly, LPS-induced galectin-1 release into the circulation was markedly reduced in Gsdmd−/− mice (Fig. 3g). In contrast, circulating amounts of galectin-1 in LPS-injected Nlrp3−/− mice were similar to that of LPS-injected WT mice (Fig. 3e,f), a phenotype consistent with our observations in BMDMs. Furthermore, galectin-1 release in vivo driven by cytosolic LPS sensing differs from that of IL-1β and IL-18 with regard to the requirement for NLRP3 (Fig. 3e,f). Overall, these data show that caspase-11 sensing of cytosolic LPS in vivo triggers the systemic release of galectin-1 in a GSDMD-dependent but NLRP3-independent manner.
Fig. 3: Cytosolic LPS-induced release of galectin-1 is dependent on caspase-11 and GSDMD in vivo.
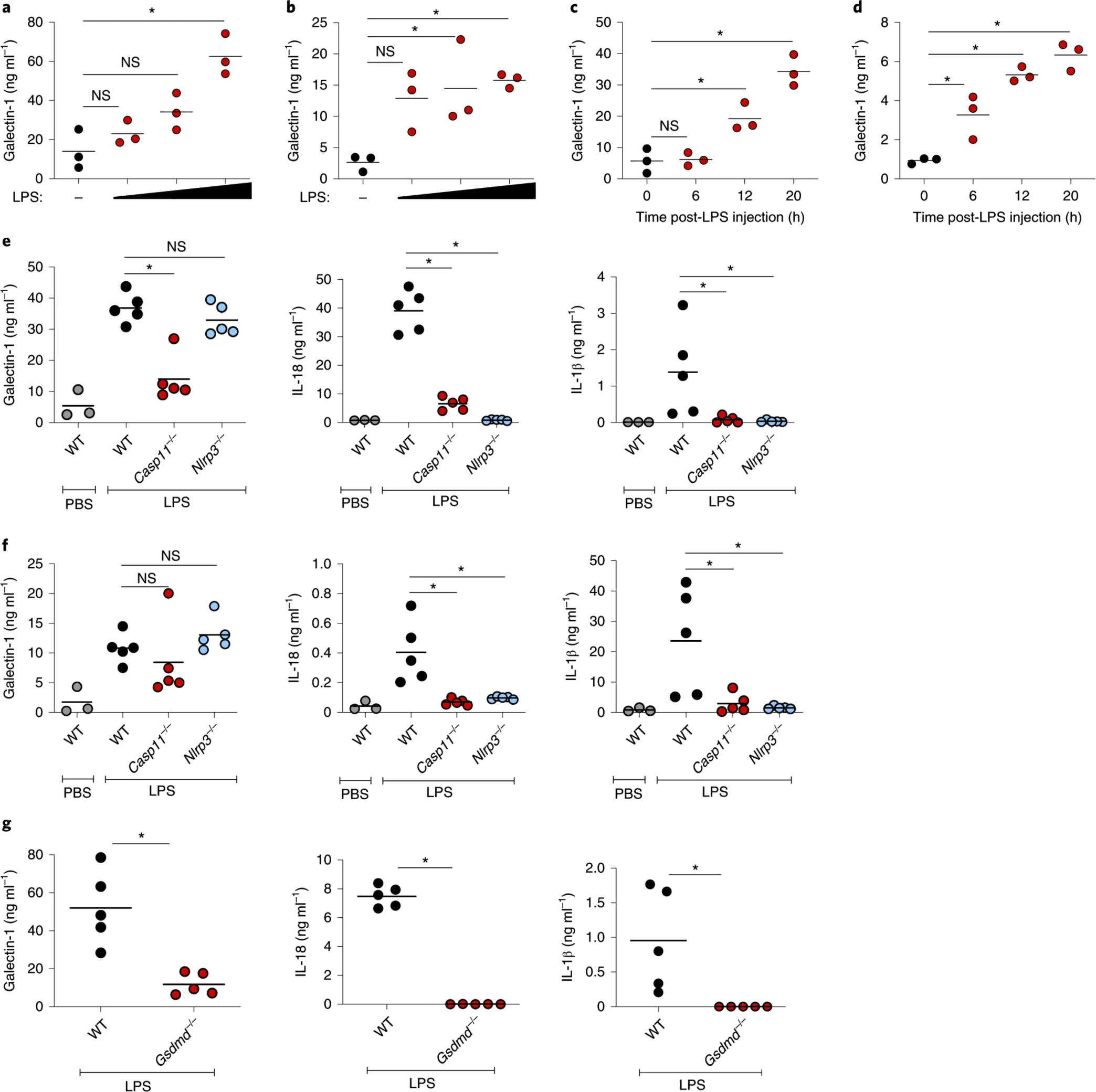
a,b, Galectin-1 levels in the plasma (a) and peritoneal lavage (b) of WT mice (n = 3) injected intraperitoneally with PBS or increasing amounts of LPS (50–200 μg) at 18 h post-injection as measured by ELISA. c,d, Galectin-1 levels in the plasma (c) and peritoneal lavage (d) of WT mice (n = 3) at 0, 6, 12 and 20 h post-LPS (200 μg) injection. e,f, Galectin-1, IL-18 and IL-1β levels in the plasma (e) and peritoneal lavage (f) of WT, Casp11−/− or Nlrp3−/− mice at 18 h post-LPS (200 μg) injection. g, Galectin-1, IL-18 and IL-1β in the plasma of WT and Gsdmd−/− mice at 18 h post-LPS (200 μg) injection. Data presented are from one experiment representative of two. a–g, Each circle represents a mouse and the horizontal lines represent the mean. *P < 0.05; one-way ANOVA followed by Šidák’s post-test (a–f) or unpaired two-tailed t-test (g).
Galectin-1 release is a common feature of inflammatory cell death programs
Since galectin-1 is released during pyroptosis downstream of noncanonical and canonical inflammasomes, we asked if other forms of lytic cell death would also lead to galectin-1 release. Necroptosis is a lytic cell death driven by receptor-interacting serine/threonine-protein kinase 3 (RIPK3) and RIPK1 upon ligation of death receptors such as Fas and tumor necrosis factor receptor 1 (TNFR). RIPK3 phosphorylates mixed lineage kinase domain-like protein (MLKL), which then translocates to the plasma membrane to cause cell lysis25. The engagement of TNFR with TNF and TIR-domain-containing adapter-inducing interferon-β (TRIF) signaling with TLR ligands, such as LPS and poly(I:C), triggers the assembly of a signaling complex comprising Fas-associated protein with a death domain, RIPK1, RIPK3 and caspase-8, which leads to apoptosis. However, if caspase-8 is inhibited, then RIPK1 and RIPK3 trigger MLKL-mediated necroptosis26. Additionally, the inhibition of inhibitor of apoptosis proteins, which normally prevent apoptosis by regulating caspase activation, with second mitochondrial-derived activator of caspases (SMAC) mimetics under the condition of caspase-8 inhibition can induce necroptosis26. To test if galectin-1 is released from cells undergoing necroptosis, we stimulated L929 fibroblasts and RAW 264.7 macrophages with necroptosis-inducing ligands and assessed galectin-1 release. L929 cells stimulated with the death ligands—TNF or poly(I:C) to activate TNFR and TRIF signaling, respectively—in combination with the pan-caspase inhibitor zVAD underwent necroptosis as evidenced from MLKL phosphorylation and LDH release and released increased amounts of galectin-1 (Fig. 4a). Galectin-1 release was blocked by the pretreatment of cells with RIPK3 inhibitors GSK872 and GSK843, indicating it was indeed mediated by necroptosis (Fig. 4a). Similarly, RAW 264.7 macrophages stimulated with LPS, poly(I:C), or a SMAC mimetic (LCL-161) in combination with zVAD underwent necroptosis and released galectin-1; this effect was also inhibited by RIPK3 inhibitors (Fig. 4b). To test if galectin-1 release occurs during necroptotic cell death in vivo, TNF was injected into WT mice and galectin-1 release was monitored over time. We found that high amounts of galectin-1 were released into the plasma and peritoneal cavity upon TNF challenge (Fig. 4c). Together these findings show that necroptosis, like pyroptosis, leads to galectin-1 release in vitro and in vivo, suggesting that galectin-1 release is a common feature of inflammatory cell death.
Fig. 4: Necroptosis triggers galectin-1 release.

a, Galectin-1 and LDH release by L929 cells stimulated with DMSO, TNF + zVAD and poly(I:C) + zVAD in the presence or absence of the RIPK3 inhibitors GSK872 or GSK843 (1 μM). Cells were treated with inhibitors 1 h before stimulation and supernatants were collected at 18 h. The immunoblot of phospho-MLKL and total MLKL in the lysates of L929 cells stimulated as above is shown. b, Galectin-1 and LDH release by RAW 264.7 macrophages stimulated with DMSO, LPS + zVAD, poly(I:C) + zVAD and the SMAC mimetic LCL-161 + zVAD in the presence or absence of the RIPK3 inhibitors GSK872 or GSK843 (2.5 μM). Cells were treated with inhibitors 1 h before stimulations and supernatants were collected at 18 h. Immunoblot of phospho-MLKL and total MLKL in the lysates of RAW 264.7 macrophages stimulated as above. c, Galectin-1 levels in the plasma (left) and peritoneal cavity (right) of WT mice (n = 6) injected intraperitoneally with 500 μg kg−1 TNF at 6, 12 and 20 h. Combined data from three independent experiments are shown as the mean ± s.e.m. a,b, Immunoblots are representative of two independent experiments. c, Combined data from two independent experiments are shown; each circle represents a mouse and the horizontal lines represent the mean (n = 6). *P < 0.05; two-way (a,b) or oneway (c) ANOVA followed by Šidák’s post-test.
Galectin-1 levels are elevated in human patients with sepsis
Sepsis is a highly lethal disease with 270,000 deaths annually in the USA alone; it is the leading cause of death for patients in the intensive care unit (ICU)27. Dysregulated cell death processes are implicated in extensive inflammation, tissue damage and organ failure during sepsis28,29,30. Therefore, we assessed the relevance of galectin-1 release as a consequence of inflammatory cell death in macrophages and other cell types to human sepsis. For this, we measured galectin-1 levels in the sera of human patients with sepsis and compared it to that of a cohort without sepsis (patients admitted for other critical conditions) in the same ICU under similar critical care as well as to that of healthy volunteers. Strikingly, the serum from patients with sepsis had significantly increased levels of galectin-1 compared to the serum of patients without sepsis and healthy volunteers (Fig. 5a; see the patient characteristics in Supplementary Table 1). These data suggest that galectin-1 release into the systemic circulation occurs during human sepsis.
Fig. 5: Galectin-1 plays a detrimental role during LPS septic shock.
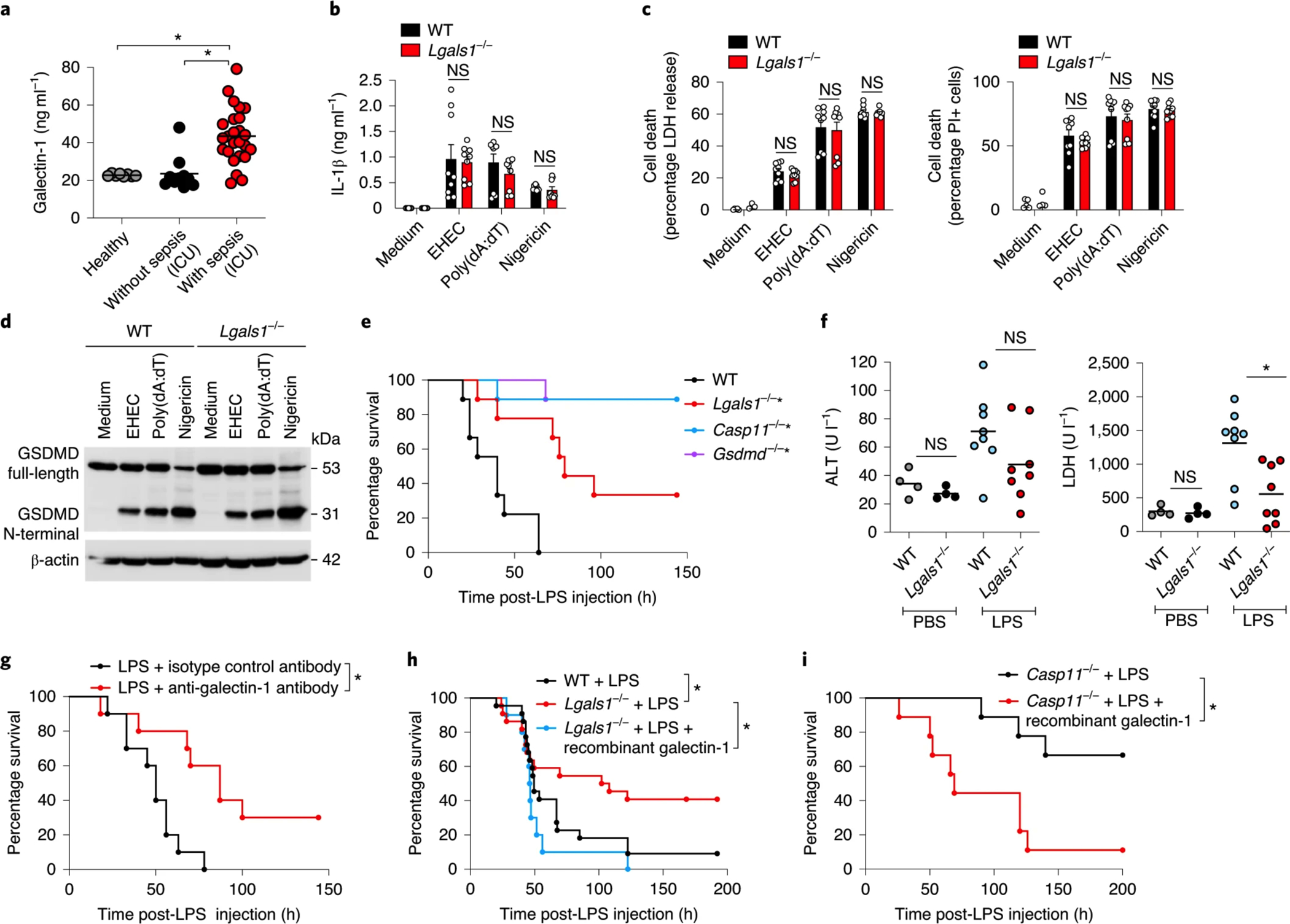
a, Galectin-1 levels in the serum of humans (healthy volunteers (n = 8), patients without sepsis in the ICU (n = 10) or patients with sepsis in the ICU (n = 25)). b,c, IL-1β release (b), LDH release and PI uptake (c) in Pam3CSK4-primed WT and Lgals1−/− BMDMs stimulated with EHEC (MOI = 50) or poly(dA:dT) for 16 h or 10 μM nigericin for 1 h. d, Immunoblot of GSDMD in the lysates of WT and Lgals1−/− BMDMs stimulated as in b,c. e, Survival of WT, Lgals1−/−, Casp11−/− and Gsdmd−/− mice (n = 9) injected intraperitoneally with 5 mg kg−1 LPS. f, Plasma levels of ALT and LDH in WT and Lgals1−/− mice injected intraperitoneally with PBS (n = 4) or 5 mg kg−1 LPS (n = 8) for 18 h. g, Survival of WT mice injected intraperitoneally with 5 mg kg−1 LPS followed by intraperitoneal injection of 250 μg of anti-galectin-1 antibody or an isotype control antibody (n = 10). h, Survival of WT mice (n = 22) and Lgals1−/− mice (n = 22) injected intraperitoneally with 3–5 mg kg−1 LPS followed by PBS or 100 μg of recombinant galectin-1 (n = 10) 1 h later. i, Survival of Casp11−/− mice injected intraperitoneally with LPS (20 mg kg−1) followed by intraperitoneal injection of PBS (n = 9) or 100 μg of recombinant galectin-1 (n = 9) 1 h later. The combined data from three (b,c) or two (e–i) independent experiments are shown. a, Each circle represents a human patient or healthy volunteer as indicated, and the horizontal lines represent the mean. b,c, Data are presented as the mean ± s.e.m. f, Each circle represents a mouse and the horizontal lines represent the mean. d, Immunoblots are representative of two independent experiments. *P < 0.05; one-way (a) or two-way ANOVA (b,c) followed by Šidák’s post-test, unpaired two-tailed t-test (f) or Mantel–Cox test (e,g–i). e, *P < 0.05 for WT versus each genotype.
Galectin-1 is a DAMP playing a detrimental role during LPS-induced lethality
Since intracellular molecules released by damaged or dying cells can act as DAMPs or alarmins in the extracellular milieu regulating the immune and inflammatory responses31, we explored the functional significance of galectin-1 release. To test if galectin-1 release upon inflammasome activation acts as a feedback mechanism to regulate noncanonical or canonical inflammasomes in vitro, we stimulated BMDMs from WT and Lgals1−/− mice with EHEC, poly(dA:dT) transfection and nigericin and assessed inflammasome responses. GSDMD processing, LDH release, PI uptake and IL-1β release induced by EHEC, poly(dA:dT) and nigericin were comparable between WT and Lgals1−/− BMDMs (Fig. 5b–d). These data ruled out a direct role for galectin-1 in the regulation of noncanonical, AIM2 and NLRP3 inflammasome responses in BMDMs.
Although the hyperactivation of caspase-11 by intracellular LPS is pathological and lethal, the underlying effector mechanisms are not clear. Caspase-11-mediated pyroptosis and release of DAMPs are considered to play a central role in driving lethality5,11. In this context, we asked whether galectin-1 released upon caspase-11 activation acts as a DAMP and contributes to the lethal manifestations in LPS shock. Toward this end, WT and Lgals1−/− mice were injected with LPS and their survival was monitored. We found that Lgals1−/− mice were more resistant to LPS shock and had a significantly better survival rate compared to WT mice injected with LPS (Fig. 5e). Casp11−/− and Gsdmd−/− mice were expectedly even more resistant than Lgals1−/− mice to LPS lethality (Fig. 5e). Consistent with this improved survival, mice lacking galectin-1, like Casp11−/− and Gsdmd−/− mice, experienced less severe organ damage as evident from lower plasma levels of alanine aminotransferase (ALT) and LDH (Fig. 5f and Extended Data Fig. 2a). Remarkably, a single dose of anti-galectin-1 neutralizing antibody significantly protected WT mice from LPS shock reinforcing the proposed DAMP-like role of galectin-1 during LPS shock (Fig. 5g). Mirroring our findings in the LPS shock model, Lgals1−/− mice, like Casp11−/− mice, were less susceptible than WT mice to E. coli infection-induced sepsis (Extended Data Fig. 2b). Importantly, the administration of recombinant galectin-1 to LPS-injected Lgals1−/− mice reversed the phenotype and made them succumb to LPS shock like WT mice, further indicating that galectin-1 enhances lethality (Fig. 5h). Notably, the failure of a control recombinant protein (green fluorescent protein (GFP)) and heat-inactivated recombinant galectin-1 to sensitize Lgals1−/− mice to LPS shock (Extended Data Fig. 2c,d) as well as the lack of immunostimulatory or lethal effects of recombinant galectin-1 on naïve BMDMs and mice, respectively (Extended Data Fig. 2e,f), rule out the confounding effects from any microbial contaminants in recombinant galectin-1 preparation. Casp11−/− mice were highly resistant to LPS-induced lethality5,11 and had low circulating levels of galectin-1 after LPS stimulation (Fig. 3e). If galectin-1 plays a detrimental role downstream of caspase-11 activation as the data suggest, the administration of recombinant galectin-1 to Casp11−/− mice would make them susceptible to LPS. Indeed, Casp11−/− mice that received LPS followed by recombinant galectin-1 succumbed to LPS shock unlike the Casp11−/− mice that received only LPS (Fig. 5i). Taken together, our findings obtained with anti-galectin-1 neutralizing antibody, recombinant galectin-1 and Lgals1−/− mice provide strong evidence that galectin-1 functions as a DAMP enhancing LPS-induced lethality.
Galectin-1 promotes systemic inflammatory responses to LPS in vivo
To gain mechanistic insights into the role of galectin-1 in systemic inflammatory responses, we profiled the pro-inflammatory cytokine and chemokine levels in the plasma of WT and Lgals1−/− mice injected with LPS. Remarkably, we found a broad impairment in the production of pro-inflammatory cytokines, including IL-3, IL-6, TNF, interferon-γ, granulocyte-colony stimulating factor, granulocyte-macrophage colony-stimulating factor, IL-1α, IL-1β and IL-12 in Lgals1−/− mice (Fig. 6a). Additionally, Lgals1−/− mice had significantly reduced circulating levels of chemokines, such as MCP-1, MIP-1α, MIP-1β, RANTES, keratinocyte chemoattractant and eotaxin (Fig. 6a). A similar reduction in inflammatory mediators, such as IL-3, IL-6, IL-12p40 and MCP1 was also observed in the lungs and spleens of Lgals1−/− mice (Extended Data Fig. 3a,b). Furthermore, the blunted cytokine response of Lgals1−/− mice to LPS was similar to that of Casp11−/− and Gsdmd−/− mice (Extended Data Fig. 4a). To assess the global impact of galectin-1 on the LPS-induced transcriptomic response, we performed RNA sequencing (RNA-seq) analysis of total RNA from the spleen and lungs of WT and Lgals1−/− mice injected with LPS. The transcriptome of Lgals1−/− spleen and lungs was broadly altered compared to that of WT mice (Fig. 6b). A pathway analysis of expression profiles revealed that many pathways related to inflammation and immune responses were downregulated in Lgals1−/− spleen and lungs (Fig. 6c,d). Of these, notable pathways included acute phase response signaling, chemokine and cytokine signaling and death receptor signaling. Furthermore, the pathways generally considered to be anti-inflammatory, such as aryl hydrocarbon and peroxisome proliferator-activated receptor signaling, were upregulated in the spleen and lungs of Lgals1−/− mice (Fig. 6c,d). Taken together, a broad attenuation of LPS-induced systemic pro-inflammatory responses that are known to be associated with sepsis was evident at the transcript and protein levels in Lgals1−/− mice. This reduced inflammatory state likely contributes to the increased survival of Lgals1−/− mice during LPS-induced shock. Thus, global gene expression and multiplex cytokine profiling indicate that galectin-1 functions as a DAMP to enhance LPS-elicited inflammation.
Fig. 6: Galectin-1 amplifies systemic inflammatory responses during LPS shock.

a, Cytokines and chemokines in the plasma of WT and Lgals1−/− mice injected intraperitoneally with PBS or 5 mg kg−1 LPS for 20 h. b, Scatter plots of differentially expressed genes (red) in the spleens and lungs of WT and Lgals1−/− mice at 8 h post-LPS (5 mg kg−1) injection as identified by RNA-seq analysis. The RNA from the spleens and lungs of two mice was pooled and subjected to RNA-seq. c, Bar graphs of 39 canonical pathways (z-score and the −log P values > 2) that were differentially regulated in both the spleens and lungs of WT and Lgals1−/− mice treated as above. d, Heatmap of genes included in 39 canonical pathways that were differentially regulated in both the spleens and lungs of WT and Lgals1−/− mice treated as above. Only genes that were differentially expressed in both organs are shown. a, Combined data from two independent experiments are shown. Each circle represents a mouse and the horizontal lines represent the mean. *P < 0.05; unpaired two-tailed t-test.
Galectin-1 inhibition of CD45 underlies its detrimental role in endotoxin shock
We next sought to identify the mechanism underlying the detrimental role of galectin-1 in LPS shock. We asked whether galectin-1 functions in endotoxemia by binding to N- and/or O-linked glycans on glycoproteins. N-acetylglucosaminyltransferase 5 (MGAT5) is an enzyme responsible for the formation of β1,6-branched complex N-glycans, while core 2 β1,6-N-acetylglucosaminyltransferase 1 (C2GNT1) catalyzes the generation of core 2 O-glycans. The addition of N-acetyllactosamine residues to these glycan structures creates potential galectin-1 ligands32. We subjected Mgat5−/− and C2gnt1−/− mice to LPS shock and assessed their survival. Both Mgat5−/− and C2gnt1−/− mice phenocopied Lgals1−/− mice and were more resistant to LPS shock compared to their WT counterpart (Fig. 7a). Administration of recombinant galectin-1 failed to sensitize these knockout mice to LPS shock (Extended Data Fig. 5a). We observed a broad attenuation of inflammatory responses in Mgat5−/− and C2gnt1−/− mice characterized by reduced circulating levels of cytokines, such as IL-1β, IL-12 and interferon-γ (Fig. 7b). Moreover, Mgat5−/− mice had lower levels of pro-inflammatory cytokines than C2gnt1−/− mice, which is consistent with their better survival than C2gnt1−/− mice. Furthermore, plasma ALT and LDH levels, indicators of organ injury, were slightly reduced in Mgat5−/− and C2gnt1−/− mice compared to WT mice (Extended Data Fig. 5b). Plasma galectin-1 levels in these mutant mice after LPS challenge were comparable to that of WT mice (Extended Data Fig. 5c). This dataset is consistent with a model where galectin-1 binds to glycosylated ligands generated by MGAT5 and C2GNT1 and mediate inflammatory responses during endotoxemia.
Fig. 7: Galectin-1 inhibition of CD45 underlies its detrimental role in endotoxin shock.

a, Survival of WT, Mgat5−/− and C2gnt1−/− mice (n = 10) injected intraperitoneally with 5 mg kg−1 LPS. b, Cytokines and chemokines in the plasma of WT, Mgat5−/− and C2gnt1−/− mice injected intraperitoneally with 5 mg kg−1 LPS for 18 h. c, CD45-specific phosphatase activity in the spleen homogenates of WT and Lgals1−/− mice injected intraperitoneally with 5 mg kg−1 LPS for 20 h (normalized using a CD45 phosphatase inhibitor). d, Phosphatase activity of CD45 immunoprecipitated from the spleen homogenates of WT and Lgals1−/− mice injected intraperitoneally with 5 mg kg−1 LPS for 20 h (normalized using a CD45 phosphatase inhibitor). The immunoblot of CD45 that was immunoprecipitated from the spleen homogenates of WT and Lgals1−/− mice, treated as described above, with anti-CD45 antibody or IgG isotype control is shown. e, Immunoblot of phospho-Src and non-phospho-Src family proteins in the spleen lysates of WT and Lgals1−/− mice injected intraperitoneally with 5 mg kg−1 LPS for 20 h. Densitometric analysis of phospho-Src in WT versus Lgals1−/− spleens normalized to non-phospho-Src. f, Survival of Lgals1−/− mice injected intraperitoneally with 5 mg kg−1 LPS followed by intraperitoneal injection of either neutralizing anti-CD45 antibody (n = 6) or an isotype control antibody (n = 6) 1 h later. b, Each circle represents a mouse and the horizontal lines represent the mean. c–e, Each circle represents a mouse and the data are presented as the mean ± s.e.m. (n = 5). d,e, Immunoblots are representative of two independent experiments. *P < 0.05; Mantel–Cox test (a,f), one-way ANOVA followed by Šidák’s post-test (b) and unpaired two-tailed t-test (c–e).
We then set out to identify the glycoprotein that could act as a possible receptor for galectin-1 during LPS shock. Galectin-1 crosslinks several cell surface and matrix glycoproteins including integrins, vascular endothelial growth factor receptor 2 and fibronectin16. Of these, CD45 or protein tyrosine phosphatase receptor type C (PTPRC) is an established receptor for galectin-1. CD45 is a heavily glycosylated cell surface protein and studies have shown that galectin-1 binds to the glycan branches on CD45 and regulates its function33,34. CD45 is highly expressed by immune cells including lymphocytes and macrophages and is a well-known regulator of inflammatory responses35,36. Taking all this into account, we hypothesized that the detrimental function of galectin-1 in LPS shock is mediated by its modulation of CD45. We first examined if galectin-1 regulates CD45 phosphatase activity during endotoxemia and observed an increase in CD45 phosphatase activity in the total spleen lysates of Lgals1−/− mice compared to that of WT mice (Fig. 7c). Furthermore, CD45 was immunoprecipitated from the spleen lysates of WT and Lgals1−/− mice injected with LPS and the phosphatase assay was performed on the immunoprecipitates to assess CD45 phosphatase activity directly and more specifically; the CD45-specific phosphatase activity was higher in Lgals1−/− mice compared to WT mice (Fig. 7d). Immunoblotting confirmed equivalent amounts of CD45 immunoprecipitation from WT and Lgals1−/− spleen lysates (Fig. 7d and Extended Data Fig. 5d). In line with the results from the phosphatase assays, Lgals1−/− mice had significantly less phosphorylation of Src proteins, which are CD45 substrates, in the spleen compared to WT mice, indicating that CD45 phosphatase activity was increased in Lgals1−/− mice (Fig. 7e). Lastly, considering that CD45 negatively regulates inflammatory responses35,36, the suppressive effect of galectin-1 on CD45 may underlie its inflammatory role in endotoxemia. If this is the case, then reducing the elevated activity of CD45 in Lgals1−/− mice should render them more susceptible to LPS. To test this, Lgals1−/− mice were injected with LPS, followed by either an anti-CD45 neutralizing antibody or an isotype control antibody and monitored for survival. Remarkably, Lgals1−/− mice that received anti-CD45 antibody died at a significantly faster rate compared to Lgals1−/− mice that received the isotype control (Fig. 7f). Taken together, these data indicate that intracellular LPS-induced galectin-1 release facilitates lethal inflammation by negatively regulating CD45.
Discussion
While the collective release of DAMPs from dying cells is considered to regulate pathophysiological processes ranging from inflammation to tissue repair31, the identity and function of the individual DAMPs in the context of cytosolic LPS sensing are poorly defined. This work establishes galectin-1 as a bona fide DAMP that functions in a glycan-dependent manner to enhance the inflammatory response elicited by intracellular LPS. This conclusion is based on the following independent lines of evidence: (1) the extracellular release of galectin-1 upon cytosolic LPS sensing in multiple cell types as well as in vivo; (2) the impairment of inflammatory responses in Lgals1−/− mice; (3) the attenuation of LPS-induced lethality by galectin-1 neutralizing antibody and by the deficiency of galectin-1 or its glycan ligands; (4) the enhancement of LPS-induced lethality in Lgals1−/− and Casp11−/− mice by recombinant galectin-1. Thus, galectin-1 represents a new effector mechanism shaping the inflammatory aftermath of intracellular LPS sensing.
Terminal cell lysis during the pyroptotic cascade is not a prerequisite for galectin-1 release, which is consistent with its smaller size of 14.5 kDa. Although cells ultimately undergo lysis upon inflammasome activation in most scenarios, cells can tolerate subthreshold GSDMD activation and become hyperactive22,37,38. In these nonlytic contexts, DAMPs, such as galectin-1, which are small enough for release via GSDMD pores, are likely to dictate the inflammatory consequence of inflammasome activation. Galectin-1 is involved in a variety of cellular processes, such as adhesion, proliferation, apoptosis and angiogenesis16,17. Since Mgat5−/− and C2Gnt1−/− mice phenocopy the enhanced resistance of Lgals1−/− mice to LPS, our work suggests that released galectin-1 functions in a glycan-dependent fashion during LPS shock. Indeed, it appears that galectin-1 targets CD45 and suppresses its anti-inflammatory signaling, which contributes to the lethal manifestations of LPS shock. CD45 is a known glycoprotein receptor for galectin-1; binding of CD45’s glycan branches by galectin-1 induces the clustering and segregation of CD45 on activated T cells into distinct microdomains resulting in diminished phosphatase activity33,34. Therefore, it is likely that galectin-1 limits the phosphatase activity of CD45 during endotoxemia and minimizes the inhibitory effects of this receptor on inflammatory pathways such as cytokine signaling35,36, thus licensing a stronger inflammatory response as evident from our RNA-seq and multiplex analysis. Nonetheless, given the broad repertoire of galectin-1 receptors, additional glycoprotein target(s) may be involved in galectin-1 function in LPS shock.
Consistent with the findings presented in this study, galectin-1 has been shown to play a pro-inflammatory role in certain diseases, such as experimental autoimmune orchitis and osteoarthritis39,40,41. On the other hand, a number of studies have also reported anti-inflammatory activity of galectin-1 in other autoimmune and inflammatory diseases16,42,43,44. All these findings are consistent with the pleiotropic nature of galectin-1 function and suggest that the dual role of galectin-1 in the amplification versus resolution of inflammatory responses is context-dependent and dictated by several factors including the stage of inflammatory response (acute versus chronic), glycosylation and activation status of cells, the cytokine milieu and intrinsic biochemical parameters (for example, monomeric/dimeric and redox balance) that control the structure-function of this protein45,46,47. Moreover, it is possible that while galectin-1 functions as a pro-inflammatory alarmin at the early stages of inflammation, it may serve as an anti-inflammatory mediator during the later stages, similar to cytokines with dual pro- and anti-inflammatory effects including IL-6 and transforming growth factor-β16.
It would be of interest to determine if galectin-1 can be used as a biomarker in sepsis and if galectin-1-glycan interactions can be targeted for therapeutic benefit in sepsis. It should be noted that although Lgals1−/− mice were less susceptible than WT mice to LPS shock, they were more susceptible than Casp11−/− and Gsdmd−/− mice. This pattern of susceptibility to LPS lethality in Lgals1−/−, Casp11−/− and Gsdmd−/− mice is expected given that caspase-11- and GSDMD-mediated pyroptosis leads to the release of a number of DAMPs and that these additional pathogenic DAMPs and mechanisms, besides galectin-1, contribute to endotoxin shock. Further studies are needed to elucidate the involvement of those hitherto unknown DAMPs in sepsis. In conclusion, cytosolic LPS sensing triggers the release of galectin-1, which serves as a functional DAMP, promoting inflammatory responses.
Methods
Mice
The animal protocols were carried out in accordance with the guidelines set forth by the University of Connecticut Health Institutional Animal Care and Use Committee. C57BL/6J and Nlrp3−/− mice obtained from The Jackson Laboratory and Gsdmd−/−2,19 and Casp11−/− (refs. 7,11) mice obtained from Genentech were bred and maintained in specific pathogen-free conditions in the animal facilities of University of Connecticut Health. All mice used in this study were housed at an ambient temperature of approximately 22 °C, a humidity of 40–60% and a light–dark cycle of 12 h. Lgals1−/− mice originally generated by F. Poirier48 (Institut Jacques Monod) were provided by J. Conejo-Garcia (Moffitt Cancer Center). Lgals1−/− mice were bred and maintained in specific pathogen-free conditions in the animal facilities of University of Connecticut Health. This mutant strain develops normally and does not have any overt abnormalities or phenotypes48. However, aged Lgals1−/− mice (>1 year old) have recently been shown to develop spontaneous salivary gland autoimmunity49. Casp1−/−, Mgat5−/− and C2gnt1−/− mice were described previously11,46. Mgat5−/− and C2gnt1−/− (obtained from The Jackson Laboratory) mice were bred at the animal facilities of the Instituto de Biología y Medicina Experimental (Buenos Aires) according to institutional guidelines. Both male and female 8–24-week-old mice were used.
Primary BMDMs
Primary BMDMs were generated by culturing bone marrow cells from WT and various mutant mice in DMEM with 10% FCS and 20% L929 supernatant at 37 °C and 5% CO2. On days 3 and 6, the cell culture medium was replaced with fresh DMEM with 10% FCS and 20% L929 supernatant. Cells were collected for experiments on days 8–11 of culture.
Bacterial strains and growth conditions
EHEC (EDL933), S. flexneri and F. novicida were used in this study to infect macrophages (mean multiplicity of infection (MOI) = 50) and E. coli BL21 was used (5 × 108 colony-forming units (CFUs) per mouse) for in vivo infection. Bacteria were grown and used as described previously7,19,50.
PF2D proteomics
WT and Casp11−/− BMDMs were infected with EHEC (MOI = 50) and the serum-free supernatants were collected after 16 h. The supernatants were centrifuged at 400g for 5 min to remove cell debris and concentrated using a 3K Macrosep Advance Centrifugal Device (Pall Corporation). PF2D was performed as described previously51. Briefly, the concentrated supernatants were subjected to buffer exchange with Sigma PD-10 columns pre-equilibrated with Beckman Coulter PF2D start buffer (catalog no. 391110; Beckman Coulter). Protein concentration was estimated using the bicinchoninic acid assay (BCA) (Thermo Fisher Scientific) and 2 mg of WT or Casp11−/− samples were loaded on a Beckman Coulter ProteomeLab PF2D platform. Samples were fractionated in the first dimension by a pH gradient from pH 8.0 to pH 4.0 and fractions were collected every 0.3 pH units. Fractions were further separated in the second dimension on an HPRP column using a gradient of acetonitrile from 0 to 100%. Fractions were monitored for protein content via ultraviolet absorbance at 214 nm and collected at 0.5-min intervals into 96-well Deepwell plates and stored at −80°C. Fractions were compiled and integrated with the ProteoVue software v.1.0.0.6 (Beckman Coulter) to create a 2D map (pH versus hydrophobicity) of proteins in the supernatants of WT and Casp11−/− BMDMs (Fig. 1c). A 2D protein map was generated from the 214 nm absorbance profiles of the fractions by the ProteoVue software and the fractions with differential fingerprints were identified using the DeltaVue software v.1.0.0.2 (Beckman Coulter). Fractions of interest were then lyophilized in a SpeedVac centrifuge and resolved in ammonium bicarbonate buffer and subjected to liquid chromatography–tandem mass spectrometry (LC–MS/MS) at the Yale MS & Proteomics Resource core facility.
Generation of CRISPR–Cas9 knockout cell lines
Single-guide RNAs targeting caspase-11 (GGCCAATGGCCGTACACGAA) and caspase-4 (GGTGGTCCAGCCTCCATATT) were cloned into the LentiCRISPRv2 plasmid (Addgene) using the GeCKO protocol from the Zhang laboratory. LentiCRISPRv2, pSPAX and pMD2 plasmids (4, 3 and 1 μg, respectively) were cotransfected into 293T cells; lentivirus-containing supernatants were collected at 48 and 72 h post-transfection. The lentivirus-containing supernatants were filtered using a 0.22-μm syringe filter and centrifuged at 20,000g for 2 h at 4 °C to pellet the virus. MS1 endothelial cells and HeLa cells were transduced with the virus in complete DMEM containing 2 μg ml−1 of polybrene in a 6-well plate by centrifuging at 2,000g for 45 min at 37 °C. Cells were selected using 2 μg ml−1 of puromycin.
Cell stimulation
BMDMs, MS1, HeLa, RAW 264.7 macrophages and L929 cells were cultured in complete DMEM supplemented with 10% FCS at 37 °C and 5% CO2. Primary BMDMs were generated as described above. BMDMs used to assess the inflammasome and cell death responses were primed with Pam3CSK4 (0.5 μg ml−1; InvivoGen) for 3 h. Cells were infected with bacteria at an MOI of 50 for 1 h and medium was replaced with fresh complete DMEM containing gentamicin (100 μg ml−1). Supernatants were collected 16 h poststimulation. Cells were stimulated with 10 μM of nigericin (Sigma-Aldrich) and supernatants were collected 1 h later, or cells were transfected with Lipofectamine 2000 (2 μl μg−1 of DNA)-complexed poly(dA:dT) (1μg 10−6 cells) and supernatants were collected 16 h later. In some experiments, glycine (50 mM) was added to BMDMs 1 h after infection at the time of gentamicin addition. HeLa and MS1 cells primed with LPS were electroporated with 1 or 5 μg (for 1 × 106 cells) of LPS using the Neon Transfection System (Thermo Fisher Scientific) according to the manufacturer’s protocol and supernatants were collected 2 h later. To induce necroptosis, L929 and RAW 264.7 macrophages were stimulated with either 10 ng ml−1 of TNF, 40 μg ml−1 of poly(I:C), 1 μg ml−1 of LPS or 100 nM of the SMAC mimetic LCL-161 in combination with 25 μM of zVAD and supernatants were collected 18 h later. In these experiments, cells were also pretreated for 1 h with the RIPK3 inhibitors GSK872 and GSK843 (1 or 2.5 μM) or the vehicle control dimethylsulfoxide (DMSO). WT BMDMs were stimulated with 5 μM of recombinant galectin-1 protein or 1 μg ml−1 of LPS as a positive control for 16 h and cell culture supernatants were collected and analyzed for IL-6 and TNF cytokines by ELISA.
Cytokine analysis and cell death assays
Galectin-1 levels were assessed with the DuoSet ELISA kit (R&D Systems) according to the manufacturer’s protocol. IL-1β, IL-1α, TNF and IL-6 levels in the cell culture supernatant or mouse plasma were measured by Ready-Set-Go! ELISA kits (eBioscience) according to the manufacturer’s protocol. IL-18 levels were assessed by ELISA as described previously7,19. All other cytokines and chemokines were analyzed using the Bio-Plex Pro Mouse Cytokine 23-Plex assay (Bio-Rad Laboratories) according to the manufacturer’s protocol. Cell death was analyzed by measuring LDH release into the supernatant with the LDH cytotoxicity kit (Clontech) according to the manufacturer’s protocol. Additionally, plasma membrane integrity was monitored by measuring PI (1 μg ml−1; Sigma-Aldrich) uptake. Microplates were read using BMG Labtech CLARIOstar and the BMG Labtech MARS software v.3.10 R6.
Immunoblotting
Cell lysates and supernatants were collected for immunoblot analysis. For lysates, cells were collected after stimulation and lysed with 1% NP-40 lysis buffer containing protease inhibitor cocktail (Thermo Fisher Scientific). For supernatants, protein was precipitated using the methanol-chloroform extraction method. Spleens were dissected and homogenized in PBS with protease inhibitor cocktail (Thermo Fisher Scientific). The BCA assay was performed to determine protein concentration and 50 μg of total protein was loaded into the gel. All samples were added to NuPAGE LDS sample buffer (Invitrogen) and run on polyacrylamide gels and then transferred onto nitrocellulose membranes using the Trans-Blot Turbo Transfer System (Bio-Rad Laboratories). Membranes were blocked in 2.5% milk and probed with the appropriate primary and secondary antibodies. Blots were visualized for proteins using the Bio-Rad Clarity ECL horseradish peroxidase substrate on a Syngene gel documentation box and the GeneSnap program v.7.12.06. Densitometry was done using ImageJ v.1.52. Immunoblot analysis was done with antibodies to mouse galectin-1 (1:1,000 dilution; catalog no. GTX101566; GeneTex), mouse caspase-11 (1:1,000 dilution; catalog no. 17D9; Cell Signaling Technology), human caspase-4 (clone 4B9; 1:1,000 dilution; Medical & Biological Laboratories), phospho-Src family (Tyr416) (1:1,000 dilution; clone D49G4; Cell Signaling Technology), non-phospho-Src family (1:1,000 dilution; clone 7G9; Cell Signaling Technology), mouse phospho-MLKL (Ser345) (1:1,000 dilution; clone D6E3G; Cell Signaling Technology), mouse total MLKL (1:1,000 dilution; clone D6W1K; Cell Signaling Technology), mouse recombinant GSDMD (1:1,000 dilution; catalog no. ab209845; Abcam), mouse CD45 (1:500 dilution; clone D3F8Q; Cell Signaling Technology) and β-actin (1:5,000 dilution; clone 8H10D10; Cell Signaling Technology). Secondary antibodies were peroxidase-conjugated goat anti-mouse IgG, donkey anti-rabbit IgG and donkey anti-rat IgG (1:5,000 dilution; Jackson ImmunoResearch). Uncropped immunoblot images are included in the supplementary information.
Liposome leakage assay
The liposome leakage assay was performed as described previously24. Liposomes were prepared with 126 μl of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (25 mg ml−1) and 84 μl of synthetic 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (10 mg ml−1) dissolved in chloroform (Avanti Polar Lipids) and mixed in a glass tube. The lipid mixture was dried under a stream of N2 gas for 30 min and rehydrated with 300 μl of recombinant galectin-1 solution (0.23 mg ml−1) in buffer A (20 mM of HEPES at pH 7.5, 150 mM of NaCl) to yield a final lipid concentration of 17 mM. The lipid/protein suspension was kept on ice for 30 min and then vortexed continuously for 5 min to encapsulate the recombinant galectin-1 into large liposomes. To obtain unilamellar vesicles, liposomes were extruded by 21 passes through a mini-extruder device (Avanti Polar Lipids) with a membrane (Whatman Nuclepore Track-Etched Membrane; catalog no. WHA800319; Merck Millipore) having a pore size of 1 μm. To remove the unencapsulated protein, the obtained liposomes were centrifuged at 21,000g for 20 min at 4 °C and washed three times with buffer A. The liposome pellet was finally resuspended in 900 μl of buffer A. For the galectin-1 release experiment, 2 μg of recombinant GSDMD was added into 96 μl of liposomes and release was initiated by adding 1 μg of active caspase-11 for 1 h at room temperature. As a positive control, 0.1% Triton X-100 was added to liposomes to achieve complete release of galectin-1. The release of galectin-1 was detected by ELISA.
In vivo stimulation
Age- and sex-matched C57BL/6 (WT), Lgals−/−, Casp11−/−, Gsdmd−/−, Nlrp3−/−, Mgat5−/− or C2Gnt1−/− mice were injected intraperitoneally with E. coli O111:B4 LPS (5 mg per kg body weight (mg kg−1) unless otherwise indicated; Sigma-Aldrich) to induce septic shock. WT, Lgals1−/− and Casp11−/− mice were injected intraperitoneally with E. coli (5 × 108 CFU) to induce bacterial septic shock. WT mice were injected intraperitoneally with 500 μg kg–1 of recombinant murine TNF (PeproTech) to induce necroptosis. In some experiments, WT and the indicated strains of mice were also injected intraperitoneally with 100 μg of murine recombinant galectin-1 1 h post-LPS injection. The murine recombinant galectin-1 was expressed and purified from E. coli and endotoxin-tested as described previously52. To test if any bacterial contaminants in the recombinant galectin-1 protein preparation contributed to the effect of recombinant galectin-1 administration, LPS-challenged Lgals1−/− mice were injected intraperitoneally with 100 μg of recombinant galectin-1, heat-inactivated recombinant galectin-1 or recombinant GFP and monitored for survival. Recombinant galectin-1 was heat-inactivated by incubating at 65 °C for 20 min. Additionally, naïve WT and Casp11−/− mice were injected intraperitoneally with 100 μg of recombinant galectin-1 alone and monitored for survival. In some experiments, WT mice were injected intraperitoneally with 250 μg of an anti-galectin-1 antibody (clone Gal-1-mAb3 (ref. 53)) or an isotype control antibody (clone MPC-11; Bio X Cell) 1 h after LPS injection. In some experiments, Lgals1−/− mice were injected intraperitoneally with 250 μg of an anti-CD45 antibody (clone 104.2; Bio X Cell) or an isotype control antibody (clone C1.18.4; Bio X Cell) 1 h after LPS injection. Cytokine levels in the plasma and peritoneal lavage at the indicated times, and mice survival, were analyzed as described above. Plasma levels of ALT and LDH were analyzed at the IDEXX laboratories.
CD45 phosphatase activity assay
WT and Lgals1−/− mice were injected intraperitoneally with 5 mg kg−1 LPS for 20 h. Spleens were dissected and homogenized in 500 μl of PBS with protease inhibitor cocktail (Thermo Fisher Scientific). The BCA assay was performed to determine total protein concentration. To determine phosphatase activity, 2 μg of total protein from the spleen lysates was incubated with 2 mM p-nitrophenyl phosphate (New England Biolabs) in a phosphatase assay buffer (100 mM of HEPES, 2 mM of EDTA and 2 mM of dithiothreitol) at 37 °C for 18 h with or without a specific CD45 phosphatase inhibitor (10 μg ml−1; Calbiochem). The enzymatic reaction generated a color change that was measured in a plate reader at an optical density (OD) of 410 nm. The resulting OD values ‘with the inhibitor’ were subtracted from the OD values ‘without the inhibitor’ for each lysate sample to determine the phosphatase activity specific to CD45. For the CD45 immunoprecipitation, 500 μg of total protein from spleen lysates was mixed with either 2 μg of biotin-conjugated anti-CD45 antibody (clone 104) or biotin-conjugated IgG2a isotype control antibody (clone eBM2a) and streptavidin agarose beads and incubated overnight at 4 °C. The beads were washed three times with PBS containing 0.5% NP-40 and two times with phosphatase assay buffer. After washing, the beads were split for the phosphatase assay and immunoblot analysis. The beads for the phosphatase assay were resuspended in the phosphatase assay buffer and incubated with 2 mM of p-nitrophenyl phosphate with or without 10 μg ml−1 CD45 phosphatase inhibitor as described above. The normalized phosphatase activity was calculated similarly by subtracting the ‘with the inhibitor’ OD value from the ‘without the inhibitor’ one for each immunoprecipitate. The beads for the immunoblot analysis were resuspended in NuPAGE LDS sample buffer, boiled and subjected to immunoblotting for CD45 to determine immunoprecipitation efficiency.
RNA-seq analysis
Total RNA was extracted from freshly isolated spleen or lungs from WT or Lgals1−/− mice treated with LPS (5 mg kg−1 for 8 h) using the RNeasy Mini Kit (QIAGEN). After quality checking, RNA samples were subjected to library generation using the NEBNext Ultra RNA Library Prep Kit with polyA selection followed by high-throughput sequencing using the Illumina HiSeq 4000 System at Genewiz. Illumina basecall files (BCL) were converted to FASTQ files with the bcl2fastq program v.2.20. Sequence reads were mapped to the mouse genome mm10 using Histat2 v.2.1.0 followed by StringTie for normalization and quantification. Gene expression analyses were performed using R v.3.6.1. The heatmap was generated using R or Cluster 3.0 and visualized using Java TreeView v.3.0. Bar graphs and scatter plots were generated using R or Prism v.8 (GraphPad Software). Signaling pathway enrichment analyses were conducted using Ingenuity Pathway Analyses v.01–16 (QIAGEN). RNA-seq data have been deposited with the Gene Expression Omnibus (accession no. GSE140892).
Galectin-1 analysis in human samples
Blood samples were collected from a clinical cohort at the multidisciplinary ICU of Jena University Hospital. After approval by the local ethics committee, all patients or legal surrogates gave informed consent for blood collection, analyses and data evaluation. The patient characteristics with regard to age, sex, APACHE score and SOFA score are shown in Supplementary Table 1. The blood samples were collected from patients with sepsis, patients without sepsis (patients admitted for other conditions including surgery) in the same ICU under similar critical care and healthy volunteers. All patients admitted to the ICU were screened within 24 h after admission for clinical signs of sepsis and systemic inflammatory response syndrome resulting from possible or proven infection. Adult patients with clinically diagnosed sepsis with organ dysfunction according to the American College of Chest Physicians/Society of Critical Care Medicine criteria54 were eligible for inclusion in the study. All samples were collected in accordance with the Sepsis-3 definition55. Blood samples were collected after clinical diagnosis of severe sepsis/septic shock. Galectin-1 in serum samples was measured using the galectin-1 DuoSet ELISA kit (R&D Systems).
Statistical analysis
Data were analyzed for statistical significance by unpaired two-tailed t-test, one-way or two-way analysis of variance (ANOVA) followed by Šidák’s post-test or Mantel–Cox test with the Prism software as indicated in the figure legends. P < 0.05 was considered statistically significant.
Data availability
The RNA-seq data have been deposited with the Gene Expression Omnibus (accession number GSE140892). Source data and uncropped immunoblot images are included in the paper as supplementary information. All other data supporting the findings of the paper are available from the corresponding author upon reasonable request.
Extended Data
Extended Data Fig. 1. Galectin-1 is released as a consequence of inflammasome activation.

a, Experimental design for profiling caspase-11-mediated release of DAMPs by PF2D-based proteomics. WT and Casp11−/− BMDMs were infected with EHEC (MOI=50) for 16 h and supernatants were harvested and concentrated and processed on the Beckman Coulter ProteomeLab PF2D platform. b, Galectin-1 secretion by Pam3CSK4-primed WT and Lgals1−/− BMDMs stimulated with EHEC (MOI=50) or poly(dA:dT) transfection for 16 h or 10 μM nigericin for 1 h. c, Immunoblot of galectin-1 (14.5 kDa) in the supernatant and lysates of BMDMs stimulated as in (b). d, IL-1α secretion by Pam3CSK4-primed WT, Casp11−/−, and Casp1−/− BMDMs stimulated with 10 μM nigericin for 1 h. e, Immunoblot of caspase-11 and β-actin in lysates of MS1 endothelial cells transduced with LentiCRISPR V2 plasmid expressing control (GFP) sgRNA or caspase-11 sgRNA. f, Immunoblot of caspase-4 and β-actin in lysates of HeLa cells transduced with LentiCRISPR V2 plasmid expressing control (GFP) sgRNA or caspase-4 sgRNA. g, Immunoblot of GSDMD in the lysates of Pam3CSK4-primed WT BMDMs infected with EHEC or S. flexneri (MOI=50) for 16 h or 10 μM nigericin for 1 h in the presence or absence of 50 mM glycine. h, Galectin-1 release from liposomes packaged with galectin-1 and incubated with either 1 μg recombinant active caspase-11, 2 μg recombinant gasdermin D, both caspase-11 and gasdermin D, or 0.1% Triton X-100. Data are presented as mean ± SEM of one experiment representative of two (b,h; h is the replicate for the Fig. 2i). Combined data from two independent experiments (d) are shown as mean ± SEM. Immunoblots (c,e–g) are representative of two independent experiments.
Extended Data Fig. 2. Galectin-1 contributes to lethality during sepsis.
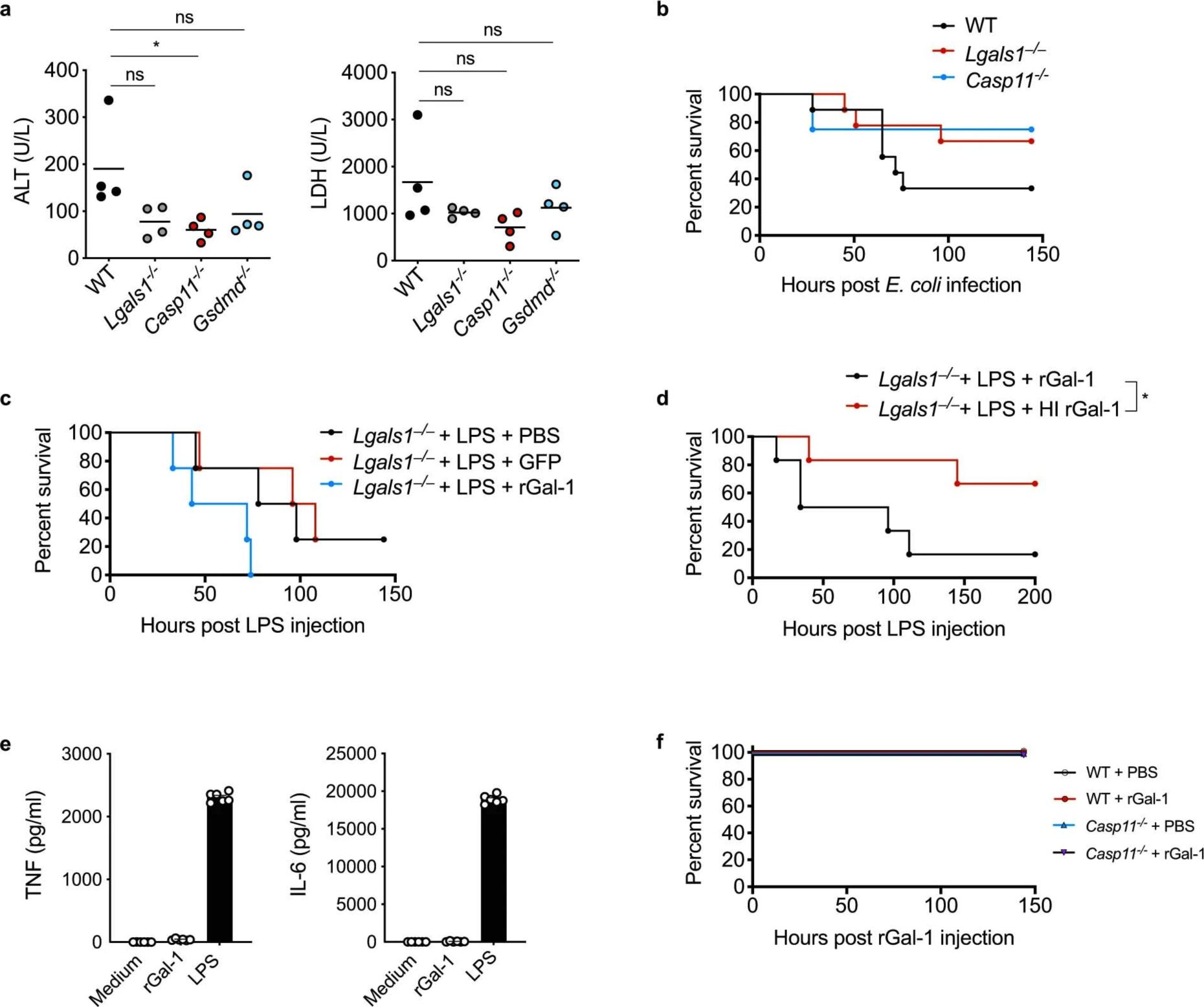
a, Plasma amounts of ALT and LDH in WT, Lgals1−/−, Casp11−/−, and Gsdmd−/− mice injected i.p. with 5 mg/kg LPS for 18 h. b, Survival of WT (n=9), Lgals1–/− (n=9), and Casp11−/− mice (n=4) injected i.p. with E. coli (5×108 CFU). c, Survival of Lgals1–/– mice injected i.p. with LPS (3 mg/kg) followed by i.p. injection of PBS (n=4), 100 μg of recombinant GFP (n=4), or 100 μg rGal-1 (n=4) 1 h later. d, Survival of Lgals1−/− mice injected i.p. with LPS (3 mg/kg) followed by i.p. injection of 100 μg of rGal-1 (n=6) or heat inactivated rGal-1 (HI rGal-1) (n=6) 1 h later. e, TNF and IL-6 secretion by WT BMDMs stimulated with 5 μM rGal-1 or 1 μg LPS for 16 h. f, Survival of WT and Casp11−/− mice injected i.p. with PBS (n=3) or 100 μg of rGal-1 (n=3). Combined data from two independent experiments are shown (b,d, and e). In (e) data are presented as mean ± SEM. In (a), each circle represents a mouse and the horizontal lines represent mean. ns, not significant (one-way ANOVA). *p < 0.05, one-way ANOVA (a); Mantel-Cox test (d).
Extended Data Fig. 3. Galectin-1 amplifies systemic inflammatory responses during LPS shock.
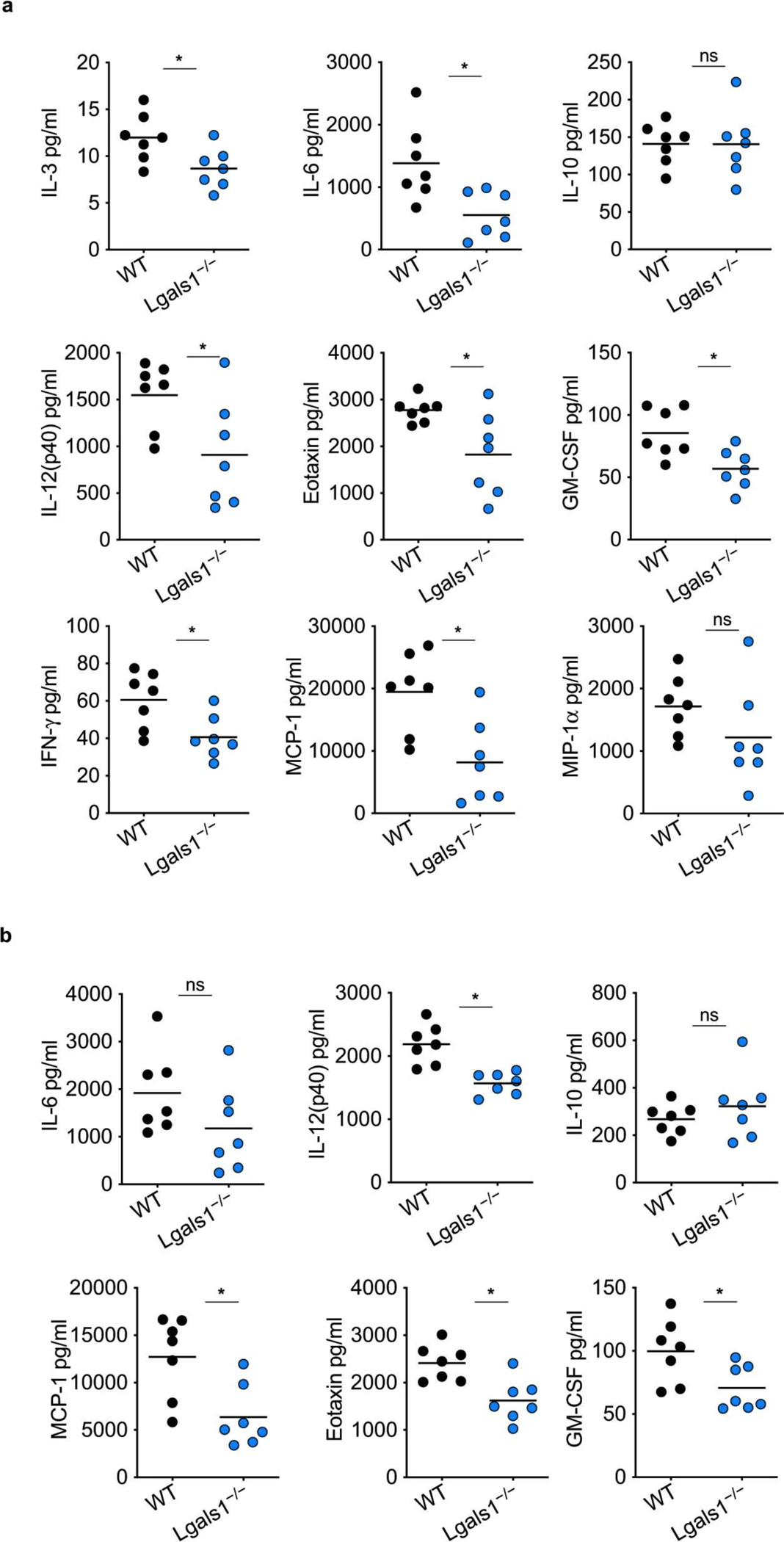
a,b, Cytokine and chemokine levels in the lung (a) and spleen (b) homogenates of WT and Lgals1−/− mice injected i.p. with 5 mg/kg LPS for 20 h. Combined data from two independent experiments are shown. Each circle represents a mouse and the horizontal lines represent mean. *p < 0.05; unpaired two-tailed t test (a–b); ns, not significant.
Extended Data Fig. 4: Galectin-1 amplifies systemic inflammatory responses during endotoxemia.

a, Indicated cytokine and chemokine levels in the plasma of WT (n=5), Lgals1−/− (n=5), Casp11−/− (n=5), and Gsdmd−/− (n=5) mice injected i.p. with 5 mg/kg LPS for 20 h. Each circle represents a mouse and the horizontal lines represent mean. *p < 0.05; one-way ANOVA followed by Sidak’s post-test.
Extended Data Fig. 5: Galectin-1 functions in a glycan-dependent manner during endotoxemia.

a, Survival of WT, Mgat5−/−, and C2gnt1−/− mice injected i.p. with 5 mg/kg LPS followed by i.p. injection of PBS or 100 μg of rGal-1 1 h later (WT LPS PBS n=6, WT LPS rGal-1 n=7, Mgat5−/− LPS PBS n=5, Mgat5−/− LPS rGal-1 n=5, C2gnt1−/− LPS PBS n=9 and C2gnt1−/− LPS rGal-1 n=11). b, Plasma amounts of ALT and LDH in WT (n=6), Mgat5−/− (n=5), and C2gnt1−/− (n=9) mice injected i.p. with 5 mg/kg LPS for 18 h. c, Galectin-1 levels in the plasma of WT (n=6), Mgat5−/− (n=5), and C2gnt1−/− (n=9) mice injected i.p. with 5 mg/kg LPS for 18 h. d, Immunoblot of CD45 that was immunoprecipitated with anti-CD45 antibody or IgG control antibody from the spleen homogenates of WT mice. Each circle represents a mouse and the horizontal lines represent mean (b,c). ns, not significant; one-way ANOVA followed by the Sidak’s post-test. Immunoblots (d) are representative of two independent experiments (d).
Supplementary Material
Acknowledgements
We thank J. Conejo-Garcia for the Lgals1−/− mice and V. Dixit (Genentech) and K. Fitzgerald (University of Massachusetts Medical School) for the Casp11−/− and Gsdmd−/− mice, S. Yeung and K. Khanna for help with the multiplex analysis and G. Fong for the MS1 cell line (University of Connecticut Health). This work was supported by the National Institutes of Health (grant nos. AI119015 and AI135528 to V.A.R.); Agencia Nacional de Promoción Científica y Tecnológica, Fundación Sales and Fundación Bunge y Born to G.A.R.; the Federal Ministry of Education and Research, Germany (grant no. 01EO1502) and Deutsche Forschungsgemeinschaft (German Research Foundation) under Germany’s Excellence Strategy-EXC 2051, project ID 390713860 (to M.B.).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Rathinam VAK & Fitzgerald KA Inflammasome complexes: emerging mechanisms and effector functions. Cell 165, 792–800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Shi J et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Kayagaki N et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Hagar JA, Powell DA, Aachoui Y, Ernst RK & Miao EA Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanaja SK et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell 165, 1106–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rathinam VAK, Zhao Y & Shao F Innate immunity to intracellular LPS. Nat. Immunol.20, 527–533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng KT et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Invest.127, 4124–4135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang R et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe 24, 97–108.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kayagaki N et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Song F et al. Sphingosine-1-phosphate receptor 2 signaling promotes caspase-11–dependent macrophage pyroptosis and worsens Escherichia colisepsis outcome. Anesthesiology 129, 311–320 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Lamkanfi M et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol.185, 4385–4392 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Keller M, Rüegg A, Werner S & Beer H-D Active caspase-1 is a regulator of unconventional protein secretion. Cell 132, 818–831 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Sundblad V, Morosi LG, Geffner JR & Rabinovich GA Galectin-1: a jack-of-all-trades in the resolution of acute and chronic inflammation. J. Immunol.199, 3721–3730 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Camby I, Le Mercier M, Lefranc F & Kiss R Galectin-1: a small protein with major functions. Glycobiology 16, 137R–157R (2006). [DOI] [PubMed] [Google Scholar]
- 18.Lee H-J et al. Application of a peptide-based PF2D platform for quantitative proteomics in disease biomarker discovery. Proteomics 8, 3371–3381 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Banerjee I et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity 49, 413–426.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carta S, Lavieri R & Rubartelli A Different members of the IL-1 family come out in different ways: DAMPs vs. cytokines? Front. Immunol.4, 123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piccioli P & Rubartelli A The secretion of IL-1β and options for release. Semin. Immunol.25, 425–429 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Evavold CL et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48, 35–44.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y et al. Gasdermin D drives the nonexosomal secretion of galectin-3, an insulin signal antagonist. J. Immunol.203, 2712–2723 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Liu X et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frank D & Vince JE Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ.26, 99–114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galluzzi L, Kepp O, Chan FK-M & Kroemer G Necroptosis: mechanisms and relevance to disease. Annu. Rev. Pathol.12, 103–130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delano MJ & Ward PA Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J. Clin. Invest.126, 23–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W & Coopersmith CM Dying as a pathway to death in sepsis. Anesthesiology 129, 238–240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandal P et al. Caspase-8 collaborates with caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity 49, 42–55.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schenck EJ et al. Circulating cell death biomarker TRAIL is associated with increased organ dysfunction in sepsis. JCI Insight 4, e127143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gong T, Liu L, Jiang W & Zhou R DAMP-sensing receptors in sterile inflammation and inflammatory diseases.Nat. Rev. Immunol.20, 95–112 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Rabinovich GA & Toscano MA Turning ‘sweet’ on immunity: galectin–glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol.9, 338–352 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Earl LA, Bi S & Baum LG N- and O-glycans modulate galectin-1 binding, CD45 signaling, and T cell death. J. Biol. Chem.285, 2232–2244 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walzel H, Schulz U, Neels P & Brock J Galectin-1, a natural ligand for the receptor-type protein tyrosine phosphatase CD45. Immunol. Lett. 67, 193–202 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Irie-Sasaki J et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 409, 349–354 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Piercy J, Petrova S, Tchilian EZ & Beverley PCL CD45 negatively regulates tumour necrosis factor and interleukin-6 production in dendritic cells. Immunology 118, 250–256 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanoni I et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf AJ et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166, 624–636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lei T et al. Galectin-1 enhances TNFα-induced inflammatory responses in Sertoli cells through activation of MAPK signalling. Sci. Rep. 8, 3741 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pérez CV et al. Dual roles of endogenous and exogenous galectin-1 in the control of testicular immunopathology. Sci. Rep. 5, 12259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toegel S et al. Galectin-1 couples glycobiology to inflammation in osteoarthritis through the activation of an NF-κB-regulated gene network. J. Immunol. 196, 1910–1921 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Starossom SC et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity 37, 249–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang X-T et al. Galectin-1 ameliorates lipopolysaccharide-induced acute lung injury via AMPK-Nrf2 pathway in mice. Free Radic. Biol. Med. 146, 222–233 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Ge XN et al. Regulation of eosinophilia and allergic airway inflammation by the glycan-binding protein galectin-1. Proc. Natl Acad. Sci. USA 113, E4837–E4846 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodrigues LC et al. Galectin-1 modulation of neutrophil reactive oxygen species production depends on the cell activation state. Mol. Immunol. 116, 80–89 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Croci DO et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156, 744–758 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Di Lella S et al. When galectins recognize glycans: from biochemistry to physiology and back again. Biochemistry 50, 7842–7857 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poirier F & Robertson EJ Normal development of mice carrying a null mutation in the gene encoding the L14 S-type lectin. Development 119, 1229–1236 (1993). [DOI] [PubMed] [Google Scholar]
- 49.Martínez Allo VC et al. Suppression of age-related salivary gland autoimmunity by glycosylation-dependent galectin-1-driven immune inhibitory circuits. Proc. Natl Acad. Sci. USA 117, 6630–6639 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rathinam VAK et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by Gram-negative bacteria. Cell 150, 606–619 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ménoret A et al. The oxazolidinone derivative locostatin induces cytokine appeasement. J. Immunol. 183, 7489–7496 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fettis MM & Hudalla GA Engineering reactive oxygen species-resistant galectin-1 dimers with enhanced lectin activity. Bioconjug. Chem. 29, 2489–2496 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Pérez Sáez JM et al. Characterization of a neutralizing anti-human galectin-1 monoclonal antibody with angioregulatory and immunomodulatory activities. Angiogenesis 10.1007/s10456-020-09749-3 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Bone RC et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 101, 1644–1655 (1992). [DOI] [PubMed] [Google Scholar]
- 55.Singer M et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315, 801–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data have been deposited with the Gene Expression Omnibus (accession number GSE140892). Source data and uncropped immunoblot images are included in the paper as supplementary information. All other data supporting the findings of the paper are available from the corresponding author upon reasonable request.